

Abstract
Rationale.
Calcific Aortic Valve Disease (CAVD) affects more than 5.2 million people in the US. The only effective treatment is surgery and this comes with complications and no guarantee of long-term success.
Objective.
Outcomes from pharmacological initiatives remain unsubstantiated and therefore the aim of this study is to determine if repurposing a selective XPO1 inhibitor drug (KPT-330) is beneficial in the treatment of CAVD.
Methods and Results.
We show that KPT-330 prevents, attenuates and mitigates calcific nodule formation in heart valve interstitial cells (VICs) in vitro, and prevents CAVD in Klotho−/− mice. Using RNA-sequencing and Mass Spectrometry we show that KPT-330’s beneficial effect is mediated by inhibiting nuclear export of the transcription factor C/EBPβ in VICs, leading to repression of canonical Wnt signaling, in part through activation of the Wnt antagonist Axin1, and a subsequent decrease in pro-osteogenic markers and cell viability.
Conclusions.
Our findings have met a critical need to discover alterative, pharmacological-based therapies in the treatment of CAVD.
Keywords: Heart valve, calcification, interstitial cells, signaling pathways, signal transduction, cell signaling, cardiovascular research
Subject Terms: Translational Studies
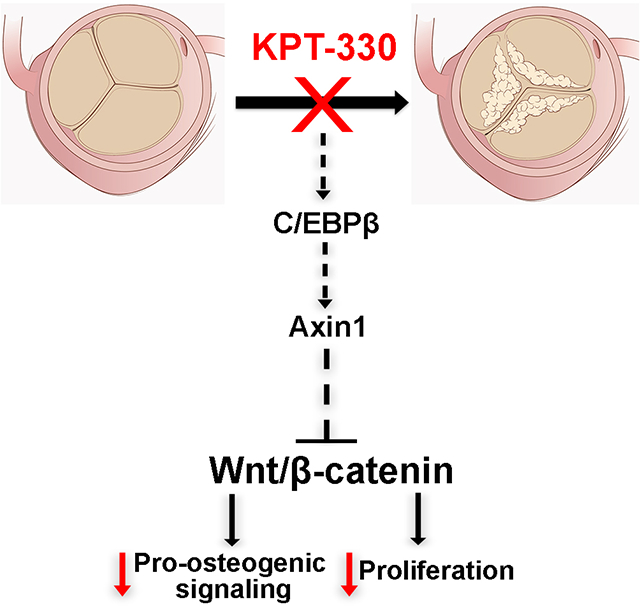
Graphical Abstract
INTRODUCTION
Calcific aortic valve disease (CAVD) is a growing public health burden affecting up to 27% of the aging population, and pediatrics with congenital valve malformations.1 CAVD is characterized by formation of calcific nodules on the surface of the aortic valve leading to stenosis, secondary myocardial dysfunction and eventual heart failure. Currently, there are no pharmacological treatments for CAVD, and despite initiatives to target known risk factors, outcomes of statin therapy remain unsubstantiated.2–15 As a result, transcatheter aortic valve replacement (TAVR) therapy and surgical intervention remain the most effective options which comes with complications and no guarantee of long-term success, and therefore the field continues to explore better alternatives.
Calcification of the aortic valve is characterized by overall thickening of the valve cusp and the presence of fibrotic and calcium-rich nodules on the aortic valve surface and/or within the annulus region, leading to functional stiffening and stenosis.16 The onset of CAVD is slow and progressive, and in human pathology, early stages are associated with endothelial dysfunction and infiltration of immune cells.17–20 In contrast, studies of calcified aortic valves at end-stage disease have shown histological and molecular similarities with endochondral and heterotopic ossification.21 This includes expression of transcription factors important for bone development (Runx2), and detection of molecular markers of mineralized tissue (Matrix Gla Protein, Osteopontin, Bone Sialoprotein).22 As a result of these observations, recent efforts to develop pharmacotherapies to treat CAVD have taken the approach of targeting these osteogenic signaling pathways to halt disease progression. However, to date no suitable therapeutic targets have been identified with clinical potential.
In our previous work, we showed that treatment of porcine aortic VICs (pAVICs) with the nuclear export inhibitor compound, Leptomycin B prevents calcification under osteogenic stimulus in vitro.23 As a result of these studies, we have drawn our attention to the potential of targeting protein shuttling for CAVD treatment. However, due to the cytotoxicity of Leptomycin B, the therapeutic application in vivo is limited. Therefore, we sought to explore alternative nuclear export inhibitor drugs that are currently FDA approved or in clinical trials, and determine their potential in attenuating CAVD phenotypes. Exportin-1 (XPO1), also known as Chromosome region maintenance 1 (CRM1) is the protein transporter responsible for the nucleo-cytoplasmic shuttling of ~200 proteins including tumor suppressors and regulatory growth factors.24 Its inhibition has been a target for therapy, and as a result, selective inhibitors of nuclear transport (SINE) compounds have been developed as a novel class of anti-cancer agents. The most well-known SINE agent is KPT-330 which has been widely tested in phase I and II clinical trials for the treatment of solid tumors and hematologic malignancies, and is thought to inhibit the nuclear export of XPO1-dependent tumor suppressor proteins including p53 and p27 in cancerous cells (reviewed 24). Based on the efficacy of KPT-330 in humans and mode of action in targeting nuclear export inhibition, the goal of this current study is to determine the potential of re-purposing KPT-330 (Selinexor) to treat CAVD phenotypes in mice and in vitro.
METHODS
Data Availability.
The authors declare that all supporting data are available within the article (and its online supplementary files), or can be made available from the corresponding author upon reasonable request. Materials included in this study will be made available upon request to the corresponding author.
RESULTS
In vitro model of VIC-mediated calcific nodule formation.
As a means of stimulating calcification of pAVICs in culture, cells were plated on a glass (stiff) substrate in complete media (CM) for up to 14 days, until the appearance of calcific nodules were observed.25 During the progression of this process, three notable stages were observed based on distinct cell morphologies. At Stage I (~day 5), pAVICs show their characteristic fibroblast appearance (elongated with cytoplasmic extensions) (arrows, Figure 1Ai) with undetectable calcium deposition as noted by an absence of Alizarin Red staining (Figures 1Aiv, B), and minimal Alkaline Phosphatase (ALP) activity (Figure 1C). Based on these data, Stage I pAVICs are referred to as quiescent and non-calcified. By ~day 8 of culture, pAVIC confluency increases and ‘swirling’ patterns are first noted to form foci (arrows, Figure 1Aii) with low levels of Alizarin Red reactivity (Figures 1Av, B) and ALP activity (Figure 1C). Cells exhibiting this morphology are categorized as being at Stage II and termed pre-calcific. By Stage III (days 12–14), mineralized lesions referred to as calcific nodules are present and positive for Alizarin Red and ALP (arrows, Figures 1Aiii, Avi, B, C). Immunohistochemistry detected expression of known pro-osteogenic proteins including Runx2 (Figures 1Di, Div), Osteopontin (Opn) (Figures 1Dii, Dv) and Cadherin-11 (Figures 1Diii, Dvi)26, 27 in calcific nodules at Stage III (calcified), but not Stage I (non-calcified). Simultaneously, cell viability as an indirect measure of proliferation was determined using the MTT assay, and shown to increase in pAVICs cultured on glass and harvested at Stage III, compared to Stage I (Figure 1E).
Figure 1. In vitro model of VIC calcific nodule formation.

(A) pAVICs were plated on glass to stimulate the formation of calcific nodules. Brightfield (i-iii) and Alizarin Red stained images (iv-vi) of pAVICs at Stage I (~5 days), Stage II (~8–9 days), Stage III (~12–14 days). Arrows depict quiescent (Stage I), pre-calcific (Stage II), and calcific nodules (Stage III) (n=3). (B) Graph showing quantification of the area positive for Alizarin Red staining/microscopic field. The Y axis indicates calibrated arbitrary units of area based on selected montages in Image J. Quantification was performed using 3 biological replicates, with 3 different field of views from each replicate. P-value by one-way ANOVA = 6.3E-08. Adjusted p-values using post-hoc pairwise analysis: *Stage I vs Stage II 7.7E-06, *Stage I vs Stage III 1.8E-06, #Stage II vs Stage III 0.0004. (C) Graph showing area of positive Alkaline Phosphatase (ALP) reactivity/field at Stages I-III (n=3). P-value by one-way ANOVA = 0.0003. Adjusted p-values using post-hoc pairwise analysis: *Stage I vs Stage II 0.004, *Stage I vs Stage III 0.0003, #Stage II vs Stage III 0.02. (D) Immunofluorescence images to detect Runx2 (green), Osteopontin (red), and Cadherin-11 (red) protein expression at Stages I and III. DAPI highlight nuclei in blue (n=3). (E) Percent change in MTT assay (absorbance) of pAVICs harvested at Stages I and III (n=3). *P-value using corrected Mann-Whitney: 0.03. (F) Heat map and (G) Venn diagram of RNA-Seq analysis (n=3). (H) Immunofluorescence images to detect Osteomodulin (red) and Sclerostin (green) protein expression in pAVICs fixed at Stages I, II and III (n=3). (I) qPCR analysis of osteogenic markers: Sclerostin (Sost), Osteomodulin, Osteoglycin, and Osteoclast Stimulating Factor 1 (Ostf1) (n=3). P-values for datasets by one-way ANOVA: Sclerostin n/s, Osteomodulin 0.008, Osteoglycin 0.004, Ostf1 0.027. Adjusted p-values using post-hoc pairwise analysis: Sclerostin: Stage I vs Stage II 0.6, Stage I vs Stage III 0.2, #Stage II vs Stage III 0.03. Osteomodulin: *Stage I vs Stage II 0.0006, *Stage I vs Stage III 0.01, Stage II vs Stage III 0.09. Osteoglycin : *Stage I vs Stage II 9.4E-05, *Stage I vs Stage III 0.001, Stage II vs Stage III 0.2,. Ostf1: *Stage I vs Stage II 0.004, Stage I vs Stage III 0.5, Stage II vs Stage III 0.09.
To examine differential transcriptomes at Stages I, II and III, RNA-sequencing was performed. Heat map hierarchical clustering (Figure 1F) and principal component analyses (Supplementary Figures IA–C) analyses molecularly distinguishes the three stages, and the Venn diagram (Figure 1G) indicates common and unique mRNAs at each Stage (see Supplementary Table I for unique gene lists). Pathway analysis between Stages I and II reveals enrichment of biological processes including ‘osteogenic differentiation’, ‘cell adhesion’, and ‘cell proliferation’ (Supplementary Figures IB–D). Similar analysis comparing Stages II and III also identifies ‘osteogenic differentiation’-related processes (Supplementary Figures IIB–D). This includes increased expression of the CAVD biomarker Sclerostin (Sost),28 and Osteoglycin (Ogn) present in stenotic aortic valves.29 The analysis also identified other osteogenic genes including Osteomodulin (Omd) and Osteoclast Stimulating Factor 1 (Ostf1), that have yet to be extensively explored in human aortic valve disease. Additional validation shows that Sost is most detectable at Stage III (Figures 1Hiv–Hvi, 1I), Omd and Ogn peak at Stage II and remain high (Figures 1Hi–Hiii, 1I), while Ostf1 (Figure 1I) is most enriched at Stage II. These studies highlight the molecular and cellular profiles of calcific nodule formation in pAVICs in vitro and show strong enrichment with osteogenic processes at each stage.
The pharmacological XPO1 inhibitor KPT-330 prevents, attenuates and mitigates calcific nodule formation in vitro.
As discussed above, our previous work has shown that treatment of pAVICs with Leptomycin B, a known nuclear export inhibitor compound, prevents calcific nodule formation when cultured on a stiff (glass) substrate.23 However the known cytotoxicity of this compound hinders its pharmacological potential in humans. Therefore, we re-purposed the selective XPO1 inhibitor drug, KPT-330 and determined its ability to have the same beneficial effects as Leptomycin B in the absence of overt toxicity. To do this, we first performed a dose-response curve (10nM, 50nM, 100nM) to examine pAVIC survivability in the presence of increasing KPT-330 doses (10nM, 50nM, 100nM), and determined 100mM as the most effective concentration (data not shown). Therefore, as in Figure 1, pAVICs were plated on glass to stimulate calcification and treated with 100nM KPT-330 (or DMSO as a vehicle control) at Stage I, and harvested when control-treated cells reached Stage III and calcific nodules were observed (Figure 2A). Compared to DMSO controls that exhibited calcification, Alizarin Red-positive calcific nodules (Figures 2B, C), ALP activity (Figure 2D), and Sclerostin and Omd expression (Figure 2E) were all reduced following KPT-330 treatment at Stage I.
Figure 2. KPT-330 prevents calcific nodule formation and reduces proliferation.

(A) Experimental timeline. (B) Brightfield (i,ii) and Alizarin Red (iii,iv) images from pAVICs cultured on glass and treated with DMSO or 100nM KPT-330 at Stage I. Arrows, Alizarin Red positive nodules (n=3). (C) Quantification of positive Alizarin Red stained area/field in pAVICs treated with 100nM KPT-330 at Stage I (n=3). *P-value using corrected Mann-Whitney: 0.03. The Y axis indicates calibrated arbitrary units of area based on selected montages in Image J. (D) Area of positive ALP reactivity in pAVICs cultured in complete media (CM), osteogenic media (OM), or treated with KPT-330 in the presence of OM (n=3). The Y axis indicates calibrated arbitrary units of area based on selected montages in Image J. P-values using corrected Mann-Whitney: *CM vs OM 0.03, #OM vs OM+KPT 0.03, CM vs OM+KPT 0.3. (E) Immunofluorescence of pAVICs to detect Sclerostin (green) or Osteomodulin (red) protein expression (arrows showing calcified nodules) after DMSO treatment (n=3). (F) Western Blot analysis and densitometric quantitation (G) to confirm Xpo1 knockdown by Xsi compared to Csi (n=3). P-values using corrected Mann-Whitney: *CM+Csi vs OM+Csi 0.03, *CM+Csi vs CM+Xsi 0.03, #OM+Csi vs OM+Xsi 0.03. (H) Alizarin Red staining of pAVICs cultured in CM or OM, and transfected with control (Csi) or siRNA against Xpo1 (Xsi) (n=3). (I) Quantification of Alizarin Red stained area/field from H (n=3). The Y axis indicates calibrated arbitrary units of area based on selected montages in Image J. P-values using corrected Mann-Whitney: *CM+Csi vs OM+Csi 0.03, *CM+Xsi vs OM+Xsi 0.03, *CM+Csi vs CM+Xsi 0.03, #OM+Csi vs OM+Xsi 0.03. (J) Percent change in Caspase-glo 3/7 luminosity (cell death) in pAVICs treated with DMSO or 100nM KPT-330 plated on glass (n=3). P-value using corrected Mann-Whitney 0.1. (K) Percent change in MTT assay performed in pAVICs treated with DMSO or 100nM KPT-330 plated on glass (n=3). *P-value using corrected Mann-Whitney 0.03. (L) Immunofluorescence of pHH3 expression in pAVICs cultured in OM and treated with DMSO or 100nM KPT-330 (n=3). (M) Quantification of data presented in U, V. *P-value using corrected Mann-Whitney: 0.03.
To examine the potential of KPT-330 in preventing calcification of human aVICs, cells were cultured in osteogenic media (OM)30 as an alternative to glass-induced calcific nodule formation, and treated with KPT-330 at the time of plating. Worthy of mention, the onset of calcification using OM is more rapid, and by ~day 5, positive Alizarin Red staining is observed, however the developmental Stages of I-III, and the formation of defined calcific nodules are not distinguishable using this method. For this study, human VICs were obtained from two healthy donors (Supplementary Figures IIIA, B) and two aortic stenosis patients (Supplementary Figures IIIC, D) and cultured in OM for ~5 days and treated with KPT-330 from day 1. Interestingly, the dose-response curve for human aVIC viability was lower than that of porcine cells (data not shown), and therefore these cells were treated with 20nM KPT-330. In parallel, pAVICs were also cultured in OM and treated with 100nM KPT-330 (Supplementary Figures IIIE, F). Similar to glass-induced calcification, KPT-330 prevented Alizarin Red reactivity in both human and porcine cells cultured under OM conditions. Furthermore, KPT-330 treatment had similar beneficial effects on pAVICs treated with 6mM inorganic phosphate to create hyperphosphatemic conditions as an alternative stimulus of calcification (Supplementary Figures IIIG, H).31
To demonstrate that the beneficial effects of KPT-330 are mediated through XPO1 inhibition, experiments were performed in pAVICs using siRNA against Xpo1 (Xsi) in the presence of OM. As shown in Figures 2F and 2G, Xsi treatment significantly reduced Xpo1 expression both in the presence of CM and OM. In association with Xpo1 knockdown, like KPT-330, Xsi significantly attenuated Alizarin Red staining compared to controls (Csi) (Figures 2H, I). Therefore supporting the hypothesis that the beneficial effects of KPT-330 in preventing calcification are mediated by Xpo1.
Important to note, 100nM KPT-330 treatment does not significantly affect apoptosis of treated pAVICs at Stage III (Figure 2J), but significantly reduces cell viability suggestive of proliferation as indicated by the MTT assay at Stage III (Figures 2K–M).
Figure 2 suggests that KPT-330 treatment at Stage I prevents calcific nodule formation. To further determine if KPT-330 treatment attenuates progression of calcific nodule formation, pAVICs were cultured on glass and treated with KPT-330 at Stage II (Figure 3A). When DMSO-controls reached Stage III as indicated by the presence of calcific nodules, all cells were harvested and stained for Alizarin Red. As indicated in Figure 3Biv, KPT-330 treatment at Stage II reduced Alizarin Red staining compared to DMSO controls, suggesting attenuated calcific nodule formation. In addition, pAVICs were also treated with KPT-330 at Stage III when calcific nodules were observed (Figure 3C). This approach not only mitigated existing calcific nodules after 72 hours (Figures 3D, E) but also decreased expression of pro-osteogenic markers Sost, Omd, Ogn and Ostf1 (Figure 3F) previously identified as being increased through Stages I-III by RNA-seq analysis (Figure 1). These data suggest that KPT-330 has beneficial effects in preventing, attenuating and mitigating calcific nodule formation, associated with reduced cell viability and expression of pro-osteogenic markers.
Figure 3. KPT-330 attenuates and mitigates nodule formation in vitro.

(A) Schematic of the experimental timeline to show pAVICs plated on glass and treated with KPT-330 at Stage II. (B) Brightfield (i,ii) and Alizarin Red (iii,iv) images from DMSO or 100nM KPT-330 treated cells at Stage II, plated on glass. Arrows indicate calcific nodules (n=3). (C) Schematic of the experimental timeline utilized in D to show pAVICs plated on glass and treated with KPT-330 at Stage III. (D) Brightfield (i,ii) and Alizarin Red (iii,iv) images from DMSO or KPT-330 treated cells at Stage III, plated on glass (n=6). (E) Quantification of Alizarin red positive area/field in pAVICs before and after KPT-330 treatment (post 72 hours) (n=6). *P-value using corrected Mann-Whitney: 0.0004. (F) qPCR data to show changes in expression of osteogenic markers (Sost, Osteomodulin, Osteoglycin, and Ostf1) between Stage III and Stage III+KPT-330 (n=3). *P-value using corrected Mann-Whitney: Sost 0.03, Osteomodulin 0.03, Osteoglycin 0.03, Osteoclast Stimulating Factor 1 0.03. (G) Schematic of the experimental timeline and accompanying Alizarin Red staining H (n=1). (H) Alizarin Red images of pAVICs from KPT-330 added at Stage III (i) and replated after 72 hours with DMSO treatment (ii) or KPT-330 treatment (iii).
To further examine the molecular mechanisms underlying mitigation, or treatment of calcific nodules following KPT-330 treatment at Stage III, RNA-seq analysis was performed and compared to DMSO treated controls. Heat map analysis clustered biological replicates and showed further variance in the transcriptome of KPT-330 treated cells (Supplementary Figure 4A). GO pathway analysis of differentially expressed mRNAs associated with KPT-330 treatment included ‘cell adhesion’, ‘regulation of cell cycle’ and ‘cell division’ (Supplementary Figures IVC, D). Furthermore, pathways related to ‘in utero embryonic development’ ‘heart development’ and ‘outflow tract morphogenesis’ were apparent (Supplementary Figures IVC, D, Table I). To determine the potency of KPT-330 in preventing the return of calcific nodules, pAVICs were treated with KPT-330 at Stage III as above, but following the mitigation of calcific nodules (following ~72 hours of treatment), cells were re-plated and further treated with DMSO or KPT-330 for an additional 14 days (Figure 3G). Interestingly, calcific nodules did not return in cells that were initially treated with KPT-330 and subsequently DMSO (Figure 3Hii). However, pAVICs that received two rounds of KPT-330 treatment did not survive this long-term treatment strategy (Figure 3Hiii). These data suggest that short term exposure to KPT-330 prevents the recurrence of calcific nodule formation.
KPT-330 treatment prevents aortic valve calcification in Klotho−/− mice.
Hyperphosphatemia, as in the setting of chronic kidney disease, is a known risk factor of cardiovascular calcification affecting both the vasculature and aortic valves.32,33 To explore the therapeutic potential of KPT-330 in hyperphosphatemic-induced calcification, pAVICs were cultured in 1mM (Figure 4Aii, vi), 2.5mM (Figures 4iii, 4vii) and 6mM (Figures 4iv, viii) concentrations of inorganic phosphate (Pi) (Sodium phosphate dibasic salt) for 4–5 days, with (Figures 4Av–viii) and without (Figures 4Ai–iv) 100nM KPT-330. As shown, Alizarin Red, indicating calcification is detected under 2.5mM conditions, and increases are further enhanced at 6mM (Figure 4B). Worthy of mention, Pi treatment alone did not affect cell viability (Figure 4C), however KPT-330 treatment in the presence of high phosphate, reduced levels.
Figure 4. KPT-330 prevents aortic valve calcification in Klotho−/− mice.

(A) Alizarin Red staining of pAVICs treated with no phosphate (Pi) (i,v), 1mM Pi (ii,vi) 2.5mM Pi (iii,vii) and 6mM Pi (iv,viii) in the absence (i-iv) and presence of 100nM KPT-330 (v-viii) for 4–5 days. (B) Quantification of positive Alizarin Red stained area/field in untreated and treated pAVICs, *indicates comparison to indicated Pi concentration, # indicates comparison to DMSO-treated at the same Pi concentration (n=3). The Y axis indicates calibrated arbitrary units of area based on selected montages in Image J. P-values using corrected Mann-Whitney: *No Pi vs 2.5 mM Pi 0.03, *No Pi vs 6 mM Pi 0.03, *1 mM Pi vs 2.5 mM Pi 0.03, *2.5 mM Pi vs 6 mM Pi 0.03, #2.5 mM Pi vs 2.5 mM Pi+KPT 0.03, #6 mM Pi vs 6 mM Pi+KPT 0.03. (C) Percent change in MTT assay of untreated and treated pAVICs, *indicates comparison to indicated Pi concentration, #indicates comparison to DMSO-treated at the same Pi concentration (n=3). *P-values using corrected Mann-Whitney: No Pi vs No Pi+KPT 0.03, 1 mM Pi vs 1 mM Pi+KPT 0.03, 2.5 mM Pi vs 2.5 mM Pi+KPT 0.03, 6 mM Pi vs 6 mM Pi+KPT 0.03, No Pi+KPT vs 2.5 mM Pi+KPT 0.03, No Pi+KPT vs 6 mM Pi+KPT 0.03. (D) Schematic showing experimental treatment design in Klotho−/− mice. (E) Low and high magnification of Alizarin Red stained tissue sections from Klotho−/− mice treated with vehicle. Arrows show presence of annular calcification and arrow heads show vascular calcification. (F) Low and high magnification of Alizarin Red tissue sections from Klotho−/− mice treated with 30 mg/kg KPT-330. Arrows show absence of annular calcification and arrow heads show vascular calcification (n=6). (G) Quantification of positive Alizarin Red stained area/field within the aortic valve annular region of vehicle and KPT-330 treated Klotho−/− mice (n=5). *P-value using corrected Mann-Whitney: 0.002. The Y axis indicates calibrated arbitrary units of area based on selected montages in Image J.
To further explore the therapeutic potential of KPT-330 treatment in vivo, we utilized the Klotho−/− mouse model of aging34 and hyperphosphatemia,35 that have previously been shown to develop calcification of the vessels and aortic valve annulus similar to humans36 by 6 weeks,34 before dying prematurely at 8 weeks.37 While these mice do not exhibit significant valve dysfunction prior to premature death limiting recapitulation of human disease,34 they consistently develop calcific nodules at the base on the aortic valve annulus making them suitable for prevention studies. Here, Klotho−/− and Klotho+/+ mice were treated with vehicle, or 30mg/kg KPT-330 at 4 weeks of age, and then again at 5, and 7 weeks, and tissue was harvested prior to their expected premature lethality by 8 weeks (Figure 4D). Alizarin Red staining of cardiac tissue at 7 weeks of age did not detect calcification in DMSO or KPT-330-treated Klotho+/+ littermates (data not shown). As expected, the aortic valve annulus (arrows, Figures 4Ei, Eii) and aorta (arrowhead, Figures 4Ei, Eii) were calcified in Klotho−/− mice treated with DMSO. However, annular calcification was not detected in any Klotho−/− mice administered with KPT-330 (Figures 4Fi, Fii, 4G), while vascular calcification persisted. In our hands, KPT-330 treated Klotho−/− mice did not exhibit any overt phenotypes beyond those expected by the underlying genotype. This includes no change in body weight with KPT-330 treatment, and cardiac function was not affected by the Klotho−/− genotype,34 or drug treatment. These observations suggest that KPT-330 can prevent phosphate-mediated valve calcification in vitro and in Klotho−/− mice.
KPT-330 prevents the nuclear export of C/EBPβ in valve interstitial cells under calcific stimulus.
KPT-330 is a selective XPO1 inhibitor drug, however XPO1-dependent target proteins in the setting of preventing calcification in VICs are not known. To identify proteins affected by KPT-330 treatment, Mass Spectrometry was performed on nuclear and whole cell lysates isolated from pAVICs plated on glass at Stages I, II and III, and pAVICs treated with KPT-330 at Stage I (prevention) and Stage III (treatment). Principal component (Supplementary Figure IA) and clustering (Supplementary Figure IB) analyses highlight homogeneity of biological replicates within experimental groups, and heterogeneity between these groups. Potential target proteins of KPT-330 in nuclear lysates were considered if they were shown to 1) leave the nuclei of pAVICs during calcific nodule formation (Stages I vs. III), and 2) be retained in the nuclei of pAVICs by KPT-330, and 3) harbor an XPO1-dependent nuclear export signal recognition site. The transcription factor C/EBPβ fit these criteria (Figures 5A–C).
Figure 5. KPT-330 prevents the nuclear export of C/EBPβ in VICs.

C/EBPβ protein intensity from Mass Spectrometry at Stages I vs III (A), Stages III vs Stage I+KPT-330 (prevention) (B) and Stages III vs Stage III+KPT-330 (treatment) (C) n=3. *P-value using corrected Mann-Whitney: Stage I vs Stage III 0.03, Stage III vs Stage I 0.03, Stage III vs Stage III+ KPT 0.03. (D) Western Blot analysis and quantitation (E) of C/EBPβ in cytoplasmic and nuclear lysates from pAVICs cultured on glass and harvested at Stages I (n=3), III (n=5), and III+KPT (n=5). *P-value using corrected Mann-Whitney: 0.08. (F) Western Blot analysis and quantitation (G) of C/EBPβ in cytoplasmic and nuclear lysates from pAVICs cultured in complete media (CM), osteogenic media (OM) or OM + 100nM KPT-330 (n=5). *P-value using corrected Mann-Whitney: 0.006. (H) Western Blot analysis and quantitation (I) of C/EBPβ in nuclear lysates from pAVICs cultured in 2.5mM Phosphate (Pi) in the absence and presence of 100nM KPT-330 (n=3). *P-value using corrected Mann-Whitney: 0.03. (J) Western Blot of nuclear/cytoplasmic fractions of pAVICs in OM, or OM + 100nM KPT-330 with DMSO or MG132 (n=3). (K) Quantification of data presented in (J). P-value using corrected Mann-Whitney: DMSO vs KPT 0.13, DMSO+MG132 vs KPT+MG132 0.2, DMSO vs DMSO+MG132 0.03, KPT vs KPT+MG132 0.3. Molecular weights (MW) are indicated in kDa.
Independent studies validating Mass Spectrometry findings show decreased C/EBPβ in the nuclei of pAVICs cultured on glass (Figures 5D, E), and in OM (with no glass) (Figures 5F, G). In addition, KPT-330 treatment at Stage I prevented nuclear export of C/EBPβ (retained higher levels in the nucleus) under these conditions, in addition to high phosphate treatments (Figures 5H–I). The addition of the proteasome inhibitor, MG132 under OM culture conditions supports the notion that reductions in nuclear C/EBPβ during later stages of calcific nodule formation is not due to proteosomal degradation in the nucleus or cytoplasm, as MG132 did not lead to a further increase in protein levels (Figures 5J, K).
C/EBPβ overexpression in valve interstitial cells is sufficient to prevent calcification.
To determine if C/EBPβ is sufficient to prevent calcification similar to KPT-330 treatment, pAVICs were cultured in OM and transfected with plasmid DNA to overexpress full-length C/EBPβ (pC/EBPβ). As expected, pC/EBPβ led to increased C/EBPβ (Figures 6A, B) predominantly in the nuclear compartment (Figures 6C, D). Similar to KPT-330, pC/EBPβ-transfection led to significantly less Alizarin Red staining after 5 days (Figures 6E, F). Alternative to pC/EBPβ, C/EBPβ was also overexpressed using recombinant protein (rC/EBPβ) in the presence of OM (Figures 6G, H), which had similar beneficial effects on calcification (Figures 6I, J). Together, these data suggest that KPT-330’s beneficial mechanism of action is in part via preventing nuclear export of C/EBPβ in VICs.
Figure 6. C/EBPβ overexpression prevents calcification of VICs.

(A) Western Blot analysis and densitometric analysis (B) of Western Blot from pAVICs transfected with pcDNA3-C/EBPβ (pC/EBPβ) or empty pcDNA3 (pEmpty) (n=3). *P-value using corrected Mann-Whitney: 0.03. (C) Immunofluorescence and quantitation (D) of nuclear/cytoplasmic localization of C/EBPβ in pAVICs following transfection with pC/EBPβ or empty pcDNA3 in complete media (CM), (n=3). *P-value using corrected Mann-Whitney: empty pcDNA3 vs pC/EBPβ cytoplasmic 0.03, empty pcDNA3 vs pC/EBPβ nuclear 0.03, empty pcDNA3 vs pC/EBPβ cytoplasmic+nuclear 0.03. (E) Alizarin Red stained images and quantification (F) from pAVICs cultured in OM and transfected with pEmpty or pC/EBPβ (n=3). *P-value using corrected Mann-Whitney: 0.03. The Y axis indicates calibrated arbitrary units of area based on selected montages in Image J. (G) Western Blot and densitometric analysis (H) of C/EBPβ levels from pAVICs treated with recombinant C/EBPβ protein (rC/EBPβ) (n=3). *P-value using corrected Mann-Whitney: CM vs 50 0.03, CM vs 100 0.03, CM vs 200 0.03. (I) Alizarin Red images and quantification (J) of positive Alizarin red stained area/field of pAVICs cultured in OM alone, or OM + rC/EBPβ (n=3). *P-value using corrected Mann-Whitney: 0.03. The Y axis indicates calibrated arbitrary units of area based on selected montages in Image J. Molecular weights (MW) are indicated in kDa.
KPT-330 and C/EBPβ repress activated canonical Wnt signaling in pAVICs and Klotho−/− mice.
Previous studies have shown that C/EBPβ is a negative regulator of Wnt signaling,38–40 and activated Wnt signaling has previously been shown to be present in excised calcified aortic valves.41–46 Here, RNA-seq analysis identified a significant increase in mRNAs associated with positively regulating Wnt signaling between Stages I-III, while treatment with KPT-330 decreased their expression (Lrp5, Wnt10B, Lef1, Wisp1, Wisp2). Conversely KPT-330 increased levels of Wnt inhibitors (Wif1, Axin1) (Figure 7A). In support of KPT-330 inhibiting Wnt signaling in VICs, nuclear β-catenin expression is reduced in pAVICs plated on glass and treated with KPT-330 at Stage I (Figures 7B–C). Similar observations are also made using an antibody to detect the active form (dephosphorylated at Ser37 or Thr41) of β-catenin in pAVICs cultured in OM (Figures 7D, E). LiCl is a Wnt agonist, and activity of the signaling pathway can be measured in pAVICs treated with 10mM LiCl using the TOP-FLASH plasmid containing three copies of TCF binding sites. Here we show that KPT-330 treatment reduces LiCl-induced TOP-Flash activity in the presence of OM (Figure 7F), and this is abolished when TCF sites are mutated (FOP-FLASH). Wnt activity, as indicated by β-catenin expression is also reduced in the annular valve region of Klotho−/− mice treated with KPT-330 compared to vehicle-treated controls (Figures 7G, H), and 2.5mM Pi-treated pAVICs (Figures 7I, J). These data strongly suggest that KPT-330 treatment suppresses canonical Wnt signaling in VICs, potentially by inhibiting the nuclear export of C/EBPβ.
Figure 7. KPT-330 represses canonical Wnt signaling in pAVICs.

(A) RPKM values obtained from RNA-Seq data for Wnt signaling mediator and regulatory genes, *indicates comparison to previous time point, #indicates comparison to Stage III (Wnt activators or downstream effectors – Lrp5, Wnt10b, Lef1, Wisp1, Wisp2; Wnt inhibitors- Wif1, Axin1) (n=3). P-values: ≤ 0.01–0.05. (B) Western Blot analysis and quantitation (C) of β-catenin expression in pAVICs harvested at Stage I, or Stage III in the presence and absence of 100nM KPT-330 (n=3). P-value using corrected Mann-Whitney: *Stage I vs Stage III 0.03, #Stage III vs Stage III+KPT 0.03, *Stage I vs Stage III + KPT 0.03. (D) Western Blot to show active β-catenin expression in whole pAVIC lysates cultured in complete media (CM), osteogenic media (OM), or OM in the presence of KPT-330. (E) Quantitation of Western Blot shown in (D) (n=3). *P-value using corrected Mann-Whitney: 0.07. (F) Luminescence as %RLU/mg in pAVICs transfected with TOP/FOP Flash plasmids and Renilla plasmid treated with DMSO or 100nM KPT-330 in OM. LiCl treatment was performed for 24 hours to activate Wnt signaling, *indicates comparison to TOP-Flash, #indicates comparison to DMSO (n=3). *P-value using corrected Mann-Whitney: OM vs OM+KPT (TOP-Flash) 0.03, OM (FOP-Flash) vs OM (TOP-Flash) 0.03, OM+KPT (FOP-Flash) vs OM+KPT (TOP-Flash) 0.03. (G) Immunofluorescent images of aortic valve annulus (highlighted) stained with β-catenin (red) and DAPI (blue) from Klotho−/− mice treated with vehicle or KPT-330. (H) Quantitation of β-catenin immunoreactivity (CTCF unit) in the annulus region of Klotho−/− mice treated with vehicle or KPT-330 (n=3). *P-value using corrected Mann-Whitney: 0.03. The Y axis indicates calibrated arbitrary units of area based on selected montages in Image J. (I) Western blot and densitometric analysis (J) of nuclear β-catenin expression in pAVICS treated with 2.5mM Pi in the absence and presence of 100nM KPT-330 (n=3). *P-value using corrected Mann-Whitney: 0.03. Molecular weights (MW) are indicated in kDa.
To further explore the molecular relationship between C/EBPβ and Wnt suppression, pAVICs were cultured in OM and treated with rC/EBPβ. As indicated, this approach also reduced active β-catenin expression (Figures 8A, B). Axin1 and Wif1 are Wnt antagonists, and by RNA-seq analysis were shown to increase in response to KPT-330 treatment (Figure 7A). Axin1 harbors C/EBPβ binding sites (CCAAT), and has been shown to be transcriptionally regulated by C/EBPβ in 3T3 cells.38 To examine if C/EBPβ regulates Axin1 in pAVICs, cells were cultured in OM and treated with pC/EBPβ. Similar to other cell types, C/EBPβ was sufficient to increase Axin1 expression in pAVICs under osteogenic stimulus (Figures 8C, D). However, using this approach, consistent increases in Wif1 were not observed (data not shown) and therefore this candidate was eliminated from our studies. Next, we sought to explore if Axin1 transcription is regulated by C/EBPβ in pAVICs. For this, luciferase assay was performed using firefly luciferase-tagged Axin1 containing a known C/EBPβ binding site (CCAAT), in the presence of pC/EBPβ or empty vector, in addition to either DMSO, or KPT-330. As shown in Figure 8E, pC/EBPβ is sufficient to increase the transcriptional activity of Axin1. Furthermore, KPT-330 treatment alone (without pC/EBPβ) significantly increases Axin1 over DMSO controls, suggesting that KPT-330 promotes endogenous C/EBPβ-mediated Axin1 activation. While KPT-330 in the presence of pC/EBPβ was not able to further increase Axin1, increasing trends were observed, however saturation of Axin1 activity by pC/EBPβ may be a limiting factor. To further examine the beneficial effects of inhibiting Wnt signaling in preventing calcification, canonical signaling was directly targeted by treating pAVICs in OM with the antagonist XAV939 (XAV, 10μM). As shown, this approach prevented calcific nodule formation (Figures 8F, G) and reduced cell proliferation (Figure 8H, I), as indicated by phospho-histone H3 expression. Interestingly, direct activation of Wnt signaling using 10mM LiCl or 50ng/mL Wnt3a for up to 12 days (without additional osteogenic stimuli) was not sufficient to cause calcific nodule formation (data not shown), suggesting that inhibiting Wnt signaling prevents calcification, but activating the pathway is not sufficient to cause the process. Overall, the above data shows that KPT-330-mediated retention of C/EBPβ in the nuclei of VICs prevents calcification in part, by repressing Wnt signaling (via Axin1 activation) to reduce pro-osteogenic signaling and cell proliferation.
Figure 8. C/EBPβ overexpression represses canonical Wnt signaling in pAVICs.

(A) Western Blot to show active β-catenin expression in whole pAVIC lysates cultured in OM, and in the presence of 400ng/μl rC/EBPβ (n=2). (B) Quantitation of Western Blot shown in (A). (C) Western blot and densitometric analysis (D) of Axin1 from pAVICs transfected with pcDNA3-C/EBPβ (pC/EBPβ) or empty pcDNA3 (n=3). *P-value using corrected Mann-Whitney: 0.03. (E) Luminescence as %RLU/mg in pAVICs transfected with pC/EBPβ or empty pcDNA controls, along with Axin1-Luciferase plasmid, in presence of DMSO or 100nM KPT-330 (n=3). *P-value using corrected Mann-Whitney: empty pcDNA3+DMSO vs empty pcDNA3+KPT 0.03, empty pcDNA3+DMSO vs pC/EBPβ+DMSO 0.03, empty pcDNA3+KPT vs pC/EBPβ+KPT 0.03, pcDNA3+DMSO vs pC/EBPβ+KPT 0.2. (F) Alizarin Red images and quantification (G) of positive Alizarin Red stained area/field in pAVICs cultured in OM or OM with XAV-939 (XAV) (n=3). *P-value using corrected Mann-Whitney: 0.03. (H) Immunofluorescence staining images and quantitation (I) of pHH3 positive pAVICs cultured in OM or OM with XAV-939 (n=3). *P-value using corrected Mann-Whitney: 0.03.
DISCUSSION
KPT-330 is currently in clinical trials as a promising anti-cancer drug causing cell cycle arrest resulting in apoptosis and cell death by retaining tumor suppressor and growth regulator proteins in the nucleus.47, 48 In this study, we, for the first time demonstrate the therapeutic value of administrating KPT-330 to treat CAVD, and use high throughput approaches to define its beneficial mechanism of action in VICs.
Molecular observations emanating from this study are consistent with previous studies21 showing that VIC-mediated calcification shares similarities with osteogenic-related processes (Figure 1, Supplementary Figures I, II). However, this study additionally identifies temporal mRNAs profiles that could serve as molecular biomarkers of early (Stage I), mid (Stage II) and late (Stage III) stages of calcification progression. This includes Omd and Ogn as indicators of calcific nodule formation progression, and Ostf11 as a marker of pre-calcific (Stage II) but not mature (Stage III) nodules in our system. However, parallel studies of human pathogenesis are needed to fully determine if these stages align with mild, moderate and severe calcific aortic valve disease in vivo.
Our data also shows that KPT-330 treatment of pAVICs under osteogenic stimulus is sufficient to prevent (Figures 2B, 4A, Supplementary Figure III), attenuate (Figure 3B) and mitigate (Figure 3D) calcific nodules in vitro. Interestingly, phenotypic changes in VIC fate towards the osteogenic lineage is thought to underline the pathological development of CAVD, and we demonstrate that these beneficial effects of KPT-330 are associated with attenuation of these markers (Figure 2). However, while these data suggest that KPT-330 prevents VIC differentiation towards the osteogenic lineage, the mRNA profile of pAVICs treated with KPT-330 at Stage III (nodule mitigation, Figures 3D, G, H) does not mimic that of Stages I or II, at least short-term (72 hours post treatment). Therefore suggesting that KPT-330 is a promising drug for resolving existing nodules, although treatment is not reversing cell fate but rather promoting mRNA profiles associated with ‘heart development’ and ‘outflow tract morphogenesis’ and expression of mesoderm markers including GATA4, GATA5, GATA6, MEF2C and HEY2. However further work is required to determine the phenotypic effects of KPT-330 treatment on pAVIC phenotypes long-term.
Our analysis indicates that prior to calcific nodule formation, pAVICs undergo proliferation in vitro (Figure 1, Supplementary Figure I), similar to previous in vivo findings.49–51 It is not clear if an increase in cell number is required for calcification, but preventing proliferation by KPT-330 (Figure 2) in this study, or HMG-CoA reductase inhibitors (statins) shown by others,49 does prevent the process. Furthermore, it is considered that increased proliferation supports the need for cell-cell contact in the formation of calcific nodules as suggested by published work showing sufficiency of Cadherin-11; a known regulator of cell-cell and cell-matrix interactions, to promote calcification in mice.52 This study identifies several KPT-330 responsive cell cycle (and osteogenic) proteins in pAVICs, but it is not yet known if these are XPO1-dependent, or effectors of C/EBPβ nuclear retention and subsequent Wnt inhibition.53–56
KPT-330’s ability to prevent calcification is largely mediated by Xpo1 as direct silencing has similar beneficial effects (Figure 2), and Mass Spectrometry analysis suggests that C/EBPβ is a likely downstream target, consistent to a report in kidney cell lines.57 In Figure 5, we show that C/EBPβ abundance is reduced in the nuclei of pAVICs under osteogenic stimulus, which is likely independent of proteosomal degradation. Interestingly it is noted that while nuclear C/EBPβ is reduced, cytoplasmic localization is not significantly increased, and the reasons for this are unclear but may be related to the poor stability of the protein outside of the nucleus. In contrast, increased nuclear abundance, or export inhibition by KPT-330 prevents, attenuates and treats calcific nodule formation. However worthy of mention, C/EBPβ mRNA is increased x2.3-fold in KPT-330-treated nodules versus DMSO-treatments (data not shown but included in RNA-seq analysis), and therefore it cannot be ruled out that C/EBPβ protein abundance may in part by a result of increased transcript levels.
C/EBPβ overexpression had similar beneficial effects to KPT-330, and its high nuclear expression in Stage I VICs suggests an anti-osteogenic role to maintain homeostasis of healthy valves. This is in contrast to studies showing that inflammatory signaling promotes bone regeneration during fracture repair via C/EBPβ, suggesting a more pro-osteogenic role.58 It is recognized that C/EBPβ is likely not the only target of KPT-330 in this system and the utilization of KPT compounds has received mixed results largely related to off-target effects in non-pathological cell types that express XPO1-dependent proteins. However, our high throughout analysis has provided mechanistic insights for the development of more specific molecular mechanism to promote C/EBPβ nuclear retention to treat CAVD by inhibiting the previously predicted putative NES sequence,59 or preventing phosphorylation at Serine 240.60
In VICs treated with KPT-330 or C/EBPβ overexpression, the resulting increase in nuclear C/EBPβ leads to decreased canonical Wnt activity (Figure 7) associated with a notable increase in expression and transactivation of the Wnt antagonist, Axin1, and attenuated calcification. This is particularly relevant as increased canonical Wnt signaling has been reported in calcified valves from human patients, mouse models and cultured VICs.41–46 In our hands and other studies, direct activation of Wnt by LiCl or Wnt3a treatment is not sufficient to promote calcification of VICs (data not shown),61 but inhibition is necessary to reduce calcific nodule formation and cell proliferation of pAVICs under calcific stimulus (Figure 7). In contrast to VICs, activated Wnt can induce calcification of vascular smooth muscle cells (VSMCs),55, 56 suggesting potentially divergent mechanisms of calcification between these two cell types.62 This is further supported by the inability of KPT-330 to attenuate vascular calcification in Klotho−/− mice, despite 100% prevention in aortic valves. This discrepancy may also be dependent on the differential pool of XPO1-dependent cargo proteins located in the nuclei of VICs versus VSMCs at the time of treatment and differences in mechanisms involved in vascular v/s valve calcification.63
From the data generated in this study, we postulate that C/EBPβ nuclear retention mediated by KPT-330 treatment benefits calcification by suppressing canonical Wnt signaling. More specifically, we show that KPT-330 treatment and C/EBPβ overexpression increases expression, and in part, transactivation of the Wnt antagonist Axin1 (Figure 8), consistent with previous studies showing the same molecular interaction in other cell types.38–40 However, it is also considered that similar to hepatic cells, C/EBPβ may also mediate increases in Axin1 by enhancing its molecular interaction with β-catenin to promote its degradation.38 While we propose here that KPT-330 exerts its beneficial effects on calcification by suppressing Wnt signaling by increasing Axin1, it is also known in other systems that Axin1 interacts with glycogen synthase kinase 3 (Gsk3) and casein kinase-1 (CK-1) at the plasma membrane to conversely promote β-catenin stabilization and propagate Wnt signaling.64 However, our studies indicating decreased active β-catenin signaling in the presence of KPT-330 or C/EBPβ overexpression favor Axin1 as a negative regulator of Wnt signaling in preventing calcification mediated by VICs.
To our knowledge, this is the first study to report inhibition of aortic valve calcification using a pharmacological nuclear export inhibitor, and define the molecular mechanisms underlying this beneficial effect, however we recognize limitations to the study and the translational perspective. Experimentally, our study utilized pAVICs from otherwise healthy pigs in our study and initiated the calcification response by exposing to a variety of stimuli (glass, osteogenic media, phosphate); however this process of calcification may not represent the pathobiology of acquired CAVD seen in humans, which furthermore may be different again from calcification in the setting of a primary congenital malformation (BAV). In addition, KPT-330 (Karyopharm) is currently seeking FDA approval as a chemotherapeutic compound (Selinexor) to treat relapsed refractory multiple myeloma (RRMM). Given the widespread distribution of XPO1-dependent proteins throughout most tissues, it is interesting that reported side effects to date, are limited to the gastrointestinal (nausea) and hematological (thrombocytopenia) systems of these immunocompromised patients and most are reversible and without evidence of major organ toxicities not cumulative toxicity.65 While these observations have been made in RRMM patients, the pathogenesis of CAVD is different, and any new medical therapy would need to be chronic with a low risk to benefit ratio. As the effects of chronic administration of KPT-330 have not yet been tested in cardiovascular disease, the integrated safety profile in this patient population is uncertain, and it is likely that the effects of KPT-330 are disease- and even cell-dependent. Future studies specifically targeting C/EBPβ nuclear export may circumvent potential off-target effects of using a global XPO1 inhibitor.
We show here that in VICs under osteogenic stimulus, KPT-330 inhibits the nuclear export of several XPO1-dependent (and -independent) proteins including the transcription factor C/EBPβ which we demonstrate is important for this process. This study further contributes to emerging discoveries highlighting the pre-clinical application of KPT-based compounds in the treatment of several pathological disorders by targeting biological processes including cell proliferation, inflammation and fibrosis.66–68 Findings from this current study pave the foundation for future mechanistic-based therapeutic interventions in the treatment of CAVD.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Calcific Aortic Valve Disease affects more than 5.2 million Americans and has limited treatment options.
Calcific Aortic Valve Disease shares molecular phenotypes with endochondral and heterotopic ossification.
Previous studies have shown that nuclear export inhibition by Leptomycin B treatment prevents calcific nodule formation in heart valve interstitial cells in vitro.
What New Information Does This Article Contribute?
KPT-330, a pharmacological XPO1 inhibitor, prevents, attenuates and mitigates calcific nodule formation in vitro, in the absence of cell toxicity, by reducing cell proliferation and attenuating osteogenic gene expression.
KPT-330 prevents aortic valve annular calcification in Klotho−/− mice.
Under calcific stimulus, KPT-330 prevents the nuclear export of C/EBPβ which in turn reduces Wnt activity by activating the antagonist, Axin1.
Direct overexpression of C/EBPβ has similar beneficial effects in preventing calcific nodule formation in culture valve interstitial cells
Calcific Aortic Valve Disease (CAVD) is highly prevalent affecting up to 27% of the population, particularly those over the age of 65, and young adults born with congenital valve malformations. At present, there are no pharmacological treatments for CAVD, and therefore transcatheter aortic valve replacement (TAVR) therapy and surgical intervention remain the most effective options, but these have limitations, and therefore there is a need to discover alternatives. Here we re-purpose the selective XPO1 inhibitor drug, KPT-330 and show that it has beneficial effects in preventing and treating CAVD in vitro and in vivo using an established mouse model. We further illustrate that the beneficial effects of KPT-330 is due to nuclear export inhibition of the transcription factor C/EBPβ, which consequently leads to activation of the Wnt antagonist Axin1, to repress signaling, and shut down pro-osteogenic signaling associated with calcification. Our novel findings have met a critical need to discover alterative, pharmacological-based therapies in the treatment of CAVD.
ACKNOWLEDGEMENTS
We thank Anna Ashbrook at Nationwide Children’s Hospital for assistance with animals.
SOURCES OF FUNDING
This work was supported by NIH/NHLBI R01HL132801, R01HL142685 (JL), Advancing a Healthier Wisconsin (JL), Peter Sommerhauser Endowment Fund for Quality, Outcomes and Research (The Herma Heart Institute) (JL), The Therapeutic Acceleration Program (TAP) at Medical College of Wisconsin (JL), and American Heart Association 18POST33990408 (PD).
Nonstandard Abbreviations and Acronyms:
- ALP
Alkaline Phosphatase
- CAVD
Calcific aortic valve disease
- Ogn
Osteoglycin
- Omd
Osteomodulin
- Osft1
Osteogenic stimulating factor-1
- pAVICs
Porcine aortic valve interstitial cells
- Sost
Sclerostin
- SINE
Selective inhibitors of nuclear transport
- TAVR
Transcatheter aortic valve replacement
- VICs
Valve interstitial cells
Footnotes
DISCLOSURES
The authors have no perceived conflicts of interest.
Publisher's Disclaimer: This article is published in its accepted form. It has not been copyedited and has not appeared in an issue of the journal. Preparation for inclusion in an issue of Circulation Research involves copyediting, typesetting, proofreading, and author review, which may lead to differences between this accepted version of the manuscript and the final, published version.
REFERENCES
- 1.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, et al. Heart disease and stroke statistics−−2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–e220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antonini-Canterin F, Hirsu M, Popescu BA, Leiballi E, Piazza R, Pavan D, Ginghina C and Nicolosi GL. Stage-related effect of statin treatment on the progression of aortic valve sclerosis and stenosis. Am J Cardiol. 2008;102:738–42. [DOI] [PubMed] [Google Scholar]
- 3.Aronow WS, Ahn C KI and Goldman ME. Association of coronary risk factors and use of statins with progression of mild valvular aortic stenosis in older persons. Am J Cardiol. 2001;88:693–5. [DOI] [PubMed] [Google Scholar]
- 4.Moura LM, Ramos SF, Zamorano JL, Barros IM, Azevedo LF, Rocha-Gonçalves F and Rajamannan NM. Rosuvastatin affecting aortic valve endothelium to slow the progression of aortic stenosis. J Am Coll Cardiol. 2007;49:554–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Novaro GM, Tiong IY, Pearce GL, Lauer MS, Sprecher DL and Griffin BP. Effect of hydroxymethylglutaryl coenzyme a reductase inhibitors on the progression of calcific aortic stenosis. Circulation. 2001;104:2205–9. [DOI] [PubMed] [Google Scholar]
- 6.Rosenhek R, Rader F, Loho N, Gabriel H, Heger M, Klaar U, Schemper M, Binder T, Maurer G and Baumgartner H. Statins but not angiotensin-converting enzyme inhibitors delay progression of aortic stenosis. Circulation. 2004;110:1291–5. [DOI] [PubMed] [Google Scholar]
- 7.Shavelle DM, Takasu J, Budoff MJ, Mao S, Zhao XQ and O’Brien KD. HMG CoA reductase inhibitor (statin) and aortic valve calcium. Lancet. 2002;359:1125–6. [DOI] [PubMed] [Google Scholar]
- 8.Bellamy MF, Pellikka PA, Klarich KW, Tajik AJ and Enriquez-Sarano M. Association of cholesterol levels, hydroxymethylglutaryl coenzyme-A reductase inhibitor treatment, and progression of aortic stenosis in the community. J Am Coll Cardiol. 2002;40:1723–30. [DOI] [PubMed] [Google Scholar]
- 9.Chan KL, Teo K, Dumesnil JG, Ni A, Tam J and Investigators A. Effect of Lipid lowering with rosuvastatin on progression of aortic stenosis: results of the aortic stenosis progression observation: measuring effects of rosuvastatin (ASTRONOMER) trial. Circulation. 2010;121:306–14. [DOI] [PubMed] [Google Scholar]
- 10.Dichtl W, Alber HF, Feuchtner GM, Hintringer F, Reinthaler M, Bartel T, Sussenbacher A, Grander W, Ulmer H, Pachinger O and Muller S. Prognosis and risk factors in patients with asymptomatic aortic stenosis and their modulation by atorvastatin (20 mg). Am J Cardiol. 2008;102:743–8. [DOI] [PubMed] [Google Scholar]
- 11.Cowell SJ, Newby DE, Prescott RJ, Bloomfield P, Reid J, Northridge DB, Boon NA, Scottish Aortic S and Lipid Lowering Trial IoRI. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N Engl J Med. 2005;352:2389–97. [DOI] [PubMed] [Google Scholar]
- 12.Mohler ER 3rd, Wang H, Medenilla E and Scott C. Effect of statin treatment on aortic valve and coronary artery calcification. J Heart Valve Dis. 2007;16:378–86. [PubMed] [Google Scholar]
- 13.Panahi Y, Sahebkar A, Taghipour HR, Dadjou Y, Pishgoo B and Rakhshankhah AS. Atorvastatin therapy is not associated with slowing the progression of aortic stenosis: findings of a randomized controlled trial. Clin Lab. 2013;59:299–305. [DOI] [PubMed] [Google Scholar]
- 14.Pohle K, Maffert R, Ropers D, Moshage W, Stilianakis N, Daniel WG and Achenbach S. Progression of aortic valve calcification: association with coronary atherosclerosis and cardiovascular risk factors. Circulation. 2001;104:1927–32. [DOI] [PubMed] [Google Scholar]
- 15.Rossebo AB, Pedersen TR, Boman K, Brudi P, Chambers JB, Egstrup K, Gerdts E, Gohlke-Barwolf C, Holme I, Kesaniemi YA, et al. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med. 2008;359:1343–56. [DOI] [PubMed] [Google Scholar]
- 16.Chen JH and Simmons CA. Cell-matrix interactions in the pathobiology of calcific aortic valve disease: critical roles for matricellular, matricrine, and matrix mechanics cues. Circ Res. 2011;108:1510–24. [DOI] [PubMed] [Google Scholar]
- 17.Rajamannan NM. Mechanisms of aortic valve calcification: the LDL-density-radius theory: a translation from cell signaling to physiology. Am J Physiol Heart Circ Physiol. 2010;298:H5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farrar EJ, Huntley GD and Butcher J. Endothelial-derived oxidative stress drives myofibroblastic activation and calcification of the aortic valve. PLoS One. 2015;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Towler DA. Oxidation, inflammation, and aortic valve calcification peroxide paves an osteogenic path. J Am Coll Cardiol. 2008;52:851–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee SH and Choi JH. Involvement of Immune Cell Network in Aortic Valve Stenosis: Communication between Valvular Interstitial Cells and Immune Cells. Immune Netw. 2016;16:26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mohler ER 3rd, Gannon F, Reynolds C, Zimmerman R, Keane MG and Kaplan FS. Bone formation and inflammation in cardiac valves. Circulation. 2001;103:1522–8. [DOI] [PubMed] [Google Scholar]
- 22.Dutta P and Lincoln J. Calcific Aortic Valve Disease: a Developmental Biology Perspective. Curr Cardiol Rep. 2018;20:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huk DJ, Austin BF, Horne TE, Hinton RB, Ray WC, Heistad DD and Lincoln J. Valve Endothelial Cell-Derived Tgfbeta1 Signaling Promotes Nuclear Localization of Sox9 in Interstitial Cells Associated With Attenuated Calcification. Arterioscler Thromb Vasc Biol. 2016;36:328–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang AY and Liu H. The past, present, and future of CRM1/XPO1 inhibitors. Stem Cell Investig. 2019;6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yip CY, Chen JH, Zhao R and Simmons CA. Calcification by valve interstitial cells is regulated by the stiffness of the extracellular matrix. Arterioscler Thromb Vasc Biol. 2009;29:936–42. [DOI] [PubMed] [Google Scholar]
- 26.Rajamannan NM, Subramaniam M, Rickard D, Stock SR, Donovan J, Springett M, Orszulak T, Fullerton DA, Tajik AJ, Bonow RO and Spelsberg T. Human aortic valve calcification is associated with an osteoblast phenotype. Circulation. 2003;107:2181–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bowen CJ, Zhou J, Sung DC and Butcher JT. Cadherin-11 coordinates cellular migration and extracellular matrix remodeling during aortic valve maturation. Dev Biol. 2015;407:145–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koos R, Brandenburg V, Mahnken AH, Schneider R, Dohmen G, Autschbach R, Marx N and Kramann R. Sclerostin as a potential novel biomarker for aortic valve calcification: an in-vivo and ex-vivo study. J Heart Valve Dis. 2013;22:317–25. [PubMed] [Google Scholar]
- 29.Deckx S, Heggermont W, Carai P, Rienks M, Dresselaers T, Himmelreich U, van Leeuwen R, Lommen W, van der Velden J, Gonzalez A, et al. Osteoglycin prevents the development of age-related diastolic dysfunction during pressure overload by reducing cardiac fibrosis and inflammation. Matrix Biol. 2018;66:110–124. [DOI] [PubMed] [Google Scholar]
- 30.Osman L, Yacoub MH, Latif N, Amrani M and Chester AH. Role of human valve interstitial cells in valve calcification and their response to atorvastatin. Circulation. 2006;114:I547–52. [DOI] [PubMed] [Google Scholar]
- 31.Cui L, Rashdan NA, Zhu D, Milne EM, Ajuh P, Milne G, Helfrich MH, Lim K, Prasad S, Lerman DA, et al. End stage renal disease-induced hypercalcemia may promote aortic valve calcification via Annexin VI enrichment of valve interstitial cell derived-matrix vesicles. Journal of cellular physiology. 2017;232:2985–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Linefsky JP, O’Brien KD, Katz R, de Boer IH, Barasch E, Jenny NS, Siscovick DS and Kestenbaum B. Association of serum phosphate levels with aortic valve sclerosis and annular calcification: the cardiovascular health study. J Am Coll Cardiol. 2011;58:291–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hutcheson JD, Blaser MC and Aikawa E. Giving Calcification Its Due: Recognition of a Diverse Disease: A First Attempt to Standardize the Field. Circ Res. 2017;120:270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheek JD, Wirrig EE, Alfieri CM, James JF and Yutzey KE. Differential activation of valvulogenic, chondrogenic, and osteogenic pathways in mouse models of myxomatous and calcific aortic valve disease. J Mol Cell Cardiol. 2012;52:689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakatani T, Sarraj B, Ohnishi M, Densmore MJ, Taguchi T, Goetz R, Mohammadi M, Lanske B and Razzaque MS. In vivo genetic evidence for klotho-dependent, fibroblast growth factor 23 (Fgf23) -mediated regulation of systemic phosphate homeostasis. FASEB J. 2009;23:433–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Philip F, Faza NN, Schoenhagen P, Desai MY, Tuzcu EM, Svensson LG and Kapadia SR. Aortic annulus and root characteristics in severe aortic stenosis due to bicuspid aortic valve and tricuspid aortic valves: implications for transcatheter aortic valve therapies. Catheter Cardiovasc Interv. 2015;86:E88–98. [DOI] [PubMed] [Google Scholar]
- 37.Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. [DOI] [PubMed] [Google Scholar]
- 38.Park S, Lee MS, Gwak J, Choi TI, Lee Y, Ju BG, Kim CH and Oh S. CCAAT/enhancer-binding protein-beta functions as a negative regulator of Wnt/beta-catenin signaling through activation of AXIN1 gene expression. Cell Death Dis. 2018;9:1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang QQ, Gronborg M, Huang H, Kim JW, Otto TC, Pandey A and Lane MD. Sequential phosphorylation of CCAAT enhancer-binding protein beta by MAPK and glycogen synthase kinase 3beta is required for adipogenesis. Proc Natl Acad Sci U S A. 2005;102:9766–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chung SS, Lee JS, Kim M, Ahn BY, Jung HS, Lee HM, Kim JW and Park KS. Regulation of Wnt/beta-catenin signaling by CCAAT/enhancer binding protein beta during adipogenesis. Obesity (Silver Spring). 2012;20:482–7. [DOI] [PubMed] [Google Scholar]
- 41.Caira FC, Stock SR, Gleason TG, McGee EC, Huang J, Bonow RO, Spelsberg TC, McCarthy PM, Rahimtoola SH and Rajamannan NM. Human degenerative valve disease is associated with up-regulation of low-density lipoprotein receptor-related protein 5 receptor-mediated bone formation. J Am Coll Cardiol. 2006;47:1707–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miller JD, Weiss RM, Serrano KM, Castaneda LE, Brooks RM, Zimmerman K and Heistad DD. Evidence for active regulation of pro-osteogenic signaling in advanced aortic valve disease. Arterioscler Thromb Vasc Biol. 2010;30:2482–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Askevold ET, Gullestad L, Aakhus S, Ranheim T, Tonnessen T, Solberg OG, Aukrust P and Ueland T. Secreted Wnt modulators in symptomatic aortic stenosis. J Am Heart Assoc. 2012;1:e002261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gu GJ, Chen T, Zhou HM, Sun KX and Li J. Role of Wnt/beta-catenin signaling pathway in the mechanism of calcification of aortic valve. J Huazhong Univ Sci Technolog Med Sci. 2014;34:33–6. [DOI] [PubMed] [Google Scholar]
- 45.Gao X, Zhang L, Gu G, Wu PH, Jin S, Hu W, Zhan C, Li J and Li Y. The effect of oxLDL on aortic valve calcification via the Wnt/ beta-catenin signaling pathway: an important molecular mechanism. J Heart Valve Dis. 2015;24:190–6. [PubMed] [Google Scholar]
- 46.Albanese I, Yu B, Al-Kindi H, Barratt B, Ott L, Al-Refai M, de Varennes B, Shum-Tim D, Cerruti M, Gourgas O, Rheaume E, Tardif JC and Schwertani A. Role of Noncanonical Wnt Signaling Pathway in Human Aortic Valve Calcification. Arterioscler Thromb Vasc Biol. 2017;37:543–552. [DOI] [PubMed] [Google Scholar]
- 47.Senapedis WT, Baloglu E and Landesman Y. Clinical translation of nuclear export inhibitors in cancer. Semin Cancer Biol. 2014;27:74–86. [DOI] [PubMed] [Google Scholar]
- 48.Mahipal A and Malafa M. Importins and exportins as therapeutic targets in cancer. Pharmacol Ther. 2016;164:135–43. [DOI] [PubMed] [Google Scholar]
- 49.Rajamannan NM, Evans FJ, Aikawa E, Grande-Allen KJ, Demer LL, Heistad DD, Simmons CA, Masters KS, Mathieu P, O’Brien KD, et al. Calcific aortic valve disease: not simply a degenerative process: A review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: Calcific aortic valve disease-2011 update. Circulation. 2011;124:1783–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chakraborty S, Combs MD and Yutzey KE. Transcriptional regulation of heart valve progenitor cells. Pediatr Cardiol. 2010;31:414–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lerman DA, Prasad S and Alotti N. Calcific Aortic Valve Disease: Molecular Mechanisms and Therapeutic Approaches. Eur Cardiol. 2015;10:108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sung DC, Bowen CJ, Vaidya KA, Zhou J, Chapurin N, Recknagel A, Zhou B, Chen J, Kotlikoff M, Butcher JT. Cadherin-11 Overexpression Induces Extracellular Matrix Remodeling and Calcification in Mature Aortic Valves. Arterioscler Thromb Vasc Biol. 2016;36:1627–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kahlert UD, Suwala AK, Koch K, Natsumeda M, Orr BA, Hayashi M, Maciaczyk J and Eberhart CG. Pharmacologic Wnt Inhibition Reduces Proliferation, Survival, and Clonogenicity of Glioblastoma Cells. J Neuropathol Exp Neurol. 2015;74:889–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cai T, Sun D, Duan Y, Wen P, Dai C, Yang J and He W. WNT/beta-catenin signaling promotes VSMCs to osteogenic transdifferentiation and calcification through directly modulating Runx2 gene expression. Exp Cell Res. 2016;345:206–17. [DOI] [PubMed] [Google Scholar]
- 55.Kratchmarova I, Blagoev B, Haack-Sorensen M, Kassem M and Mann M. Mechanism of divergent growth factor effects in mesenchymal stem cell differentiation. Science. 2005;308:1472–7. [DOI] [PubMed] [Google Scholar]
- 56.Gaur T, Lengner CJ, Hovhannisyan H, Bhat RA, Bodine PV, Komm BS, Javed A, van Wijnen AJ, Stein JL, Stein GS and Lian JB. Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J Biol Chem. 2005;280:33132–40. [DOI] [PubMed] [Google Scholar]
- 57.Tan M, Wettersten HI, Chu K, Huso DL, Watnick T, Friedlander S, Landesman Y and Weiss RH. Novel inhibitors of nuclear transport cause cell cycle arrest and decrease cyst growth in ADPKD associated with decreased CDK4 levels. Am J Physiol Renal Physiol. 2014;307:F1179–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Y, Kim J, Chan A, Whyne C and Nam D. A two phase regulation of bone regeneration: IL-17F mediates osteoblastogenesis via C/EBP-beta in vitro. Bone. 2018;116:47–57. [DOI] [PubMed] [Google Scholar]
- 59.Williams SC, Angerer ND and Johnson PF. C/EBP proteins contain nuclear localization signals imbedded in their basic regions. Gene Expr. 1997;6:371–85. [PMC free article] [PubMed] [Google Scholar]
- 60.Buck M, Zhang L, Halasz NA, Hunter T and Chojkier M. Nuclear export of phosphorylated C/EBPbeta mediates the inhibition of albumin expression by TNF-alpha. EMBO J. 2001;20:6712–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hulin A, Moore V, James JM and Yutzey KE. Loss of Axin2 results in impaired heart valve maturation and subsequent myxomatous valve disease. Cardiovasc Res. 2017;113:40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ferdous Z, Jo H and Nerem RM. Differences in valvular and vascular cell responses to strain in osteogenic media. Biomaterials. 2011;32:2885–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Etchin J, Montero J, Berezovskaya A, Le BT, Kentsis A, Christie AL, Conway AS, Chen WC, Reed C, Mansour MR, et al. Activity of a selective inhibitor of nuclear export, selinexor (KPT-330), against AML-initiating cells engrafted into immunosuppressed NSG mice. Leukemia. 2016;30:190–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.MacDonald BT, Tamai K and He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gavriatopoulou M, Chari A, Chen C, Bahlis N, Vogl DT, Jakubowiak A, Dingli D, Cornell RF, Hofmeister CC, Siegel D, et al. Integrated safety profile of selinexor in multiple myeloma: experience from 437 patients enrolled in clinical trials. Leukemia. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hightower RM, Reid AL, Gibbs DE, Wang Y, Widrick JJ, Kunkel LM, Kastenschmidt JM, Villalta SA, van Groen T, Chang H, et al. The SINE Compound KPT-350 Blocks Dystrophic Pathologies in DMD Zebrafish and Mice. Mol Ther. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Walker CJ, Oaks JJ, Santhanam R, Neviani P, Harb JG, Ferenchak G, Ellis JJ, Landesman Y, Eisfeld AK, Gabrail NY, et al. Preclinical and clinical efficacy of XPO1/CRM1 inhibition by the karyopherin inhibitor KPT-330 in Ph+ leukemias. Blood. 2013;122:3034–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schmidt J, Braggio E, Kortuem KM, Egan JB, Zhu YX, Xin CS, Tiedemann RE, Palmer SE, Garbitt VM, McCauley D, et al. Genome-wide studies in multiple myeloma identify XPO1/CRM1 as a critical target validated using the selective nuclear export inhibitor KPT-276. Leukemia. 2013;27:2357–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all supporting data are available within the article (and its online supplementary files), or can be made available from the corresponding author upon reasonable request. Materials included in this study will be made available upon request to the corresponding author.