

Abstract
Cardiac fibrosis is characterized by excessive deposition of extracellular matrix proteins and myofibroblast differentiation. Our previous findings have implicated resistin in cardiac fibrosis; however, the molecular mechanisms underlying this process are still unclear. Here we investigated the role of resistin in fibroblast-to-myofibroblast differentiation and elucidated the pathways involved in this process. Fibroblast-to-myofibroblast transdifferentiation was induced with resistin or TGFβ1 in NIH-3T3 and adult cardiac fibroblasts. mRNA and protein expression of fibrotic markers were analyzed by qPCR and immunoblotting. Resistin-knockout mice, challenged with a high-fat diet (HFD) for 20 weeks to stimulate cardiac impairment, were analyzed for cardiac function and fibrosis using histologic and molecular methods. Cardiac fibroblasts stimulated with resistin displayed increased fibroblast-to-myofibroblast conversion, with increased levels of αSma, col1a1, Fn, Ccn2 and Mmp9, with remarkable differences in the actin network appearance. Mechanistically, resistin promotes fibroblast-to-myofibroblast transdifferentiation and fibrogenesis via JAK2/STAT3 and JNK/c-Jun signaling pathways, independent of TGFβ1. Resistin-null mice challenged with HFD showed an improvement in cardiac function and a decrease in tissue fibrosis and reduced mRNA levels of fibrogenic markers. These findings are the first to delineate the role of resistin in the process of cardiac fibroblast-to-myofibroblast differentiation via JAK/STAT3 and JNK/c-Jun pathways, potentially leading to stimulation of cardiac fibrosis.
Keywords: Resistin, Fibroblast/Myofibroblast differentiation, Myocardial Fibrosis, JAK2/STAT3, JNK/c-Jun, WP1066: JAK2 inhibitor, SP600125: JNK inhibitor
Graphical Abstract
INTRODUCTION
Cardiac fibrosis, one of the most common features of failing hearts [1, 2], is characterized by myofibroblast differentiation and excessive deposition of extracellular matrix (ECM) proteins in the cardiac muscle. It leads to profound changes in ventricular architecture and geometry and ultimately reduction in cardiac function [3]. Typically, cardiac fibroblasts are quiescent, maintaining a normal turnover of ECM. However, under disease conditions, such as diabetes or following an ischemic insult, they transdifferentiate to myofibroblasts and promote the formation of a protective fibrotic scar that prevents wall rupture [4]. The persistent presence of myofibroblasts results in the loss of the carefully controlled ECM levels in the heart leading to chronic cardiac fibrosis and consequently cardiac dysfunction [4].
The TGF-β pathway appears to be a key contributor to many fibrotic diseases [5–8]. However, other fibrogenic mediators are also elevated in diseased heart tissues and participate in the pathogenesis of cardiac fibrosis. Overwhelming evidence has implicated Janus kinases (JAKs) and c-Jun N-terminal kinase (JNK) signaling in fibrosis [9]. The receptor-associated tyrosine kinases JAKs are known to regulate cytokines and growth factors signaling by recruiting and phosphorylating the signal transducer and activator of transcription (STAT) proteins. Phosphorylated STAT3 translocates to the nucleus, and through interaction with other transcription activators, promotes the transcription of several target genes. TGF-β1 signaling also contributes to the activation of JAK2/STAT3 leading to fibrotic responses [10–13]. Additionally, a large number of animal studies have implicated JNK activation in pathological insults, such as ischemia/reperfusion injury and myocardial infarction [14]. However, the role of JNK signaling in cardiac remodeling during heart failure and fibrosis remains unclear [15–17] and conflicting results have been reported [18, 19]
Resistin, initially discovered as an adipocyte-derived hormone, has been implicated in obesity, insulin resistance, and diabetes [20, 21]. Clinical studies have shown a positive association between the elevated circulating concentrations of resistin and incident of heart failure and hypertension with prevalent coronary heart disease [22–24], suggesting a link of this protein with cardiac remodeling [25]. We have previously demonstrated that hyper-resistinemia contributes to cardiomyocyte hypertrophy and impairment of cardiac contractility and function [26].
Furthermore, we found that long-term cardiac-specific overexpression of resistin triggered oxidative stress, cardiac fibrosis and apoptosis, leading to myocardial remodeling in normal rats [27]. However, the role of resistin in fibroblast-to-myofibroblast transdifferentiation and how it regulates this process has not been explored before.
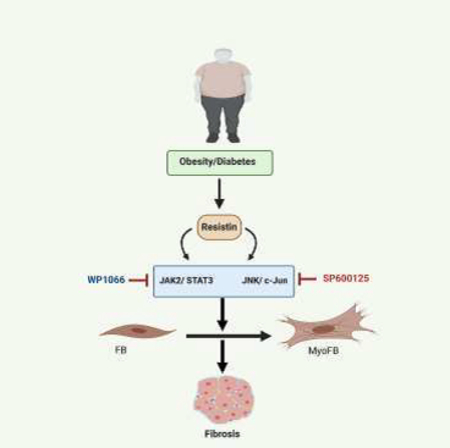
The present study documents resistin as a critical determinant of fibroblast-to-myofibroblast differentiation. Resistin stimulation of immortalized mouse embryonic fibroblasts (NIH-3T3 cells) and adult mouse heart fibroblasts resulted in the proliferation of fibroblasts and up-regulation of fibrosis-related genes via activation of JAK2/STAT3 and JNK/cJun pathways. Specific inhibition of these pathways attenuated resistin’s induction of fibrotic markers. Moreover, in the absence of resistin, high-fat diet failed to activate JAK2/STAT3 and JNK/cJun signaling and the associated fibrotic changes in the mouse hearts. Collectively, this study demonstrates a previously unexplored stimulatory role of resistin in the process of cardiac fibroblast-to-myofibroblast conversion through JAK2/STAT3 and JNK/cJun signaling.
METHODS AND MATERIALS
AAV9-resistin recombinant virus production-
AAV9 virus production was carried out using 293T cells as described elsewhere [27]. Briefly, 293T cells were co-transfected with pTR-Resistin and pDG-9 (expressing AAV9 capsid and helper functions under the control of CMV promoter). Three days after transfection, cells were washed with PBS, resuspended in 150 mM NaCl, 50 mM Tris, and lysed by 3 cycles of freeze/thaw to release the virus. Subsequently, cell lysate was incubated for 30 min at 37 °C after the addition of Benzonase and MgCl2 to a final concentration of 150 units/ml and 2 μM respectively, and then centrifuged at 3400g for 20 min. The supernatant was subjected to iodixanol gradient centrifugation for 1 hour at 69,000 rpm at 18°C. The virus-enriched iodixanol fractions were collected from the 40% to 60% interfaces. The fractions containing the virus were concentrated and dialyzed using Lactated Ringer’s solution, and then filtered with a 0.2 μm filter. Stocks were stored at −80°C, and genome-containing particles were determined by qPCR.
Animals-
Wild type 8 to 10-weeks old male and female mice (C57BL/6J; 000664) were obtained from Jackson Laboratory and injected with AAV9.Empty or AAV9-Resistin (3×1012 viral particles) via tail-vein. Samples were harvested from these mice after 10 weeks of injection. C57BL/6 (WT) and Resistin knockout mice (8–10 weeks old, male and female; n=10/group) were fed either high fat diet (HFD) or normal chow diet (NCD) for 20–22 weeks. All experimental procedures were approved by the Mount Sinai Institutional Animal Care and Use Committee in accordance with the Principles of Laboratory Animal Care by the National Society for Medical Research and Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication no. 86–23, revised 1996).
Echocardiography
Echocardiography was performed under sedation with ketamine 80–100 mg/kg injected intraperitoneally. Sedation was optimized by (1) giving the lowest dose of ketamine needed to restrain the animal and prevent motion artifact and (2) maintain the heart rate as close as possible to 550 beats/min. Two-dimensional images and M-mode tracings were recorded on the short-axis at the level of the mid-papillary muscle to determine percent fractional shortening and ventricular dimensions (GE Vivid Vision) as recommended by the American Society of Echocardiography.
Isolation of mouse cardiac fibroblasts, cell culture, and differentiation-
Cardiac fibroblasts were enzymatically isolated from mouse hearts using retrograde perfusion technique with collagenase and hyaluronidase. After perfusion, left ventricle was dissected and used for the fibroblasts preparation. Fibroblasts were separated from myocytes by three cycles of differential centrifugation (1×50g and 2×300g). The final pellet was resuspended in DMEM (Gibco, Invitrogen) supplemented with 5% fetal bovine serum (FBS) (Gibco, Invitrogen) and 1% of penicillin/streptomycin cocktail and seeded onto 10 cm2 culture plates. Thirty minutes later, plates were washed with PBS to remove non-attached cells, and culture media was re-added. The quality and purity of the fibroblast preparations were verified by immunostaining and western blots of fibroblast-enriched protein markers, such as periostin, fibronectin and vimentin.
Mouse embryonic fibroblast cells (NIH-3T3) were grown in DMEM supplemented with 10% FBS and 1X cocktail of penicillin/streptomycin antibiotics. For cell differentiation, fibroblasts were serum-starved or cultured in 1% FBS for 24 hours and treated with recombinant resistin (100ng/ml) (#1069-RN-050/CF, R&D System) or TGFβ1 (10ng/ml) (#7666-MB-005/CF, R&D System) for additional 24 hours or 48 hours as indicated. For JAK and JNK inhibition, cells were incubated with JAK2 inhibitor (WP1066, # sc-203282, Santa Cruz Biotechnology) and JNK inhibitor (SP600125, # sc-200635, Santa Cruz Biotechnology) at the indicated concentrations selected as per earlier studies [28]. Harvested cells were used for F-actin staining, total RNA isolation, and protein lysates.
Western Blotting -
Isolated heart tissues and cultured fibroblasts were homogenized in whole cells lysis buffer (#9803 Cell Signaling Technology) containing protease and phosphatase inhibitors (Roche). Proteins lysates (20–40 μg) were separated by SDS-PAGE (4–12%) and transferred onto PVDF membrane (Bio Rad). Antibodies used at 1:1000 dilution were phospho-Smad3 #9520, Smad3 #9523, phospho-JAK2 #3771, JAK2 #3230, phospho-Stat3 #9131, Stat3 #9132, phospho-JNK #4668, JNK #9252, phospho-c-Jun #3270, c-Jun #9165, β actin # 12262, α-smooth muscle actin #19245, (Cell Signaling Technology), LOX #ab31238, CTGF #ab6992 (Abcam), resistin # 5997 (Biovision), Alexa Fluor 488-conjugated phalloidin (#A12379, Life Technologies), and anti-SF2/ASF (#MA5–35441 Invitrogen). Densities of the immunoreactive bands from at least three independent experiments were evaluated using NIH ImageJ. Protein loading was verified against β-actin densities.
Real-Time-PCR -
Total RNA was prepared from mouse hearts or cultured cells using TRIzol reagent (Ambion, Life technologies) and 200–500 ng were reverse transcribed by using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) according to the manufacturer’s instructions. The cDNAs were quantified in triplets using the PerfeCTa SYBR Green FastMix, Low ROX (cat no. 95074–012, Quanta Biosciences) on a 7500 Real-time PCR (Applied Biosystems) using mouse-specific primers or 18S rRNA as an internal control and after adjusting the threshold cycle (Ct). Fold changes in gene expression were determined using the ΔΔCT method with normalization to endogenous 18S rRNA controls. Primer used in the study are as follow: Resistin-F: CCAGCTGCAATGAAGAACAC, Resistin-R: CCGCTGTCCAGTCTATGCTT; αSma-F: GCTGGACTCTGGAGATGG, αSma-R: GCAGTAGTCACGAAGGAATAG; Fibronectin-F: AGGC AATGGACGCATCAC, Fibronectin-R: TTCCTCGGTTGTCCTTCTTG; Col1a1-F: GTATGCTTGATCTGTATCTG, Col1a1-R: CGACTCCTACATCTTCTG; Ccn2-F: CTGCGAGGAGTGGGTGTG, Ccn2-R: ATGTGTCTTCCAGTCGGTAGG; Timp-1-F: GAAACTCGGACCTGGTCATAA, Timp-1-R: CGGCCCGTGATGAGAAACT; Lox-F: ACCTGGTACCCGATCCCTACT, Lox-R: GAGGAAATCGTAGCAGTACCC; Mmp9-F: TCAAGGACGGTCGGTATTGG, Mmp9-R: ACGTGCGGGCAATAAGAAAG; 18S-F: AGTCCCTGCCCTTTGTACACA, 18S-R: CGATCCGAGG GCCTCACTA.
Histology and Fluorescent staining -
To assess cardiac fibrosis, harvested hearts were perfused with 10 ml of PBS followed by 10% buffered formalin solution (Sigma) for 24 hours then embedded in paraffin. Sections (8-μm) were cut from left ventricular tissue, stained with standard Masson’s trichrome as per the manufacturer’s instructions (Sigma). Cultured fibroblasts were fixed in 3.7% formaldehyde solution (Sigma) in PBS for 10 minutes and permeabilized using 0.1% Triton-X (Sigma). To detect the polymerized actin cytoskeleton, cells were incubated in 0.5 μM solution of Alexa Fluor 488-conjugated phalloidin (Life Technologies) in PBS for 1 hour at room temperature. DAPI (Life Technologies) was used to stain nuclei. Image Quantifications: NIH Image J software was used to quantify % fibrosis and myofibroblast cell size. Blue colored area positively stained with Masson trichrome was measured in the entire cross-sectional area of ventricular tissue using Image J software. % blue area to the total area gives an index of % fibrosis as we described previously [29].
Immunofluorescence staining of fibroblasts –
Cardiac fibroblasts were isolated as described. Fibroblasts were serum-starved or cultured in 1% or 10% FBS for 24 hours to stimulate differentiation. Cells were fixed with 4% paraformaldehyde for 15 minutes, and then washed 3 times with 1X PBS. Fibroblasts were then permeabilized with triton X-100 (0.1%) for 10 minutes, blocked with 1% BSA in PBS for 1 hour, and then incubated with the following antibodies (1/100): Vimentin (#5741, Cell signaling), Perisotin (#14041, Abcam), fibronectin (#FBN11, Thermofisher), and F-actin/phalloidin (#49409, Sigma) for 2 hours. Cells were next washed with 1X PBS and incubated with fluorescent tagged-secondary antibodies (Aexa Fluor-488, ALEXA-555, Molecular Probes) diluted (1/500) for 1 hour, washed and mounted with DAPI. Cells were screened under Zeiss epifluorescence microscope.
Gelatin zymography –
Gelatin zymography was performed as described in the Abcam protocol (Abcam). Briefly, cardiac fibroblasts were serum-starved and cultured in 1% FBS (control), TGFβ1 (10 ng/ml) or resistin (Retn, 100 ng/ml) for 48 hours to stimulate differentiation. Conditioned media were collected, centrifuged to eliminate dead cells and concentrated using Amicon® Ultra-4 Centrifugal Filter Units Ultracel −3K (EMD Millipore). Concentrated conditioned media (10ug) were subjected to SDS-PAGE through 7.5% polyacrylamide gels containing 4 mg/ml gelatin under non-reducing conditions. Gel was rinsed 2 × 30 min with washing buffer (50 mM Tris-HCl, pH 7.5, 2.5% Triton X-100, 5 mM CaCl2 and 1 μM ZnCl2) at room temperature and incubated for 24 hours at 37°C in incubation buffer (50 mM Tris-HCl, pH 7.5, 1% Triton X-100, 5 mM CaCl2, 1 μM ZnCl2). Gel was fixed and stained with 0.1% Coomassie blue R250. After destaining, gelatinolytic activity was visualized as a clear band against a blue background of stained gelatin.
Statistical analysis –
Data is expressed as mean ± SEM. Comparison of paired individual values is performed by paired t-test, respectively. Multiple comparisons are performed by analysis of variance (ANOVA) with post hoc Bonferroni’s test where appropriate. P<0.05 was considered statistically significant; GraphPad Prism 5 was used as the statistical software.
RESULTS
Resistin modulates the expression of fibrotic markers in NIH-3T3 fibroblast cells-
To investigate whether resistin directly influences the fibrotic response, we first examined the expression of fibrotic genes in NIH-3T3 cells stimulated with resistin or TGFβ1 for 48 hours. TGFβ1, a well-recognized growth factor that drives differentiation of fibroblasts into myofibroblasts leading to tissue fibrosis, was used as a positive control. A significant increase in the mRNA expression of profibrotic genes such as α-smooth muscle actin (αSma), collagen type 1 alpha 1 chain (Col1a1), connective tissue growth factor (Ctgf) (also known as cellular communication network factor 2 (Ccn2)), fibronectin (Fn), matrix metallopeptidase 9 (Mmp9) and tissue inhibitor of metalloproteinases 1 (Timp1) was observed in NIH-3T3 cells treated with resistin or TGFβ1 as compared to control (Fig. 1A). Similarly, increased protein expression of αSMA, CTGF/CCN2, lysyl oxidase (LOX) and splicing factor (SF)-2 was observed in resistin or TGFβ1 stimulated cells as compared to control (Fig. 1B). Interestingly, resistin-driven fibrotic gene expression pattern highly resembled that of TGFβ1-induced expression, suggesting that resistin-induced upregulation may be a result of an aberrant increase in molecular signaling, which may transcriptionally regulate fibrotic gene expression.
Figure -1: Resistin upregulates fibrosis marker genes expression in vitro -.

A) NIH-3T3 cells were non-treated (cont) or treated with resistin (Retn) 100ng/ml or TGFβ1 10ng/ml for 48 hours. The mRNA expression of αSma, Col1a1, Ccn2, Fn, Mmp 9, and Timp1 were analyzed by q-PCR. 18S rRNA was used as an internal control; the data are mean ± SEM of at least three experiments in triplicates. p**< 0.01, ***p < 0.001. B) Western blotting analysis and densitometry quantifications of αSMA, LOX, CTGF/CCN2 and SF-2 protein expression are shown. β-actin was used as an internal control. The data are mean ± SEM of at least three experiments in triplicates. *p < 0.05, **p < 0.01, ***p < 0.001.
Resistin promotes cardiac fibroblast-to-myofibroblast conversion in vitro-
Our observation that resistin increased profibrotic gene expression in NIH-3T3 cells prompted us to examine whether resistin also activates the process of cardiac fibroblast-to-myofibroblast conversion. Adult mouse cardiac fibroblasts were isolated as indicated in the methods section and the quality and purity of the fibroblast culture preparations were verified by immunostaining (Supplemental Figure 1A) and western blots (Supplemental Figure 1B) of active fibroblast-enriched protein markers, such as periostin, α-smooth muscle actin, fibronectin and vimentin. Adult mouse cardiac fibroblasts were treated with resistin or TGFβ1 for 48 hours as indicated. Stimulated fibroblasts exhibited an early increase in cell size and appearance of stress fibers. These features are indicative of fibroblast transdifferentiation into myofibroblasts (Fig. 2A). To determine the molecular changes involved in the resistin-induced fibroblast-to-myofibroblast conversion, we determined the expression of fibrotic gene markers. We observed an up-regulation of mRNA expression of αSma, Col1a1, Ccn2, and Mmp9 in resistin-treated fibroblasts, parallel to TGFβ1 (Fig. 2B). Likewise, protein expression of αSMA, LOX, CTGF/CCN2, and SF-2 were also increased in resistin or TGFβ1 stimulated fibroblasts compared to control cells (Fig. 2C). Furthermore, the activation of fibroblast to myofibroblast was also functionally confirmed by measuring MMP6 activity using gel zymography assay which demonstrated increased MMP9 enzymatic activity (Supplemental figure 2). Taken together, these data indicate that resistin induces profound changes in fibroblasts leading to the acquisition of an early myofibroblast phenotype in vitro.
Figure -2: Resistin promotes cardiac fibroblasts to myofibroblasts conversion in vitro -.

A) Representative microscopic images of differentiated cardiac fibroblasts non-treated (cont) or treated with resistin 100ng/ml or TGFβ1 10ng/ml for 48 hours and then stained for F actin to visualize stress fibers. B) The mRNA expression of αSma, Col1a1, Ccn2 and Mmp 9 was analyzed by q-PCR. 18S rRNA was used as an internal control; the data are mean ± SEM of at least three experiments in triplicates. p*< 0.05, p**< 0.01, ***p < 0.001. C) Western blotting analysis and densitometry quantifications of resistin, αSMA, LOX, CTGF/CCN2, and SF-2 are shown. The data are mean ± SEM of at least three experiments in triplicates. p**< 0.01, ***p < 0.001.
Resistin cardiac overexpression modulates fibrotic markers expression in vivo-
We then investigated if resistin also promotes the expression of fibrotic genes in vivo. Resistin was overexpressed in the hearts of adult mice using adeno-associated virus serotype 9 (AAV9-Retn) which confers high cardiac tropism. We then examined the expression of fibrotic genes in AAV9-Retn or AAV9-Empty mouse hearts at the mRNA and protein levels. The overexpression of resistin significantly enhanced mRNA expression of αSma, Fn, Mmp9, and Timp1 compared to AAV9-Empty control mice (Fig. 3A). Similarly, protein expression levels of αSMA, CTGF/CCN2, LOX, and SF-2 were significantly enhanced in resistin overexpressing hearts (Fig. 3B). These in vivo findings are nicely in line with the in vitro results following resistin’s treatment of cardiac fibroblast or NIH-3T3 cells. Altogether, these findings point to an important role of resistin in the process of fibroblast activation in vitro and in vivo, similar to TGFβ1 effects.
Figure -3: Resistin overexpression increases fibrosis markers expression in adult mice -.

C57B6 adult mice were infected with AAV9-resistin (AAV9-Retn) or AAV9-empty vectors for 10 weeks. A) mRNA expression of αSma, Fn, Mmp9 and Timp1 were analyzed by q-PCR; 18S rRNA was used as an internal control; p*< 0.05 and p*** < 0.01 vs AAV9-Empty. B) Protein expression levels of resistin, αSMA, CTGF/CCN2, LOX and SF-2 were analyzed by western blotting and densitometry quantification were determined. p*< 0.05, **p < 0.01 and ***p < 0.001 vs AAV9-Empty.
Resistin promotes fibrogenesis via JAK2/STAT3 and JNK/c-Jun signaling in cardiac fibroblasts, independent of TGFβ pathway-
We next attempted to determine the potential mechanism(s) underlying resistin effects on the molecular signaling associated with the fibrotic response. Isolated cardiac fibroblasts were stimulated with resistin or TGFβ1 for various time points and phosphorylation of Smad3, a well-known downstream effector of TGFβ1 signaling and mediator of fibrosis, was measured. We found that Smad3 phosphorylation is significantly elevated in TGFβ1-treated cells compared to untreated control cells, however, resistin did not affect Smad3 phosphorylation in this setting (Fig. 4A), suggesting that resistin mediates fibrotic genes transcription independent of canonical TGFβ1 signaling.
Figure -4: Resistin promotes fibrogenesis via activating JAK-STAT and JNK-c-Jun signaling pathways in cardiac fibroblasts, independently of the TGFβ1 pathway -.

Cardiac fibroblasts were treated with resistin (100ng/ml) and TGFβ1 (10ng/ml) for the indicated times. The phosphorylation of Smad3, JAK2, STAT3, JNK, c-Jun was analyzed by western blotting (A), and densitometry quantifications were performed (B). β-actin was used as internal controls. The data are mean ± SEM of at least three experiments in triplicates. p*< 0.05, **p < 0.01, ***p < 0.001, 0 min vs indicated minutes in figure.
Mechanisms that regulate organs fibrosis are linked through numerous intricate molecular pathways [8, 30, 31], raising the possibility that besides TGFβ1, resistin may emerge as a new mediator utilizing other signaling pathways to promote cardiac tissue fibrosis. This assumption was verified by our observation that resistin stimulated the phosphorylation of JAK2 and JNK in fibroblasts in a time-dependent manner (Fig. 4 A and B). Signaling pathways such as JAK2/STAT3, JNK/c-Jun are well-known drivers of fibrosis [9]. Increased activation of JAK2 and JNK correlated with increased p-STAT3 and p-c-Jun phosphorylation, respectively, in resistin-stimulated fibroblasts compared to untreated cells (Fig. 4A and B). These findings underscore the biological importance of resistin in the transcriptional regulation of profibrotic genes via the JAK2/STAT3 and JNK/c-Jun pathways and ultimately in myocardial fibrosis.
Resistin modulates cardiac JAK2/STAT3 and JNK/c-Jun signaling pathways in vivo-
The in vitro study suggested that resistin induces profibrotic gene expression through JAK2/STAT3 and JNK/c-Jun pathways; we then wanted to know whether resistin can also activate these pathways in vivo. We examined the phosphorylation of JAK2/STAT3 and JNK/c-Jun in mouse hearts overexpressing AAV9-Resistin or the AAV9.Empty vector. Resistin overexpression significantly increased the phosphorylation of JAK2/STAT3 (Fig. 5A), and JNK/c-Jun (Fig. 5B) compared to hearts that received the AAV9.Empty vector. These findings provide strong evidence that resistin regulates the fibrotic pathway through a tight network and dysregulation in resistin levels may result in cardiac fibrosis.
Figure -5: Resistin overexpression induces fibrosis signaling pathways in adult mice-.

C57B6 adult mice were infected with AAV9-resistin (AAV9-Retn) or AAV9-empty vectors for 10 weeks. The phosphorylation of JAK2, STAT3 (A) and JNK, c-Jun (B) was analyzed by western blotting with densitometry quantification shown below the blots. β-actin was used as internal controls. The data are mean ± SEM of n=5. p*< 0.05, **p < 0.01 vs AAV9-Empty.
JAK2 and JNK inhibition attenuates resistin-induced fibroblast activation-
To further demonstrate the significance of the JAK2 pathway in resistin-induced fibrosis, we examined the phosphorylation level of JAK2 in cardiac fibroblasts treated with JAK2 inhibitor (WP1066) for 48 hours. As shown in Fig. 6A, resistin-mediated phosphorylation of JAK2 and STAT3 significantly decreased in the presence of the JAK2 inhibitor. This observation is paralleled by the ability of the JAK2 inhibitor to effectively prevent resistin-induced phenoconversion of fibroblasts into myofibroblasts. After 48 hours of culture, resistin treated fibroblasts significantly increased their differentiation with increased well-organized actin fibers, compared to control untreated fibroblasts, whereas this was not observed in fibroblasts receiving JAK2 inhibitor (Fig. 6B). To further assess the impact of this inhibition, JAK2 inhibited and resistin stimulated fibroblasts were analyzed for fibrosis markers gene expression. Resistin treatment significantly induced the expression of profibrotic markers, such as αSma, Col1a1, Ctgf/Ccn2, Fn, Mmp9, and Timp1, but failed to induce these genes in JAK2 inhibitor pre-treated fibroblasts (Fig. 6C).
Figure -6: Resistin fails to induce expression of fibrotic genes in JAK-inhibited and JNK-inhibited cells –

A) Cardiac fibroblasts were treated with JAK inhibitor WP1066 (5μM) for 30 min and stimulated with resistin (100ng/ml) for an additional 2 hours. The phosphorylation of JAK2 and STAT3 was analyzed by western blotting. B) Representative fluorescence microscopic images of cardiac fibroblasts treated with JAK2 inhibitor for 2 hours and stimulated with resistin in presence of JAK2 inhibitor for an additional 48 hours and then stained for F actin to visualize stress fibers. C) mRNA expression of αSma, Col1a1, Ccn2, Fn, Mmp9, and Timp1 was analyzed by q-PCR in cardiac fibroblasts treated with JAK2 inhibitor for 2 hours and stimulated with resistin for an additional 48 hours. D) Cardiac fibroblasts were treated with JNK inhibitor SP600125 (25μM) for 30 min and stimulated with resistin (100ng/ml) in the presence of JAK2 inhibitor for an additional 2 hours. The phosphorylation of JNK and c-Jun was analyzed by western blotting. E) Representative fluorescence microscopic images of cardiac fibroblasts treated with JNK inhibitor for 2 hours and stimulated with resistin for an additional 48 hours and then stained for F actin to visualize stress fibers; bar size = 200μM. F) mRNA expression of αSma, Col1a1, Ccn2, Fn, Mmp9, and Timp1 was analyzed by q-PCR in cardiac fibroblasts treated with JAK2 inhibitor for 2 hours and stimulated with resistin for additional 48 hours. 18S rRNA was used as an internal control. The data are mean ± SEM of three experiments in triplicates, p*< 0.05, ***p < 0.001.
To further determine the role of resistin in JNK activation, cardiac fibroblasts were treated with resistin in the presence or absence of the JNK inhibitor SP600125. Resistin treatment failed to activate JNK and its downstream effector c-Jun in fibroblasts pretreated with the JNK inhibitor as evidenced by a significant decrease in their phosphorylation status (Fig. 6D). We also investigated the direct involvement of JNK activation in resistin induced activation of fibroblasts. After 48 hours of culture, resistin treated fibroblasts exhibited remarkable differences in the appearance of the actin network with parallel bundles, compared to control untreated fibroblasts, whereas this was not observed in fibroblasts receiving the JNK inhibitor (Fig. 6E). As expected, in JNK-inhibited fibroblasts resistin failed to induce the expression of profibrotic genes αSma, Col1a1, Ctgf/Ccn2, Fn, Mmp9, and Timp1 (Fig. 6F). This suggests that resistin regulates the expression of genes involved in fibrotic response by recruiting the JNK/-cJun pathway. Collectively, these results indicate that signaling via JAK2 and JNK is critical for resistin’s activation of the fibrotic response.
Resistin knockout mice are protected from HFD-induced cardiac remodeling-
To obtain further insight into how resistin impacts heart muscle remodeling, we examined the effect of high-fat diet (HFD) in resistin knockout (Retn-KO) and wild-type (WT) mice. Echocardiographic analysis showed a significant decrease in contractility as assessed by fractional shortening in WT:HFD, which was improved in Retn-KO:HFD mice (FS; 45.26 ± 1.19 % vs 50.26 ± 0.25% P=0.0003) (Fig. 7A). This improvement in contractility was accompanied by a reduction in ventricular geometry, characterized by a significant decrease in left ventricular internal dimension at end-systole (LVIDs) and end-diastole (LVIDd) in Retn-KO:HFD mice compared to WT:HFD mice (Fig. 7A).
Figure -7: Resistin knockout mice on high-fat diet exhibit lower fibrosis gene expression and decreased fibrosis in the heart-.

Eight to 10 weeks old resistin knockout mice (Retn-KO) and age-matched WT (n=10/group) were fed normal chow diet (NCD) or high-fat diet (HFD) for 20–22 weeks. A) Analysis of echocardiographic parameters such as fractional shortening, and left ventricular internal dimension at end-systole (LVIDs) and diastole (LVIDs) are shown. **p < 0.01 and ***p < 0.001. B) Representative histological micrographs of heart tissue sections fixed in OCT and analyzed for fibrosis with Masson’s Trichrome staining; fibrotic areas were quantified using Image J software; bar size =50μM. C) The mRNA expression of αSma, Col1a1, Ccn2, Mmp9, and Lox was analyzed by q-PCR with 18S rRNA as an internal control. p*< 0.05, **p < 0.01 ***p < 0.001. D) Western blotting analysis and densitometry quantification of resistin, αSMA, CTGF/CCN2, LOX, and SF-2 are shown. The data are mean ± SEM of three experiments in triplicates. p*< 0.05, **p < 0.01, ***p < 0.001.
Histological analysis showed that HFD induced deposition of interstitial collagen in WT:HFD mice, whereas Retn-KO:HFD showed less fibrosis, very much similar to normal chow diet mice (WT:NCD; Retn-KO:NCD) (Fig. 7B). We then analyzed the expression of fibrotic genes, αSma, Col1a1, Ctgf/Ccn2, Mmp9 and Lox. A significant increase in the mRNA expression of these genes was observed in the heart of WT:HFD but the expression of these genes was markedly reduced in the heart tissues of Retn-KO:HFD mice (Fig. 7C). No change in the expression of these genes was observed in either group of mice on NCD (Fig. 7C). Interestingly, HFD also failed to induce the protein expression of the ECM matricellular proteins; αSMA, CTGF/CCN2, LOX, and SF-2 in Retn-KO:HFD mouse hearts compared to WT:HFD (Fig. 7D). Collectively, these results showed that mice with resistin deletion were resistant to HFD-induced cardiac fibrosis. The results also suggest that the absence of resistin prevents cardiac fibroblasts transdifferentiation to myofibroblasts, resulting in reduced ECM deposition.
High-fat diet failed to induce JAK2/STAT3 and JNK/c-Jun signaling in Retn-KO mice-
We next attempted to determine the effects of HFD on JAK2/STAT3 and JNK/c-Jun signaling and whether resistin deletion attenuates the activation of these pathways. We analyzed the phosphorylation of JAK2/STAT3 and JNK/c-Jun in Retn-KO and WT hearts by western blotting. We observed that in Retn-KO mice, HFD mediated activation of p-JAK2, p-STAT3, p-JNK, and p-c-Jun was significantly decreased compared to WT mouse hearts (Fig 9A and B), indicating that resistin is essential for HFD to trigger the activation of the of JAK2/STAT3 and JNK/cJun pathways. These results are consistent with our in vitro observations implicating resistin in the activation of JAK2/STAT3 and JNK/cJun signaling in cardiac fibroblasts. Altogether, these data provide strong evidence that resistin likely controls the fibrotic response through the regulation of JAK2/STAT3 and JNK/cJun pathways leading to manipulation of pathological fibrosis, highlighting new opportunities to target resistin in cardiac fibrosis.
DISCUSSION
This study identified a new regulatory mechanism underlying cardiac fibroblast differentiation, whereby resistin is a mediator in the molecular pathways orchestrating fibroblast-myofibroblast transdifferentiation and the fibrotic response. Previous work from our group demonstrated increased fibrosis in the myocardium of rats cardiac-specifically transduced with AAV9-Resistin [27]. We have also shown that resistin is expressed in cardiomyocytes and cardio-fibroblasts and its expression is enhanced following myocardial infarction injury and in response to neurohumoral stimulation [32]. However, the functional role of resistin in cardiac fibroblasts is still poorly understood. This study was designed to gain a better understanding of the role of resistin in fibroblast-myofibroblast transdifferentiation and the molecular mechanisms underlying the regulation of this process. Herein, we report that resistin is capable of stimulating the differentiation of cardiac fibroblasts into myofibroblasts. We demonstrate that resistin-stimulated primary adult cardiac fibroblasts, mouse embryonic fibroblastic cells, as well as resistin-overexpressing cardiac tissues in vivo, exhibited increased expression of ECM proteins such as αSMA, COL1a1, CTGF/CCN2, Fibronectin, LOX, and SF2, in addition to the appearance of organized stress fibers, all major markers and phenotypic features of myofibroblasts and fibrosis. These findings directly implicate resistin in the process of cardiac fibrogenesis and lend further support to our previous studies. We have shown that animal models of cardiac hypertrophy associated with fibrosis (diabetes, pressure overload, and heart failure) exhibit elevated tissue levels of resistin compared to non-fibrosing hypertrophy (volume overload) where resistin is minimally or not elevated [32]. These observations are corroborated by our finding in the current study indicating that resistin overexpression activated the fibrotic response while resistin deletion attenuated HFD-induced cardiac fibrosis in resistin-KO mice. Linking resistin to fibroblast-myofibroblast transdifferentiation is of particular prognostic and therapeutic interest since a) serum resistin is elevated in hypertrophy and heart failure, conditions in which myocardial fibrosis is emerging as a predictor of arrhythmias and as a potential criterion for device therapy; and b) the deposition of collagen and its gradual organization into irreversible fibrosis are histological hallmarks of pressure overload heart failure [33].
We went further to demonstrate how resistin regulates fibrogenesis. One of the fundamental initiators of the transdifferentiation of fibroblasts is TGF-β1, which is known to induce the expression of ECM proteins in myofibroblasts primarily through the activation of Smad3 [6, 34]. In our study, we observed that, unlike TGFβ1, resistin did not activate TGFβ/Smad3 signaling, suggesting that it may very likely activate the fibrotic response via a different pathway independent of Smad3. Indeed, resistin-induced myofibroblast differentiation and the initiation of the fibrotic response were found to be dependent on JAK and JNK signaling. Vast evidence has substantiated the role of JAKs and JNK signaling pathways in fibrosis [9]. We have observed that JAK2 and JNK - and the phosphorylation of their downstream effectors, STAT3 and c-Jun, respectively - are activated in resistin-treated cardiac fibroblasts and in AAV9-Retn transduced adult hearts. Deletion of resistin in vivo or pharmacological inhibition of JAK2 and JNK in cultured fibroblasts inactivated the phosphorylation of STAT3 and c-Jun, prevented resistin-induced differentiation of resting fibroblasts into myofibroblasts and significantly reduced the stimulatory effect of resistin on gene expression, including those of potential biomarkers of fibrosis. These results strongly suggest that resistin regulates the fibrotic response through a network of numerous signaling pathways, and impairment of this regulation by resistin results in excess ECM deposition and cardiac fibrosis.
Fibroblast differentiation coincides with the overproduction of collagen type I (COL1) and other ECM proteins. This transition requires the engagement of transcription factors such as STAT3 and c-Jun to drive the increased COL1A1, aSMA, CTGF/CCN2, FN, MMP9 and TGFβ1 expression observed in myofibroblasts. The activation of TGFβ1 feed-forwardly exacerbates this vicious cycle of myofibroblast gene expression program and ECM production. The matricellular proteins, CCN2/CTGF, LOX, SF and FN are of particular interest since they have been directly involved in fibroblast activation and fibrogenesis.
CCN2 has been implicated in a number of fibrotic disease states and has therefore been proposed to mediate fibrogenesis [35]. Targeted expression of CCN2 in fibroblasts in vivo promoted systemic tissue fibrosis [36] while CCN2 transgenic mice showed significantly increased fibrosis in response to pressure overload [37]. CCN2 may partially promote fibrosis by recruiting other matricellular proteins and enhancing expression of TIMP1 [36]. Lysyl oxidase (LOX), a copper-dependent amine oxidase, catalyzes the cross-linking of collagen and elastin fibrils in ECM, thus promoting collagen deposition. Increased expression of LOX is associated with fibrosis and cardiac dysfunction [38–40] and with enhanced oxidative stress and vascular stiffness and hypertension [41]. LOX expression is transcriptionally induced by c-Jun. The splicing factor SF2 is implicated in the generation of the extra type III domain A (EDA) fibronectin isoform (EDA-FN) [42], which is crucial for fibroblast activation and development of fibrosis [43]. Phosphorylation and activation of the SF2 protein, by kinases like Akt, results in enhanced alternative splicing of the EDA-FN exon and increased FN production [44], leading to enhanced myofibroblast differentiation and enhanced ECM production. Interestingly, EDA-FN-deficient mice exhibited less fibrosis and better heart function after myocardial infarction, highlighting the critical role of EDA as a mediator of cardiac remodeling [45]. Given the direct role of CCN2, LOX, SF2 and FN in cardiac fibrosis, their increased expression in our study further confirms this function and provides evidence that resistin implicates these matricellular proteins in its activation of fibroblasts.
Resistin triggered phosphorylation of sTAT3 is of particular interest. STAT3 is activated by phosphorylation at Tyr-705 in the STAT3 transactivation domain by kinases such as JAK2. Phosphorylation of STAT3 allows it to dimerize and translocate to the nucleus and modulates the transcription of target genes, such as Mmp9, Vimentin, Tgfβ1, Ctgf/Ccn2, Fn and Col1a1 [46–48]. Of particular interest, STAT3 has been linked to myocardial tissue responses. Recent studies have demonstrated that STAT3 activation regulates Ang II-induced cardiac remodeling and is a negative regulator of ventricular hypertrophy and fibrosis [49–51]. Ang II stimulates the direct binding of STAT3 with toll-like receptor 4 (TLR4), resulting in STAT3 activation. Blockade of either TLR4 or STAT3 prevented Ang II-induced cardiac remodeling and preserved normal cardiac functions [51]. It is, therefore, conceivable to speculate that STAT3 is a crucial mediator of the profibrotic effects of resistin and therefore inhibition of STAT3, via inhibition of JAK2, mitigates resistin-driven myofibroblast differentiation.
TLR4 is a pattern recognition receptor in innate immunity. Besides its implication in Ang II-induced cardiac remodeling [51], it has been reported to be an essential player in the phenotypic transformation of myofibroblasts as well [52]. Intriguingly, resistin has been reported to bind to TLR4 [53] and to employ TLR4 in the setting of diseases such as insulin resistance [54, 55], cancer [56], hypertension and abdominal aortic aneurysm in mice [10, 57, 58]. These studies raise the intriguing possibility that resistin may also engage TLR4 to activate JAK2 and STAT3 in cardiac tissue to regulate the conversion of fibroblasts to myofibroblasts (Fig. 8C). Furthermore, given the reports that STAT3 directly binds to TLR4 [51], it is conceivable to suggest that resistin may bind to TLR4 and directly activates STAT3 leading to its phosphorylation and subsequent activation of fibroblasts.
Figure -8: Resistin knockout mice on high-fat diet exhibit lower activation of JAK2 and JNK signaling pathways –

Mice were treated as in Figure 7. A) Protein samples were extracted from heart muscles and analyzed for the total and phosphorylated forms of JAK2, STAT3, JNK, and c-Jun by western blotting. B) Quantification of band densities in (A). β-actin was used as internal control. The data are mean ± SEM of n=5. p*< 0.05; **p < 0.01. C) Schematic diagram summarizing the role of JAK2 and JNK signaling pathways in resistin-driven regulation of fibroblast-myofibroblast conversion. Resistin potentially binds to TLR4 and activates JAK2 which in turn phosphorylates STAT3 causing it to translocate into the nucleus and activate pro-fibrotic target genes (i.e. Mmp9, Ccn2, Col1a1, vimentin and Tgfβ1). Resistin also activates JNK, potentially through ASK as we demonstrated previously [26], phosphorylating c-Jun causing it to translocate into the nucleus and activate pro-fibrotic target genes (i.e. Ccn2, Col1a1, Fn, Lox and Tgfβ1). TGFβ1 binds to TGFβ1R and activates the profibrotic genes by phosphorylating its downstream targets like Smad3, and JNK.
CONCLUSION
Here we describe a novel association between resistin and cardiac myofibroblast phenotypic differentiation. Resistin engages the JAK2/STAT3 and JNK-cJun pathways leading to enhanced expression of fibrotic genes, as well as increased matricellular proteins and stress fiber organization. Resistin deletion or blockage of JAK and JNK prevented resistin-induced activation of cardiac fibroblasts. Based on these findings, we propose inhibition of resistin as a new potential therapy for cardiac fibrosis especially in cases where aberrant resistin signaling leads to chronic fibrosis, such as obesity and diabetes.
Supplementary Material
Highlights.
Myocardial fibrosis is characterized by excessive ECM deposition and myofibroblast differentiation
Resistin stimulates fibroblast-to-myofibroblast conversion and pro-fibrotic genes
Resistin promotes myofibroblast transdifferentiation via JAK2/STAT3 and JNK/c-Jun
Resistin-null mice showed decreased tissue fibrosis and improved cardiac function
Modulation of resistin may represent a new therapeutic target in myocardial fibrosis
Abbreviations
- Retn
Resistin
- TGFβ 1
transforming growth factor beta 1
- MMP
matrix metalloproteinase
- ECM
extracellular matrix
- HFD
high-fat diet
- NCD
normal chow diet
- AAV9
adeno-associated virus, serotype 9
- FS
fractional shortening
- LVIDs
left ventricular internal dimension at end-systole
- LVIDd
left ventricular internal dimension end-diastole
Footnotes
Conflict of interest:
none declared
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Rockey DC, Bell PD, Hill JA, Fibrosis - A Common Pathway to Organ Injury and Failure Reply, New Engl J Med 373(1) (2015) 96–96. [DOI] [PubMed] [Google Scholar]
- [2].Kong P, Christia P, Frangogiannis NG, The pathogenesis of cardiac fibrosis, Cell Mol Life Sci 71(4) (2014) 549–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC, Cardiac Fibrosis The Fibroblast Awakens, Circ Res 118(6) (2016) 1021–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].van den Borne SWM, Diez J, Blankesteijn WM, Verjans J, Hofstra L, Narula J, Myocardial remodeling after infarction: the role of myofibroblasts, Nat Rev Cardiol 7(1) (2010) 30–37. [DOI] [PubMed] [Google Scholar]
- [5].Bhandary B, Meng Q, James J, Osinska H, Gulick J, Valiente-Alandi I, Sargent MA, Bhuiyan MS, Blaxall BC, Molkentin JD, Robbins J, Cardiac Fibrosis in Proteotoxic Cardiac Disease is Dependent Upon Myofibroblast TGF -beta Signaling, Journal of the American Heart Association 7(20) (2018) e010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, Vagnozzi RJ, Liu R, Huynh T, Lee SJ, Karch J, Molkentin JD, Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis, J Clin Invest 127(10) (2017) 3770–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Molkentin JD, Bugg D, Ghearing N, Dorn LE, Kim P, Sargent MA, Gunaje J, Otsu K, Davis J, Fibroblast-Specific Genetic Manipulation of p38 Mitogen-Activated Protein Kinase In Vivo Reveals Its Central Regulatory Role in Fibrosis, Circulation 136(6) (2017) 549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Valiente-Alandi I, Potter SJ, Salvador AM, Schafer AE, Schips T, Carrillo-Salinas F, Gibson AM, Nieman ML, Perkins C, Sargent MA, Huo J, Lorenz JN, DeFalco T, Molkentin JD, Alcaide P, Blaxall BC, Inhibiting Fibronectin Attenuates Fibrosis and Improves Cardiac Function in a Model of Heart Failure, Circulation 138(12) (2018) 1236–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gourdie RG, Dimmeler S, Kohl P, Novel therapeutic strategies targeting fibroblasts and fibrosis in heart disease, Nat Rev Drug Discov 15(9) (2016) 620–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Qin Z, Bagley J, Sukhova G, Baur WE, Park HJ, Beasley D, Libby P, Zhang Y, Galper JB, Angiotensin II-induced TLR4 mediated abdominal aortic aneurysm in apolipoprotein E knockout mice is dependent on STAT3, Journal of molecular and cellular cardiology 87 (2015) 160–70. [DOI] [PubMed] [Google Scholar]
- [11].Cowling RT, Kupsky D, Kahn AM, Daniels LB, Greenberg BH, Mechanisms of cardiac collagen deposition in experimental models and human disease, Transl Res 209 (2019) 138–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kong P, Christia P, Frangogiannis NG, The pathogenesis of cardiac fibrosis, Cell Mol Life Sci 71(4) (2014) 549–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC, Cardiac Fibrosis: The Fibroblast Awakens, Circ Res 118(6) (2016) 1021–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Qi D, Hu X, Wu X, Merk M, Leng L, Bucala R, Young LH, Cardiac macrophage migration inhibitory factor inhibits JNK pathway activation and injury during ischemia/reperfusion, J Clin Invest 119(12) (2009) 3807–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bogoyevitch MA, GillespieBrown J, Ketterman AJ, Fuller SJ, BenLevy R, Ashworth A, Marshall CJ, Sugden PH, Stimulation of the stress-activated mitogen-activated protein kinase subfamilies in perfused heart - p38/RK mitogen-activated protein kinases and c-Jun N-terminal kinases are activated by ischemia/reperfusion, Circ Res 79(2) (1996) 162–173. [DOI] [PubMed] [Google Scholar]
- [16].Clerk A, Fuller SJ, Michael A, Sugden PH, Stimulation of “Stress-regulated” mitogen-activated protein kinases (stress-activated protein kinases c-Jun N-terminal kinases and p38-mitogen-activated protein kinases) in perfused rat hearts by oxidative and other stresses, J Biol Chem 273(13) (1998) 7228–7234. [DOI] [PubMed] [Google Scholar]
- [17].Sadoshima J, Montagne O, Wang Q, Yang GP, Warden J, Liu J, Takagi G, Karoor V, Hong C, Johnson GL, Vatner DE, Vatner SF, The MEKK1-JNK pathway plays a protective role in pressure overload but does not mediate cardiac hypertrophy, J Clin Invest 110(2) (2002) 271–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Nemoto S, Sheng ZL, Lin AN, Opposing effects of Jun kinase and p38 mitogen-activated protein kinases on cardiomyocyte hypertrophy, Mol Cell Biol 18(6) (1998) 3518–3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hreniuk D, Garay M, Gaarde W, Monia BP, McKay RA, Cioffi CL, Inhibition of c-Jun N-terminal kinase 1, but not c-Jun N-terminal kinase 2, suppresses apoptosis induced by ischemia/reoxygenation in rat cardiac myocytes, Mol Pharmacol 59(4) (2001) 867–74. [DOI] [PubMed] [Google Scholar]
- [20].Lazar MA, Rasistion- and obasky-associated metabolic diseases, Horm Metab Res 39(10) (2007) 710–716. [DOI] [PubMed] [Google Scholar]
- [21].Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA, The hormone resistin links obesity to diabetes, Nature 409(6818) (2001) 307–312. [DOI] [PubMed] [Google Scholar]
- [22].Muse ED, Feldman DI, Blaha MJ, Dardari ZA, Blumenthal RS, Budoff MJ, Nasir K, Criqui MH, Cushman M, McClelland RL, Allison MA, The association of resistin with cardiovascular disease in the Multi-Ethnic Study of Atherosclerosis, Atherosclerosis 239(1) (2015) 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Burnett MS, Devaney JM, Adenika RJ, Lindsay R, Howard BV, Cross-sectional associations of resistin, coronary heart disease, and insulin resistance, J Clin Endocr Metab 91(1) (2006) 64–68. [DOI] [PubMed] [Google Scholar]
- [24].Zhang YX, Li YX, Yu L, Zhou L, Association between serum resistin concentration and hypertension: A systematic review and meta-analysis, Oncotarget 8(25) (2017) 41529–41537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Frankel DS, Vasan RS, D’Agostino RB, Benjamin EJ, Levy D, Wang TJ, Meigs JB, Resistin, Adiponectin, and Risk of Heart Failure The Framingham Offspring Study, J Am Coll Cardiol 53(9) (2009) 754–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kang S, Chemaly ER, Hajjar RJ, Lebeche D, Resistin Promotes Cardiac Hypertrophy via the AMP-activated Protein Kinase/Mammalian Target of Rapamycin (AMPK/mTOR) and c-Jun N-terminal Kinase/Insulin Receptor Substrate 1 (JNK/IRS1) Pathways, J Biol Chem 286(21) (2011) 18465–18473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chemaly ER, Hadri L, Zhang S, Kim M, Kohlbrenner E, Sheng J, Liang L, Chen J, Purushothaman KR, Hajjar RJ, Lebeche D, Long-term in vivo resistin overexpression induces myocardial dysfunction and remodeling in rats, Journal of molecular and cellular cardiology 51(2) (2011) 144–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW, SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase, Proceedings of the National Academy of Sciences 98(24) (2001) 13681–13686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Karakikes I, Chaanine AH, Kang S, Mukete BN, Jeong D, Zhang S, Hajjar RJ, Lebeche D, Therapeutic cardiac-targeted delivery of miR-1 reverses pressure overload-induced cardiac hypertrophy and attenuates pathological remodeling, Journal of the American Heart Association 2(2) (2013) e000078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Abe H, Takeda N, Isagawa T, Semba H, Nishimura S, Morioka MS, Nakagama Y, Sato T, Soma K, Koyama K, Wake M, Katoh M, Asagiri M, Neugent ML, Kim JW, Stockmann C, Yonezawa T, Inuzuka R, Hirota Y, Maemura K, Yamashita T, Otsu K, Manabe I, Nagai R, Komuro I, Macrophage hypoxia signaling regulates cardiac fibrosis via Oncostatin M, Nat Communications 10(1) (2019) 2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Morine KJ, Qiao X, York S, Natov PS, Paruchuri V, Zhang Y, Aronovitz MJ, Karas RH, Kapur NK, Bone Morphogenetic Protein 9 Reduces Cardiac Fibrosis and Improves Cardiac Function in Heart Failure, Circulation 138(5) (2018) 513–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chemaly ER, Kang S, Zhang S, McCollum L, Chen J, Benard L, Purushothaman KR, Hajjar RJ, Lebeche D, Differential patterns of replacement and reactive fibrosis in pressure and volume overload are related to the propensity for ischaemia and involve resistin, The Journal of physiology 591(21) (2013) 5337–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Creemers EE, Pinto YM, Molecular mechanisms that control interstitial fibrosis in the pressure-overloaded heart, Cardiovascular research 89(2) (2011) 265–72. [DOI] [PubMed] [Google Scholar]
- [34].Dobaczewski M, Bujak M, Li N, Gonzalez-Quesada C, Mendoza LH, Wang XF, Frangogiannis NG, Smad3 Signaling Critically Regulates Fibroblast Phenotype and Function in Healing Myocardial Infarction, Circulation Research 107(3) (2010) 418–U176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Leask A, Parapuram SK, Shi-Wen X, Abraham DJ, Connective tissue growth factor (CTGF, CCN2) gene regulation: a potent clinical bio-marker of fibroproliferative disease?, Journal of cell communication and signaling 3(2) (2009) 89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Tsoutsman T, Wang X, Garchow K, Riser B, Twigg S, Semsarian C, CCN2 plays a key role in extracellular matrix gene expression in severe hypertrophic cardiomyopathy and heart failure, J Molecular and Cellular Cardiology 62 (2013) 164–78. [DOI] [PubMed] [Google Scholar]
- [37].Yoon PO, Lee MA, Cha H, Jeong MH, Kim J, Jang SP, Choi BY, Jeong D, Yang DK, Hajjar RJ, Park WJ, The opposing effects of CCN2 and CCN5 on the development of cardiac hypertrophy and fibrosis, J Molecular and Cellular Cardiology 49(2) (2010) 294–303. [DOI] [PubMed] [Google Scholar]
- [38].Al-U’datt D, Allen BG, Nattel S, Role of the lysyl oxidase enzyme family in cardiac function and disease, Cardiovasc Res 115(13) (2019) 1820–1837. [DOI] [PubMed] [Google Scholar]
- [39].Galán M, Varona S, Guadall A, Orriols M, Navas M, Aguiló S, de Diego A, Navarro MA, García-Dorado D, Rodríguez-Sinovas A, Martínez-González J, Rodriguez C, Lysyl oxidase overexpression accelerates cardiac remodeling and aggravates angiotensin II-induced hypertrophy, FASEB journal 31(9) (2017) 3787–3799. [DOI] [PubMed] [Google Scholar]
- [40].López B, González A, Hermida N, Valencia F, de Teresa E, Diez J, Role of lysyl oxidase in myocardial fibrosis: from basic science to clinical aspects, American journal of physiology. Heart and circulatory physiology 299(1) (2010) H1–9. [DOI] [PubMed] [Google Scholar]
- [41].Martínez-Revelles S, García-Redondo AB, Avendaño MS, Varona S, Palao T, Orriols M, Roque FR, Fortuño A, Touyz RM, Martínez-González J, Salaices M, Rodríguez C, Briones AM, Lysyl Oxidase Induces Vascular Oxidative Stress and Contributes to Arterial Stiffness and Abnormal Elastin Structure in Hypertension: Role of p38MAPK, Antioxidants & redox signaling 27(7) (2017) 379–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].White ES, Baralle FE, Muro AF, New insights into form and function of fibronectin splice variants, The Journal of pathology 216(1) (2008) 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Muro AF, Moretti FA, Moore BB, Yan M, Atrasz RG, Wilke CA, Flaherty KR, Martinez FJ, Tsui JL, Sheppard D, Baralle FE, Toews GB, White ES, An essential role for fibronectin extra type III domain A in pulmonary fibrosis, American journal of respiratory and critical care medicine 177(6) (2008) 638–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].White ES, Sagana RL, Booth AJ, Yan M, Cornett AM, Bloomheart CA, Tsui JL, Wilke CA, Moore BB, Ritzenthaler JD, Roman J, Muro AF, Control of fibroblast fibronectin expression and alternative splicing via the PI3K/Akt/mTOR pathway, Experimental cell research 316(16) (2010) 2644–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Arslan F, Smeets MB, Riem Vis PW, Karper JC, Quax PH, Bongartz LG, Peters JH, Hoefer IE, Doevendans PA, Pasterkamp G, de Kleijn DP, Lack of fibronectin-EDA promotes survival and prevents adverse remodeling and heart function deterioration after myocardial infarction, Circulation Research 108(5) (2011) 582–92. [DOI] [PubMed] [Google Scholar]
- [46].Huang G, Yan H, Ye S, Tong C, Ying QL, STAT3 phosphorylation at tyrosine 705 and serine 727 differentially regulates mouse ESC fates, Stem cells 32(5) (2014) 1149–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Papaioannou I, Xu S, Denton CP, Abraham DJ, Ponticos M, STAT3 controls COL1A2 enhancer activation cooperatively with JunB, regulates type I collagen synthesis posttranscriptionally, and is essential for lung myofibroblast differentiation, Molecular biology of the cell 29(2) (2018) 84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zhang L, Xu X, Yang R, Chen J, Wang S, Yang J, Xiang X, He Z, Zhao Y, Dong Z, Zhang D, Paclitaxel attenuates renal interstitial fibroblast activation and interstitial fibrosis by inhibiting STAT3 signaling, Drug design, development and therapy 9 (2015) 2139–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Skoumal R, Toth M, Serpi R, Rysa J, Leskinen H, Ulvila J, Saiho T, Aro J, Ruskoaho H, Szokodi I, Kerkela R, Parthenolide inhibits STAT3 signaling and attenuates angiotensin Il-induced left ventricular hypertrophy via modulation of fibroblast activity, Journal of Molecular and Cellular Cardiology 50(4) (2011) 634–41. [DOI] [PubMed] [Google Scholar]
- [50].Zouein FA, Zgheib C, Hamza S, Fuseler JW, Hall JE, Soljancic A, Lopez-Ruiz A, Kurdi M, Booz GW, Role of STAT3 in angiotensin Il-induced hypertension and cardiac remodeling revealed by mice lacking STAT3 serine 727 phosphorylation, Hypertension research 36(6) (2013) 496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Han J, Ye S, Zou C, Chen T, Wang J, Li J, Jiang L, Xu J, Huang W, Wang Y, Liang G, Angiotensin II Causes Biphasic STAT3 Activation Through TLR4 to Initiate Cardiac Remodeling, Hypertension 72(6) (2018) 1301–1311. [DOI] [PubMed] [Google Scholar]
- [52].Yang R, Song Z, Wu S, Wei Z, Xu Y, Shen X, Toll-like receptor 4 contributes to a myofibroblast phenotype in cardiac fibroblasts and is associated with autophagy after myocardial infarction in a mouse model, Atherosclerosis 279 (2018) 23–31. [DOI] [PubMed] [Google Scholar]
- [53].Tarkowski A, Bjersing J, Shestakov A, Bokarewa MI, Resistin competes with lipopolysaccharide for binding to toll-like receptor 4, Journal of Cellular and Molecular Medicine 14(6b) (2010) 1419–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Benomar Y, Amine H, Crepin D, Al Rifai S, Riffault L, Gertler A, Taouis M, Central Resistin/TLR4 Impairs Adiponectin Signaling, Contributing to Insulin and FGF21 Resistance, Diabetes 65(4) (2016) 913–26. [DOI] [PubMed] [Google Scholar]
- [55].Benomar Y, Gertler A, De Lacy P, Crepin D, Ould Hamouda H, Riffault L, Taouis M, Central resistin overexposure induces insulin resistance through Toll-like receptor 4, Diabetes 62(1) (2013) 102–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zhang M, Yan L, Wang GJ, Jin R, Resistin effects on pancreatic cancer progression and chemoresistance are mediated through its receptors CAP1 and TLR4, Journal of Cellular Physiology 234(6) (2019) 9457–9466. [DOI] [PubMed] [Google Scholar]
- [57].Jiang Y, Lu L, Hu Y, Li Q, An C, Yu X, Shu L, Chen A, Niu C, Zhou L, Yang Z, Resistin Induces Hypertension and Insulin Resistance in Mice via a TLR4-Dependent Pathway, Scientific reports 6 (2016) 22193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Lai CH, Wang KC, Lee FT, Tsai HW, Ma CY, Cheng TL, Chang BI, Yang YJ, Shi GY, Wu HL, Toll-Like Receptor 4 Is Essential in the Development of Abdominal Aortic Aneurysm, PloS one 11(1) (2016) e0146565. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.