

Abstract
The immune system of the central nervous system (CNS) consists primarily of innate immune cells. These are highly specialized macrophages found either in the parenchyma, called microglia, or at the CNS interfaces, such as leptomeningeal, perivascular, and choroid plexus macrophages. While they were primarily thought of as phagocytes, their function extends well beyond simple removal of cell debris during development and diseases. Brain-resident innate immune cells were found to be plastic, long-lived, and host to an outstanding number of risk genes for multiple pathologies. As a result, they are now considered the most suitable targets for modulating CNS diseases. Additionally, recent single-cell technologies enhanced our molecular understanding of their origins, fates, interactomes, and functional cell states during health and perturbation. Here, we review the current state of our understanding and challenges of the myeloid cell biology in the CNS and treatment options for related diseases.
Keywords: microglia; macrophages; development; single-cell profiling; fate mapping, multiple sclerosis
CNS MACROPHAGES AS TISSUE-RESIDENT IMMUNE CELLS
The highly specialized central nervous system (CNS), comprising the brain and the spinal cord, consists of not only billions of neuroectodermal cells such as neurons, astrocytes, and oligodendrocytes but also resident immune cells, which account for 10% of all CNS cells. They play a pivotal role in the maintenance of tissue homeostasis and prevent overshooting immune reactions during development, health, and disease. Tissue-resident macrophages are ideally positioned to act as tissue watchdogs, with their relative longevity, remarkable motility, defined anatomical location, and plethora of immune sensors. Consequently, virtually all organs are endowed with this amazing innate immune cell type, with Kupffer cells in the liver, Langerhans cells in the skin, alveolar macrophages in the lung, osteoclasts in bones, and so forth (1,2).
Tissue macrophages in the CNS come in two flavors: Microglia are located in the parenchyma (their name pointing to their small cell body of around 7–10 μm), and CNS-associated macrophages (CAMs) are found in CNS interfaces including the meninges, perivascular space, and choroid plexus (Figure 1). CAMs are also known as border-associated macrophages (BAMs) and encompass perivascular macrophages (PVM), subdural leptomeningeal macrophages (MMs), and choroid plexus macrophages (3–7). PVMs have a clearly distinct location, in contrast to perivascular microglia, which are captured between two basal laminae: one from the endothelial cells and the other from the glia limitans (8). Additional neural-associated macrophages can be found in the eye as retinal, corneal, and ciliary body macrophages, and in the peripheral nervous system as endoneurial and epineurial macrophages (Figure 1). While microglia are the only immune cells located in the CNS parenchyma in close vicinity to neurons, the CAMs and additional immune cells, such as T and B cells, dendritic cells (DCs), monocytes, natural killer (NK) cells, and NKT cells, are found at the CNS borders, such as the meninges (leptomeninges, dura), and in the choroid plexus (8).
Figure 1.
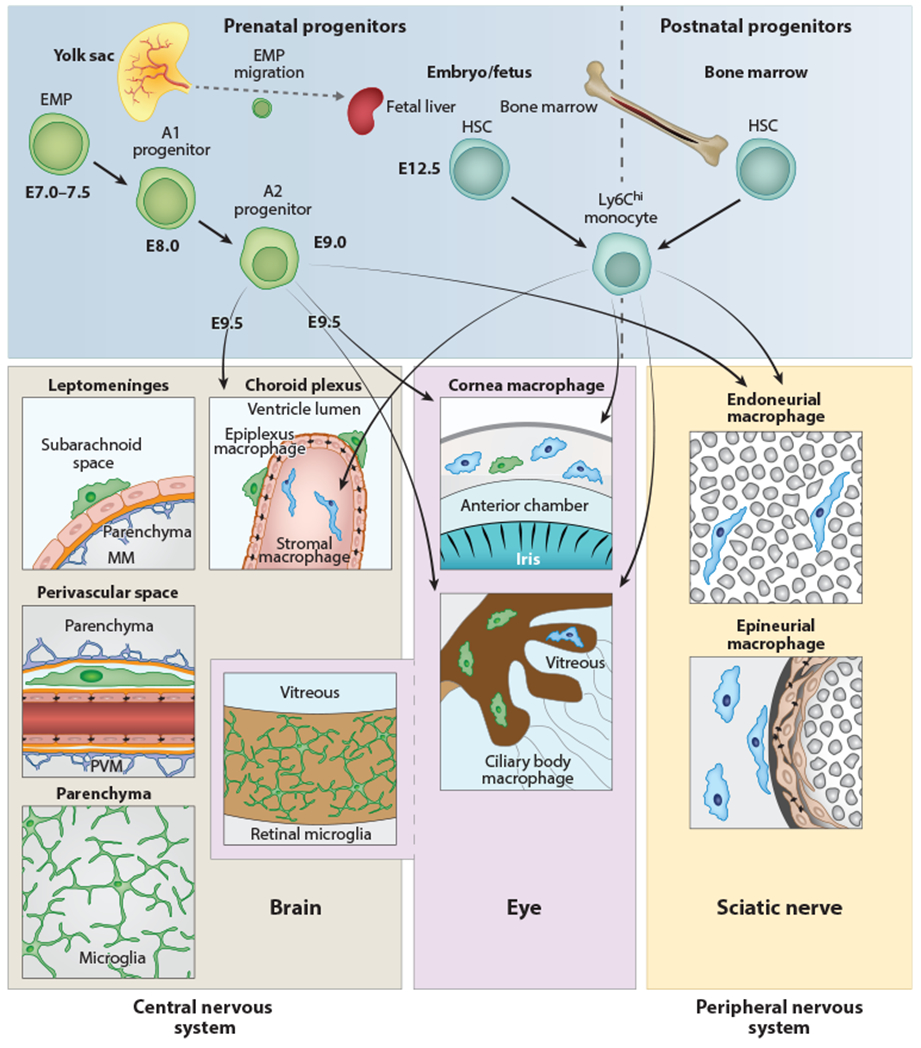
Anatomy and origin of microglia and CNS-associated macrophages. Cellular sources and anatomical location of myeloid cells found in the CNS, peripheral nervous system, or eye in the mouse. Microglial cells, PVMs, MMs, and retinal microglia are exclusively derived from prenatal sources and have no exchange with HSC-derived monocytes during postnatal stages. In contrast, choroid plexus stromal macrophages, ciliary body macrophages, corneal macrophages in the eye, endoneurial macrophages, and epineurial macrophages in the sciatic nerve have a mixed origin, from prenatal (yolk sac and fetal liver) and postnatal (bone marrow) sources. A transient early wave of myeloid cell development called primitive hematopoiesis takes place at E7.0–E7.5 (upper left). At this time, c-Kit+ EMP cells develop in the blood islands of the yolk sac. Their progeny (myeloid precursor cells) mature, expand, and migrate. CX3CR1+ A2 myeloid progenitor cells are derived from c-Kit+ CX3CR1+ A1 progenitors and seed the brain at E9.5, where they differentiate into microglial cells, PVMs, ATMs, and retinal microglia, respectively. During further development, some EMPs travel to the fetal liver (which is part of the definitive hematopoiesis), where they differentiate into monocytes. Myelopoiesis is thought to be restricted to bone marrow starting from birth (right). Abbreviations: CNS, central nervous system; EMP, erythromyeloid progenitor; HSC, hematopoietic stem cell; ATM, meningeal macrophage; PVM, perivascular macrophage.
Microglial cells are evolutionarily conserved, with a broad presence across clades. Recently, microglia were morphologically characterized and genetically profiled at the bulk and single-cell levels in numerous species covering 450 million years of evolution, ranging from leeches, axolotls, and whales to several primates, including humans (9). The basic transcriptional microglial profile was astonishingly conserved, suggesting comparable functions of microglial cells across species. Moreover, microglial density was comparable in over 33 vertebrate species (10).
To date, little information is available about the presence of CAMs across species (4). However, CAMs have been documented in various vertebrates such as the Greenland shark (11), cats (12), macaques (13), mice (14), and humans. Further studies are needed to elucidate the evolutionary origins and fates of these specialized macrophages at the CNS borders.
ONTOGENY AND FATE OF CNS MACROPHAGES
As tissue macrophages, microglia, and CAMs belong to the mononuclear phagocytic system. Tissue macrophages were previously thought to rely on circulating monocytes for their origin, replenishment, and maintenance (15). This hypothesis was largely based on the expression of immune markers shared between macrophages and monocytes, including CD45, CD11b, and CX3CR1, and the replacement of tissue macrophages and microglia by bone marrow reconstitutions following CNS irradiation in mice and humans (16–19). However, accumulating evidence has clarified that brain irradiation induces an artificial, transient impairment of the blood-brain barrier and evokes a cytokine storm, which together allow the nonphysiological engraftment of myeloid cells into the CNS (20, 21). To circumvent irradiation-induced artifacts in the CNS, scientists can also use an alternative approach called parabiosis. During parabiosis, the mice are joined so as to share circulation, allowing scientists to follow the fate of, e.g., green fluorescent protein (GFP)-labeled hematopoietic cells derived from one parabiont and transferred into a connected nontransgenic mouse. One drawback of parabiosis is the obvious limited rate of blood chimerism, which naturally is about 5 0%. An alternative hypothesis supporting the notion that circulating myeloid cells replace microglia in the adult mouse brain was recently put forward based on studies in which microglia were genetically depleted (22–24) or functionally impaired (25). Interestingly, newly engrafted blood-or bone marrow-derived microglia considerably differ from CNS-derived microglia at transcriptional and functional levels (22–24), suggesting that differences in ontogeny have long-term effects on their functional responses.
Nowadays, however, tissue macrophages in the mouse body are thought to be physiologically derived from distinct developmental pathways that differentially contribute to specific embryo and adult tissue compartments (2). In fact, most adult tissues in the mouse contain (to different extents) macrophages from two sources: long-lived macrophages from prenatal sources (presumably the yolk sac and the fetal liver) and short-lived macrophages from postnatal sources such as the bone marrow (26–32) (Figure 1).
Similar to other embryonic tissue macrophages in mice, microglia and potentially CAMs develop from c-Kit+ noncommitted erythromyeloid progenitors (EMPs) that originate around embryonic day 7.25 (E7.25) in the yolk sac (33). The idea of microglia establishment during embryogenesis has been further supported by studies in chicken and zebrafish (32, 34–36). The generation of microglia as well as CAMs through EMPs is independent of the transcription f actor c-Myb (14, 32). c-Kit+ CX3CR1− EMPs give rise, in an IRF-8-dependent manner, to c-Kit− CX3CR1+ maturating macrophages designated A1 and A2, which have been shown to invade the brain anlage at E9.5 via the vasculature without a monocyte intermediate (30, 37) (Figure 1). In contrast, most other tissue macrophages are derived from c-Myb-dependent EMPs generated at later stages, which move to the fetal liver, where they give rise to monocyte intermediates (38).
As for the prenatal origin of CAMs, a significant ontogenetic overlap with microglia in the mouse brain was recently hypothesized. CAMs have now been characterized as c-Myb-independent, monocyte (CCR2)-independent lineage cells that derive from prenatal sources (14), as confirmed in several studies (39, 40). The only exceptions to this rule are choroid plexus macrophages, which in contrast to PVMs and MMs are partially replenished and maintained by monocytes, most likely due to the presence of fenestrated capillaries in the choroid plexus (14). Distinct yolk sac progenitors have recently been proposed for microglia and CAMs, suggesting that their cell fate is determined prior to their invasion, infiltration, or seeding into the developing brain (41). Similar prenatal origins of tissue macrophages have been found in other parts of the CNS and head, such as the retina (4, 42, 43), with slight modifications in the ciliary body, in the cornea, and in peripheral nerves (nervus ischiadicus) (44) (Figure 1).
How microglia and CAMs migrate to the brain during development and which signals guide them to their predefined final destination are largely unknown. Chemokine-derived signals such as the CXCL2/CXCR4 axis (4) seem to play an important role together with extracellular matrix proteins modulated by matrix metalloproteinases, which appear necessary for microglial migration throughout the developing CNS (30). The niche hypothesis postulates that motile macrophages occupy free spatial territories during development and disease only until this compartment is filled (45). However, the start and stop signals regulating replenishment of the microglia niche are poorly understood.
Knowledge about the precise myeloid cell origin and its location is a precondition for proper distinction of myeloid cell members in the body: Monocytes are generally circulating cells derived from hematopoietic stem cells exclusively originated from the bone marrow at postnatal stages, whereas tissue macrophages (with the exception of microglia, MMs, and PVMs) are usually tissue associated and derived from two hematopoietic origins, prenatal sources such as yolk sac (or fetal liver) and, later, bone marrow cells. Thus, for accurate classification of myeloid cells, origin and place need to be considered.
In summary, microglia, CAMs, and presumably retinal microglia originate solely from prenatal EMPs, exist after birth independently from the circulation, and display long life spans of several months to years in mice (14) and humans (46). Their numbers are maintained by slow homeostatic proliferation of preexisting mature microglia in situ in a random clonal fashion, without any evidence of a noncommitted microglial precursor in the adult mouse brain (47–49).
FUNCTIONS OF MICROGLIA AND CAMs DURING DEVELOPMENT AND HOMEOSTASIS
As described above, CX3CR1+ F4/80+ yolk sac–derived A2 macrophages infiltrate the developing mouse brain at E9.5 through the pial surface and migrate along the abluminal surface of penetrating vessels to become microglia, as CNS vascularization starts. Microglia are therefore perfectly positioned to influence vasculogenesis, including vascular sprouting and the building of vessel anastomoses (Figure 2). Indeed, studies of angiogenesis revealed roles for juxtavascular microglia in vessel formation and the establishment of anastomoses in the retina (50). Similarly, mice lacking microglia due to deletion of the transcription factor PU.1 displayed an aberrant development of vasculature in the developing midbrain (51).
Figure 2.
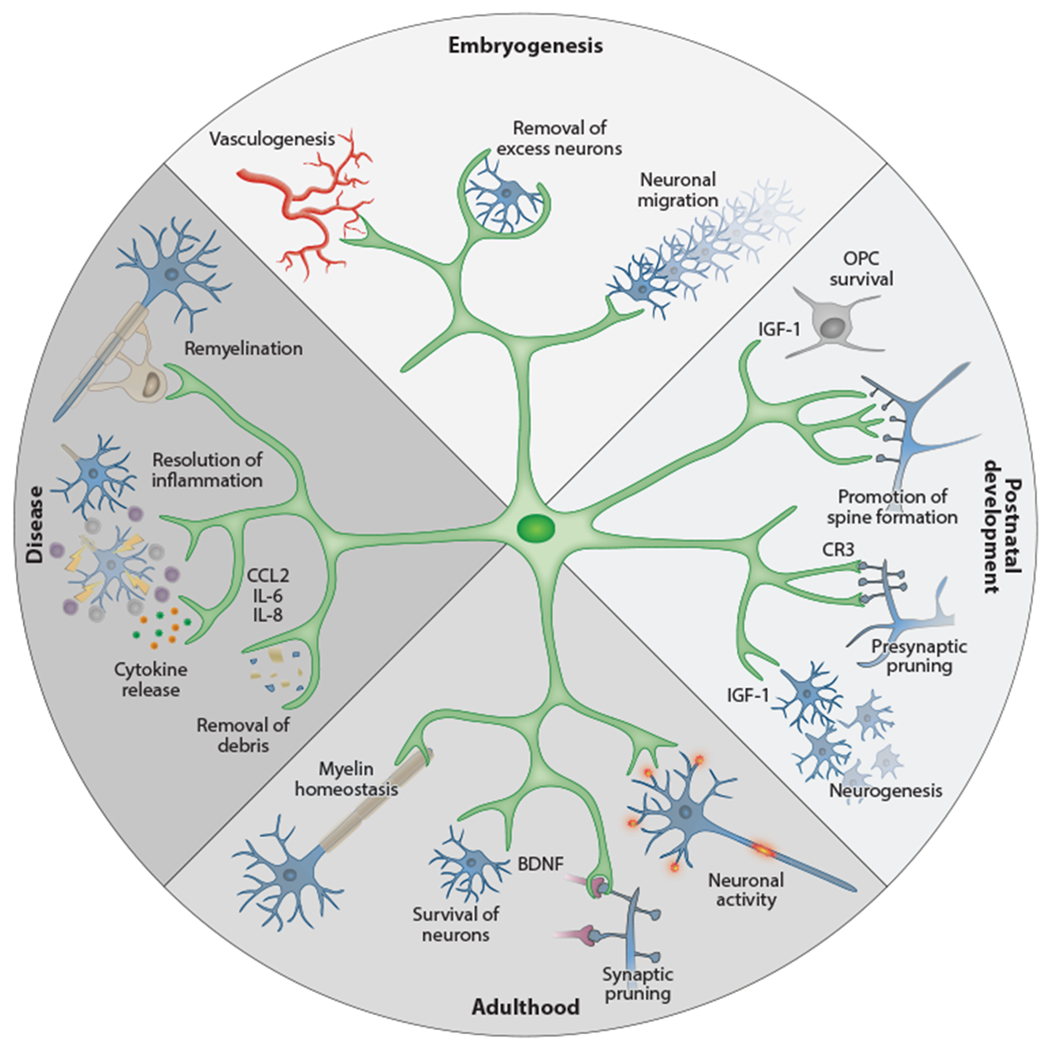
Physiological functions of microglia during development and adulthood. Microglial cells show a remarkable diversity of functions at different stages of ontogeny. During embryogenesis they are involved in promoting vessel development and in the removal of superfluous neurons, and they guide neuronal migration. At postnatal time points, however, they support OPC survival and development (e.g., by IGF-1 production), support neurogenesis in defined brain areas, and promote neuronal spine formation. After finalization of myelinization and establishment of neuronal circuits, their functions shift to ensure the survival of neurons (e.g., by production of BDNF) and oligodendrocytes. Upon perturbation, microglia transform their homeostatic program to an activated state that involves enhanced phagocytosis and production of soluble factors such as cytokines, chemokines, and surface markers. These mechanisms allow the removal of cellular debris and promote regeneration (e.g., remyelination). Abbreviation: OPC, oligodendrocyte precursor cell.
Microglia in the embryonic and early postnatal CNS differ from adult microglia. Indeed, they are characterized by a star-like morphology with numerous highly active processes. Instead, early microglia display a round amoeboid morphology reminiscent of the activated adult microglia present in CNS pathologies. Indeed, transcriptional analyses of embryonic and early postnatal microglia detected highly activated phagocytosis and an enhanced proliferating cellular state (29, 30, 52, 53). Most recently, the transcriptional transition of fetal to adult microglia has been shown to be dependent on a CD4+ T cell population traveling transiently to both the mouse and human brain (54).
Microglia engraft to the early developing embryonic brain around the same time neurons develop, and in the absence of other glial cells (astrocytes and oligodendrocytes develop later at perinatal and postnatal time points), microglia are critically involved in shaping neurogenesis and the resulting neuronal networks. Initial work in zebrafish revealed the capacity of microglia to engulf and dispose of developing neurons (55). Further detailed analyses demonstrated that microglia control the number of neuronal progenitor cells (NPCs) around E20 by active apoptosis and removal of dying, apoptotic NPCs or excess NPCs (56, 57). At later stages of embryogenesis, microglia reach deeper cortical layers and associate with distinct axonal tracts, including the dopaminergic midbrain neurons and the corpus callosum. They populate specific niches that contain neurogenic progenitors such as the subependymal region (6). Microglia can also modulate the migration of inhibitory cortical interneurons, which are essential players in the inhibition-excitation equilibrium of neurons, which is involved in autism spectrum disorders and schizophrenia (58).
The modulation of neuronal progenitors by microglial phagocytosis and induction of neurogenesis is also detected in developmental processes at postnatal stages. For example, microglia shape adolescent neuronal circuits by clearing apoptotic neurons by phagocytosis in the neurogenic area of the murine hippocampus (59). Microglial production of insulin-like growth factor-1 (IGF-1) during the first postnatal week provides a survival signal for layer V neurons in the somatosensory cortex (60). Accordingly fewer pyramidal cells are detected in Cx3cr1 null mice, which harbor reduced numbers of microglia (60).
Around this specific postnatal time window, microglia start to interact at axonal tracts with another cellular partner, oligodendrocyte progenitor cells (OPCs), the source of nascent oligodendrocytes. Amoeboid CD11c+ microglia in subcortical regions of mice were found in close proximity to OPCs, where they promoted their development directly by physical contact or through the production of IGF-1 (61,62). Similarly, white matter microglia regulate the number of mature oligodendrocytes and the homeostasis of myelin sheaths after developmental myelination concludes (61), partially by modulating myelin turnover through Rab7-mediated microglia phagocytosis (63). Interestingly, however, in a recent microglial depletion study in zebrafish microglia phagocytosed myelin sheaths during development, and unexpectedly, microglia depletion resulted in excessive oligodendrocyte production of ectopic myelin (64). These opposing results suggest multiple microglial activities in myelin homeostasis remain to be investigated. Consequently, any in vivo genetic [Csfr1−/− (33), Csf1rΔFIRE/ΔFIRE (65), etc.] or pharmacological approaches (PLX, BLZ compounds, etc.) to delete myeloid cells, including microglia as well as other macrophages and eventually monocytes, will impair normal CNS homeostasis, including myelin maintenance.
Even under homeostatic conditions, microglia continuously survey their environment, including constant surveillance of synaptic processes (66). Along these lines, Cx3cr1-deficient animals, which show reduced microglial cell numbers at postnatal day P15, display transient defects in synaptic activity (67). Moreover, microglia have been shown to contribute to activity-dependent synapse reorganization in the visual cortex (68) and the developing optical tract (69), with a role in promoting postsynaptic spine formation and presynaptic nibbling, also called trogocytosis (70, 71). Microglial synaptic pruning is closely regulated by astrocyte-and neuron-produced IL-33 (72). In addition, direct physical interactions of micro glial processes with neuronal cell bodies have recently been demonstrated in mouse and human brains, which might be important for tempering neuronal functions (73). Taken together, these studies establish microglia as essential modulators of the cross talk between neurons and glial cells, where they modulate several aspects of early brain wiring, neuronal survival, and homeostasis, in addition to fulfilling their well-known roles in synaptic modulation.
In contrast to the roles of microglia, the functions of CAMs are less understood (4). This lack of knowledge is mostly due to low numbers of CAMs in the CNS compared to parenchymal microglia, which renders them challenging to explore. Moreover, most genetic and pharmacological approaches to target myeloid cells in the brain do not distinguish between microglia and CAMs, and specific tools to manipulate CAMs are still lacking (5). PVMs promote vessel repair after microlesions in zebrafish brains (74). Due to their ability to phagocytose and migrate, CAMs found in the leptomeninges, choroid plexus, and perivascular space are thought to be important effectors and regulators of the immune response at CNS borders. However, further studies are needed to fully understand their functions during development, health, and disease.
MICROGLIA AS INDUCERS OF CNS DISEASES
Microglia are extremely versatile and as such respond rapidly to subtle changes in CNS homeostasis. Due to their ability to respond to a plethora of highly diverse stimuli, they are involved in virtually all CNS disorders, ranging from degenerative and neurodevelopmental diseases to neoplastic and autoimmune neuroinflammatory conditions (75). It can be assumed that microglial responses to various perturbations will evoke multimodal cellular changes similar to the multidimensional responses of macrophages, as described above (76). This spectrum of microglia responses will revise the oversimplified paradigm of Ml and M2 microglia that was derived from early in vitro studies (77) but is not representative of microglia in vivo.
A clear link between microglia dysfunction and CNS illness is gradually emerging. In their most extreme forms, rare monogenetic defects affect microglial cell functionality, which subsequently induces CNS disturbances. In 2011 Rademakers and colleagues (78) described a monogenetic disease affecting microglia as microgliopathy, an autosomal dominant disorder induced by various sequence variations in the gene locus encoding the tyrosine-kinase domain of the cytokine receptor CSF-1R. Patients harboring this allelic variant display typical white matter changes, with varied clinical symptoms ranging from parkinsonism to dementia to depression to seizures. The underlying pathology is characterized by diffuse demyelination, astrocyte activation, and neuronal damage evident by eponymous neurofilament-positive axonal spheroids. Vascular alterations in this pathology might be linked to the impairment of PVMs, which also require functional CSF-1R expression. Recently, homozygous CSF1R sequence variants have been identified in patients that lack microglia (79, 80). In these patients, the brain displayed major morphological changes, such as an absence of or structural alterations in the corpus callosum, pointing to essential functions of microglia for myelination in humans, as previously described in mice (61).
Other examples of microgliopathies are linked to sequence variations in the TYROBP and TREM2 genes that result in a recessive chronic neurodegenerative disease characterized by psychotic symptoms and bone cysts (81). An alternative microgliopathy involves white matter changes, polymicrogyria, intracerebral bleedings, calcifications, and tremendously elevated systemic levels of type I interferon in mouse mutants (82, 83) as well as patients (84) lacking the negative type I interferon receptor regulating protease USP18 in microglia. Furthermore, in an elegant set of experiments, a microglial mosaicism for somatic sequence variations in the RAS-MEK-ERK pathway was achieved by targeting the BRAF V600E variant exclusively in EMPs as microglial precursors, without affecting circulating myeloid cells such as monocytes (85). Mice carrying this microglia-specific variant showed histological signs of microgliosis with typical microglial nodules, demyelination, and in later stages neuronal loss, which led to ataxic and paralytic clinical symptoms.
Genome-wide association studies have predicted a key role of microglia/CAMs in several neuroinflammatory and neurodegenerative diseases in addition to these rare monogenetic disorders. Indeed, dozens of loci affecting microglial phagocytosis, activation, or immunoregulation have been linked to Parkinson disease (e.g., TREM2) (86), Alzheimer disease (e.g., CD33) (87), frontotemporal dementia (e.g., GRN) (88), schizophrenia (e.g., C4) (89), and multiple sclerosis (MS) (e.g., TNFRSF1A, IRF8) (90).
Overall, there is accumulating evidence for a pathophysiological role of microglia, and probably CAMs, in multiple CNS disorders, making them candidate targets for therapeutic intervention.
Myeloid Cells in CNS Inflammation
Microglia, monocytes, DCs, potentially CAMs, and other myeloid cells play integral roles in the initiation and resolution of CNS inflammation, such as in MS and its animal model, experimental autoimmune encephalomyelitis (EAE). Although many cell types are involved in the pathogenesis of MS, the root cause is thought to be self-reactive T cells that home to the CNS and mount an immune response against the myelin sheaths that coat neurons (91) (Figure 3). In this context, myeloid cells are highly dynamic, cross talking with multiple cell lineages including CNS-resident astrocytes, oligodendrocytes, and neurons as well as CNS-recruited peripheral immune cells such as T cells.
Figure 3.
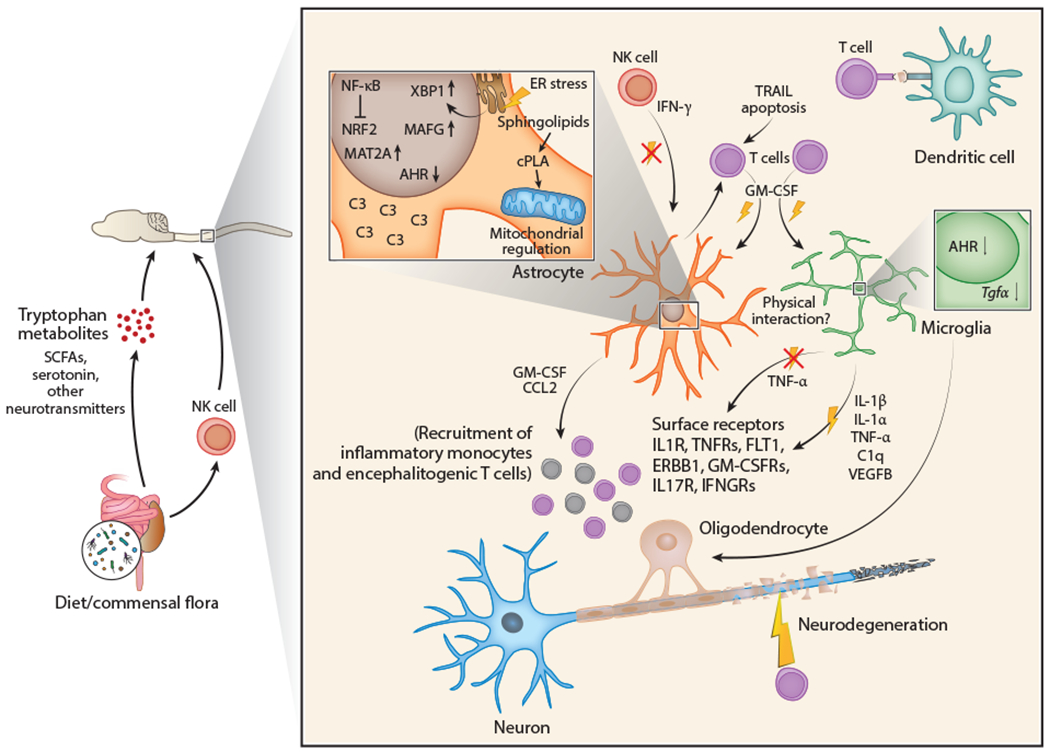
Interactions between CNS-resident cells and the immune system during CNS inflammation. T cell activation is induced by myelin autoantigen presentation via dendritic cells and leads to the production and secretion of proinflammatory cytokines such as GM-CSF and IL-17. These cytokines exert proinflammatory effects on CNS-resident glial cells that respond to them, such as microglia and astrocytes, and promote neurodegeneration. In response to proinflammatory cytokine signals, microglia and astrocytes cross talk through multiple cues. In both astrocytes and microglia, proinflammatory cytokine signaling activates the transcription factor NF-kB and downregulates expression of the transcription factor AHR. NF-kB also inhibits activation of the antioxidant transcription factor NRF2 in astrocytes. Consequently, microglia upregulate and secrete proinflammatory molecules including IL-1β, IL-1α, TNF-α, C1q, and VEGFB while downregulating expression of anti-inflammatory TGF-α. Astrocytes sense these cues through cell surface receptors (IL1R, TNFRs, FLT1, ERBB1, GM-CSFRs, IL-17R, IFNGRs) and induce the expression of pathogenic transcriptional cascades including XBP1 (activated by ER stress), MAFG, and MAT2A alongside other pathogenic molecules like sphingolipid-induced cPLA2/MAVS, and complement component C3. Once activated, pathogenic astrocytes can secrete proinflammatory molecules including GM-CSF and CCL2, which lead to the recruitment of proinflammatory monocytes and encephalitogenic T cells to further promote CNS inflammation. Microglia and astrocytes may also engage in physical interactions during CNS inflammation, yet prominent examples remain to be elucidated. IFN-γ is reported to suppress CNS inflammation in mice and may mediate these functions in astrocytes and microglia through as-yet undetermined mechanisms. Control of microglial and astrocyte activation in the CNS has been linked to molecules derived from the commensal flora such as short-chain fatty acids, tryptophan metabolites, and neurotransmitters. Please note, however, that dietary metabolites such as tryptophan can be produced by the host as well as the commensal flora. Abbreviations: AHR, aryl hydrocarbon receptor; CNS, central nervous system; cPLA2, cytosolic phospholipase A2; C3, complement component 3; ER, endoplasmic reticulum; GM-CSF, granulocyte-macrophage colony-stimulating factor; IFNGR, interferon gamma receptor; NK, natural killer; SCFA, short-chain fatty acid; TNFR, tumor necrosis factor receptor; XBP1, X-box binding protein 1.
Proinflammatory monocytes play an important role in the pathogenesis of EAE and, potentially, MS. Although effector Th1 and Th17 cells are known to contribute to pathogenesis (92,93), several groups have demonstrated the importance of proinflammatory monocytes in EAE. Over a decade ago, classical proinflammatory CCR2+ Ly-6Chi monocytes were shown to be recruited to the CNS to drive the pathology (94). This aspect of the EAE model is consistent with the current pathological criteria for lesion classification in MS (95). In another set of classic and elegant studies using parabiotic mice, it was shown that the number of proinflammatory monocytes directly correlates with EAE disease severity (96). Interestingly, CNS-recruited monocytes do not ultimately contribute to the microglial compartment. Astrocytes and other CNS-resident cells have been identified as the source of CCL2, which is involved in the recruitment of proinflammatory monocytes to the CNS (97). Indeed, the perturbation of pathogenic astrocyte-specific transcriptional programs leads to decreased proinflammatory monocyte recruitment to the inflamed CNS (98, 99).
The advent of high-dimensional single-cell technologies has led to unprecedented access to myeloid cells in the CNS and adjoining areas, as well as in-depth classification of these cells. In a comprehensive set of single-cell mass spectrometry [cytometry by time-of-flight (CyTOF)] and flow cytometry experiments, the CNS myeloid compartment, including the brain parenchyma and meninges, was analyzed in homeostasis and EAE (39). The researchers detected the activation of several conventional pathways in brain-associated myeloid cells during CNS inflammation, including MHC-II, CD44, CD11c, and PDL1 activation.
In EAE, it had been unclear which cells are primarily responsible for presenting antigen to reactivate myelin-reactive T cells. Plausible candidates included CNS-resident microglia, CAMs at CNS interfaces, and DCs. In fact, in some animal models even astrocytes have been reported to present antigen to T cells (100–102). However, two recent studies deployed high-dimensional technologies to investigate the myeloid cells that are the primarily fulfillers of this role (103, 104). In the first study, CyTOF was used to identify MHC-II+ cells in the CNS during EAE and revealed several myeloid populations. Conditional mouse models in which genes responsible for MHC-II function were deleted identified cDC2s as the antigen-presenting cells involved in T cell reactivation in the EAE model, while microglia and other brain-associated myeloid cells were dispensable for this function (104). These data were corroborated by independent, intensive single-cell RNA sequencing (scRNA-seq) investigations of several myeloid compartments in the CNS, including the leptomeninges, perivascular space, CNS parenchyma, and choroid plexus (103). In these studies, the researchers identified and localized several novel subpopulations of myeloid cells regionally and temporally. In particular, they were able to combine several approaches including scRNA-seq, fate mapping, and in vivo imaging to identify cells harboring an MHC-II transcriptional signature, which in combination with transgenic mice reinforced the idea of CD11c+ DCs as important antigen-presenting cells in the EAE model, and not microglia or CAMs.
These data are consistent with classic studies pointing to CD11c+ DCs as mediators of T cell reactivation in the CNS and not CNS-resident macrophages (105). These studies also trigger important questions regarding the therapeutic value of DC-targeting therapies in MS, for example, that are related to approaches aimed at boosting the tolerogenic activity of DCs (106).
Microglia and Their Mechanisms of Cross Talk During CNS Inflammation
Microglia play key roles in the pathogenesis of EAE and MS. As T cells are one of the primary pathogenic cell types in EAE, their cross talk with microglia has been extensively studied. One proinflammatory cytokine secreted by pathogenic T cells is granulocyte-macrophage colony-stimulating factor (GM-CSF), which acts on myeloid cells, including CNS-resident microglia, to exacerbate CNS inflammation (107). Indeed, in vitro studies showed that GM-CSF drives microglial proliferation (108). Moreover, during the analysis of the role of GM-CSF in EAE pathogenesis, it was found that the adoptive transfer of GM-CSF-deficient T cells into wild-type mice failed to induce EAE, as previously reported (109), and also prevented microglial activation (110). Intriguingly, the impaired EAE in mice adoptively transferred with GM-CSF-deficient T cells could be rescued through systemic LPS injection and microglial activation, suggesting that microglial activation by GM-CSF is required for EAE pathogenesis. Importantly, GM-CSF is detected in the cerebrospinal fluid of MS patients (111,112) and is considered a viable therapeutic target in MS (113). Thus, T cell-driven microglial activation may amplify disease-promoting microglial activities in EAE and MS, offering potential targets for therapeutic intervention.
Proinflammatory cytokines derived from effector T cells, and other cells, also control microglia cross talk with oligodendrocytes and control of remyelination. Induction of a proinflammatory microglial state limits the production of activin-A, which is an inducer of remyelination (114). Recent studies focused on the heterogeneity of oligodendrocytes in MS patients and animal models of MS suggest that oligodendrocytes can adopt multiple fates in the context of CNS inflammation (115, 116). These data, combined with recent analyses classifying the regional and functional heterogeneity of microglia in the context of CNS inflammation in humans and mice, will likely fuel the discovery of additional pathways mediating microglia-oligodendrocyte cross talk (103, 117).
T cells, however, are not the only cell types known to activate microglia. Fate-mapping studies have shown that spinal cord astrocytes can produce GM-CSF during EAE (118) and that astrocyte-derived GM-CSF activates microglia (119). Of note, astrocytes can also respond to GM-CSF, which drives astrocyte pathogenic activities and promotes EAE pathogenesis (120), suggesting a feed-forward loop that amplifies CNS pathology. Intriguingly, additional astrocyte-derived factors activate microglia in other contexts, as exemplified by microglial activation by astrocyte-produced IL-33 during neural circuit development and synaptic pruning (72). New technologies such as scRNA-seq (121) and spatial transcriptomics (122–125) in combination with the sophisticated bioinformatic tools now available to probe cell-cell cross talk (126, 127) promise to identify additional mechanisms involved in the control of microglial responses by astrocytes.
Microglia are also important regulators of astrocyte responses. Astrocytes play important roles in neurodegenerative diseases including Alzheimer disease (128), amyotrophic lateral sclerosis (129, 130), Huntington disease (131, 132), and MS (99, 119, 120, 133–136). Early studies by the Barres lab (137) used transgenic mice and microarray-based gene expression to identify transcriptional signatures associated with reactive astrocytes induced in response to LPS injection or a stroke model. Later work using in vitro and in vivo approaches showed that IL-1α, TNF, and C1q secretion by microglia induces a neurotoxic phenotype in astrocytes, characterized by C3 expression (138). Recent studies identified microglia-derived VEGF-B signaling through astrocyte FLT1 as a driver of disease-promoting astrocyte functions that contribute to pathogenesis in EAE and MS (134).
Less is known about the role of microglia in limiting disease-promoting astrocyte responses. RNA-seq and genomic analyses of ligand-receptor pairs led to the identification of microglial TGF-α, signaling via ERBB1, as a negative regulator of astrocyte pathogenic responses in EAE (134). Interestingly, TGF-α production by microglia is driven by the ligand-activated transcription factor aryl hydrocarbon receptor (AHR), reminiscent of previous studies implicating thyroid hormones and other stimuli in controlling TGF-α expression (139, 140). AHR was activated in microglia by metabolites of dietary tryptophan produced by the intestinal commensal flora. Consequently, the depletion of tryptophan from the diet or antibiotic perturbations in the commensal flora were found to modulate AHR signaling in microglia, controlling microglial transcriptional responses and TGF-α-mediated astrocyte modulation. Microbial metabolites of dietary tryptophan also act directly on astrocytes to limit their disease-promoting responses via AHR signaling (135).
While tryptophan is an important dietary metabolite derived from the commensal flora (and also host cells), the gut also produces neurotransmitters and their precursors, such as serotonin (141), that act on microglia to control their responses (142). Similarly, short-chain fatty acids were previously identified as critical regulators of microglial maturation and function (143). Thus, the microbiota produces multiple bioactive molecules that signal to the CNS and modulate microglial responses. Hence, these findings together with a growing body of literature (144–149) highlight the complexity of the gut-brain axis and its control of the activity and cross talk of CNS-resident cells, such as astrocytes and microglia.
Newly Identified Microglia States as Future Therapeutic Targets
An enthralling topic for decades has been the extent of microglial heterogeneity. Because of their wide-ranging functions during development, homeostasis, and perturbation, microglia had been thought to constitute a heterogeneous cell population in the CNS. However, until recently, microglia were simply categorized based on their cellular density, morphology, surface markers, and electrophysiological properties, which hindered comprehensive profiling of microglial diversity (150, 151). This was partially due to the limitations inherent in low-throughput analyses of microglia, such as immunohistochemistry and flow cytometry or in situ hybridization, which were limited to probing a few preselected proteins or RNA species, respectively. Thus, prior to high-throughput techniques that could comprehensively and systematically catalog the properties of individual cells, many questions regarding microglial diversity were left unanswered, such as context-dependent microglial phenotypes in development or pathology.
The advent of game-changing single-cell technologies, such as scRNA-seq and CyTOF, revolutionized the method of assessing cell identity on a single-cell level and grouping cell populations, which allowed discovering novel microglia subsets in animal models and humans, shedding new light on microglial functions during homeostasis and diseases (28, 105, 152). Of note, scRNA-seq allows sequencing of whole transcriptomes without prior knowledge of genes as well as clustering of cells based on their transcriptome profiling. Likewise, CyTOF powered by a synergistic combination of more than 50 distinct metal-bound antibodies enables profiling of the proteomic signature of single cells on a large scale. In addition, one strength of new single-cell analyses is the potential to build a comprehensive molecular landscape of microglia and identify novel markers, pathways, and regulatory factors during development, health, and disease.
In 2017, technical improvements in single-cell analysis allowed Keren-Shaul et al. (153) to identify a novel microglia cluster in a disease context related to Alzheimer disease, so-called disease-associated microglia (DAMs), which exhibited a gene signature represented by high expression of Apoe (encoding apolipoprotein E), Lpl (encoding lipoprotein lipase), Itgax (encoding CD11c), and Cst7 (encoding cystatin F). At almost the same time, another group also described an activated state of microglia with similar transcriptional characteristics of DAMs in animal models of amyotrophic lateral sclerosis and multiple sclerosis (MS), in addition to that for Alzheimer disease; they thus named them neurodegenerative microglia (MGnDs) (154). In the same year, another scRNA-seq study identified several microglia subclasses that appear during Alzheimer disease progression (155).
Are DAMs/MGnDs always identical across distinct disease conditions? The answer seems to be no. Using a mouse model of Alzheimer disease, Mathys and colleagues (155) identified a different activation state of microglia with high expression of interferon-related genes. Furthermore, distinct context-dependent activation states of microglia were clearly confirmed by another study using scRNA-seq in combination with different disease models (117), where microglia related to demyelination or remyelination were shown to be transcriptionally distinct from activated microglia after facial nerve axotomy. These results strongly suggest that microglia undergo changes in gene expression, by virtue of their plasticity, in response to alterations in the surrounding tissue microenvironment (156). More recently, another reactive state of microglia was characterized in the aging brains of mice, in which microglia massively accumulate lipid droplets and are thereby called lipid-droplet-accumulating microglia (LDAMs) (157). These LDAMs present a unique transcriptional profile with defects in phagocytosis and enhanced proinflammatory capacity with massive production of reactive oxide species, suggesting detrimental roles of LDAMs contributing to neurodegenerative diseases. Yet, the LDAMs have not been assessed on a single-cell level, which might provide another intriguing aspect of LDAM subclasses. On the other hand, CyTOF analyses identified a comprehensive atlas of the CNS immune cells, including microglia, on a single-cell level during homeostasis and diseases (39, 158), leading to the identification of microglia states.
Recent scRNA-seq studies also highlighted the complexity of microglia with highly diverse phenotypes during normal development and aging (Figure 4). In particular, the microglia states clearly differ between embryonic, early postnatal, adult, and aged brains (117, 159, 160). For instance, a time-and region-specific appearance of one microglial state that was enriched in Igf1, Spp1, Clec7a, and Lgal3 in the white matter regions was evident at early postnatal stages, when amoeboid microglia have been shown to contribute to proper maintenance and maturation of oligodendrocyte progenitors (61, 62). In addition, microglia in the Clec7a+ state phagocytose newly formed caspase-3+ oligodendrocytes during physiological myelination (160). On the other hand, another microglial state with an immunogenic profile (e.g., high levels of inflammatory or type 1 interferon-responsive genes) that is rarely observed in adolescent and adult brains expands in aged brains (159).
Figure 4.
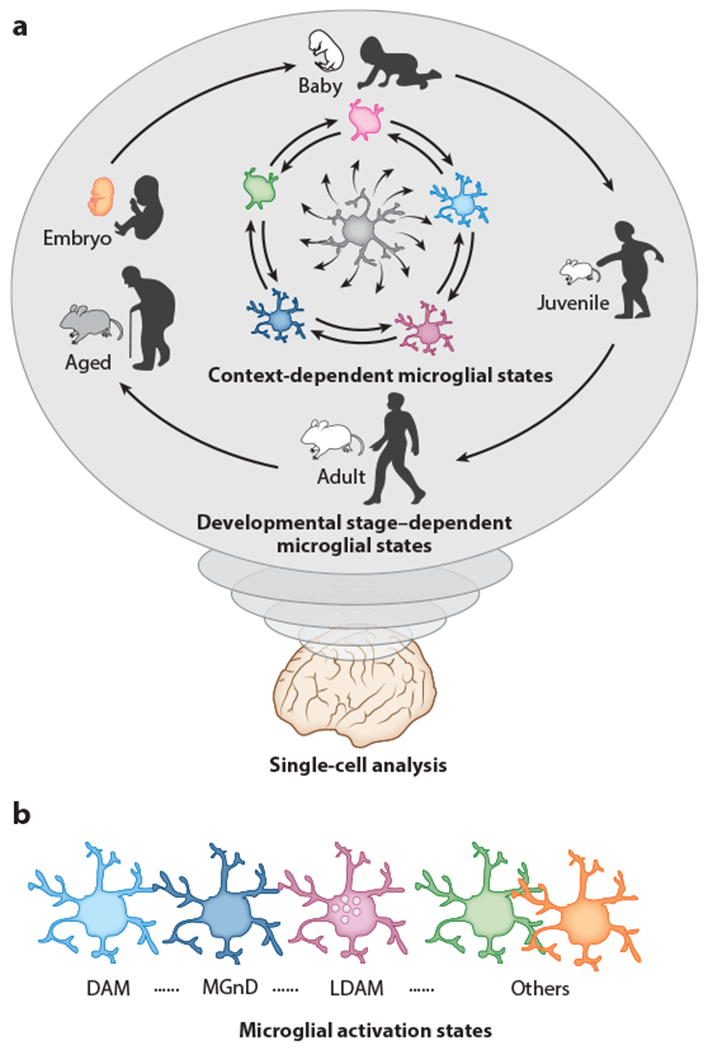
Developmental and context-dependent diverse states of microglia in mice and humans, (a) Different single-cell analyses (scRNA-seq, CyTOF, etc.) revealed that microglia possess highly plastic capacity, thereby altering the molecule (RNA, protein, etc.) expression profile during the course of homeostatic development, aging, or disease progression in a context-dependent manner, (b) Activated microglia states are highly divergent. For instance, scRNA-seq analyses deciphered that DAMs (153) or MGnDs (154) found at sites of neurodegeneration represent transcriptionally distinct profiles with high expression levels of Apoe, Clec7a, Itgax, Cst7, and Spp1 and reduced expression of P2ry12 and Tmem 119. In contrast, LDAMs (157) enriched in the central nervous system of aged mice, in which they accumulate lipid droplets, are defective in phagocytosis with massive production of reactive oxygen species and secretion of proinflammatory cytokines and chemokines such as IL-10, IL-6, CCL3, CCL4, and TNF-α. Abbreviations: CyTOF, cytometry by time-of-flight; DAM, disease-associated microglia; LDAM, lipid-droplet-accumulating microglia; MGnD, neurodegenerative microglia; scRNA-seq, single-cell RNA sequencing.
The spatial diversity of adult microglia under homeostatic conditions was addressed using microarray assays, which detected considerable regional multiplicity of microglia with a biased gene expression pattern in the cerebellum, compared to the cortex, hippocampus, and striatum (152). However, recent scRNA-seq-based clustering failed to separate microglia based on distinct brain regions but instead defined a relatively homogeneous microglia population that revealed a transcriptional continuum between microglia states across the CNS regions with few preexisting states (117, 159, 160). Nonetheless, an exiguous enrichment of distinct microglia clusters in the cerebellum compared to other regions was still observed (117). Furthermore, an enhanced epigenetic regulation for high-clearance activities in cerebellar microglia compared to those of the striatum may be generated locally by prominent neuronal turnover (161). Thus, further studies are needed to reevaluate the spatial diversity of adult microglia under homeostatic conditions.
In contrast, increased heterogeneity has been observed in microglia in the context of aging and gives rise to immunogenic types with inflammatory and interferon-responsive profiles. Those aging-related states of microglia may be implicated in cognitive decline (162). It remains unclear whether the appearance of the aging-related microglia clusters occurs because of sensing changes in the CNS environment or alternatively because of loss of intrinsic molecular machinery that controls proper functionality during senescence. This is an important question, since dysfunctional microglia have been recently recognized as a therapeutic target for CNS pathology.
Only a limited number of studies have explored microglial diversity in humans. The scarcity of studies can be attributed to the difficulties in obtaining fresh microglia from human CNS tissue. Recently, single-nucleus RNA sequencing (snRNA-seq) with frozen tissue samples allowed profiling of the human CNS cells on a single-cell level (163), but technical limitations remain, such as a limited number of collected cells or an inability to capture cytosolic transcripts. By contrast, a recent scRNA-seq study overcame this disadvantage and shed new light on the existence of microglial states in humans (117). This study identified distinct homeostatic human microglial clusters with some similarity to mouse homeostatic microglia, such as Cst3 expression. In addition, small subclasses were identified in human microglia, which showed high expression of chemokine genes (e.g., those encoding CCL4 and CCL2) and zinc finger transcription factors (e.g., EGR2 and EGR3), indicating a slightly activated state. Furthermore, another recent study took full advantage of scRNA-seq and CyTOF to assess transcriptional states of microglia across different anatomical regions of the CNS (164), identifying a microglia state in the white matter characterized by high expression of CD68 and MHC-II-related HLA-DR. Such microglial diversity in the white matter was previously observed in mice (82). Furthermore, a CyTOF-based comprehensive analysis of human microglia provided valuable insight into regional heterogeneity, comparing microglia from five brain regions and identifying distinct microglia states enriched in the white matter-rich subventricular zone (165). Of note, no region-specific microglia subset was observed in the human cerebellum. In general, microglia seem to be more heterogeneous in humans than in mice, which might reflect the complexity of the human CNS microenvironment or the longevity of human beings.
No studies have explored CAM diversity in healthy or diseased human brains. This is mostly due to the difficulty associated with the isolation of these less-numerous CNS macrophages. However, it will be interesting to see whether context-specific CAM clusters show features distinct from or overlapping those of microglial clusters.
Hereafter, advanced multiplex single-cell technologies will help uncover further novel states of microglia and CAMs in mice and humans, but it should be kept in mind that newly appearing clusters would presumably reflect specific activation states of preexisting cells rather than the appearance of distinct subpopulations. Nevertheless, the identification of mechanisms of disease pathogenesis and novel therapeutic interventions requires an in-depth analysis of the functional consequences of microglia states for each pathological condition.
Genetic Tools to Investigate CNS Macrophage Biology in Rodents
A long list of exciting discoveries about microglia biology (e.g., ontogeny, fate, gene expression, and dynamics) has been achieved with experimental rodent models. In particular, genetic tools have facilitated precise monitoring and manipulation of microglia, enabling in-depth analyses to uncover novel aspects of microglia physiology. Among the several tools available to target microglia, the mouse line in which the CX3C chemokine receptor 1 gene (Cx3cr1) is replaced by a green fluorescent protein (GFP) reporter gene (Cx3cr1GFP) is one of the most widely used mouse models to study microglia (166). In fact, the strong GFP signal allows the monitoring of microglia in various conditions, ranging from in vitro culture systems to intravital imaging (167). Likewise, the Cx3cr1 locus was knocked in with the CreERT2 cassette (Cx3cr1CreERT2) (168, 169), which allows Cre-loxP-mediated gene targeting of CX3CR1-expressing cells including microglia in a tamoxifen-dependent manner, opening new avenues for the study of microglia (170) (Table 1). Finally, recent studies have shown that in addition to microglia, long-lived CAMs also express Cx3cr1 (14); thus, the aforementioned tools fail to discriminate between microglia and CAMs (171). The same is true for Csf1r, Itgam (encoding CD11b), and Aif1, whose promoters have been used for constitutive microglia targeting.
Table 1.
Existing tamoxifen-inducible mouse lines for fate mapping and genetic manipulation of microglia and CNS-associated macrophages in adult mice
| Line | References | Recombination in microglia | Recombination in CAMs | Recombination in nonmyeloid CNS cells | Recombination in blood cells |
|---|---|---|---|---|---|
| Cx3crlCnERT2 | 168–170 | High | High | No | Transient, high in blood monocytes |
| Csf1rMer-iCre-Mer | 28,32 | High | Unknown | In a small subset of embryonic endothelial cells, EMPs | Transient, high in blood monocytes |
| Sall1CreERT2 | 40,171, 176, 177, 190 | High | No | Relatively high in astrocytes and oligodendrocytes, lower in neurons | No |
| Tmeml19CreERT2 | 173 | High | No | CD31+ endothelial cells, perivascular fibroblasts, meningeal fibroblasts | Transient (?), very low in CD45+ cells |
| HexbCreERT2 | 171 | High | Very low | No | Transient, very low in monocytes, granulocytes |
| P2ryl2CreERT2 | 175 | High | Very low (dura) | No | No |
This table does not include all mouse lines ever used in microglial research. Lines with constitutive Cre expression (e.g., Cx3cr1GFP, Siglec1Cre, VaviCre), poor recombination in microglia in general (e.g., LysmCre, Flt3Cre), or applications in early hematopoietic fate mapping during embryogenesis (e.g., Runx1CreERT2, Cdh5CreERT2) are not included. Abbreviations: CAM, CNS-associated macrophage; CNS, central nervous system; EMP, erythromyeloid precursor.
Recent technological advances have helped identify differentially expressed genes in microglia and CAMs, providing candidate genes to specifically target microglia. For instance, transmembrane protein 119 (TMEM119) has been characterized as specifically expressed in microglia (172). Thus, novel mouse lines with P2A-EGFP or P2A-CreERT2 inserted into the locus of Tmem119 were generated to specifically target microglia (173). A reporter mouse line in which Tmem119-expressing microglia concomitantly express tdTomato has also been generated (174). In these mice, microglia are nicely separated from CAMs, although early postnatal endothelial cells and adult vessel-associated fibroblasts in the brain can also be targeted (173). In addition, one group used the promoter region of P2Y purinoceptor 12 (P2RY12), which is known to be restricted to microglia in the CNS, to generate a P2ry12-CreERT2 mouse line, which in fact provides high Cre recombination in microglia with minor effects on CAMs, but other peripheral myeloid cells can also be targeted (175). In addition to these lines, Sall1-CreERT2 targets microglia without affecting CAMs (176), but this construct may also target other CNS cells, including astrocytes and oligodendrocytes (171, 177).
Researchers taking advantage of scRNA-seq recently compared microglia core signature genes in different disease conditions; many of these genes, such as Tmem119, P2ry12, and Sall1, were downregulated in the context of disease (89). These studies identified the gene encoding the beta subunit of hexosaminidase (Hexb) as a gene persistently expressed by microglia in various disease models, but much less in CAMs. These data encouraged the generation of new mouse lines in which either T2A-tdTomato or T2A-CreERT2 was inserted into the Hexb locus to specifically target microglia in vivo (171). In these mice, microglia can be targeted with only minor effects on CAMs, and some kidney macrophages are also affected (171). Together, these recently generated mouse lines will facilitate the transcriptional and functional analysis of microglia during homeostasis and in response to perturbation.
In contrast, genetic tools to target CAMs are scarce, despite the fact that CAM signature genes have already been defined. A recent study showed that a transgenic mouse line in which Cre expression is constitutively regulated under the control of the Siglec1 promoter is useful for targeting CAMs without affecting microglia (41). Although other mouse lines may target CAMs, their utility has yet to be experimentally proven. These mouse lines will certainly provide new insights into the nature of CAMs.
Novel Approaches to Study Microglia in Human Diseases and Their Therapeutic Potential
Fueled by the technological advancements described above, we are currently facing an important problem: Microglia functions in animal models have been intensively investigated, but few findings have translated to humans (165,178–180). A feasible option to overcome these difficulties is to utilize human microglia-like cells generated from embryonic stem cells or from induced pluripotent stem cells (iPSCs) (181–184), for which specialized protocols have been optimized to ensure reproducibility and simplicity In fact, recent studies have shown cell-autonomous dysfunction of microglia-like cells generated using iPSCs derived from patients with neurologic diseases (181, 184). Furthermore, microglia-like cells develop in cerebral organoids derived from iPSCs (185), identifying them as a valuable tool for studying the role of microglia in human brain development and disease. However, considering the plasticity of microglia, the functionality of iPSC-derived microglia may greatly vary with the culture environment, which requires further optimization to establish a translatable and representative model for the characterization of human microglia in the CNS tissue environment.
As an alternative, humanized mouse models in which iPSC-derived human microglia are transplanted to replace resident host-derived microglia are increasingly important as preclinical animal models for the study of human diseases (22). Importantly, xenografted iPSC-derived microglia-like cells retain human microglial identity in chimeric mouse brains, recapitulating the heterogeneity of adult human microglia, and even respond to cuprizone-induced demyelination (186). Moreover, it is of interest that host-derived (mouse) and iPSC-derived (human) microglia present species-specific differences in the expression of CNS-disease-risk genes, which might provide unique opportunities for better understanding the biology of human microglia in health and disease.
Recently, microglia have gained attention as a therapeutic target for several neurological disorders (187). Moreover, the gut microbiome may be an important modulator of microglial phenotype, as the commensal microbiota affects the host immune system throughout the body, including CNS microglia (143). Indeed, several studies identified an altered microglial state in mice housed under germ-free conditions that can be reproduced in specific-pathogen-free mice treated with antibiotics (188), suggesting that microglial phenotypes can be artificially controlled through the microbiota per se or microbial by-products. Furthermore, a recent study identified CD22, a canonical receptor typically expressed in B cells, as a potential target for reactivating the phagocytic activity of microglia (189). Microglia in aged mice upregulate CD22 while showing decreased phagocytic activity. Interestingly, the blockade of CD22 with a specific antibody or by genetic ablation enhanced the clearance of myelin debris, amyloid-β oligomers, and a-synuclein fibrils in vivo while reprogramming microglia toward a homeostatic transcriptional state and improving hippocampus-dependent learning and memory (157). However, the antiphagocytic role of CD22 has been shown only in mouse models. Thus, to evaluate the therapeutic potential of CD22, it is important to determine whether its function is conserved in humans.
CONCLUDING REMARKS
The field of neuroimmunology is gaining momentum, with research on microglia and CAMs evolving at a rapid pace. However, the extensive use of animal models to study microglia biology combined with the absence of consistent replication of findings in humans creates important translatability issues. Consequently, future microglial research should aim for better model systems that allow a detailed understanding of the pathophysiology of CNS diseases as well as feasible readouts for therapeutic approaches.
From a therapeutic perspective, new approaches should be established to target more specifically the disease-associated microglia and CAM subsets, while preserving the homeostatic ones (187). These approaches could be based on molecular signatures identified in each subset in the context of various neuropathologies. However, these targetable signatures should combine several levels of microglial functionality, from genetic to epigenetic to metabolic molecules. Microglia, and potentially CAMs, are highly dependent on environmental signals to maintain their functionality. Notably, important signals that instruct microglial cells are also provided by the host microbiome. It is desirable to identify those signals and to exploit them therapeutically. However, the main challenge is to dynamically modulate microglial function in time and in space, which requires integrating the complex and multilevel interactions with other immune cells as well as neurons, astrocytes, and oligodendrocytes.
SUMMARY POINTS.
Myeloid cells are the major immune cells in the healthy CNS, with microglia as the only immune cells populating the parenchyma and CNS-associated macrophages (CAMs) strategically positioned at central nervous system (CNS) borders.
Both microglia and CAMs develop exclusively from prenatal sources (e.g., the yolk sac) and are long-lived cells with a low self-renewal rate in the frame of homeostatic proliferation.
Microglia and CAMs are watchdogs of the brain that hold important functions in the maintenance of CNS homeostasis.
In rare monogenetic disorders called microgliopathies, microglia (and potentially CAMs) are the primary drivers of pathology, whereas in other CNS disorders microglia-linked risk factors contribute to disease outcome.
Microglia reciprocally interact with other CNS-resident cells during development, homeostasis, and disease (e.g., neurons, oligodendrocytes, astrocytes, and endothelial cells).
In the context of pathology, microglia acquire characteristic disease-linked transcriptional profiles.
FUTURE ISSUES.
Identify the mechanisms used by yolk sac-derived cells to migrate and infiltrate the developing brain, and find their final anatomical destinations.
Define the cell type-specific functions of non-microglia macrophages in the CNS (CAMs), and compare them to microglia during development, health, and perturbation.
Understand how disease-associated-risk genes expressed by CNS myeloid cells contribute to neurodegenerative, neuroinflammatory, and neuropsychiatric disorders.
Establish humanized model systems of CNS diseases that allow the study of pathogenesis and therapeutic screens.
Develop approaches to specifically target disease-linked microglia states and treat CNS diseases.
ACKNOWLEDGMENTS
We thank A. Dumas for critical proofreading and L. Amann for excellent figure preparations. M.P. was supported by the Sobek Foundation; the Ernst-Jung Foundation, the DFG (SFB 992, SFB1160, SFB/TRR167, Reinhart-Koselleck-Grant, Gottfried Wilhelm Leibniz-Prize); and the Ministry of Science, Research and Arts, Baden-Wuerttemberg (Sonderlinie “Neuroinflammation”). This study was supported by the DFG under Germany’s Excellence Strategy (CIBSS - EXC-2189 - Project ID390939984). M.A.W. was supported by a K99/R00 Pathway to Independence Award from the NIH (1K99NS114111), a postdoctoral fellowship from the NIH (F32NS101790), a training grant from the NIH and Dana-Farber Cancer Institute (T32CA207201), a traveling neuroscience fellowship from the Program in Interdisciplinary Neuroscience at Brigham and Women’s Hospital, and the Women’s Brain Initiative at Brigham and Women’s Hospital. F.J.Q. was supported by the NIH (NS102807, ES02530, ES029136, AI126880), the National Multiple Sclerosis Society (RG4111A1) and the International Progressive MS Alliance (PA-1604–08459).
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Colonna M, Butovsky O 2017. Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 35:441–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ginhoux F, Guilliams M 2016. Tissue-resident macrophage ontogeny and homeostasis. Immunity 44(3):439–49 [DOI] [PubMed] [Google Scholar]
- 3.Herz J, Filiano AJ, Smith A, Yogev N, Kipnis J 2017. Myeloid cells in the central nervous system. Immunity 46(6):943–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kierdorf K, Masuda T, Jordao MJC, Prinz M 2019. Macrophages at CNS interfaces: ontogeny and function in health and disease. Nat. Rev. Neurosci. 20(9): 547–62 [DOI] [PubMed] [Google Scholar]
- 5.Prinz M, Erny D, Hagemeyer N 2017. Ontogeny and homeostasis of CNS myeloid cells. Nat. Immunol. 18(4): 3 85–92 [DOI] [PubMed] [Google Scholar]
- 6.Prinz M, Jung S,Priller J 2019. Microglia biology: one century of evolving concepts. Cell 179(2):292–311 [DOI] [PubMed] [Google Scholar]
- 7.Ransohoff RM, Cardona AE 2010. The myeloid cells of the central nervous system parenchyma. Nature 468(7321):2 53–62 [DOI] [PubMed] [Google Scholar]
- 8.Prinz M, Priller J 2017. The role of peripheral immune cells in the CNS in steady state and disease. Nat. Neurosci. 20(2):136–44 [DOI] [PubMed] [Google Scholar]
- 9.Geirsdottir L, David E, Keren-Shaul H, Weiner A, Bohlen SC, et al. 2019. Cross-species single-cell analysis reveals divergence of the primate microglia program. Cell 179(7): 1609–22.e16 [DOI] [PubMed] [Google Scholar]
- 10.Dos Santos SE, Medeiros M, Porfirio J, Tavares W, Pessoa L, et al. 2020. Similar microglial cell densities across brain structures and mammalian species: implications for brain tissue function. J. Neurosci. 40(24):4622–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erny D, Jakobsdóttir KB, Prinz M 2020. Neuropathological evaluation of a vertebrate brain aged ~245 years. Acta Neuropathol. 141:133–36. 10.1007/s00401-020-02237-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fleisehhauer K 1964. [On fluorescence of perivascular cells in the cat brain]. Z. Fur Zellforsch. Und Mikrosk. Anat. 64:140–52 (In German) [PubMed] [Google Scholar]
- 13.Soulas C, Donahue RE, Dunbar CE, Persons DA, Alvarez X, Williams KC 2009. Genetically modified CD34+ hematopoietic stem cells contribute to turnover of brain perivascular macrophages in long-term repopulated primates. Am. J. Pathol. 174(5): 1808–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldmann T, Wieghofer P, Jordao MJ, Prutek F, Hagemeyer N, et al. 2016. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 17(7):797–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Furth R, Cohn ZA, Hirsch JG, Humphrey JH, Spector WG, Langevoort HL 1972. The mononuclear phagocyte system: a new classification of macrophages, monocytes, and their precursor cells. Bull. World Health Organ. 46(6): 845–52 [PMC free article] [PubMed] [Google Scholar]
- 16.Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, et al. 2009. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 326(5954):818–23 [DOI] [PubMed] [Google Scholar]
- 17.Hickey WE, Kimura H 1988. Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science 239(4837):290–92 [DOI] [PubMed] [Google Scholar]
- 18.Hickey WE, Vass K, Lassmann H 1992. Bone marrow-derived elements in the central nervous system: an immunohistochemical and ultrastructural survey of rat chimeras. J. Neuropathol. Exp. Neurol. 51(3):246–56 [DOI] [PubMed] [Google Scholar]
- 19.Priller J, Flugel A, Wehner T, Boentert M, Haas CA, et al. 2001. Targeting gene-modified hematopoietic cells to the central nervous system: Use of green fluorescent protein uncovers microglial engraftment. Nat. Med. 7(12): 1356–61 [DOI] [PubMed] [Google Scholar]
- 20.Mildner A, Schlevogt B, Kierdorf K, Bottcher C, Erny D, et al. 2011. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer’s disease. J. Neurosci. 31(31):11159–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, et al. 2007. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat. Neurosci. 10(12): 1544–53 [DOI] [PubMed] [Google Scholar]
- 22.Bennett FC, Bennett ML, Yaqoob F, Mulinyawe SB, Grant GA, et al. 2018. A combination of ontogeny and CNS environment establishes microglial identity. Neuron 98(6): 1170–83.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cronk JC, Filiano AJ, Louveau A, Marin I, Marsh R, et al. 2018. Peripherally derived macrophages can engraft the brain independent of irradiation and maintain an identity distinct from microglia. J. Exp. Med. 215(6): 1627–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shemer A, Grozovski J, Tay TL,Tao J, Volaski A, et al. 2018. Engrafted parenchymal brain macrophages differ from microglia in transcriptome, chromatin landscape and response to challenge. Nat. Commun 9(1): 5206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Varol D, Mildner A, Blank T, Shemer A, Barashi N, et al. 2017. Dicer deficiency differentially impacts microglia of the developing and adult brain. Immunity 46(6): 1030–4.e8 [DOI] [PubMed] [Google Scholar]
- 26.Ensan S, Li A, Besla R, Degousee N, Cosme J, et al. 2016. Self-renewing resident arterial macrophages arise from embryonic CX3CR1+ precursors and circulating monocytes immediately after birth. Nat. Immunol. 17(2): 159–68 [DOI] [PubMed] [Google Scholar]
- 27.Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, et al. 2014. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40(1):91–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, et al. 2015. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518(7540): 547–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hagemeyer N, Kierdorf K, Frenzel K, Xue J, Ringelhan M, et al. 2016. Transcriptome-based profiling of yolk sac-derived macrophages reveals a role for Irf8 in macrophage maturation. EMBO J. 35(16): 1730–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, et al. 2013. Microglia emerge from erythromyeloid precursors via Pu.1-and Irf8-dependent pathways. Nat. Neurosci. 16(3):273–80 [DOI] [PubMed] [Google Scholar]
- 31.Molawi K, Wolf Y, Kandalla PK, Favret J, Hagemeyer N, et al. 2014. Progressive replacement of embryo-derived cardiac macrophages with age. J. Exp. Med. 211(11): 2151–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, et al. 2012. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336(6077):86–90 [DOI] [PubMed] [Google Scholar]
- 33.Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, et al. 2010. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330(6005): 841–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alliot F, Godin I, Pessac B 1999. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res. Dev. Brain Res. 117(2): 145–52 [DOI] [PubMed] [Google Scholar]
- 35.Cuadros MA, Martin C, Coltey P, Almendros A, Navascues J 1993. First appearance, distribution, and origin of macrophages in the early development of the avian central nervous system. J. Comp. Neurol. 3 30(1): 113–29 [DOI] [PubMed] [Google Scholar]
- 36.Herbomel P, Thisse B, Thisse C 2001. Zebrafish early macrophages colonize cephalic mesenchyme and developing brain, retina, and epidermis through a M-CSF receptor-dependent invasive process. Dev. Biol. 238(2):274–88 [DOI] [PubMed] [Google Scholar]
- 37.Stremmel C, Schuchert R, Wagner F, Thaler R, Weinberger T, et al. 2018. Yolk sac macrophage progenitors traffic to the embryo during defined stages of development. Nat. Commun. 9(1): 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, et al. 2015. c-Myb+ erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 42(4):665–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mrdjen D, Pavlovic A, Hartmann FJ, Schreiner B, Utz SG, et al. 2018. High-dimensional single-cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity 48(2):380–95.e6 [DOI] [PubMed] [Google Scholar]
- 40.Van Hove H, Martens L, Scheyltjens I, De Vlaminck K, Pombo An tunes AR, et al. 2019. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat. Neurosci. 22(6): 1021–35 [DOI] [PubMed] [Google Scholar]
- 41.Utz SG, See P, Mildenberger W, Thion MS, Silvin A, et al. 2020. Early fate defines microglia and non-parenchymal brain macrophage development. Cell 181(3):557–73.e18 [DOI] [PubMed] [Google Scholar]
- 42.O’Koren EG, Yu C, Klingeborn M, Wong AYW, Prigge CL, et al. 2019. Microglial function is distinct in different anatomical locations during retinal homeostasis and degeneration. Immunity 50(3):723–37.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wieghofer P, Hagemeyer N, Sankowski R, Schlecht A, Staszewski O, et al. 2020. Mapping the origin and fate of myeloid cells in distinct compartments of the eye by single-cell profiling. EMBO J. In press. 10.15252/embj.2020105123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ydens E, Amann L, Asselbergh B, Scott CL, Martens L, et al. 2020. Profiling peripheral nerve macrophages reveals two macrophage subsets with distinct localization, transcriptome and response to injury. Nat. Neurosci. 23(5):676–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guilliams M, Scott CL 2017. Does niche competition determine the origin of tissue-resident macrophages? Nat. Rev. Immunol. 17(7):451–60 [DOI] [PubMed] [Google Scholar]
- 46.Reu P, Khosravi A, Bernard S, Mold JE, Saleh pour M, et al. 2017. The lifespan and turnover of microglia in the human brain. Cell Rep. 20(4): 779–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM 2007. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 10(12): 1538–43 [DOI] [PubMed] [Google Scholar]
- 48.Fuger P, Hefendehl JK, Veeraraghavalu K, Wendeln AC, Schlosser C, et al. 2017. Microglia turnover with aging and in an Alzheimer’s model via long-term in vivo single-cell imaging. Nat. Neurosci. 20(10): 1371–76 [DOI] [PubMed] [Google Scholar]
- 49.Tay TL, Mai D, Dautzenberg J, Fernandez-Klett F, Lin G, et al. 2017. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat. Neurosci. 20(6):793–803 [DOI] [PubMed] [Google Scholar]
- 50.Checchin D, Sennlaub F, Levavasseur E, Leduc M, Chemtob S 2006. Potential role of microglia in retinal blood vessel formation, lnvestig. Ophthalmol. Vis. Sci. 47(8):3595–602 [DOI] [PubMed] [Google Scholar]
- 51.Fantin A, Vieira JM, Gestri G, Denti L, Schwarz Q, et al. 2010. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VE.GF-mediated endothelial tip cell induction. Blood 116(5):829–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Matcovitch-Natan O, Winter DR, Giladi A, Vargas Aguilar S, Spinrad A, et al. 2016. Microglia development follows a stepwise program to regulate brain homeostasis. Science 353(6301):aad8670. [DOI] [PubMed] [Google Scholar]
- 53.Kracht L, Borggrrewe M, Eskandar S, Brouwer N, de Sousa Chuva Lopes SM, et al. 2020. Human fetal microglia acquire homeostatic immune-sensing properties early in development. Science 369(6503):aba5906. [DOI] [PubMed] [Google Scholar]
- 54.Pasciuto E, Burton OT, Roca CP, Lagou V, Rajan WD, et al. 2020. Microglia require CD4 T cells to complete the fetal-to-adult transition. Cell 182(3):625–40.e24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peri F, Nusslein-Volhard C. 2008. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase al in phagosomal fusion in vivo. Cell 133(5):916–27 [DOI] [PubMed] [Google Scholar]
- 56.Marin-Teva JL, Dusart I, Colin C, Gervais A, van Rooijen N, Mallat M. 2004. Microglia promote the death of developing Purkinje cells. Neuron 41 (4): 535–47 [DOI] [PubMed] [Google Scholar]
- 57.Sedel F, Bechade C, Vyas S, Triller A. 2004. Macrophage-derived tumor necrosis factor alpha, an early developmental signal for motoneuron death. J. Neurosci. 24(9):2236–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Squarzoni P, Oiler G, Hoeffel G, Pont-Lezica L, Rostaing P, et al. 2014. Microglia modulate wiring of the embryonic forebrain. Cell Rep. 8(5): 1271–79 [DOI] [PubMed] [Google Scholar]
- 59.Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, et al. 2010. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 7(4):483–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ueno M, Fujita Y, Tanaka T, Nakamura Y, Kikuta J, et al. 2013. Layer V cortical neurons require microglial support for survival during postnatal development. Nat. Neurosci. 16(5): 543–51 [DOI] [PubMed] [Google Scholar]
- 61.Hagemeyer N, Hanft KM, Akriditou MA, Unger N, Park ES, et al. 2017. Microglia contribute to normal myelinogenesis and to oligodendrocyte progenitor maintenance during adulthood. Acta Neuropathol. 134(3):441–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wlodarczyk A, Holtman IR, Krueger M, Yogev N, Bruttger J, et al. 2017. A novel microglial subset plays a key role in myclinogenesis in developing brain. EMBO J. 36(22):3292–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Safaiyan S, Kannaiyan N, Snaidero N, Brioschi S, Biber K, et al. 2016. Age-related myelin degradation burdens the clearance function of microglia during aging. Nat. Neurosci. 19(8):995–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hughes AN, Appel B. 2020. Microglia phagocytose myelin sheaths to modify developmental myelination. Nat. Neurosci. 23 (9): 1055–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rojo R, Raper A, Ozdemir DD, Lefevre L, Grabert K, et al. 2019. Deletion of a Csf1r enhancer selectively impacts CSF1R expression and development of tissue macrophage populations. Nat. Commun. 10(1):3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nimmerjahn A, Kirchhoff F, Helmchen F. 2005. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308(5726): 1314–18 [DOI] [PubMed] [Google Scholar]
- 67.Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, et al. 2011. Synaptic pruning by microglia is necessary for normal brain development. Science 333(6048): 1456–58 [DOI] [PubMed] [Google Scholar]
- 68.Wake H, Moorhouse AJ, Miyamoto A, Nabekura J. 2013. Microglia: actively surveying and shaping neuronal circuit structure and function. Trends Neurosci. 3 6(4):209–17 [DOI] [PubMed] [Google Scholar]
- 69.Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, et al. 2007. The classical complement cascade mediates CNS synapse elimination. Cell 131(6): 1164–78 [DOI] [PubMed] [Google Scholar]
- 70.Miyamoto A, Wake H, Ishikawa AW, Eto K, Shibata K, et al. 2016. Microglia contact induces synapse formation in developing somatosensory cortex. Nat. Commun. 7:12540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weinhard L, di Bartolomei G, Bolasco G, Machado P, Schieber NL, et al. 2018. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun. 9(1): 1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vainchtein ID, Chin G, Cho FS, Kelley KW, Miller JG, et al. 2018. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 359(6381): 1269–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cserep C, Posfai B, Lenart N, Fekete R, Laszlo ZI, et al. 2020. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science 367(6477):528–37 [DOI] [PubMed] [Google Scholar]
- 74.Liu C, Wu C, Yang Q, Gao J, Li L, et al. 2016. Macrophages mediate the repair of brain vascular rupture through direct physical adhesion and mechanical traction. Immunity 44(5): 1162–76 [DOI] [PubMed] [Google Scholar]
- 75.Prinz M, Priller J. 2014. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat. Rev. Neurosci. 15(5):300–12 [DOI] [PubMed] [Google Scholar]
- 76.Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, et al. 2014. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40(2): 274–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ransohoff RM. 2016. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 19(8):987–91 [DOI] [PubMed] [Google Scholar]
- 78.Rademakers R, Baker M, Nicholson AM, Rutherford NJ, Finch N, et al. 2011. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat. Genet. 44(2):200–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Guo L, Bertola DR, Takanohashi A, Saito A, Segawa Y, et al. 2019. Bi-allelic CSF1R mutations cause skeletal dysplasia of dysosteosclerosis-pyle disease spectrum and degenerative encephalopathy with brain malformation. Am. J. Hum. Genet. 104(5):925–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Oosterhof N, Chang IJ, Karimiani EG, Kuil LE, Jensen DM, et al. 2019. Homozygous mutations in CSF1R cause a pediatric-onset leukoencephalopathy and can result in congenital absence of microglia. Am. J. Hum. Genet. 104(5):936–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Paloneva J, Manninen T, Christman G, Hovanes K, Mandelin J, et al. 2002. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 71(3):656–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Goldmann T, Zeller N, Raasch J, Kierdorf K, Frenzel K, et al. 2015. USP18 lack in microglia causes destructive interferonopathy of the mouse brain. EMBO J. 34(12): 1612–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schwabenland M,Mossad O, Peres AG, Kessler F, Maron FJM, et al. 2019. Loss of USP18 in microglia induces white matter pathology. Acta Neuropathol. Commun. 7(1): 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Meuwissen ME, Schot R, Buta S, Oudesluijs G,Tinschert S, et al. 2016. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J. Exp. Med. 213(7): 1163–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mass E, Jacome-Galarza CE, Blank T, Lazarov T, Durham BH, et al. 2017. A somatic mutation in erythro-myeloid progenitors causes neurodegenerative disease. Nature 549(7672):389–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, et al. 2013. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368(2): 117–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, et al. 2011. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, COM and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 43(5):429–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, et al. 2017. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet. 49(9)1373–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, et al. 2016. Schizophrenia risk from complex variation of complement component 4. Nature 530(7589)177–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.De Jager PL, Jia X, Wang J, de Bakker PI, Ottoboni L, et al. 2009. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat. Genet. 41(7):776–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Reich DS, Lucchinetti CF, Calabresi PA. 2018. Multiple sclerosis. N. Engl. J. Med. 378(2)169–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Korn T, Bettelli E,Oukka M, Kuchroo VK. 2009. IL-17 and Thl7 cells. Annu. Rev. Immunol. 27:485–517 [DOI] [PubMed] [Google Scholar]
- 93.Spath S, Komuczki J, Hermann M, Pelczar P, Mair F, et al. 2017. Dysregulation of the cytokine GM-CSF induces spontaneous phagocyte invasion and immunopathology in the central nervous system. Immunity 46(2): 245–60 [DOI] [PubMed] [Google Scholar]
- 94.Mildner A, Mack M, Schmidt H, Brack W, Djukic M, et al. 2009. CCR2+Ly-6Chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. Brain 132(Part 9):2487–500 [DOI] [PubMed] [Google Scholar]
- 95.Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, et al. 2011. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann. Neurol. 69(2):292–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM. 2011. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat. Neurosci. 14(9): 1142–19 [DOI] [PubMed] [Google Scholar]
- 97.Kim RY, Hoffman AS, Itoh N, Ao Y, Spence R, et al. 2014. Astrocyte CCL2 sustains immune cell infiltration in chronic experimental autoimmune encephalomyelitis. J. Neuroimmunol. 274(1—2):53–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Locatelli G, Theodorou D, Kendirli A, Jordao MJC, Staszewski O, et al. 2018. Mononuclear phagocytes locally specify and adapt their phenotype in a multiple sclerosis model. Nat. Neurosci. 21(9)1196–208 [DOI] [PubMed] [Google Scholar]
- 99.Wheeler MA, Jaronen M, Covacu R, Zandee SEJ, Scalisi G, et al. 2019. Environmental control of astrocyte pathogenic activities in CNS inflammation. Cell 176(3):581–96.el8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fontana A, Fierz W, Wekerle H. 1984. Astrocytes present myelin basic protein to encephalitogenic T-cell lines. Nature 307(5948):273–76 [DOI] [PubMed] [Google Scholar]
- 101.Gold R, Schmied M, Tontsch U, Hartung HP, Wekerle H, et al. 1996. Antigen presentation by astrocytes primes rat T lymphocytes for apoptotic cell death: a model for T-cell apoptosis in vivo. Brain 119(Part 2):651–59 [DOI] [PubMed] [Google Scholar]
- 102.Sun D, Wekerle H. 1986. Ia-restricted encephalitogenic T lymphocytes mediating EAE lyse autoantigen-presenting astrocytes. Nature 320(6057):70–72 [DOI] [PubMed] [Google Scholar]
- 103.Jordao MJC, Sankowski R, Brendecke SM, Sagar Locatelli G, et al. 2019. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 363(6425):eaat7554. [DOI] [PubMed] [Google Scholar]
- 104.Mundt S, Mrdjen D, Utz SG, Greter M, Schreiner B, Becher B. 2019. Conventional DCs sample and present myelin antigens in the healthy CNS and allow parenchymal T cell entry to initiate neuroinflammation. Sci. Immunol. 4(31):eaau8380. [DOI] [PubMed] [Google Scholar]
- 105.Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, et al. 2005. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat. Med. 11(3):328–34 [DOI] [PubMed] [Google Scholar]
- 106.Comabella M, Montalban X, Munz C, Lunemann JD. 2010. Targeting dendritic cells to treat multiple sclerosis. Nat. Rev. Neurol. 6(9):499–507 [DOI] [PubMed] [Google Scholar]
- 107.Becher B, Tugues S, Greter M. 2016. GM-CSF: from growth factor to central mediator of tissue inflammation. Immunity 45(5):963–73 [DOI] [PubMed] [Google Scholar]
- 108.Lee SC, Liu W, Brosnan CF, Dickson DW. 1994. GM-CSF promotes proliferation of human fetal and adult microglia in primary cultures. Glia 12(4):309–18 [DOI] [PubMed] [Google Scholar]
- 109.McQualter JL, Darwiche R, Ewing C, Onuki M, Kay TW, et al. 2001. Granulocyte macrophage colony-stimulating factor: a new putative therapeutic target in multiple sclerosis. J. Exp. Med. 194(7): 873–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ponomarev ED, Shriver LP, Maresz K, Pedras-Vasconcelos J, Verthelyi D, Dittel BN. 2007. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J. Immunol. 178(1):39–48 [DOI] [PubMed] [Google Scholar]
- 111.Al-Saffar N, Khwaja HA, Kadoya Y, Revell PA. 1996. Assessment of the role of GM-CSF in the cellular transformation and the development of erosive lesions around orthopaedic implants. Am. J. Clin. Pathol. 105(5):628–39 [DOI] [PubMed] [Google Scholar]
- 112.Carrieri PB, Provitera V, De Rosa T, Tartaglia G, Gorga F, Perrella O. 1998. Profile of cerebrospinal fluid and serum cytokines in patients with relapsing-remitting multiple sclerosis: a correlation with clinical activity. Immunopharmacol. Immunotoxicol. 20(3):373–82 [DOI] [PubMed] [Google Scholar]
- 113.Filippi M, Bar-Or A, Piehl F, Preziosa P, Solari A, et al. 2018. Multiple sclerosis. Nat. Rev. Dis. Primers 4(1):43. [DOI] [PubMed] [Google Scholar]
- 114.Miron VE, Boyd A, Zhao J, Yuen TJ, Ruckh JM, et al. 2013. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 16(9): 1211–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Falcao AM, van Bruggen D, Marques S, Meijer M, Jaekel S, et al. 2018. Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat. Med. 24(12): 1837–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jaekel S, Agirre E, Falcao AM, van Bruggen D, Lee KW, et al. 2019. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 566(7745):543–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Masuda T, Sankowski R, Staszewski O, Bottcher C, Amann L, et al. 2019. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 566(7744):388–92 [DOI] [PubMed] [Google Scholar]
- 118.Komuczki J, Tuzlak S, Friebel E, Hartwig T, Spath S, et al. 2019. Fate-mapping of GM-CSF expression identifies a discrete subset of inflammation-driving T helper cells regulated by cytokines IL-23 and IL-1β. Immunity 50(5): 1289–304.e6 [DOI] [PubMed] [Google Scholar]
- 119.Mayo L, Trauger SA, Blain M, Nadeau M, Patel B, et al. 2014. Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat. Med. 20(10): 1147–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wheeler MA, Clark IC, Tjon EC, Li Z, Zandee SEJ, et al. 2020. MAFG-driven astrocytes promote CNS inflammation. Nature 578(7796):593–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Prakadan SM, Shalek AK, Weitz DA. 2017. Scaling by shrinking: empowering single-cell ‘omics’ with microfluidic devices. Nat. Rev. Genet. 18(6):345–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Eng CL, Lawson M, Zhu Q, Dries R, Koulena N, et al. 2019. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 568(7751):235–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Moffitt JR, Bambah-Mukku D, Eichhorn SW, Vaughn E, Shekhar K, et al. 2018. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 362(6416):eaau5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, et al. 2019. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 363(6434):1463–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wang X, Allen WE, Wright MA, Sylwestrak EL, Samusik N,et al. 2018. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 361 (6400):eaat5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Efremova M, Vento-Tormo M, Teichmann SA, Vento-Tormo R. 2020. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 15(4): 1484–506 [DOI] [PubMed] [Google Scholar]
- 127.Vento-Tormo R, Efremova M, Botting RA, Tureo MY, Vento-Tormo M, et al. 2018. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature 563(7731):347–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Habib N, McCabe C, Medina S, Varshavsky M, Kitsberg D, et al. 2020. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 23(6):701–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K. 2007. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat. Neurosci. 10(5):608–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, et al. 2007. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10(5):615–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Khakh BS, Beaumont V, Cachope R, Munoz-Sanjuan I, Goldman SA, Grantyn R. 2017. Unravelling and exploiting astrocyte dysfunction in Huntington’s disease. Trends Neurosci. 40(7):422–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, et al. 2014. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat. Neurosci. 17(5):694–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chao CC, Gutierrez-Vazquez C, Rothhammer V, Mayo L, Wheeler MA, et al. 2019. Metabolic control of astrocyte pathogenic activity via cPLA2-MAVS. Cell 179(7): 1483–98.e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rothhammer V, Borucki DM, Tjon EC, Takenaka MC, Chao CC, et al. 2018. Microglial control of astrocytes in response to microbial metabolites. Nature 557(7707):724–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rothhammer V, Mascanfroni ID, Bunse L, Takenaka MC, Kenison JE, et al. 2016. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 22(6): 586–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wheeler MA, Quintana FJ. 2019. Regulation of astrocyte functions in multiple sclerosis. Cold Spring Harb. Perspect. Med. 9(l):a029009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, et al. 2012. Genomic analysis of reactive astrogliosis. J. Neurosci. 32(18):6391–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, et al. 2017. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541(7638):481–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fernandez-Pol JA, Klos DJ, Hamilton PD. 1989. Modulation of transforming growth factor alpha-dependent expression of epidermal growth factor receptor gene by transforming growth factor beta, triiodothyronine, and retinoic acid. J. Cell. Biochem. 41 (3): 159–70 [DOI] [PubMed] [Google Scholar]
- 140.Raja RH, Paterson AJ, Shin TH, Kedlow JE. 1991. Transcriptional regulation of the human transforming growth factor-α gene. Mol. Endocrinol. 5(4): 514–20 [DOI] [PubMed] [Google Scholar]
- 141.Abdel-Haq R, Schlachetzki JCM, Glass CK, Mazmanian SK. 2019. Microbiome-microglia connections via the gut-brain axis.y. Exp. Med. 216(1):41–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Glebov K, Lächner M, Jabs R, Lau T, Merkel O, et al. 2015. Serotonin stimulates secretion of exosomes from microglia cells. Glia 63(4):626–34 [DOI] [PubMed] [Google Scholar]
- 143.Erny D, Hrab de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, et al. 2015. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 18(7):965–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Morais LH, Schreiber HL 4th, Mazmanian SK. 2020. The gut microbiota-brain axis in behavior and brain disorders. Nat. Rev. Microbiol. In press, 10.1038/s41579-020-00460-0 [DOI] [PubMed] [Google Scholar]
- 145.Needham BD, Kaddurah-Daouk R, Mazmanian SK. 2020. Gut microbial molecules in behavioural and neurodegenerative conditions. Nat. Rev. Neurosci. 21:717–31 [DOI] [PubMed] [Google Scholar]
- 146.Dalile B, van Oudenhove L, Vervliet B, Verbeke K. 2019. The role of short-chain fatty acids in microbiota-gut-brain communication. Nat. Rev. Gastroenterol. Hepatol. 16(8):461–78 [DOI] [PubMed] [Google Scholar]
- 147.Rothhammer V, Quintana FJ. 2019. The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 19(3): 184–97 [DOI] [PubMed] [Google Scholar]
- 148.Johnson KVA, Foster KR. 2018. Why does the microbiome affect behaviour? Nat. Rev. Microbiol. 16(10):647–55 [DOI] [PubMed] [Google Scholar]
- 149.Linnerbauer M, Wheeler MA, Quintana FJ. 2020. Astrocyte crosstalk in CNS inflammation. Neuron 108(4):608–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.De Biase LM, Schuebel KE, Fusfeld ZH, Jair K, Hawes IA, et al. 2017. Local cues establish and maintain region-specific phenotypes of basal ganglia microglia. Neuron 95(2):341–56.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lawson LJ, Perry VH, Dri P, Gordon S. 1990. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39(1): 151–70 [DOI] [PubMed] [Google Scholar]
- 152.Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, et al. 2016. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 19(3):504–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, et al. 2017. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169(7):1276–90.e17 [DOI] [PubMed] [Google Scholar]
- 154.Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, et al. 2017. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47(3): 566–81.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mathys H, Adaikkan C, Gao F, Young JZ, Manet E, et al. 2017. Temporal tracking of microglia activation in neurodegeneration at single-cell resolution. Cell Rep. 21(2):366–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Masuda T, Sankowski R, Staszewski O, Prinz M. 2020. Microglia heterogeneity in the single-cell era. Cell Rep. 30(5):1271–81 [DOI] [PubMed] [Google Scholar]
- 157.Marschallinger J, Iram T, Zardeneta M, Lee SE, Lehallier B, et al. 2020. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci. 23(2): 194–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ajami B, Samusik N, Wieghofer P, Ho PP, Crotti A, et al. 2018. Single-cell mass cytometry reveals distinct populations of brain myeloid cells in mouse neuroinflammation and neurodegeneration models. Nat. Neurosci. 21 (4): 541–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, et al. 2019. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity 50(1):253–71.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Li Q, Cheng Z, Zhou L, Darmanis S, Neff NF, et al. 2019. Developmental heterogeneity of microglia and brain myeloid cells revealed by deep single-cell RNA sequencing. Neuron 101 (2):207–23.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ayata P, Badimon A, Strasburger HJ, Duff MK, Montgomery SE, et al. 2018. Epigenetic regulation of brain region-specific microglia clearance activity. Nat. Neurosci. 21(8)1049–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Deczkowska A, Matcovitch-Natan O, Tsitsou-Kampeli A, Ben-Hamo S, Dvir-Szternfeld R, et al. 2017. Mef2C restrains microglial inflammatory response and is lost in brain ageing in an IFN-I-dependent manner. Nat. Commun. 8(1):717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, et al. 2019. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570(7761):332–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sankowski R, Bottcher C, Masuda T, Geirsdottir L, Sagar, et al. 2019. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat. Neurosci. 22(12):2098–110 [DOI] [PubMed] [Google Scholar]
- 165.Bottcher C, Schlickeiser S, Sneeboer MAM, Kunkel D, Knop A, et al. 2019. Human microglia regional heterogeneity and phenotypes determined by multiplexed single-cell mass cytometry. Nat. Neurosci. 22(1):78–90 [DOI] [PubMed] [Google Scholar]
- 166.Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, et al. 2000. Analysis of fractalkine receptor CX3CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol. 20(11):4106–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, et al. 2005. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8(6):752–58 [DOI] [PubMed] [Google Scholar]
- 168.Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, et al. 2013. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155(7)4596–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Yona S, Kim KW, Wolf Y, Mildner A, Varol D, et al. 2013. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38(1):79–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Goldmann T, Wieghofer P, Muller PF, Wolf Y, Varol D, et al. 2013. A new type of microglia gene targeting shows TAKl to be pivotal in CNS autoimmune inflammation. Nat. Neurosci. 16(11)1618–26 [DOI] [PubMed] [Google Scholar]
- 171.Masuda T, Amann L, Sankowski R, Staszewski O, Lenz M, et al. 2020. Novel Hexb-based tools for studying microglia in the CNS. Nat. Immunol. 21(7):802–15 [DOI] [PubMed] [Google Scholar]
- 172.Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, et al. 2016. New tools for studying microglia in the mouse and human CNS. PNAS 113(12):E1738M6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kaiser T, Feng G. 2019. Tmem 119-EGFP and Tmem 119-CreERT2 transgenic mice for labeling and manipulating microglia. eNeuro 6(4). 10.1523/ENEURO.0448-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ruan C, Sun L, Kroshilina A, Beckers L, De Jager P, et al. 2020. A novel Tmem 119-tdTomato reporter mouse model for studying microglia in the central nervous system. Brain Behav. Immun. 83:180–91 [DOI] [PubMed] [Google Scholar]
- 175.McKinsey GL, Lizama CO, Keown-Lang AE, Niu A, Santander N, et al. 2020. A new genetic strategy for targeting microglia in development and disease. eLife 9:e54590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Buttgereit A, Lelios I, Yu X, Vrohlings M, Krakoski NR, et al. 2016. Sall 1 is a transcriptional regulator defining microglia identity and function. Nat. Immunol. 17(12): 1397–406. Erratum. 2017. Nat. Immunol. 18(2):246 [DOI] [PubMed] [Google Scholar]
- 177.Chappell-Maor L, Kolesnikov M, Kim JS, Shemer A, Haimon Z, et al. 2020. Comparative analysis of CreER transgenic mice for the study of brain macrophages: a case study. Eur. J. Immunol. 50(3):353–62 [DOI] [PubMed] [Google Scholar]
- 178.Darmanis S, Sloan SA, Croote D, Mignardi M, Chernikova S, et al. 2017. Single-cell RNA-seq analysis of infiltrating neoplastic cells at the migrating front of human glioblastoma. Cell Rep. 21(5): 1399–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, et al. 2017. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355(6332):eaai8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zhong S, Zhang S, Fan X, Wu Q, Yan L, et al. 2018. A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex. Nature 555(7697): 524–28 [DOI] [PubMed] [Google Scholar]
- 181.Abud EM, Ramirez RN, Martinez ES, Healy LM, Nguyen CHH, et al. 2017. iPSC-derived human microglia-like cells to study neurological diseases. Neuron 94(2):278–93.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Beutner C, Roy K, Linnartz B, Napoli I, Neumann H. 2010. Generation of microglial cells from mouse embryonic stem cells. Nat. Protoc. 5(9): 1481–94 [DOI] [PubMed] [Google Scholar]
- 183.Muffat J, Li Y, Yuan B, Mitalipova M, Omer A, et al. 2016. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med. 22(11): 1358–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Pandya H, Shen MJ, Ichikawa DM, Sedlock AB, Choi Y, et al. 2017. Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nat. Neurosci. 20(5):753–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Ormel PR, Vieira de Sá R, van Bodegraven EJ, Karst H, Harschnitz O, et al. 2018. Microglia innately develop within cerebral organoids. Nat. Commun. 9(1):4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Xu R, Li X, Boreland AJ, Posyton A, Kwan K, et al. 2020. Human iPSC-derived mature microglia retain their identity and functionally integrate in the chimeric mouse brain. Nat. Commun. 11(1): 1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Priller J, Prinz M. 2019. Targeting microglia in brain disorders. Science 365(6448):32–33 [DOI] [PubMed] [Google Scholar]
- 188.Mezo C, Dokalis N, Mossad O, Staszewski O, Neuber J, et al. 2020. Different effects of constitutive and induced microbiota modulation on microglia in a mouse model of Alzheimer’s disease. Acta Neuropathol. Commun. 8(1): 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Pluvinage JV, Haney MS, Smith BAH, Sun J, Iram T, et al. 2019. CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Nature 568(7751): 187–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Inoue S, Inoue M, Fujimura S, Nishinakamura R. 2010. A mouse line expressing Sall 1-driven inducible Cre recombinase in the kidney mesenchyme. Genesis 48(3):207–12 [DOI] [PubMed] [Google Scholar]