

Abstract
Background:
Heart failure is a leading cause of death worldwide and is associated with the rising prevalence of obesity, hypertension and diabetes. O-GlcNAcylation is a post-translational modification of intracellular proteins and serves as a metabolic rheostat for cellular stress. The total levels of O-GlcNAcylation are determined by nutrient and metabolic flux, in addition to the net activity of two enzymes, O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA). Failing myocardium is marked by increased O-GlcNAcylation, but it is unknown if excessive O-GlcNAcylation contributes to cardiomyopathy and heart failure.
Methods:
We developed two new transgenic mouse models with myocardial overexpression of OGT and OGA to control O-GlcNAcylation independent of pathological stress.
Results:
We found that OGT transgenic hearts showed increased O-GlcNAcylation, and developed severe dilated cardiomyopathy, ventricular arrhythmias and premature death. In contrast, OGA transgenic hearts had lower O-GlcNAcylation but identical cardiac function to wild type littermate controls. Additionally, OGA transgenic hearts were resistant to pathological stress induced by pressure overload with attenuated myocardial O-GlcNAcylation levels after stress and decreased pathological hypertrophy compared to wild type controls. Interbreeding OGT with OGA transgenic mice rescued cardiomyopathy and premature death, despite persistent elevation of myocardial OGT. Transcriptomic and functional studies revealed disrupted mitochondrial energetics with impairment of complex I activity in hearts from OGT transgenic mice. Complex I activity was rescued by OGA transgenic interbreeding, suggesting an important role for mitochondrial complex I in O-GlcNAc mediated cardiac pathology.
Conclusions:
Our data provide evidence that excessive O-GlcNAcylation causes cardiomyopathy, at least in part, due to defective energetics. Enhanced OGA activity is well tolerated and attenuation of O-GlcNAcylation is beneficial against pressure overload induced pathologic remodeling and heart failure. These findings suggest attenuation of excessive O-GlcNAcylation may represent a novel therapeutic approach for cardiomyopathy.
Keywords: Dilated cardiomyopathy, O-GlcNAcylation, mouse model, mitochondrial energetics, pressure overload
Introduction
Failing myocardium from model animals, and patients, is marked by increased protein O-GlcNAcylation1. However, it is unknown if excessive O-GlcNAcylation is a cause or consequence of cardiomyopathy. The hexosamine biosynthesis pathway is a metabolic sensor that utilizes glucose, amino and fatty acids to synthesize UDP-GlcNAc (Uridine Diphosphate N-acetylglucosamine), the substrate for OGT (O-GlcNAc transferase). O-GlcNAc is cycled on and off proteins by the activity of two enzymes: OGT, which adds GlcNAc from UDP-GlcNAc to proteins, and OGA (O-GlcNAcase), which removes UDP-GlcNAc from proteins2. Dynamic changes in O-GlcNAc are important components of the stress response and appear essential given constitutive OGT knock out is embryonically lethal3. Furthermore, inducible loss of myocardial OGT in adult mice causes increased susceptibility to myocardial injury2, indicating that O-GlcNAcylation is necessary, and suggesting that elevated O-GlcNAcylation can be beneficial. Alternatively, excessive O-GlcNAcylation is also suspected to contribute to myocardial dysfunction in diabetes and hyperglycemia1, and OGT inhibitors can reverse or prevent pathological myocardial hypertrophy1. Indeed, recent work has highlighted the role of O-GlcNAcylation in cardiac injury via interaction with CaMKII and Stim1, and a beneficial role in heart failure via HDAC44–7. Thus, major unresolved questions remain regarding the potential role of excessive O-GlcNAcylation to cause or contribute to cardiomyopathy.
The complexity of myocardial responses to pathological stress, and lack of genetic tools to control O-GlcNAcylation levels in vivo, independent of glucose or pathological stress, has limited understanding of the role of increased O-GlcNAcylation in cardiomyopathy. We developed novel mouse models to independently control O-GlcNAcylation levels in myocardium, and directly test the hypothesis that excessive O-GlcNAcylation causes or contributes to cardiomyopathy. Here, we report transgenic myocardial OGT overexpression (OGT TG) causes increased O-GlcNAcylation, dilated cardiomyopathy, and premature death. In contrast, transgenic myocardial OGA overexpression (OGA TG) does not cause cardiomyopathy. However, OGA TG mice had reduced myocardial O-GlcNAcylation, and were protected against cardiomyopathy due to transverse aortic constriction (TAC) surgery, a model of acquired pathological myocardial hypertrophy8, 9. Interbreeding of OGT TG with OGA TG mice reduced cardiac O-GlcNAcylation toward WT levels, rescued dilated cardiomyopathy, and prevented premature death seen in the OGT TG mice. We identified reduced expression of genes and proteins important for oxidative phosphorylation, and impaired energetics in OGT TG hearts; these patterns were restored to near WT levels in hearts from OGT TG x OGA TG interbred mice. Taken together, these data show that excessive O-GlcNAcylation is sufficient to cause severe cardiomyopathy, heart failure, and premature mortality. Our findings identify novel targets likely to explain, at least in part, the deleterious effects of excessive myocardial O-GlcNAcylation, and suggest reducing myocardial O-GlcNAcylation could be a successful therapeutic approach for cardiomyopathy and heart failure.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Animal Models
All animal studies were carried out in accordance with the guidelines of the Johns Hopkins Institutional Animal Care and Use Committee (IACUC) under protocol # M017M290. No human studies were performed. Mice used in these studies were a mixture of male and female animals on a C57BL/6J background. Animals used in the majority of studies were 7–12 weeks of age unless otherwise noted in the figure legend.
OGT TG mice
Human cDNA encoding the nucleocytoplasmic variant of the human OGT gene was fused with a C-terminal Myc epitope tag. The resulting construct was cloned into the pBS-αMHC-script-hGH vector for myocardial expression. Pronuclear injections of linearized DNA (digested with NotI) were performed in the Johns Hopkins Transgenic Mouse Core Facility and embryos implanted into pseudo-pregnant females to generate C57BL/6J F1 mice. Insertion of the transgene into the mouse genome was confirmed by PCR analysis (supplement) using the forward primer, 5’-GGA CTT CAC ATA GAA GCC TAG C-3’, and reverse primer, 5’-CAC TGC GAA CAC AGT ACA AAT C--3’, producing a product of 500 base pairs.
OGA TG mice
Human cDNA encoding the long form of the Meningioma Expressed Antigen 5 gene (O-GlcNAcase, approved symbol OGA) was fused with a N-terminal HA epitope tag. The resulting construct was cloned into the pBS-αMHC-script-hGH vector for myocardial expression. Pronuclear injections of linearized DNA (digested with Not1) were performed in the Johns Hopkins Transgenic Mouse Core Facility and embryos implanted into pseudo-pregnant females to generate C57BL/6J F1 mice. Insertion of the transgene into the mouse genome was confirmed by PCR analysis (supplement) using the forward primer, 5’- TGGTCAGGATCTCTAGATTGGT-3’ and reverse primer, 5’-TCATAAGTTGCTCAGCTTCCTC-3’, producing a product of 850 base pairs.
AC3-I TG and CaMKIIδ S280A knock in mice
Experimental studies were performed on male and female mice with C57BL/6J background. C57BL/6J and mice lacking a functional NADPH oxidase (p47−/−) were purchased from The Jackson Laboratory. Our lab previously described the generation of AC3-I10 transgenic mice.
CaMKIIδ-S280A knock-in mice harboring a point mutation in the mouse CaMKIIδ gene to substitute Serine 280 with Alanine (S280A) were generated on a C57BL/6J background using the CRISPR/Cas9 technology. A single-guide RNA (target sequence: 5’-CTGTTGCCTCCATGATGCACAGG-3’) was designed to target CaMKIIδ. Synthetic single-stranded DNA for CRISPR-homology repair was designed to harbor mutations including S280A (TCC --> GCG) and NsiI recognition site (ATGCAT). Genotyping of founder mice and generations of offspring was performed initially by both direct sequencing of PCR amplified fragments and PCR genotyping from tail DNA with the following primers: Forward, 5’-AGGAAATGCTTGCCAAAGTAGTG-3’; Reverse, 5’-CCAGCACATACTGCCCTAGC-3’.
Statistical Analysis
Statistical analyses were performed using Graph Pad Prism 8 software. Sample size and information about statistical tests are reported in the figure legends. Data are presented as mean ± SEM. Pairwise comparisons were performed using a two-tailed Student’s t test. For experiments with more than 2 groups, data were analyzed by 1 way ANOVA followed by Tukey’s post-hoc multiple comparisons test. For Kaplan-Meier survival analysis data are represented means ± SEM, significance was determined using the log rank (Mantel-Cox) test.
Procedures used for transverse aortic constriction, murine echocardiography, arrhythmia monitoring by electrocardiographic telemetry implant, western blot, OGT and OGA activity assays, mitochondrial isolation, complex I, II and IV activity assays, Seahorse mitochondrial bioenergetics measurements and ventricular myocyte intracellular Ca2+ measurements were performed according to published methods11–16 and are detailed in the Extended Methods Supplement.
Results
Myocardial-targeted OGA overexpression protects against left ventricular hypertrophy and heart failure
Elevated myocardial O-GlcNAcylation has been reported in multiple models of cardiomyopathy1, 17–21. We first asked if TAC, a validated model of pathologically increased left ventricular afterload8, resulted in augmented myocardial O-GlcNAcylation (see Methods). We found robust elevation in total O-GlcNAcylation in hearts from C57BL/6J mice with TAC compared to sham operated mice (Fig 1a and 1b). These results were consistent with previous reports of increased O-GlcNAcylation in hearts subjected to pathological stress1, 21. Based on these findings, we next asked if attenuation of O-GlcNAcylation during sustained cardiac stress could be beneficial.
Figure 1. Myocardial OGA over-expression decreases total O-GlcNAc modified protein levels but does not cause cardiomyopathy.
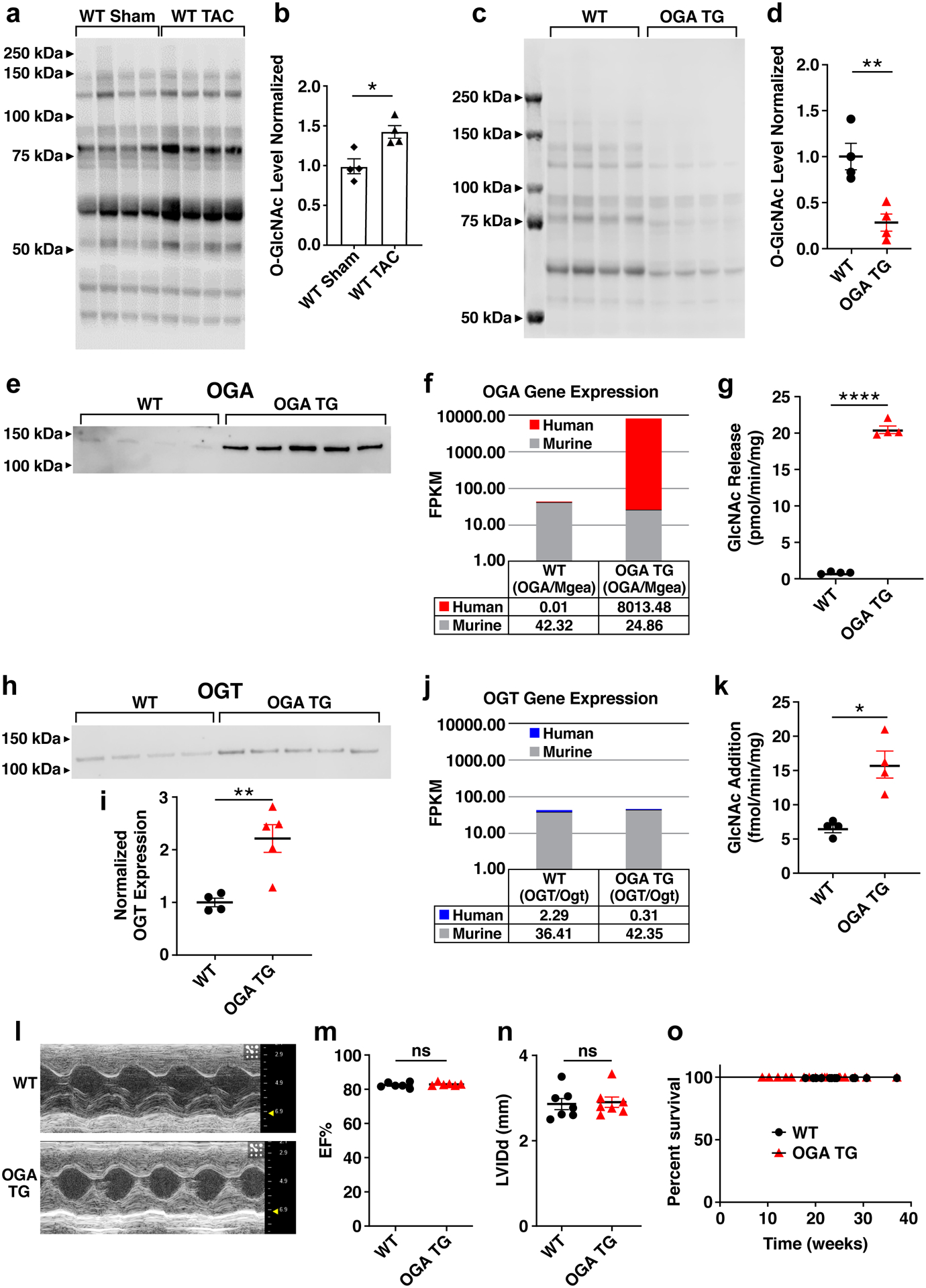
a. Representative western blot and b. summary data for total O-GlcNAc modified protein levels (OGN) from whole heart lysates of 8–12 week old mice. Hearts were removed 9 weeks after transverse aortic constriction(TAC) or sham surgery (n = 4 mice/group). c. Western blot of O-GlcNAc levels and d. summary data from cardiac lysates using WT (n = 4) and OGA TG (n = 4) mice. e. Western blot of OGA, 25 ug WT (n=4) protein, 0.25 ug OGA (n=5) TG protein loaded f. human (OGA) and murine (Mgea) transcript levels in WT (n=6) and OGA TG (n=6) mice g. OGA activity assay measuring GlcNAc release in WT (n = 4) and OGA TG (n = 4) mouse hearts at 8–10 weeks. h. OGT protein expression and i. summary data from whole heart lysates from OGA TG (n = 5) and WT (n = 4) mice. j. Human (OGT) and murine (Ogt) transcript levels WT (n=6) and OGA TG (n=6) mice. k. OGT activity assay measuring O-GlcNAc addition in WT (n =4) and OGA TG(n = 4) animals. l. Example images of left ventricular M-mode echocardiograms from WT and OGA TG mice. Summary echocardiographic data for m. left ventricular ejection fraction (EF) and n. left ventricular end-diastolic internal diameter (LVIDd) acquired at 8–10 weeks of age (WT n = 7, OGA TG n = 7). o. Kaplan-Meier survival analysis for OGA TG (n = 11) and WT littermates (n = 9). Data are represented as means ± SEM, significance was determined using the log rank (Mantel-Cox) test. (****P<0.0001, **P<0.01, *P<0.05, ns=not significant for all panels).
In order to test the effect of reducing myocardial O-GlcNAcylation under conditions of pathological stress, we developed OGA TG mice where OGA expression was under control of the α-myosin heavy chain promoter (Figure Ia in the Supplement) (see Methods)22. The OGA TG mice were born in normal Mendelian ratios. The O-GlcNAcylation levels in OGA TG hearts were lower compared to WT hearts at baseline (Fig 1c and 1d). Notably, the antibody used to detect OGA by western blot preferentially recognizes human OGA such that the transgenic (human OGA) is over-represented when compared to mouse OGA. Underpinning this observation, while human and mouse OGA homologs are 97% identical, the antibody used in this study was raised against the first 50 amino acids in which the identity between these homologs is 78% identical. These observations preclude a quantitative comparison. A representative western blot is shown comparing OGA levels in WT and OGA TG mice (Fig 1e) where OGA TG protein levels were loaded at 1/100th that of WT. Coomassie loading is shown in Figure Ib in the Supplement (upper panel) for blots assayed for OGA in WT and OGA TG animals. Consistent with the decreased O-GlcNAcylation measured in OGA TG mice (Fig 1d), we detected significant increases in the abundance of human OGA mRNA in OGA TG (Fig 1f) and in OGA activity (~20 fold) in OGA TG heart lysates compared to WT heart lysates (Fig 1g). OGA and OGT expression are coupled by mechanisms that include changes in transcription and splicing23, 24 so we examined the protein expression levels of OGT. We did find increased OGT protein expression (Fig 1h and 1i, Coomassie loading is shown in Figure Ib in the Supplement, lower panel), non-significant changes in OGT transcript levels (Fig 1j) and increased OGT activity (Fig 1k) in OGA TG lysates. Localization of OGA expression to the heart in OGA TG mice was confirmed by analyzing different tissue types (Figure Ic in the Supplement). The OGA TG mice had modestly, but significantly increased heart weight/body weight ratios compared to WT littermate controls (Figure IIa in the Supplement), but no difference from WT mice in left ventricular function by echocardiography (Fig 1l and 1m), left ventricular dimensions (Fig 1n), or Nppa expression (Figure IIb in the Supplement), a marker of pathological hypertrophy25. The OGA TG mice exhibited no premature mortality (Fig 1o). We interpreted these findings to suggest myocardial OGA over-expression is effective at decreasing baseline O-GlcNAcylation but this change is well tolerated without deleterious effect on cardiac structure, function or mortality.
Elevated cardiac O-GlcNAcylation is associated with pathological hypertrophy and heart failure in animal models, and is well described in patients with hypertension and aortic stenosis, conditions of increased left ventricular afterload1. Based on these associations, we next challenged OGA TG and WT littermate control mice with transverse aortic constriction (TAC) or sham surgery (Fig 2a). The OGA TG mice showed significantly attenuated myocardial O-GlcNAcylation (Fig 2b and 2c) after sham and TAC surgery compared to WT mice after TAC surgery. Protein loading was similar between groups assessed for total O-GlcNAc levels, as analyzed by Coomassie total protein staining (Figure IIc in the Supplement). OGA protein levels were assessed with 1:1 protein loading in Fig 2d and 2e comparing WT Sham and WT TAC (25 μg of protein loaded per sample) and with 1:1 protein loading comparing OGA TG Sham and TAC (0.25 μg protein loaded per sample) in Fig 2f. Coomassie is shown for OGA levels (Figure IId in the Supplement). OGT protein levels were increased in WT TAC mice compared to WT sham but not significantly different in OGA TG sham versus TAC animals (Fig 2g). Coomassie is shown for OGT levels (Figure IIe in the Supplement). OGA TG mice had less severe hypertrophy following TAC compared to WT mice after TAC (Fig 2h). OGA TG mice following TAC had no significant change in systolic function unlike WT mice after TAC (Fig 2i). Both pathological hypertrophy and heart failure following TAC have been associated with perturbation of mitochondrial energetics, and increased O-GlcNAc is linked to changes in Complex I26, so we assayed complex I function but found no significant difference between WT and OGA TG groups undergoing sham or TAC surgery (Fig 2j). Marker genes for hypertrophic myocardial remodeling, Nppa and Myh7, that were normal at baseline in OGA TG mice (Figure IIb and Figure IIf in the Supplement), showed reduced expression (Fig 2k and 2l), compared to WT littermate controls after TAC. In contrast, left ventricular wall thickness and left ventricular ejection fraction were similar in both sham groups (Fig 2h and 2i). Taken together, we interpreted these results to support a view that excessive myocardial O-GlcNAcylation is associated with pathological stress, and that increased OGA expression can reduce excessive O-GlcNAcylation and protect against myocardial hypertrophy after TAC surgery.
Figure 2. Myocardial-targeted OGA over-expression protects against left ventricular hypertrophy and contractile dysfunction after transverse aortic constriction (TAC) surgery.
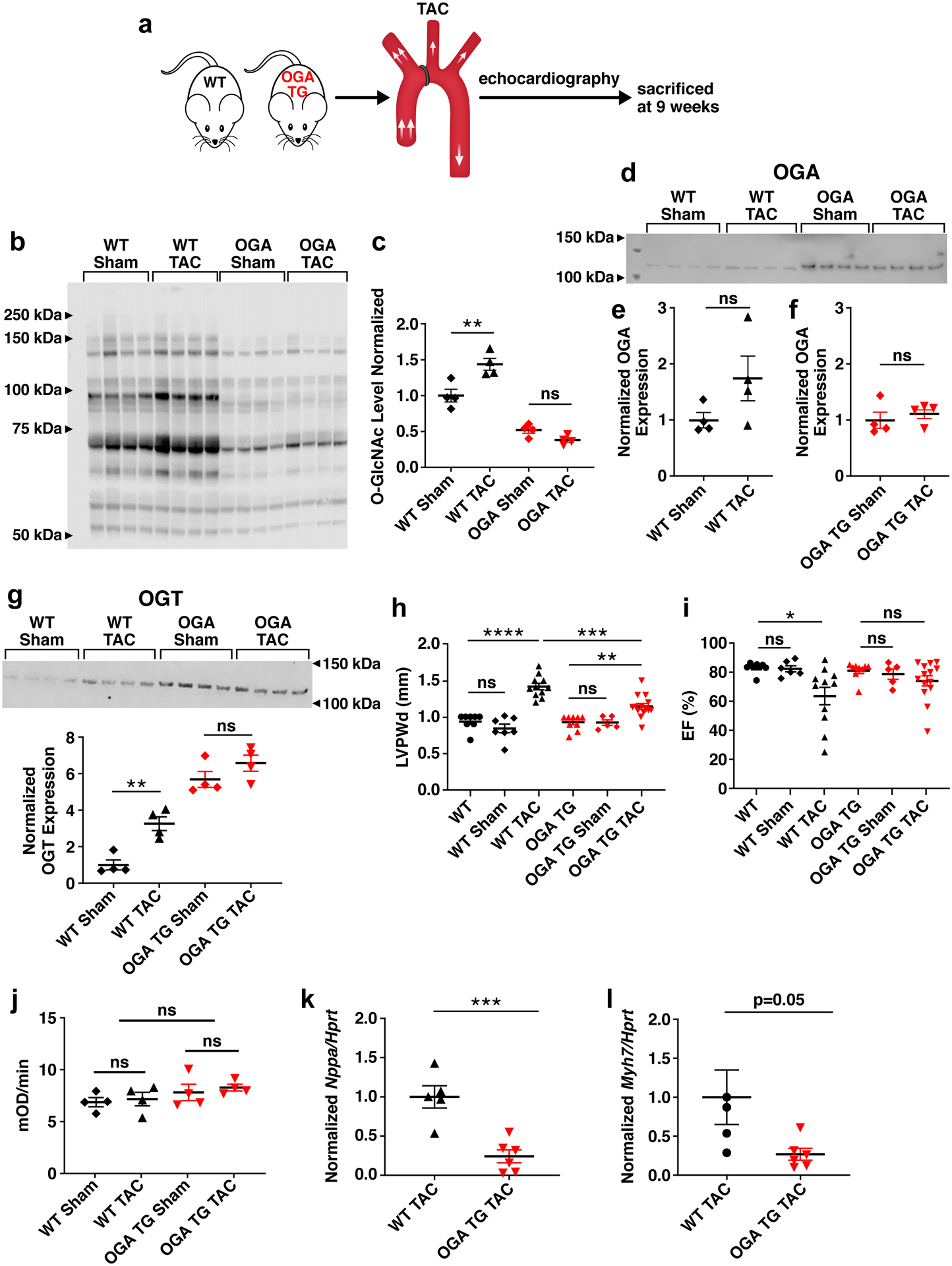
a. Schematic of the TAC left ventricular pressure-overload model performed in 8–10 week old WT and OGA TG mice. b. Western blot and c. summary data of total O-GlcNAc levels from WT sham (n=4), WT TAC (n=4), OGA TG Sham (n=4) and OGA TG TAC (n=4) whole heart lysates 9 weeks after intervention. d. Western blot for OGA from whole heart lysates from OGA TG (Sham n =4, TAC n=4) and WT (Sham n=4, TAC n=4) mice, protein loading 25 ug WT mice, 0.25 ug OGA TG mice. e. Quantification OGA levels in WT sham and TAC. f. Quantification of OGA levels in OGA TG Sham and TAC. g. OGT protein expression and quantification h. left ventricular posterior wall thickness measured at end-diastole (LVPWd) in WT(baseline n=8, sham n =7, TAC n=11) and OGA TG (baseline n=9, sham n=5, TAC n=11) and i. Left ventricular ejection fractions (EF) in OGA TG and WT littermate hearts 9 weeks after TAC surgery. j. Complex I activity spectrophotometer assay summary data in WT (sham n=4, TAC n =4) and OGA TG (sham n=4, TAC n=4). k. Quantification of Nppa and l. Myh7 mRNA expression normalized to Hypoxanthine Peroxidase Reductase Transferase (Hprt) in OGA TG (n=6) and WT (n=5) hearts 9 weeks after TAC. Data are represented as mean ± SEM. Significance was determined using a two-tailed Student’s t test or 1 way ANOVA with Tukey’s multiple comparisons test, as appropriate (****P<0.0001, ***P<0.001, **P<0.01, *P<0.05, ns=not significant).
Increased O-GlcNAcylation, dilated cardiomyopathy, arrhythmias, and premature death in OGT TG mice
Our findings up to this point showed reducing O-GlcNAcylation may be beneficial to protect against pathological hypertrophy due to TAC surgery. We next used an orthogonal approach to assess the role of increased O-GlcNAcylation in cardiomyopathy by developing an OGT TG mouse model. OGT TG mice were designed for myocardial-targeted OGT over-expression, using the α-myosin heavy chain promoter, similar to our OGA TG mice (Figure IIIa in the Supplement) (see Methods). OGT TG and WT littermate mice were sacrificed at 8 weeks of age; OGT protein overexpression was exclusively localized to heart (Figure IIIb in the Supplement). Cardiac O-GlcNAcylation (Fig 3a and 3b), and OGT protein expression were increased compared to WT littermates (Fig 3c and 3d). As expected, OGT transcripts were increased in OGT TG mice compared to WT (Fig 3e). OGT activity in OGT TG hearts was significantly increased compared to WT (Fig 3f). Cardiac OGA expression (Fig 3g and 3h) was not significantly increased over WT littermates. Coomassie protein loading controls are shown in Figure IIIc and Figure IIId in the Supplement. Transcript expression of murine OGA was slightly increased in the OGT TG vs WT (Fig 3i) with OGA activity mildly increased in OGT TG compared to WT (Fig 3j). The OGT TG mice developed dilated cardiomyopathy (Fig 3k) with reduced left ventricular ejection fraction (Fig 3l), and increased left ventricular diameter (Fig 3m). Nppa transcript levels obtained from RNA sequencing were increased in OGT TG mice compared to OGA TG (Figure IIf in the Supplement). Left ventricular function began to decline after 6 weeks of age (Figure IIIe in the Supplement). By 8 weeks OGT TG mice had significantly increased left ventricular mass, assessed by echocardiography (Figure IIIf in the Supplement), and by morphometric analysis (Figure IIIg in the Supplement). Echocardiography measurements demonstrated a dilated cardiomyopathy phenotype in OGT TG mice (Figure IIIh in the Supplement). Ages of mice used in these experiments are noted in the figure legends. These findings illustrate increased myocardial OGT with concomitant increase in O-GlcNAcylation were sufficient to cause progressive cardiomyopathy.
Figure 3. OGT TG mice have increased O-GlcNAc, dilated cardiomyopathy, and premature death.
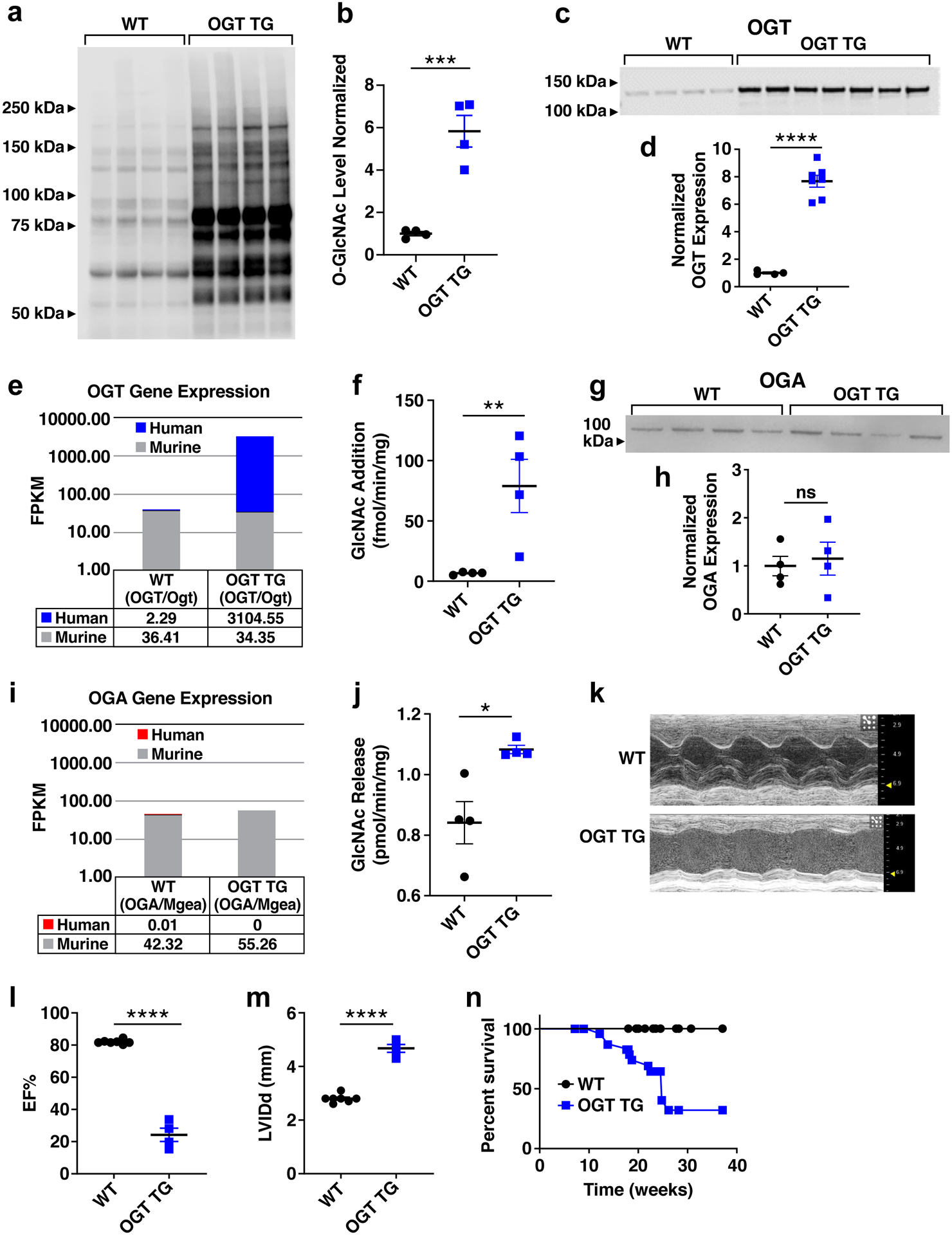
a. Western blot and b. summary data for total O-GlcNAc levels in cardiac lysates from OGT TG and WT littermates (WT n = 4, OGT TG n = 4 hearts). c. OGT protein expression and d. summary data (WT n = 4, OGT TG n = 7) and e. human (OGT) and murine (Ogt) transcript levels WT (n=6) and OGT TG (n=6) mice. f. OGT activity assay measuring O-GlcNAc addition in WT (n =4) and OGT TG (n = 4) animals g. OGA protein expression and h. summary data in cardiac lysates (WT n = 4 and OGT TG n = 4 hearts). i. human (OGA) and murine(Mgea) transcript levels in WT (n=6) and OGT TG (n=6) mice. j. OGA activity assay measuring GlcNAc release in WT (n = 4) and OGT TG (n = 4) mouse hearts at 8–10 weeks. k. Example M-mode left ventricular echocardiograms from WT and OGT TG mice. l. Summary echocardiographic data for left ventricular ejection fraction (EF) and m. left ventricular internal diameter in diastole (LVIDd) in 8–12 week old mice (WT n = 7, OGT TG n = 5). n. Kaplan-Meier survival analysis for OGT TG and WT littermate mice (n = 9 WT, n = 14 OGT TG). Data are represented as mean ± SEM; significance was determined using a two-tailed Student’s t test (****P<0.0001, ***P<0.001, **P<0.01, *P<0.05, ns=not significant).
Sudden death is a major complication of many types of heart failure and OGT TG mice exhibited a striking pattern of premature mortality (Fig 3n). Arrhythmias are an important cause of premature mortality in heart failure27. In order to determine if arrhythmias contributed to premature death in OGT TG mice, we surgically implanted electrocardiographic monitors in OGT TG and WT littermate mice (mice 20–22 weeks of age were used to determine cardiac electrical activity preceding death). We detected bradycardia, spontaneous ventricular tachycardia and ventricular fibrillation leading to death in OGT TG, but not in WT control mice (Figure IVa in the Supplement, left panel). The OGT TG mice exhibited a higher arrhythmia score (see Methods)11 (Figure IVa in the Supplement, right panel), confirming an increased arrhythmia burden in OGT TG compared to WT controls. These findings suggested that premature death in OGT TG mice was related, at least in part, to arrhythmias. An important proarrhythmic feature of failing heart muscle is increased intracellular Ca2+ leak from the sarcoplasmic reticulum to the cytoplasm28. Leak of Ca2+ from the intracellular Ca2+ stores can be detected as Ca2+ sparks via cytosolic Ca2+ activated fluorescent indicators29. We measured intracellular Ca2+ sparks (see Supplementary Methods) in ventricular myocytes isolated from OGT TG and WT hearts, and found increased Ca2+ spark frequency and size in the OGT TG ventricular myocytes compared to WT (Fig 4a–g). We next measured phospholamban (PLN) and SERCA2a (sarcoplasmic-endoplasmic reticulum Ca2+ ATPase) expression in OGT TG and WT control hearts and found increased PLN in OGT TG hearts and lower levels of SERCA2a compared to WT hearts (Fig 4h and 4i). Excessive CaMKII activity can cause increased intracellular Ca2+ sparks30, cardiomyopathy31 and arrhythmias31. Furthermore, CaMKII may be activated by O-GlcNAcylation of S280, a residue at the intersection of the regulatory and catalytic domains4. Based on these concepts we initially hypothesized that CaMKII was a critical cardiomyopathic signal for OGT TG cardiomyopathy. In order to test this idea, we first interbred OGT TG mice with mice engineered with a knock in replacement of S280 (S280A) on CaMKIIδ32, the predominant CaMKII isoform in myocardium. The S280A mice are born in Mendelian ratios, develop normally, and have heart morphometry and function indistinguishable from WT littermates32. The OGT TG x S280A interbred mice had similar left ventricular dysfunction and dilation compared to OGT TG mice in a WT CaMKIIδ background (Figure IVb and Figure IVc in the Supplement). We next considered the possibility that CaMKII was activated by excessive O-GlcNAcylation in OGT TG mice independent of S280. To test this idea, we interbred OGT TG mice with an established mouse model of myocardial CaMKII inhibition, due to transgenic expression of a CaMKII inhibitory peptide AC3-I that inhibits all CaMKII isoforms10. Mirroring the S280A phenotype, the interbred OGT x AC3-I mice exhibited severe left ventricular dysfunction and dilation (Figure IVb and Figure IVc in the Supplement), similar to age-matched OGT TG mice (Fig 3l and 3m). Taken together, these findings did not support our initial hypothesis that CaMKII was an important contributor to cardiomyopathy in OGT TG mice.
Figure 4. OGT TG mice have increased calcium sparks and impaired mitochondrial energetics.
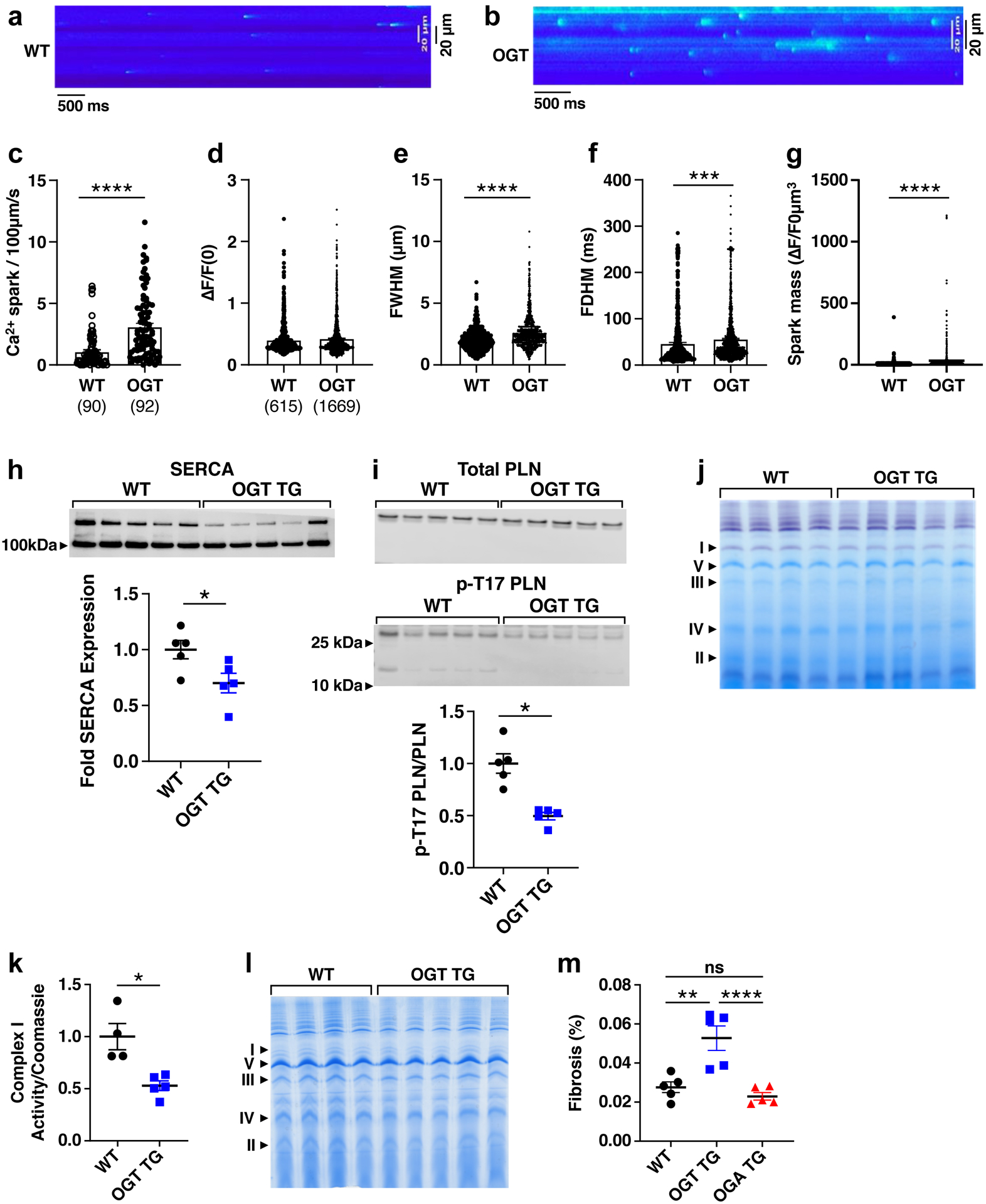
Representative images of calcium sparks recorded from ventricular myocytes from a. WT and b. OGT mice. c. Frequency of calcium sparks from ventricular myocytes of WT (n=90 cells, m=3 mice) and OGT (n=92 cells, m=4 mice) mice. d. Amplitude of calcium sparks from ventricular myocytes of WT (n=615 sparks) and OGT (n=1669 sparks) mice. e. Full width at half-maximum amplitude (FWHM) of calcium sparks from ventricular myocytes of WT and OGT mice. f. Full duration at half-maximum amplitude (FDHM). g. Spark mass based on equation: spark amplitudeX1.206XFWHM^3. h. SERCA and i. phospho-PLN/PLN levels measured by western blot in WT(n=5) and OGT TG (n=5) mice j. A blue native gel with WT (n = 4) and OGT TG (n = 5) mitochondrial isolates from heart stained for complex I activity. k. Summary data for complex I activity normalized to total mitochondrial protein expression. l. Total mitochondrial protein (1 heart/lane for panels j and l). m. Percent fibrosis of the left ventricular cavity in WT, OGT TG, OGA TG (all n=5) via Masson’s Trichrome stain. Data are represented as mean ± SEM, significance was determined using a Student’s two-tailed t test (****P<0.0001, ***P<0.001, **P<0.01, *P<0.05, ns=not significant).
Reduced oxidative phosphorylation in OGT TG cardiomyopathy
O-GlcNAcylation can modify thousands of proteins,23, 24, 26, 33 many of which could potentially contribute to cardiomyopathy. However, we focused on mitochondrial proteins because excessive O-GlcNAcylation has been implicated in compromising oxidative phosphorylation26 and failing myocardium is marked by depressed energetics34, 35. We evaluated mitochondrial respiration and tested complexes I, II and IV with a focus on complex I, based on previous work highlighting complex I as a target for O-GlcNAcylation and altering mitochondrial respiration26, 36, 37. We evaluated the activity of complex I (Fig 4j and 4k), using blue native page gel electrophoresis38, and normalized its activity to complex V protein expression (Fig 4l). We found marked reduction in complex I activity in OGT TG hearts compared to WT controls (Fig 4j and 4k). In contrast, the activity of complexes II and IV showed no significant change in activity between WT and OGT TG (Figure Va and Figure Vb in the Supplement). These data show that OGT TG overexpression leads to increased O-GlcNAcylation, and loss of complex I expression and activity, suggesting that OGT TG cardiomyopathy is due, at least in part, to depressed energetics.
No evidence for increased myocardial death or deterioration of intracellular Ca2+ transients in OGT TG hearts
Many types of acquired and genetic cardiomyopathies exhibit increased myocardial cell death39 and/or depressed intracellular Ca2+ concentration transients28. Surprisingly, we did not find differences in myocardial cell death (Figure Vc in the Supplement) and only a small amount of fibrosis (Fig 4m). These findings suggested that the notable loss of myocardial performance in OGT TG hearts was not due to loss of myocardium, or myocardial scarring. Heart muscle cells contract and relax under control of intracellular Ca2+ concentration transients, grading myofilament interactions to regulate muscle shortening40. We found that mechanically unloaded OGT TG ventricular myocytes had modest, but significantly reduced resting (diastolic) cytosolic Ca2+ concentrations (Figure VIa and Figure VIb in the Supplement), and faster decay of peak (systolic) cytosolic Ca2+ (Figure VIc in the Supplement) compared to WT controls. OGT TG and WT ventricular myocytes had similar intracellular Ca2+ transient amplitudes (Figure VId in the Supplement), and caffeine-releasable intracellular, sarcoplasmic reticulum, Ca2+ stores (Figure VIe in the Supplement). The findings suggested that, despite increased intracellular Ca2+ sparks (Fig 4a–g), systolic function in OGT TG was not impaired due to defective intracellular Ca2+ transients or paucity of sarcoplasmic reticulum Ca2+ reserve.
Rescue of dilated cardiomyopathy and premature mortality in OGT TG and OGA TG interbred mice
We interpreted our findings in the OGT TG mice to suggest that excessive O-GlcNAcylation levels were a direct cause of cardiomyopathy. However, we also considered that OGT over-expression could have unanticipated pathological actions, independent of O-GlcNAcylation. To further test these possibilities, we interbred OGT TG and OGA TG mice. O-GlcNAcylation levels from OGT TG x OGA TG interbred hearts were similar to O-GlcNAcylation levels in OGA TG hearts, and significantly less than in OGT TG heart lysates (Fig 5a and 5b). Double transgenic mouse heart weights, adjusted for body weight, were less than OGT TG, and were not different than WT littermates (Fig 5c). Echocardiography at 6–8 weeks of age revealed significant improvement in left ventricular ejection fraction (Fig 5d and 5e) and left ventricular dilation (Fig 5f). Due to the robust expression of OGT in mice overexpressing the OGT transgene signal intensity between WT and transgenic mice exceed the linear range of the imaging system. A representative western blot is shown in Fig 5g comparing OGT levels in WT, OGT TG, OGA TG and OGT x OGA TG mice where OGT TG and OGT x OGA TG protein levels were loaded at 1/10th that of WT. Quantification is approximate and OGT expression is significantly elevated in the interbred mice. Notably, the rescue of O-GlcNAcylation levels and myocardial function in double transgenic mice occurred despite elevated levels of cardiac OGT in OGT x OGA TG mice (Fig 5g and h). OGT transcript levels in OGT x OGA TG mice were increased when compared to OGT TG and WT mice (Fig 5i). OGA protein expression loaded 1:1 in WT and OGT TG (Fig 5j and 5k) was not significantly different. OGA protein expression levels loaded 1:1 between OGA TG and OGT x OGA TG mice were not significantly different (Fig 5j and 5l). Transcript analysis is notable for increased murine and decreased human OGA transcripts in OGT x OGA TG mice compared to OGA TG hearts (Fig 5m). Protein loading, assessed by Coomassie stain of heart lysates for OGT is shown in Fig 5n and for OGA in Fig 5o. We interpreted these data to suggest that OGT TG cardiomyopathy was due to significantly elevated O-GlcNAcylation levels, rather than a non-specific consequence of transgenic protein over-expression. Remarkably, OGT TG x OGA TG mice were protected from the increased premature death seen in OGT TG mice (Fig 5p). Similar to our experiments in OGT TG mice (Fig 4j – l), we evaluated mitochondrial function using blue native gel activity assays for complex I. We conducted mitochondrial functional studies in OGT TG isolated mitochondria prior to detectable reduction in heart function, using mice ages 6–8 weeks, in order to examine mitochondrial function and activity responses to OGT over-expression independent of overt cardiomyopathy. We found similar complex I activity in the interbred and WT mice (Fig 6a) normalized to complex V protein expression (Fig 6b). We then performed a complex I activity assay utilizing spectrophotometric quantification and confirmed that complex I activity was decreased, by approximately half, in OGT TG compared to WT and recovered to WT levels in double transgenic mice (Fig 6c). Finally, we assessed oxygen consumption rates in isolated mitochondria in the four experimental groups (WT, OGT TG, OGA TG, OGT x OGA TG), utilizing the Seahorse XF96 Analyzer, and confirmed decreased complex I - linked respiration in OGT TG versus WT (Fig 6d). We did not detect differences between WT and OGA TG, and between OGA TG and OGT x OGA TG mitochondria in the absence of ADP (state 2, Fig 6e). In contrast, we measured larger differences in state 3 respiration (i.e., in the presence of ADP) between OGT TG, WT and OGT x OGA TG cardiac mitochondria (Fig 6f). We did not detect a difference in the state 3 oxygen consumption rates between WT and OGT TG x OGA TG mitochondria, suggesting that complex I activity is equivalent in these groups. Taken together, these results support a concept that OGT TG cardiomyopathy is driven, at least in part, as a consequence of impaired metabolism and energetics due to loss of complex I expression and activity.
Figure 5. Rescue of OGT TG cardiomyopathy and premature mortality by OGA TG interbreeding.
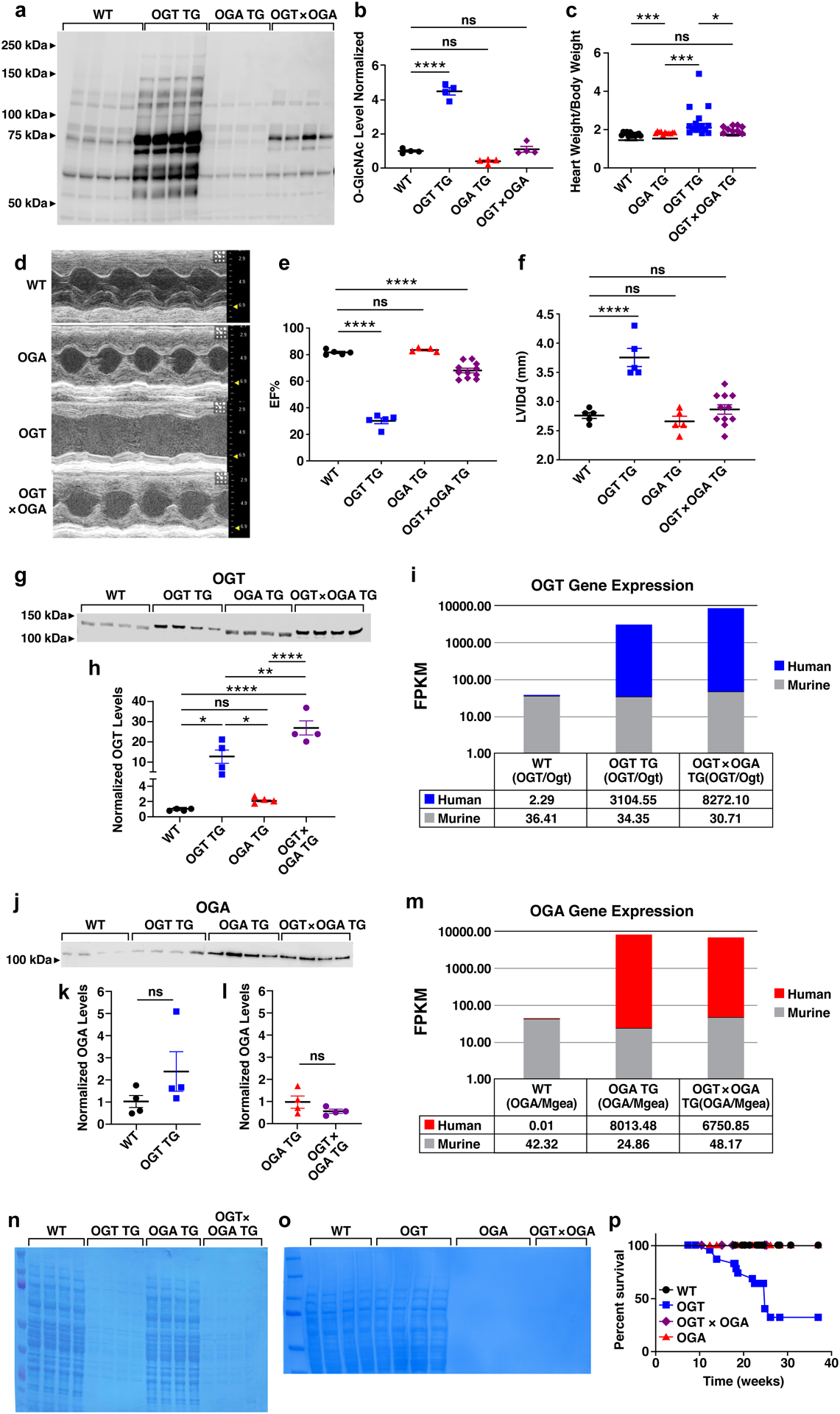
a. Western blot of total O-GlcNAcylation modified proteins in cardiac lysates from WT (n = 4), OGT TG (n = 4), OGA TG (n = 4), and OGT x OGA TG (n = 4) mice, and b. summary data. c. heart weight/body weight from WT (n = 12), OGA TG (n = 7), OGT TG (n = 15), OGT x OGA TG (n = 9) d. example images of left ventricular M - mode echocardiography of WT, OGA TG, OGT TG and OGT x OGA TG mice at 8–10 weeks of age. e. left ventricular ejection fraction (EF) and f. left ventricular internal diameter in diastole (LVIDd) g. western blot of OGT protein expression and summary data h. in hearts from WT (n = 4), OGT TG (n = 4), OGA TG (n = 4), and OGT x OGA TG (n = 4) mice (15 ug protein loaded in WT, OGA TG, 1.5 ug protein loaded in OGT TG, OGT x OGA TG) i. human (OGT) and murine (Ogt) transcript levels WT (n=6), OGT TG (n=6) and OGT x OGA TG (n=6) mice j. OGA protein expression in WT (n=4), OGT TG (n=4), OGA TG (n=4) and OGT x OGA TG (n=4) protein loading 25 ug WT and OGT TG mice, 0.25 ug OGA TG and OGT x OGA TG mice . k. summary data from WT (n = 4), OGT TG (n = 4) and l. OGA TG (n = 4), OGT x OGA TG (n = 4). m. human (OGA) and murine(Mgea) transcript levels in WT (n=6), OGT TG (n=6) and OGT x OGA TG (n=6) mice. n. Coomassie total protein loading control for OGT (15 ug protein loaded in WT, OGA TG, 1.5 ug protein loaded in OGT TG, OGT x OGA TG lanes) and o. OGA levels in WT, OGT TG, OGA TG and OGT x OGA TG, WT and OGT TG sample loaded at 25 ug protein, OGA TG and OGT x OGA TG samples loaded at 0.25 ug. p. Kaplan-Meier survival analysis for WT (n = 9), OGA TG (n = 11), OGT TG (n = 14) and OGT TG x OGA TG mice (n = 9). Data are represented as mean ± SEM, significance was determined using a two- tailed Student’s t test or log-rank test (survivorship). (****P<0.0001, ***P<0.001, **P<0.01 , *P<0.05, ns=not significant for all panels).
Figure 6. Reduced oxidative phosphorylation through impaired Complex I activity in OGT TG cardiomyopathy is rescued in OGT x OGA TG mice.

Blue Native gel WT (n=4), OGT TG (n=4), OGT x OGA TG (n=4) stained for a. complex I activity, and b. Coomassie stain for total mitochondrial protein expression from heart (1 heart/lane), and c. Complex I activity spectrophotometer assay summary data. d. Measurement of isolated mitochondria OCR after sequential addition of ADP, oligomycin, FCCP and rotenone/ antimycin A in the presence of substrates pyruvate, glutamate, and malate from all genotypes (WT, OGT TG, OGA TG n = 6, OGT x OGA TG n = 5). e. OCR measurement before ADP addition (the first time point in 6d) f. OCR measurement after ADP addition (the third time point in 6d). Data are represented as mean ± SEM, significance was determined using a 1 way ANOVA with Tukey’s multiple comparisons test. (**P<0.01, *P<0.05, ns=not significant).
Transcriptional reprogramming in OGT TG hearts
Given the known role of O-GlcNAcylation modification in modulating transcriptional pathways23 and the complexity of targets and pathways potentially under the influence of pathological O-GlcNAcylation, we hypothesized that multiple gene programs were affected in OGT TG cardiomyopathy. We compared polyA transcriptomes by RNA sequencing (RNA Seq) using hearts from age and gender matched WT, OGT TG, OGA TG and OGT TG x OGA TG interbred mice. Principle component analysis showed that each of these groups exhibited gene expression patterns that were more similar within than between groups (Fig 7a). In order to gain further insight into genes with the potential to drive O-GlcNAcylation cardiomyopathy, we focused on RNA Seq data gene sets that were significantly altered in OGT TG compared to WT hearts, and where these genes were repaired toward WT expression in the OGT x OGA TG interbred hearts. Gene and transcript expression levels (FPKM values) were computed with the tool Cuffdiff2 v.2.2.1 and imported into Partek GS v7.0 where we performed two-tailed Student’s t-test analyses comparing gene expression changes (multiple-test false discovery rate-adjusted q-value < 0.05) between the OGT TG and WT, and the OGT x OGA TG group versus WT. We found 2813 genes were significantly changed in the OGT TG versus WT hearts and 1798 individual genes showed significant expression differences between the WT and the OGT x OGA TG hearts. We applied QIAGEN Ingenuity Pathway Analysis and identified the top 5 significant canonical pathways and biological functions in OGT TG and OGT x OGA TG hearts using a regulation z-score and an overlap p-value (Fisher’s exact test; p < 0.05). The most prominent functional pathways identified included inhibition of oxidative phosphorylation pathways (Fig 7b) in the OGT TG hearts compared to hearts from OGT x OGA interbred mice, and activation of pathways involved in inflammation, inflammatory cell signalling and transcription (Figure VIIa in the Supplement).
Figure 7. Metabolic gene expression defects in OGT TG mice are recovered by OGA TG interbreeding.
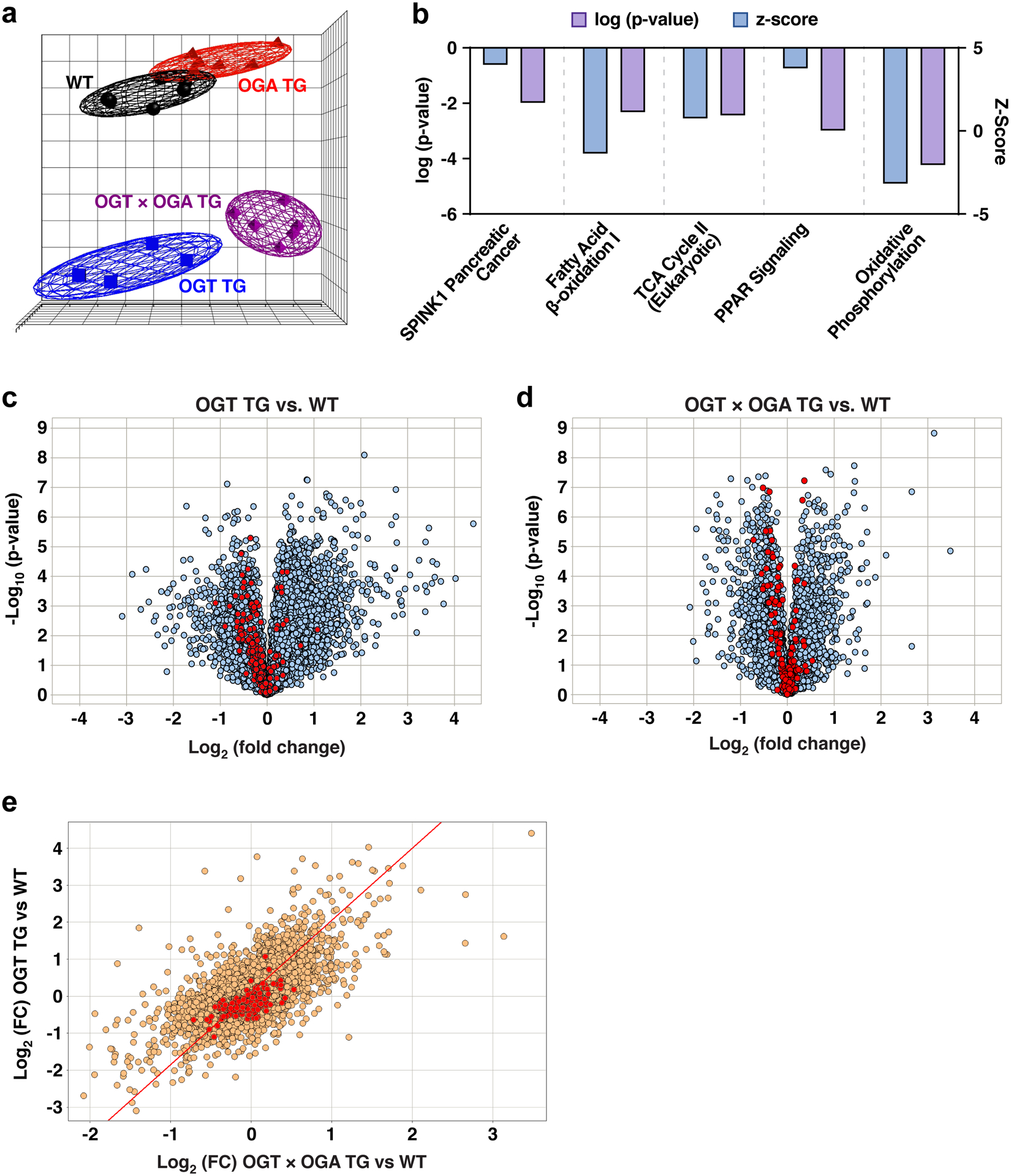
a. Principal component analysis of RNA Seq data from WT, OGT TG, OGA TG and OGT x OGA TG hearts (n = 6 in all groups, M = F) demonstrating clustering by similarity of transcriptome. b. The top 5 significant canonical pathways and biological functions identified by Ingenuity Pathway analysis with a regulation z-score and an overlap p-value (Fisher’s exact test; p < 0.05) for the comparison of significant differentially expressed genes in the dataset comparing OGT TG vs OGT x OGA TG. The Z-score represents the observed up or down regulation compared to known changes that are either activating or inhibiting, as derived from the literature and compiled in the Ingenuity® Knowledge Base. Pathways represented here are overall inhibited. c. Volcano plots representing gene set enrichment analysis of hallmark genes (as derived from the Molecular Signature Database) for oxidative phosphorylation comparing OGT TG versus WT and d. expression of oxidative phosphorylation genes in OGT x OGA TG versus WT. All genes are represented in grey and hallmark oxidative phosphorylation genes are represented in red. e. Scatter plot comparing overlapping genes between OGT TG versus WT and OGT x OGA TG versus WT (all genes in gold and hallmark gene set oxidative phosphorylation genes in red, FC = fold change).
The prominent changes exhibited in oxidative phosphorylation pathways align with our data from blue native gels and complex I activity assays (Fig 6) that suggested impairment in mitochondrial energetics, with subsequent recovery in the interbred OGT TG x OGA TG mice. We compared the hallmark oxidative phosphorylation gene set defined by Liberzon et al20. to genes identified by our RNA sequencing study, and identified 177 genes out of 200 genes listed in this gene set. We found that the majority of these genes (highlighted in red) are downregulated in OGT TG hearts compared to WT hearts (Fig 7c) and less gene downregulation in OGT x OGA TG compared to WT hearts (Fig 7d), suggesting that OGT TG hearts are deficient in expression of oxidative phosphorylation genes, and OGT x OGA TG interbreeding recovered expression of critical oxidative phosphorylation related genes towards WT levels. This observation was further supported by hierarchical clustering analysis (Figure VIIb in the Supplement) and scatter plot analysis (Fig 7e). These data appeared to confirm our experimental findings, and support published work41, highlighting defective mitochondrial energetics in response to excessive O-GlcNAcylation.
Discussion
The association between increased O-GlcNAcylation with diverse forms of cardiac stress is well established42–44. However, to our knowledge, no direct causal relationship has been established between increased O-GlcNAcylation and cardiomyopathy. Notably, mouse models with near elimination of myocardial OGT exhibited exaggerated responses to injury2. We interpret this important finding to indicate some increase in O-GlcNAcylation, likely within an acute or subacute timeframe, is required for optimal cardiac responses to stress. However, our results show therapeutic benefit from modest reduction of O-GlcNAcylation in OGA TG mice after transverse aortic constriction, and severe cardiomyopathy, heart failure and sudden death resulting from massive and chronic O-GlcNAcylation increases in OGT TG mice. Collectively, we interpret these data to strongly suggest excessive myocardial O-GlcNAcylation contributes to cardiomyopathy. In contrast to our new data, most work supporting a connection between excessive O-GlcNAcylation and cardiomyopathy has focused on hyperglycemic conditions, including in models of type I diabetes26. Our study provides new evidence that exposure to chronic, excessive O-GlcNAcylation is sufficient to cause dilated cardiomyopathy and premature death, even in the absence of hyperglycemia, diabetes or metabolic disease. This finding may be broadly important because excessive O-GlcNAcylation could add to other established cardiomyopathy mechanisms, and because it suggests that reversing or preventing excessive myocardial O-GlcNAcylation could be an innovative and effective therapeutic strategy for cardiomyopathy and heart failure.
We recognize that transgenic models may imperfectly represent the pathological potential of pathways linked to protein over-expression, and this caveat could apply to the OGT TG mouse. To our knowledge, all published data supporting a contribution of OGT overexpression to cardiomyopathy are from in vitro experiments performed using adeno-viral expression of OGT on ventricular myocytes45, or the inhibition of OGA utilizing PUGNAc or Thiamet G46. No studies have examined the effect of increased OGT protein levels on the heart in vivo. However, the findings that OGT TG cardiomyopathy and excessive myocardial O-GlcNAcylation were rescued by interbreeding with OGA TG mice, resulting in very high levels of transgenic protein over-expression, strongly suggests that OGT TG cardiomyopathy was a specific consequence of excessive OGT enzymatic activity. Intriguingly, some investigators have proposed a non-enzymatic role for OGT, acting as a scaffold or chaperone47, and our results do not exclude this possibility. We do not yet know if O-GlcNAcylation modified proteins are different in OGT TG hearts compared to WT hearts with increased O-GlcNAcylation due to pathological stress. The OGA TG mice have very mild myocardial hypertrophy, but no other measured phenotypic differences compared to WT littermate controls. Our finding that OGA TG mouse hearts had less stress induced O-GlcNAcylation and were resistant to TAC surgery suggests that transgenic overexpression of OGA is well tolerated, and is capable of reversing pathological O-GlcNAcylation.
Our RNA Seq studies showed a wide variety of transcripts were affected by increased O-GlcNAcylation through OGT overexpression. Expression of genes involved in metabolism was reordered toward WT levels in OGT TG x OGA TG hearts. The role of depressed energetics and deranged metabolism is a consistent finding in dilated cardiomyopathy in patients19, 35, 39 and, excessive O-GlcNAcylation is associated with defects in oxidative phosphorylation, in part, by actions in mitochondria13, 20, 26, 41, 48. We found reduced expression of a number of genes encoding complex I proteins, loss of complex I proteins, and decreased complex I activity in OGT TG hearts. Complex I gene expression, complex I proteins and complex I activity were remodeled toward WT levels in the OGT TG x OGA TG interbred hearts, suggesting that defective metabolism contributes to OGT TG cardiomyopathy.
In contrast to myocardial OGT overexpression, we found that myocardial OGA overexpression did not cause cardiomyopathy, and that OGA TG hearts were partly resistant to TAC induced cardiomyopathy. These results suggested to us that reducing chronic O-GlcNAcylation elevation by OGA activation could be a novel therapeutic strategy for cardiomyopathy. The cardiomyopathy phenotypes in OGT TG hearts were notable for an absence of large amounts of fibrosis, or myocardial death, features often associated with irreversible disease49. Thus, it may be that O-GlcNAcylation contributes to heart disease by mechanisms, including depressed energetics, that are reversible. An intriguing finding is enhanced arrhythmogenicity in the OGT TG mice with decreased levels of SERCA, phosphorylated PLN, and increased intracellular Ca2+ sparks, all features associated with failing hearts. It has been previously observed that prolonged periods of diabetes resulted in decreased SERCA protein levels46. Yokoe et al50 have described inhibition of PLN phosphorylation by O-GlcNAcylation in a diabetic model of cardiomyopathy and our findings are complementary to these data. Heart failure of diverse etiologies are often co-morbid with arrhythmia, wherein the regulation of SERCA plays a central role. Our findings highlight the relevance of developing O-GlcNAc based therapies to serve as novel regulators of SERCA, with the potential to become potent heart failure and anti-arrhythmic therapies. Given the public health consequences of cardiomyopathy and heart failure despite current treatments, the quest for improved therapeutic options remains an important goal for patients. Our findings are suggestive that attenuation of excessive O-GlcNAcylation could improve cardiomyopathy, heart failure and arrhythmias. These findings may serve as a springboard for the development of future therapies.
Supplementary Material
Clinical Perspective.
What is new?
Cardiomyopathy from diverse causes is marked by increased O-GlcNAcylation. Here we provide new genetic mouse models to control myocardial O-GlcNAcylation independent of pathological stress.
Genetically increased myocardial O-GlcNAcylation causes progressive dilated cardiomyopathy and premature death, while genetic reduction of myocardial O-GlcNAcylation is well tolerated at baseline and protective against pathological hypertrophy caused by transverse aortic constriction.
Excessive myocardial O-GlcNAcylation decreases activity and expression of mitochondrial complex I.
What are the clinical implications?
Increased myocardial O-GlcNAcylation is associated with a diverse range of clinical heart failure including aortic stenosis, hypertension, ischemia and diabetes.
Using novel genetic mouse models, we have provided new proof of concept data that excessive O-GlcNAcylation is sufficient to cause cardiomyopathy.
We have shown myocardial over-expression of O-GlcNAcase, an enzyme that reverses O-GlcNAcylation, is well tolerated at baseline, and improves myocardial responses to pathological stress.
Our findings suggest attenuating excessive myocardial O-GlcNAcylation could be beneficial in heart failure.
Acknowledgements:
Grateful thanks to Teresa Ruggle at the University of Iowa for help with graphic art.
Funding Support:
GWH, NZ, PU: NIH 1K12HL141952-01, PU: NIH T32 HL7227-43, AW: MOST-107-2636-B-002-001, GWH, PSB,NA : NIH P01HL107153, NZ: NIH R01 HL139640, MEA: NIH R35 HL140034, MEA, GWH: AHA Collaborative Science Award 17CSA33610107
Non-standard Abbreviations and Acronyms:
- UDP-GlcNAc
Uridine Diphosphate N-acetylglucosamine
- OGT
O-GlcNAc transferase
- OGA
O-GlcNAcase
- CaMKII
calcium-calmodulin kinase II
- PLN
phospholamban
- SERCA2a
sarcoplasmic-endoplasmic reticulum Ca2+ ATPase
Footnotes
References
- 1.Ida GL, Jan Magnus A, Heidi K, Eirik Q, Ivar S, Theis T, Geir C, Line MG-W and Cathrine RC. Cardiac O-GlcNAc signaling is increased in hypertrophy and heart failure. Physiological Genomics. 2012;44:162–172. [DOI] [PubMed] [Google Scholar]
- 2.Watson LJ, Facundo HT, Ngoh GA, Ameen M, Brainard RE, Lemma KM, Long BW, Prabhu SD, Xuan Y-T and Jones SP. O-linked β-N-acetylglucosamine transferase is indispensable in the failing heart. Proceedings of the National Academy of Sciences. 2010;107:17797–17802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hill BG, Annamalai L, Readnower RD, Brainard RE, Brittian KR, Cummins TD, DeMartino AM, Watson LJ, Jones SP and Long BW. Cardiomyocyte Ogt is essential for postnatal viability. American Journal of Physiology (Consolidated). 2014;306:H142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM and Bers DM. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature. 2013;502:372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lehmann LH, Jebessa ZH, Kreusser MM, Horsch A, He T, Kronlage M, Dewenter M, Sramek V, Oehl U, Krebs-Haupenthal J, von der Lieth AH, Schmidt A, Sun Q, Ritterhoff J, Finke D, Volkers M, Jungmann A, Sauer SW, Thiel C, Nickel A, Kohlhaas M, Schafer M, Sticht C, Maack C, Gretz N, Wagner M, El-Armouche A, Maier LS, Londono JEC, Meder B, Freichel M, Grone HJ, Most P, Muller OJ, Herzig S, Furlong EEM, Katus HA and Backs J. A proteolytic fragment of histone deacetylase 4 protects the heart from failure by regulating the hexosamine biosynthetic pathway. Nat Med. 2018;24:62–72. [DOI] [PubMed] [Google Scholar]
- 6.Zhu-Mauldin X, Marsh SA, Zou L, Marchase RB and Chatham JC. Modification of STIM1 by O-linked N-acetylglucosamine (O-GlcNAc) attenuates store-operated calcium entry in neonatal cardiomyocytes. 2012;287:39094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kronlage M, Dewenter M, Grosso J, Fleming T, Oehl U, Lehmann LH, Falcao-Pires I, Leite-Moreira AF, Volk N, Grone HJ, Muller OJ, Sickmann A, Katus HA and Backs J. O-GlcNAcylation of Histone Deacetylase 4 Protects the Diabetic Heart From Failure. Circulation. 2019;140:580–594. [DOI] [PubMed] [Google Scholar]
- 8.Rockman HA, Ross RS, Harris AN, Knowlton KU, Steinhelper ME, Field LJ, Ross J and Chien KR. Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:8277–8281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.deAlmeida AC, van Oort RJ and Wehrens XHT. Transverse aortic constriction in mice. Journal of Visualized Experiments: JoVE. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE Jr., Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ and Anderson ME. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005;11:409–17. [DOI] [PubMed] [Google Scholar]
- 11.Khoo MSC, Li J, Singh MV, Yang Y, Kannankeril P, Wu Y, Grueter CE, Guan X, Oddis CV, Zhang R, Mendes L, Ni G, Madu EC, Yang J, Bass M, Gomez RJ, Wadzinski BE, Olson EN, Colbran RJ and Anderson ME. Death, cardiac dysfunction, and arrhythmias are increased by calmodulin kinase II in calcineurin cardiomyopathy. Circulation. 2006;114:1352–1359. [DOI] [PubMed] [Google Scholar]
- 12.Heinen A, Raupach A, Behmenburg F, Hölscher N, Flögel U, Kelm M, Kaisers W, Nederlof R, Huhn R and Gödecke A. Echocardiographic Analysis of Cardiac Function after Infarction in Mice: Validation of Single-Plane Long-Axis View Measurements and the Bi-Plane Simpson Method. Ultrasound in Medicine & Biology. 2018;44:1544–1555. [DOI] [PubMed] [Google Scholar]
- 13.Zhao L, Feng Z, Yang X and Liu J. The regulatory Roles of O-GlcNAcylation in mitochondrial Homeostasis and metabolic Syndrome. Free radical research. 2016;50:1080–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu Y and Anderson ME. Reduced repolarization reserve in ventricular myocytes from female mice. Cardiovascular Research. 2002;53:763–769. [DOI] [PubMed] [Google Scholar]
- 15.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R and Salzberg SL. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology. 2013;14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT and Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature biotechnology. 2015;33:290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ducheix S, Magré J, Cariou B and Prieur X. Chronic O-GlcNAcylation and Diabetic Cardiomyopathy: The Bitterness of Glucose. Frontiers in Endocrinology. 2018;9:642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gélinas R, Mailleux F, Dontaine J, Bultot L, Demeulder B, Ginion A, Daskalopoulos EP, Esfahani H, Dubois-Deruy E, Lauzier B, Gauthier C, Olson AK, Bouchard B, Rosiers CD, Viollet B, Sakamoto K, Balligand J-L, Vanoverschelde J-L, Beauloye C, Horman S and Bertrand L. AMPK activation counteracts cardiac hypertrophy by reducing O-GlcNAcylation. Nature Communications. 2018;9:374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luptak I, Sverdlov AL, Panagia M, Qin F, Pimentel DR, Croteau D, Siwik DA, Ingwall JS, Bachschmid MM, Balschi JA and Colucci WS. Decreased ATP production and myocardial contractile reserve in metabolic heart disease. Journal of Molecular and Cellular Cardiology. 2018;116:106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov Jill P and Tamayo P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Systems. 2015;1:417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wende AR. Post-translational modifications of the cardiac proteome in diabetes and heart failure. Proteomics Clinical Applications. 2016;10:25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rindt H, Subramaniam A and Robbins J. An in vivo analysis of transcriptional elements in the mouse alpha-myosin heavy chain gene promoter. Transgenic Research. 1995;4:397–405. [DOI] [PubMed] [Google Scholar]
- 23.Hardivillé S and Hart GW. Nutrient Regulation of Signaling, Transcription, and Cell Physiology by O-GlcNAcylation. Cell Metabolism. 2014;20:208–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zachara N, Akimoto Y and Hart GW. The O-GlcNAc Modification. In: Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH, Schnaar RL and Seeberger PH, eds. Essentials of Glycobiology. 3rd ed. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2015. [PubMed] [Google Scholar]
- 25.Man J, Barnett P and Christoffels V. Structure and function of the Nppa–Nppb cluster locus during heart development and disease. Cellular and Molecular Life Sciences. 2018;75:1435–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Banerjee PS, Ma J and Hart GW. Diabetes-associated dysregulation of O-GlcNAcylation in rat cardiac mitochondria. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:6050–6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Santangeli P, Rame JE, Birati EY and Marchlinski FE. Management of Ventricular Arrhythmias in Patients With Advanced Heart Failure. Journal of the American College of Cardiology. 2017;69:1842–1860. [DOI] [PubMed] [Google Scholar]
- 28.Luo M and Anderson M. Mechanisms of Altered Ca2+ Handling in Heart Failure. Circulation Research. 2013;113:690–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guatimosim S, Guatimosim C and Song LS. Imaging calcium sparks in cardiac myocytes. Methods Mol Biol. 2011;689:205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schulman H and Anderson ME. Ca/Calmodulin-dependent Protein Kinase II in Heart Failure. Drug Discov Today Dis Mech. 2010;7:e117–e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He BJ, Joiner M-LA, Singh MV, Luczak ED, Swaminathan PD, Koval OM, Kutschke W, Allamargot C, Yang J, Guan X, Zimmerman K, Grumbach IM, Weiss RM, Spitz DR, Sigmund CD, Blankesteijn WM, Heymans S, Mohler PJ and Anderson ME. Oxidation of CaMKII determines the cardiotoxic effects of aldosterone. Nature Medicine. 2011;17:1610–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mesubi OO, Rokita AG, Abrol N, Wu Y, Chen B, Wang Q, Granger JM, Tucker-Bartley A, Luczak ED, Murphy KR, Umapathi P, Banerjee PS, Boronina T, Cole RN, Maier LS, Wehrens XH, Pomerantz JL, Song LS, Ahima R, Hart GW, Zachara NE and Anderson ME. Oxidized-CaMKII and O-GlcNAcylation cause increased atrial fibrillation in diabetic mice by distinct mechanisms. Journal of Clinical Investigation. 2021;131(2):e95747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hart GW, Slawson C, Ramirez-Correa G and Lagerlof O. Cross Talk Between O-GlcNAcylation and Phosphorylation: Roles in Signaling, Transcription, and Chronic Disease. Annual Review of Biochemistry. 2011;80:825–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murphy E, Ardehali H, Balaban R, DiLisa F, Dorn IIG, Kitsis R, Otsu K, Ping P, Rizzuto R, Sack M, Wallace D and Youle R. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association. Circulation Research. 2016;118:1960–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown DA, Perry JB, Allen ME, Sabbah HN, Stauffer BL, Shaikh SR, Cleland JGF, Colucci WS, Butler J, Voors AA, Anker SD, Pitt B, Pieske B, Filippatos G, Greene SJ and Gheorghiade M. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nature Reviews Cardiology. 2017;14:238–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma J, Liu T, Wei A-C, Banerjee P, O’Rourke B and Hart GW. O-GlcNAcomic Profiling Identifies Widespread O-Linked β-N-Acetylglucosamine Modification (O-GlcNAcylation) in Oxidative Phosphorylation System Regulating Cardiac Mitochondrial Function. The Journal of biological chemistry. 2015;290:29141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu Y, Suarez J, Fricovsky E, Wang H, Scott BT, Trauger SA, Han W, Hu Y, Oyeleye MO and Dillmann WH. Increased Enzymatic O-GlcNAcylation of Mitochondrial Proteins Impairs Mitochondrial Function in Cardiac Myocytes Exposed to High Glucose. The Journal of Biological Chemistry. 2009;284:547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nijtmans LGJ, Henderson NS and Holt IJ. Blue Native electrophoresis to study mitochondrial and other protein complexes. Methods (San Diego, Calif). 2002;26:327–334. [DOI] [PubMed] [Google Scholar]
- 39.Harvey PA and Leinwand LA. The cell biology of disease: cellular mechanisms of cardiomyopathy. The Journal of Cell Biology. 2011;194:355–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. [DOI] [PubMed] [Google Scholar]
- 41.Ma J, Banerjee P, Whelan SA, Liu T, Wei A-C, Ramirez-Correa G, McComb ME, Costello CE, O’Rourke B, Murphy A and Hart GW. Comparative Proteomics Reveals Dysregulated Mitochondrial O-GlcNAcylation in Diabetic Hearts. Journal of Proteome Research. 2016;15:2254–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chatham JC and Marchase RB. Protein O-GlcNAcylation: A critical regulator of the cellular response to stress. 2010;5:49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ding F, Yu L, Wang M, Xu S, Xia Q and Fu G. O-GlcNAcylation involvement in high glucose-induced cardiac hypertrophy via ERK1/2 and cyclin D2. Amino Acids. 2013;45:339–349. [DOI] [PubMed] [Google Scholar]
- 44.Jensen RV, Zachara NE, Nielsen PH, Kimose HH, Kristiansen SB and Bøtker HE. Impact of O-GlcNAc on cardioprotection by remote ischaemic preconditioning in non-diabetic and diabetic patients. Cardiovascular Research. 2013;97:369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clark RJ, McDonough PM, Swanson E, Trost SU, Suzuki M, Fukuda M and Dillmann WH. Diabetes and the accompanying hyperglycemia impairs cardiomyocyte calcium cycling through increased nuclear O-GlcNAcylation. The Journal of Biological Chemistry. 2003;278:44230–44237. [DOI] [PubMed] [Google Scholar]
- 46.Hu Y, Belke D, Suarez J, Swanson E, Clark R, Hoshijima M and Dillmann WH. Adenovirus-mediated overexpression of O-GlcNAcase improves contractile function in the diabetic heart. Circulation Research. 2005;96:1006–1013. [DOI] [PubMed] [Google Scholar]
- 47.Ong Q, Han W and Yang X. O-GlcNAc as an Integrator of Signaling Pathways. Frontiers in Endocrinology. 2018;9:599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tan EP, Villar MT, E L, Lu J, Selfridge JE, Artigues A, Swerdlow RH and Slawson C. Altering O-Linked β-N-Acetylglucosamine Cycling Disrupts Mitochondrial Function. The Journal of Biological Chemistry. 2014;289:14719–14730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Berlo JH, Maillet M and Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. 2013;123:37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yokoe S, Asahi M, Takeda T, Otsu K, Taniguchi N, Miyoshi E and Suzuki K. Inhibition of phospholamban phosphorylation by O-GlcNAcylation: implications for diabetic cardiomyopathy. Glycobiology. 2010;20:1217–26. [DOI] [PubMed] [Google Scholar]
- 51.Khoo MSC, Li J, Singh MV, Yang Y, Kannankeril P, Wu Y, Grueter CE, Guan X, Oddis CV, Zhang R, Mendes L, Ni G, Madu EC, Yang J, Bass M, Gomez RJ, Wadzinski BE, Olson EN, Colbran RJ and Anderson ME. Death, cardiac dysfunction, and arrhythmias are increased by calmodulin kinase II in calcineurin cardiomyopathy. Circulation. 2006;114:1352–1359. [DOI] [PubMed] [Google Scholar]
- 52.Heinen A, Raupach A, Behmenburg F, Hölscher N, Flögel U, Kelm M, Kaisers W, Nederlof R, Huhn R and Gödecke A. Echocardiographic Analysis of Cardiac Function after Infarction in Mice: Validation of Single-Plane Long-Axis View Measurements and the Bi-Plane Simpson Method. Ultrasound in Medicine & Biology. 2018;44:1544–1555. [DOI] [PubMed] [Google Scholar]
- 53.Groves JA and Zachara NE. Characterization of tools to detect and enrich human and mouse O-GlcNAcase. Glycobiology. 2017;27:791–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luczak ED, Wu Y, Granger JM, Joiner MA, Wilson NR, Gupta A, Umapathi P, Murphy KR, Reyes Gaido OE, Sabet A, Corradini E, Tseng WW, Wang Y, Heck AJR, Wei AC, Weiss RG and Anderson ME. Mitochondrial CaMKII causes adverse metabolic reprogramming and dilated cardiomyopathy. Nat Commun. 2020;11:4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu Y and Anderson ME. Reduced repolarization reserve in ventricular myocytes from female mice. Cardiovascular Research. 2002;53:763–769. [DOI] [PubMed] [Google Scholar]
- 56.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R and Salzberg SL. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology. 2013;14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT and Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature biotechnology. 2015;33:290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shen S, Park JW, Lu Z-x, Lin L, Henry MD, Wu YN, Zhou Q and Xing Y. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.