

Abstract
Defects in PTEN, a critical tumor suppressor, are associated with tumorigenesis and aberrant organ sizes. It has been shown that heterozygous PTEN loss increases brains and neuron size, while the specific loss of nuclear PTEN has the opposite effect. Here, we investigate the impact of a combination of heterozygous PTEN loss and nuclear PTEN loss on the size of various organs, including the brain, liver, thymus, spleen, and inguinal lymph node. We found that the effect of the combination varies among organs. Notably, the combination of heterozygous PTEN loss and nuclear PTEN loss restored the normal size of brains and neurons. In contrast, the liver’s size was unaffected by either single PTEN defects or their combination. Strikingly, the size of the inguinal lymph node was greatly increased due to lymphoma by the combination of the two PTEN defects. These data suggest that nuclear PTEN and non-nuclear PTEN function in an antagonistic manner in the brain while acting synergistically in the inguinal lymph node.
Keywords: PTEN, Nuclear PTEN, Brain, Neuron, Lymph Node, Lymphoma
1. Introduction
The tumor suppressor PTEN (phosphatase and tensin homolog deleted on chromosome ten) is a multi-function protein with lipid and protein phosphatase activities [1–3]. Somatic PTEN mutations have been found in various cancers, such as high-grade glioblastoma, prostate, breast, endometrial, and thyroid carcinoma. Germline mutations of PTEN also cause Cowden’s syndrome, which shows hamartoma of various tissues, such as the brain, breast, and intestine, and has a high risk of cancer in the thyroid and breast [4]. Like human patients, heterozygous loss of PTEN in mice leads to hamartoma-like features and high incidences of epithelial tumors [5–8]. These mice display a high penetrance of lymphadenopathy and splenomegaly, leading to a lethal autoimmune disease [9].
Besides tumor suppression, PTEN plays a vital role in brain development and function [10,11]. Mutations of PTEN are linked to autism spectrum disorder accompanied by macrocephaly and epilepsy in human patients [12–15]. In mouse models, brain-specific homozygous knockout of PTEN increases the size of the brain and the soma of neurons, hypertrophic dendrites, axonal terminus with increased synapses, and granule cell dysplasia in the cerebellum and dentate gyrus [10,16–18]. These mice show seizures, ataxia, and behavior abnormalities, resembling human patients with Lhermitte-Duclos disease, a cerebellar tumor associated with Cowden’s syndrome.
In cells, PTEN is located at multiple locations, including the plasma membrane and nucleus. At the plasma membrane, PTEN dephosphorylates phosphatidylinositol (3,4,5)-triphosphate to negatively regulate the intracellular signal transduction pathway mediated by phosphatidylinositol 3-kinase (PI3K), AKT, and mTORC1 [19–21]. It has been suggested that macrocephaly results from enhanced PI3K-AKT-mTOR signaling. In the nucleus, PTEN controls DNA repair, genome maintenance, and cell cycle progression [22,23]. Ubiquitination of lysine at residue 13 (K13) is critical for the nuclear localization of PTEN, and its arginine substitution (K13R) blocks the accumulation in the nucleus, leaving most of PTEN in the cytoplasm [24–26]. By CRISPR/Cas9 genome editing, we generated nuclear PTEN deficient mutant mice in which the nuclear localization of PTEN is specifically inhibited without affecting its plasma membrane localization [24,25]. In contrast to systemic PTEN knockout mice, nuclear PTEN deficient mice display microcephaly with smaller soma of neurons in the cerebellum, cerebral cortex, and hippocampus [24].
These data suggest that PTEN differently regulates the size of brains and neurons at different intracellular locations. Furthermore, unlike systemic PTEN knockout mice, nuclear PTEN deficient mice do not display spontaneous tumors [24,25,27]. Instead, nuclear PTEN deficient mice produce liver cancers when exposed to mutagen and hepatotoxin, suggesting that nuclear PTEN loss enhances the sensitivity to liver carcinogenesis [24,25,27]. It is currently unknown how the functional relationship between PTEN at the plasma membrane and nucleus affects organ size control and tumorigenesis. In the current work, we address this outstanding question by crossing heterozygous PTEN knockout and nuclear PTEN deficient mice.
2. Materials and Methods
2.1. Mice
All animal work was performed according to the guidelines established by the Johns Hopkins University Committee on Animal Care. PTENK13R,D384V/K13R,D384V mice have been previously described [24,25,27]. PTEN+/− mice were generated by crossing PTENflox/flox (006440) and CMV-Cre transgenic mice (006054), both of which were obtained from Jackson Laboratory. To produce PTENK13R,D384V/− mice, PTENK13R,D384V/K13R,D384V mice were crossed to PTEN+/− mice.
2.2. Immunofluorescence microscopy
Mice were anesthetized by intraperitoneal injection of Avertin and fixed by cardiac perfusion of ice-cold 4% paraformaldehyde in PBS [24,25,27]. The tissues were dissected and further fixed in 4% paraformaldehyde in PBS for 3 h at 4°C. The samples were incubated in PBS containing 30% sucrose overnight and frozen in optimal cutting temperature compound (Fisher Scientific; 23–730-571,) in a Tissue-Tek Cryomold (Sakura Finetek USA; 4566). Frozen tissue blocks were sectioned and mounted on Superfrost Plus Microscope Slides (12–550-15; Fisher Scientific). Sections were subjected to antigen retrieval with 10 mM citrate buffer using a microwave oven and incubated with the primary antibodies to Car8 (1:500, Frontier Institute, Car8-Go-Af780–1), NeuN (1:200, Proteintech; 26975–1-AP), and PTEN (1:100, Cell Signaling Technology; 9559) at 4°C overnight. After washes, the samples were incubated with appropriate fluorescently-labeled secondary antibodies at room temperature for 1 h. DAPI (1 μg/ml) was used to stain nuclear DNA. Samples were viewed by Zeiss LSM780 confocal scanning microscope equipped with 10× or 63× objectives. The nuclear and cytosolic fluorescent intensities were measured and averaged from three different positions in each neuron using NIH ImageJ software.
2.3. Immunohistochemistry and histology
For immunohistochemistry, cryosections were stained with antibodies to B220 (1:100, Invitrogen; 14–0452-82) and CD3 (1:100, Sigma; SAB5500058). After washes with PBS, the samples were incubated with HRP-conjugated secondary antibody for 1 h at room temperature and stained with 3,3'-diaminobenzidine. For histology, paraformaldehyde-fixed tissues were embedded in paraffin at the Johns Hopkins School of Medicine Pathology Core. Paraffin sections were cut, and stained with hematoxylin and eosin. The samples were viewed using an Olympus BX51 microscope equipped with a DP-70 color camera.
2.4. Western blotting
Tissues were harvested from mice, flash-frozen in liquid nitrogen, and homogenized in RIPA buffer (Cell Signaling Technology; 9806) containing phosphatase inhibitor cocktail 2 and 3 (Sigma; P5726 and P0044, respectively) [24,25,27]. Lysates were centrifuged at 14,000 × g for 10 min, and the supernatants were collected. Proteins were separated by SDS-PAGE and transferred onto Immobilon-FL (Millipore; IPFL00010). After blocking in 3% BSA/PBS/Tween-20 for 1 h at room temperature, the blots were incubated with primary antibodies: PTEN (Cell Signaling Technology; 9559), NeuN (Proteintech; 26975–1-AP), AldhL1 (EMD Millipore; MABN495), GFAP (Sigma; G3893), and GAPDH (Thermo Fisher; MA5–15738). Immunocomplexes were visualized using appropriate fluorescent-labeled secondary antibodies and detected using a PharosFX Plus molecular imager (Bio-Rad).
3. Results
3.1. Antagonistic effects of PTEN+/− and PTENK13R,D384V/K13R,D384V on brain size and neuronal morphology
Nuclear PTEN deficient mice carry two mutations, K13R and D384V, which together block the accumulation of PTEN effectively in the nucleus [24,25,27]. Heterozygous knockout mice for PTEN were produced by crossing PTENflox/flox mice to CMV-Cre mice [28,29]. CMV-Cre was then crossed out. By breeding, we generated wild-type mice (WT), heterozygous PTEN knockout mice (PTEN+/−), nuclear PTEN deficient mice (PTENK13R,D384V/K13R,D384V), and PTENK13R,D384V/− mice. These mice showed similar body sizes (Fig. 1A and B). In contrast, the size of the brain was altered. Brains were larger in PTEN+/− mice and smaller in PTENK13R,D384V/K13R,D384V mice (Fig. 1C and D). Notably, PTENK13R,D384V/− mice restored the brain’s normal size (Fig. 1C and D). In contrast, the size of livers was not affected in these PTEN mutant mice (Fig. 1C). Further analysis of the brain’s sub-regions showed that the cerebrum and cerebellum were significantly larger in PTEN+/− mice and smaller in PTENK13R,D384V/K13R,D384V mice (Fig. 1D). Again, the size of both cerebrum and cerebellum were restored to normal in PTENK13R,D384V/− mice (Fig. 1D). These data suggest that nuclear PTEN loss and heterozygous PTEN loss antagonistically change the size of the brain, cerebrum, and cerebellum.
Figure 1. Brain size in PTEN mutant mice.
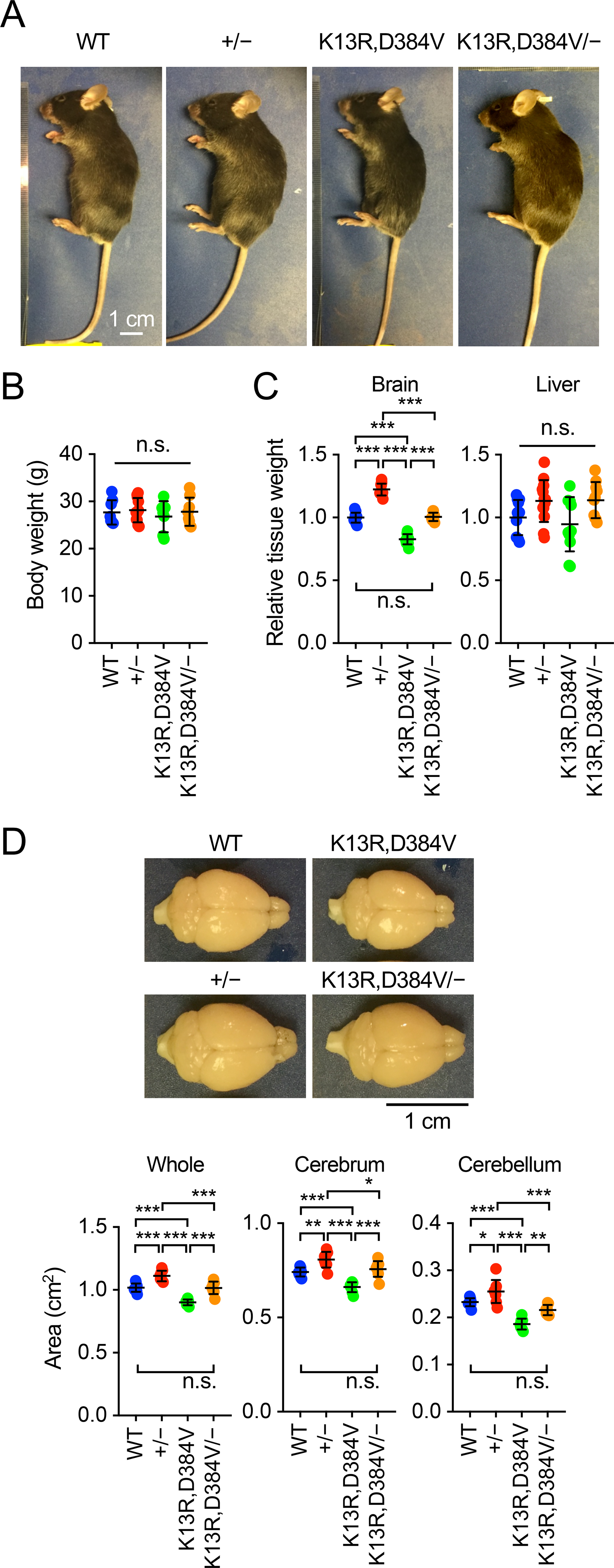
(A) General appearance of WT, PTEN+/−, PTENK13R,D384V/K13R,D384V, and PTENK13R,D384V/− male mice at 8–14 weeks old. (B) Bodyweight at 7–15 weeks old. Values are mean ± SD (n = 7–11 male mice). (C) Weight of the brain and liver relative to body weight at 7–15 weeks old. Values are mean ± SD (n = 10–13 male mice). (D) The area of the whole brain, cerebrum, and cerebellum was determined from the top view at 7–15 weeks old. Values are mean ± SD (n = 8–10 mice). Statistical analysis was performed using one-way ANOVA with post-hoc Tukey: *p < 0.05, **p < 0.01, ***p < 0.001.
The cerebellum consists of two layers, the molecular and granular layers. The molecular layer mainly contains the soma of large Purkinje cells and their highly extended and branched dendrites. The granular layer has small granule cells. Consistent with the changes observed in the whole cerebellum (Fig. 1D), immunofluorescence microscopy of sagittal sections using antibodies to Car8 (a Purkinje cell marker) and NeuN (a neuron marker used to visualize granular layer) showed that the area of the cerebellum was increased in PTEN+/− mice, decreased in PTENK13R,D384V/K13R,D384V mice, and restored in PTENK13R,D384V/− mice (Fig. 2A and B). These changes result from combined changes in both molecular and granular layers, which were increased in PTEN+/− mice, decreased in PTENK13R,D384V/K13R,D384V, and restored in PTENK13R,D384V/− mice (Fig. 2C-E). Furthermore, PTEN+/− mice showed an increased frequency of Purkinje neurons that extend two primary dendrites from the soma (Fig. 2F and G). This increase was compensated in PTENK13R,D384V/− mice (Fig. 2F and G). PTENK13R,D384V/K13R,D384V mice showed the normal frequency (Fig. 2F and G).
Figure 2. Size of neurons and dendritic branching in PTEN mutant cerebellum.
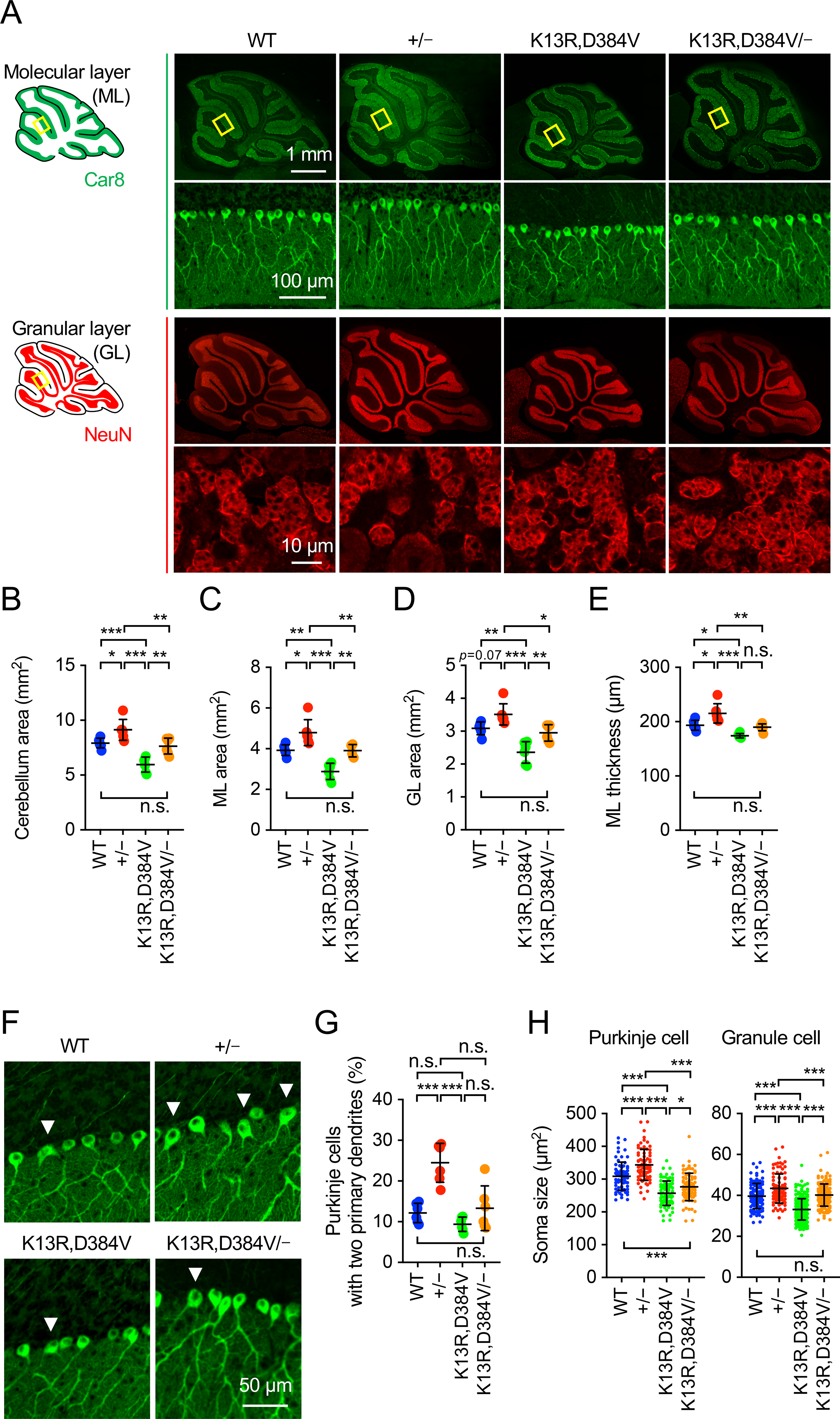
(A) Sagittal sections of the cerebellum cut in the midline were immunostained with anti-Car8 and anti-NeuN antibodies. Boxed regions are enlarged. (B-E) Quantification of the size of the cerebellum: area of the cerebellum (B), area of the molecular layer (C), area of the granular layer (D), and the thickness of the molecular layer (E). Values are mean ± SD (n = 6 mice). (F and G) Sagittal sections of the cerebellum in the midline were immunostained with anti-Car8 antibodies. The percentage of Purkinje cells that extend more than one dendrite from the soma was quantified. Values are mean ± SD (n = 6 mice). (H) Soma area of Purkinje cells (n = 73–85) and granule cells (n = 93–261) was quantified using Car8 and NeuN staining, respectively. Values are mean ± SD. Statistical analysis was performed using one-way ANOVA with post-hoc Tukey: *p < 0.05, **p < 0.01, ***p < 0.001.
To examine the size of individual neurons, we quantified the soma of Purkinje cells by Car8 staining and granule cells by NeuN staining. We found that the soma size of both neurons was increased in PTEN+/− mice, decreased in PTENK13R,D384V/K13R,D384V mice, and returned to WT levels in PTENK13R,D384V/− mice (Fig. 2H). These data suggest that the loss of nuclear PTEN and the heterozygous loss of PTEN show opposite effects on the sizes of neurons.
3.2. Intracellular distribution of PTEN in PTEN+/−, PTENK13R,D384V/K13R,D384V, and PTENK13R,D384V/− neurons
To examine the localization of PTEN in PTEN+/−, PTENK13R,D384V/K13R,D384V, and PTENK13R,D384V/neurons, we performed quantitative immunofluorescence microscopy of sagittal sections of the cerebellum using antibodies to PTEN and the Purkinje cell marker Car8 (Fig. 3A). In WT Purkinje cells, the quantification of PTEN signals showed approximately equal intensity in the nucleus and cytosol (Fig. 3A and B). In PTEN+/− mice, although the total intensity is decreased, the ratio of PTEN signal in the nucleus relative to that in the cytosol was unaffected (Fig. 3A and B). In contrast, as expected [24], PTENK13R,D384V/K13R,D384V mice showed a dramatic decrease in the relative intensity of PTEN in the nucleus (Fig. 3A and B). This decrease remained unchanged in PTENK13R,D384V/− mice (Fig. 3A and B). As a control, we analyzed the signal intensity of Car8. Distinct from PTEN, Car8 was predominantly located in the cytosol, and the signal ratio between the nucleus and cytosol were similar in WT, PTEN+/−, PTENK13R,D384V/K13R,D384V, and PTENK13R,D384V/− Purkinje cells (Fig. 3A and B). These data indicate that the K13R and D384V mutations block the localization of PTEN in the nucleus, and that decreasing the total amount of PTEN does not change the relative distribution of PTEN in the nucleus.
Figure 3. PTEN localization and neuron size in PTEN mutant mice.
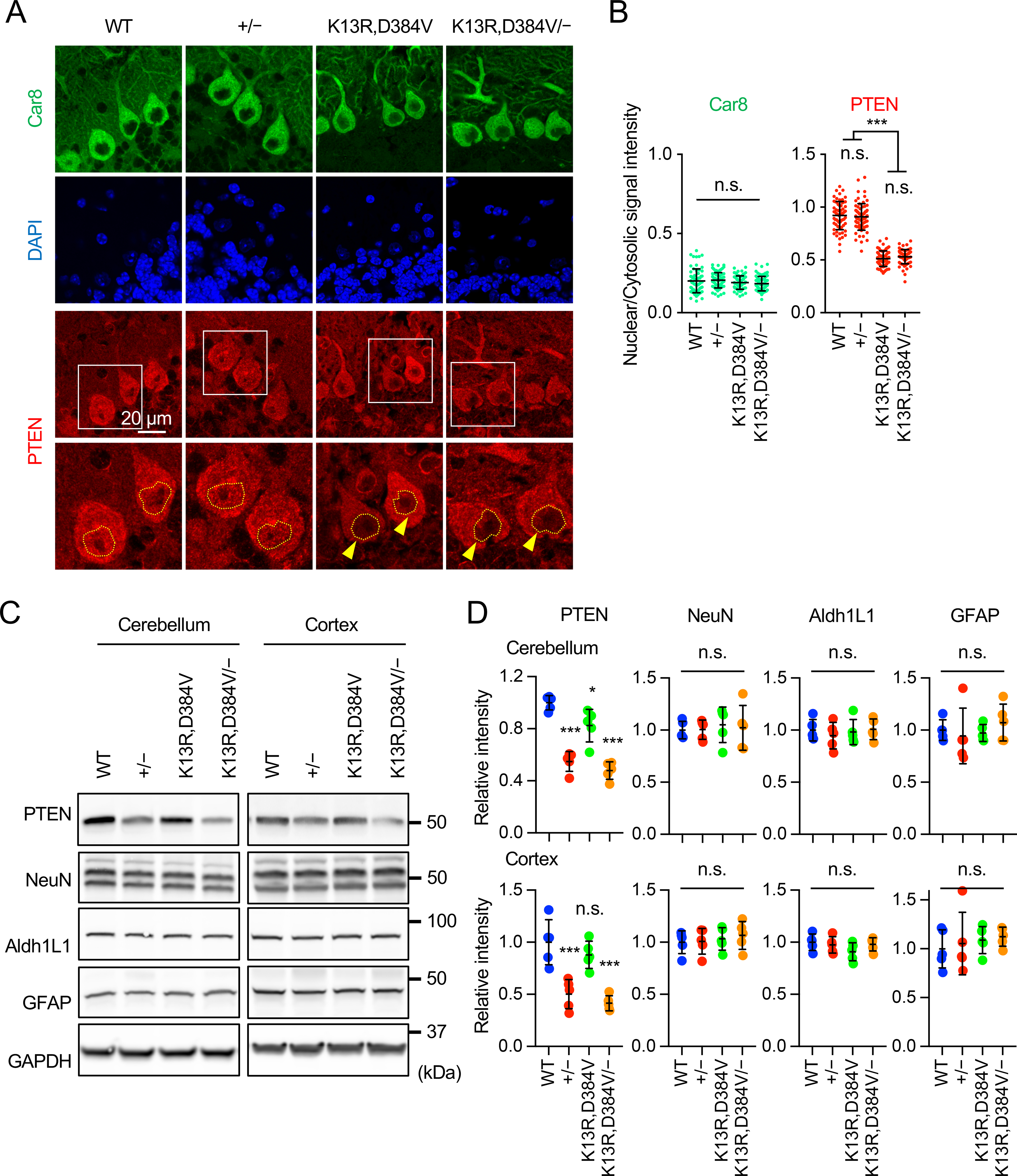
(A) Sagittal sections of the cerebellum in the midline of the indicated mice were immunostained with antibodies to Car8 and PTEN along with DAPI. Boxed regions are enlarged. (B) The signal intensity of Car8 and PTEN in the nucleus relative to the cytosol was determined. Values are mean ± SD (n = 63–73 Purkinje cells). (C) Western blotting of the cerebellum and cerebral cortex from the indicated mice at 7–15 weeks old using antibodies PTEN, NeuN, Aldh1L1, GFAP, and GAPDH. (D) Quantification of band intensity. Values are mean ± SD (n = 5 mice). Statistical analysis was performed using one-way ANOVA with post-hoc Tukey: *p < 0.05, ***p < 0.001.
To examine the effects of decreases in levels of nuclear PTEN and total PTEN on the ratio of neurons and astrocytes in the brain, we analyzed levels of NeuN (a neuron marker), Aldh1L1 (an astrocyte marker), and GFAP (an astrocyte marker), along with PTEN in the cerebellum and cerebral cortex (Fig. 3C and D). We found that PTEN levels were dramatically decreased in PTEN+/− and PTENK13R,D384V/− mice. In contrast, levels of NeuN, Aldh1L1, and GFAP were equivalent in WT, PTEN+/−, PTENK13R,D384V/K13R,D384V, and PTENK13R,D384V/− mice (Fig. 3C and D). Therefore, it appears that the ratio of neurons and astrocytes is not grossly affected in the PTEN mutant mice.
Figure 4. Neoplastic lymph nodes in PTENK13R,D384V/− mice.
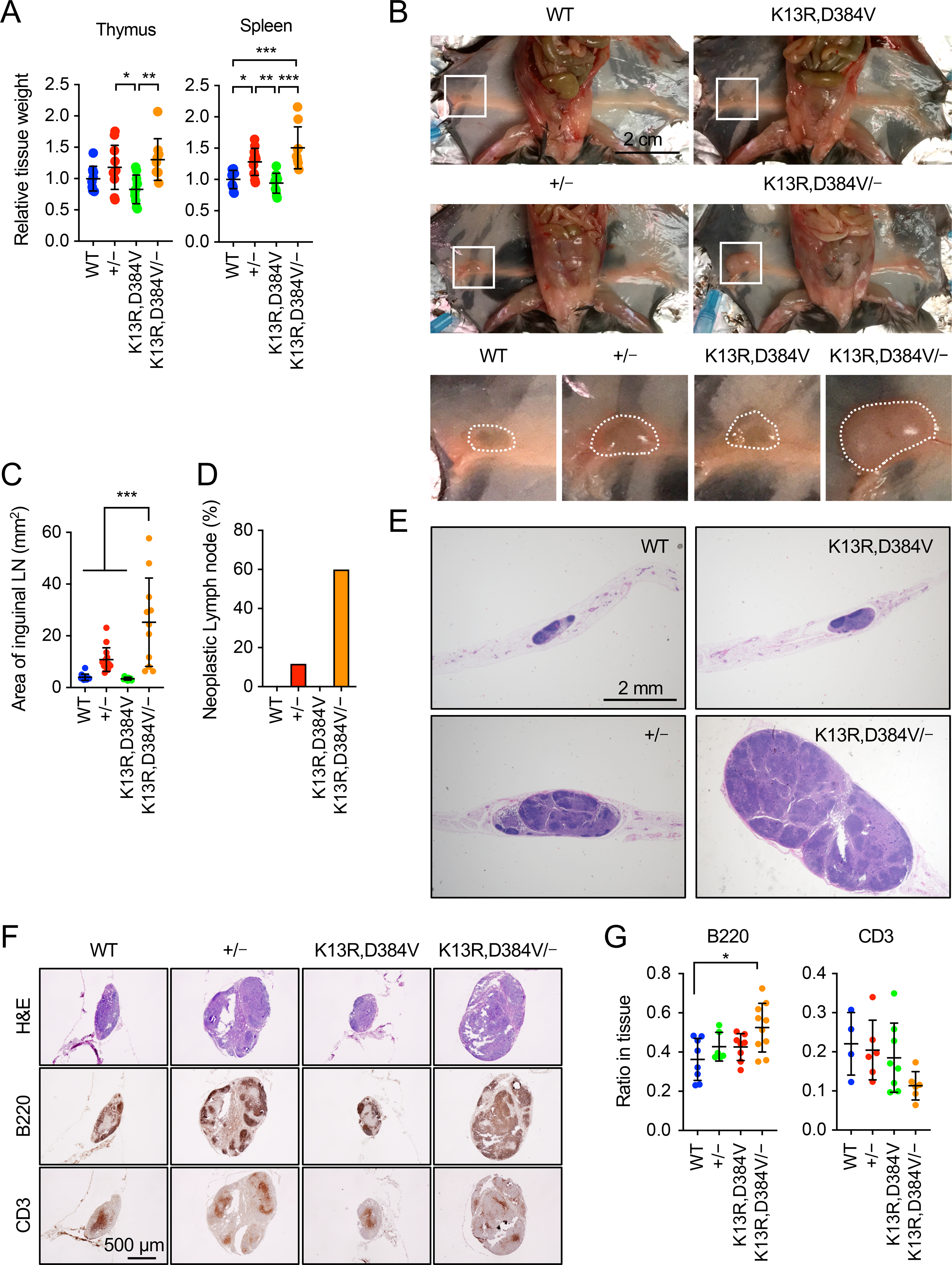
(A) Weight of the thymus and spleen relative to body weight at 7–15 weeks old. Values are mean ± SD (n = 10–13 male mice). (B) Inguinal lymph nodes in WT, PTEN+/−, PTENK13R,D384V/K13R,D384V, and PTENK13R,D384V/− mice at 9–16 weeks old. Boxed regions are enlarged. (C) Areas of inguinal lymph nodes were quantified. Values are mean ± SD (n = 9–14). (D) The frequency of mice with neoplastic lymph nodes. Values are mean (n = 12–17). (E) H&E staining of lymph node sections. (F) Serial sections of inguinal lymph nodes were immunostained with antibodies to B220 and CD3, and H&E stained. (G) The relative area positive for B220 or CD3 in lymph nodes was determined. Values are mean ± SD (n= 6–10 for B220, n= 4–8 for CD3). Statistical analysis was performed using one-way ANOVA with post-hoc Tukey: *p < 0.05, **p < 0.01, ***p < 0.001.
3.3. Synergistic effects of PTEN+/− and PTENK13R,D384V/K13R,D384V on the size of immune organs
In addition to the brain, we found that the sizes of the immune organs — the thymus, spleen, and inguinal lymph node — were also altered in the PTEN mutant mice (Fig. 4A). PTEN+/− mice showed increased weights of the thymus and spleen, while PTENK13R,D384V/K13R,D384V mice displayed normal weight. In contrast to the brain, the average weight of the thymus and spleen was modestly increased in PTENK13R,D384V/− mice, although the differences were not significant (Fig. 4A).
Notably, we found a dramatic change in the inguinal lymph node of PTENK13R,D384V/− mice. Consistent with previous studies [30], PTEN+/− mice displayed hypertrophy of inguinal lymph nodes (Fig. 4B and C). This hypertrophy was greatly enhanced in PTENK13R,D384V/− mice (Fig. 4B and C). PTENK13R,D384V/K13R,D384V mice showed a normal size of inguinal lymph node (Fig. 4B and C). Also, the frequency of lymph node hypertrophy was higher in PTENK13R,D384V/− mice than PTEN+/mice (Fig. 4D). Hematoxylin and eosin (H&E) staining revealed that lymphoma is formed in PTEN+/− and PTENK13R,D384V/− mice (Fig. 4E). Consistent with a further increased size of the inguinal lymph node, lymphoma was larger in PTENK13R,D384V/− mice compared with PTEN+/− mice (Fig. 4E). WT and PTENK13R,D384V/K13R,D384V mice did not develop lymphoma. To test what types of cells are increased in the lymph node, we performed immunohistochemistry using antibodies to B220 (a marker for B-cells) and CD3 (a marker for T-cells). The area positive for these antibodies was normalized relative to the total area of inguinal lymph nodes. We found that the relative CD3-positive area did not change in WT, PTEN+/−, PTENK13R,D384V/K13R,D384V, and PTENK13R,D384V/mice (Fig. 4F and G). In contrast, the relative B220-positive area was increased in PTENK13R,D384v/− mice (Fig. 4F and G). These data suggest the enhanced proliferation of B-cell lineages in PTENK13R,D384V/− mice (Fig. 4F and G).
4. Discussion
PTEN is a master regulator of the size and morphology of the brain and neurons. Heterozygous loss of PTEN in PTEN+/− mice induces macrocephaly through enhanced PI3K-mTORC1 signaling, leading to excess cell growth [16,17,31–34]. In contrast, we have shown that decreasing levels of PTEN in the nucleus in PTENK13R,D384V/K13R,D384V mice results in microcephaly with reduced sizes of multiple types of neurons in the cerebellum, cerebral cortex, and hippocampus [24]. In the current work, we found that normal brain and neuron size is restored in PTENK13R,D384V/− mice. Our data suggest that the heterozygous loss of PTEN and the decrease in nuclear PTEN levels antagonistically change cell growth in the brain. We do not yet know how such opposing effects are generated. Although one possible mechanism is regulation of the PI3K-mTORC1 pathway, we found that this pathway is not affected in PTENK13R,D384V/K13R,D384V brains [24]. It is possible that other signaling mechanisms that control cell growth contribute to the brain phenotypes in PTENK13R,D384V/− mice. For example, nuclear PTEN may control cell growth through another crucial tumor suppressor, p53, which functions together with PTEN in the nucleus [27,35]. It would be of great importance to further investigate the molecular basis underlying these physiological changes in the brain.
PTEN+/− mice develop a lethal polyclonal autoimmune disease due to decreased Fasmediated apoptosis in T lymphocytes in which PI3K signaling is elevated [9]. In humans, heterozygous loss of PTEN has been reported in lymphoma [7,36,37]. Consistent with these previous studies, we confirmed that heterozygous loss of PTEN leads to lymphoma. Furthermore, we found that PTENK13R,D384V/K13R,D384V mice have a normal thymus, spleen, and inguinal lymph node. To our surprise, the combination of heterozygous PTEN loss and nuclear PTEN deficiency synergistically increased the size of the inguinal lymph node. This synergistic effect in the lymph node is in sharp contrast to the antagonistic effects in the brain. In PTENK13R,D384V/− lymph nodes, a modest increase was found in the population of B220-positive B-cells compared with CD3-positive T-cells. Therefore, in these mutant lymph nodes, enhanced growth of B-cells may be preferentially induced. We have previously shown that nuclear PTEN deficiency increases DNA damage in response to oxidative stress [25,27]. In highly mitotic B- and T-cells, a compromised DNA damage response may synergistically increase the frequency of hyperplasia and tumorigenesis in the immune organs.
Our data support the notion that PTEN has multiple tumor suppressor activities in different intracellular locations. PTEN has been described at other locations beyond the nucleus and plasma membrane, such as endosomes and ER-mitochondria contact sites. It will be necessary to further decipher tumor suppressor activities and targeting mechanisms at each of these compartments to fully understand the pathogenesis of PTEN-associated human cancers.
Highlights.
Nuclear and non-nuclear PTEN play distinct roles in organ growth and tumorigenesis
Nuclear PTEN and non-nuclear PTEN antagonistically regulate brain and neuron size
Nuclear PTEN controls dendritic shape in Purkinje neurons
Lymphoma is synergistically suppressed by nuclear PTEN and non-nuclear PTEN
Acknowledgments
We thank past and current members of the Iijima and Sesaki labs for helpful discussions and technical assistance. This work was supported by NIH grants to MI (GM131768 and NS114458) and HS (GM123266), Sol Goldman grant to MI, and Robert J. Kleberg, Jr. and Helen C. Kleberg Foundation Medical Research grant to HS.
Abbreviation:
- PTEN
phosphatase and tensin homolog deleted on chromosome ten
- PI3K
phosphatidylinositol 3-kinase
- H&E
hematoxylin and eosin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Biochemical and Biophysical Research Communications
Declaration of competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Lee YR, Chen M, Pandolfi PP, The functions and regulation of the PTEN tumour suppressor: new modes and prospects, Nat Rev Mol Cell Biol 19 (2018) 547–562. [DOI] [PubMed] [Google Scholar]
- [2].Milella M, Falcone I, Conciatori F, Cesta Incani U, Del Curatolo A, Inzerilli N, Nuzzo CM, Vaccaro V, Vari S, Cognetti F, Ciuffreda L, PTEN: Multiple Functions in Human Malignant Tumors, Front Oncol 5 (2015) 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Worby CA, Dixon JE, Pten, Annual review of biochemistry 83 (2014) 641–669. [DOI] [PubMed] [Google Scholar]
- [4].Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, Bose S, Call KM, Tsou HC, Peacocke M, Eng C, Parsons R, Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome, Nat Genet 16 (1997) 64–67. [DOI] [PubMed] [Google Scholar]
- [5].Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP, Pten is essential for embryonic development and tumour suppression, Nat Genet 19 (1998) 348–355. [DOI] [PubMed] [Google Scholar]
- [6].Di Cristofano A, De Acetis M, Koff A, Cordon-Cardo C, Pandolfi PP, Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse, Nat Genet 27 (2001) 222–224. [DOI] [PubMed] [Google Scholar]
- [7].Podsypanina K, Ellenson LH, Nemes A, Gu J, Tamura M, Yamada KM, Cordon-Cardo C, Catoretti G, Fisher PE, Parsons R, Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems, Proc Natl Acad Sci U S A 96 (1999) 1563–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Suzuki A, de la Pompa JL, Stambolic V, Elia AJ, Sasaki T, del Barco Barrantes I, Ho A, Wakeham A, Itie A, Khoo W, Fukumoto M, Mak TW, High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice, Curr Biol 8 (1998) 1169–1178. [DOI] [PubMed] [Google Scholar]
- [9].Di Cristofano A, Kotsi P, Peng YF, Cordon-Cardo C, Elkon KB, Pandolfi PP, Impaired Fas response and autoimmunity in Pten+/− mice, Science 285 (1999) 2122–2125. [DOI] [PubMed] [Google Scholar]
- [10].Endersby R, Baker SJ, PTEN signaling in brain: neuropathology and tumorigenesis, Oncogene 27 (2008) 5416–5430. [DOI] [PubMed] [Google Scholar]
- [11].Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT, The PI3K Pathway in Human Disease, Cell 170 (2017) 605–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hobert JA, Embacher R, Mester JL, Frazier TW 2nd, Eng C, Biochemical screening and PTEN mutation analysis in individuals with autism spectrum disorders and macrocephaly, Eur J Hum Genet 22 (2014) 273–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Klein S, Sharifi-Hannauer P, Martinez-Agosto JA, Macrocephaly as a clinical indicator of genetic subtypes in autism, Autism Res 6 (2013) 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Marchese M, Conti V, Valvo G, Moro F, Muratori F, Tancredi R, Santorelli FM, Guerrini R, Sicca F, Autism-epilepsy phenotype with macrocephaly suggests PTEN, but not GLIALCAM, genetic screening, BMC Med Genet 15 (2014) 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Conti S, Condo M, Posar A, Mari F, Resta N, Renieri A, Neri I, Patrizi A, Parmeggiani A, Phosphatase and tensin homolog (PTEN) gene mutations and autism: literature review and a case report of a patient with Cowden syndrome, autistic disorder, and epilepsy, J Child Neurol 27 (2012) 392–397. [DOI] [PubMed] [Google Scholar]
- [16].Backman SA, Stambolic V, Suzuki A, Haight J, Elia A, Pretorius J, Tsao MS, Shannon P, Bolon B, Ivy GO, Mak TW, Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease, Nat Genet 29 (2001) 396–403. [DOI] [PubMed] [Google Scholar]
- [17].Knobbe CB, Lapin V, Suzuki A, Mak TW, The roles of PTEN in development, physiology and tumorigenesis in mouse models: a tissue-by-tissue survey, Oncogene 27 (2008) 5398–5415. [DOI] [PubMed] [Google Scholar]
- [18].Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF, Pten regulates neuronal arborization and social interaction in mice, Neuron 50 (2006) 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chalhoub N, Baker SJ, PTEN and the PI3-kinase pathway in cancer, Annu Rev Pathol 4 (2009) 127–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Iijima M, Devreotes P, Tumor suppressor PTEN mediates sensing of chemoattractant gradients, Cell 109 (2002) 599–610. [DOI] [PubMed] [Google Scholar]
- [21].Iijima M, Huang YE, Devreotes P, Temporal and spatial regulation of chemotaxis, Dev Cell 3 (2002) 469–478. [DOI] [PubMed] [Google Scholar]
- [22].Kreis P, Leondaritis G, Lieberam I, Eickholt BJ, Subcellular targeting and dynamic regulation of PTEN: implications for neuronal cells and neurological disorders, Front Mol Neurosci 7 (2014) 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gil A, Andres-Pons A, Pulido R, Nuclear PTEN: a tale of many tails, Cell Death Differ 14 (2007) 395–399. [DOI] [PubMed] [Google Scholar]
- [24].Igarashi A, Itoh K, Yamada T, Adachi Y, Kato T, Murata D, Sesaki H, Iijima M, Nuclear PTEN deficiency causes microcephaly with decreased neuronal soma size and increased seizure susceptibility, J Biol Chem 293 (2018) 9292–9300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kato T, Yamada T, Nakamura H, Igarashi A, Anders RA, Sesaki H, Iijima M, The Loss of Nuclear PTEN Increases Tumorigenesis in a Preclinical Mouse Model for Hepatocellular Carcinoma, iScience 23 (2020) 101548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yang J-M, Schiapparelli P, Nguyen H, Igarashi A, Zhang Q, Abbadi S, Amzel L, Sesaki H, Quiñones-Hinojosa A, Iijima M, Characterization of PTEN mutations in brain cancer reveals that pten mono-ubiquitination promotes protein stability and nuclear localization, Oncogene 36 (2017) 3673–3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kato T, Murata D, Anders RA, Sesaki H, Iijima M, Nuclear PTEN and p53 suppress stress-induced liver cancer through distinct mechanisms, Biochem Biophys Res Commun 549 (2021) 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Schwenk F, Baron U, Rajewsky K, A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells, Nucleic Acids Res 23 (1995) 5080–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lesche R, Groszer M, Gao J, Wang Y, Messing A, Sun H, Liu X, Wu H, Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene, Genesis 32 (2002) 148–149. [DOI] [PubMed] [Google Scholar]
- [30].Alimonti A, Carracedo A, Clohessy JG, Trotman LC, Nardella C, Egia A, Salmena L, Sampieri K, Haveman WJ, Brogi E, Richardson AL, Zhang J, Pandolfi PP, Subtle variations in Pten dose determine cancer susceptibility, Nat Genet 42 (2010) 454–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kwon CH, Zhu X, Zhang J, Knoop LL, Tharp R, Smeyne RJ, Eberhart CG, Burger PC, Baker SJ, Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease, Nat Genet 29 (2001) 404–411. [DOI] [PubMed] [Google Scholar]
- [32].Groszer M, Erickson R, Scripture-Adams DD, Dougherty JD, Le Belle J, Zack JA, Geschwind DH, Liu X, Kornblum HI, Wu H, PTEN negatively regulates neural stem cell self-renewal by modulating G0-G1 cell cycle entry, Proc Natl Acad Sci U S A 103 (2006) 111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cupolillo D, Hoxha E, Faralli A, De Luca A, Rossi F, Tempia F, Carulli D, Autistic-Like Traits and Cerebellar Dysfunction in Purkinje Cell PTEN Knock-Out Mice, Neuropsychopharmacology 41 (2016) 1457–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Marino S, Krimpenfort P, Leung C, van der Korput HA, Trapman J, Camenisch I, Berns A, Brandner S, PTEN is essential for cell migration but not for fate determination and tumourigenesis in the cerebellum, Development 129 (2002) 3513–3522. [DOI] [PubMed] [Google Scholar]
- [35].Chang CJ, Mulholland DJ, Valamehr B, Mosessian S, Sellers WR, Wu H, PTEN nuclear localization is regulated by oxidative stress and mediates p53-dependent tumor suppression, Mol Cell Biol 28 (2008) 3281–3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Suzuki K, Nishihata J, Arai Y, Honma N, Yamamoto K, Irimura T, Toyoshima S, Molecular cloning of a novel actin-binding protein, p57, with a WD repeat and a leucine zipper motif, FEBS Lett 364 (1995) 283–288. [DOI] [PubMed] [Google Scholar]
- [37].Stambolic V, Tsao MS, Macpherson D, Suzuki A, Chapman WB, Mak TW, High incidence of breast and endometrial neoplasia resembling human Cowden syndrome in pten+/mice, Cancer Res 60 (2000) 3605–3611. [PubMed] [Google Scholar]