

Abstract
Although many components of the cell division machinery in bacteria have been identified, the mechanisms by which they work together to divide the cell remain poorly understood. Key among these components is the tubulin FtsZ, which forms a Z ring at midcell. FtsZ recruits the other cell division proteins, collectively called the divisome, and the Z ring constricts as the cell divides. We applied live-cell single molecule imaging to describe the dynamics of the divisome in detail, and to evaluate the individual roles of FtsZ-binding proteins, specifically FtsA and the ZBPs (EzrA, SepF, and ZapA), in cytokinesis. We show that the divisome comprises two subcomplexes that move differently: stationary ZBPs that transiently bind to treadmilling FtsZ filaments, and a moving complex that includes cell wall synthases. Our imaging analyses reveal that ZBPs bundle FtsZ filaments together and condense them into Z rings, and that this condensation is necessary for cytokinesis.
The mechanism by which bacteria divide remains poorly understood. In Bacillus subtilis, as in most other bacteria, division begins when filaments of FtsZ, a tubulin homolog, form a “Z ring” at midcell1. The Z ring recruits other cell division proteins, collectively called the divisome (Fig. 1a). The first group of these proteins (early proteins) arrives concurrently with FtsZ and includes the actin homolog FtsA and several other FtsZ binding proteins (ZBPs): the cytoplasmic protein ZapA, the integral membrane protein EzrA, and the peripheral membrane protein SepF. The second group of integral membrane proteins (late proteins) is then recruited, including DivIB, DivIC, and FtsL, and the cell wall synthesis enzymes Pbp2B and FtsW2,3. During cytokinesis, the Z ring constricts while the associated cell wall synthesis enzymes build a septum that divides the cell in half4. Recent work has shown that FtsZ filaments treadmill around the division plane, moving at the same rate as the transpeptidase Pbp2B5,6 (Supplementary Video 1). FtsZ treadmilling dynamics are critical for cell division: In B. subtilis, the rate of treadmilling limits Pbp2B motion, the rate of septal cell wall synthesis, and the overall rate of septation5.
Figure 1: The divisome consists of two dynamically distinct subcomplexes.
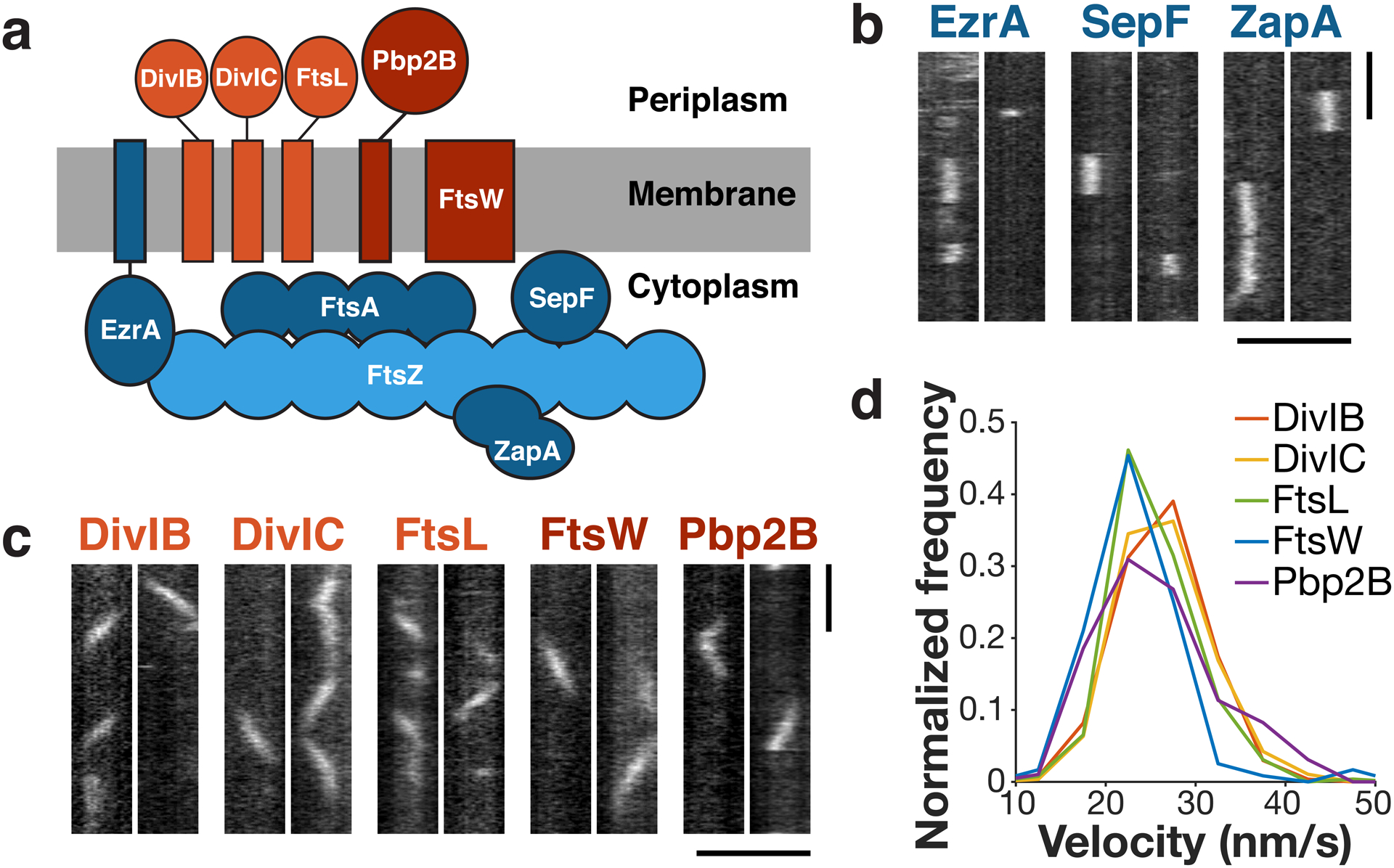
a Schematic of divisome proteins in B. subtilis, with early-arriving proteins in blue and late-arriving proteins in red. Light blue: FtsZ filament, dark blue: FtsZ binding proteins, light red: trimeric complex, dark red: cell wall synthesis enzymes. All blue proteins are stationary, and all red proteins move directionally with the same velocity. b Kymographs of single molecules of stationary ZBPs at division sites, from two replicates for each condition. c Kymographs of single molecules of directionally-moving proteins at division sites, from at least two replicates for each condition. d Velocity distributions of all directionally-moving proteins, measured from kymographs. Scale bars: horizontal: 2 μm, vertical: 1 min.
To understand how these proteins work to divide cells, we sought to build a dynamic characterization of how this multi-component machine functions in B. subtilis. We first worked to identify groups of divisome proteins that move together, then investigated how the FtsZ-associated proteins modulate FtsZ filaments, cell wall synthesis, and the overall process of cell division.
First, in order to understand which of the divisome proteins in B. subtilis associate with each other and work together, we characterized their dynamics using single-molecule imaging, as associated proteins should have similar motions. We expressed HaloTag fusions of each protein either as a functional sole copy or, in the case of SepF, at low levels from an ectopic site (Extended Data Fig.1). Cells were sparsely labelled with JF549-HaloTag Ligand7 and imaged with Total Internal Reflection Fluorescence Microscopy (TIRFM). Just as single molecules of FtsZ and FtsA are immobile5, single molecules of the ZBPs were stationary (Fig. 1b, Supplementary Video 2), consistent with their binding to stationary FtsZ subunits within treadmilling filaments. In contrast, the late proteins all moved directionally, with velocity distributions similar to Pbp2B (Fig. 1c–d, Supplementary Video 3). The divisome-associated cell wall synthesis enzymes are known to function together8,9, and these data also show that the DivIB-DivIC-FtsL trimeric complex10 remains persistently associated with these enzymes as they move around the division site. Thus, the divisome is composed of two distinct dynamic subcomplexes: 1) a directionally-moving group of periplasmic-facing membrane proteins that includes the cell wall synthesis enzymes, and 2) a group of cytoplasmic-facing proteins that bind to the stationary subunits within treadmilling FtsZ filaments.
Next, we investigated the function of the stationary subcomplex. We first examined FtsZ treadmilling, both to build a detailed characterization of its in vivo polymer dynamics and to provide a baseline for further investigation of how the ZBPs might affect its dynamics. In addition to visualizing FtsZ filament motion, we also measured FtsZ’s subunit lifetimes: While filament motion reports only treadmilling velocity, subunit lifetime measurements reflect both the treadmilling velocity and filament length11,12 (Fig. 2a). Single-molecule imaging of FtsZ (Fig. 2b, Extended Data Fig. 2, Supplementary Video 4) gave a mean lifetime of 8.1 seconds (Fig. 2c), corresponding to t½ = 5.6 s, similar to measurements of Z ring recovery after photobleaching (t½ = 8 ± 3 s)13. Interestingly, FtsZ subunit lifetimes were higher at the division site than elsewhere (Fig. 2d), possibly due to proteins such as Min and Noc that destabilize FtsZ filaments away from the division site14. Confirming that our lifetime measurements report on FtsZ treadmilling, lifetimes increased with FtsZ(T111A), a mutant that treadmills more slowly, and decreased when treadmilling was accelerated by MciZ expression5 (Fig. 2e). We also characterized the dynamics of the rest of the stationary subcomplex and found that the lifetimes of FtsA and the ZBPs were similar to or slightly shorter than that of FtsZ (Fig. 2f, Supplementary Table 4).
Figure 2: FtsZ lifetime reports treadmilling dynamics in vivo.
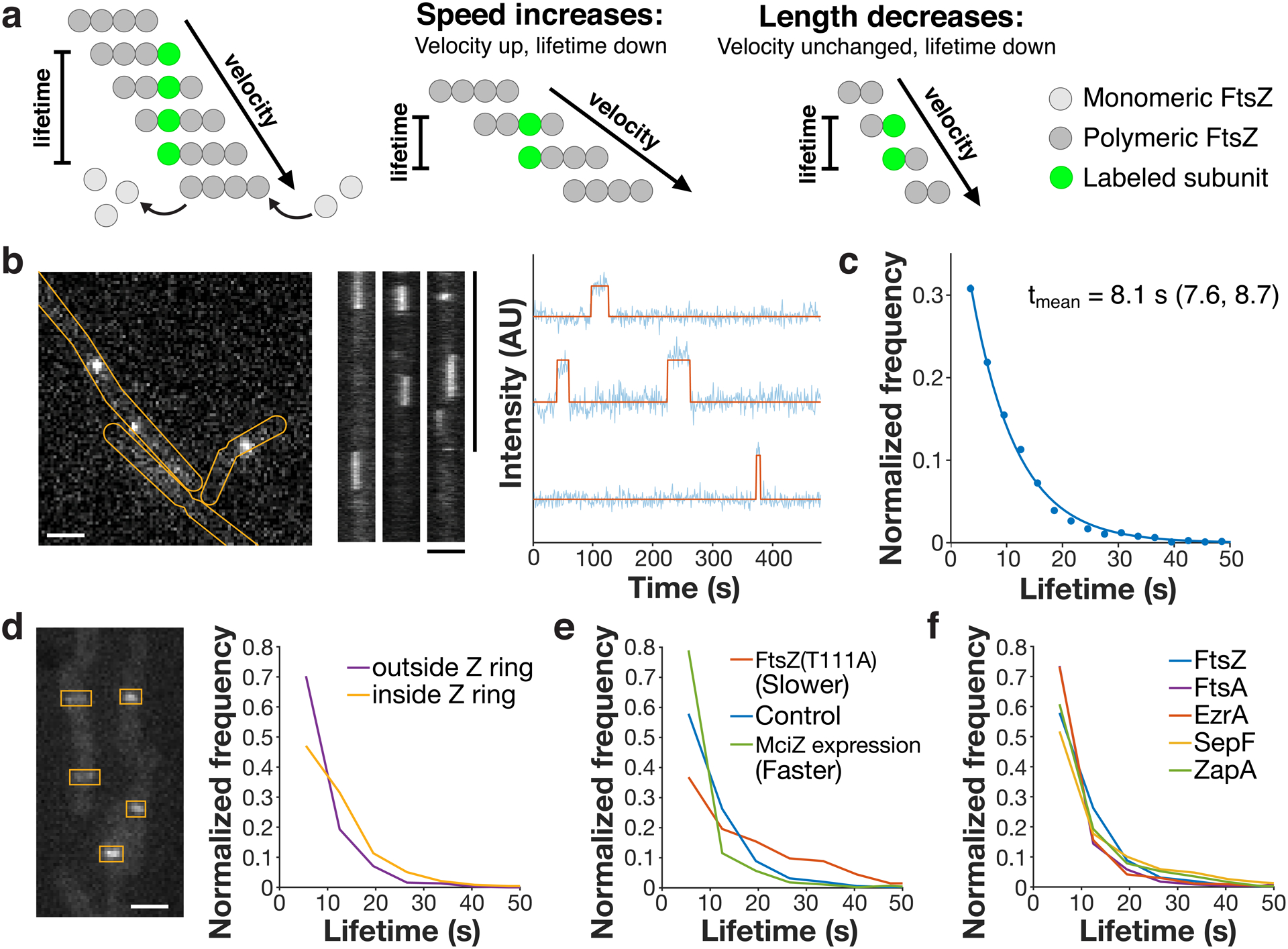
a Left: Velocity and lifetime can be measured independently for a treadmilling filament. Velocity is measured by imaging the motion of FtsZ filaments (dark grey circles), whereas lifetime is measured from the dwell time of a single labelled subunit (green circle) in the filament. Centre: If the speed of a FtsZ filament changes, this changes both the measured velocity and lifetime. Right: If the length of a FtsZ filament changes, the lifetime will change, but velocity will not. Thus, lifetime reflects both treadmilling speed and filament length, whereas velocity is insensitive to filament length. b Lifetimes were measured by live-cell single-molecule TIRFM (left) of stationary FtsZ subunits (kymographs, centre). Representative images from 17 replicates. Intensity traces were fit to a hidden Markov model to measure single-molecule lifetimes (right). c FtsZ subunit lifetime distribution, fit to a single exponential f(t) = Ae−τt. tmean: mean lifetime and 95% confidence interval, measured from this fit. d Left: mNeonGreen-Pbp2B was imaged by epifluorescence microscopy, and ROIs were drawn around Z rings. Representative image from 4 replicates. Right: Lifetime distributions of FtsZ inside and outside of Z rings, classified by colocalization with ROIs. e FtsZ subunit lifetime distributions in conditions that change treadmilling speed. f Lifetime distributions of FtsZ, FtsA, and the ZBPs. Scale bars: horizontal: 2 μm, vertical: 1 min.
Combining our lifetime measurements with treadmilling velocity gives insight into the in vivo properties of FtsZ filaments. First, we estimate that the average FtsZ filament is ~230 nm long, similar to the lengths of purified FtsZ filaments measured by electron microscopy (200 ± 75 nm)15. Second, assuming that treadmilling filaments elongate with a diffusion-limited on-rate of 5 μM−1 s−1 16,17, the concentration of free FtsZ monomer in cells can be estimated to be ~1.3 μM, similar to the critical concentration of FtsZ measured in vitro (1–1.5 μM)15. This agreement between the critical concentration in vitro and in vivo suggests that the dynamics of FtsZ polymers in vivo are intrinsic to the polymer itself and unaffected by other factors.
However, past work has suggested that FtsZ filament dynamics are modulated by other proteins in vivo, including the ZBPs ZapA, SepF, and EzrA2. Both ZapA and SepF have been shown in vitro to promote FtsZ filament formation, stability and bundling, and to decrease FtsZ’s GTPase activity18–25. In vivo, ZapA has been shown to promote the formation of a coherent Z ring22,26,27, while SepF is involved in both tethering FtsZ to the membrane and modulating septum morphology28,29. EzrA, meanwhile both increases FtsZ’s critical concentration and decreases filament bundling in vitro30–32 and inhibits Z ring formation and modulates the rate of Z ring recovery after photobleaching in vivo13,33,34. Thus, ZapA and SepF have both been described broadly as FtsZ stabilizing proteins, and EzrA as a FtsZ destabilizer.
We next investigated whether these ZBPs affected FtsZ dynamics in vivo. We measured FtsZ’s treadmilling velocity and subunit lifetime in strains as we changed the levels of individual ZBPs by deletion or overexpression (Fig. 3a, Extended Data Figs. 3–4, Supplementary Tables 3–4). None of these ZBP perturbations had any effect on FtsZ’s treadmilling velocity (Fig. 3a, Supplementary Table 3). Knockouts of ZapA or SepF also did not change FtsZ subunit lifetime, and overexpression had only subtle effects (Fig. 3a, Extended Data Fig. 4, Supplementary Table 4). However, ΔezrA cells had increased lifetimes, consistent with previous FRAP measurements of FtsZ-GFP13, and EzrA overexpression decreased lifetime in a dose-dependent manner (Fig. 3a–b, Extended Data Fig. 4, Supplementary Table 4). EzrA’s effect of decreasing subunit lifetime without changing treadmilling speed suggest that it shortens FtsZ filaments, which we confirmed using Structured Illumination-TIRF (SIM-TIRF) (Fig. 3c, Supplementary Video 5); however, it does so without changing FtsZ’s treadmilling dynamics, likely by binding to FtsZ monomers and sequestering them from the polymerizing pool (Supplementary Discussion 1). Thus, the ZBPs individually do not affect FtsZ treadmilling in vivo.
Figure 3: The ZBPs affect Z ring morphology but not dynamics.
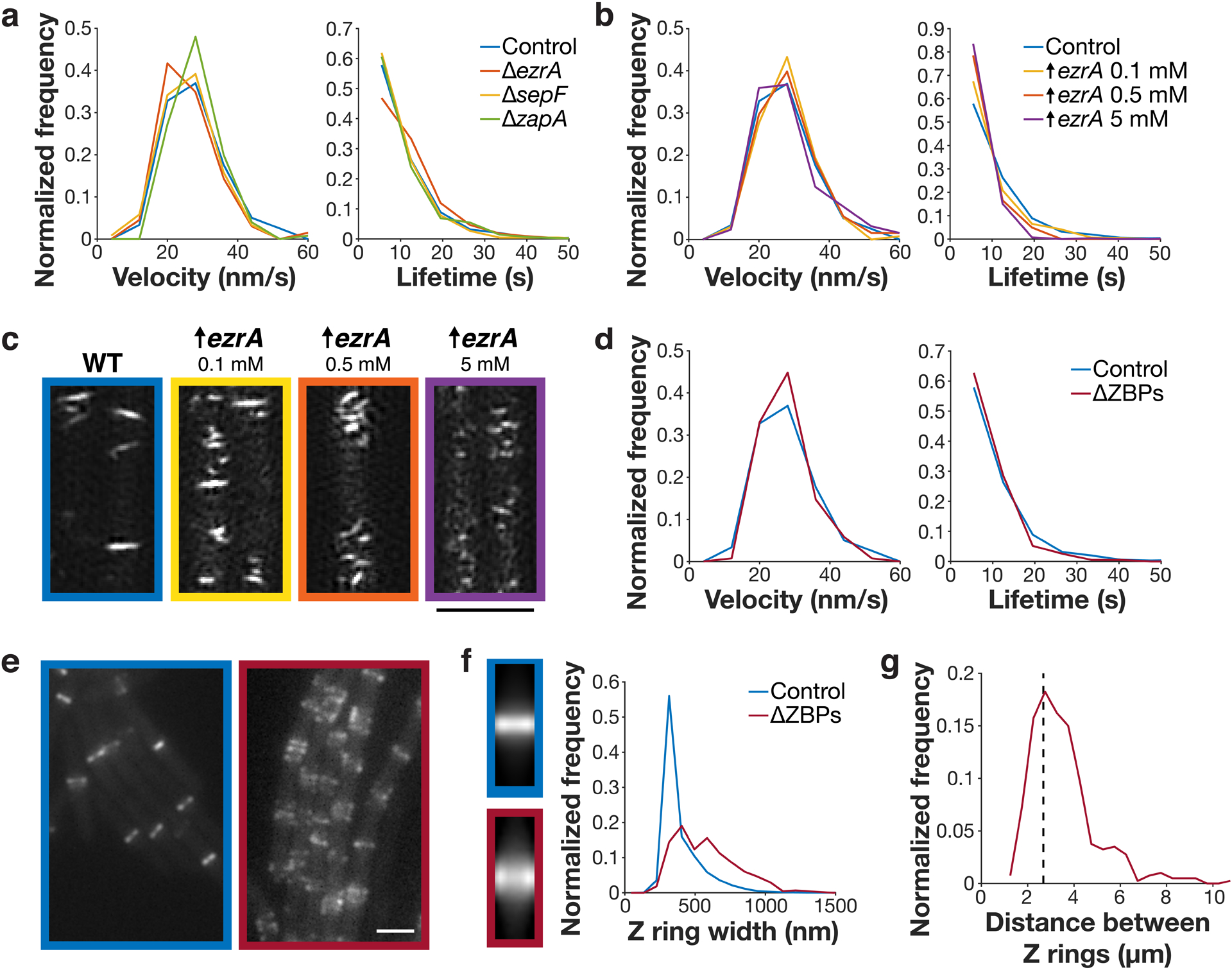
a FtsZ filament velocity distributions (left) and subunit lifetime distributions (right) in cells with individual ZBPs deleted. b FtsZ filament velocity distributions (left) and subunit lifetime distributions (right) in cells with EzrA overexpressed. c FtsZ filaments in cells overexpressing EzrA, visualized by SIM-TIRF imaging. 4 replicates were obtained for control cells, and one replicate was obtained for each EzrA overexpression condition (4 replicates total). d Left: FtsZ filament velocity distributions (left) and subunit lifetime distributions (right) in cells with all ZBPs removed. ΔZBPs cells had sepF and zapA deleted and ezrA depleted. e Z rings in control (left) and ΔZBPs cells (right), from at least two replicates for each condition. f Left: average intensity projections of Z rings in control and ΔZBPs cells, created by averaging N>400 Z ring images for each strain. Right: Z ring width distributions in control and ΔZBPs cells. g Distances between neighbouring Z rings in ΔZBPs cells. Dashed line: estimated spacing between Z rings in non-dividing B. subtilis cells, based on cell length (see Methods). Scale bars: 2 μm.
Next, we examined how the ZBPs in combination affect FtsZ dynamics. While none of the ZBPs are individually essential, ΔsepF and ΔzapA are each synthetically lethal with ΔezrA2. We created a ΔZBPs strain that lacked all ZBPs by knocking out sepF and zapA and depleting ezrA using a xylose-inducible promoter. We depleted EzrA for 7 hours, at which point cells were filamented, indicating that division was blocked. We additionally repeated this for all other synthetically lethal combinations of ZBPs (Extended Data Fig. 5). In all cases, FtsZ treadmilling velocity and subunit lifetime were unchanged from the control (Fig. 3d, Extended Data Fig. 6, Supplementary Tables 3–4, Supplementary Video 6). We note that, although ΔezrA cells have longer FtsZ subunit lifetimes, the lifetimes under these synthetic lethal conditions are statistically indistinguishable from the control. This suggests that EzrA’s roles in bundling and in filament length modulation are separate from one another (Supplementary Discussion 1). Regardless, together these data indicate that ZBPs do not affect FtsZ treadmilling in vivo.
Next, we investigated whether the ZBPs instead mediated filament bundling. ZBPs have been shown to mediate FtsZ filament bundling in vitro18,21,23–25,31,32, and lateral interactions between FtsZ filaments have been proposed to play a functional role in cytokinesis26,35. We therefore investigated how each ZBP knockout, individually and in combination, affected Z ring morphology. Overall Z ring morphology was normal in single ZBP knockouts, in the only viable double knockout (ΔsepF ΔzapA), and in all overexpression conditions except EzrA (Extended Data Figs. 3–4, 7).
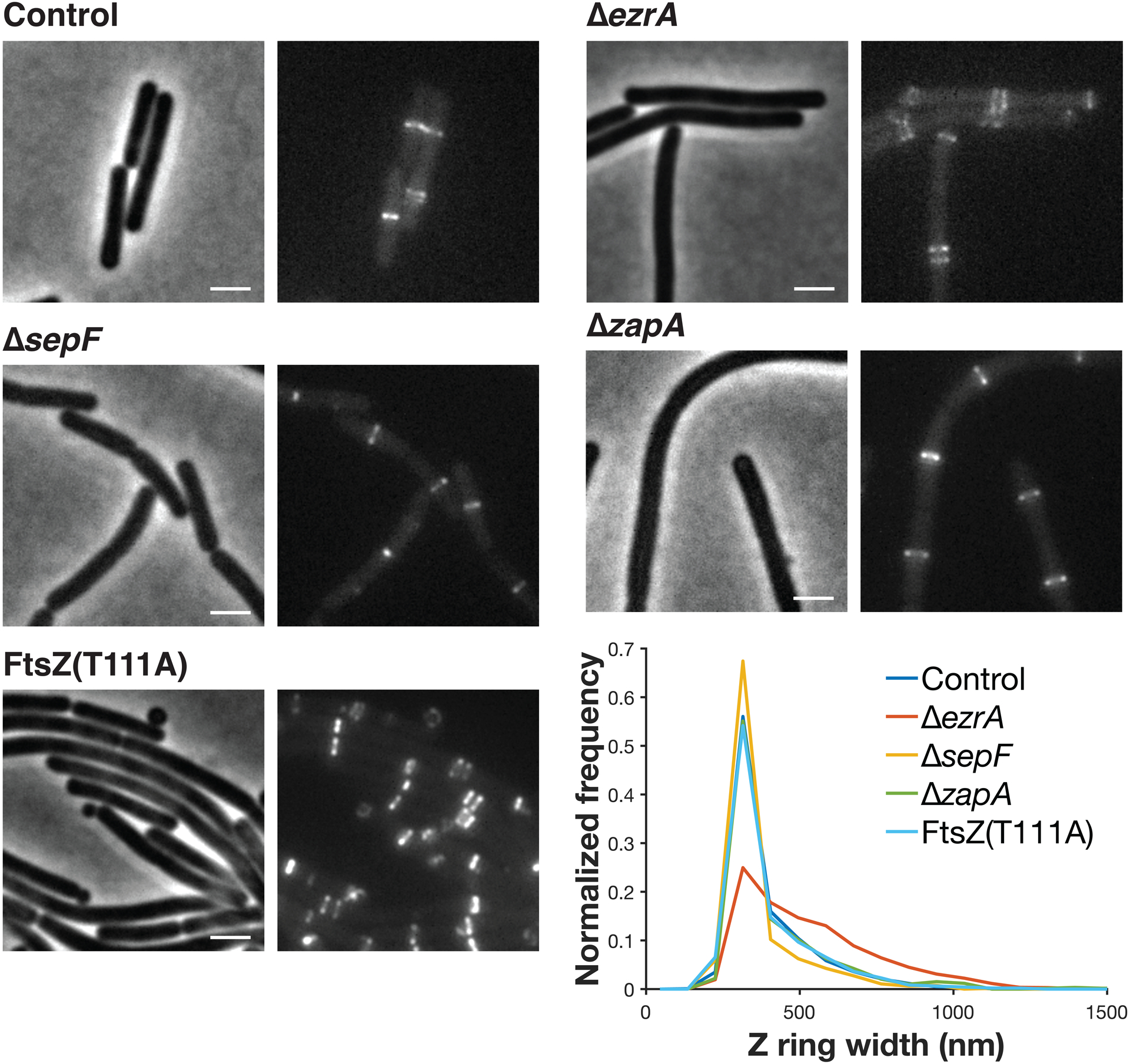
However, in the absence of synthetically lethal combinations of ZBPs, cells showed severely altered Z rings. Filaments no longer condensed, instead forming bands: regions of loosely-organized FtsZ filaments ~1.6x as wide as control Z rings (Fig. 3e–f, Extended Data Figs. 5–6). These FtsZ bands were still regularly spaced apart from one another, indicating that FtsZ was still able to localize to the division site under these conditions (Fig. 3g). These loosely-organized filaments resemble the transient FtsZ structures that occur when FtsZ first arrives at the division site (Fig. 4a). Over time under normal conditions, the width occupied by these FtsZ bands decreases, ultimately condensing into a Z ring (Fig 4b). Simultaneously, FtsZ recruitment increases, further concentrating FtsZ into a smaller area (Fig 4b). However, without ZBPs Z ring condensation never occurs (Fig. 3e–f). These results agree with previous observations that ZapA and SepF promote FtsZ bundling, whereas EzrA has previously been described as an inhibitor of Z ring formation and bundling (Supplementary Discussion 1). Here, we find that the ZBPs work collectively to promote Z ring condensation. Thus, without ZBPs, FtsZ filaments treadmill normally and localize correctly, but cannot condense into Z rings or divide the cell.
Figure 4: Z ring condensation is required for cell division.
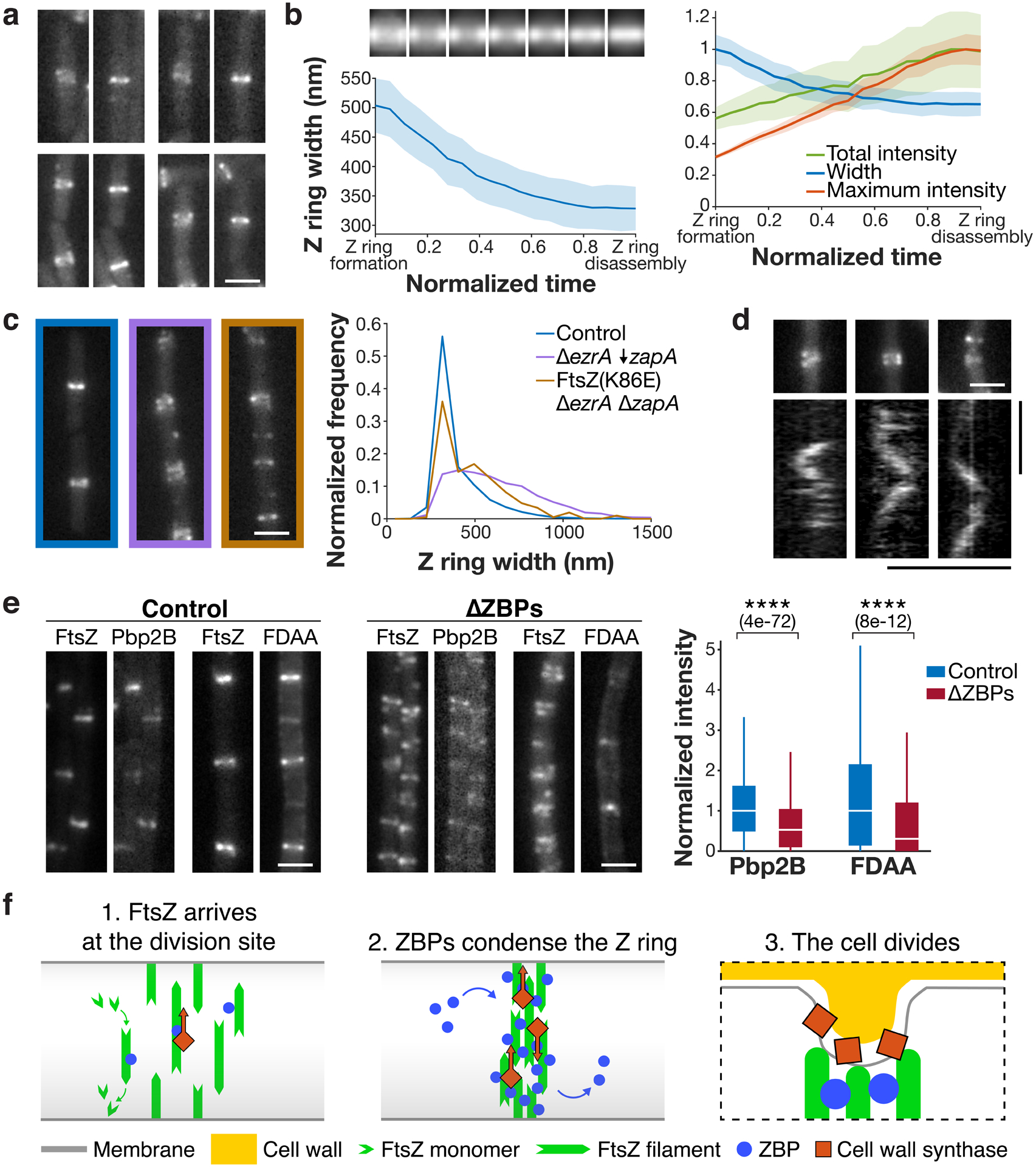
a Z ring condensation in control cells. Each pair of images shows a newly formed Z ring that has not yet condensed (left), and the same Z ring after condensation (right). Representative images from 4 replicates. b Left: Z ring condensation during the cell cycle. Top: Average intensity projections of Z rings from normalized time points over the cell cycle. Bottom: Z ring width over the cell cycle, measured as the full width at half maximum of the average intensity projections. Time from Z ring formation to Z ring disassembly (defined as the first and last frames in which the Z ring could be detected) was normalized for each cell. Shading: bootstrapped standard error. Right: Z ring width, total intensity, and maximum intensity over the cell cycle, normalized and plotted on the same axis. N = 760 cell cycles. c The FtsZ(K86E) mutant rescues the ΔezrA ΔzapA synthetic lethal condition and partially restores Z ring morphology, as seen in representative Z ring images from at least two replicates each (left) and width distributions (right). ↓ indicates depletion. d Pbp2B dynamics in ΔZBPs. Kymographs were drawn at the Z rings indicated above. 4 replicates were obtained. e Left and centre: Colocalization of Pbp2B with FtsZ and colocalization of FDAA labelling with FtsZ in control cells (left) and ΔZBPs cells (centre), from at least two replicates for each condition. Right: Amount of Pbp2B and FDAA labelling at the division site, measured by fluorescence intensity. N > 1000 for each condition. For each box plot, the white line indicates the median, the box extends to the 25th and 75th percentiles, and the whiskers indicate 1.5x interquartile range. P-values were obtained from a two-sided t-test; **** indicates p<0.0001, and p-values are included in parenthesis. f Left: At the start of the cell division process, FtsZ filaments treadmill around the cell circumference at midcell. Centre: Stationary ZBPs transiently bind to FtsZ filaments to condense the Z ring. Right: ZBP-driven bundling of FtsZ filaments may also function during cytokinesis, where crowding may induce inward membrane deformations, both concentrating cell wall synthesis to the Z ring and orienting it to divide the cell in two. Scale bars: horizontal: 2 μm, vertical: 1 min.
We next sought to clarify whether the Z ring condensation is specifically due to lateral bundling of FtsZ filaments by ZBPs. If this were the case, we might expect to isolate mutations that promote lateral bundling of FtsZ filaments in cells lacking ZBPs. Thus, we conducted a suppressor screen in the ΔZBPs strain (see Supplemental Methods for details). Whole-genome sequencing of the resulting suppressor candidates revealed a charge-inverting mutation (K86E) in helix H3 of FtsZ; both this helix and the homologous residue have been shown to affect lateral FtsZ filament interactions in E. coli35,36. We hypothesized that this mutation might restore viability in the absence of ZBPs by enhancing filament interactions. Indeed, FtsZ(K86E) restored viability and partially restored Z ring condensation in ΔezrA ΔzapA cells (Fig. 4c, Extended Data Fig. 8). Thus, Z ring condensation occurs due to bundling of FtsZ filaments by ZBPs.
Interestingly, the FtsZ(K86E) suppressor mutant can rescue the ΔezrA ΔzapA cells but not other synthetic lethal combinations. Although the ZBPs work collectively to bundle FtsZ filaments, they may each affect bundling differently. Beyond their role as bundlers, the ZBPs have been shown to have distinct functions2. Thus, the fact that FtsZ(K86E) can replace EzrA and ZapA but not SepF may reflect that each ZBP has different effects on FtsZ superstructure.
Next, we investigated whether ΔZBPs cells were unable to divide due to decreased septal cell wall synthesis, which is required for cell division. In ΔZBPs cells, we investigated the localization and motion of the division-specific cell wall synthesis enzyme Pbp2B, as well as septal cell wall synthesis activity (Fig. 4d–e). Pbp2B recruitment to the Z ring decreased by 50% in ΔZBPs relative to control cells (Fig. 4e). We found that in ΔZBPs cells, Pbp2B continued to move directionally at midcell; because the directional motion of Pbp2B reflects its activity, this suggests that it remains active under these conditions (Fig. 4d, Extended Data Fig. 9). To more directly assay the activity of cell wall synthesis enzymes, we measured the incorporation of fluorescent D-amino acids (FDAAs) into the division site5; FDAA incorporation was still present in ΔZBPs but reduced 40% relative to the control (Fig. 4e, Extended Data Fig. 9). Thus, septal cell wall synthesis enzymes are still active in the absence of the ZBPs. Next, to understand whether the decrease in Pbp2B recruitment was due to FtsZ’s inability to condense in ΔZBPs cells, we asked whether the FtsZ(K86E) suppressor mutant restored Pbp2B localization to midcell. Although this mutant allowed ΔezrA ΔzapA cells to divide and partially rescued Z ring condensation, it did not rescue Pbp2B recruitment (Extended Data Fig. 8). This indicates that the failure of ΔZBPs cells to divide is not due to defects in Pbp2B recruitment. This also suggests that the ZBPs may play a role in recruiting the late proteins to the division site that is independent of their effects on FtsZ.
Finally, we investigated the effects of FtsA on FtsZ filaments. FtsA is an actin homolog that serves as FtsZ’s primary membrane tether37, and B. subtilis ΔftsA cells are less viable and have a strong division defect and altered Z ring morphology38,39. FtsA has been shown in vitro and in vivo to modulate FtsZ treadmilling5,11. Indeed, ΔftsA cells showed a decrease in the fraction of directionally-treadmilling FtsZ filaments (Extended Data Fig. 10, Supplementary Video 7). Thus, unlike the ZBPs, FtsA modulates FtsZ filament treadmilling. It has also been suggested that FtsA might regulate FtsZ bundling40. We observe Z ring morphology defects in the ΔftsA strain, but these defects are distinct from the condensation defect observed in the ΔZBPs strain (Extended Data Fig. 10). Because ΔftsA cells have severely perturbed FtsZ filaments, it will be difficult to decouple these effects from any possible higher-order effects on their bundling state.
Combined with past work, these experiments provide new insights into the mechanisms underlying bacterial cell division. The cell division process begins with short treadmilling FtsZ filaments that are restricted to midcell by negative regulators. Our data reveal that FtsZ filaments treadmill at their biochemical steady state; their dynamics are not modulated by other factors. However, FtsZ cannot form a functional Z ring on its own: ZBPs are also required to bundle FtsZ filaments into a condensed Z ring, transiently interacting with stationary FtsZ subunits without affecting filament dynamics. Z ring condensation increases the recruitment of cell wall synthesis enzymes to the division site, which move around the division site as part of a directionally-moving complex. This condensation is ultimately necessary for cell division (Fig. 4f). FtsZ bundling proteins have been identified across the bacterial tree and even in archaea, suggesting that Z ring condensation may be an important process across diverse organisms41–46.
These results also yield new insights into the role of Z ring condensation in bacterial cytokinesis. Why is FtsZ filament bundling required for division, and what role does it play in the process? In contrast to previous models35,47, FtsZ bundling does not modulate FtsZ treadmilling dynamics, but rather condenses the Z ring. While condensation is not required for the activity of division-associated cell wall synthesis enzymes, it may be necessary to concentrate their activity in a small enough region to allow for productive septation. It is also possible that lateral filament association serves to inwardly deform the membrane. FtsZ has been seen to deform liposomes when filaments coalesce48,49, and crowding of membrane-associated proteins is sufficient to deform membranes50. Such deformations may be easier if the periplasm is iso-osmotic with the cytoplasm51,52, which would reduce the force required for membrane deformation. This membrane deformation could then direct circumferential septal wall synthesis inward to divide the cell5,53.
Methods
Culture growth:
Strains were stored as glycerol stocks at −80°C. At the start of each experiment, strains were streaked onto LB agar plates containing the appropriate antibiotic and incubated overnight at 37°C. For strains whose survival was dependent on the induction of a promoter, these plates were additionally top spread with xylose or IPTG at the appropriate concentration. For imaging, single colonies were inoculated into 1 mL casein hydrolysate (CH) media and grown on a roller at 37°C until they reached mid-exponential-phase growth (OD600 around 0.2). Cells were back diluted 1:10 and again grown until mid-exponential phase; this process was repeated until cells were ready for imaging. Alternately, cells were grown overnight in CH on a roller at 25°C. These cultures were grown in a 1:10 dilution series out to 1:10,000; the next day, the culture whose OD600 was nearest to 0.2 was back diluted 1:10 and grown in CH at 37°C as above.
Microscopy: sample preparation:
Cells were grown in 1 mL CH media at 37°C to OD600 around 0.2 as described above. Cultures were concentrated approximately 10-fold by centrifugation for 2 minutes at 7,000 RPM and resuspended in CH. Agarose pads were prepared using square plastic frames with inner dimensions 1.5 cm × 1.5 cm × 1 mm. Frames were placed on a cleaned glass pane, molten CH + 2% agarose was poured into the frames, and a second glass plane was placed on top to form a mould. Pads were allowed to solidify at room temperature, and excess agarose was cut away from the outside of the frame. To prepare slides for imaging, 2 μL of concentrated cells were pipetted onto a base-washed coverslip, and an agarose pad was placed on top. For multi-hour acquisitions, glass-bottom dishes (MatTek) were used instead of coverslips; these were also base-washed before use, and a moist KimWipe was wound around the edge of the dish to retain humidity.
Microscopy: phase contrast, epifluorescence, and TIRFM:
Phase contrast, epifluorescence, and Total Internal Reflection Fluorescence Microscopy (TIRFM) images were collected on a Nikon Ti-E microscope using a Nikon CFI Plan Apochromat DM Lambda 100X Oil objective, 1.45 NA, phase ring Ph3. Cameras used were an ORCA-Flash4.0 V2 sCMOS (Hamamatsu) and an iXon Ultra 897 EMCCD (Andor). Fluorescence excitation was achieved using a MLC4008 laser launch (Agilent), with a 488 nm laser used for mNeonGreen imaging and a 561 nm laser used for JF549 imaging. For fluorescence emission, a C-NSTORM QUAD filter cube was used, along with an additional ET525/50m filter for green emission and ET600/50m filter for red emission (Chroma). The microscope was enclosed in a chamber heated to 37°C. Images were acquired using NIS-Elements version 5.02.01.
Microscopy: SIM-TIRF:
Live-cell SIM data were acquired as described previously54 on a Zeiss Axio Observer.Z1 inverted microscope outfitted for structured illumination. An Olympus ×100/1.49NA objective was used instead of the Zeiss 1.45 NA objective because the slightly larger NA of the Olympus objective gives higher tolerance for placing the excitation beams inside the TIRF annulus. Data were acquired at 1 s frame rates for 2 minutes with 20 ms exposures from a 488 nm laser for each rotation. SIM-TIRF images were reconstructed as described previously54. Samples were enclosed in a chamber heated to 37°C.
Microscopy: Confocal:
Confocal images were collected on a Nikon TI microscope with Yokogawa CSU-10 spinning disk confocal unit using a Nikon 100X NA 1.45 TIRF objective and an ImagEM EM-CCD camera (Hamamatsu). A 494 nm excitation laser and a 609/57 nm bandpass emission filter were used for imaging of FM5-95. Images were acquired using MetaMorph version 7.8.1.0.
Induction, depletion, and HaloTag labelling:
Specific conditions for each experiment are listed in Supplementary Table 6. For FtsZ imaging, FtsZ-mNeonGreen or FtsZ-HaloTag were expressed from the IPTG-inducible pHyperSpank promoter. In all cases except the ΔftsA strain, FtsA was co-expressed from the same promoter, preserving the native operon structure. These constructs were merodiploid, meaning that the inducible FtsZ constructs were cloned into the chromosome at an ectopic site; the native untagged operon remained intact. Labelled FtsZ was induced by adding 20 μM IPTG to the growth media for 1 hour before imaging. For single-molecule imaging, strains containing FtsZ-HaloTag were labelled by adding 20 pM JF549-HaloTag Ligand (JF549-HTL) to the growth media for 1 hour before imaging7. For total labelling, 5 nM JF549-HTL was used. For overexpression, the FtsZ-mNeonGreen construct was induced with 100 μM IPTG for 2 hours before imaging. For MciZ expression, 5 μL of a 3 M xylose stock was added to the top of the agarose pad during imaging (final concentration ~65 mM), allowing comparison of the same sample before and after imaging.
For the ΔftsA strain, FtsZ was induced continuously from an ectopic locus under the pHyperSpank promoter induced with 10 μM IPTG; IPTG concentrations above or below this did not permit cell growth. To image FtsZ, FtsZ-mNeonGreen was expressed at a different ectopic locus from the pXyl promoter and induced by adding 30 mM xylose to the growth media for 1 hour prior to imaging.
Single-molecule imaging of other divisome proteins was conducted as follows. FtsA, EzrA, and ZapA HaloTag constructs were expressed as a sole copy from their native promoters and labelled with 50 pM, 300 pM, and 600 pM JF549-HTL, respectively. SepF-HaloTag was expressed as a merodiploid under an IPTG-inducible promoter; no IPTG was added, as leaky expression from the promoter was sufficient for single-molecule imaging. SepF-HaloTag was labelled with 200 pM JF549-HTL. DivIB, DivIC, and FtsW HaloTag constructs were expressed as sole copies from xylose-inducible promoters. They were induced continuously with 1 mM, 5 mM, and 8 mM xylose, and labelled with 400 pM, 500 pM, and 300 pM JF549-HTL, respectively. FtsL-HaloTag and Pbp2B-HaloTag were expressed as a sole copy from an IPTG-inducible promoter, induced continuously with 30 μM and 20 μM IPTG, and labelled with 400 pM and 200 pM JF549-HTL, respectively. All JF549-HTL labelling was performed for 15 minutes before imaging; when JF549-HTL concentrations were sufficiently high, cells were washed once in 1 mL CH media before imaging to remove excess dye.
For overexpression of ZBPs, xylose was added at the indicated concentration for 2 hours before imaging. For depletion of ZBPs, cells were grown initially in 1 mM xylose; xylose was then withdrawn, and cells were imaged at the point when they had filamented but were largely still alive, approximately 7 hours after xylose withdrawal. We judged whether cells were alive based on their appearance by phase contrast microscopy and whether or not they contained fluorescent signal. For imaging of Pbp2B dynamics in these mutants, cells were grown overnight in 100 μM IPTG and 1 mM xylose; 7 hours before imaging, xylose was withdrawn and the concentration of IPTG was reduced to 20 μM. Pbp2B-HaloTag was labelled with 100 pM JF549-HaloTag Ligand for 15 minutes before imaging.
Cell length measurements:
To determine whether or not the fluorescent fusions in this study impacted the functioning of the division machinery, we measured the cell length in each strain; cells with division defects show an increase in cell length. In B. subtilis, simple imaging of the cells by e.g. phase contrast microscopy cannot be reliably used to measure cell length because of cell chaining, so a membrane stain was used. We grew cells for imaging as described above. Cell membranes were labelled by staining with 1 μg/mL FM5-95 for 1 minute at room temperature, washed once with 1mL CH, and were immediately imaged at room temperature. Images were taken by spinning disk confocal microscopy at a 1 s exposure. Cell lengths were measured manually using ImageJ, with images anonymized and shuffled before analysis. Violin plots were generated in MATLAB using violin.m55. N>149 for each sample.
Velocity measurements:
To measure FtsZ treadmilling velocity, cells expressing FtsZ-mNeonGreen were imaged by TIRFM. Time lapses were taken using the sCMOS camera with 1 s exposures for 4 minutes total; after each time lapse, a phase-contrast image was taken to visualize cells. Velocity was measured by kymograph analysis5. Kymographs were created from these time lapses of fluorescently labelled FtsZ filaments by manually drawing ROIs along the short axis of cells in ImageJ. Regions of these kymographs containing diagonal bands of fluorescence, representing directional treadmilling, were selected and their slopes were measured manually in ImageJ.
Velocity measurements of the single-molecule motions of DivIB, DivIC, FtsL, FtsW, and Pbp2B were taken similarly; cells were labelled for single-molecule imaging as described above and imaged by TIRFM. Each of these cells additionally expressed FtsZ-mNeonGreen to visualize the division site. Time lapses were taken using the sCMOS camera with 1-second exposures for 2–4 minutes total; before and after each time lapse, a phase-contrast image was taken to visualize cells, and a green epifluorescence image was taken to visualize the division site. Kymograph analysis of velocities was performed as summarized above; in this case, molecules that colocalized with the division site were specifically selected for analysis. A summary of these results is provided in Supplementary Table 3.
To characterize the stationary behaviour of EzrA, SepF, and ZapA, cells were labelled for single-molecule imaging and imaged by TIRFM as above. These cells also expressed FtsZ-mNeonGreen to visualize the division site. The microscopy protocol was identical to that in the previous paragraph; molecules that colocalized with the division site were selected for analysis.
Cell segmentation:
Phase-contrast images of cells were segmented using DeepCell, a deep learning-based cell segmentation platform56. A different custom net was trained for each combination of objective and camera used. Training sets were manually generated and varied in size but were generally around 20 images each. For cells in synthetic lethal conditions, masks were refined manually to omit dead cells.
Single-molecule lifetime measurements:
To measure the single-molecule lifetimes of FtsZ and the ZBPs, HaloTag fusions were grown and labelled for single-molecule imaging as described above. TIRFM time lapses were taken using the emCCD camera, with 500 ms exposures for 4 minutes total; after each time lapse, a phase-contrast image was taken to visualize cells. To analyse the data, first, the phase images of cells were segmented using DeepCell to generate cell masks. Next, TrackMate was used to identify single particles in the image and preliminarily link them together57. Spots were detected with a 1.5-pixel radius and an intensity threshold that was manually selected for each data set. Spots were then linked roughly into tracks, with a 3-pixel linking distance and a maximum gap of 10 frames; in this way, only localizations with at least one other spot detected nearby were considered for further analysis, which decreased computational load in the next stage. These data were exported into MATLAB for further analysis.
The track list from TrackMate was then filtered and converted to intensity traces. First, the spot positions in each track were averaged to generate a mean position of each spot. Next, spots that were not inside cells were excluded using the cell masks generated by DeepCell. Spots within 3 pixels of one another were then combined, and a new average position was calculated, weighted based on the length of each track. Then, for each spot, an intensity trace was generated: intensity was averaged in a 5 × 5 pixel window around the mean spot position, and the local background was averaged in a 2-pixel frame around the window and subtracted to generate a background-subtracted trace. Finally, intensity traces were filtered; only traces with a maximum background-subtracted intensity above 500 counts were included for further analysis.
To measure each single molecule lifetime, these intensity traces were fit to a Hidden Markov Model (HMM) using the MATLAB package vbFRET58. To rule out spots which contained no single-molecule fluorescence events, and to exclude cases where multiple single molecules overlap, models were fit with 1, 2, 3, and 4 states. Bayesian model selection was used to select the best-fitting model, and only traces in which the 2-state model fit best were included. Traces for which the difference between state 1 (no fluorescence) and state 2 (single-molecule fluorescence) was less than 60 counts were also discarded. The duration of each state 2 event was measured; dwell times less than 2 seconds (4 frames) were discarded, as were events that overlapped with the start or end of the trace since they cannot be measured accurately. Traces containing more than 2 events were also excluded. The resulting single-molecule lifetimes were fit to a single exponential distribution and the mean lifetime was computed. We measured the contribution of photobleaching to our lifetimes by repeating the experiment at 1 second imaging intervals rather than 500 ms intervals without changing the exposure time; the measured lifetime did not change, indicating that the photobleaching contribution was negligible. To compare lifetimes, p-values were calculated using a Wilcoxon rank-sum test. Results are summarized in Supplementary Table 4.
Single-molecule lifetime measurements: inside versus outside of the Z ring:
To compare the single-molecule lifetime of FtsZ inside and outside of the Z ring, cells expressing FtsZ-HaloTag were prepared for imaging as described previously. These strains also expressed mNeonGreen-Pbp2B to visualize the division site. TIRFM time lapses were taken using the emCCD camera, with 500 ms exposures for 4 minutes total; before and after each time lapse, a phase-contrast image was taken to visualize cells, and a green epifluorescence image was taken to visualize the division site. To compare single-molecule lifetimes inside and outside of the Z ring, ROIs were manually drawn around each division site, as seen in Figure 2d; lifetimes were analysed as described above and assigned to be inside or outside of the Z ring based on the drawn ROIs. 38% of single molecules were identified as being inside Z rings, consistent with quantitative fluorescence measurements in B. subtilis that found 30–35% of FtsZ in the Z ring13.
Single-molecule lifetime measurements: manual:
To confirm that the automated single-molecule lifetime measurements described above were accurate, single-molecule lifetimes were also measured by hand. FtsZ-HaloTag was imaged as described in the Single-molecule lifetime measurements section above, although the sCMOS camera was used instead of the emCCD camera for ease of visualization. Kymographs were drawn manually in ImageJ. These kymographs were then examined for single-molecule events, and the duration of these events was measured manually in ImageJ.
Z ring identification, spacing, width, and average projections:
To visualize the Z ring, cells expressing FtsZ-mNeonGreen under an IPTG-inducible promoter were grown for imaging as usual. Cells were imaged using the sCMOS camera; at each position, one phase-contrast image to visualize cells and 1 green epifluorescence image to visualize Z rings were taken. To identify Z rings in the image, cells were segmented using DeepCell to generate binary masks. The pill mesh function in Morphometrics was then used to generate midlines down the long axis of each cell52. Using custom MATLAB code, the fluorescence intensity of FtsZ was averaged along each cell midline by taking the average along each mesh spline, these intensity traces were smoothed, and Z rings were identified by peak detection.
To measure the spacing between Z rings, the distances between neighbouring peaks were measured. The predicted spacing between Z rings was calculated as follows. Z rings assemble around 25% of the way through the cell cycle3, and because ΔZBPs cells do not divide, these division sites remain indefinitely available for division protein localization. Thus, the expected Z ring spacing is equal to the cell length for cells at or below the 25th percentile for length and is ½ the cell length for cells above the 25th percentile. To calculate this, we used the cell length distribution shown for WT cells in Extended Data Fig. 1.
To plot the Z ring width distributions, the full width at half maximum of each Z ring peak was calculated To measure Z ring width, a 1 μm region around each peak was sub-selected from each intensity trace, and these traces were averaged to create an average intensity trace. The Z ring width was measured by calculating the full width at half maximum of the Z ring peak in these intensity traces. Results are summarized in Supplementary Table 5. To display average projections of Z rings, regions of each corresponding cell were sub-selected around each peak, using the meshes to align and straighten cells and normalize cell width; these images were averaged to create an average projection.
Z ring features across the cell cycle:
To quantify the appearance of the Z ring over the cell cycle, FtsZ-mNeonGreen cells were grown as above. Cells were then imaged in phase and epifluorescence as above, repeated every minute for 2 hours. Time lapses were registered in ImageJ, and phase images were segmented using DeepCell. Morphometrics was used to generate midlines down each cell, as well as to track cells over time. Fluorescent images and cell meshes were then imported into MATLAB for further analysis.
First, cell tracks from Morphometrics were filtered for quality control. Cell tracks with a duration less than 20 frames (20 minutes) were discarded. Additionally, the total cell length for each frame in the track was then fit to a line, and tracks for which the R2 of this fit was less than 0.99 were also discarded.
Bleach correction was applied by measuring the average intensity Iavg in each cell over time in all cells which had appreciable photobleaching, defined as cells whose final intensity was at least 40 counts lower than their initial intensity. For each cell, we fit this to Iavg(t) = Io*ekt+Ibg where Io is the initial intensity, k is the photobleaching coefficient, Ibg is the background intensity, and t is time. We plotted the distributions of Ibg and k for all of our fits, used gaussian fitting to extract the peak value for each, and assigned these as our final Ibg and k values. We then computed the corrected intensities for each cell in each frame Icorr = (Im–Ibg)/ekt where Im is the measured intensity.
Next, Z rings in the cell in each frame were identified as described above. These Z rings were then linked together between frames by particle tracking to create Z ring tracks: Z rings were linked if they were within 5 pixels (325 nm) and 5 frames (5 minutes) of one another. Only Z ring tracks between 20 and 40 minutes in duration were considered for further analysis. Time was normalized for each track, and average Z ring intensity traces and projections were computed at each time point as described above. Z ring peak width, peak height, and total intensity were computed by measuring the full width at half maximum, height above baseline, and peak area of each average Z ring intensity trace. N = 760 Z rings.
Pbp2B localization:
To quantify the localization of Pbp2B at the division site, cells were grown containing FtsZ-HaloTag under an IPTG-inducible promoter and Pbp2B-mNeonGreen under its native promoter. The ZBP depletion strain was depleted for 7 hours before imaging as usual. FtsZ-HaloTag was induced with 20 μM IPTG and labelled with 5 nM JF549 for 1 hour before imaging. Cells were imaged at 20 positions using the sCMOS camera; at each position, in order, 1 phase contrast image, 1 red epifluorescence image, and 1 green epifluorescence image were taken. Images were background-subtracted in ImageJ with rolling ball radius 50. To analyse, Z rings were identified in each cell as described above. These same peak regions were selected from the corresponding Pbp2B image, to visualize Pbp2B intensity at the Z ring. Finally, the area under these peak regions was calculated to estimate the amount of Pbp2B at midcell in each strain. N>1000 division sites analyzed in each condition. Box plots were generated using the boxplot function in MATLAB.
Cell wall synthesis labelling:
For live-cell fluorescent D-amino acid (FDAA) labelling of cell wall synthesis, cells were grown for imaging as normal; the ZBP depletion strain was depleted for 7 hours before imaging as usual. All FDAA labelling experiments were performed in a ΔdacA background. To visualize the division site, these cells also expressed FtsZ-HaloTag, which was induced with 20 μM IPTG and labelled with 5 nM JF549 for 1 hour before imaging. Cells were pelleted at 8000 RPM for 30 seconds and resuspended in 10 μL CH + 1 mM fluorescent D-lysine (FDL). Cells were incubated for 3 minutes to allow labelling to occur, after which 1 mL CH was added to the tube to halt FDL labelling. Cells were pelleted at 8000 RPM for 30 seconds, resuspended in 100 μL CH, and immediately placed under an agarose pad for imaging.
The average time between the end of FDL labelling and the start of imaging was 3 minutes and 20 seconds. Image acquisition was automated to increase speed and took an additional 1 minute. Cells were imaged at 10 positions using the sCMOS camera; at each position, in order, 1 phase contrast image, 1 red epifluorescence image, and 1 green epifluorescence image were taken. There is in total a ~4 minute delay between FDAA labelling and imaging, and in some cases the positions of Z rings may have changed during this time. For instance, if a Z ring constricted and disassembled during this delay, we would observe FDAA labelling without a Z ring, and vice versa for a newly assembled Z ring. However, we expect these events to be relatively rare because the cell cycle duration is roughly 30 minutes under these conditions.
Images were background-subtracted in ImageJ with a rolling ball radius of 50. To analyse colocalization, Z rings were identified in each cell as described above. These same regions were selected from the corresponding FDAA image. To correct for differences in labelling efficiency between cells, the FDAA signal at midcell was normalized to signal in the nearby sidewall. Finally, the area under these peak regions was calculated to estimate the amount of FtsZ and FDAA at midcell in each strain. N > 1000 division sites analyzed in each condition. Box plots were generated using the boxplot function in MATLAB.
ΔftsA analysis:
To characterize FtsZ dynamics in the ΔftsA strain, FtsZ dynamics were imaged in each strain as described above. FtsZ filaments were tracked using the FIJI plugin TrackMate with the following parameters: spots with a diameter of 210 nm were identified using the Laplacian of Gaussians (LoG) detector; these spots were tracked over a 140 nm search radius using the Sparse LAP Tracker with no frame gaps. The resulting tracks were filtered to exclude tracks whose duration was less than 10 or greater than 25 frames (10 and 25 seconds, respectively) using a custom MATLAB script, as described previously5. Mean squared displacement (MSD) vs time interval (Δt) was computed for each track and fit to MSD(Δt) = D*Δtα (D: diffusion coefficient) to obtain the α coefficient. N>6000 tracks analysed in each condition.
Pbp2B dynamics:
To visualize Pbp2B dynamics at the division site, cells expressed both Pbp2B-HaloTag from an inducible promoter to visualize single-molecule Pbp2B dynamics and FtsZ-mNeonGreen from an ectopic site under its native promoter to visualize the Z ring. Cells were plated overnight on LB plates top spread with 100 μM IPTG; 1 mM xylose was also added to the plate for the synthetic lethal depletion strain. The following day, single colonies were inoculated into 1 mL CH + 1 mM xylose + 100 μM IPTG cultures and grown overnight at room temperature with shaking. The next morning, cells were washed once in 1 mL CH media and then grown for 7 hours in CH media + 20 μM IPTG without xylose. This both began the depletion process for the synthetic lethal strain and decreased Pbp2B expression to a level suitable for single-molecule analysis. 15 minutes before imaging, cells were labelled with 100 pM JF549-HaloTag Ligand. Cells were imaged by TIRFM: time lapses were taken using the sCMOS camera with 1-second exposures for 4 minutes total; before and after each time lapse, a phase-contrast image was taken to visualize cells, and a green epifluorescence image was taken to visualize the division site. Kymographs were created manually as described above. Z ring images were assigned to these kymographs by extracting a 61 × 61 pixel region from the FtsZ epifluorescence image taken before TIRFM, cantered on the midpoint of the kymograph.
To characterize Z rings at which Pbp2B moved directionally, directionally moving Pbp2B particles were identified by particle tracking. Particles were tracked using the FIJI plugin TrackMate with the following parameters: spots with a diameter of 400 nm were identified using the Laplacian of Gaussians (LoG) detector; these spots were tracked over a 100 nm search radius using the Sparse LAP Tracker with no frame gaps. The resulting tracks were further filtered to obtain a selection of tracks with clear directional motion using a custom MATLAB script, as described previously5. Tracks between 10 and 25 seconds and with end-to-end displacement above 225 nm were included for further analysis; further quality control was achieved by selecting tracks well fit (r2 >0.95) linearly to a log-log plot of mean squared displacement (MSD) vs time interval (Δt). Any remaining diffusing particles were omitted by ensuring a nonzero velocity from the fit MSD(Δt) = 4*D*Δt + (v*Δt)2 (v: velocity; D: diffusion coefficient). Tracks were assigned to cells using phase images segmented as described above. Z rings were identified in these cells (described above) and tracks were assigned to the nearest Z ring up to a maximum distance of 1 μm.
Suppressor screen:
To isolate potential mutations that could suppress the synthetic lethality of the ZBPs, cells of strain bGS308 (ΔsepF, ΔzapA, xylose-inducible ezrA) were plated overnight on LB plates without xylose. Colonies grew overnight, as expected since EzrA depletion is slow and cells that are inhibited for division can grow for some time before death. The following day, colonies were inoculated into 3 mL LB cultures in triplicate and grown for 8 hours at 37°C with shaking. During the 8 hours of growth, the OD first increased, then decreased as cell death occurred, and then began to increase again. After 8 hours, 200 μL of each culture was plated on LB plates and grown overnight. The following day, 5 colonies from each plate were restruck for single colonies. Each colony was also patched onto LB plates top-spread with 25 mM xylose: because EzrA overexpression is lethal, candidates with an intact xylose-inducible promoter were expected to die on high xylose plates. Of the 15 candidate colonies, 4 did not grow after restreaking, and 3 had become insensitive to high xylose; the remaining 8 were submitted for whole-genome sequencing.
All 8 candidates had mutations in the xylose-inducible promoter; 4 candidates also had additional mutations. Of these, strain bGS390 (containing the FtsZ(K86E) mutation) was selected for further analysis. To verify that this mutation was capable of suppressing ZBP synthetic lethality, the FtsZ(K86E) mutation was introduced into a WT background. Individual ΔezrA, ΔsepF, and ΔzapA mutations were introduced into this background and combined by crossing. As expected, the ΔsepF ΔzapA double mutant was viable, since these proteins are not synthetically lethal even in the WT background. Additionally, the normally synthetically lethal ΔezrA ΔzapA mutants could be combined in the FtsZ(K86E) background, verifying that this mutation is a bona fide suppressor; the presence of the FtsZ(K86E) mutation was confirmed in this strain by Sanger sequencing. As a control, a ΔezrA ΔzapA cross was attempted in parallel in the WT background; as expected, although colonies appeared on the transformation plate after overnight incubation, these colonies could not be further grown in liquid culture and became transparent after an additional day of incubation, verifying that these mutations are indeed synthetically lethal.
To rule out the possibility of additional suppressors arising during cloning, a FtsZ(K86E) ΔzapA xylose-inducible ezrA strain was constructed. This strain was maintained in 1 mM xylose during the cloning process, conditions under which cells containing WT FtsZ are viable; this same process was used to generate the synthetic lethal depletion mutants. After cloning, xylose was withdrawn; unlike WT FtsZ-containing cells, which die after xylose is withdrawn, FtsZ(K86E) mutant cells remained viable after xylose was withdrawn.
Interestingly, neither the ΔezrA ΔsepF nor the ΔezrA ΔsepF ΔzapA mutants could be constructed in the FtsZ(K86E) background. We suspect that the ability of this mutant to survive in the ↓ezrA ΔsepF ΔzapA condition during the initial suppressor screen was due to some leaky expression from the xylose promoter, which may have been enhanced by the mutations in the promoter that arose during the screen.
Strain construction:
All strains were constructed in B. subtilis strain PY79; strains used in this study are listed in Supplementary Table 1. Constructs were assembled by PCR amplification and Gibson cloning. These Gibson products were transformed directly into competent B. subtilis, where they were integrated into the chromosome by homologous recombination with homology regions that were engineered at each end of the construct. Each construct was initially transformed into the WT background except for the ΔftsAZ construct, which was transformed into a strain containing an inducible FtsAZ operon at an ectopic locus. Transformants were selected by growth on LB plates containing the appropriate antibiotic. The resulting strains were verified by PCR and, when appropriate, by sequencing. Constructs used in this study, as well as the plasmids used to create each construct, are listed in Supplementary Table 2.
To combine constructs in the same strain, parent strains containing the constructs to be combined were crossed by transforming genomic DNA from one strain into the other. When two strains to be combined contained the same antibiotic marker, the marker was removed from one of the parent strains. All antibiotic resistance cassettes used were engineered with loxP sites flanking the cassette, and so these cassettes could be removed by transforming cells with a plasmid that expresses Cre recombinase (plasmid pDR244, a gift from David Rudner). This plasmid also has a temperature-sensitive origin of replication, so after incubating cells at 30°C for 24 hours to remove the antibiotic cassette, cells were shifted to 45°C and incubated overnight to remove the plasmid. The removal of the cassette was verified by lack of growth on antibiotic selective plates.
Many of the strains used to investigate synthetic lethal conditions, namely bGS204, bGS206, bGS290, bGS293, bGS297, bGS298, bGS306, bGS308, bGS316, and bGS331, were additionally verified by whole genome sequencing to confirm that no suppressor mutations had arisen during cloning.
Statistical analysis:
Single-molecule lifetime distributions were compared using a two-sided Wilcoxon rank sum test, with p < 0.05 considered significant. Results are presented in Supplementary Table 4. FDAA and Pbp2B intensities were compared using a two-sided t-test, with p < 0.05 considered significant. Results are presented in Figure 4e and Extended Data Figure 8c.
Code availability:
Custom MATLAB code is available at: https://bitbucket.org/garnerlab/squyres-2020
Data availability:
Numerical source data that underlie the graphs shown in Figs. 1–4 and Extended Data Figs. 1–4 and 6–10 are provided with the paper. The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
Extended Data
Extended Data Fig. 1. Cell lengths with fusions for fluorescence microscopy.
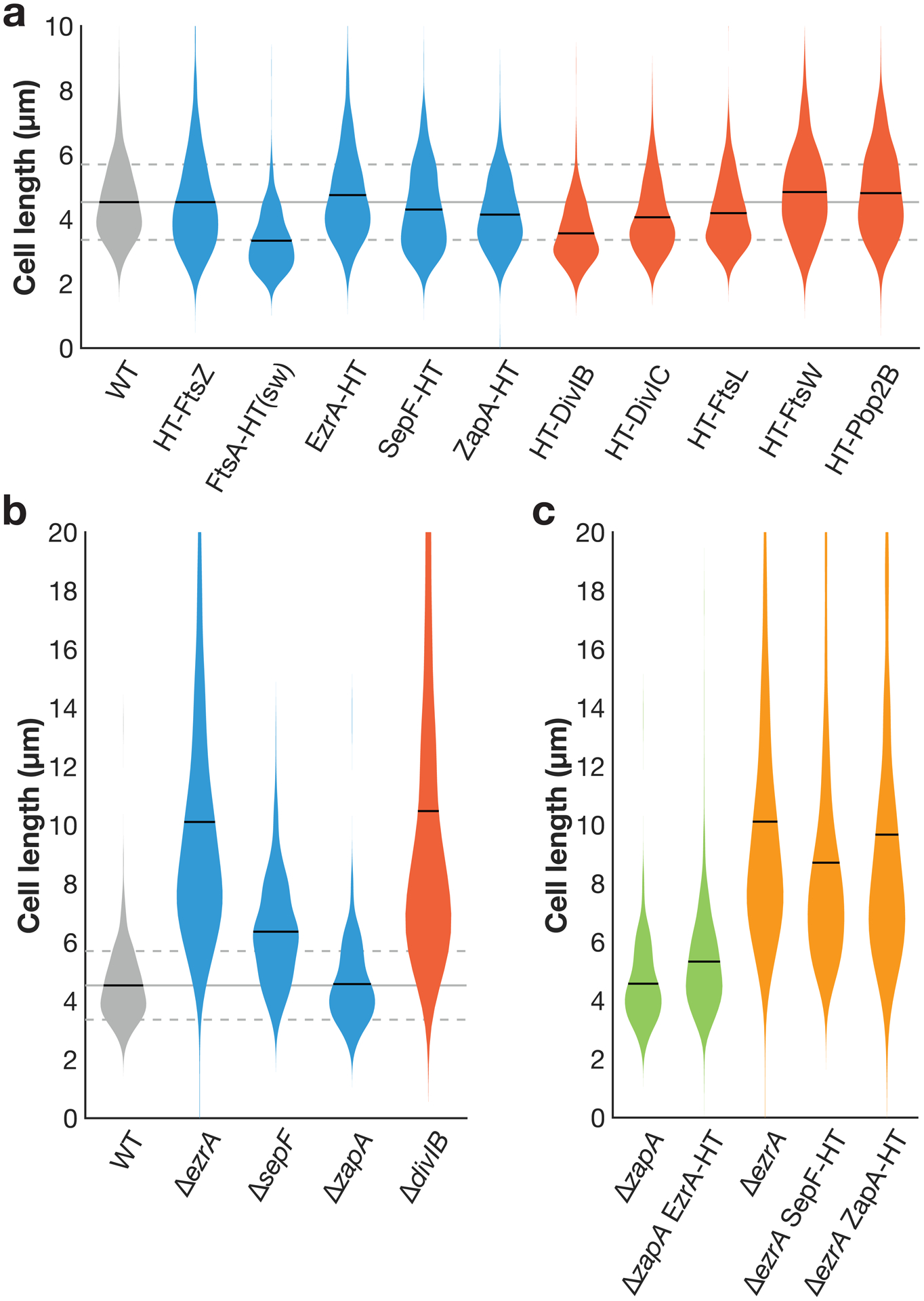
a Cell lengths in strains expressing HaloTag (HT) fusions used in this study. (sw) indicates a sandwich fusion. Cell lengths were measured from confocal microscopy of FM5-95 membrane stained cells. When cell division is inhibited, cell length increases; that cell lengths in each strain are equal to or less than that of wild type (WT) cells indicates that these fluorescent fusions do not strongly inhibit cell division. In some cases where the fluorescent fusion is merodiploid or expressed under inducible control, cells are shorter than WT, as might be expected when components of the cell division machinery are overexpressed. Blue: fusions to early-arriving division proteins, red: fusions to late-arriving division proteins. Gray lines: mean (solid line) ± standard deviation (dashed lines) for WT cell lengths. N>400 cells for each sample. b Lengths of cells with various division proteins knocked out, for comparison with a. We include all knockouts whose lengths can be measured in a straightforward way: ΔftsA cells have severe division defects and are highly elongated (see Extended Data Fig. 10), and the remaining division proteins are essential to avoid lethal filamentation59–62. N>140 cells for each sample. c EzrA and ZapA HT fusions are functional and SepF HT fusion expressed at an ectopic site does not disrupt SepF function. EzrA is synthetically lethal with SepF and ZapA18,29. We therefore knocked out one of these proteins and then expressed our HT fusion to the other protein; if HT fusion induced a critical defect in protein function, this combination will be lethal. Instead, in each case cells remained viable, with comparable lengths to the knockout alone. N>200 cells for each sample.
Extended Data Fig. 2. Controls for FtsZ lifetime measurements.

a FtsZ subunit lifetime is consistent across experimental replicates. To measure lifetimes, cells expressing FtsZ-HaloTag were induced with 20 μM IPTG for 2 hours, labelled with 40 pM JF549-HTL, and then imaged by TIRFM. Light curves: 4 experimental replicates for which N>200. Points represent combined data from all experimental replicates (17 replicates). Error bars: weighted standard deviation of distributions for all replicates. b FtsZ subunit lifetime is consistent across measurement techniques. Lifetime distributions were measured using an automated hidden Markov model (HMM) based analysis pipeline and manually for N=265 particles (dashed line). c FtsZ subunit lifetime is not affected by Pbp2B tagging. The 1-colour strain (bAB309) contains labelled FtsZ-HaloTag, induced as a second copy with 20 μM IPTG for 2 hours; the 2-colour strain (bGS104) contain both this FtsZ-HaloTag construct and a native mNeonGreen-Pbp2B fusion, which was used to localize the division site. d FtsZ subunit lifetime is not affected by photobleaching. If the measurements were affected by photobleaching, the measured lifetimes would increase when we decrease the imaging interval; however, we see that the lifetime distributions are consistent for images taken at 0.5-second intervals and 1-second intervals. For images at 0.5-second intervals, images were acquired continuously with 0.5-second exposures. For images at 1-second intervals, images were acquired with the same settings, with 0.5 seconds of exposure and 0.5-second intervals without illumination. e Co-overexpression of FtsA and FtsZ increases the number of FtsZ filaments in the cell (left) but does not change FtsZ subunit lifetime (right). The increased number of filaments that form upon FtsAZ overexpression is consistent with steady-state treadmilling of FtsZ. The fact that the subunit lifetime does not change when FtsAZ is overexpressed further indicates that the additional FtsZ forms new filaments rather than elongating existing filaments. A second copy of ftsAZ is expressed from an IPTG-inducible promoter with 100 μM IPTG for 2 hours. Filament density is visualized by TIRFM for at least two replicates of each condition. Scale bar: 2 μm.
Extended Data Fig. 3. Effects of individual ZBP knockouts on cell and Z ring morphology.
Each pair of images shows cell morphology (phase-contrast imaging, left), and Z ring morphology (epifluorescence images of cells expressing FtsZ-mNeonGreen induced with 20 μM IPTG for 2 hours, right) in control cells, compared to cells with individual ZBPs deletions. ΔezrA cells less condensed Z rings, along with the expected Z rings near their poles33; ΔsepF and ΔzapA cells have normal Z rings. FtsZ(T111A) mutant cells have excess Z rings and form mini-cells. The distribution of Z ring widths in each strain is plotted at bottom left. Representative images from at least two replicates of each condition. Scale bars: 2 μm.
Extended Data Fig. 4. Effects of ZBP overexpression on FtsZ.
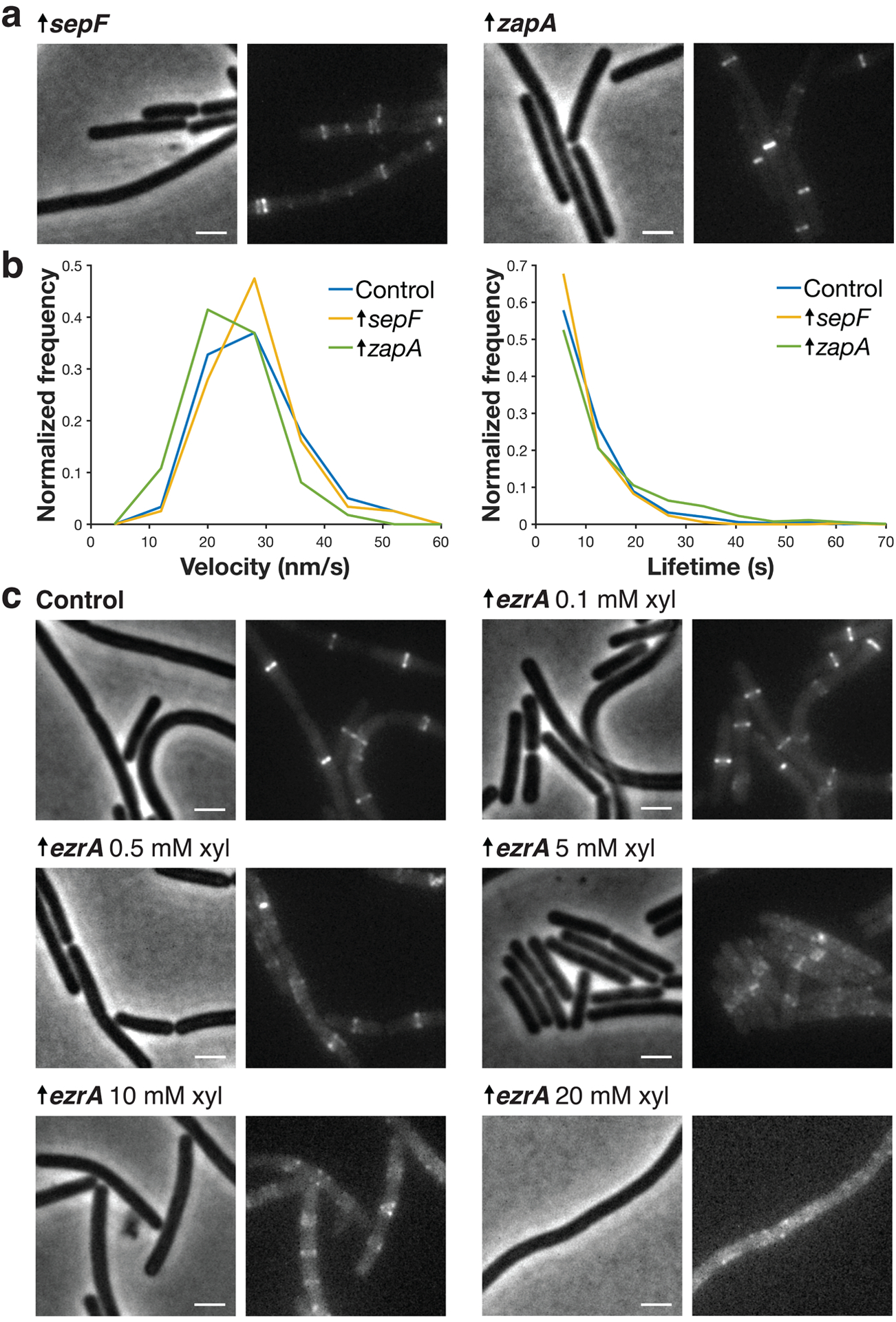
a Each pair of images shows cell morphology (phase-contrast imaging, left), and Z ring morphology (epifluorescence images of cells expressing FtsZ-mNeonGreen induced with 20 μM IPTG for 2 hours, right), in cells overexpressing SepF and ZapA. These cells have normal Z ring morphology except for some polar Z rings in SepF-overexpressing cells. Second copies of sepF and zapA were expressed from a xylose-inducible promoter with 30 mM xylose for 2 hours. Representative images from at least two replicates of each condition. Scale bars: 2 μm. b sepF- and zapA-overexpressing cells have similar FtsZ treadmilling velocities (left) and subunit lifetimes (right) to control cells. For velocity measurements, FtsZ-mNeonGreen was induced with 20 μM IPTG for 2 hours, imaged by TIRFM, and analysed from kymographs. For lifetime measurements, FtsZ-HaloTag was induced with 20 μM IPTG for 2 hours and labelled with 40 pM JF549-HTL. c Each pair of images shows cell morphology (phase-contrast imaging, left), and Z ring morphology (epifluorescence images of cells expressing FtsZ-mNeonGreen induced with 20 μM IPTG for 2 hours, right), in control cells and cells with EzrA overexpressed. EzrA-overexpressing cells have perturbed Z ring morphology, as expected30, a phenotype exacerbated with increasing induction. A second copy of ezrA was expressed from a xylose-inducible promoter by adding xylose at the indicated mM concentration. The 0.1, 0.5, and 5 mM concentrations were selected for quantitative analysis as the 10 and 20 mM xylose overexpression yielded unstable FtsZ filaments whose lifetimes were too short to be measured accurately. Representative images from at least two replicates of each condition. Scale bars: 2 μm.
Extended Data Fig. 5. Effects of removing synthetically lethal combinations of ZBPs on cell and Z ring morphology.
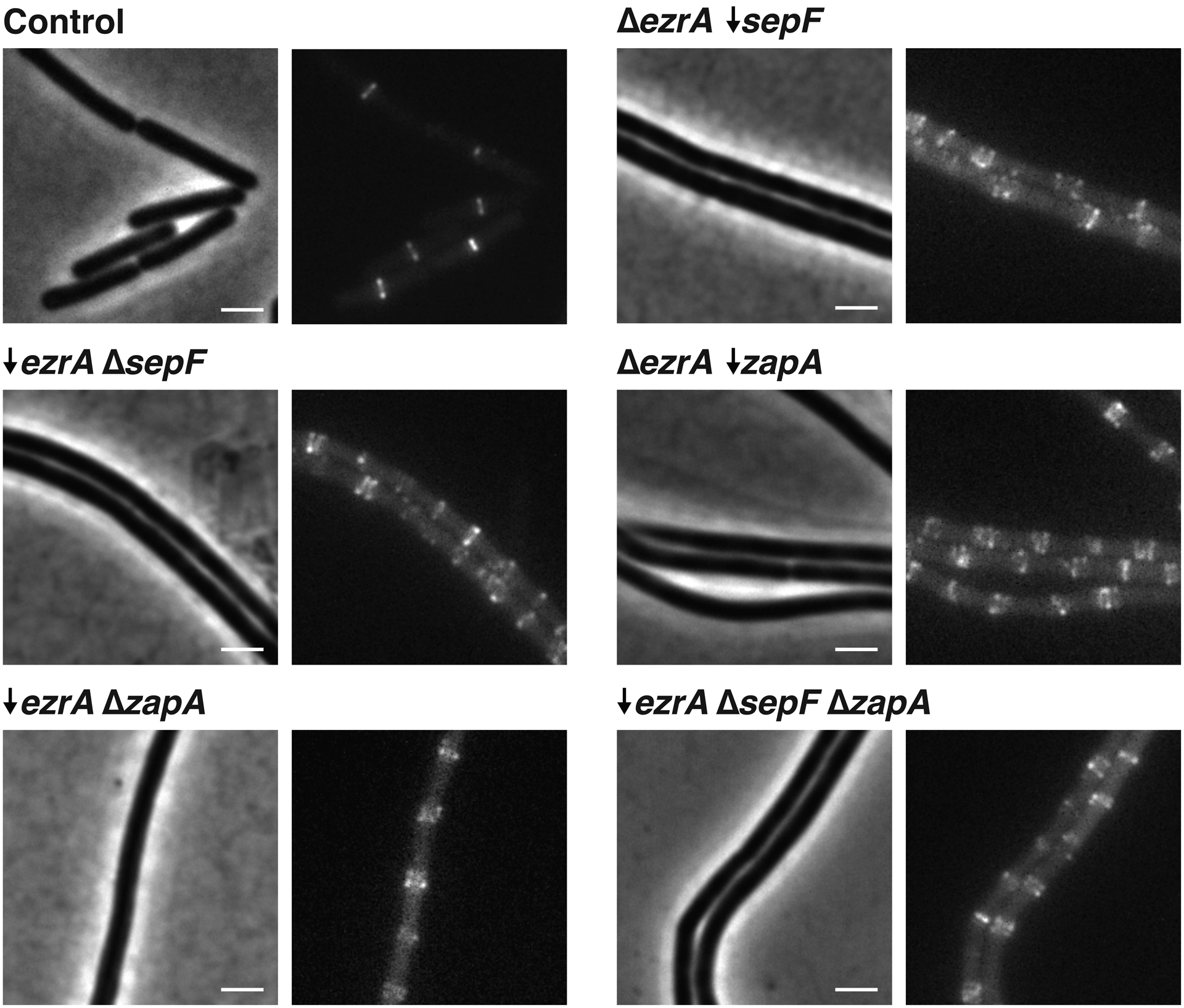
Each pair of images shows cell morphology (phase-contrast imaging, left), and Z ring morphology (epifluorescence images of cells expressing FtsZ-mNeonGreen induced with 20 μM IPTG for 2 hours, right), in control cells and cells lacking synthetically lethal combinations of ZBPs. To achieve this, a combination of knockouts (indicated by Δ) and depletions (indicated by ↓) were used; depletions were performed by expressing each gene under an inducible promoter until the start of the experiment, then withdrawing the inducer for 7 hours. This was repeated for all permutations of synthetically lethal combinations of ZBPs; all of these combinations result in elongated cells and disrupted Z ring architecture. Representative images from at least two replicates of each condition. Scale bars: 2 μm.
Extended Data Fig. 6. Effects of removing synthetically lethal combinations of ZBPs on FtsZ.
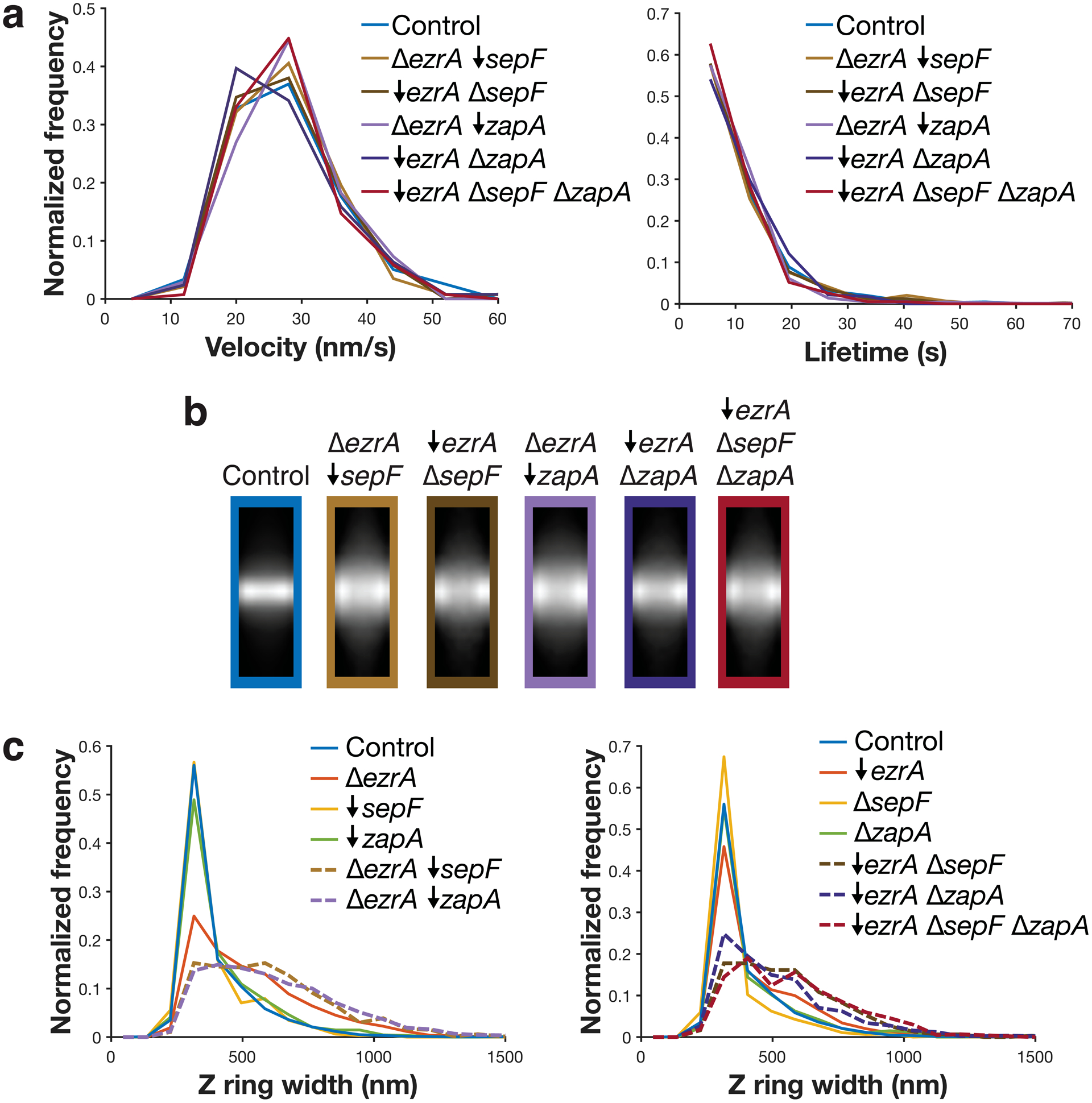
Velocity, lifetime, and Z ring morphology measurements for cells missing each synthetic lethal combination of ZBPs. All synthetic lethal combinations were investigated by a combination of knockouts (indicated by Δ) and depletions (indicated by ↓); depletions were performed by expressing the gene under an inducible promoter until the start of the experiment, then withdrawing the inducer for 7 hours. a Velocity (left) and lifetime (right) of cells missing synthetically lethal combinations of ZBPs are unchanged from control. For velocity measurements, FtsZ-mNeonGreen was induced with 20 μM IPTG for 2 hours, imaged by TIRFM, and then analysed from kymographs. For lifetime measurements, FtsZ-HaloTag was induced with 20 μM IPTG for 2 hours and labelled with 40 pM JF549-HTL. bc Z rings in cells missing synthetically lethal combinations of ZBPs are wider than control cells and cells missing individual ZBPs. Average intensity projections (b) and widths (c) of Z rings in each condition. Z rings were visualized using epifluorescence images of cells expressing FtsZ-mNeonGreen, induced with 20 μM IPTG for 2 hours. Z ring projections were created by averaging >100 Z ring images for each strain. Because ZBPs can be removed by either knockout or depletion, for each strain we compare to the equivalent single mutant knockouts and depletions.
Extended Data Fig. 7. Effects of a ΔsepF ΔzapA dual knockout.
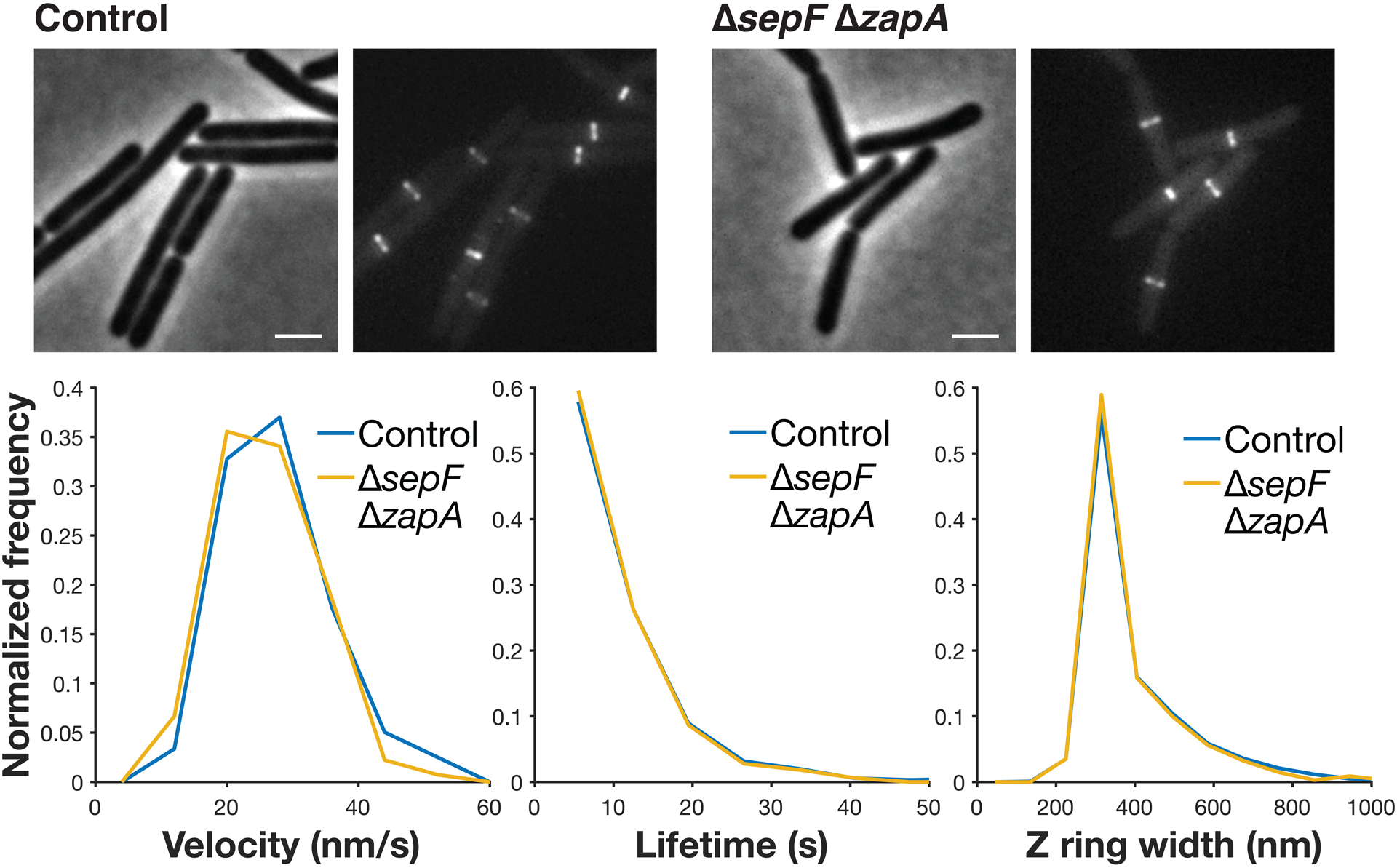
top Each pair of images shows cell morphology (phase-contrast imaging, left), and Z ring morphology (epifluorescence images of cells expressing FtsZ-mNeonGreen induced with 20 μM IPTG for 2 hours, right), in control cells and cells with both sepF and zapA knocked out: ΔsepF ΔzapA is the only combination of ZBP deletions that is not synthetically lethal and their Z ring morphology is normal. Representative images from at least two replicates of each condition. Scale bars: 2 μm. bottom ΔsepF ΔzapA cells have similar FtsZ treadmilling velocities (left), subunit lifetimes (centre), and Z ring widths (right) to control cells. For velocity measurements, FtsZ-mNeonGreen was induced with 20 μM IPTG for 2 hours, imaged by TIRFM, and analysed from kymographs. For lifetime measurements, FtsZ-HaloTag was induced with 20 μM IPTG for 2 hours and labelled with 40 pM JF549-HTL. For Z ring width measurements, FtsZ-mNeonGreen was induced with 20 μM IPTG for 2 hours and imaged by epifluorescence.
Extended Data Fig. 8. Characterization of the FtsZ(K86E) suppressor mutant.
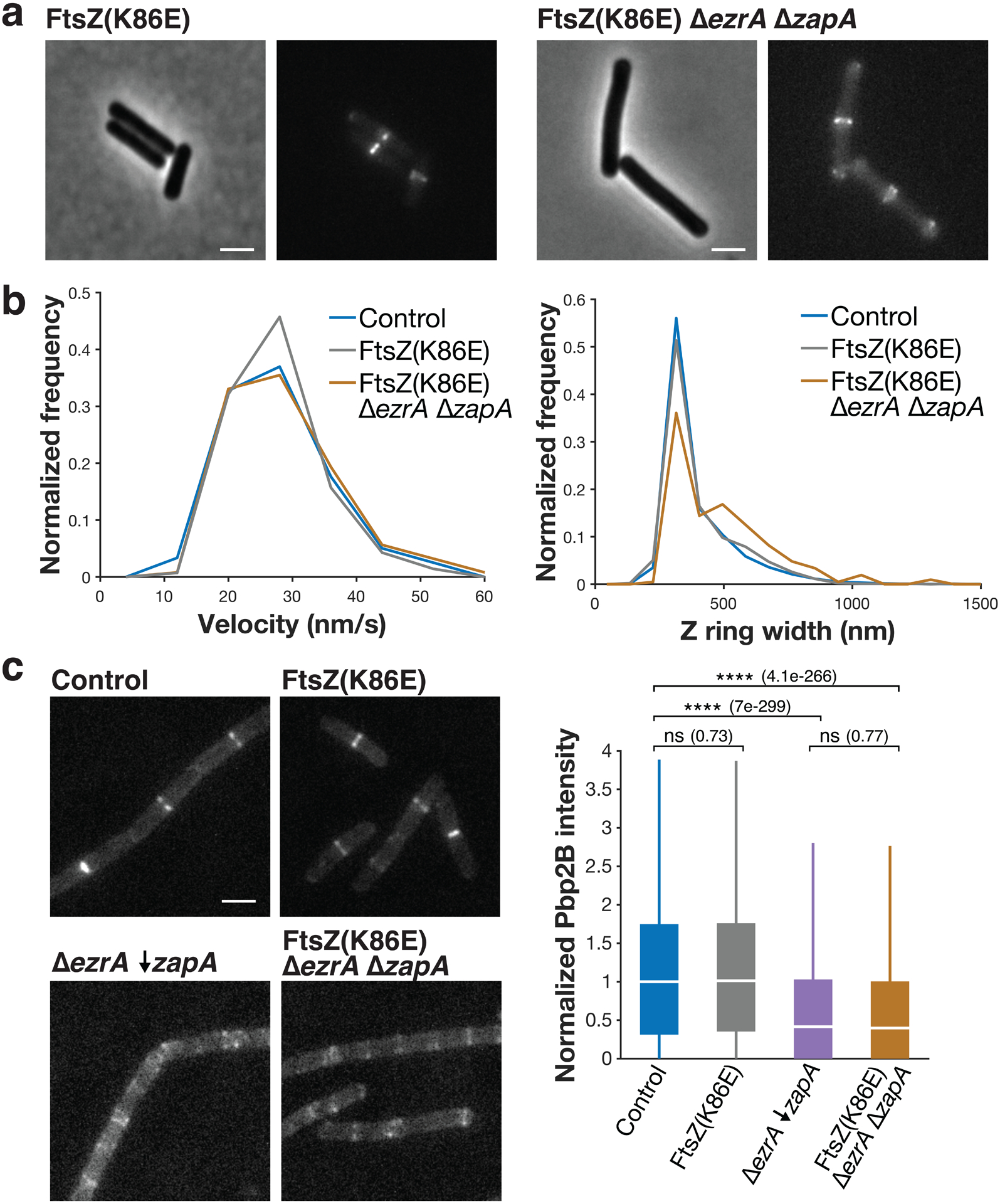
a Each pair of images shows cell morphology (phase-contrast imaging, left), and Z ring morphology (epifluorescence images of cells expressing FtsZ-mNeonGreen induced with 20 μM IPTG for 2 hours, right) in FtsZ(K86E) and FtsZ(K86E) ΔezrA ΔzapA cells. FtsZ(K86E) Z rings look similar to the control. Z rings in FtsZ(K86E) ΔezrA ΔzapA are somewhat perturbed, but less so than typical cells missing synthetically lethal combinations of ZBPs; they also have polar Z rings, as is typical for ΔezrA strains. Representative images from at least two replicates of each condition. Scale bars: 2 μm. b FtsZ(K86E) and FtsZ(K86E) ΔezrA ΔzapA have similar FtsZ treadmilling velocities to control (left), and FtsZ(K86E) Z rings are identical in width to control, while FtsZ(K86E) ΔezrA ΔzapA are wider (right). For velocity measurements in each strain, FtsZ-mNeonGreen was induced with 20 μM IPTG for 2 hours, imaged by TIRFM, and analysed from kymographs. For Z ring width measurements, FtsZ-mNeonGreen was induced with 20 μM IPTG for 2 hours and imaged by epifluorescence. c Pbp2B intensity at midcell in FtsZ(K86E) mutant cells. Left: Representative images of Pbp2B in the indicated strains, visualized by epifluorescence imaging of cells expressing Pbp2B-mNeonGreen, from at least 4 replicates of each condition. Right: Pbp2B intensity at the division site in each strain. Although the FtsZ(K86E) restores viability in a ΔezrA ΔzapA strain, it does so without rescuing Pbp2B recruitment to midcell. For each box plot, the white line indicates the median, the box extends to the 25th and 75th percentiles, and the whiskers indicate 1.5x interquartile range. P-values were obtained from a two-sided t-test; ns indicates p > 0.5, **** indicates p<0.0001, and p-values are included in parenthesis. N>5000 division sites for each condition. Scale bars: 2 μm.
Extended Data Fig. 9. Pbp2B localization and FDAA incorporation in ΔZBPs cells.
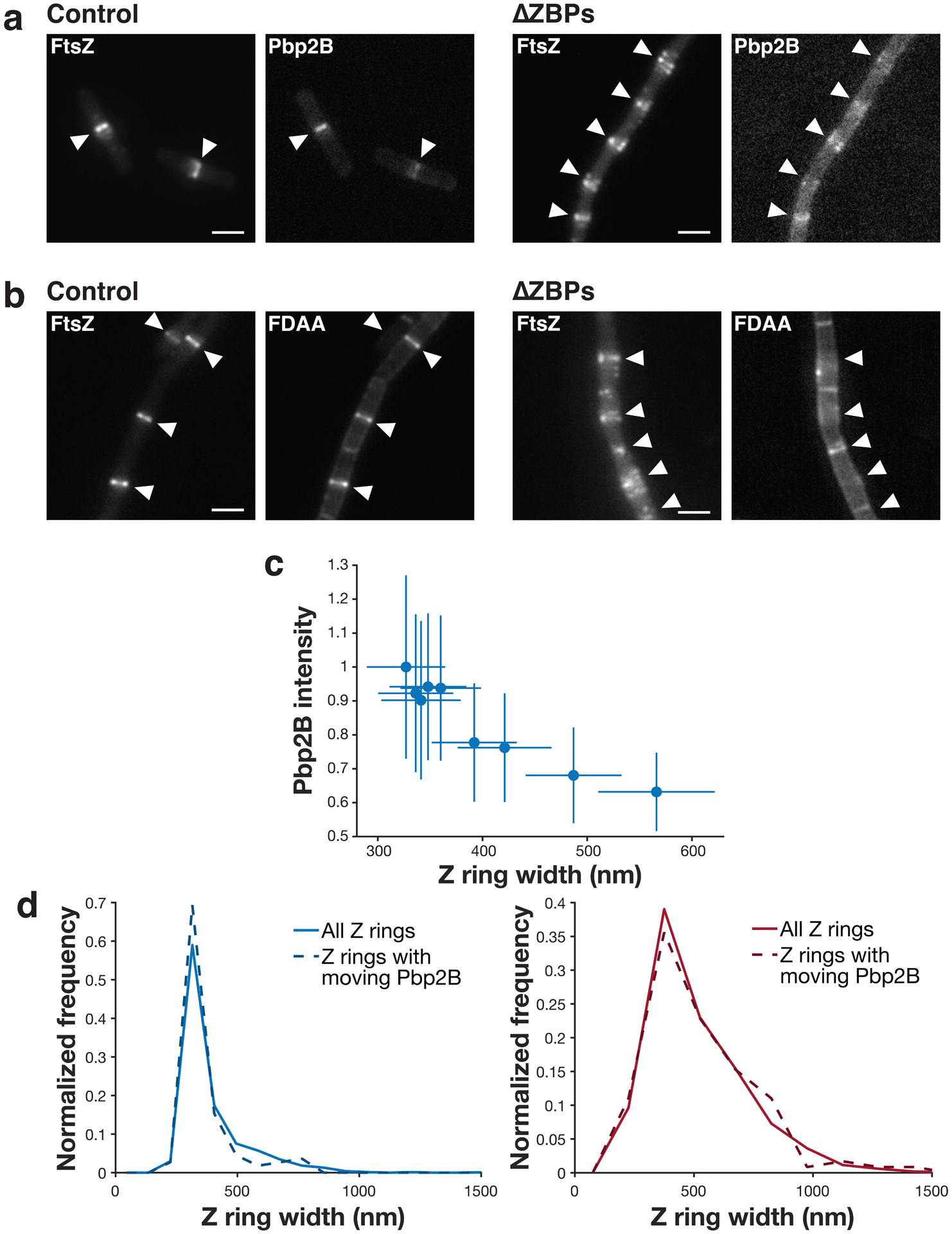
a Z rings (left) and Pbp2B localization (right) (epifluorescence images of cells expressing both Pbp2B-mNeonGreen and FtsZ-HaloTag induced with 20 μM IPTG for 2 hours and labelled with 5 nM JF549-HTL) in control and ΔZBPs cells. White arrows indicate the Z ring positions in each image. Representative images from at least two replicates of each condition. Scale bars: 2 μm. b Z rings and fluorescent D-amino acid (FDAA) incorporation (epifluorescence images of cells labelled with 1 mM fluorescent D-lysine (FDL) for 30 seconds, right) in control and ΔZBPs cells. White arrows indicate the Z ring positions in each image. Representative images from at least 4 replicates of each condition. Scale bars: 2 μm. c Z ring width versus Pbp2B recruitment. Pbp2B intensity at midcell is higher when Z rings are more condensed; this is expected given that Pbp2B recruitment and Z ring condensation both increase over time. N = 2761 Z rings. d Pbp2B directional motion is seen at Z rings of all widths. Z ring width distributions of all Z rings (solid lines) and the Z rings at which Pbp2B moves directionally (dashed lines) for either control cells (left) or ΔZBPs cells (right) are similar. This confirms that the Pbp2B motion seen in ΔZBPs is present at decondensed rings. N>100 Z rings in each distribution. Shaded area: SEM.
Extended Data Fig. 10. FtsA modulates FtsZ dynamics and Z ring formation.
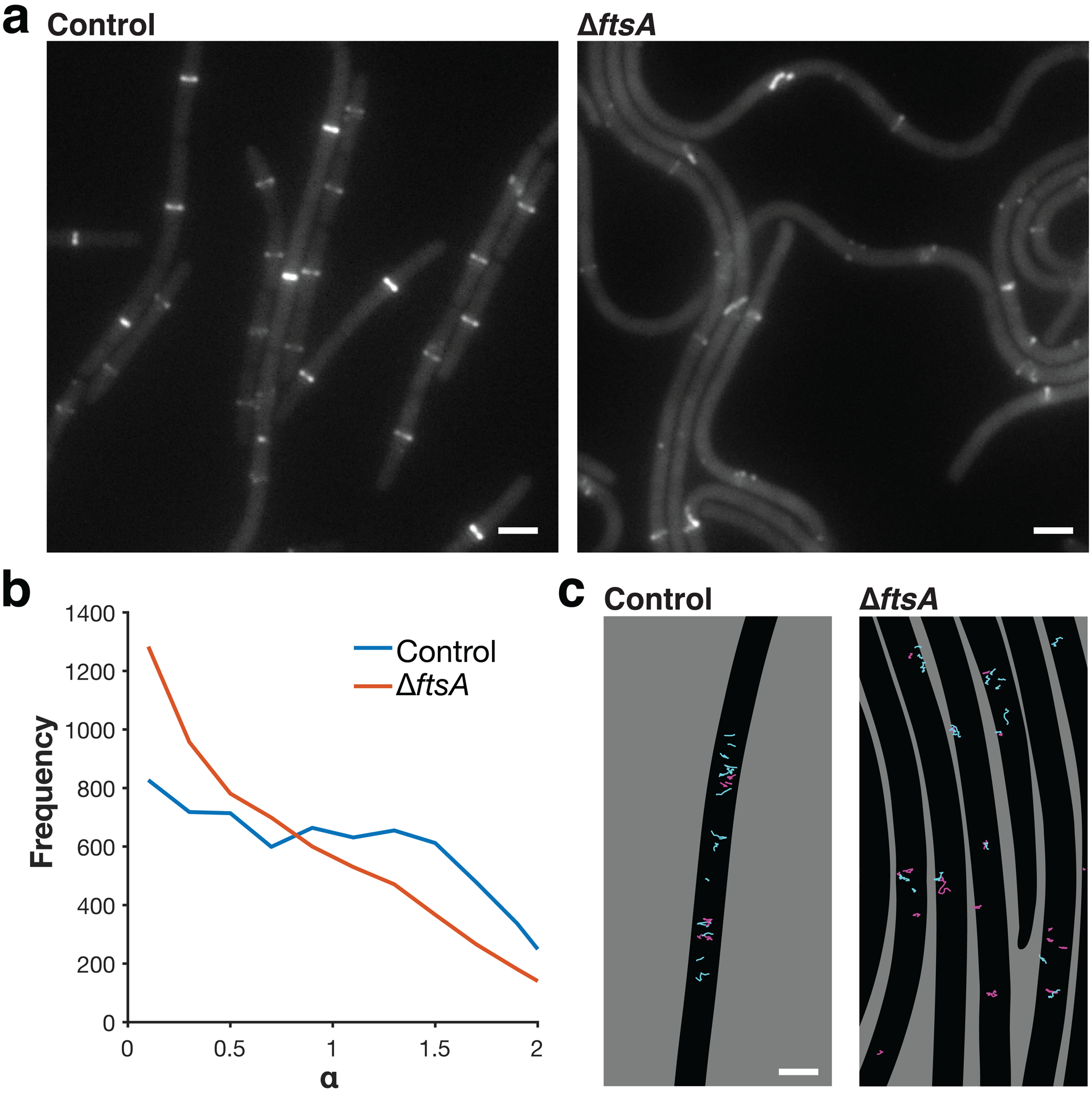
a Z ring morphology (epifluorescence images of cells expressing FtsZ-mNeonGreen induced with 30 mM xylose for 2 hours) in control cells and ΔftsA cells. ΔftsA cells have highly altered Z rings. In ΔftsA cells, FtsZ is expressed with 10 μM IPTG from the pHyperSpank promoter; higher or lower expression levels do not allow for cell survival. Representative images from three replicates of each condition. Scale bars: 2 μm. b Distributions of α values for FtsZ motion in control and ΔftsA cells, obtained by tracking FtsZ filament motion and fitting each track to MSD(Δt) = D*Δtα. α > 1 indicates directional motion, so FtsZ filaments in ΔftsA cells exhibit less directional treadmilling compared to control cells. N>6000 tracks for each condition. c Tracks of FtsZ filament motion in control and ΔftsA cells. Tracks with α > 1 are cyan, tracks with α ≤ 1 are magenta. In control cells, FtsZ filaments often treadmill directionally along the short axis of the cell. In ΔftsA cells, directional motion occurs less frequently and follows the short axis of the cell less consistently. Segmented cells are shown in black on a grey background. Scale bars: 2 μm.
Supplementary Material
Supplementary Video 6 FtsZ filament treadmilling is unaltered in the absence of ZBPs. ∆ZBPs cells have sepF and zapA knocked out, and ezrA depleted from a xylose-inducible promoter for 7 hours prior to imaging. Cells expressing FtsZ-mNeonGreen were imaged at 1-second intervals for 100 seconds by TIRFM. The video is displayed at 30 frames per second (30x actual speed). Scale bar: 5 µm.
Supplementary Video 5 EzrA overexpression decreases FtsZ filament length, visualized by SIM-TIRF microscopy. A second copy of ezrA was expressed from a xylose-inducible promoter. Panel labels indicate the amount of xylose added in each experiment. Cells expressed FtsZ-mNeonGreen to visualize FtsZ filaments and were imaged at 1-second intervals for 100 seconds by SIM-TIRF microscopy. The video is displayed at 30 frames per second (30x actual speed). Scale bar: 5 µm.
Supplementary Video 7 FtsA filaments show less directional treadmilling in a ∆ftsA strain. ∆ftsA cells express FtsZ under the pHyperSpank promoter induced with 10 µM IPTG. Cells expressing FtsZ-mNeonGreen were imaged at 1-second intervals for 100 seconds by TIRFM. The video is displayed at 30 frames per second (30x actual speed). Scale bar: 5 µm.
Supplementary Video 1 FtsZ filaments treadmill around the cell. Cells expressing FtsZ-mNeonGreen (strain bAB219) were induced with 20 µM IPTG for 2 hours, then imaged at 1-second intervals for 100 seconds by TIRFM. The video is displayed at 30 frames per second (30x actual speed). Scale bar: 5 µm.
Supplementary Video 2 Single molecules of EzrA, SepF, and ZapA are stationary. Each protein was expressed as a HaloTag fusion and labelled with JF549-HTL dye for single-molecule imaging using TIRFM. Videos begin with an image of the Z rings in each cell, visualized by FtsZ-mNeonGreen. Cells were imaged at 1-second intervals for 200 seconds; every other frame is displayed here. The video is displayed at 30 frames per second (60x actual speed). Scale bar: 5 µm.
Supplementary Video 4 Single-molecule imaging of FtsZ for lifetime analysis. Cells expressing FtsZ-HaloTag were imaged at 500-ms intervals for 50 seconds by TIRFM. The video is displayed at 30 frames per second (15x actual speed). Scale bar: 5 µm.
Supplementary Video 3 Single molecules of DivIB, DivIC, FtsL, FtsW, and Pbp2B move directionally around the division site. Each protein was expressed as a HaloTag fusion and labelled with JF549-HTL dye for single-molecule imaging using TIRFM. Videos begin with an image of the Z rings in each cell, visualized by FtsZ-mNeonGreen. Cells were imaged at 1-second intervals for 200 seconds; every other frame is displayed here. The video is displayed at 30 frames per second (60x actual speed). Scale bar: 5 µm.
Acknowledgements:
We thank the Garner lab, especially A. Bisson-Filho, Y. Sun, and S. Wilson, for discussions and strains, and L. Lavis for JF dyes. TIRF-SIM was performed at the Advanced Imaging Center at the Janelia Research Campus, a facility jointly supported by the Gordon and Betty Moore Foundation and Howard Hughes Medical Institute. This work was funded by National Institutes of Health Grants DP2AI117923-01 to ECG, as well as support from the Volkswagen Foundation, NSF GRFP (DGE1144152) to GRS, and the Physiology course at the Marine Biological Laboratory at Woods Hole. This work was supported by the NSF-Simons Center for Mathematical and Statistical Analysis of Biology at Harvard (1764269) and the Harvard Quantitative Biology Initiative.
Footnotes
Competing Interests Statement: Authors declare no competing interests.
Supplementary Information is available for this paper.
References:
- 1.Erickson HP, Anderson DE & Osawa M FtsZ in bacterial cytokinesis: cytoskeleton and force generator all in one. Microbiol. Mol. Biol. Rev 74, 504–528 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adams DW & Errington J Bacterial cell division: assembly, maintenance and disassembly of the Z ring. Nature Publishing Group 7, 642–653 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Gamba P, Veening J-W, Saunders NJ, Hamoen LW & Daniel RA Two-step assembly dynamics of the Bacillus subtilis divisome. J. Bacteriol 191, 4186–4194 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meier EL & Goley ED Form and function of the bacterial cytokinetic ring. Curr. Opin. Cell Biol 26, 19–27 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Bisson-Filho AW et al. Treadmilling by FtsZ filaments drives peptidoglycan synthesis and bacterial cell division. Science 355, 739–743 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang X et al. GTPase activity–coupled treadmilling of the bacterial tubulin FtsZ organizes septal cell wall synthesis. Science 355, 744–747 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grimm JB et al. A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat. Methods 12, 244–250 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perez AJ et al. Movement dynamics of divisome proteins and PBP2x:FtsW in cells of Streptococcus pneumoniae. Proceedings of the National Academy of Sciences 116, 3211–3220 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taguchi A et al. FtsW is a peptidoglycan polymerase that is functional only in complex with its cognate penicillin-binding protein. Nat Microbiol 4, 587–594 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noirclerc-Savoye M et al. In vitro reconstitution of a trimeric complex of DivIB, DivIC and FtsL, and their transient co-localization at the division site in Streptococcus pneumoniae. Molecular Microbiology 55, 413–424 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Loose M & Mitchison TJ The bacterial cell division proteins FtsA and FtsZ self-organize into dynamic cytoskeletal patterns. Nat. Cell Biol 16, 38–46 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramirez-Diaz DA et al. Treadmilling analysis reveals new insights into dynamic FtsZ ring architecture. PLoS Biol 16, e2004845 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anderson DE, Gueiros-Filho FJ & Erickson HP Assembly Dynamics of FtsZ Rings in Bacillus subtilis and Escherichia coli and Effects of FtsZ-Regulating Proteins. J. Bacteriol 186, 5775–5781 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodrigues CDA & Harry EJ The Min system and nucleoid occlusion are not required for identifying the division site in Bacillus subtilis but ensure its efficient utilization. PLoS Genet 8, e1002561 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bisson-Filho AW et al. FtsZ filament capping by MciZ, a developmental regulator of bacterial division. Proc. Natl. Acad. Sci. U.S.A 112, E2130–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Northrup SH & Erickson HP Kinetics of protein-protein association explained by Brownian dynamics computer simulation. Proceedings of the National Academy of Sciences 89, 3338–3342 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Romberg L, Simon M & Erickson HP Polymerization of Ftsz, a bacterial homolog of tubulin. is assembly cooperative? J. Biol. Chem 276, 11743–11753 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Gueiros-Filho FJ & Losick R A widely conserved bacterial cell division protein that promotes assembly of the tubulin-like protein FtsZ. Genes & Development 16, 2544–2556 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Low HH, Moncrieffe MC & Löwe J The crystal structure of ZapA and its modulation of FtsZ polymerisation. J. Mol. Biol 341, 839–852 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Small E et al. FtsZ polymer-bundling by the Escherichia coli ZapA orthologue, YgfE, involves a conformational change in bound GTP. J. Mol. Biol 369, 210–221 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Singh JK, Makde RD, Kumar V & Panda D SepF increases the assembly and bundling of FtsZ polymers and stabilizes FtsZ protofilaments by binding along its length. J. Biol. Chem 283, 31116–31124 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dajkovic A, Pichoff S, Lutkenhaus J & Wirtz D Cross-linking FtsZ polymers into coherent Z rings. Molecular Microbiology 78, 651–668 (2010). [DOI] [PubMed] [Google Scholar]
- 23.Gündoğdu ME et al. Large ring polymers align FtsZ polymers for normal septum formation. The EMBO Journal 30, 617–626 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pacheco-Gómez R et al. Tetramerization of ZapA is required for FtsZ bundling. Biochem. J 449, 795–802 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Caldas P et al. Cooperative ordering of treadmilling filaments in cytoskeletal networks of FtsZ and its crosslinker ZapA. Nat Commun 10, 5744–13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Monahan LG, Robinson A & Harry EJ Lateral FtsZ association and the assembly of the cytokinetic Z ring in bacteria. Molecular Microbiology 74, 1004–1017 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Buss J et al. In vivo organization of the FtsZ-ring by ZapA and ZapB revealed by quantitative super-resolution microscopy. Molecular Microbiology 89, 1099–1120 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duman R et al. Structural and genetic analyses reveal the protein SepF as a new membrane anchor for the Z ring. Proc. Natl. Acad. Sci. U.S.A 110, E4601–10 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hamoen LW, Meile J-C, de Jong W, Noirot P & Errington J SepF, a novel FtsZ-interacting protein required for a late step in cell division. Molecular Microbiology 59, 989–999 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Haeusser DP, Schwartz RL, Smith AM, Oates ME & Levin PA EzrA prevents aberrant cell division by modulating assembly of the cytoskeletal protein FtsZ. Molecular Microbiology 52, 801–814 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh JK, Makde RD, Kumar V & Panda D A membrane protein, EzrA, regulates assembly dynamics of FtsZ by interacting with the C-terminal tail of FtsZ. Biochemistry 46, 11013–11022 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Cleverley RM et al. Structure and function of a spectrin-like regulator of bacterial cytokinesis. Nat Commun 5, 5421 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levin PA, Kurtser IG & Grossman AD Identification and characterization of a negative regulator of FtsZ ring formation in Bacillus subtilis. Proceedings of the National Academy of Sciences 96, 9642–9647 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haeusser DP, Garza AC, Buscher AZ & Levin PA The Division Inhibitor EzrA Contains a Seven-Residue Patch Required for Maintaining the Dynamic Nature of the Medial FtsZ Ring. J. Bacteriol 189, 9001–9010 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guan F et al. Lateral interactions between protofilaments of the bacterial tubulin homolog FtsZ are essential for cell division. Elife 7, 30 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shin JY, Vollmer W, Lagos R & Monasterio O Glutamate 83 and arginine 85 of helix H3 bend are key residues for FtsZ polymerization, GTPase activity and cellular viability of Escherichia coli: lateral mutations affect FtsZ polymerization and E. coli viability. BMC Microbiol 13, 26 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pichoff S & Lutkenhaus J Tethering the Z ring to the membrane through a conserved membrane targeting sequence in FtsA. Molecular Microbiology 55, 1722–1734 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Beall B & Lutkenhaus J Impaired cell division and sporulation of a Bacillus subtilis strain with the ftsA gene deleted. J. Bacteriol 174, 2398–2403 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jensen SO, Thompson LS & Harry EJ Cell division in Bacillus subtilis: FtsZ and FtsA association is Z-ring independent, and FtsA is required for efficient midcell Z-Ring assembly. J. Bacteriol 187, 6536–6544 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krupka M & Margolin W Unite to divide: Oligomerization of tubulin and actin homologs regulates initiation of bacterial cell division. F1000Res 7, 235 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang K-H, Durand-Heredia J & Janakiraman A FtsZ ring stability: of bundles, tubules, crosslinks, and curves. J. Bacteriol 195, 1859–1868 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bhattacharya D, Kumar A & Panda D WhmD promotes the assembly of Mycobacterium smegmatis FtsZ: A possible role of WhmD in bacterial cell division. Int J Biol Macromol 95, 582–591 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Eswara PJ et al. An essential Staphylococcus aureus cell division protein directly regulates FtsZ dynamics. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sogues A et al. Essential dynamic interdependence of FtsZ and SepF for Z-ring and septum formation in Corynebacterium glutamicum. Nat Commun 11, 1641 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rahman MU et al. Assembly properties of bacterial tubulin homolog FtsZ regulated by the positive regulator protein ZipA and ZapA from Pseudomonas aeruginosa. Sci Rep 10, 21369 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pende N et al. SepF is the FtsZ-anchor in Archaea: implications for cell division in the Last Universal Common Ancestor. bioRxiv 11, 2020.10.06.328377 (2020). [Google Scholar]
- 47.Mateos-Gil P, Tarazona P & Vélez M Bacterial cell division: modeling FtsZ assembly and force generation from single filament experimental data. FEMS Microbiol. Rev 43, 73–87 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Osawa M, Anderson DE & Erickson HP Reconstitution of contractile FtsZ rings in liposomes. Science 320, 792–794 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Szwedziak P, Wang Q, Bharat TAM, Tsim M & Löwe J Architecture of the ring formed by the tubulin homologue FtsZ in bacterial cell division. Elife 3, e04601 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guigas G & Weiss M Effects of protein crowding on membrane systems. Biochim. Biophys. Acta 1858, 2441–2450 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Cayley DS, Guttman HJ & Record MT Biophysical characterization of changes in amounts and activity of Escherichia coli cell and compartment water and turgor pressure in response to osmotic stress. Biophysj 78, 1748–1764 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Erickson HP How bacterial cell division might cheat turgor pressure - a unified mechanism of septal division in Gram-positive and Gram-negative bacteria. Bioessays 39, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nguyen LT et al. Simulations suggest a constrictive force is required for Gram-negative bacterial cell division. Nat Commun 10, 1259–11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kner P, Chhun BB, Griffis ER, Winoto L & Gustafsson MGL Super-resolution video microscopy of live cells by structured illumination. Nat. Methods 6, 339–342 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hoffmann H Violin Plot https://www.mathworks.com/matlabcentral/fileexchange/45134-violin-plot (2015)
- 56.Van Valen DA et al. Deep Learning Automates the Quantitative Analysis of Individual Cells in Live-Cell Imaging Experiments. PLoS Comput Biol 12, e1005177–24 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tinevez J-Y et al. TrackMate: an open and extensible platform for single-particle tracking. Methods 1–29 (2016). doi: 10.1016/j.ymeth.2016.09.016 [DOI] [PubMed] [Google Scholar]
- 58.Bronson JE, Fei J, Hofman JM, Gonzalez RL Jr & Wiggins CH Learning Rates and States from Biophysical Time Series: A Bayesian Approach to Model Selection and Single-Molecule FRET Data. Biophysj 97, 3196–3205 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Levin PA & Losick R Characterization of a cell division gene from Bacillus subtilis that is required for vegetative and sporulation septum formation. J. Bacteriol 176, 1451–1459 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gamba P, Hamoen LW & Daniel RA Cooperative Recruitment of FtsW to the Division Site of Bacillus subtilis. Front Microbiol 7, 1808 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Daniel RA, Harry EJ, Katis VL, Wake RG & Errington J Characterization of the essential cell division gene ftsL(yIID) of Bacillus subtilis and its role in the assembly of the division apparatus. Molecular Microbiology 29, 593–604 (1998). [DOI] [PubMed] [Google Scholar]
- 62.Daniel RA, Harry EJ & Errington J Role of penicillin‐binding protein PBP 2B in assembly and functioning of the division machinery of Bacillus subtilis. Molecular Microbiology 35, 299–311 (2000). [DOI] [PubMed] [Google Scholar]
- 63.Youngman PJ, Perkins JB & Losick R Genetic transposition and insertional mutagenesis in Bacillus subtilis with Streptococcus faecalis transposon Tn917. Proceedings of the National Academy of Sciences 80, 2305–2309 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hussain S et al. MreB filaments align along greatest principal membrane curvature to orient cell wall synthesis. Elife 7, 1239 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Levin PA, Schwartz RL & Grossman AD Polymer Stability Plays an Important Role in the Positional Regulation of FtsZ. J. Bacteriol 183, 5449–5452 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chung K-M, Hsu H-H, Yeh H-Y & Chang B-Y Mechanism of regulation of prokaryotic tubulin-like GTPase FtsZ by membrane protein EzrA. J. Biol. Chem 282, 14891–14897 (2007). [DOI] [PubMed] [Google Scholar]
- 67.Land AD, Luo Q & Levin PA Functional Domain Analysis of the Cell Division Inhibitor EzrA. PLoS ONE 9, e102616 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Feucht A, Lucet I, Yudkin MD & Errington J Cytological and biochemical characterization of the FtsA cell division protein of Bacillus subtilis. Molecular Microbiology 40, 115–125 (2001). [DOI] [PubMed] [Google Scholar]
- 69.Kawai Y & Ogasawara N Bacillus subtilis EzrA and FtsL synergistically regulate FtsZ ring dynamics during cell division. Microbiology (Reading, Engl.) 152, 1129–1141 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Video 6 FtsZ filament treadmilling is unaltered in the absence of ZBPs. ∆ZBPs cells have sepF and zapA knocked out, and ezrA depleted from a xylose-inducible promoter for 7 hours prior to imaging. Cells expressing FtsZ-mNeonGreen were imaged at 1-second intervals for 100 seconds by TIRFM. The video is displayed at 30 frames per second (30x actual speed). Scale bar: 5 µm.
Supplementary Video 5 EzrA overexpression decreases FtsZ filament length, visualized by SIM-TIRF microscopy. A second copy of ezrA was expressed from a xylose-inducible promoter. Panel labels indicate the amount of xylose added in each experiment. Cells expressed FtsZ-mNeonGreen to visualize FtsZ filaments and were imaged at 1-second intervals for 100 seconds by SIM-TIRF microscopy. The video is displayed at 30 frames per second (30x actual speed). Scale bar: 5 µm.
Supplementary Video 7 FtsA filaments show less directional treadmilling in a ∆ftsA strain. ∆ftsA cells express FtsZ under the pHyperSpank promoter induced with 10 µM IPTG. Cells expressing FtsZ-mNeonGreen were imaged at 1-second intervals for 100 seconds by TIRFM. The video is displayed at 30 frames per second (30x actual speed). Scale bar: 5 µm.
Supplementary Video 1 FtsZ filaments treadmill around the cell. Cells expressing FtsZ-mNeonGreen (strain bAB219) were induced with 20 µM IPTG for 2 hours, then imaged at 1-second intervals for 100 seconds by TIRFM. The video is displayed at 30 frames per second (30x actual speed). Scale bar: 5 µm.
Supplementary Video 2 Single molecules of EzrA, SepF, and ZapA are stationary. Each protein was expressed as a HaloTag fusion and labelled with JF549-HTL dye for single-molecule imaging using TIRFM. Videos begin with an image of the Z rings in each cell, visualized by FtsZ-mNeonGreen. Cells were imaged at 1-second intervals for 200 seconds; every other frame is displayed here. The video is displayed at 30 frames per second (60x actual speed). Scale bar: 5 µm.
Supplementary Video 4 Single-molecule imaging of FtsZ for lifetime analysis. Cells expressing FtsZ-HaloTag were imaged at 500-ms intervals for 50 seconds by TIRFM. The video is displayed at 30 frames per second (15x actual speed). Scale bar: 5 µm.
Supplementary Video 3 Single molecules of DivIB, DivIC, FtsL, FtsW, and Pbp2B move directionally around the division site. Each protein was expressed as a HaloTag fusion and labelled with JF549-HTL dye for single-molecule imaging using TIRFM. Videos begin with an image of the Z rings in each cell, visualized by FtsZ-mNeonGreen. Cells were imaged at 1-second intervals for 200 seconds; every other frame is displayed here. The video is displayed at 30 frames per second (60x actual speed). Scale bar: 5 µm.
Data Availability Statement
Numerical source data that underlie the graphs shown in Figs. 1–4 and Extended Data Figs. 1–4 and 6–10 are provided with the paper. The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.