

Abstract
Intensive cropping degrades soil quality and disrupts the soil microbiome. To understand the effect of rice monocropping on soil-microbiome, we used a comparative 16S rRNA metagenome sequencing method to analyze the diversity of soil microflora at the genomic level. Soil samples were obtained from five locations viz., Chamarajnagara, Davangere, Gangavathi, Mandya, and Hassan of Karnataka, India. Chemical analysis of soil samples from these locations revealed significant variations in pH (6.00–8.38), electrical conductivity (0.17–0.69 dS m−1), organic carbon (0.51–1.29%), available nitrogen (279–551 kg ha−1), phosphorous (57–715 kg ha−1) and available potassium (121–564 kg ha−1). The 16S metagenome analysis revealed that the microbial diversity in Gangavathi soil samples was lower than in other locations. The soil sample of Gangavathi showed a higher abundance of Proteobacteria (85.78%) than Mandya (27.18%). The Firmicutes were more abundant in Chamarajnagar samples (36.01%). Furthermore, the KEGG pathway study revealed enriched nitrogen, phosphorus, and potassium metabolism pathways in all soil samples. In terms of the distribution of beneficial microflora, the decomposers were more predominant than the nutrient recyclers such as nitrogen fixers, phosphorous mineralizers, and nitrifiers. Furthermore, we isolated culturable soil microbes and tested their antagonistic activity in vitro against a fungal pathogen of rice, Magnaporthe oryzae strain MG01. Six Bacillus sp. and two strains of Trichoderma harzianum showed higher antagonistic activity against MG01. Our findings indicate that metagenome sequencing can be used to investigate the diversity, distribution, and abundance of soil microflora in rice-growing areas. The knowledge gathered can be used to develop strategies for maintaining soil quality and crop conservation to increase crop productivity.
Supplementary Information
The online version contains supplementary material available at 10.1007/s13205-021-02783-y.
Keywords: Rice, Soil, Metagenomics, Microbial diversity, Biocontrol, Blast disease
Introduction
In recent times, soil regeneration is gaining importance due to reduced crop yields due to loss of soil health and fertility (Zhang and He 2004). The soil has an enormous diversity of microbial communities affected by geographical location (Dubey et al. 2019) and cropping pattern (Wieland et al. 2001). The soil surrounding the rhizosphere zone is a hot spot for microbial interactions, as the exudates released from plant roots serve as a primary food source for microbes. Such interactions may have deleterious or beneficial effects on the plant (Bais et al. 2006). While India is among the leading rice producers globally, Karnataka state contributes significantly to India’s rice production. Rice productivity is influenced by both abiotic and biotic factors. Rice blast disease caused by the fungus Magnaporthe oryzae is the most severe and widespread rice disease and reduces crop yields up to 70%. Rice blast disease is more frequent and severe in areas with high agro-fertilizer use, particularly the high nitrogenous fertilizer-based rice cultivation, wherein it has been reported previously that the microbial diversity is poor in the rhizosphere (Long et al. 2000; Dai et al. 2018). The rhizospheric region allows both parasitic and mutualistic plant-microbe interactions (Raaijmakers et al. 2009).
Intensive monocropping and excessive use of agrochemicals reduces diversity and facilitates the prevalence of one or few microbial communities (Geisseler and Scow 2014; Xuan et al. 2012; Wu et al. 2013). However, the Indian subcontinent is known for the rice-based cropping system, the information on rice-soil-microbial community structure and diversity is limited. Furthermore, the comparative data on soil microbial diversity and the physicochemical properties of diverse rice soil samples are limited. Understanding soil microbial diversity and species abundance are essential in modifying the agro-inputs recommendations in terms of quantity, frequency of application for enhancing crop productivity, and maintaining overall soil health. Recent advances in genomics and extensive data analysis have facilitated the study of soil metagenomics more comprehensively. This approach has made it possible to identify untapped microbial reservoirs of soil in terms of abundance and diversity of both culturable and unculturable microbes (Arjun and Harikrishnan 2011; Anderson et al. 2018).
This study was undertaken to generate soil metagenomic data. Variation across the five selected rice-growing regions and wet-lab experiments were also carried out to identify putative antagonistic and biocontrol agents against the M. oryzae pathogen. This study highlights the Spatio-temporal variability in microbial communities, which in turn correlated with soil chemical properties. The genomic resources developed in this study will provide valuable information regarding the impact of soil chemical properties on soil microbial communities. Furthermore, culturable microbes with potential antagonistic action against M. oryzae pathogen were also identified, and they can be employed as a biocontrol agent in managing rice blast disease of rice.
Materials and methods
Study area
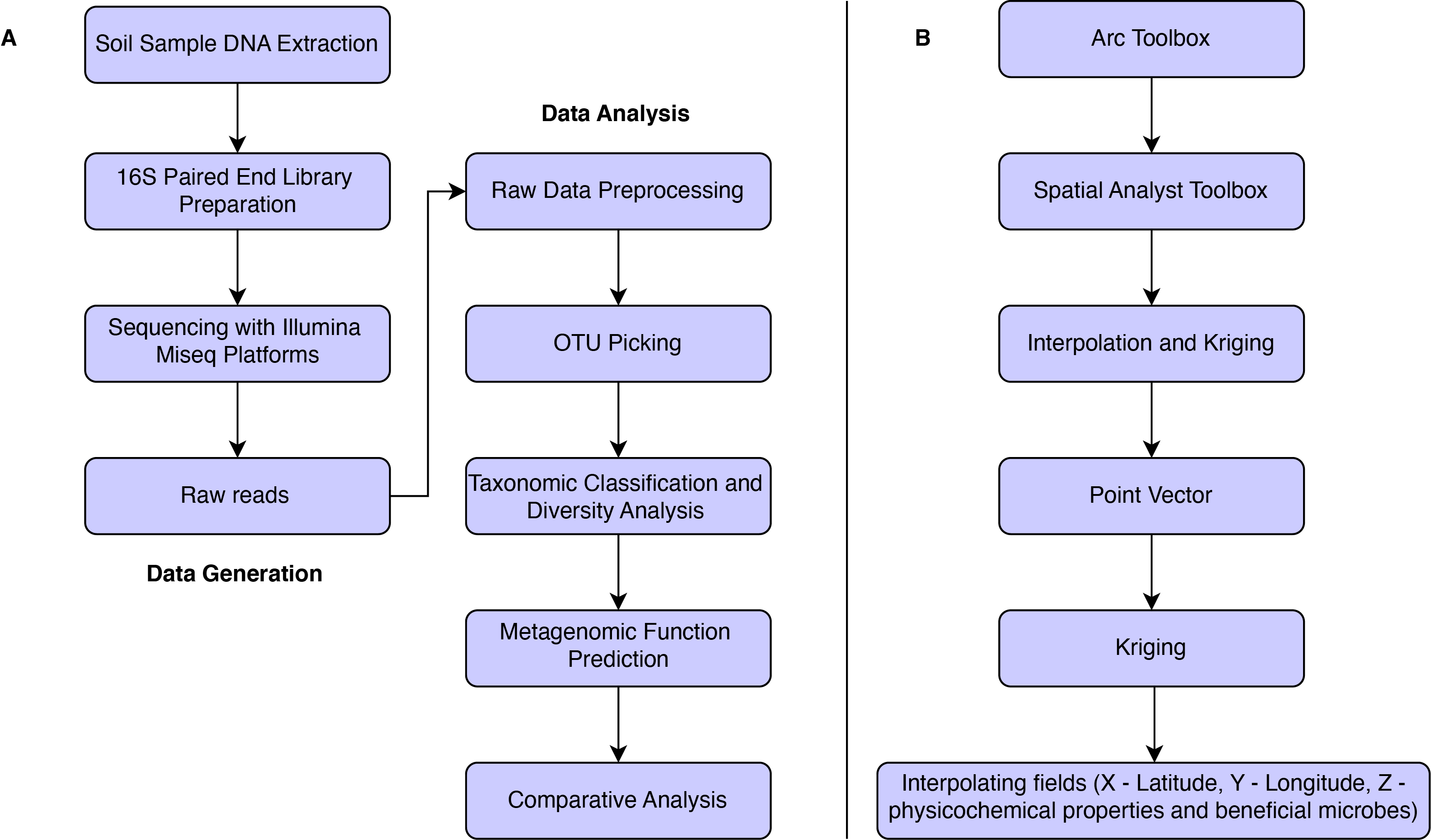
In India, Karnataka state is located at 15.3173° N and 75.7139° E. We selected five districts across Karnataka viz., Chamarajanagar (CHM) (12.142634° N and 77.081752° E), Davanagere (DVG) (14.557998° N and 75.821886° E), Hassan (HSN) (12.774781° N and 76.319256° E), Mandya (MND) (12.665881° N and 76.955810° E) and Gangavathi (GVT) (15.335556° N and 76.305555° E) where rice is being cultivated since many years. All these locations are geographically separated from each other with different agro-climatic conditions. Hence, variations in the microbial diversity and size at a genomic level across these different locations can be determined (Fig S1).
Soil sampling and analysis of soil chemical properties
Approximately, 50 g of soil from each of the five rice-growing regions was collected by randomly selecting 10–12 spots near the rhizosphere zone; then, the 10–12 sample of each location was pooled, from which a representative sample of 50 g was collected as described previously (https://cals.arizona.edu/backyards/sites/cals.arizona.edu.backyards/files/p16-17). Half of the collected sample (25 g) was used for soil metagenomic analysis. The remaining half was used to analyze the soil chemical properties and isolate antagonistic microbes. All the five samples were analyzed following the previously described protocol for their chemical characteristics viz., pH, electrical conductivity (EC) (Jackson 1973), percent organic carbon (OC) (Walkley and Black 1934), available nitrogen (N) (Subbiah and Asija 1956) available phosphorus (P) (Dickman and Bray 1940) and total potassium (K) (Barnes et al. 1945).
Genomic DNA extraction and 16S metagenome sequencing
The total genomic DNA was extracted from five soil samples using the Soil DNA isolation kit (DNeasy Power Soil Kit, QIAGEN, Hamburg, Germany) following the manufacturer’s instructions. The quality and quantity of the DNA were determined by 0.8% agarose gel and Nanodrop (DeNovix, USA). The genomic DNA was fragmented to obtain ≈ 500 bp fragments using Covaris M220 (Covaris, Inc. USA), and resultant fragments were end-repaired. The paired-end libraries were prepared using TruSeq DNA PCR-Free High Throughput Library Prep Kit (Illumina, San Diego, California, USA). Libraries were further enriched by nine PCR cycles and purified with Agencourt® AMPure® XP beads (Beckman Coulter Life Sciences, USA). The paired-end library was sequenced on the Illumina MiSeq platform using 2 × 300 bp sequencing chemistry (Illumina, California, USA).
Pre-processing of sequence data, taxonomic classification, and diversity assessment
The raw FastQ paired-end reads were initially assessed for quality using the FastQC program (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). With the parameters such as PHRED score cut off ≥ 30 and read length cut off of a minimum of 20 bases, the raw reads were trimmed using TrimGalore (https://github.com/FelixKrueger/TrimGalore). After trimming, the high-quality reads were merged into a single file using pandaseq (https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-13-31) with parameters “-B -A simple_bayesian” followed by the creation of a mapping file, and later qiime labels were added using add_qiime_labels.py. The Operational Taxonomic Units (OTUs) were identified using pick_closed_reference_otus.py wrapper available as a part of the Quantitative Insights Into Microbial Ecology, QIIME v1.9.1 (Caporaso et al. 2010) with qiime default reference (gg_13_8_97_outs.fa). Furthermore, taxa summaries were obtained till the species level using summarize_taxa.py script with otu_table_mc2_w_tax_no_pynast_failures.biom as input and “-L 2, 3, 4, 5, 6, 7” as option. Later, the summarized taxa were visualized using plot_taxa_summary.py with the option “-c pie, bar.” Alpha rarefactions were also analyzed to observe the sampling-depth versus species identification across all the samples. Beta diversity was analyzed using UniFrac (https://github.com/biocore/unifrac).
Function prediction
The resulting biom file from QIIME pick_open_reference_otus.py was further normalized using normalize_by_copy_number.py script, which is part of PICRUSt (https://picrust.github.com/picrust/), and function predictions were carried out using predict_metagenome.py and metagenome_contribution.py available in the same PICRUSt package. The total number of pathways to each nitrogen, phosphorus, and potassium metabolism was ranged from 150 to 300 KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways per sample, whereas the topmost abundant KEGG IDs were considered and analyzed. Furthermore, custom Python scripts were used to extract particular KEGG IDs using the KEGG database.
Mapping of microbial community and soil chemical properties
Computerized geostatistical tools such as Kriging (Krige 1951) was employed using ArcGIS 10.4 version software (https://www.arcgis.com/index.html) to provide the maps which pictorially represents the abundance and diversity of the microbial community and soil chemical properties at a scale of several hundreds of meters to kilometers and revealing the heterogeneous and spatially structured distribution of microbial biomass (Constancias et al. 2015) (Fig S2B).
Isolation and in vitro antagonistic assay
Isolation and identification of the potential biocontrol agents were carried out using the serial dilution method for all five soil samples. The dilutions of 10–3–10–7 were used to isolate the microbes on potato dextrose agar (10–3 and 10–4) and nutrient agar (10–5, 10–6, and 10–7) for fungi and bacteria, respectively. Based on morphological features, unique genera/species/strains were selected and stored at − 80 °C as glycerol stocks (50% v/v) for further in vitro studies. The bio-efficacy of all the isolated bacterial and fungal strains was tested against rice blast pathogen M. oryzae strain MG01 (Accession no- AYPX01000000) using the dual culture technique (Vincent et al. 1947). The best antagonistic genera/species/strain was identified based on percent inhibition calculated as per the formula below.
where, I = percentage inhibition of mycelia growth; C = growth of the pathogen in the control plate (cm); T = growth of the pathogen in the treatment plate (cm)
Taxonomic identification of potential antagonistic microbes
Molecular identification of bacteria and fungi was carried out by amplification of genomic DNA using 16S rDNA universal primers (for bacteria) 5′-GAGTTTGATCCTGGTCA-3′ (forward primer, fD1) and 5′-ACGGCTACCTTGTTACGACTT-3′ (reverse primer, rP2) and ITS regions (for fungi) ITS-1: 5′-CTTGGTCATTTAGAGGAAGTAA-3′ and ITS-4 5′-TCCTCCGCTTATTGATATGC-3′). The PCR amplification was performed using Eppendorf Thermal Cycler (Eppendorf-AG 22331, Eppendorf, Hamburg, Germany), and amplification was checked on 1% agarose gel. The amplified PCR product was purified using QiaQuick reagents (Qiagen, Germany) and sequenced by the Sanger sequencing method at a commercial facility (Eurofins Genomics India Pvt. Ltd, Bengaluru, India). The low-quality bases were trimmed using the BioEdit platform. The identity of culturable soil microbes was identified by matching (> 98% similarity) sequences to the NCBI-nr database using BLAST.
Results
Correlating the soil chemical properties and weather parameters with taxonomic classifications across the five selected rice-growing regions
Analysis of soil chemical properties, such as pH, EC (ds m−1), OC (%), N (kg ha−1), P (kg ha−1), and K (kg ha−1), revealed that soil pH varied between acidic (6.00) and alkaline (8.38) in the five samples studied. At CHM, high EC (0.69) and low OC (0.51%) were recorded; similar variation was also seen in the DVG location. Available N and available K were found to be lowest in CHM (279 and 121 kg ha−1), and the lowest available P was found in DVG (57 kg ha−1). In contrast, high N (551 kg ha−1) and P (714 kg ha−1) were recorded in GVT rice soil samples, and a high amount of total K was recorded in HSN (564 kg ha−1) (Table 1).
Table 1.
The results of soil chemical properties and the top five organisms identified at each location through metagenomics
| Soil sampled regions | pH | EC (dS/m) |
OC (%) |
Mean N (kg ha−1) |
Mean P (kg ha−1) |
Mean K (kg ha−1) |
Top five microbes |
|---|---|---|---|---|---|---|---|
| CHM | 8.38 | 0.69 | 0.51 | 279 | 82 | 121 | Bacillus boroniphilus, B. selenatarsenatis, B. aquimaris, Vibrio rotiferianus, B. niacini |
| DVG | 6.00 | 0.17 | 1.29 | 379 | 57 | 158 |
B. selenatarsenatis, B. boroniphilus V. rotiferianus, B. aquimaris, Nitrosococcus halophilus |
| HSN | 6.52 | 0.26 | 1.11 | 329 | 518 | 564 |
V. rotiferianus, B. selenatarsenatis B. aquimaris, B. boroniphilus, B. thioparans |
| MND | 7.80 | 0.62 | 0.60 | 360 | 311 | 252 |
B. selenatarsenatis, B. boroniphilus N. halophilus, B. thioparans |
| GVT | 7.11 | 0.35 | 0.84 | 551 | 715 | 182 |
V. rotiferianus, V. parahaemolyticus V. owensii, V. xuii, B. selenatarsenatis |
CHM Chamarajnagara, DVG davangere, HSN hassan, MND mandya, GVT gangavathi, EC electrical conductivity, OC organic carbon, N available nitrogen, P available phosphorous, K total potassium
Furthermore, high genomic read contents were found at CHM and GVT. Single-tailed Pearson’s correlation indicated that plant growth-promoting rhizobacteria (PGPR) abundance was correlated positively with pH and N (Table 3; Table S1).
Table 3.
Correlation of soil chemical properties with different beneficial microbial abundance across selected rice growing regions of Karnataka
| Soil properties | CDM | HDM | OMD | Nfixers | Nfiers | PS | PM | PGPR |
|---|---|---|---|---|---|---|---|---|
| pH | 0.55 | 0.54 | 0.54 | 0.31 | 0.43 | 0.81 | 0.26 | 0.82 |
| EC | 0.58 | 0.58 | 0.58 | 0.32 | 0.40 | 0.83 | 0.39 | 0.84 |
| OC | − 0.46 | − 0.46 | − 0.46 | − 0.21 | − 0.33 | − 0.73 | − 0.36 | − 0.73 |
| N | − 0.08 | − 0.86 | − 0.86 | − 0.41 | − 0.37 | − 0.69 | 0.04 | − 0.69 |
| P | 0.35 | 0.36 | 0.36 | − 0.74 | − 0.70 | − 0.38 | − 0.20 | − 0.37 |
| K | 0.35 | 0.36 | 0.36 | − 0.74 | − 0.70 | − 0.38 | − 0.20 | − 0.37 |
CDM Cellulose degrading microbes, HDM hemicellulose degrading microbes, OMD organic matter decomposers, Nfixers nitrogen fixers, Nfiers nitrifiers, PS phosphorous solubilizers, PM phosphorous mobilizers, PGPR Plant growth producing rhizobacteria
16S Metagenome sequencing and Taxonomic classification
From a summarized count of 1,12,116 reads across samples, a total of 1880 operational taxonomic units (OTUs) were identified as a result of closed reference OTU picking.
Variations in the microbial diversity
The Shannon diversity index, alpha diversity index, and beta diversity index were calculated to determine the species richness and evenness in the samples. The alpha diversity of the GVT samples (which had the highest N, P, and K) showed the lowest species richness and diversity at each rarefying depth and among the soil samples from all the rice-growing areas. The Shannon diversity index was used to estimate the microbial diversity within the samples and showed a significantly lower (p ≤ 0.05) microbial diversity at GVT (1.650) compared to other locations, followed by MND (2.980), CHM (3.02), HSN (3.06), and DVG (3.06) (Table S2). The beta diversity index indicated that the highest UniFrac distance (far clustering) was observed in the MND, HSN, CHM, DVG, and GVT samples, indicating variations in genomic diversity among the five samples from different rice-growing regions (Fig. 1a, b).
Fig. 1.

a Alpha diversity and b Beta diversity of the soil sample locations
Bacterial community structure
The sequence analysis indicated that Proteobacteria was abundant in all the five soil samples and ranged from 85.78% in GVT to 27.18% in MND; the exception was CHM, where Firmicutes (36.01%) were recorded as abundant. However, Bacilli in CHM (25.78%) and HSN (19.34%) and Gammaproteobacteria (82.83%) in GVT, followed by Alphaproteobacteria in DVG (14.41%) and Halobacteria in MND (14.18%) were the most abundant classes of microflora. Hence, the abundant orders and families were Bacillales and Bacillaceae in CHM (24.39% and 21.50%) and HSN (17.72% and 16.81%), followed by Pseudomonadales and Moraxellaceae in GVT (70.39% and 70.38%) and Flavobacteria and Flavobacteriaceae in DVG (12.50% and 12.50%), whereas in the MND location, an abundance of Halobacteriales (14.18%) and Halobacteriaceae (14.18%) were recorded. The most abundant genus was Bacillus in CHM (15.72%) and HSN (14.96%), followed by Lawinella, Psychrobacter, and Balneola in DVG (4.38%), GVT (69.98%), and MND (4.44%), respectively. Bacillus boronophilus, Methylonatrum sp, Psychrobacter celery, Vibrio rotiferianus, and Balneola vulgaris were abundant at CHM (3.31%), DVG (2.52%), GVT (66.71%), HSN (3.99%), and MND (3.33%), respectively (Table S3, Fig. 2).
Fig. 2.

Chromo chart representing the hierarchical microbial abundance in five rice soil
Core functional gene analysis
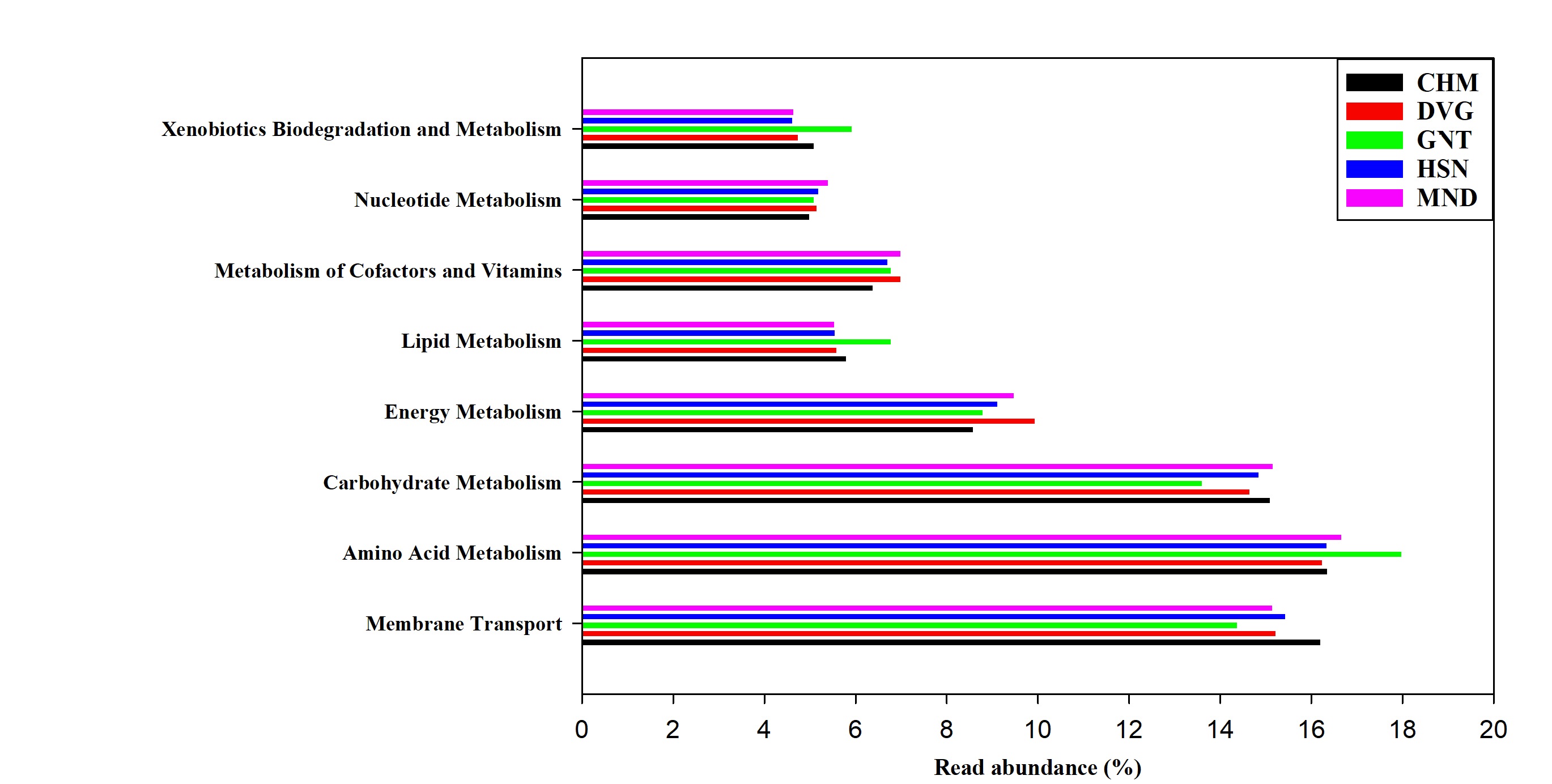
The KEGG pathway predictions of metagenome-assembled contigs revealed the OTUs of metabolic pathways such as amino acid metabolism (16.24–17.97% of OTUs), carbohydrate metabolism (13.60–15.15% of OTUs), and energy metabolism (8.58–9.94% of OTUs). Among other pathways, membrane transporters were abundant. The remaining pathways, such as cell motility, cell growth/cell death, transport/catabolism, signaling molecules, and their interactions, were least abundant (Table S4, Fig S3).
Pathways linked to the availability of major soil nutrients
The KEGG analysis revealed that the pathways, viz., the nitrogen fixation protein NifU and related proteins (K04488), nitrogen regulatory protein P-II 2 (K04752), two-component system, NtrC family, nitrogen regulation sensor histidine kinase GlnL (K07708) were abundant in GVT. The remaining four rice soil samples, CHM, DVG, HSN, and MND, had abundances in two pathways, viz., the PTS system nitrogen regulatory IIA component (K02806) and nitrogen regulatory protein P-II 1 (K04751), but these two pathways were found to be less abundant at GVT (4.37 and 6.03%) (Table S5, Fig. 3a).
Fig. 3.

Nitrogen metabolism pathways a Phosphate metabolism pathways, b Potassium metabolism pathways, c diversity and abundance across selected five rice regions
The KEGG pathway analysis for phosphate metabolism revealed that all the pathways were abundant at the GVT location except for geranylgeranyl diphosphate synthase, type I (K13787), and adenosylcobinamide kinase (K02231), whereas geranylgeranyl diphosphate synthase, type I (K13787) was abundant at MND (13.21%) and DVG (10.77%). The abundance of the remaining pathways was less than 10% (Table S6, Fig. 3b). Similarly, KEGG analysis of potassium metabolism pathways revealed that Trk system potassium uptake protein TrkH (K03498) was abundant in DVG (40.24%), and Trk system potassium uptake protein TrkA (K03499) was abundant in MND (56.32%). The potassium efflux system protein KefB (K05802) was abundant in GVT (24.70%), whereas the abundance of the other pathways was insignificant (Table S7; Fig. 3c).
Categorization of beneficial microbes
The comparison of eight different beneficial microbes indicated that cellulose (15.47–27.87%) and hemicellulose decomposing microbes (12.90–27.26%), organic matter decomposers (12.92–27.26%), and phosphorus solubilizers (5.62–14.73%) were abundant, while the remaining microbes, nitrogen fixers (0.84–10.80%), nitrifiers (0.03–1.29%), and phosphate mineralizers (0.10–1.67%) were less abundant at all five rice-growing locations (Table 2). A similar comparison was made across the five rice-growing locations and revealed that GVT was enriched with cellulose- and hemicellulose-degrading microbes and organic matter decomposers. The MND and DVG locations were found to harbor nitrogen fixers (Rhodospirillum rubrum), nitrifiers (Nitrosococcus halophilus), and phosphate solubilizers (Leucothrix mucor). Moreover, phosphate mineralizers (B. selenatarsenatis and B. boroniphilus) and PGPR were abundant in CHM and DVG, respectively.
Table 2.
Beneficial microbial categorization in five rice soil samples of Karnataka
| Sampled district | CDM (%) | HDM (%) | OMD (%) | Nfixers (%) | Nfiers (%) | PS (%) | PM (%) | PGPR (%) |
|---|---|---|---|---|---|---|---|---|
| CHM | 17.12 (8155) | 16.60 (7905) | 16.60 (7905) | 3.22 (1532) | 0.44 (208) | 14.73 (7018) | 0.10 (50) | 14.29 (6807) |
| DVG | 14.87 (1663) | 13.77 (1540) | 13.78 (1541) | 10.71 (1198) | 1.00 (112) | 12.57 (1405) | 0.80 (90) | 11.56 (1293) |
| HSN | 20.07 (5303) | 19.69 (5203) | 19.70 (5204) | 2.04 (540) | 0.11 (29) | 12.46 (3291) | 0.90 (238) | 12.14 (3207) |
| MND | 15.47 (686) | 12.90 (572) | 12.92 (573) | 10.80 (479) | 1.29 (57) | 14.09 (625) | 1.67 (74) | 11.95 (530) |
| GVT | 27.87 (6967) | 27.26 (6815) | 27.26 (6815) | 0.84 (211) | 0.03 (8) | 5.62 (1404) | 0.13 (32) | 5.43 (1358) |
CHM Chamarajnagara, DVG davangere, HSN hassan, MND mandya, GVT gangavathi, CDM cellulose degrading microbes, HDM hemicellulose degrading microbes, OMD organic matter decomposers, Nfixers nitrogen fixers, Nfiers nitrifiers, PS phosphorous solubilizers, PM phosphorous mobilizers, PGPR plant growth producing rhizobacteria
Linking soil chemical properties and the microbial community
Cellulose, hemicellulose, and organic matter decomposers were abundant in GVT, followed by HSN and CHM, which recorded the neutral soil pH (6.50–7.00), moderate EC (< 0.5 dSm−1), and high OC content (0.5–1%). A high abundance of nitrogen fixers and nitrifiers was found in MND, followed by DVG, which had N contents of 360 and 379 kg ha−1, respectively. A low abundance of nitrogen fixers and nitrifiers was found in GVT, where N content was observed (N: 552 kg ha−1). The phosphorus solubilizers and phosphorous mobilizers were indirectly proportional to each other relative to the available phosphorus content; hence, phosphorous mobilizers were highly abundant, and phosphorus solubilizers were least abundant in MND (P: 311 kg ha−1) followed by HSN (P: 518 kg ha−1). Phosphorus solubilizers were highly abundant in CHM (P: 81 kg ha−1) and GVT (P: 714 kg ha−1). The PGPR abundance was high in CHM (pH: 8.38; EC: 0.69 dsm−1; OC: 0.51%; N: 279.10 kg ha−1; P: 81.85 kg ha−1; K: 121.08 kg ha−1), followed by MND (pH: 8.02; EC: 0.62; OC: 0.60; N: 360.64 kg ha−1; P: 311.05 kg ha−1; K: 252.00 kg ha−1) (Fig. 4). Single-tailed Pearson’s correlation was performed using SPSS 16.0 at significance levels of p < 0.05 and p < 0.01 for the five rice-growing locations and indicated that pH and EC were positively correlated with all the beneficial microbial categories, whereas these two parameters have a highly positive correlation with P solubilizers and PGPR. The P and K were positively correlated with only CDM, HDM, and OMD. Although the N has a negative correlation with all beneficial microbes, it positively correlates with P mobilizers. The OC showed a negative correlation with all the beneficial microbes. The results indicated that high N (through inorganic fertilizers) impacts the most beneficial soil microbes (Table 3).
Fig. 4.

Distribution of a available nitrogen, b nitrifiers, c nitrogen fixers, d organic carbon, e organic matter decomposers and f plant growth-promoting rhizobacteria in rice soils
In vitro assay of culturable microbes against the M. oryzae strain MG01
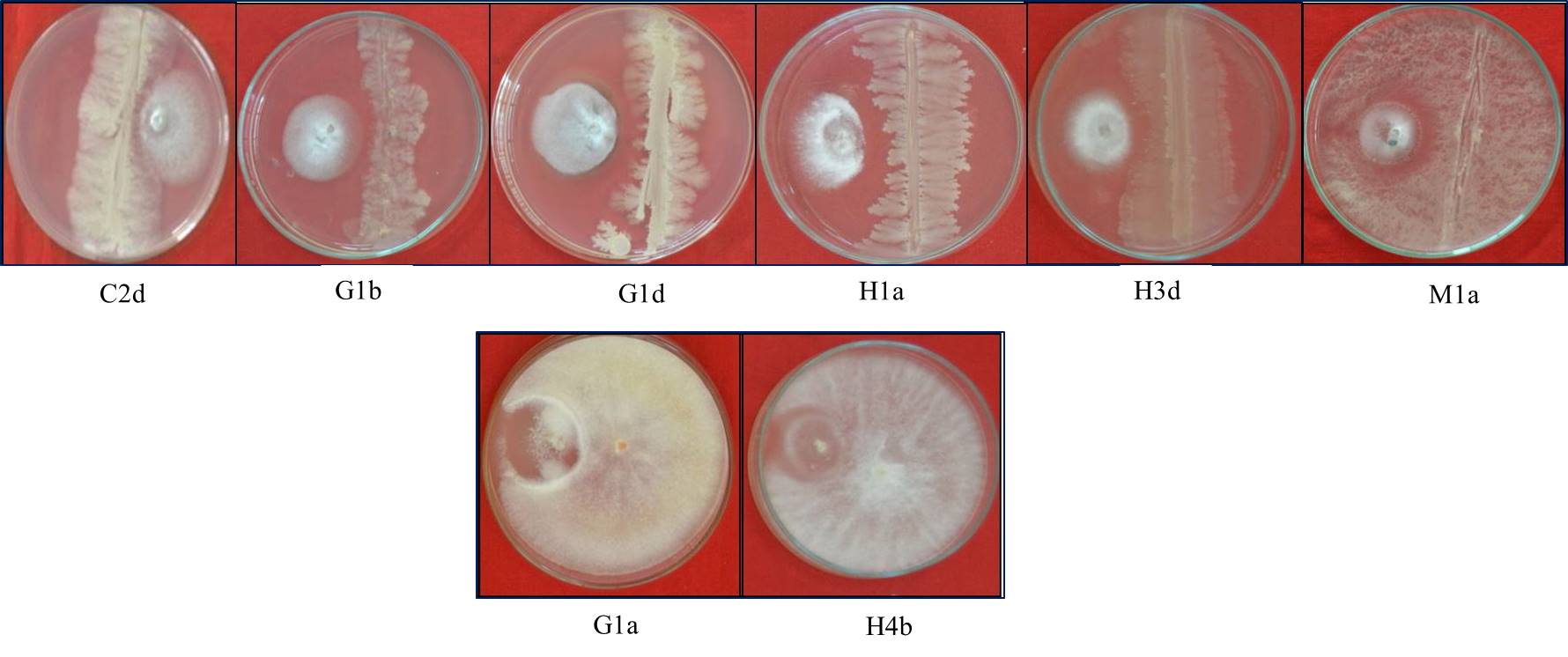
Culturable bacteria and fungi were isolated from the soil samples collected in five rice-growing areas. Following isolation, we selected 40 microbes from all 5 regions. All the selected microbes were tested in vitro against MG01. The molecular identification indicated that the best antagonistic microbes were Bacillus wiedmannii strain C2d, B. siamensis strain H1a and B. wiedmannii strain M1a, which showed high antagonistic activity. Among eight fungal isolates, Trichoderma harzianum strain G1a and T. harzianum strain H4b were highly antagonistic to MG01 (Tables S8 and S9; Figs S4 and S5).
Discussion
Soil chemical characteristics, cropping patterns or systems, and the microbiome is interlinked factors, and these factors influence crop productivity in every cropping system. Therefore, their management is important to maintain overall soil health and sustain crop productivity (Song et al. 2018). In the present study, a systematic investigation was carried out to understand rice monocropping effects on soil chemical properties and microbial diversity and abundance across five selected regions of Karnataka, India.
In line with previous studies, reports from the current study suggest that the chemical properties associated with soil and cropping patterns contribute significantly to enhancing soil microbial diversity. We observed differences in microbiota within the same cropping pattern, which could be attributed to variations in the soil physicochemical characteristics. We collected and studied three different types of soils from rice-growing areas, viz., red soil with a low concentration of ionic bases and less water-holding capacity at MND (http://cgwb.gov.in/District_Profile/karnataka/Mandya_brouchere.pdf), HSN (http://cgwb.gov.in/District_Profile/karnataka/2012/HASSAN-2012.pdf) and DVG (http://cgwb.gov.in/District_Profile/karnataka/DAVANAGERE_BROCHURE.pdf); brownish-black forest soil with high water holding capacity at CHM (http://cgwb.gov.in/District_Profile/karnataka/CHAMARAJNAGARA_BROCHURE.pdf); and a black soil at GVT (http://cgwb.gov.in/District_Profile/karnataka/KOPPAL_BROCHURE.pdf). Previous studies have reported pH as a key parameter influencing the soil microbiome (Zhalnina et al., 2014). In the present study, the pH of the samples varied from acidic to basic, based on which the soil microbiota also varied. Proteobacteria was abundant in slightly acidic (DVG and HSN) to neutral pH (GVT and MND) soil samples, whereas Firmicutes were abundant in the soil with an alkaline pH (CHM). This finding is in line with those of an earlier report (Anderson et al. 2018), which showed that rapid increases in soil pH strongly stimulate microorganisms of Firmicutes phylum.
The variations in the total carbon mineralizers were correlated with the pH of the soil samples. Total carbon mineralizers, viz., cellulose degraders, and organic matter decomposers were more abundant in the neutral pH soil (GVT) than in the acidic soil samples of MND. The rice soil samples from CHM were brownish, with a pH of 8.38 (alkaline) and low N and P (which could be due to lower usage of inorganic fertilizers by the farmers) and were found to have high GC content (56%), gene diversity, and abundance of PGPR and P solubilizers such as B. boronophilus. The higher abundance of P solubilizers, such as B. boronophilus, in soil samples with pH values of 6.3–9.0 have been reported in earlier studies (Ahmed et al. 2007; Baldrian 2017). Crop husbandry at CHM was still mainly based on small quantities of inorganic fertilizers; therefore, this location harbors a larger microbial population. In contrast, the microbial population had low abundance in GVT, where the ecosystem is known for rice followed by rice monocropping and a higher dose of inorganic fertilizers. This finding is also reflected in the soil chemical analysis in the present study, wherein available N and P of the GVT sample were high compared to samples of other rice-growing areas. In our findings, low GC content, low abundance of nitrogen fixers and nitrifiers, and low gene diversity were observed in the GVT soil samples. Our results were well supported by previous reports (Orr et al. 2011). The KEGG pathway analysis revealed that nitrogen metabolism in the rice soil samples of GVT had a low abundance of nitrogen metabolism-related pathways mapped to Cyanobacteria, Azospirillum, and Azotobacter (nitrogen fixers), which could be due to the impact of excessive nitrogenous fertilizers, monocropping, and lack of good crop management practices, as reported previously (Liu et al. 2009; Shen et al. 2010). Excessive application of nitrogenous fertilizers also led to the accumulation of only one kind of cyanobacteria, Synechocystis sp., which is nondiazotrophic and originally possessed the ability to fix nitrogen gas. However, it has lost the genes required to regulate the nitrogen fixation (nif gene cluster). Interestingly, we did not identify Synechocystis sp. in high-nitrogen soil samples such as GVT. Furthermore, we report that nitrogen fixers and nitrifiers are abundant at the DVG and MND locations in soils with moderate levels of available nitrogen, a condition related to the abundance of nitrogen-associated microbial diversity (Ollivier et al. 2011).
Plants absorb phosphorus primarily as orthophosphate (H2PO4−), predominantly found in soil samples with pH values below 7.2, and secondarily as (HPO4−2), which is prevalent in soil samples with pH values above 7.2. The pH of the soil samples from CHM and MND ranged from 7.8 to 8.3 and may have facilitated the rapid availability of H2PO4- to plants and soil microbes, which could be the reason for the abundant phosphorus-related metabolic pathways in these locations (Barrow 2017). The abundant phosphorus mineralizers/solubilizers in these locations were B. boronophilus, followed by B. selenatarsenatis. Interestingly, B. selenatarsenatis, a facultative anaerobe and selenate- and arsenate-reducing bacterium that inhabits the soil with a pH of 7.5–9.0, was also found in the soil samples from the CHM and HSN locations. In a report by Yamamura et al., this organism was isolated from selenium-contaminated sediment formed by the effluent drain of a glass-manufacturing industry in Japan (Yamamura et al. 2007).
Soil potassium pathway analysis revealed that among eight selected pathways, the trk system potassium uptake protein TrkH and the trk system potassium uptake protein TrkA were abundant across all locations except GVT. The trk system potassium uptake protein TrkA was abundant at MNDs where the pH was acidic (7.84) and, hence, the soil harbored halophilic bacteria, as reported earlier (Kraegeloh et al. 2005; Oren 2013). Compared to other nitrogen fixers, R. rubrum, a phototrophic nitrogen fixer and anaerobic bacteria (Nordlund et al. 1997), was abundant in the MND and DVG locations. Furthermore, nitrifiers, such as N. halophilus, an ammonia-oxidizing halophilic bacterium (Bano and Hollibaugh 2000), were predominant in the soil samples of all the rice-growing areas (CHM, DVG, MND, and HSN) except GVT. The phosphorus solubilizer Leucothrix mucor, a marine algal epiphyte (Bland and Brock 1973), was abundant in acidic soil (DVG).
In the present investigation, an attempt was made to identify antagonistic soil microbes for application against plant fungal pathogens. We found that Bacillus was the predominant bacteria, followed by Trichoderma harzianum, in the rice soil samples, as described previously as biocontrol agents (Ding et al. 2019). Among the Bacillus genera, B. wiedmannii, B. siamensis, and B. subtilis were found in the soil samples and confirmed by metagenomics and validated through isolation and plating. The B. licheniformis and B. methylotrophicus were confirmed only through isolation. Their antagonism was confirmed in vitro against one of the major rice pathogens, M. oryzae, and all the species tested exhibited a relatively high potential to be employed as biopesticides for controlling M. oryzae in field conditions.
Conclusion
In the present study, we have validated the results obtained by conventional methods, which are otherwise time-consuming, cumbersome, and less accurate in enumerating microbial diversity and abundance analysis. We explored five major rice cultivation areas of Karnataka by employing metagenomics. Our results suggest that the monocropping of rice with improper use of fertilizers and poor land management practices reduces the beneficial microbial diversity and prevalence. In addition, we found variations in the accumulation of nitrogen, phosphorus, and potassium metabolism pathways in all five rice monocropping regions. The microbes identified by conventional methods were compared with the microbes identified through metagenomics, and we found similarities between the results obtained by these two methods. We identified several microbes that showed antagonistic potential against M. oryzae, which could serve as potential biocontrol agents for effective management of rice blast disease.
Supplementary Information
Below is the link to the electronic supplementary material.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Authors’ contributions
Conceptualization- MKP, HBM; Data curation- AJD, BSR; Data analysis- LMN, BP, MEP, DP, AS; Fund acquisition- MKP; Project Supervision- MKP and HBM; Data Validation- MKP, HBM, DP, AJD and AS; Writing manuscript- LMP, HBM, MKP and AJD; Manuscript review and editing: HBM and MKP.
Funding
This project was funded by Watershed Development Department, Government of Karnataka, India. Also, supported by Directorate of Research, University of Agricultural Sciences, Bangalore under Varietal Development Project ID. DR/VTDP/2017–18. The funding agency had no role in study design, data collection, analysis and interpretation, decision to publish, or preparation of the manuscript.
Declarations
Conflict of interests
The authors declare no conflict of interests.
References
- Ahmed I, Yokota A, Fujiwara T. A novel highly boron tolerant bacterium, Bacillus boroniphilus sp. nov., isolated from soil, that requires boron for its growth. Extremophiles. 2007;11:217–224. doi: 10.1007/s00792-006-0027-0. [DOI] [PubMed] [Google Scholar]
- Anderson CR, Peterson ME, Frampton RA, et al. Rapid increases in soil pH solubilise organic matter, dramatically increase denitrification potential and strongly stimulate microorganisms from the Firmicutes phylum. PeerJ. 2018 doi: 10.7717/peerj.6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arjun JK, Harikrishnan K (2011) Metagenomic analysis of bacterial diversity in the rice rhizosphere soil microbiome. Biotechnol Bioinf Bioeng 1(3):361–367
- Bais HP, Weir TL, Perry LG, et al. The role of root exudates in rhizosphere interactions with plants and other organisms. Annu Rev Plant Biol. 2006;57:233–266. doi: 10.1146/annurev.arplant.57.032905.105159. [DOI] [PubMed] [Google Scholar]
- Baldrian P. Forest microbiome: diversity, complexity and dynamics. FEMS Microbiol Rev. 2017;41:109–130. doi: 10.1093/femsre/fuw040. [DOI] [PubMed] [Google Scholar]
- Bano N, Hollibaugh JT. Diversity and distribution of DNA sequences with affinity to ammonia-oxidizing bacteria of the beta subdivision of the class proteobacteria in the Arctic Ocean. Appl Environ Microbiol. 2000;66(5):1960–1969. doi: 10.1128/aem.66.5.1960-1969.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes RB, Richardson D, Berry JW, Hood RL (1945) Flame photometry a rapid analytical procedure. Indus Eng Chem Anal Edn 17(10):605–611
- Barrow NJ. The effects of pH on phosphate uptake from the soil. Plant Soil. 2017;410:401–410. doi: 10.1007/s11104-016-3008-9. [DOI] [Google Scholar]
- Bland JA, Brock TD. The marine bacterium Leucothrix mucor as an algal epiphyte. Mar Biol. 1973;23:283–292. doi: 10.1007/BF00389335. [DOI] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7(5):335–336 [DOI] [PMC free article] [PubMed]
- Constancias F, Terrat S, Saby NPA, et al. Mapping and determinism of soil microbial community distribution across an agricultural landscape. Microbiologyopen. 2015;4:505–517. doi: 10.1002/mbo3.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Z, Su W, Chen H, et al. Long-term nitrogen fertilization decreases bacterial diversity and favors the growth of Actinobacteria and Proteobacteria in agro-ecosystems across the globe. Glob Chang Biol. 2018;24:3452–3461. doi: 10.1111/gcb.14163. [DOI] [PubMed] [Google Scholar]
- Dickman SR, Bray RH. Colorimetric determination of phosphate. Ind Eng Chem Anal Ed. 1940;12:665–668. doi: 10.1021/ac50151a013. [DOI] [Google Scholar]
- Ding LJ, Cui HL, Nie SA, Long XE, Duan GL, Zhu YG. Microbiomes inhabiting rice roots and rhizosphere. FEMS Microbiol Ecol. 2019;95(5):fiz040. doi: 10.1093/femsec/fiz040. [DOI] [PubMed] [Google Scholar]
- Dubey A, Malla MA, Khan F, et al. Soil microbiome: a key player for conservation of soil health under changing climate. Biodivers Conserv. 2019;28:2405–2429. doi: 10.1007/s10531-019-01760-5. [DOI] [Google Scholar]
- Geisseler D, Scow KM. Long-term effects of mineral fertilizers on soil microorganisms—a review. Soil Biol Biochem. 2014;75:54–63. doi: 10.1016/j.soilbio.2014.03.023. [DOI] [Google Scholar]
- Jackson M (1973) Soil chemical analysis. Prentice-Hall of India Private Limited, New Delhi, India, pp 498
- Kraegeloh A, Amendt B, Kunte HJ. Potassium transport in a halophilic member of the Bacteria domain: Identification and characterization of the K+ uptake systems TrkH and TrkI from Halomonas elongata DSM 2581T. J Bacteriol. 2005;187:1036–1043. doi: 10.1128/JB.187.3.1036-1043.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krige DG. Journal of the chemical metallurgical & mining society of South Africa. J Chem Metall Soc South Min Africa. 1951;52:119–139. [Google Scholar]
- Liu M, Hu F, Chen X, Huang Q, Jiao J, Zhang B, Li H (2009) Organic amendments with reduced chemical fertilizer promote soil microbial development and nutrient availability in a subtropical paddy field: the influence of quantity, type and application time of organic amendments. Appl Soil Ecol 42(2):166–175
- Long DH, Lee FN, TeBeest DO. Effect of nitrogen fertilization on disease progress of rice blast on susceptible and resistant cultivars. Plant Dis. 2000;84:403–409. doi: 10.1094/PDIS.2000.84.4.403. [DOI] [PubMed] [Google Scholar]
- Nordlund S, Johansson M, Lindblad A, Norén A (1997) Nitrogen Fixation in Rhodospirillum rubrum: Regulation of Activity and Generation of Reductant. In: Biological Fixation of Nitrogen for Ecology and Sustainable Agriculture. Springer, pp 151–154
- Ollivier J, Töwe S, Bannert A, et al. Nitrogen turnover in soil and global change. FEMS Microbiol Ecol. 2011;78:3–16. doi: 10.1111/j.1574-6941.2011.01165.x. [DOI] [PubMed] [Google Scholar]
- Oren A (2013) Life at High Salt Concentrations. In: Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F (eds) The Prokaryotes. Springer, Berlin, Heidelberg. 10.1007/978-3-642-30123-0_57
- Orr CH, James A, Leifert C, et al. Diversity and activity of free-living nitrogen-fixing bacteria and total bacteria in organic and conventionally managed soils. Appl Environ Microbiol. 2011;77:911–919. doi: 10.1128/AEM.01250-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raaijmakers JM, Paulitz TC, Steinberg C, Alabouvette C, Moënne-Loccoz Y (2009) The rhizosphere: a playground and battlefield for soilborne pathogens and beneficial microorganisms. Pl Soil 321(1–2):341–361
- Shen W, Lin X, Shi W, Min J, Gao N, Zhang H, Yin R, He X (2010) Higher rates of nitrogen fertilization decrease soil enzyme activities, microbial functional diversity and nitrification capacity in a Chinese polytunnel greenhouse vegetable land. Pl Soil 337(1–2):137–150
- Song X, Tao B, Guo J, et al. Changes in the microbial community structure and soil chemical properties of vertisols under different cropping systems in northern China. Front Environ Sci. 2018;6:1–14. doi: 10.3389/fenvs.2018.00132. [DOI] [Google Scholar]
- Subbiah BV, Asija GL (1956) A rapid procedure for the estimation of available nitrogen in soils. Current Sci 25:259
- Vincent JM. Distortion of fungal hyphæ in the presence of certain inhibitors [20] Nature. 1947;159:850. doi: 10.1038/159850b0. [DOI] [PubMed] [Google Scholar]
- Walkley A, Black IA. An examination of the degtjareff method for determining soil organic matter, and a proposed modification of the chromic acid titration method. Soil Sci. 1934;37:29–38. doi: 10.1097/00010694-193401000-00003. [DOI] [Google Scholar]
- Wieland G, Neumann R, Backhaus H. Variation of microbial communities in soil, rhizosphere, and rhizoplane in response to crop species, soil type, and crop development. Appl Environ Microbiol. 2001;67:5849–5854. doi: 10.1128/AEM.67.12.5849-5854.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Li Z, Li J, et al. Assessment of shifts in microbial community structure and catabolic diversity in response to Rehmannia glutinosa monoculture. Appl Soil Ecol. 2013;67:1–9. doi: 10.1016/j.apsoil.2013.02.008. [DOI] [Google Scholar]
- Xuan DT, Guong VT, Rosling A, Alström S, Chai B, Högberg N (2012) Different crop rotation systems as drivers of change in soil bacterial community structure and yield of rice, Oryza sativa. Biol Fertil Soils 48(2):217–225
- Yamamura S, Yamashita M, Fujimoto N, et al. Bacillus selenatarsenatis sp. nov., a selenate- and arsenate-reducing bacterium isolated from the effluent drain of a glass-manufacturing plant. Int J Syst Evol Microbiol. 2007;57:1060–1064. doi: 10.1099/ijs.0.64667-0. [DOI] [PubMed] [Google Scholar]
- Zhalnina K, Dias R, de Quadros PD, et al. Soil pH determines microbial diversity and composition in the park grass experiment. Microb Ecol. 2014;69:395–406. doi: 10.1007/s00248-014-0530-2. [DOI] [PubMed] [Google Scholar]
- Zhang M, He Z. Long-term changes in organic carbon and nutrients of an Ultisol under rice cropping in southeast China. Geoderma. 2004;118:167–179. doi: 10.1016/S0016-7061(03)00191-5. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.