

Abstract
Increased population movement has increased the risk of reintroducing parasites to elimination areas and also dispersing drug-resistant parasites to new regions. Therefore, reliable and repeatable methods to trace back to the source of imported infections are essential. The recently developed 23-single-nucleotide polymorphism (SNP) barcode from organellar genomes of mitochondrion (mt) and apicoplast (apico) provides a valuable tool to locate the geographic origin of Plasmodium falciparum. This study aims to explore the feasibility of using the 23-SNP barcode for tracking P. falciparum by polymerase chain reaction and sequencing, while providing geographical haplotypes of isolates that originated from Central Africa. Based on 23-SNP barcode analysis, SNPs were found at seven loci; 27 isolates were confirmed to have originated in West Africa, and this study also showed four isolates from Central Africa (Equatorial Guinea, 3; Republic of Congo, 1) that originated in East Africa. This study provides the sequence data from Central Africa and fills 23-SNP barcode data gaps of sample origins.
Keywords: Plasmodium falciparum, mitochondrion, apicoplast, SNP, barcode
Introduction
Malaria is a serious public health problem in tropical and subtropical areas, with an estimated 228 million cases of malaria occurring worldwide [(1), available online at: https://www.who.int/publications/i/item/9789241565721]. Among the five species of Plasmodium that infect humans, Plasmodium falciparum is the most dangerous one, causing high levels of mortality and morbidity worldwide, particularly in sub-Saharan Africa. With today's ease of transmissibility, the emergence and spread of artemisinin-resistant P. falciparum threatens malaria eradication (2–4). Increased population movement has increased the risk of reintroducing parasites to elimination areas and dispersing drug-resistant parasites to new regions. To facilitate a better response for this new challenge and to understand it well, except drug sensitivity monitoring, the geographic origins of imported malaria need be tracked accurately and in good time. Reliable and repeatable methods to trace back to the source of imported infections are therefore essential.
In the past decade, the single-nucleotide polymorphism (SNP) barcode has been developed as a forceful genotyping technique to investigate the origin of Plasmodium (5). The first P. falciparum molecular barcode was composed of 24 SNPs, which in combination created a unique and concise signature to differentiate recrudescence from reinfection from malaria parasites or to monitor distribution and frequency of specific parasites in patients (6). However, these nuclear SNPs are constrained by a lack of geographic specificity and frequent recombination. The establishment of a 23-SNP barcode using polymorphisms from mitochondrion (mt) and apicoplast (apico) genomes provides evidence that they are non-recombining and coinherited and are capable of identifying the geographic origin of P. falciparum strains (7). The 23-SNP barcoding strategy was 92% accurate in identifying the continental origin of P. falciparum samples from West and East Africa, Southeast Asia, Oceania, and South America (7). However, feasible and conventional methods for the 23-SNP barcode are lacking, and information about the haplotypes of isolates from Central Africa is rare. To conquer such a plight, we designed a simple 23-SNP barcode based on polymerase chain reaction (PCR) and sequencing, and assayed the samples from P. falciparum malaria cases imported from sub-Saharan Africa.
Materials and Methods
Study Sites and Participants
The study was carried out in Jiangsu Province, China, where only imported malaria cases have been reported since 2013. The study was approved by the institutional review board of Jiangsu Institute of Parasitic Diseases (IRB00004221), Wuxi, China. Samples were collected from imported malaria cases. Imported cases were identified based on the travel history of the patients (travel to a malaria-endemic country within the previous month of illness onset); the last country visited with ongoing malaria transmission was taken as the potential location of infection (8). All of the malaria cases were routinely confirmed by microscopy and PCR through the malaria diagnosis reference laboratory in Jiangsu Institute of Parasitic Diseases. The 32 samples included in this study are a subset of the 765 imported malaria samples analyzed earlier (3); 21 mutated isolates were selected for this study. In addition, 11 isolates with wild-type Pf kelch13 from Equatorial Guinea were also random selected for this barcode assay. Together, 32 samples, including 30 samples from Central Africa (29 samples from Equatorial Guinea, 1 from Guinea), and 2 samples from West Africa (Republic of Congo, Sierra Leone) as reference, were enrolled in this study (Figure 1). Figure 1 was produced using Microsoft Excel and SPSS software 26.0 for Windows.
Figure 1.
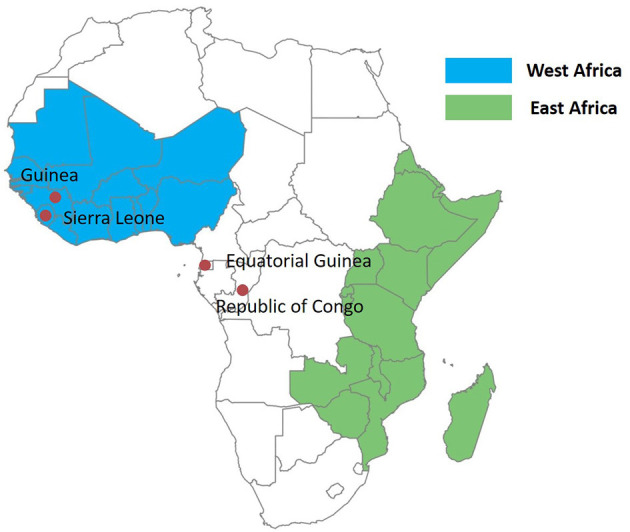
Map of Africa showing the countries where Plasmodium falciparum isolates were imported from: Equatorial Guinea (n = 29), Guinea (n = 1), the Republic of Congo (n = 1), Sierra Leone (n = 1).
PCR and Sequencing of the Targets
Genomic DNA of P. falciparum isolates was extracted from the whole blood samples on the QIAcube Connect platform, using a QIAamp DNA Blood Kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. From the 200 μL of input whole blood sample, a final 100 μL of elution volume was obtained. The gDNA was used for PCR amplification with gene-specific primers to amplify SNP loci targets within the mt and apico P. falciparum genomes. Primers were designed manually on the basis of geographically informative barcoding; the reference sequences used in this study were from the database of GenBank (https://www.ncbi.nlm.nih.gov/nucleotide/); AY282930 (5,949 bp) was selected as the reference sequence for mt, and X95276 (1–14,009 bp) combined the reverse sequence of X95275 (14,010–29,430 bp) for apico (9) (Appendix 1 in Supplementary Material).
A total of 10 sets of primers that covered the sites of the 23-SNP barcode were included in this study; detailed primer sequences are shown in Table 1, and the target sequences are shown (Figure 2 and Appendix 2 in Supplementary Material). Amplifications were performed in a reaction mixture that contained 25 μL of 2 × PCR buffer for KOD FX, 10 μL of 2 mM dNTPs, 1.5 μL each of 10 μM Primer-F and Primer-R, 3 μL of template DNA, 1 μL of 1.0 U/μL KOD FX, and 8 μL of double-distilled water. The PCR was performed with initial denature at 98°C for 2 min, and 35 cycles of 98°C for 10 s, 58°C for 30 s, and 68°C for 2 min and ending with a final extension of 5 min at 68°C. The PCR products were purified and sequenced. Double-strand capillary sequencing of PCR products was performed on an Applied Biosystems 3,730 sequence analyzer with the sequencing primers (Table 1). The deduced amino acid sequences were aligned and analyzed with the Lasergene® software (DNASTAR, Madison, WI).
Table 1.
Primers used for polymerase chain reaction (PCR) amplification and sequencing of mt/apico.
| Primer sequence (5′ → 3′) | Amplified position | Amplicon | |
|---|---|---|---|
| mt | F: AAGCTTTTGGTATCTCGTAATGTAGAA R*: TTAGCAATAACATTCCTGATGTAATGA SF1: CCTTCTCGCCATTTGATAGC SR1: GGCGAACCTTCTTACCGTTAT |
mt772, mt853, mt973, mt1283, mt2383 | 1–2,650 |
| apico | F: TTAGTTAATAATCCAGAAACCCATTT | apico15131 | 14,392–15,247 |
| 1 | R*: GTGGATATTCTTTACATACAGA | ||
| F: TGCCTGAGTGGTTAAAAGGAA | apico2122 | 1,473–2,278 | |
| 2 | R*: Ccctgtttacatacaggtg | ||
| F: TCTTTGACCCCCTTGTTTTG | apico20831, apico21188 | 19,599–21,277 | |
| 3 | R*: ctaatgaaggattaacttgtgg | ||
| F*: Gaagctgtacatccttct | apico23803 | 23,595–24,104 | |
| 4 | R: Tatatttaaattacctgattggaa | ||
| F*: AAGTACATCCTTTAATATTTAGAGG | apico4370, apico4878, apico4945, apico5005 | 3,857–5,257 | |
| 5 | R*: CCTATAACTATATCACCATATTTAGG | ||
| F*: GTAGGTACTAAAAGTAATAATTATATATC | apico5715, apico6361, apico6832 | 5,415–7,054 | |
| 6 | R*: TATTATTTTACAATTAAAATAACCTAAAC | ||
| F*: CAACATATAAATTTAGGTACTATAGG | apico9003, apico9096 | 8,785–9,778 | |
| 7 | R: GAGGTTTATATCCAATATTAAAAGG | ||
| 8 | F: CCAAAATTTATAATAAAGGTTCG R: ATCCAACTTCCTATAGGTTTA SF2: ACTAGAAGGGGTAAATGATA Sr2: TATCATTTACCCCTTCTAGT |
apico11066, apico11619, apico11671 | 10,497–12,414 |
| F*: AATTCGATTGGCATTTCACC | apico26659 | 26,347–27,183 | |
| 9 | R: GGATTCATGCTCCGAAGGTA |
F, forward; R, reverse; S, sequencing. F and R pairs were used in PCR amplifications; SF, SR, and primers with
labels were used in the sequencing reactions.
Figure 2.

Schematic representation of the apicoplast genome (A) and mitochondrion genome (B) in Plasmodium falciparum. Protein coding genes are boxed and color-coded. tRNAs are represented by vertical lines. Genes shown above the horizontal line in each genome are transcribed left to right, and those below are transcribed from right to left.
Results
The reference mt and apico sequences were selected from the complex database and were shown to be well-correlated to the loci of the barcode designed (7). With the designed primers, the 32 isolates were amplified and sequenced successfully (Figure 3); however, the sequencing peak chromatograms showed one sample with a double peak at the two special loci indicated in mixed haplotype infections. For the five loci of mt genomes, including mt772, mt853, mt973, mt1283, and mt2383, the wild-type was shown except in one isolate with a double peak at mt772 mentioned previously. A total of 18 loci of the apico genomes were selected for the barcode, with six loci including apico2122, apico6832, apico20831, apico21188, apico23803, and apico26659 with different alleles (Figures 2, 3). The haplotype analysis of mt/apico among all 32 samples in this study revealed seven distinct haplotypes. Figure 4 and Appendix 3 (Supplementary Material) show the number of haplotypes identified in samples from each country for the seven haplotypes identified therein. A single mutation at G26659 of mt/apico (haplotype 9) was the most prevalent haplotype (22/29) in Equatorial Guinea, which is unique to West Africa. In addition, haplotype 9 was also found in the isolates from Guinea and Sierra Leone. Among isolates from Equatorial Guinea, the C23803 single mutation (haplotype 8) was found in two isolates, and the A21188G26659 double mutations (haplotype 7) were observed in one isolate, corresponding to the barcodes published for West Africa. Meanwhile, the isolate CWX, with 23-SNP barcode identified in this study was consisted with the results analyzed with whole-genome sequencing (3).
Figure 3.

Agarose gel electrophoresis of polymerase chain reaction (A) and sequencing wave of the target loci with wild-type (B), mutant type (C), and mixed type (D) compared to the reference Plasmodium falciparum 3D7. Gray shading marked in the sequencing waves indicates the target loci.
Figure 4.

Geographically informative barcode of Plasmodium falciparum mitochondrion and apicoplast genome single-nucleotide polymorphism (SNPs). The sample sequence results from the 23-SNP loci were compared to the geographically informative haplotype barcodes previously defined (7). WAF, West Africa; EAF, East Africa. Sample information: 18: Guinea, 19: Republic of Congo, 20: Sierra Leone; others originated from Equatorial Guinea. CWX and 1–20 samples marked with color were isolates with PfK13 mutation. *Haplotypes are based on the reference (7).
The A20831G26659 double mutations (haplotype 12), the T6832G26659 double mutations (haplotype 13), and the C2122 single mutation (haplotype 14) identified in some of the isolates correspond to the barcodes published for East Africa. Haplotype 12 and haplotype 14 were observed in two isolates and one isolate from Equatorial Guinea, respectively. Haplotype 13 was found in only one isolate from the Republic of Congo. In addition, the mixed genotype of the wild-type and position mt772, apico26659 mutations, was found in one sample from Equatorial Guinea.
Discussion
The Greater Mekong Subregion is the cradle of now widespread resistance to previous frontline antimalarial drugs (10). There is a significant risk that the artemisinin-resistant phenotypes within the Greater Mekong Subregion may similarly spread to other endemic regions. Therefore, the geographic origin of the parasites is important for monitoring the global emergence and spread of resistance and may prevent or delay the spread of artemisinin resistance from South East Asia to sub-Saharan Africa (11, 12).
With the establishment of sequence-based genome-wide polymorphisms in P. falciparum parasites, it is becoming feasible to design panels of SNP-based genotyping assays in tracing parasite geographic origin. Highly polymorphic microsatellites are useful to characterize community-, country-, or region-level genetic diversity over relatively short periods of time, as often required in outbreak investigations. However, it is difficult to standardize the interpretation of microsatellite assays across laboratories (13). The genomes of mt and apico, which have uniparental inheritance and do not recombine with each other, could provide more stable region-specific genotypes than nuclear genome SNPs. As a result, Preston et al. (7) developed a 23-SNP barcode using polymorphisms from mt/apico to identify the geographic origin of P. falciparum strains. In the study, 151 SNPs in mt and 488 in apico were identified from 711 P. falciparum samples in five geographic regions: West Africa, East Africa, Southeast Africa, Southeast Africa, Oceania, and South America, and a 23-SNP barcode was designed to trace the origin and dispersal of parasite strains across and between continents (7).
PCR-based approaches are inexpensive ways of extracting genomic data from samples containing very small quantities of parasite DNA, excluding the interference of host DNA. PCR-based approaches that genotype small collections of SNPs or a limited number of amplicons are of great value for timely genetic analysis of clinical samples collected directly from sporadic cases. In this study, we chose the method that used a 23-SNP barcode based on single-step PCR (ssPCR) and sequencing because it is a low-cost, rapid, and easy method to perform in the laboratory. This study presented the feasible application of PCR and sequencing in examining the 23-SNP barcode from mt/apico, effectively identifying the geographic origin of P. falciparum strains imported to China.
In this study, 10 fragments were amplified with 10 sets of primers by ssPCR, and the amplification efficiency of mt was much higher than apico (Figure 3A). Molecular targets are involved in malaria diagnosis assays; 18S rRNA genes are the most common; meanwhile, mt and apico are also an obvious target for malaria diagnosis (14, 15). Normally, the 18S rRNA gene of P. falciparum has 7-fold the amount of nuclear genomes, while mt and apico are ~20-fold and between 1- and 15-fold, respectively. All of them can be used to classify samples with mixed infections; however, ssPCR targeting 18S rRNA still showed similar sensitivity with targeting mt and even higher than the apico genome-based classification. Of the 23-SNP barcode, a total of 13 loci were polymorphic and composed of African haplotypes (7), and only seven loci were found to be polymorphic in this study. Then, it could also select specific loci for amplification and attempt to predict the geographic source. Our results confirm the relatively low efficiency of the amplification of apico targets. Although the limit of detection was not tested, we recommend using whole blood, high-efficiency enzymes, and DNA enrichment if necessary.
We not only supported a feasible method for the 23-SNP barcode assay, but we also listed the clear sequence references of the targets and genome of apico (Appendices 1, 2 in Supplementary Material). For the accurate demand of the assay, the reference sequences must contain the original design (7); even a single deletion/insertion will make the analysis change completely. The genome of P. falciparum 3D7 was the first reference genome published in Plasmodium research; however, the apico was not sequenced in the original genome project, and the apico from the P. falciparum isolate C10 was used (GenBank X95275.2, X95276.2) (9) until version 3.1 of the P. falciparum genome including a complete apico genome was uploaded (16, 17). We checked the database carefully and found X95276 (1–14,009 bp) combined with the reverse sequence of X95275 (14,010–29,430 bp) was well-confirmed with the loci selected in the design. Our sorted data supplied good reference for the later relative study.
In the present study, parasites were screened from origins that are not yet included in the network (WAF: Burkina Faso, Gambia, Ghana, and Mali; EAF: Kenya, Malawi, and Uganda) (7). Most of the malaria imported cases in this study were acquired in the central–western part of sub-Saharan Africa (Equatorial Guinea and Republic of Congo), and two isolates from West Africa were selected as control (Guinean and Sierra Leonean). Based on 23-SNP barcode analysis, West African isolates as well as the 25 isolates from Equatorial Guinea were confirmed to have the same haplotypes as those originating in West Africa. Surprisingly, the geographic origins of three isolates from Equatorial Guinea and one isolate from the Republic of Congo were East Africa. In addition, one isolate from Equatorial Guinea showed mixed haplotypes, indicating either heteroplasmy or multiclonal infections, in which it was difficult to infer the geographical origin for the parasites with multiple mt and apico genomes. The confused results consisted of the prediction that the 23-SNP barcode may lack the genetic resolution to distinguish between ongoing autochthonous transmission and malaria infections imported from one or more nearby locations (7). However, they are better suited to tracing the origin and dispersal of parasite strains across and between continents. With limited samples, this study still could provide the sequence data from Central Africa and fill data gaps in sample origins that are not yet included in the network.
Previous studies revealed the highly conserved structure of mt and apico in the genus Plasmodium (18, 19). The ratio of NS/S substitutions, which can be used to gauge the intensity and directionality of natural selection, are generally quite low for both organelle genomes, indicative of the strong purifying selection on NS sites, but see Preston et al. (7) and Wicke et al. (20) for exceptions to this trend (21). In this study, NS/S ratios were quite low for mt; this points to the highly conserved structure of mt in P. falciparum. In addition, apico genes had a higher NS/S ratio than mt, corresponding to the previously published study by Preston et al. (7). Pressure from antimalarial drugs may account for the high NS/S ratio of the apico SNPs. As translationally active organelles, Plasmodium mt and apico have been validated as important drug targets.
The apico genome encodes ~30 proteins, and most are ribosome subunits. Apicoplast has been identified in interactions with the delayed death phenotype caused by antibiotics, which may inhibit the apico housekeeping functions (22). Ribosomes of the Plasmodium apico and mt have been validated as targets for antibiotic action (23). It has also been reported that the artemisinin-exposed persistent forms restructured the mitochondrial–nuclear associations in P. falciparum (24). In contrast, rapid killing of malaria parasites by artemisinin is thought to result from depolarization of the mitochondrial membrane (25). The Plasmodium mt genome encodes only three protein-coding genes; however, each parasite has multiple genomes in mt, which may allow suboptimal mutant genes to preadapt to drug resistance even without strong drug-selective pressure. Studies into the mutant genes of mt and apico with drug pressure are rare; conclusive evidence is still lacking. In this study, the selected 32 isolates were from Chinese travelers who had returned from African countries. CWX has been reported as an artemisinin-resistant strain (3), and the 1–20 isolates have been confirmed to have the Pfkelch13 mutant genotype. The other 11 isolates (21-31) showed Pfkelch13 wild-type in another study. However, for the limited number of Kelch13 mutant samples, drug assays were not included and also out of the focus of the study. Multiple surveys of populations subjected to drug pressure are necessary to confirm the haplotypes and drug pressure.
In conclusion, this study provides a practical and highly valuable method to trace back to the geographic origins of P. falciparum malaria based on PCR and sequencing using the 23-SNP barcode from mt/apico. As population mobility has increased, the risk of reintroducing parasites to elimination areas and dispersing drug-resistant parasites to new regions has also increased, malaria control programs should be prepared to respond to this.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
The studies involving human participants were reviewed and approved by The Institutional Review Board of Jiangsu Institute of Parasitic Diseases. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
FH and QZ performed the experiments and wrote the manuscript. GZ, HZ, and MZ helped with the sample collection and conformation. YLi and FT designed the experiments and analyzed the data. FL and YLiu conceived the experiments, provided advice on data interpretation, and edited the paper. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This work was supported by the National Key R&D Program of China (2020YFC1200105), the National Natural Science Foundation of China (82072297 and 81601790), the Six Talent Peaks Project in Jiangsu Province (2019-YY-065), and the high level talent support project of Yangzhou University.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpubh.2021.649170/full#supplementary-material
References
- 1.World Health Organazation . World Malaria Report 2019 (2019). Available online at: https://www.who.int/publications/i/item/9789241565721 (accessed December 4, 2019).
- 2.Fairhurst RM, Dondorp AM. Artemisinin-resistant Plasmodium falciparum malaria. Microbiol Spectrum. (2016) 4:EI10-0013-2016. 10.1128/9781555819453.ch22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu F, Culleton R, Zhang M, Ramaprasad A, von Seidlein L, Zhou H, et al. Emergence of indigenous artemisinin-resistant Plasmodium falciparum in Africa. N Engl J Med. (2017) 376:991–3. 10.1056/NEJMc1612765 [DOI] [PubMed] [Google Scholar]
- 4.Ménard D, Khim N, Beghain J, Adegnika AA, Shafiul-Alam M, Amodu O, et al. A worldwide map of Plasmodium falciparum K13-propeller polymorphisms. N Engl J Med. (2016) 374:2453–64. 10.1056/NEJMoa1513137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manske M, Miotto O, Campino S, Auburn S, Almagro-Garcia J, Maslen G, et al. Analysis of Plasmodium falciparum diversity in natural infections by deep sequencing. Nature. (2012) 487:375–9. 10.1038/nature11174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rachel D, Volkman SK, Danny DA, Mahesh N, Neafsey DE, Park DJ, et al. A general SNP-based molecular barcode for Plasmodium falciparum identification and tracking. Malaria J. (2008) 7:223. 10.1186/1475-2875-7-223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Preston MD, Campino S, Assefa SA, Echeverry DF, Ocholla H, Amambua-Ngwa A, et al. A barcode of organellar genome polymorphisms identifies the geographic origin of Plasmodium falciparum strains. Nat Commun. (2014) 5:4052. 10.1038/ncomms5052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lai S, Sun J, Ruktanonchai NW, Zhou S, Yu J, Routledge I, et al. Changing epidemiology andchallenges of malaria in China towards elimination. Malar J. (2019) 18:107. 10.1186/s12936-019-2736-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilson RJ, Denny PW, Preiser PR, Rangachari K, Roberts K, Roy A, et al. Complete gene map of the plastid-like DNA of the malaria parasite Plasmodium falciparum. J MolBiol. (1996) 261:155–72. 10.1006/jmbi.1996.0449 [DOI] [PubMed] [Google Scholar]
- 10.Tulloch J, David B, Newman R, Meek SJL. Artemisinin-resistant malaria in the Asia-Pacific region. Lancet. (2013) 381:e16–7. 10.1016/S0140-6736(12)61820-0 [DOI] [PubMed] [Google Scholar]
- 11.Nag S MD, Dalgaard PE, Kofoed J, Ursing M, Crespo, Andersen LO, et al. High throughput resistance profiling of Plasmodium falciparum infections based on custom dual indexing and Illumina next generation sequencing-technology. Sci Rep. (2017) 7:2398. 10.1038/s41598-017-02724-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taylor SM, Parobek CM, DeConti DK, Kayentao K, Coulibaly SO, Greenwood BM, et al. Absence of putative artemisinin resistance mutations among Plasmodium falciparum in Sub-Saharan Africa: a molecular epidemiologic study. JID. (2015) 211:680–8. 10.1093/infdis/jiu467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baniecki ML, Faust AL, Schaffner SF, Park DJ, Galinsky K, Daniels RF, et al. Development of a Single Nucleotide Polymorphism Barcode to Genotype Plasmodium vivax Infections. PLoS Negl Trop Dis. (2015) 9:e0003539. 10.1371/journal.pntd.0003539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haanshuus CG, Mohn SC, Mørch K, Langeland N, Blomberg B, Hanevik K, et al. A novel, single-amplification PCR targeting mitochondrial genome highly sensitive and specific in diagnosing malaria among returned travellers in Bergen, Norway. Malar J. (2013) 12:26. 10.1186/1475-2875-12-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oriero CE, van Geertruyden JP, Jacobs J, D'Alessandro U, Nwakanma D. Validation of an apicoplast genome target for the detection of Plasmodium species using polymerase chain reaction and loop mediated isothermal amplification. Clin Microbiol Infect. (2015) 21:686.e1–7. 10.1016/j.cmi.2015.02.025 [DOI] [PubMed] [Google Scholar]
- 16.Hunt M, Silva ND, Otto TD, Parkhill J, Keane JA, Harris SR, et al. Circlator: automated circularization of genome assemblies using long sequencing reads. Genome Biol. (2015) 16:294. 10.1186/s13059-015-0849-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Böhme U, Otto TD, Sanders M, Newbold CI, Berriman M, Llinas M, et al. Progression of the canonical reference malaria parasite genome from 2002-2019. Wellcome Open Res. (2019) 4:58. 10.12688/wellcomeopenres.15194.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hikosaka K, Watanabe Y, Kobayashi F, Waki S, Kita K, Tanabe K. Highly conserved gene arrangement of the mitochondrial genomes of 23 Plasmodium species. Parasitol Int. (2011) 60:175–80. 10.1016/j.parint.2011.02.001 [DOI] [PubMed] [Google Scholar]
- 19.Nobuko A, Tetsuo H, Hideya M, Palacpac NMQ, Akira K, Kawai S, et al. The Plasmodium apicoplast genome: conserved structure and close relationship of P. ovale to rodent malaria parasites. Mol Biol Evol Lett. (2012) 29:2095–9. 10.1093/molbev/mss082 [DOI] [PubMed] [Google Scholar]
- 20.Wicke S, Schäferhoff B, dePamphilis CW, Müller KF. Disproportional plastome-wide increase of substitution rates and relaxed purifying selection in genes of carnivorous Lentibulariaceae. Mol Biol Evol. (2014) 31:529–45. 10.1093/molbev/mst261 [DOI] [PubMed] [Google Scholar]
- 21.Smith DR. Mutation rates in plastid genomes: they are lower than you might think. Genome Biol. (2015) 7:1227–34. 10.1093/gbe/evv069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yeh E, DeRisi JL. Chemical rescue of malaria parasites lacking an apicoplast defines organelle function in blood-stage Plasmodium falciparum. PLoS Biol. (2011) 9:e1001138. 10.1371/journal.pbio.1001138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gupta A, Shah P, Haider A, Gupta K, Siddiqi MI, Ralph SA, et al. Reduced ribosomes of the apicoplast and mitochondrion of Plasmodium spp. and predicted interactions with antibiotics. Open Biol. (2014) 45:140045. 10.1098/rsob.140045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Connelly SV, Manzella-Lapeira J, Levine ZC, Brzostowski J, Krymskaya L, Rahman RS, et al. Restructured mitochondrial-nuclear interaction in P. falciparum dormancy and persister survival of artemisinin exposure. bioRxiv [Preprint]. (2020). 10.1101/2020.09.28.314435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antoine T, Fisher N, Amewu R, O'Neill PM, Ward SA, Biagini GA, et al. Rapid kill of malaria parasites by artemisinin and semi-synthetic endoperoxides involves ROS-dependent depolarization of the membrane potential. J Antimicrob Chemother. (2014) 694:1005–16. 10.1093/jac/dkt486 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.