

This case-control study investigates the association between nasal microbial profiles and odds of infection after elective surgical procedures.
Key Points
Question
Can characterization of microbiota present on preoperative nasal swab estimate risk for postoperative infection in patients undergoing elective surgical procedures?
Findings
In this case-control study, 167 patients were classified to nasal microbial profile cluster 1 and 30 patients to cluster 2 based on 16S ribosomal RNA gene sequencing. Classification to cluster 2 was associated with statistically significantly higher odds of infection (ie, deep surgical site infection, bacteremia, or pneumonia) after surgical procedure, independent of covariates, including nasal carriage of Staphylococcus aureus on preoperative culture and intrasample microbial diversity (ie, α diversity).
Meaning
These findings suggest that the nasal microbiome is an independent risk factor associated with infectious outcomes after elective surgical procedures.
Abstract
Importance
The association of the nasal microbiome with outcomes in surgical patients is poorly understood.
Objective
To characterize the composition of nasal microbiota in patients undergoing clean elective surgical procedures and to examine the association between characteristics of preoperative nasal microbiota and occurrence of postoperative infection.
Design, Setting, and Participants
Using a nested matched case-control design, 53 individuals who developed postoperative infection were matched (approximately 3:1 by age, sex, and surgical procedure) with 144 individuals who were not infected (ie, the control group). The 2 groups were selected from a prospective cohort of patients undergoing surgical procedures at 2 tertiary care university hospitals in Baltimore, Maryland, who were at high risk for postoperative infectious complications. Included individuals were aged 40 years or older; had no history of autoimmune disease, immunocompromised state, immune-modulating medication, or active infection; and were scheduled to undergo elective cardiac, vascular, spinal, or intracranial surgical procedure. Data were analyzed from October 2015 through September 2020.
Exposures
Nasal microbiome cluster class served as the main exposure. An unsupervised clustering method (ie, grades of membership modeling) was used to classify nasal microbial samples into 2 groups based on features derived from 16S ribosomal RNA gene sequencing. The microbiome cluster groups were derived independently and agnostic of baseline clinical characteristics and infection status.
Main Outcomes and Measures
Composite of surgical site infection, bacteremia, and pneumonia occurring within 6 months after surgical procedure.
Results
Among 197 participants (mean [SD] age, 64.1 [10.6] years; 63 [37.7%] women), 553 bacterial taxa were identified from preoperative nasal swab samples. A 2-cluster model (with 167 patients in cluster 1 and 30 patients in cluster 2) accounted for the largest proportion of variance in microbial profiles using grades of membership modeling and was most parsimonious. After adjusting for potential confounders, the probability of assignment to cluster 2 was associated with 6-fold higher odds of infection after surgical procedure (odds ratio [OR], 6.18; 95% CI, 3.33-11.7; P < .001) independent of baseline clinical characteristics, including nasal carriage of Staphylococcus aureus. Intrasample (ie, α) diversity was inversely associated with infectious outcome in both clusters (OR, 0.57; 95% CI, 0.42-0.75; P < .001); however, probability of assignment to cluster 2 was associated with higher odds of infection independent of α diversity (OR, 4.61; 95% CI, 2.78-7.86; P < .001).
Conclusions and Relevance
These findings suggest that the nasal microbiome was an independent risk factor associated with infectious outcomes among individuals who underwent elective surgical procedures and may serve as a biomarker associated with infection susceptibility in this population.
Introduction
The human nares in healthy individuals contains a rich diversity of microorganisms, including commensal, opportunistic, and pathogenic taxa.1 Environmental and genetic factors are reported to be associated with interindividual variability in the composition of nasal microbiota; however, the association of this variability with health and disease is poorly understood.1,2 Decreased diversity levels within the microbial niche (ie, α diversity) of the gut are associated with clinical disease outside the gut, including obesity and diabetes, and with death3,4; however, the association between features of nasal microbiota and clinical outcomes not involving the nose or sinuses has not been reported.
The presence of Staphylococcus aureus among the microbiota of the anterior nares has garnered substantial attention because of this microorganism’s pathogenic potential and known association with clinical infection at non-nasal sites.5 For example, patients who test positive for S aureus on preoperative nasal culture are at 2-fold to 9-fold increased risk of postoperative surgical site infection (SSI),6 and nasal colonization is associated with increased risk of blood stream infection7 and pneumonia8 in patients admitted to the hospital. S aureus decolonization before surgical procedure is associated with decreased risk of postoperative SSI; however, protection is incomplete.9,10 Numerous bacteria compete for the ecologic niche of the anterior nares, and species other than S aureus may contribute, either directly or indirectly, to the association between S aureus and infectious risk.
This study had 3 main aims: to thoroughly characterize the microbiota present on nasal swab samples obtained before elective surgical procedure using state of the art bacterial gene profiling; to classify individuals into cluster groups, independent and agnostic of postoperative outcomes, based solely on preoperative microbial profiles; and to evaluate the association between microbial clusters and development of postoperative infection. In this report, we describe the microbiologic characteristics that define the cluster groups, the association of cluster with baseline clinical characteristics and S aureus nasal colonization, and the associations between microbial features of the clusters and the occurrence of non-nasal infectious complications after surgical procedure.
Methods
This case-control study was approved by the institutional review boards at Johns Hopkins Hospital and the University of Maryland, Baltimore. All participants signed written informed consent. This study is reported following the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline.
Study Design and Participants
We conducted a nested matched case-control study involving participants selected from a prospective cohort study of elective high-risk clean surgical procedures (ie, cardiac, vascular, spinal fusion, and craniotomy procedures).11 Among 802 participants, 53 individuals with serious postoperative infection were identified and matched approximately 3 to 1 by age (ie, older or younger by 5 years), sex, and surgical procedure with 144 individuals who were not infected who served as controls; financial constraints limited microbial analyses to a small subset of the entire cohort. Participants were accrued into the original study cohort at the Johns Hopkins Hospital and University of Maryland Medical Center. Full details regarding setting, participants, and protocol of the original cohort are described in detail elsewhere11 and in the eAppendix in the Supplement.
Identification of S aureus and Other Microbiota From the Anterior Nares
Nasal swab of the anterior nares was obtained from each participant at baseline before surgical procedure and use of antibiotics. The presence or absence of S aureus was determined in the clinical microbiology laboratory by standard culture. Isolation of bacterial DNA from nasal swabs,12,13 16S ribosomal RNA (rRNA) gene profiling of the V3V4 hypervariable region,14,15 read processing,13 and taxonomic classification16,17 of microbiota were performed as previously described (eAppendix in the Supplement).
Main Exposure
Nasal microbiome cluster class served as the main exposure. We used an unsupervised clustering method (ie, grades of membership model18,19) to classify nasal microbial samples based solely on features derived from 16S rRNA gene sequencing. Using this method, microbiome cluster classification was derived independently and agnostic of baseline clinical characteristics and infection status. The grades of membership model allows each sample to have some proportion of its membership, or partial membership, in each cluster. We used these partial membership weights to assign each sample to a cluster and estimated the optimal number of clusters using log Bayes factor. We implemented our analysis using the CountClust package (version 1.4.1) in R statistical software version 3.4.1 (R Project for Statistical Computing)18 (eAppendix in the Supplement).
Outcomes and Covariates
The primary outcome was a composite of deep SSI, pneumonia, or bacteremia, as defined by Centers for Disease Control and Prevention surveillance criteria,20 occurring within 6 months postoperatively. Secondary outcomes were SSI, pneumonia, and bacteremia separately; the composite outcome at 30 days; and death at 6 months. Baseline covariates and outcomes were determined by participant interview and medical record review. A full list of variables and definitions is available in the eAppendix in the Supplement.
Statistical Analysis
Differential abundance of microbial taxa at the aggregate and individual levels was determined from counts of rRNA sequences annotated to the species level. We accounted for variability21 and sparsity22 in sequence data as previously described. Differential abundance results are reported in terms of false discovery rate with q < .05 considered significant. The association of taxa abundance with nasal carriage of S aureus was determined by logistic regression using log-transformed taxa counts as an independent variable. Within-sample diversity (ie, α) and between-sample diversity (ie, β) were calculated (eAppendix in the Supplement).
Baseline categorical covariates were expressed as percentages; differences between cluster classes were compared using the χ2 or Fisher exact tests. Continuous variables were expressed using mean (SD), and cluster classes were compared using t or Kruskal-Wallis tests. A series of generalized linear regression models were performed to assess the association of the primary exposure (ie, probability of assignment to cluster 2) and covariates with the primary and secondary outcomes unadjusted and adjusted for potential confounding baseline covariates. Adjusted model 1 was adjusted for demographic covariates: age, sex, and race. Adjusted model 2 was adjusted for baseline comorbidities: congestive heart failure, peripheral vascular disease, chronic obstructive pulmonary disease, history of smoking, history of cancer, and Charlson Comorbidity Index score. Adjusted model 3 was adjusted for surgical procedure–associated variables: study site, inpatient or outpatient status, and surgical procedure. Adjusted model 4 was adjusted for nasal culture results for S aureus and for methicillin-resistant S aureus. Adjusted model 5 was adjusted for inverse probability of treatment weighting (IPTW) using propensity score for assignment to microbiome cluster 2. The propensity score for assignment to cluster 2 was generated by incorporating all baseline covariates listed in Table 1. We used IPTW (with treatment defined as assignment to cluster 2) to adjust for all baseline covariates using the propensity score. Propensity score analyses were bootstrapped using 500 subsamples from approximately 70% of all samples comprising the study group (eAppendix in the Supplement). We examined the association of infection with α diversity and abundance of individual microbial taxa using logistic regression.
Table 1. Baseline Characteristics of Study Participants by Microbiome Cluster Class.
| Characteristic | No. (%) | P value | |
|---|---|---|---|
| Cluster 1 (n = 167) | Cluster 2 (n = 30) | ||
| Demographic characteristic | |||
| Age, mean (SD), y | 64.10 (10.91) | 63.83 (9.04) | .88 |
| Sex | |||
| Women | 63 (37.7) | 9 (30.0) | .54 |
| Men | 104 (62.3) | 21 (70.0) | |
| Race | |||
| White | 142 (85.0) | 26 (87.7) | .99 |
| Black | 24 (14.4) | 4 (13.3) | |
| Asian | 1 (0.6) | 0 | |
| Comorbidity | |||
| Obesity | 67 (40.1) | 9 (30.0) | .39 |
| Diabetes | 29 (17.4) | 6 (20.0) | .86 |
| Hypertension | 117 (70.1) | 19 (63.3) | .60 |
| Myocardial Infarction | 26 (15.6) | 6 (20.0) | .59 |
| Congestive heart failure | 14 (8.3) | 6 (20.0) | .09 |
| Peripheral vascular disease | 15 (9.0) | 5 (16.7) | .19 |
| Cerebrovascular disease | 17 (10.2) | 5 (16.7) | .34 |
| COPD | 11 (6.6) | 1 (3.3) | .69 |
| History of smoking | 108 (64.7) | 20 (66.7) | .99 |
| Gastric ulcer | 12 (7.2) | 2 (6.7) | .99 |
| Chronic liver disease | 2 (1.2) | 0 | .99 |
| Dialysis dependency | 2 (1.2) | 0 | .99 |
| History of cancer | 17 (10.2) | 7 (23.3) | .06 |
| Infection treated with antibiotics in previous year | 54 (32.3) | 11 (36.7) | .79 |
| Hospitalization in previous year | 54 (32.3) | 13 (43.3) | .36 |
| American Society of Anesthesiologists class | |||
| 2 | 32 (19.2) | 6 (20.0) | .96 |
| 3 | 98 (58.7) | 18 (60.0) | |
| 4 | 37(22.2) | 6 (20.0) | |
| Charlson Comorbidity Index score | |||
| 0-2 | 65 (38.9) | 8 (26.7) | .16 |
| 3-4 | 53 (31.7) | 8 (26.7) | |
| >5 | 49 (29.3) | 14 (46.7) | |
| Surgical factors associated with risk of infection | |||
| Inpatient at the time of surgical procedure | 41 (24.6) | 9 (30.0) | .68 |
| Surgical procedure | |||
| Cardiac | 73 (43.7) | 12 (40.0) | .73 |
| Vascular | 18 (10.8) | 5 (16.7) | |
| Spinal | 52 (31.1) | 8 (26.7) | |
| Intracranial | 24 (14.4) | 5 (16.7) | |
| Study site | |||
| Johns Hopkins Hospital | 149 (89.2) | 30 (100) | .08 |
| University of Maryland Medical Center | 18 (10.8) | 0 | |
| Nasal culture for S aureus | |||
| S aureus positive | 35 (21.0) | 6 (20.0) | .99 |
| Methicillin-resistant S aureus positive | 7 (4.2) | 2 (6.7) | .62 |
Abbreviations: COPD, chronic obstructive pulmonary disease; S aureus, Staphylococcus aureus.
Statistical significance was set at P < .05, and all tests were 2-sided. Benjamini-Hochberg false discovery procedure was used to correct for multiple comparisons. Data analysis was conducted from October 2015 through September 2020.
Results
Among 197 included patients, 53 individuals had a postoperative infection (29.7%) and 144 individuals did not have infections (ie, the control group; 73.1%). Mean (SD) age was 64.1 (10.6) years, 63 (37.7%) were women, and 24 individuals were Black (14.4%). Among all participants, 41 individuals (20.8%) tested positive for S aureus on preoperative nasal culture, and 9 of these individuals (22.0%) tested positive for methicillin-resistant S aureus. A total of 4423 operational taxonomic units were identified by 16S rRNA gene sequencing from the 197 nasal swab samples obtained before surgical procedure. These were organized into 477 distinct taxa to the genus level and 553 taxa to the species level (eFigure 1 in the Supplement).
Figure 1 part A shows the aggregate proportions of the top 20 most abundant taxa. Corynebacterium was the most abundant taxa detected from the anterior nares, constituting 41.0% of all sequences organized to the genus level, followed by Propionibacterium (7.6%), Alloiococcus (5.9%), Planococcaceae (5.1%), Enterobacterales (formerly Enterobacteriaceae) (4.2%), and Staphylococcus (3.7%). There was variability among study participants in proportions of the various taxa present in the anterior nares (Figure 1, part B). We detected 16S rRNA gene sequences for S aureus in 194 samples (98.5%). There was a positive association between S aureus relative abundance by 16S rRNA gene sequencing and nasal carriage of S aureus by standard clinical culture (odds ratio [OR], 1.93; 95% CI, 1.54-2.50; q < .0001). None of the other taxa, including other Staphylococcal species (ie, epidermidis, pettenkoferi, or sciuri) were associated with nasal carriage of S aureus. Pairwise comparisons of relative abundances of S aureus with other Staphylococcal species and with non-Staphylococcal taxa showed no significant associations after correcting for multiple testing.
Figure 1. Relative Abundance of Nasal Microbiota Before Surgical Procedure.
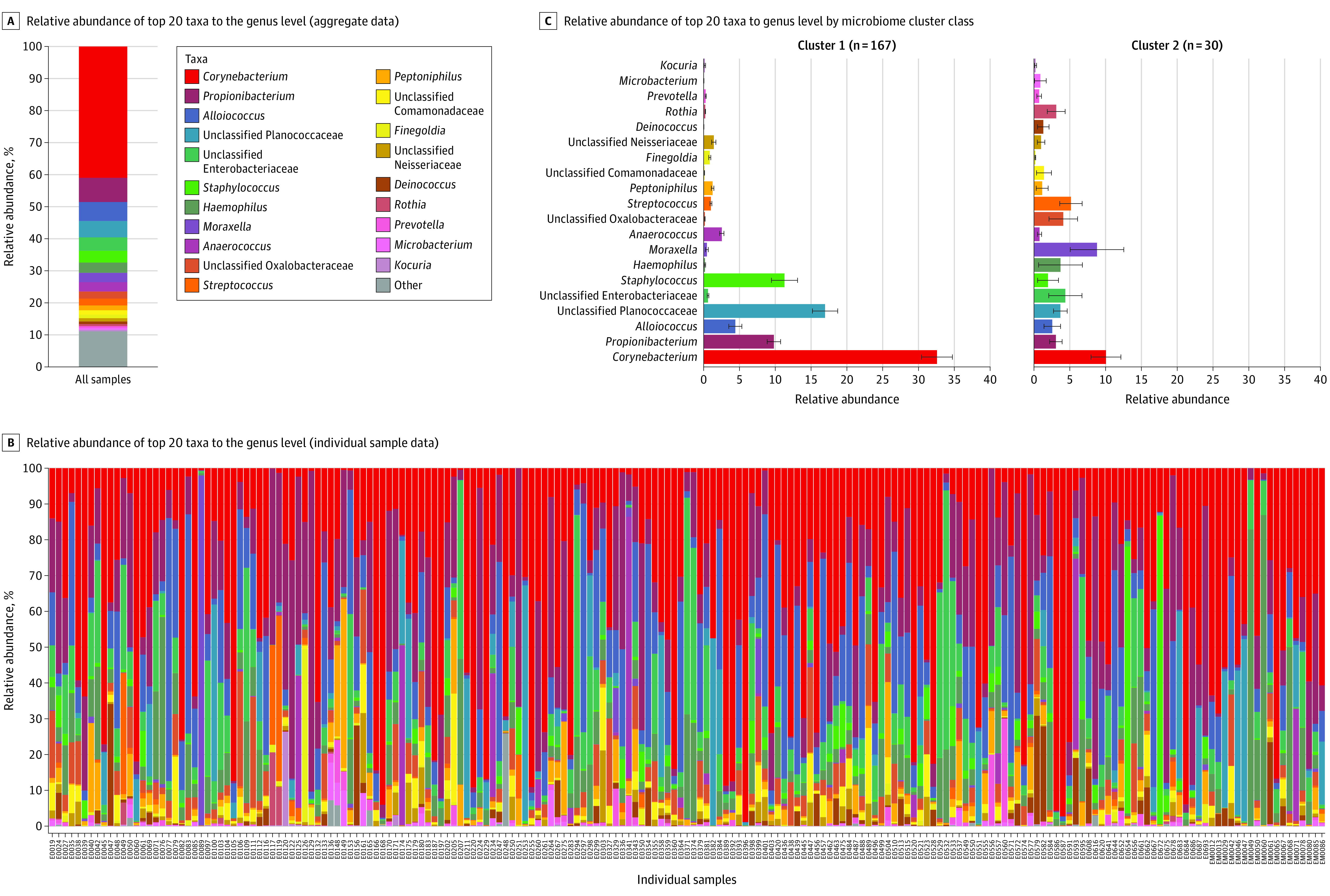
C, Bars indicate mean; whiskers, SE.
Unsupervised clustering using grades of membership modeling, which was independent and agnostic of each participant’s clinical characteristics and infection status, classified participants into groups based solely on features derived from 16S rRNA gene sequencing of nasal microbiota. The clustering model with 2 groups (167 samples in cluster 1 and 30 samples in cluster 2) accounted for the greatest proportion of variance in the nasal microbiome and was most parsimonious (eFigure 2 in the Supplement), and it was thus selected for further characterization and hypothesis testing. Proportions of the top 20 most abundant genera differed between cluster 1 and cluster 2 (Figure 1, part C). Among the 553 distinct taxa identified to the species level, 67 taxa were significantly different between cluster 1 and cluster 2 (eTable 1 in the Supplement). Cluster 2 had greater α and β diversity than cluster 1 (eFigure 3 in the Supplement). Results from principal components analysis suggested that the factors that distinguished cluster 1 from cluster 2 were dispersed over a continuum rather than behaving as discrete categories (eFigure 3 in the Supplement).
Comparison of baseline clinical characteristics of the 197 study participants by the main exposure (ie, nasal microbiome cluster class) is shown in Table 1. There were no significant differences by microbial cluster class for any of the measured covariates, including demographic characteristics, comorbidities, surgical risk factors, and preoperative nasal carriage of S aureus. Of the 53 infections, 19 infections (35.8%) were SSI, 19 infections were bacteremia, and 27 infections (50.9%) were pneumonia (some participants experienced more than 1 infectious complication). S aureus was the most common organism to be isolated, at 23 (43.4%) infections, followed by Klebsiella species, at 11 infections (20.8%), and Streptococcus species, at 9 infections (17.0%); in some instances, more than 1 bacteria species was recovered from the infected site, and in others none were recovered (Table 2).
Table 2. Bacteria Isolated From Infected Individuals by Site of Infection.
| No. (%) | ||||
|---|---|---|---|---|
| Any infection (n = 53) | Deep SSI (n = 19) | Bacteremia (n = 19) | Pneumonia (n = 27) | |
| Staphylococcus epidermidis | 5 (9.4) | 5 (26.3) | 0 | 0 |
| Staphylococcus aureus | 23 (43.4) | 9 (47.4) | 6 (31.6) | 8 (29.6) |
| Streptococcus species | 9 (17.0) | 3 (15.8) | 2 (10.5) | 4 (14.8) |
| Pseudomonas species | 3 (5.7) | 0 | 1 (5.3) | 2 (7.4) |
| Enterococcus species | 4 (7.6) | 4 (21.1) | 0 | 0 |
| Klebsiella species | 11 (20.8) | 0 | 5 (26.3) | 6 (22.2) |
| Proteus species | 2 (3.8) | 1 (5.3) | 1 (5.3) | 0 |
| Enterobacter species | 5 (9.4) | 2 (10.5) | 0 | 3 (11.1) |
| Bacteroides species | 1 (1.9) | 1 (5.3) | 0 | 0 |
| Escherichia coli | 6 (11.3) | 1 (5.3) | 3 (15.8) | 2 (7.4) |
| Haemophilus species | 1 (1.9) | 0 | 0 | 1 (3.7) |
| Serratia species | 4 (7.6) | 0 | 0 | 4 (14.8) |
| Morganella species | 1 (1.9) | 0 | 0 | 1 (3.7) |
| No organism isolated | 12 (22.6) | 4 (21.1) | 0 | 8 (29.6) |
Abbreviation: SSI, surgical site infection.
The probability of assignment to cluster 2 was associated with an unadjusted 5-fold higher odds of composite postoperative infectious outcome (OR, 5.41; 95% CI, 1.81-16.54; P = .002) (Table 3). There was a dose-response association between probability of assignment to cluster 2 and infectious outcome (Figure 2). Similarly, categorical assignment to cluster 2 was associated with higher odds of infection (OR, 2.87; 95% CI, 1.27- 6.42; P = .009) compared with assignment to cluster 1.
Table 3. Association of Microbiome Cluster Class and Covariates With Composite Infectious Outcomea.
| Variable | Unadjusted | Model 1b | Model 2c | Model 3d | Model 4e | Model 5f | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OR (95% CI) | P value | OR (95% CI) | P value | OR (95% CI) | P value | OR (95% CI) | P value | OR (95% CI) | P value | OR (95% CI) | P value | |
| Cluster 2 probability | 5.41 (1.81-16.54) | .002 | 5.73 (1.90-17.74) | .002 | 5.47 (1.63-19.00) | .006 | 5.56 (1.75-18.23) | .003 | 6.08 (1.92-19.72) | .002 | 6.18 (3.33-11.70) | <.001 |
| Age | 1.01 (0.97-1.03) | .60 | 1.01 (0.97-1.04) | .52 | NA | NA | NA | NA | NA | NA | NA | NA |
| Women | 0.85 (0.43-1.64) | .64 | 0.95 (0.47-1.88) | .90 | NA | NA | NA | NA | NA | NA | NA | NA |
| Race | ||||||||||||
| White | 1 [Reference] | 1 [Reference] | NA | NA | NA | NA | NA | NA | NA | NA | NA | |
| Black | 0.72 (0.25-1.79) | .51 | 0.76 (0.26-1.96) | .99 | NA | NA | NA | NA | NA | NA | NA | NA |
| Congestive heart failure | 0.44 (0.10-1.40) | .21 | NA | NA | 0.10 (0.03-0.69) | .02 | NA | NA | NA | NA | NA | NA |
| Peripheral vascular disease | 1.53 (0.54-3.98) | .39 | NA | NA | 1.11 (0.34-3.34) | .85 | NA | NA | NA | NA | NA | NA |
| COPD | 2.93 (0.87-9.81) | .07 | NA | NA | 3.05 (0.81-11.60) | .09 | NA | NA | NA | NA | NA | NA |
| Smoking | 2.23 (1.11-4.78) | .21 | NA | NA | 2.04 (0.92-4.77) | .08 | NA | NA | NA | NA | NA | NA |
| Cancer | 2.63 (1.08-6.34) | .02 | NA | NA | 1.59 (0.53-4.74) | .40 | NA | NA | NA | NA | NA | NA |
| Charlson Comorbidity Index score | ||||||||||||
| 0-2 | 1 [Reference] | NA | NA | NA | 1 [Reference] | NA | NA | NA | NA | NA | NA | NA |
| 3-4 | 1.65 (0.71-3.94) | .24 | NA | NA | 1.58 (0.64-3.94) | .32 | NA | NA | NA | NA | NA | NA |
| >5 | 3.57 (1.64-8.15) | .001 | NA | NA | 3.49 (1.34-9.39) | .01 | NA | NA | NA | NA | NA | NA |
| Study site | ||||||||||||
| Johns Hopkins Hospital | 1 [Reference] | NA | NA | NA | NA | NA | 1 [Reference] | NA | NA | NA | NA | NA |
| University of Maryland Medical Center | 0.51 (0.11-1.64) | .31 | NA | NA | NA | NA | 0.75 (0.15-2.74) | .69 | NA | NA | NA | NA |
| Inpatient at the time of surgical procedure | 2.03 (1.01-4.04) | .04 | NA | NA | NA | NA | 2.17 (0.92-5.20) | .08 | NA | NA | NA | NA |
| Surgical procedure | ||||||||||||
| Cardiac | 1 [Reference] | NA | NA | NA | NA | 1 [Reference] | NA | NA | NA | NA | NA | |
| Vascular | 1.10 (0.38-2.96) | .83 | NA | NA | NA | NA | 0.96 (0.31-2.76) | .95 | NA | NA | NA | NA |
| Spinal | 0.44 (0.18-1.02) | .06 | NA | NA | NA | NA | 0.56 (0.20-1.48) | .25 | NA | NA | NA | NA |
| Intracranial | 2.06 (0.85-4.95) | .10 | NA | NA | NA | NA | 2.98 (1.07-8.40) | .04 | NA | NA | NA | NA |
| Nasal culture for S aureus | ||||||||||||
| S aureus positive | 2.70 (1.30-5.57) | .006 | NA | NA | NA | NA | NA | NA | 2.47 (1.05-5.68) | .03 | NA | NA |
| Methicillin-resistant S aureus positive | 6.00 (1.52-29.31) | .01 | NA | NA | NA | NA | NA | NA | 2.69 (0.55-15.30) | .220 | NA | NA |
Abbreviations: COPD, chronic obstructive pulmonary disease; NA, not applicable; OR, odds ratio; S aureus, Staphylococcus aureus.
The association between nasal microbiome cluster class and the composite infectious outcome was assessed in a series of regression models, including an unadjusted model and models adjusted (ie, models 1-5) for potential confounding baseline covariates from Table 1.
Model 1 adjusted for demographic covariates: age, sex, and race.
Model 2 adjusted for baseline comorbidities: congestive heart failure, peripheral vascular disease, COPD, history of smoking, history of cancer, and Charlson Comorbidity Index score.
Model 3 adjusted for surgical risk factors: study site, inpatient or outpatient status, and surgical procedure.
Model 4 adjusted for nasal culture results for S aureus and for methicillin-resistant S aureus.
Model 5 adjusted for inverse probability of treatment weighting using propensity score for assignment to microbiome cluster 2. The propensity score for assignment to cluster 2 incorporated all baseline covariates listed in Table 1.
Figure 2. Association of Microbiome Cluster Class With Infectious Outcome After Surgical Procedure.
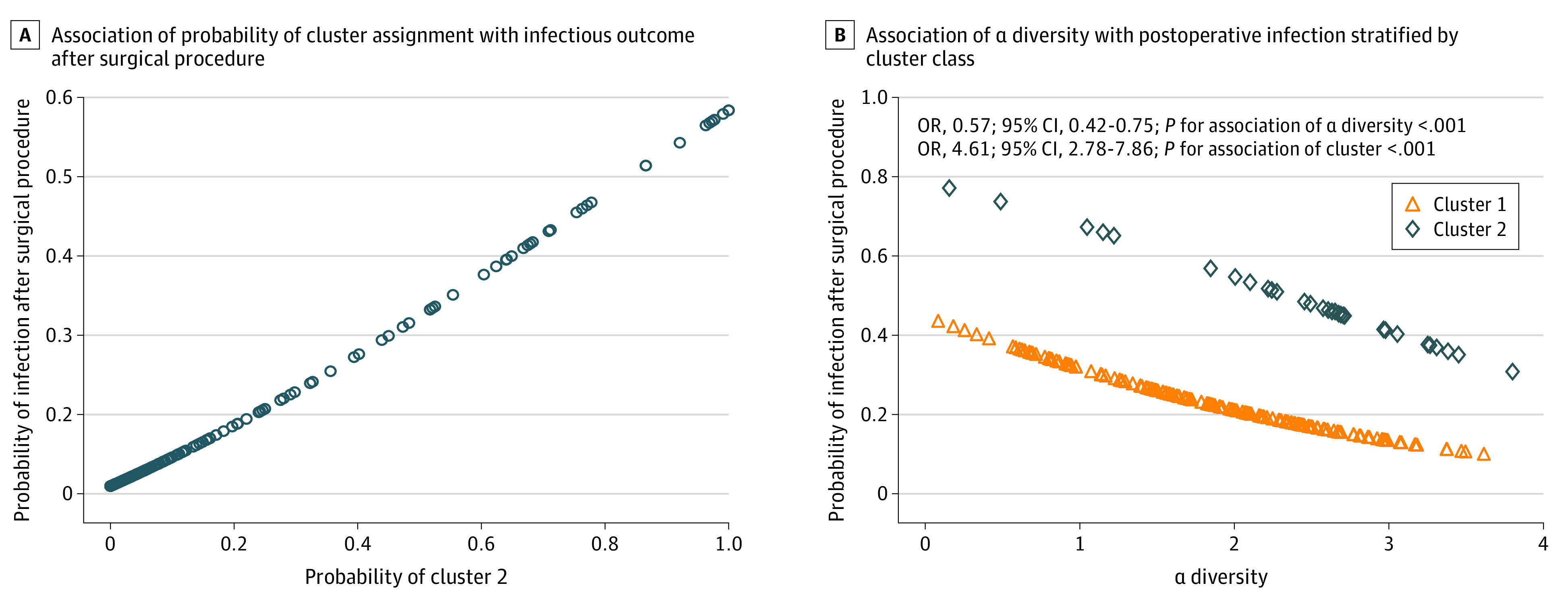
We sought to determine if the association between probability of assignment to cluster 2 and infectious outcome was independent of baseline clinical covariates in a series of adjusted regression models. After adjustment in regression models, the odds of infection remained 5-fold to 6-fold higher for participants in cluster 2, ranging from an OR of 5.47 (95% CI, 1.90-17.74; P = .002) for model 2 to an OR of 6.18 (95% CI, 3.33-11.70; P < .001) for model 5 (Table 3). These results suggest that none of the baseline covariates, including demographic characteristics, relevant comorbidities, surgical risk factors, or nasal carriage of S aureus, were confounding covariates for the association between probability of cluster 2 assignment and infectious outcome. In model 5 (Table 3), with simultaneous adjustment for all baseline covariates using IPTW of the propensity score for assignment to cluster 2, odds of infection remained 6-fold higher. In IPTW-adjusted analyses, probability of assignment to cluster 2 was also associated with statistically significantly higher odds of the secondary outcomes of SSI only (OR, 2.90; 95% CI, 1.14-7.62; P = .03), pneumonia only (OR, 5.22; 95% CI, 2.56-10.94; P < .001), and composite infection within 30 days of surgical procedure (OR, 6.64; 95% CI, 3.36-13.48; P < .001). Odds increases for bacteremia (OR, 1.47; 95% CI, 0.56-3.79) and death (OR, 1.49; 95% CI, 0.59-3.70) were not statistically significant.
In evaluations of adjusted analyses using bootstrapping of subsamples, the mean (range) number of participants in each subsample was 134 (122-144) individuals. Grades of membership classified a mean (SD) 118.3 (12.7) participants into a major cluster 1 and 16.0 (11.8) participants into a minor cluster 2 in each iteration. In IPTW-adjusted analyses, the probability of assignment to cluster 2 was associated with 8-fold higher odds of infection after surgical procedure (OR, 7.91; 95% CI, 3.61-19.29; P < .001); categorical assignment to cluster 2 was associated with 4-fold higher odds of infection (OR, 4.20; 95% CI, 2.28-8.51; P < .001).
Given the association of microbiome cluster class with infectious outcome, we sought to identify characteristics of cluster 2 that might account for its association with infectious outcome. We found that α diversity was inversely associated with infectious outcome in both cluster groups (OR, 0.57; 95% CI, 0.42-0.75; P for main association of α diversity < .001); however, this association was independent of the association between cluster 2 and outcome (OR, 4.61, 95% CI, 2.78-7.86; P < .001). At any given level of α diversity, odds of infection were higher for individuals in cluster 2 than those in cluster 1 (Figure 2). We also examined the association of the 553 species-level taxa identified by 16S rRNA gene sequencing with infectious outcome. We found that 43 taxa were significantly associated with the composite infectious outcome after adjusting for multiple comparisons (eTable 2 in the Supplement), and of these, 7 taxa were also significantly associated with cluster. However, none of these taxa were recovered from a clinical site of infection. In adjusted analysis that included cluster 2 as a covariate, Moraxella (OR, 1.16, 95% CI, 1.00-1.34; P = .04), Novosphingobium (OR, 1.13, 95% CI, 1.05-1.23; P = .001), Anaerococcus (OR, 0.43, 95% CI, 0.31-0.57; P < .001), and Atopobium (OR, 0.69, 95% CI, 0.53-0.89; P = .005) were independently associated with clinical infectious outcome; however, these genera were not associated with changes in the association of cluster 2 with infectious outcome (eTable 3 in the Supplement).
Discussion
To our knowledge, this case-control study is the first report finding an association between preoperative nasal microbial profiles and development of postoperative infectious complications, an association that was independent of the well-known connection between nasal colonization with S aureus and clinical infection. We found that microbial features derived solely from 16S rRNA gene sequencing classified individuals into groups by risk associated with development of SSI and pneumonia after surgical procedure.
This study provides new detail regarding composition of nasal microbiota in a population receiving surgical procedures for whom preoperative culture of the nares is common clinical practice. Similar to previous reports, this study found variability in the composition of nasal microbiota within and between individuals.1 Taxa representing common skin commensals, including Corynebacterium, Propionibacterium (also known as Cutibacterium), and Staphylococcus, were found in relatively high abundance,1,23,24 and we found several additional taxa, including Alloiococcus, Anaerococcus, Planococcaceae, and Enterobacteriaceae, to be present at high abundance levels. Differences between our study and others may be due to differences in sequenced amplicons, sequencing methods, bioinformatics methods used to classify operational taxonomic units, and participant populations.
Staphylococcus accounted for 3.7% of all observed sequences, and the prevalence of S aureus in the nasal microbiome was greater using 16S rRNA gene sequencing (98.5%) than standard clinical culture (20.8%). Other studies have reported higher detection rates of S aureus from concurrent samples of the anterior nares when using sequencing approaches vs when using culture methods,2,25 reflecting the greater sensitivity of sequencing for microbial detection. We found a positive association between relative abundance of S aureus as detected by 16S rRNA gene sequencing and nasal colonization with S aureus by clinical culture; however, we found no associations with abundances of other Staphylococcal species or non-Staphylococcal genera.
In our unsupervised grades of membership approach18,19 to classify samples based on 16S rRNA gene sequencing results, assignment of samples to clusters was independent and agnostic of baseline clinical characteristics or infection status. In our analysis, we found that a 2-cluster model accounted for the largest portion of variance while being most parsimonious. The approach used with grades of membership differs from popular methods for clustering sample-level microbiome data, such as hierarchical clustering26 and partition around medoids.27,28 The grades of membership method is probabilistic and based on the idea that each sample can have partial membership in multiple clusters rather than being forced into categorical assignment. Principal component analyses were consistent with partial membership of samples within clusters, given that samples were dispersed across vectors as a continuum rather than as discrete groupings. We did not find an association between nasal microbiome cluster assignment and several preoperative demographic, clinical, or surgical covariates.
Other investigators have used clustering methods to classify microbial composition of samples obtained from the anterior nares and nasal sinuses. In Liu et al,2 microbiome cluster class was associated with abundance of S aureus in the anterior nares, and in Abreu et al,29 cluster class discriminated between individuals with and without a diagnosis of chronic sinusitis. In Lehtinen et al,24 nasal microbiome class at baseline was associated with subsequent viral load, host inflammatory response, and symptom severity after experimental challenge with rhinovirus.
A major novel finding from our study was a temporal, dose-response association between preoperative nasal microbiome cluster class and subsequent development of infection at non-nasal sites. This association was independent of all measured covariates, including nasal carriage of S aureus, and was robust to iterative subsampling and bootstrapping analyses. Importantly, the odds of infection associated with nasal microbiome class were as large as or larger than those associated with nasal carriage of S aureus.
We found an inverse and independent association between α diversity of the preoperative nasal microbiome and odds of infection after surgical procedure, which is consistent with the well-described association of decreased α diversity with adverse clinical outcomes in a variety of disease states.3 A 2020 study4 reported that decreased α diversity of gut microbiota was associated with higher risk of death during 2-year follow-up in an observational cohort of patients who underwent allogeneic hematopoietic cell transplantation. Our observations extend previous reports by finding an association between nasal microbiome cluster class and adverse outcome that is independent of α diversity.
To our surprise, there was virtually no concordance between the taxa distinguishing microbiome cluster 1 from cluster 2 and those that caused clinical infection. Several potentially pathogenic taxa, including Moraxella,30 Novosphingobium (also known as Sphingomonas),31 Anaerococcus, and Atopobium,32 were associated with cluster and postoperative infectious outcome; however, none of these accounted for the association between cluster class and infection. These findings suggest that the taxa that distinguished cluster 1 from cluster 2 are not in the direct causal pathway to infectious outcome.
The mechanisms underlying the association between nasal microbiome cluster class and postoperative infection remain unclear. A possibility is that the aggregate composition of nasal microbiota signifies a latent phenotype of the host that reflects its responsiveness to infectious challenge and susceptibility to clinical infection. This possibility is supported by prior work from Lehtinen et al24 that demonstrated an association between characteristics of nasal microbiota at baseline and severity of coryzal symptoms after exposure to rhinovirus. Susceptibility to infection could be associated with an immunologic state inherent to the host, to an interaction between the host and microbiota that modifies susceptibility to infection, or both.
Limitations
This study has several limitations. Our sample size was relatively small and drawn from patients undergoing a select group of surgical procedures; thus, these results may not generalize to other surgical populations. The association we observed between nasal microbiome cluster class and postoperative infection could be confounded by unmeasured covariates. Although we bootstrapped random subsamples, we lacked an independent sample to replicate our results. Additionally, we could not identify an immunologic mechanism to account for the association between nasal microbiome cluster class and postoperative infectious outcomes. Further studies are needed to replicate our findings and to examine the immunologic basis for differences in microbial profiles between clusters and their association with infection after surgical procedure.
Conclusions
These findings suggest that nasal microbiome cluster class may be a novel risk factor associated with infection after surgical procedure, with potential to improve preoperative risk stratification. The nasal microbiome may be a biomarker associated with infectious disease susceptibility beyond the niche of the anterior nares.
eAppendix. Supplemental Methods
eReferences
eTable 1. Association of Taxa Abundance With Microbiome Cluster Class
eTable 2. Association of Taxa Abundance With Composite Infectious Outcome
eTable 3. Association of Individual Taxa and Cluster Class With Composite Infectious Outcome
eFigure 1. Taxonomic Classification of 16S rRNA Gene Sequences Identified From Preoperative Nasal Swabs
eFigure 2. Violin Plots of Variance Associated With Microbiome Cluster
eFigure 3. Diversity Characterization of Nasal Microbiota by Cluster Class
References
- 1.Human Microbiome Project Consortium . Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207-214. doi: 10.1038/nature11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu CM, Price LB, Hungate BA, et al. Staphylococcus aureus and the ecology of the nasal microbiome. Sci Adv. 2015;1(5):e1400216. doi: 10.1126/sciadv.1400216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blumberg R, Powrie F. Microbiota, disease, and back to health: a metastable journey. Sci Transl Med. 2012;4(137):137rv7. doi: 10.1126/scitranslmed.3004184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peled JU, Gomes ALC, Devlin SM, et al. Microbiota as predictor of mortality in allogeneic hematopoietic-cell transplantation. N Engl J Med. 2020;382(9):822-834. doi: 10.1056/NEJMoa1900623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wenzel RP, Perl TM. The significance of nasal carriage of Staphylococcus aureus and the incidence of postoperative wound infection. J Hosp Infect. 1995;31(1):13-24. doi: 10.1016/0195-6701(95)90079-9 [DOI] [PubMed] [Google Scholar]
- 6.Kluytmans J, van Belkum A, Verbrugh H. Nasal carriage of Staphylococcus aureus: epidemiology, underlying mechanisms, and associated risks. Clin Microbiol Rev. 1997;10(3):505-520. doi: 10.1128/CMR.10.3.505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pujol M, Peña C, Pallares R, et al. Nosocomial Staphylococcus aureus bacteremia among nasal carriers of methicillin-resistant and methicillin-susceptible strains. Am J Med. 1996;100(5):509-516. doi: 10.1016/S0002-9343(96)00014-9 [DOI] [PubMed] [Google Scholar]
- 8.Rocha LA, Marques Ribas R, da Costa Darini AL, Gontijo Filho PP. Relationship between nasal colonization and ventilator-associated pneumonia and the role of the environment in transmission of Staphylococcus aureus in intensive care units. Am J Infect Control. 2013;41(12):1236-1240. doi: 10.1016/j.ajic.2013.04.009 [DOI] [PubMed] [Google Scholar]
- 9.Perl TM, Cullen JJ, Wenzel RP, et al. ; Mupirocin and the Risk of Staphylococcus Aureus Study Team . Intranasal mupirocin to prevent postoperative Staphylococcus aureus infections. N Engl J Med. 2002;346(24):1871-1877. doi: 10.1056/NEJMoa003069 [DOI] [PubMed] [Google Scholar]
- 10.Bode LG, Kluytmans JA, Wertheim HF, et al. Preventing surgical-site infections in nasal carriers of Staphylococcus aureus. N Engl J Med. 2010;362(1):9-17. doi: 10.1056/NEJMoa0808939 [DOI] [PubMed] [Google Scholar]
- 11.Faraday N, Rock P, Lin EE, et al. Past history of skin infection and risk of surgical site infection after elective surgery. Ann. Surg. 2013;257(1):150-154. doi: 10.1097/SLA.0b013e3182588abf [DOI] [PubMed] [Google Scholar]
- 12.Zupancic ML, Cantarel BL, Liu Z, et al. Analysis of the gut microbiota in the old order Amish and its relation to the metabolic syndrome. PLoS One. 2012;7(8):e43052. doi: 10.1371/journal.pone.0043052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roghmann MC, Lydecker AD, Hittle L, et al. Comparison of the microbiota of older adults living in nursing homes and the community. mSphere. 2017;2(5):e00210-17. doi: 10.1128/mSphere.00210-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fadrosh DW, Ma B, Gajer P, et al. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome. 2014;2(1):6. doi: 10.1186/2049-2618-2-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holm JB, Humphrys MS, Robinson CK, et al. Ultrahigh-throughput multiplexing and sequencing of >500-base-pair amplicon regions on the Illumina HiSeq 2500 platform. mSystems. 2019;4(1):e00029-19. doi: 10.1128/mSystems.00029-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeSantis TZ, Hugenholtz P, Larsen N, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72(7):5069-5072. doi: 10.1128/AEM.03006-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73(16):5261-5267. doi: 10.1128/AEM.00062-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dey KK, Hsiao CJ, Stephens M. Visualizing the structure of RNA-seq expression data using grade of membership models. PLoS Genet. 2017;13(3):e1006599. doi: 10.1371/journal.pgen.1006599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding T, Schloss PD. Dynamics and associations of microbial community types across the human body. Nature. 2014;509(7500):357-360. doi: 10.1038/nature13178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.National Nosocomial Infections Surveillance System . National Nosocomial Infections Surveillance (NNIS) System Report, data summary from January 1992 through June 2004, issued October 2004. Am J Infect Control. 2004;32(8):470-485. doi: 10.1016/j.ajic.2004.10.001 [DOI] [PubMed] [Google Scholar]
- 21.Paulson JN, Stine OC, Bravo HC, Pop M. Differential abundance analysis for microbial marker-gene surveys. Nat Methods. 2013;10(12):1200-1202. doi: 10.1038/nmeth.2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paulson JN, Olson ND, Braccia DJ, et al. metagenomeSeq: statistical analysis for sparse high-throughput sequencing. Bioconductor.org. Accessed March 23, 2020. http://www.cbcb.umd.edu/software/metagenomeSeq
- 23.Hang J, Zavaljevski N, Yang Y, et al. Composition and variation of respiratory microbiota in healthy military personnel. PLoS One. 2017;12(12):e0188461. doi: 10.1371/journal.pone.0188461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lehtinen MJ, Hibberd AA, Männikkö S, et al. Nasal microbiota clusters associate with inflammatory response, viral load, and symptom severity in experimental rhinovirus challenge. Sci Rep. 2018;8(1):11411. doi: 10.1038/s41598-018-29793-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu YJ, Sasaki T, Kuwahara-Arai K, Uehara Y, Hiramatsu K. Development of a new application for comprehensive viability analysis based on microbiome analysis by next-generation sequencing: insights into Staphylococcal carriage in human nasal cavities. Appl Environ Microbiol. 2018;84(11):e00517-18. doi: 10.1128/AEM.00517-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gajer P, Brotman RM, Bai G, et al. Temporal dynamics of the human vaginal microbiota. Sci Transl Med. 2012;4(132):132ra52. doi: 10.1126/scitranslmed.3003605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koren O, Knights D, Gonzalez A, et al. A guide to enterotypes across the human body: meta-analysis of microbial community structures in human microbiome datasets. PLoS Comput Biol. 2013;9(1):e1002863. doi: 10.1371/journal.pcbi.1002863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arumugam M, Raes J, Pelletier E, et al. ; MetaHIT Consortium . Enterotypes of the human gut microbiome. Nature. 2011;473(7346):174-180. doi: 10.1038/nature09944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abreu NA, Nagalingam NA, Song Y, et al. Sinus microbiome diversity depletion and Corynebacterium tuberculostearicum enrichment mediates rhinosinusitis. Sci Transl Med. 2012;4(151):151ra124. doi: 10.1126/scitranslmed.3003783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murphy TF, Parameswaran GI. Moraxella catarrhalis, a human respiratory tract pathogen. Clin Infect Dis. 2009;49(1):124-131. doi: 10.1086/599375 [DOI] [PubMed] [Google Scholar]
- 31.Ryan MP, Adley CC. Sphingomonas paucimobilis: a persistent Gram-negative nosocomial infectious organism. J Hosp Infect. 2010;75(3):153-157. doi: 10.1016/j.jhin.2010.03.007 [DOI] [PubMed] [Google Scholar]
- 32.Murphy EC, Frick IM. Gram-positive anaerobic cocci—commensals and opportunistic pathogens. FEMS Microbiol Rev. 2013;37(4):520-553. doi: 10.1111/1574-6976.12005 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eAppendix. Supplemental Methods
eReferences
eTable 1. Association of Taxa Abundance With Microbiome Cluster Class
eTable 2. Association of Taxa Abundance With Composite Infectious Outcome
eTable 3. Association of Individual Taxa and Cluster Class With Composite Infectious Outcome
eFigure 1. Taxonomic Classification of 16S rRNA Gene Sequences Identified From Preoperative Nasal Swabs
eFigure 2. Violin Plots of Variance Associated With Microbiome Cluster
eFigure 3. Diversity Characterization of Nasal Microbiota by Cluster Class