

Abstract
The discovery of tetrazine click-induced secondary interactions is reported as a promising new tool for polymeric biomaterial synthesis. This phenomenon is first demonstrated as a tool for poly(ethylene glycol) (PEG) hydrogel assembly via purely non-covalent interactions and is shown to yield robust gels with storage moduli 1–2 orders of magnitude higher than other non-covalent crosslinking methods. In addition, tetrazine click-induced secondary interactons also enhance the properties of covalently crosslinked hydrogels. A head-to-head comparison of PEG hydrogels crosslinked with tetrazine-norbornene and thiol-norbornene click chemistry revealed an approximately 6-fold increase in storage modulus and unprecedented resistance to hydrolytic degradation in tetrazine click-crosslinked gels without substantial differences in gel fraction. Molecular dynamic simulations attribute these differences to the presence of secondary interactions between the tetrazine-norbornene cycloaddition products, which are absent in the thiol-norbornene crosslinked gels.
Keywords: hydrogels, supramolecular chemistry, non-covalent gelation, tetrazine click chemistry
Graphical Abstract
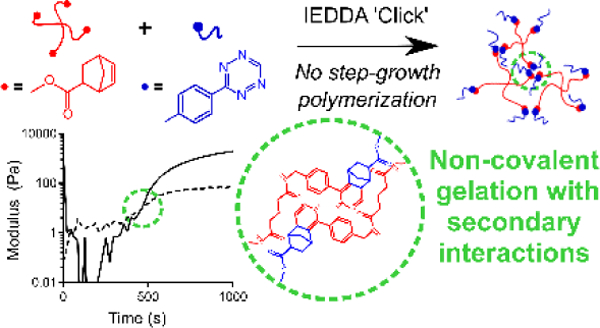
Herein, the discovery of secondary interactions between the products of the inverse-electron demand Diels Alder (IEDDA) tetrazine-norbornene click reaction are leveraged to produce purely non-covalently crosslinked hydrogels. In covalently crosslinked poly(ethylene glycol) (PEG) hydrogels, the presence of these secondary interactions results in increased storage modulus and resistance to hydrolytic degradation compared to gels crosslinked with radical-mediated thiol-norbornene chemistry.
Chemical reactions fitting the click chemistry paradigm have become an indispensable tool for the synthesis and functionalization of hydrogel biomaterials. Specifically, inverse electron demand Diels-Alder (IEDDA) click reactions between s-tetrazines and electron rich dienophiles like norbornene and trans-cyclooctene are attractive because of their bio-orthogonality. To date, tetrazine-norbornene IEDDA click reactions have been used to covalently crosslink poly(ethylene glycol) (PEG),[1] alginate,[2] and gelatin hydrogels[3] for 3D cell culture and tissue engineering. In addition, tetrazine-trans-cyclooctene IEDDA reactions have been used for spatial and temporal patterning and creating hydrogel microchannels and microfibers.[4] In all of this work, hydrogel formation has been achieved through formation of IEDDA cycloaddition products.
Herein, we report a new feature of tetrazine click chemistry that can be leveraged for engineering polymeric materials. Specifically, we report that the IEDDA click reaction between polymers functionalized with aryl 1,2,4,5-tetrazine and norbornene end-groups results in strong secondary interactions between the cycloaddition products that can be leveraged for non-covalent assembly. This phenomenon, which is distinctly different from hydrogel crosslinking through the IEDDA products, was discovered by reacting a tetrafunctional and monofunctional precursors. Specifically, 20 kDa PEG-tetra-norbornene at a concentration of 10 wt.% was reacted for 30 minutes at room temperature with 5 kDa methoxy-PEG-tetrazine at a 1:1 ratio of tetrazine to norbornene in water (Figure 1a). Methoxy-PEG-tetrazine was synthesized by functionalizing methoxy-PEG-amine with 5-(4-(1,2,4,5-tetrazin-3-yl)-benzylamino)-5-oxopentanoic acid, as previously described.[1] According to Flory-Stockmayer theory,[5] which is routinely used to predict functional group conversion required for step-growth crosslinking of polymer networks,[6, 7] a tetrafunctional component reacted with a monofunctional component would have an infinite critical conversion. Thus, gelation due to covalent bonds between the tetrazine and norbornene is not possible. Nevertheless, in situ oscillatory rheology showed the crossover of the storage modulus (G’) and the loss modulus (G”) at approximately 425 seconds, indicating a sol-gel transition (Figure 1b). Moreover, the kinetics of G’ evolution over time followed those of the tetrazine-norbornene reaction, indicating that gelation was an emergent property that followed the formation of the cycloaddition product (Figure S1). Notably, these gels exhibited a storage modulus of around 8 kPa (n=3) (Table S1). This modulus is much higher than that of other non-covalently crosslinked, multi-arm PEG hydrogels. For example, hydrogels assembled from 10 kDa tetrafunctional PEG and bifunctional polymer components with “Dock-and-Lock” peptide-peptide interactions exhibited a maximum storage modulus of ~1000 Pa at 10 wt%.[8] Similarly, Mixing-Induced Two-Component Hydrogels (MITCH) made with 8-arm, 20 kDa PEG exhibited a storage modulus of 15 Pa, at a concentration of 10% w/v, while similar experiments with 4-arm PEG failed to gel.[9] Additionally, these gels exhibited a high gel fraction of 82 ± 2.1%, and swelled considerably when placed in deionized water (Table S1).
Figure 1.
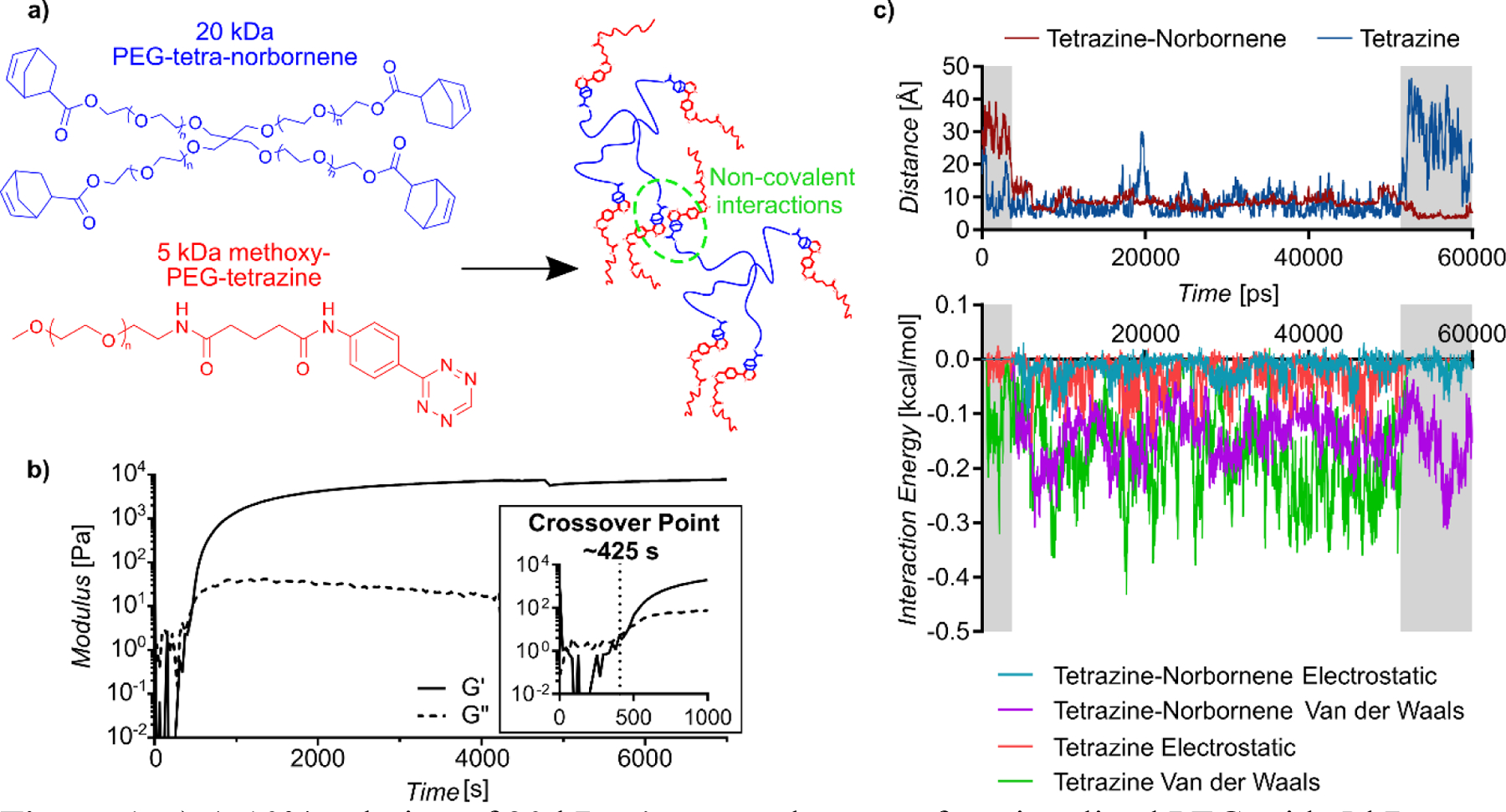
a) A 10% solution of 20 kDa 4-arm norbornene-functionalized PEG with 5 kDa monofunctional PEG-tetrazine added in a 1:1 ratio of tetrazine to norbornene b) exhibits crossover of the storage (G’) and loss (G”) moduli via in situ oscillatory rheology, indicating gelation, at 425 s at 21°C. c) Molecular dynamics simulations comparing interactions between unreacted tetrazine and tetrazine-norbornene cycloaddition products. A more negative value of interaction energy indicates a stronger interaction. Intially, the tetrazine-norbornene products are farther from each other than the tetrazines, and show little to no interaction (left region in gray). Then, the tetrazine-norbornene products drift together and remain so for the duration of the simulation. In contrast, the tetrazines drift apart later in the simulation (right region in gray) and lose all secondary interactions, as indicated in the interaction energy.
To better understand the tetrazine-norbornene click-induced non-covalent gelation, we performed classical molecular dynamics simulations on model systems. The simulations compared interactions between two reactant tetrazine molecules conjugated to a short length of PEG to interactions between two molecules containing the tetrazine-norbornene cycloaddition product flanked by short lengths of PEG (Figure S2). Molecular interactions were then quantified based on interaction energies as well as intermolecular distances (Figure 1c, bottom and top panels, respectively). Importantly, while unreacted tetrazine molecules exhibited some secondary interactions, which was expected due to their hydrophobicity, the molecules were able to drift apart during the simulation despite starting in close proximity. This drift is apparent at approximately 20,000 ps and 50,000 ps. In contrast, the tetrazine-norbornene cycloaddition products were observed to drift together and then maintain strong interactions. These results support that the sol-gel transition observed after reacting PEG-tetra-norbornene with methoxy-PEG-tetrazine was the result of secondary interactions between tetrazine-norbornene cycloaddition products.
Because tetrazine click-induced secondary interactions enabled hydrogel formation on their own, we hypothesized that secondary interactions between the tetrazine-norbornene cycloaddition products would significantly alter the properties of IEDDA covalently crosslinked hydrogels. Often times, IEDDA reactions and other reactions which fit the click paradigm are regarded as interchangeable, with the conditions for reaction initiation as the major consideration for its applicability. To date, only minor differences in gel properties have been observed between different crosslinking reactions and have been attributed to differences in crosslinking efficiency and network homogeneity. [10, 11] However, we believed IEDDA click chemistry to provide an exception. To test our hypothesis, we leveraged the dual reactivity of norbornene to perform a head-to-head comparison of hydrogels crosslinked via IEDDA tetrazine-norbornene click chemistry and radical-mediated thiol-norbornene (i.e., thiol-ene) click chemistry. Specifically, PEG hydrogels were synthesized by reacting PEG-di-norbornene (2 kDa) with either PEG-tetra-thiol (20 kDa) for thiol-ene crosslinking or PEG-tetra-tetrazine (20 kDa) for tetrazine-norbornene crosslinking (Figure 2a). Precautions were taken with the PEG-tetra-thiol to avoid oxidation of the thiols to disulfides. Characterization of the two four-arm molecules by dynamic light scattering showed that they exhibited similar size and degree of aggregation (Figure S3). For hydrogel synthesis, tetra-functional components were added to phosphate buffered saline (PBS) at a concentration of 7.5 wt.% and PEG-di-norbornene was added to achieve a 1:1 stoichiometric ratio of norbornene to the participating functional group of the tetra-functional PEG. Thiol-ene crosslinking was achieved by UV photopolymerization (365 nm, 10 mW/cm2, 5 min) initiated by 2 mM lithium acylphosphinate (LAP)[12]. Tetrazine-norbornene crosslinked gels were allowed to react for 30 minutes at room temperature.
Figure 2.
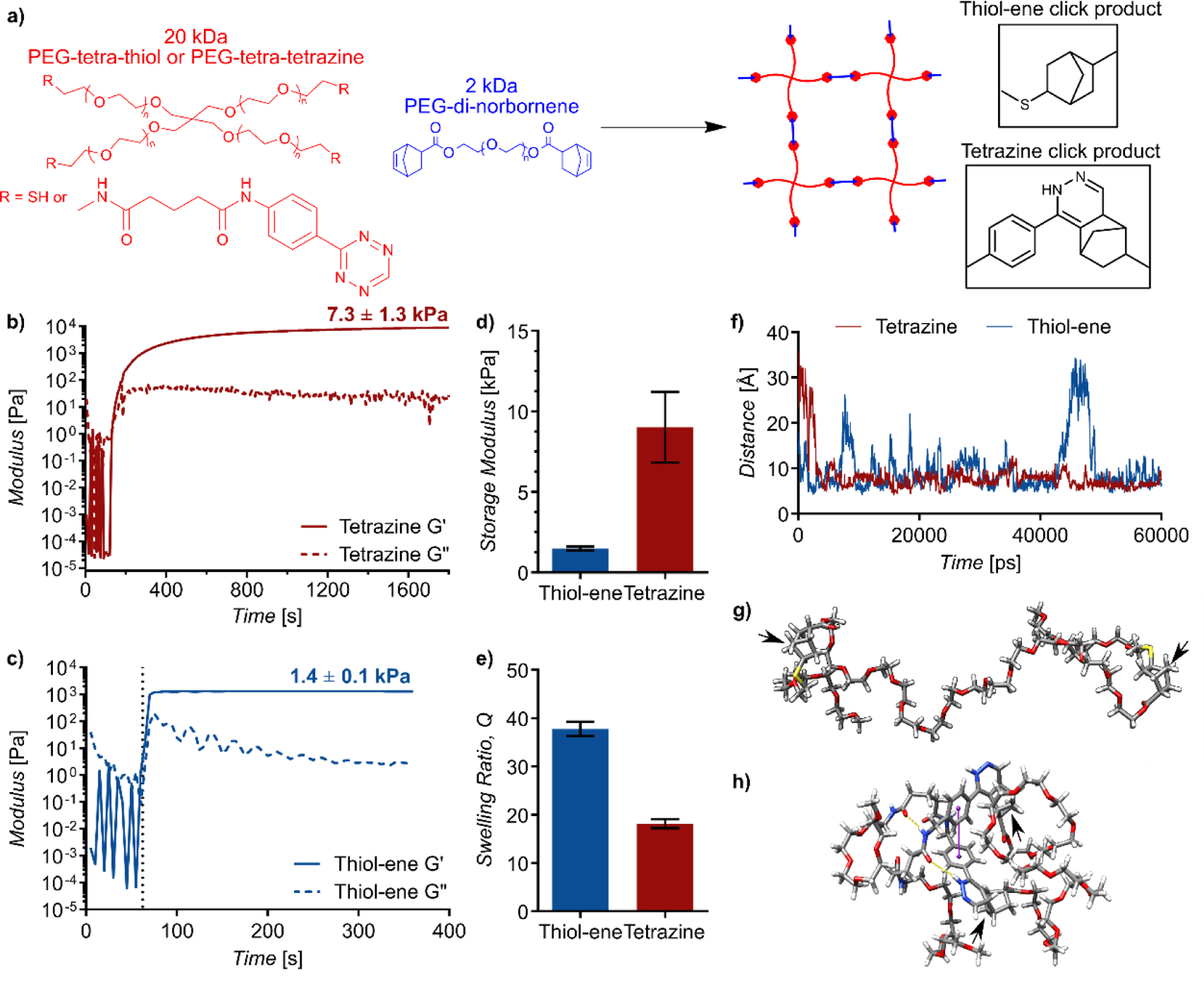
(a) Either 20 kDa PEG-tetra-thiol (thiol-ene gels) or 20 kDa PEG-tetra-tetrazine (tetrazine gels) was reacted with 2 kDa PEG-di-norbornene at a 1:1 ratio of norbornene to thiol or tetrazine. b) Modulus evolution of tetrazine crosslinked hydrogels via in situ oscillatory rheology. c) Modulus evolution of thiol-ene crosslinked hydrogels via in situ rheology. d) Average storage modulus of equilibrium swelled gels. e) Swelling ratio of equilibrium swelled gels. (f) Distance in Ångstroms over time between apical carbons (indicated with arrows) of the (g) thiol-norbornene products and (h) tetrazine-norbornene products. Yellow lines indicate hydrogen bonds and purple lines indicate pi-pi stacking.
Although the two gel formulations used the same molecular weight components with the same functionality at the same concentrations, the tetrazine click crosslinked gels exhibited markedly higher moduli. Rheological characterization of modulus evolution during in situ polymerization showed a five-fold increase in shear storage modulus for tetrazine crosslinked gels compared to the thiol-ene crosslinked gels (7.3± 1.3 kPa vs. 1.4±0.1 kPa) (Figure 2b,c). Notably, the post-polymerization modulus of the covalently crosslinked gels is closer to that of the gels formed via secondary interactions than to those formed via thiol-ene covalent crosslinking. The difference between the two chemistries persisted at equilibrium swelling, with the shear storage modulus of the tetrazine crosslinked gels being more than 6 times that of the thiol-ene crosslinked gels (9.0±1.8 kPa vs. 1.4±0.1 kPa, p < 0.0001) (Figure 2d). In addition, the swelling ratio of the tetrazine click crosslinked gels was about half that of the thiol-ene crosslinked gels (18±0.7 vs. 38±1.2, p = 0.004) (Figure 2e). We also measured the gel fraction of both gel formulations and found there was no statistically significant difference in gel fraction between the thiol-ene crosslinked and tetrazine crosslinked gels (95±2.8% and 96±0.5 %, respectively, p = 0.43). Additionally, 1H NMR of hydrolytically degraded thiol-ene crosslinked gels showed complete conversion of the norbornene alkene (Figure S4). These results, which are consistent with previous reports on the efficiency of thiol-ene crosslinking,[7, 11, 13] suggest that the apparent differences in crosslink density we observed cannot be attributed to a deficiency in the thiol-ene crosslinking reaction and are instead due to enhancements resulting from tetrazine-norbornene click-induced secondary interactions.
Molecular dynamic simulations of polymer chains containing products of the thiol-ene or tetrazine-norbornene click reactions supported this interpretation and our hypothesis (Figure S5). While both click products started in an extended conformation, the distance between the apical carbon of the bridged cyclohexanes became and stayed small in the tetrazine-norbornene products, but varied greatly over time for the thiol-ene products (Figure 2f,g,h). The random drift between thiol-ene products, which lack the capacity for hydrogen bonding or pi-pi interactions, is similar to what was observed in the prior simulation. In contrast, the tetrazine-norbornene products showed interactions via hydrogen bonding, parallel displaced and T-shaped pi-pi stacking, and hydrophobic interactions between the relatively electrostatic neutral region around the bridged cyclohexane (Figure S6). Analysis of the interaction energy per atom of the click products showed stronger interactions between the tetrazine-norbornene products than between the thiol-ene products, with the van der Waals interactions dominant for the tetrazine-norbornene products (Figure S7).
Finally, we tested whether secondary interactions between tetrazine-norbornene cycloaddition products would improve hydrogel stability against degradation. For this experiment, we exploited the susceptibility of the ester bonds between each norbornene group and the PEG backbone of the PEG-di-norbornene crosslinker to base-catalyzed hydrolysis. Tetrazine and thiol-ene click crosslinked gels were incubated in a 0.1 N sodium hydroxide solution (pH=13) at 37°C and the wet mass of the gels was recorded over 24 hours (Figure 3a). Unexpectedly, while the thiol-ene crosslinked gels degraded completely in a matter of minutes, the tetrazine crosslinked gels exhibited no significant mass loss over the period of 24 hours. Shear storage modulus measurements revealed a decrease from 9.0 ± 1.8 kPa to 2.0 ± 0.5 kPa (Figure 3b) in tetrazine crosslinked gels post-sodium hydroxide treatment. Remarkably, however, the storage modulus after hydrolysis was still higher than the initial storage modulus of the thiol-ene crosslinked gels. Additionally, tetrazine crosslinked gel samples kept in 0.1 N sodium hydroxide retained their shape and were solid enough to handle even after four weeks (not shown). The remarkable stability of these gels can be attributed to robust non-covalent interactions between the tetrazine-norbornene cycloaddition products, which was again supported by molecular dynamic simulations of the hydrolysis products (Figure S8). Simulated distances between the apical carbon of the bridged cyclohexanes showed that the two tetrazine-norbornene products stayed in close proximity after associating, despite not being connected by a PEG tether (Figure 3c). In contrast, the thiol-ene products drifted together and apart randomly and had a significantly greater distance between apical carbons on average over the course of the simulation (26.5 Å vs. 18.9 Å, p < 0.0001).
Figure 3.
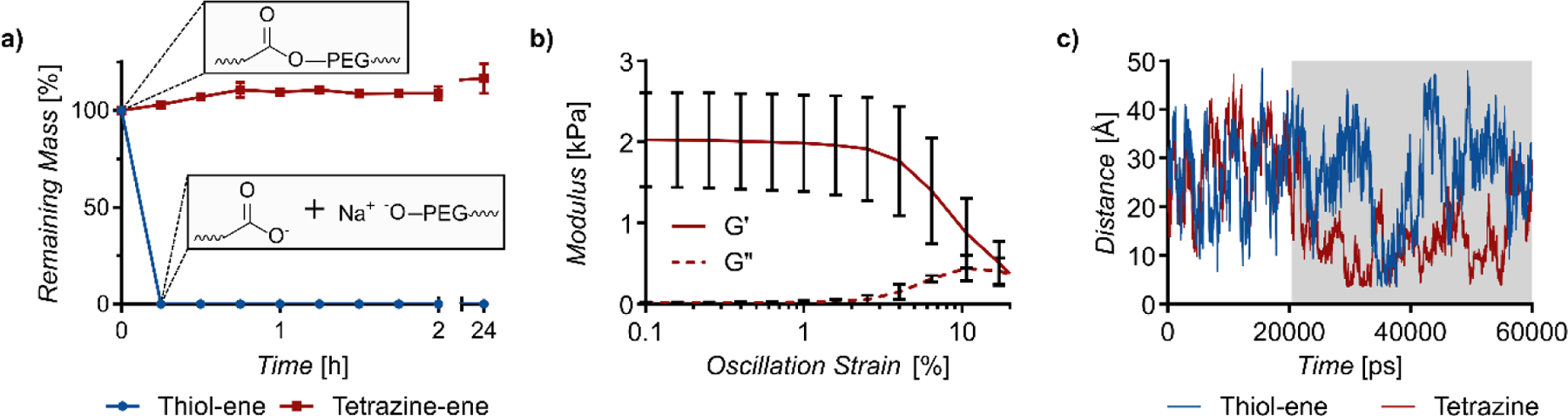
a) Thiol-ene crosslinked and tetrazine-crosslinked hydrogels gels were treated with 0.1 N NaOH for up to 24 h and mass loss over time was monitored. b) Storage (G’) and loss (G”) modulus of tetrazine-crosslinked gels after base catalyzed hydrolysis via oscillatory rheology. c) Simulated distance between the apical carbons of model hydrolyzed thiol-ene products and tetrazine-norbornene products (Figure S6). The tetrazine-norbornene products drift together and stay together in the region in gray.
In summary, our experimental and molecular dynamics simulations results demonstrate tetrazine-norbornene click-induced secondary interactions as a new tool for materials engineering. This tool is useful for non-covalent assembly, which we demonstrated via an experiment with hydrogels. However, we envision this phenomenon being useful for other applications too, such as polymer films and nanoparticulate drug delivery systems. Our finding that tetrazine-norbornene click-induced secondary interactions enhance the properties of covalently crosslinked networks, as shown in our head-to-head comparison of tetrazine and thiol-ene click crosslinked PEG hydrogels, is also significant. Mechanistically, the multiple weak, non-covalent interactions between the tetrazine-norbornene products that drive enhancement of gel stability are reminiscent of small molecule hydrogelators, which self-assemble in water to form three-dimensional supramolecular networks.[14] Though click chemistry has been used previously to synthesize small molecule hydrogelators[15] and hydrogels have been formed via metal ion coordination with pyridyl groups adjacent to unreacted tetrazines,[16] to our knowledge, this is the first demonstration of a bridging between conventional gellant systems in which hydrogels are formed via covalent crosslinking of the polymer network, and the non-covalent, supramolecular assembly of small organic molecules which characterizes hydrogelators. Combining these two disparate mechanisms of gelation is expected to open the door to the next generation of hydrogel biomaterials, designing and leveraging covalent and non-covalent assemblies as seen in naturally-occurring biopolymers.
Supplementary Material
Acknowledgements
This research was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health (R21 AR071625). Portions of this research were conducted with the advanced computing resources provided by Texas A&M High Performance Research Computing. In addition, the authors would like to thank the Gaharwar lab for the use of their rheometer, as well as Dr. A. Kristen Means, Dr. Jakrit Suriboot, and Ping Dong for their advice and assistance in gel characterization and for helpful discussions.
Acronym definitions:
- PEG
poly(ethylene glycol)
- IEDDA
inverse-electron demand Diels-Alder
Footnotes
Supporting Information
Supporting Information, including experimental procedures, NMR data, and supplemental figures, is available from the Wiley Online Library or from the author.
The authors declare no conflicts of interest.
Contributor Information
Samantha E. Holt, Department of Biomedical Engineering, Texas A&M University, College Station, TX, 77843-3120, USA
Amanda Rakoski, Department of Biomedical Engineering, Texas A&M University, College Station, TX, 77843-3120, USA.
Faraz Jivan, Department of Biomedical Engineering, Texas A&M University, College Station, TX, 77843-3120, USA.
Lisa M. Pérez, High Performance Research Computing, Texas A&M University, College Station, TX, 77843-3361, USA
Daniel L. Alge, Department of Biomedical Engineering, Texas A&M University, College Station, TX, 77843-3120, USA; Department of Materials Science and Engineering, Texas A&M University, College Station, TX, 77843-3120, USA
References
- [1].Alge DL, Azagarsamy MA, Donohue DF, Anseth KS, Biomacromolecules 2013, 14, 949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Desai RM, Koshy ST, Hilderbrand SA, Mooney DJ, Joshi NS, Biomaterials 2015, 50, 30; [DOI] [PubMed] [Google Scholar]; Lueckgen A, Garske DS, Ellinghaus A, Desai RM, Stafford AG, Mooney DJ, Duda GN, Cipitria A, Biomaterials 2018, 181, 189. [DOI] [PubMed] [Google Scholar]
- [3].Koshy ST, Desai RM, Joly P, Li JY, Bagrodia RK, Lewin SA, Joshi NS, Mooney DJ, Advanced Healthcare Materials 2016, 5, 541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zhang H, Dicker KT, Xu X, Jia X, Fox JM, ACS macro letters 2014, 3, 727; [DOI] [PMC free article] [PubMed] [Google Scholar]; Zhang H, Trout WS, Liu S, Andrade GA, Hudson DA, Scinto SL, Dicker KT, Li Y, Lazouski N, Rosenthal J, Thorpe C, Jia XQ, Fox JM, J. Am. Chem. Soc 2016, 138, 5978; [DOI] [PMC free article] [PubMed] [Google Scholar]; Dicker KT, Song J, Moore AC, Zhang H, Li Y, Burris DL, Jia X, Fox JM, Chemical Science 2018, 9, 5394; [DOI] [PMC free article] [PubMed] [Google Scholar]; Hao Y, Song J, Ravikrishnan A, Dicker KT, Fowler EW, Zerdoum AB, Li Y, Zhang H, Rajasekaran AK, Fox JM, Jia XQ, Acs Applied Materials & Interfaces 2018, 10, 26016; [DOI] [PMC free article] [PubMed] [Google Scholar]; Liu S, Moore AC, Zerdoum AB, Zhang H, Scinto SL, Zhang H, Gong L, Burris DL, Rajasekaran AK, Fox JM, Jia XQ, Biomaterials 2018, 180, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Stockmayer WH, The Journal of Chemical Physics 1943, 11, 45; [DOI] [PubMed] [Google Scholar]; Flory PJ, J. Am. Chem. Soc 1941, 63, 3083. [Google Scholar]
- [6].Sperinde JJ, Griffith LG, Macromolecules 2000, 33, 5476; [Google Scholar]; Lutolf MP, Hubbell JA, Biomacromolecules 2003, 4, 713; [DOI] [PubMed] [Google Scholar]; Lin F, Yu J, Tang W, Zheng J, Defante A, Guo K, Wesdemiotis C, Becker ML, Biomacromolecules 2013, 14, 3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fairbanks BD, Schwartz MP, Halevi AE, Nuttelman CR, Bowman CN, Anseth KS, Adv. Mater 2009, 21, 5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lu HD, Charati MB, Kim IL, Burdick JA, Biomaterials 2012, 33, 2145. [DOI] [PubMed] [Google Scholar]
- [9].Mulyasasmita W, Cai L, Dewi RE, Jha A, Ullmann SD, Luong RH, Huang NF, Heilshorn SC, J. Controlled Release 2014, 191, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Phelps EA, Enemchukwu NO, Fiore VF, Sy JC, Murthy N, Sulchek TA, Barker TH, García AJ, Adv. Mater 2012, 24, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Shih H, Lin C-C, Biomacromolecules 2012, 13, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fairbanks BD, Schwartz MP, Bowman CN, Anseth KS, Biomaterials 2009, 30, 6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lin C-C, Raza A, Shih H, Biomaterials 2011, 32, 9685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Du X, Zhou J, Shi J, Xu B, Chem. Rev 2015, 115, 13165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Tzokova N, Fernyhough CM, Topham PD, Sandon N, Adams DJ, Butler MF, Armes SP, Ryan AJ, Langmuir 2009, 25, 2479; [DOI] [PubMed] [Google Scholar]; Díaz DD, Morin E, Schön EM, Budin G, Wagner A, Remy J-S, J. Mater. Chem 2011, 21, 641; [Google Scholar]; Ramírez-López P, Torre M. C. d. l., Asenjo M, Ramírez-Castellanos J, González-Calbet JM, Rodríguez-Gimeno A, Arellano C. R. d., Sierra MA, Chem. Commun 2011, 47, 10281. [DOI] [PubMed] [Google Scholar]
- [16].Kawamoto K, Grindy SC, Liu J, Holten-Andersen N, Johnson JA, ACS Macro Letters 2015, 4, 458. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.