

Abstract
There is growing interest in PEGylation of cationic polymeric vehicles for gene delivery in order to improve vehicle stability and reduce toxicity, but little is known about the effects of PEG coatings on transfection. We used a polymer from the poly(amine-co-ester) (PACE) family blended with PEG-conjugated PACE at different ratios in order to explore the effects of polyplex PEGylation on the transfection efficiency of plasmid DNA, mRNA, and siRNA in vitro and mRNA in vivo. We discovered that concentrations of PACE-PEG as low as 0.25% by weight improved polyplex stability but also inhibited transfection in vitro. In vivo, the effect of PACE-PEG incorporation on mRNA transfection varied by delivery route; the addition of PACE-PEG improved local delivery to the lung, but PEGylation had little effect on intravenous systemic delivery. By both delivery routes, transfection was inhibited at concentrations higher than 5 wt% PACE-PEG. These results demonstrate that excess PEGylation can be detrimental to vehicle function, and suggest that PEGylation of cationic vehicles must be optimized by PEG content, cargo type, and delivery route.
Keywords: gene delivery, polymeric vehicle, polyplex, poly(ethylene glycol), biodegradable, biocompatible
Introduction
Cationic polymer vehicles have been widely studied as a promising alternative to viral vectors for gene therapy because of their non-immunogenicity, scalability and versatile chemistry, which can be tuned to deliver diverse nucleic acids such as DNA, mRNA and siRNA [1,2]. Positively charged polymers can efficiently condense nucleic acids and mediate cellular uptake through interactions with the negatively charged cell membrane [3,4]. Among cationic polymers, biodegradable polymers—such as polyesters, polycarbonates, and polyurethanes—show promise as gene delivery vectors due to their improved biocompatibility [5]. In recent years, researchers have explored a number of approaches to improve vehicle stability and gene delivery efficiency while minimizing cytotoxic effects of biodegradable polymers. These strategies include screening monomer combinations [6,7]; conjugating end-capping groups to the polymer backbone [7,8]; incorporating degradable bonds such as disulfides [5]; and adding surface coatings such as targeting ligands or poly(ethylene glycol) (PEG) [1,9].
PEG coatings have become an integral part of polymer vehicle design due to the numerous beneficial effects of PEG. PEGylation has been shown to improve vehicle stability [10,11], extend in vivo half-life [12,13], and reduce toxicity [2,14,15]. PEG coatings can also improve nanoparticle (NP) penetration through mucus [16,17]. However, several limitations of PEG-coated NPs have also been reported, such as decreased transfection efficiency [18], decreased cellular uptake [19,20], and the establishment of anti-PEG immunity after repeated doses in vivo [21,22]. There are many potential benefits to PEGylating cationic polymer NPs and polyplexes, such as decreased toxicity, improved stability, and enhanced tissue penetration; however, the effect of PEGylation on nucleic acid transfection efficiency remains unclear. In vitro, some researchers have reported unchanged or improved transfection with PEG coatings [23], while others have reported that transfection is inhibited [10,20]. Several studies have suggested that the transfection efficiency of PEG-conjugated polymers can be optimized by adjusting parameters such as PEG molecular weight (MW) and polymer to nucleic acid N:P ratio [11,24–27]. Furthermore, in vitro screens frequently correlate poorly with in vivo results. For example, Mastorakos et al. found that PEG decreased transfection efficiency of poly(β -amino ester) (PBAE) polyplexes in vitro but improved transfection via inhaled delivery in mice [28]. Williford et al. blended polyethyleneimine (PEI) with PEI-PEG and observed that transfection was inhibited at lower PEI-PEG concentrations in vivo (IV delivery in mice) as compared to in vitro experiments [29]. Apart from optimizing PEG coverage, many additional strategies have been investigated to overcome the “PEG dilemma”; these approaches include attaching targeting ligands to the vehicle surface and developing cleavable PEG coatings that detach in response to specific stimuli such as temperature, pH, enzymes, or reductive conditions [18,30]. Overall, these reports suggest that more comprehensive studies—investigating cargo type, in vivo administration route, and PEG content—are needed to understand and optimize the advantages of PEGylation for gene therapy.
We have recently described a family of cationic polymers, poly(amine-co-ester) (PACE), that can effectively deliver a variety of nucleic acids in vitro and in vivo [2,6]. PACE polymers are biodegradable and highly customizable; by varying component monomers and lactone content, we have created a library of materials with tunable characteristics, such as hydrophobicity, thermal properties, MW, toxicity, and transfection efficiency. When synthesized with lower lactone content (10–20%), PACE polymers can be used to formulate polyplexes with negatively charged nucleic acids. Synthesis of this versatile class of polymers can also be modified to incorporate PEG groups, producing PACE-PEG block copolymers. Here, we have comprehensively investigated the effects of PEG content and nucleic acid cargo type on the efficacy of gene delivery. We formulated PACE polyplexes with a range of PEG content by blending PACE with PEG-conjugated PACE (PACE-PEG) in order to examine the effect of PEGylation on the transfection efficiency, stability, and toxicity of PACE polyplexes for the delivery of different nucleic acids in vitro and in vivo both locally and systemically. PEGylation offers both advantages and disadvantages, and we explore the intersection of these effects and how they can differ in vitro and in vivo. We describe an approach to produce versatile, biocompatible, and stable polyplexes encapsulating a wide size range of nucleic acids. This study highlights the importance of testing and optimizing PEG coverage of polymeric vehicles for gene therapy applications.
Materials and Methods
Materials and Reagents
PEG, 15-pentadecanolide (PDL), N-methyl diethanolamine (MDEA), sebacic acid (SA), diethyl sebacate (DES), diphenyl ether, Novozym 435 catalyst, bovine serum albumin (BSA), Roche cOmplete EDTA-free Protease Inhibitor Cocktail, and Immobilon-FL PVDF Membranes were purchased from MilliporeSigma (Saint Louis, MO, USA). pcDNA3-EGFP plasmid was obtained from Addgene (Watertown, MA, USA). CleanCap EGFP mRNA and CleanCap FLuc mRNA were purchased from TriLink Biotechnologies (San Diego, CA, USA). siRNA against nectin-1 (GGUUAAAAGGUGAGGCAGA) was synthesized by Horizon Discovery (Waterbeach, United Kingdom). For cell culture, HeLa cells and Eagle’s Modified Essential Medium (EMEM) were purchased from ATCC (Manassas, VA, USA), and fetal bovine serum (FBS) was purchased from R&D Systems (Minneapolis, MN, USA). Lipofectamine products, TrypLE Express Enzyme, MOPS SDS running buffer, 10% NuPAGE Bis-Tris Gel, nectin-1 monoclonal antibody (clone CK8), HRP-conjugated goat anti-mouse IgG (H+L) secondary antibody, and the Pierce BCA Protein Assay were obtained from Thermo Fisher Scientific (Waltham, MA, USA). PE antihuman CD111 (nectin-1) antibody was purchased from BioLegend (San Diego, CA, USA). Beta actin monoclonal antibody was purchased from Proteintech Group (Rosemont, IL, USA). Clarity Western ECL substrate was purchased from Bio-Rad Laboratories (Hercules, CA, USA). CellTiter-Glo, Glo Lysis Buffer, and the Bright-Glo Luciferase Assay System were purchased from Promega (Madison, WI, USA). Precellys hard tissue lysing tubes were obtained from Bertin Instruments (Montigny-le-Bretonneux, France). RediJect D-Luciferin Substrate was purchased from PerkinElmer (Waltham, MA, USA).
Polymer Synthesis, Purification, and Characterization
PACE polymers were synthesized as described previously, with some modifications [2,6]. Briefly, the monomers (PDL, MDEA, and SA) were dissolved in diphenyl ether solvent with a lipase-based Novozym 435 catalyst. In the first stage, monomers were oligomerized at 1 atm under argon for 18–20 hours at 90°C. This was followed by 48–72 hours of polymerization at 90°C under vacuum (2 mmHg; Figure S1). Polymers were then purified following previously described methods [2]. For the synthesis of PACE-PEG, 5000 molecular weight (MW) PEG was added as an additional monomer and DES was substituted for SA. The synthesis was otherwise identical. 10 mol% PDL content was used for all polymers in this study. To determine their composition, polymers were dissolved in deuterated chloroform and analyzed via proton nuclear magnetic resonance (1H-NMR, Agilent DD2 400 MHz NMR Spectrometer). MW was determined by GPC using the Ultimate 3000 UHPLC system (Thermo Fisher Scientific).
Polyplex Formulation and Characterization
All PACE polyplexes were formed at a weight ratio of 100:1 polymer:nucleic acid. Polymers were dissolved 100 mg/mL in DMSO overnight at 37°C while shaking. To make blends of PACE and PACE-PEG, the dissolved PACE and PACE-PEG polymers were combined at the indicated weight ratio. Intermediate dilutions were made as necessary. Here, we refer to the PACE-PEG content of polyplex formulations, but the theoretical PEG content can be calculated (Table S1). For characterization and in vitro experiments, nucleic acids were diluted into sodium acetate buffer (25 mM, pH 6) to a final concentration of 20 μg/mL (pDNA and mRNA) or 1 μM (siRNA). Polymer was similarly diluted into sodium acetate buffer (25mM, pH 6) to a final concentration of 2 mg/mL (pDNA and mRNA) or 1.33 mg/mL (siRNA) and vortexed for 15 seconds. The polymer solution was then added to the nucleic acid solution at 1:1 volume ratio and vortexed for an additional 25 seconds. The polyplexes were incubated at room temperature for 10 minutes before use (adding directly to cell culture medium). Polyplex size and zeta potential were measured by dynamic light scattering (DLS, Zetasizer Pro, Malvern Analytical). Size and morphology were also confirmed by transmission electron microscopy (TEM, FEI Tecnai Osiris 200kV) using a tungsten stain for visualization.
Polyplex Stability Assessment
To assess their stability over time, polyplexes were formulated in sodium acetate buffer (25 mM, pH 6) to a final concentration of 2 mg/mL and placed in a 37°C incubator shaking at 200 rpm. For stability assessments in serum, 10% FBS was added to the sodium acetate buffer after polyplexes were formulated. Polyplex size was measured by DLS (Zetasizer Pro, Malvern Analytical) at each time point (0 hr, 0.5 hr, 1 hr, 2 hrs, 4 hrs, 8 hrs, 24 hrs, 48 hrs, 72 hrs) or until the PDI = 1 or multiple peaks were observed. Polyplexes were diluted to 40 μg/mL in deionized water prior to measurement.
In Vitro Toxicity Studies
CellTiter-Glo was used to assess polyplex cytotoxicity in vitro. 24 hours prior to treatment, HeLa cells were plated in 96-well tissue culture plates at a concentration of 10,000 cells/well. PACE polyplexes were delivered at varying concentrations (pDNA and mRNA: 0.313–10 μg/mL; siRNA: 6.25–200 nM). The Lipofectamine controls (Lipofectamine 3000, Lipofectamine MessengerMAX, Lipofectamine RNAiMAX) were formulated by following the manufacturer’s recommended lipid to nucleic acid ratio and delivered at the same nucleic acid concentrations. 24 hours after delivery, the cell media was refreshed, 100 μL of CellTiter-Glo reagent was added to each well, and plates were incubated at room temperature for 10 minutes. Luminescence was then measured with a plate reader using an integration time of 0.5 seconds per well.
In Vitro Transfection of pDNA, mRNA, and siRNA
HeLa cells were grown in EMEM with 10% FBS and 50 μg/mL gentamicin in a 37°C incubator under 5% CO2. For all transfection experiments, HeLa cells, were plated at 50,000 cells per well in a 24-well tissue culture plate and grown until they reached 60–80% confluency (approximately 24 hours). pcDNA3-EGFP plasmid was used for plasmid DNA transfection experiments. Cells were treated with 1 μg of plasmid per well using PACE polyplexes blended with PACE-PEG (concentrations: 0%, 0.01%, 0.025%, 0.05%, 0.1%, 0.25%, 0.5%, 1%, 2.5%), or Lipofectamine3000 according to the manufacturer’s protocol as a control, for 48 hours. For mRNA transfection experiments, cells were treated for 24 hours with 1 μg of EGFP mRNA using PACE polyplexes, or Lipofectamine MessengerMAX according to the manufacturer’s protocol as a control. An siRNA against nectin-1 was used for siRNA transfection experiments. Cells were treated for 72 hours with 100 nM siRNA with PACE polyplexes, or 10 nM siRNA with Lipofectamine RNAiMAX according to the manufacturer’s protocol as a control.
Flow Cytometry, Microscopy, and Western Blot Analysis
For flow cytometry analyses, cells transfected with EGFP pDNA and mRNA were washed once with PBS, dissociated with TrypLE Express Enzyme, resuspended in FACS buffer (2% BSA in PBS), and run directly on the flow cytometer (Attune NxT). Cells treated with siRNA were dissociated with TrypLE Express Enzyme and subsequently stained with PE-nectin-1 antibody in FACS Buffer for 30 minutes. Cells were then washed twice, resuspended in FACS buffer, and nectin-1 expression was quantified by flow cytometry. For microscopy analysis (Olympus LCPlanFl 20X, Olympus IX71), treated cells were washed three times with PBS and fixed in 4% paraformaldehyde in PBS. For western blot analysis, protein extracts were prepared by lysing cells in RIPA lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.1% SDS, 1% NP40, 0.5% sodium deoxycholate, 1 mM ß-glycerophosphate, 2.5 mM sodium pyrophosphate, 10 mM sodium fluoride, 1 mM sodium orthovanadate, 2.5 μM pepstatin A, 1 mM EDTA, and 1 mM EGTA) supplemented with complete EDTA-free Protease Inhibitor Cocktail. Samples were run on a 10% NuPAGE Bis-Tris gel in MOPS SDS running buffer and transferred to a PVDF membrane. The membrane was blocked with 5% non-fat dry milk in tris buffered saline, and incubated with a nectin-1 monoclonal antibody (clone CK8) followed by a HRP-conjugated mouse secondary antibody. The membrane was visualized using Clarity Western ECL and imaged using a ChemiDoc imaging system (Bio-Rad). A ß-actin antibody was used to probe for the loading control. Western blot quantification was performed using Image Studio Lite (LI-COR Biosciences).
In Vivo mRNA Delivery
All animal procedures were performed in accordance with the guidelines and policies of the Yale Animal Resource Center (YARC) and approved by the Institutional Animal Care and Use Committee (IACUC) of Yale University. Male BALB/c mice, age 7–12 weeks, were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). For in vivo experiments, PACE polyplexes were delivered either by intracheal instillation (IT) or intravenously (IV). Polyplexes were prepared in sodium acetate buffer (25 mM, pH 5.8) to a final concentration of 10 mg/mL polymer and 0.1 mg/mL FLuc mRNA (IT delivery) or 5 mg/mL polymer and 0.05 mg/mL FLuc mRNA (IV delivery). For IT delivery, mice were anesthetized under 3% isofluorane (Patterson Veterinary) and suspended by the incisors. The tongue was retracted with tweezers, and 50 μL of the polyplex formulation was administered to the back of the mouth. The tongue was held in the retracted position for the duration of 10 breaths while polyplexes were inhaled. For IV delivery, mice were similarly anesthetized under 3% isoflurane and then 100 μL of the polyplex formulations were delivered via retro-orbital injection. After 24 hours, mice were euthanized and heart perfused with 15 mL PBS. Organs (IT: lung, IV: lung, spleen, liver, kidney) were removed, minced, and transferred to 2 mL Precellys hard tissue lysing tubes with 1 mL Glo Lysis Buffer. Organs were homogenized at 6500 rpm twice for 30 seconds (Precellys 24) and subsequently centrifuged at 21,000 × g for 10 min to remove cell debris. 20 μL of tissue lysates were combined with 100 μL Bright-Glo luciferase substrate and luminescence was measured using an integration time of 10 seconds (Promega GloMax 20/20). The Pierce BCA Protein Assay Kit was used to measure total protein concentration following the manufacturer’s instructions. For IVIS experiments, luminesence was measure 24 hours after polyplex administration. 12 min prior to euthanasia, mice were injected intraperitoneally with RediJect D-Luciferin Substrate (150 mg/kg). Organs (heart, lung, liver, spleen, kidney) were removed and luminescent signal was imaged by IVIS (IVIS Spectrum, PerkinElmer).
Statistical Analysis
Results were analyzed using GraphPad Prism (version 7.0a for Mac OS X). Data are presented as mean ± SD of at least three independent experiments. One- and two-way ANOVA with Tukey’s multiple comparisons test were used where appropriate. Values were considered significantly different at p<0.05.
Results and Discussion
Polymer Synthesis and Characterization
PACE was synthesized by the enzymatic co-polymerzation of PDL, MDEA, and SA as described previously [2]. Briefly, the monomers dissolved in diphenyl ether were combined with lipase catalyst and oligomerized under argon for 18–20 hours. This was followed by 48–72 hours of polymerization under vacuum. For PACE-PEG synthesis, DES was substituted for SA, and 5 kDa PEG was added to the synthesis (Figure 1A). Polymer characterization, including gel permeation chromatography (GPC) analyses (Table S2) and NMR (Figure S3, Table S3) can be found in the supplementary information. Ten % PDL acid-ended PACE (referred to here as PACE) and 10% PDL PEG-conjugated PACE (PACE-PEG) were used for this study. In our experience, the synthesis of PACE polymer is a straightforward, two-step process that can be readily modified by introduction of other monomers or polymer blocks such as PEG.
Figure 1:
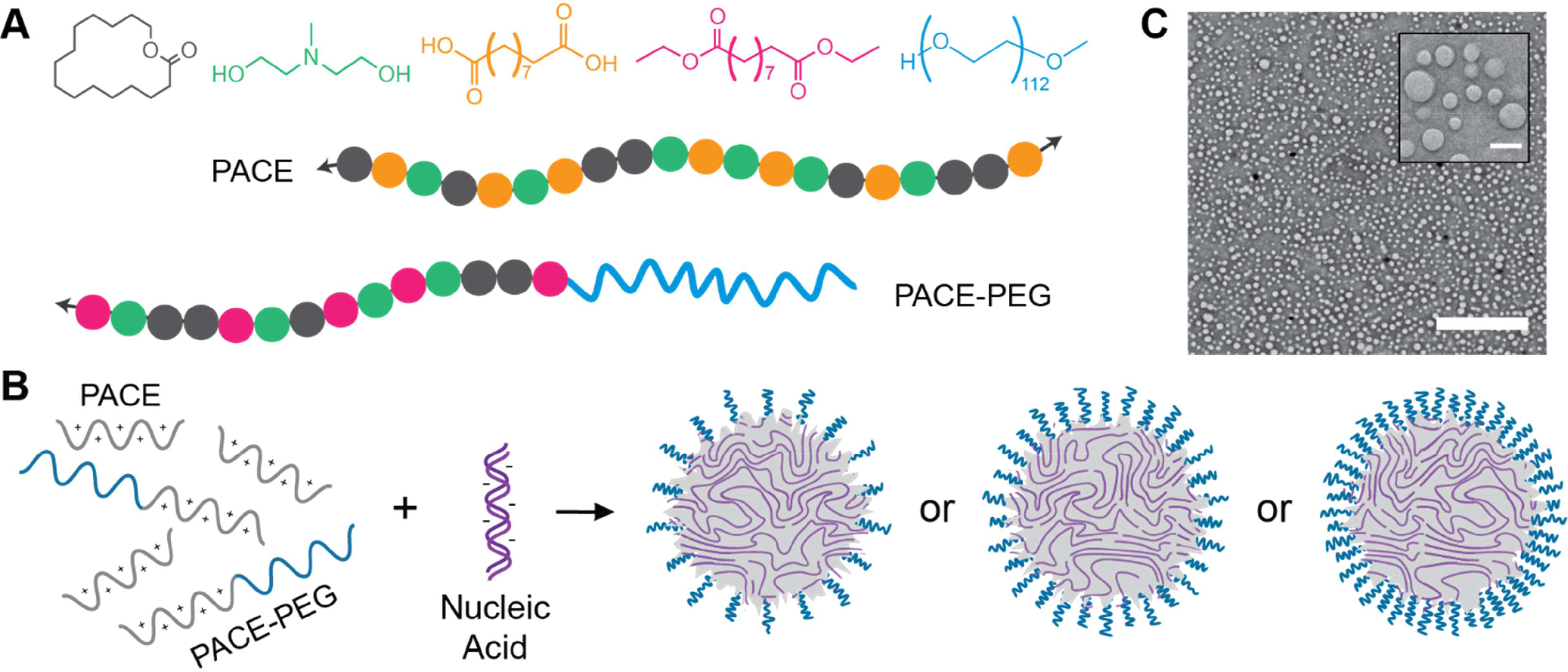
Overview of PACE and PACE-PEG synthesis and formulation. (A) Schematic of PACE and PACE-PEG polymers with monomers pentadecanolide (PDL, grey), methyldiethanolamine (MDEA, green), sebacic acid (SA, orange), diethyl sebacate (DES, pink), and polyethylene glycol (PEG, blue). (B) Schematic of PACE polyplex formulation. PACE and PACE-PEG are blended and combined with nucleic acid to form polyplexes with varied PEG content. (C) Representative TEM image of PACE blended with 0.5% PACE-PEG loaded with siRNA (scale bar, 2 μm; inset scale bar, 200 nm).
Polyplex Formulation and Characterization
We formulated PACE polyplexes to encapsulate pDNA, mRNA, or siRNA with a range of PEG content by blending PACE polymer with PACE-PEG during assembly (Figure 1B). PACE polyplexes were in a similar size range as measured by DLS (pDNA: 190–440 nm; mRNA: 160–360 nm; siRNA: 190–280 nm) (Figure 2A-C). Polyplexes with 100% PACE-PEG were much smaller and had a spherical shape and smooth morphology by TEM, whereas non-PEGylated polyplexes exhibited an uneven morphology (Figure 1C, Figure S2). Additionally, the average size tended to decrease with the addition of PACE-PEG. The polyplex zeta potentials ranged from -3.1 mV to -22 mV (Figure 2D-F), and there was no significant difference in zeta potential between non-PEGylated and PEGylated polyplexes, which is likely due to the minimal change in N:P ratio over the PACE-PEG concentrations analyzed (Table S4). PACE-PEG incorporation into polyplexes was confirmed by NMR analysis (Figure S3, Table S3). These results confirm that PACE polymer blended with PACE-PEG forms uniform polyplexes with low polydispersity indices (PDIs) (Figure S4) and without aggregates or sedimentation. At higher concentrations, PACE-PEG can also be used to decrease polyplex size.
Figure 2:
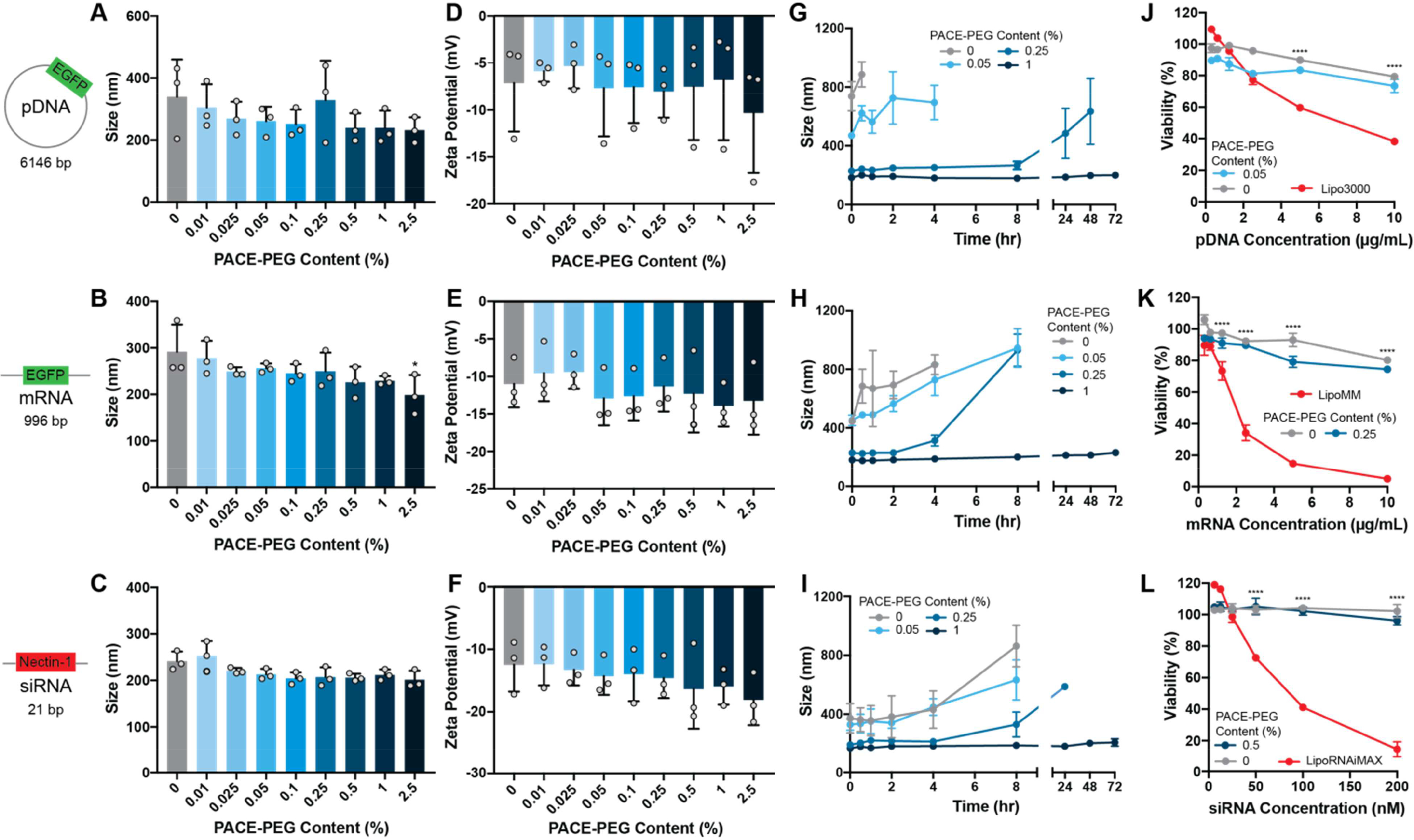
PACE blended with PACE-PEG forms nanosized, stable polyplexes in the presence of nucleic acids. (A-C) Sizes of polyplexes loaded with (A) EGFP pDNA, (B) EGFP mRNA, and(C) siRNA against nectin-1. Asterisk represents statistical difference from non-PEGylated polyplexes. (D-F) Zeta potentials of polyplexes loaded with (D) EGFP pDNA, (E) EGFP mRNA, and (F) siRNA against nectin-1. (G-I) Stability of polyplexes in sodium acetate buffer (25 mM, pH 6.0) over 72 hours loaded with (G) EGFP pDNA, (H) EGFP mRNA, and (I) nectin-1 siRNA. (J-L) Toxicity of PACE polyplexes compared to Lipofectamine for delivery of (J) EGFP pDNA, (K) EGFP mRNA, and (L) nectin-1 siRNA. Asterisks represent statistical differences between both PACE groups and Lipofectamine.* p ≤ 0.05, **** p ≤ 0.0001.
Polyplex Stability Assessment
PACE polyplexes encapsulating pDNA, mRNA, or siRNA with PACE-PEG content ranging from 0 to 1% were assessed for their stability in sodium acetate buffer (25 mM, pH 6) at 37ºC (Figure 2G-I). For all nucleic acid types, PACE polyplexes with PACE-PEG incorporation as low as 0.05% were signifcantly more stable than those without any PACE-PEG. Non-PEGylated pDNA (Figure 2G) and mRNA (Figure 2H) PACE polyplexes immediately aggregated, whearas addition of 0.25% PACE-PEG provided stabilty to the polyplexes, allowing them to remain stable over several hours—though they eventually increased in size. In contrast, the addition of 1% PACE-PEG stabilized the PACE polyplexes over 3 days for all nucleic acid types. To assess stability in the presence of negatively charged serum proteins, mRNA polyplexes with and without PACE-PEG were incubated in a solution containing 10% FBS (Figure S5). For non-PEGylated and 0.05% PACE-PEG polyplexes, the addition of serum decreased the vehicle size, but we observed several peaks and the cumulative fit error was high, suggesting that the polyplexes were unstable and too polydisperse to be measured by DLS. Polyplexes with 0.25% PACE-PEG exhibited size variations in 10% FBS, whereas 1% PACE-PEG polyplexes remained stable over 2 days. These results suggest that blending PEG-conjugated PACE with PACE polymer can produce polyplexes with tunable stability properties, and that significant stability can be achieved with PACE-PEG content as low as 0.25% to 1% by weight.
In Vitro Toxicity Studies
PACE and PACE-PEG polyplex toxicity was tested in vitro over a range of nucleic acid delivery concentrations. PACE polyplexes were compared to corresponding commercially available Lipofectamine products, prepared according to manufacturer protocols. For pDNA, mRNA, and siRNA polyplexes, cell viability of the non-PEGylated and PEGylated polyplexes was similar, and minimal toxicity was observed—even at higher concentrations (Figure 2J-L). Cell viability after PACE polyplex treatment was in the range of 70–80% at the highest concentrations of pDNA and mRNA and 90–100% at the highest concentrations of siRNA. By comparison, Lipofectamine showed significant toxicity, with viabilities of 38% (pDNA), 5% (mRNA), and 14% (siRNA) at the highest measured concentration. Overall, PACE and PACE-PEG-blended polyplexes have a favorable safety profile, likely due to the low charge density of the polymer.
In Vitro Transfection of pDNA, mRNA, and siRNA
To assess how PEG content affects PACE polyplex transfection efficiency, we treated HeLa cells with PEGylated PACE polyplexes loaded with pDNA, mRNA, and siRNA and analyzed gene expression as a readout of transfection efficiency by flow cytometry (Figure 3A-C). For pDNA polyplexes, there was a significant increase in transfection efficiency with low concentrations of PACE-PEG (0.01%, 0.025% 0.05%) compared to the non-PEGylated polyplexes (Figure 3A). We observed a drop-off in transfection efficiency starting at 0.1% PACE-PEG, and at 1% PACE-PEG no transfection was observed. For mRNA polyplexes, we observed steady transfection up to 0.25% PACE-PEG (Figure 3B). Starting at 0.5% PACE-PEG, the transfection efficiency dropped until transfection was undetectable at 2.5% PACE-PEG. The pDNA and mRNA transfection results were further confirmed by fluorescence microscopy, which revealed a noticeable decrease in EGFP expression starting at 0.05% PACE-PEG for pDNA and 0.25% PACE-PEG for mRNA (Figure 3D-E). Small differences observed between the flow cytometry and microscopy trends could be due to batch-to-batch variability in polyplex formulation or a lag between changes in EGFP expression and transfection efficiency. For example, cells might express less EGFP while still maintaining the same percentage of EGFP-positive cells. Lastly, knockdown efficiency with siRNA-loaded polyplexes remained steady from 0% PACE-PEG to 0.5% PACE-PEG and decreased starting at 1% PACE-PEG (Figure 3C). Since the flow cytometry analysis only captures changes in membrane protein expression, total nectin-1 knockdown was also tested by western blot (Figure 3F), which indicated a similar trend. Taken together, these results indicate that blended PACE-PEG polyplexes can effectively transfect a wide variety of nucleic acids as well as—or better than—non-PEGylated polyplexes when the PACE-PEG concentration is below the inhibitory concentration. However, the in vitro PACE-PEG inhibitory concentration was very low—as low as 0.1% PACE-PEG by weight in the case of pDNA. We also observed variability in PACE-PEG inhibition between different nucleic acid types, indicating that transfection inhibition by PACE-PEG is nucleic acid-dependent. The inhibitory effect of PEG on nucleic acid transfection may be due to weakened electrostatic interactions between polyplexes and cells [31], which could decrease vehicle uptake [32]. Additionally, improved vehicle stability is hypothesized to prevent polyplexes from aggregating in endosomes and facilitating endosomal escape [20].
Figure 3:
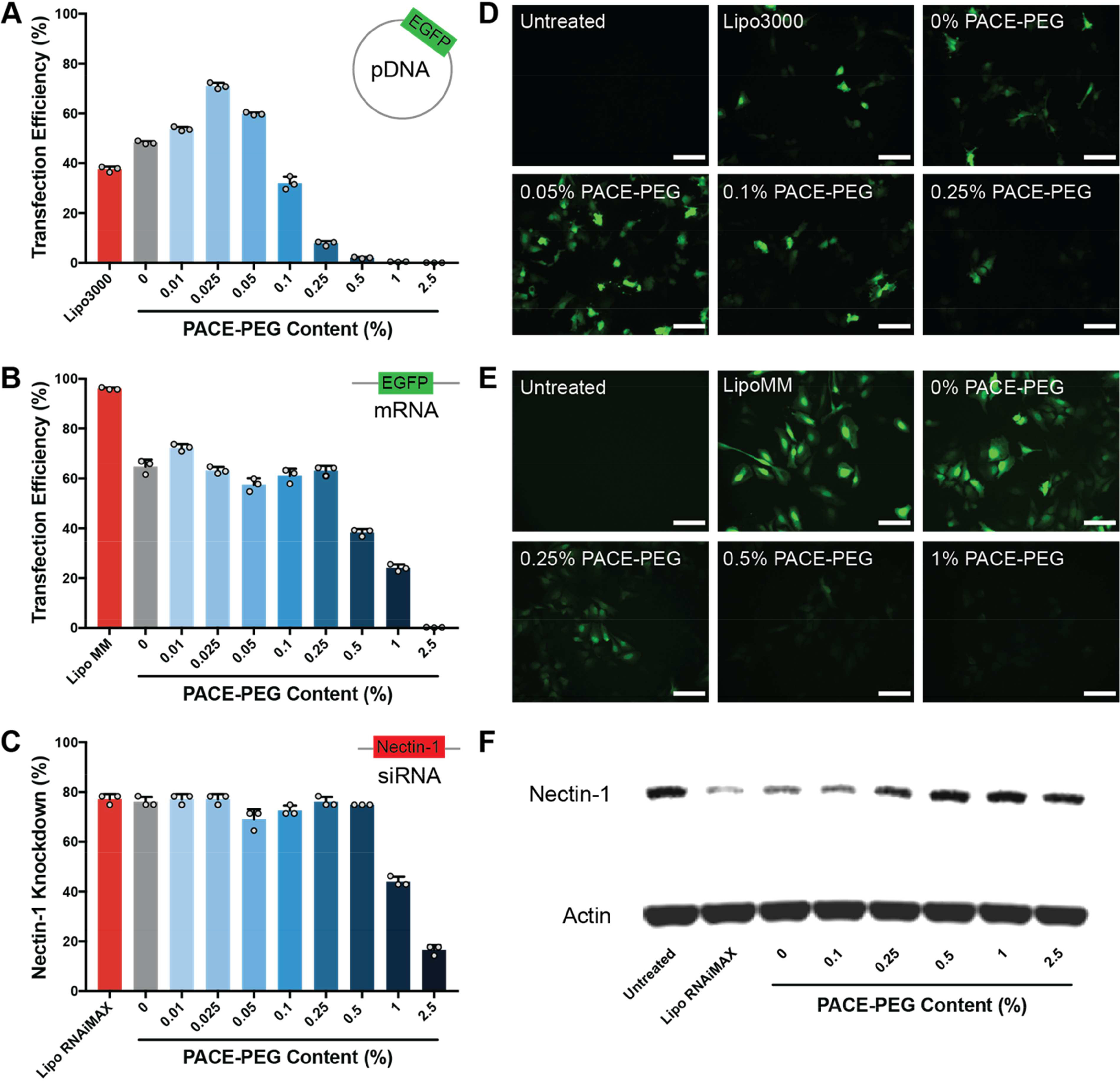
Low concentrations of PACE-PEG inhibit transfecti on of PACE polyplexes in a nucleic acid-dependent manner. (A-C) Polyplex transfection efficiency measured by flow cytometry for (A) pDNA, (B) mRNA, and (C) knockdown efficiency for siRNA. (D-E) EGFP expression in HeLa cells after transfection of EGFP (D) pDNA and (E) mRNA (scale bar, 100 μm). (F) Nectin-1 knockdown by western blot after delivery of siNectin-1 with PACE-PEG polyplexes.
In vivo mRNA Delivery
To determine the effect of PACE-PEG content on mRNA transfection in vivo, we administered FLuc mRNA using PACE polyplexes with a range of PACE-PEG concentrations by two routes: local intratracheal (IT) delivery to the lung and systemic intravenous (IV) delivery. Twenty-four hours after administration, we observed no transfection in the lung with non-PEGylated (0% PACE-PEG) PACE polyplexes by IT delivery, but luciferase expression increased with the addition of PACE-PEG, peaking at 5% PACE-PEG by weight (Figure 4A,C). When delivered IV, PACE polyplexes accumulated primarily in the spleen, which is consistent with previous findings [8]. Non-PEGylated polyplexes performed similarly to polyplexes with low levels of PACE-PEG (up to 5%), but transfection was inhibited at higher concentrations (Figure 4B,D). These results demonstrate that PACE PEGylation can improve transfection in vivo, but the effects vary by delivery route. The fact that higher PEG concentrations are effective—and even beneficial—in vivo is likely due to the additional stability PEG provides. Compared to the in vitro environment, polyplexes delivered in vivo encounter additional physiological barriers with diverse proteins, cell types, and micro-environments, which can affect their transfection efficiency. For example, PEGylation likely improves inhaled delivery to the lung by reducing polyplex aggregation in mucus and facilitating mucus penetration [16]. The observed maximum transfection point at 5% PACE-PEG is the optimal balance of the opposing effects of PEGylation such as improved stability and inhibited transfection. PEGylation is well known to improve carrier circulation times after IV administration [12]. Although we did not observe a beneficial effect on transfection of spleen cells with the addition of low amounts of PEG, we did observe a significant reduction in transfection at higher amounts, demonstrating the potential negative effects of PEG. Reducing polyplex size could improve tissue distribution, since vehicle size has been shown to have a significant effect on NP distribution after intravenous delivery [33–35]. The PACE formulations developed here successfully deliver mRNA to the lung and spleen, which are organs of interest in a broad range of disease models. For example, gene therapy to the lung has emerged as a promising treatment for cystic fibrosis [36] and a number of respiratory viruses including SARS-CoV-2 [37]. The spleen is critical in mediating the the body’s immune response, and thus gene therapies delivered to the spleen could be a promising strategy for vaccine delivery [38].
Figure 4:
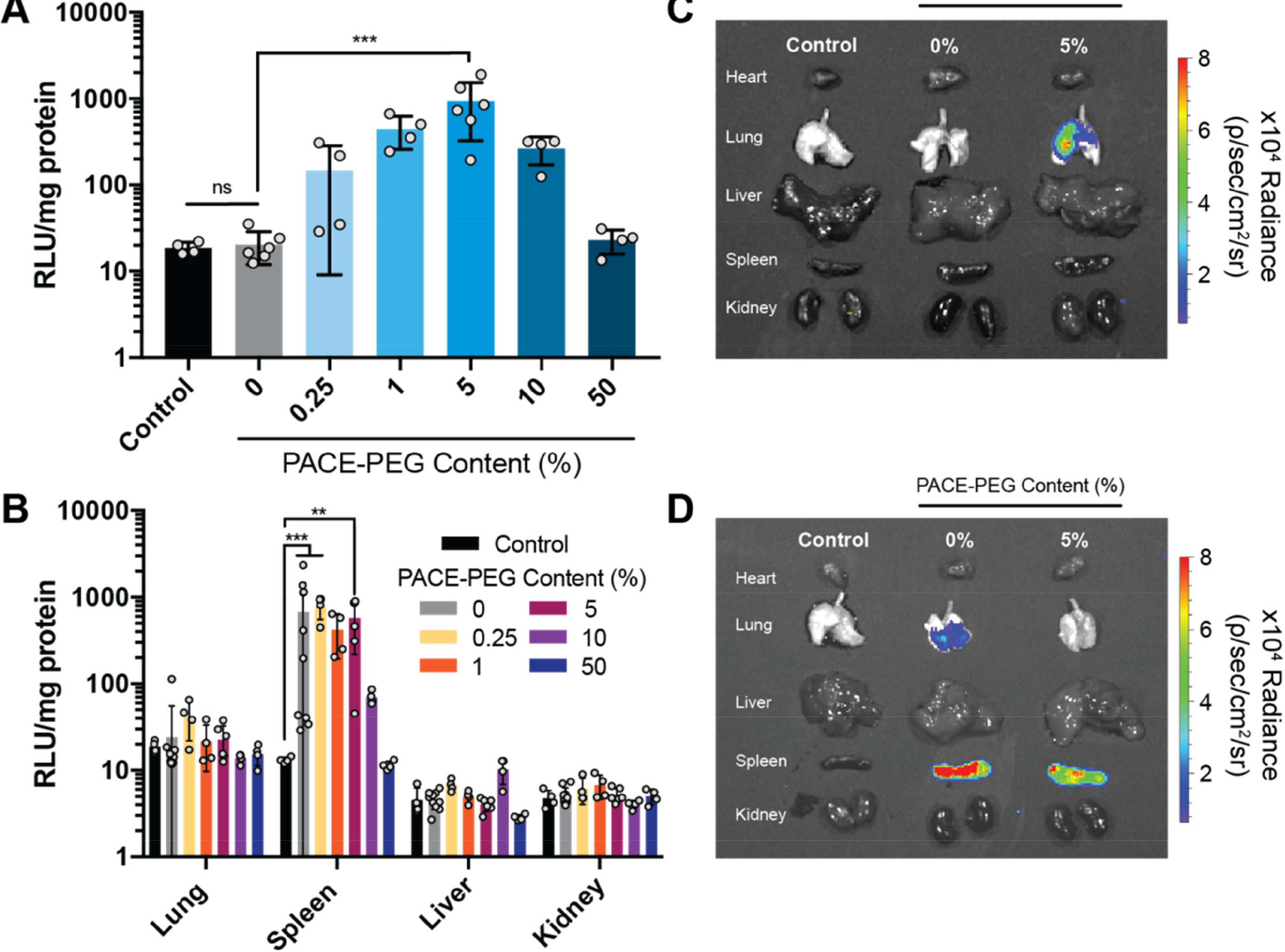
The effect of PACE-PEG content Ponin vivo mRNA delivery depends on delivery route. FLuc mRNA was delivered using PACE polyplexes blended with PACE-PEG to the lung or systemically by IV delivery. (A-B) Luciferase expression in tissue quantified by luminescence assay for (A) IT delivery (lungs) and (B) IV delivery (lung, spleen, liver, kidney). (C-D) Representative IVIS images of luminescence in organs after (C) IT and (D) IV delivery. ** p ≤ 0.01, *** p ≤ 0.001.
Conclusions
In summary, we have demonstrated a simple and robust system of producing PACE polyplexes with tunable PEG content by blending PACE with PACE-PEG in different ratios during formulation. This technique has allowed us to explore the effects of PEG on stability, biocompatibility, and transfection efficiency of PACE polyplexes in vitro and in vivo. PACE is a highly biocompatible polymer that can effectively deliver a wide variety of nucleic acid cargos, including pDNA, mRNA, and siRNA. We discovered that addition of low concentrations PACE-PEG to PACE significantly improved polyplex stability, but also completely inhibited transfection in vitro, emphasizing the importance of finding a balance between the advantages and disadvantages of PEGylation—particularly for gene delivery. Furthermore, both the stability benefits and the inhibitory effects of PACE-PEG were influenced by the type of nucleic acid cargo, suggesting that PEGylation for nucleic acid delivery should be optimized to each application. In vivo, we discovered that the effects of PACE-PEG varied by delivery route, and low concentrations of PACE-PEG significantly improved inhalation delivery to the lung. We were able use significantly higher concentrations of PACE-PEG compared to in vitro experiments before inhibitory effects were seen. These results demonstrate the poor predictive power of in vitro experiments on carrier optimization and suggest that the effects of vehicle PEGylation in vivo are influenced by additional barriers such as the presence of mucus or serum. Further optimization of this delivery system could be explored through PACE end-group conjugation [8], additional functionalization of the polyplex surface [39], and tuning the PEG MW or polymer to nucleic acid ratio [40]. However, our system is a simple and effective approach at PACE polyplex PEGylation with a wide range of potential applications.
Supplementary Material
Highlights.
Low PEG incorporation can significantly improve stability of PACE polyplexes.
Optimal PEG incorporation in vitro is not reflective of optimal in vivo efficacy.
The effect of PEG on mRNA delivery in vivo depends on the delivery route.
PEGylation at low levels improves localized mRNA delivery to the lung.
Acknowledgements
This research was supported by grants from the National Institutes of Health [R01 EB00487 and UG3 HL147352]. A.S.P. was supported by two NIH National Research Service Awards [NRSAs; a T32 GM86287 training grant and an F32 HL142144 individual postdoctoral fellowship], as well as a postdoctoral research fellowship award [PIOTRO20F0] from the Cystic Fibrosis Foundation (CFF).
Abbreviations
- DES
diethyl sebacate
- SA
sebacic acid
- MDEA
methyl diethanolamine
- PDL
pentadecanolide
- DLS
dynamic light scattering
- GPC
gel permeation chromatography
- MW
molecular weight
- NMR
nuclear magnetic resonance
- PDI
polydispersity index
- NP
nanoparticle
- PACE
poly(amine-co-ester)
- PBAE
poly(β-amino ester)
- PDI
polydispersity index
- PEG
poly(ethylene glycol)
- PEI
polyethyleneimine
- TEM
transmission electron microscopy
- FBS
fetal bovine serum
- IT
intratracheal
- IV
intravenous
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data Availability
The raw/processed data required to reproduce these findings cannot be shared at this time as the data also forms part of an ongoing study.
References
- [1].Chen J, Wang K, Wu J, Tian H, Chen X, Polycations for Gene Delivery: Dilemmas and Solutions, Bioconjugate Chem. 30 (2019) 338–349. 10.1021/acs.bioconjchem.8b00688. [DOI] [PubMed] [Google Scholar]
- [2].Kauffman AC, Piotrowski-Daspit AS, Pre-Nakazawa KH,Jiang Y,Datye A, Saltzman WM, Tunability of Biodegradable Poly(amine-co-ester) Polymers for Customized Nucleic Acid Delivery and Other Biomedical Applications, Biomacromolecules. 19 (2018) 3861–3873. 10.1021/acs.biomac.8b00997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Fröhlich E, The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles, Int J Nanomedicine. 7 (2012) 5577–5591. 10.2147/IJN.S36111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kostritskii A.Yu., Kondinskaia DA, Nesterenko AM, Gurtovenko AA, Adsorption of Synthetic Cationic Polymers on Model Phospholipid Membranes: Insight from Atomic-Scale Molecular Dynamics Simulations, Langmuir. 32 (2016) 10402–10414. 10.1021/acs.langmuir.6b02593. [DOI] [PubMed] [Google Scholar]
- [5].Chen CK, Huang PK, Law WC, Chu CH, Chen NT, Lo LW, Biodegradable Polymers for Gene-Delivery Applications, Int J Nanomedicine. 15 (2020) 2131–2150. 10.2147/IJN.S222419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zhou J, Liu J, Cheng CJ, Patel TR, Weller CE, Piepmeier JM, Jiang Z, Saltzman WM, Biodegradable poly(amine-co-ester) terpolymers for targeted gene delivery, Nature Materials. 11 (2012) 82–90. 10.1038/nmat3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Liu Y, Li Y, Keskin D, Shi L, Poly(ß-Amino Esters): Synthesis, Formulations, and Their Biomedical Applications, Advanced Healthcare Materials. 8 (2019) 1801359. 10.1002/adhm.201801359. [DOI] [PubMed] [Google Scholar]
- [8].Jiang Y, Lu Q, Wang Y, Xu E, Ho A, Singh P, Wang Y, Jiang Z, Yang F, Tietjen GT, Cresswell P, Saltzman WM, Quantitating Endosomal Escape of a Library of Polymers for mRNA Delivery, Nano Lett. 20 (2020) 1117–1123. 10.1021/acs.nanolett.9b04426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Piotrowski-Daspit AS, Kauffman AC, Bracaglia LG, Saltzman WM, Polymeric vehicles for nucleic acid delivery, Advanced Drug Delivery Reviews. (2020). 10.1016/j.addr.2020.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sung SJ, Min SH, Cho KY, Lee S, Min YJ, Yeom YI, Park JK, Effect of Polyethylene Glycol on Gene Delivery of Polyethylenimine, Biol. Pharm. Bull. 26 (2003) 492–500. 10.1248/bpb.26.492. [DOI] [PubMed] [Google Scholar]
- [11].Petersen H, Fechner PM, Martin AL, Kunath K, Stolnik S, Roberts CJ, Fischer D, Davies MC, Kissel T, Polyethylenimine-graft-Poly(ethylene glycol) Copolymers: Influence of Copolymer Block Structure on DNA Complexation and Biological Activities as Gene Delivery System, Bioconjugate Chem. 13 (2002) 845–854. 10.1021/bc025529v. [DOI] [PubMed] [Google Scholar]
- [12].Bracaglia LG, Piotrowski-Daspit AS, Lin CY, Moscato ZM, Wang Y, Tietjen GT, Saltzman WM, High-throughput quantitative microscopy-based half-life measurements of intravenously injected agents, PNAS. 117 (2020) 3502–3508. 10.1073/pnas.1915450117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gref R, Minamitake Y, Peracchia MT, Trubetskoy V, Torchilin V, Langer R, Biodegradable long-circulating polymeric nanospheres, Science. 263 (1994) 1600–1603. [DOI] [PubMed] [Google Scholar]
- [14].Petersen H, Fechner PM, Fischer D, Kissel T, Synthesis, Characterization, and Biocompatibility of Polyethylenimine-graft-poly(ethylene glycol) Block Copolymers, Macromolecules. 35 (2002) 6867–6874. 10.1021/ma012060a. [DOI] [Google Scholar]
- [15].Qi R, Gao Y, Tang Y, He RR, Liu TL, He Y, Sun S, Li BY, Li YB, Liu G, PEG-conjugated PAMAM Dendrimers Mediate Efficient Intramuscular Gene Expression, AAPS J. 11 (2009) 395. 10.1208/s12248-009-9116-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cu Y, Saltzman WM, Controlled surface modification with poly(ethylene)glycol enhances diffusion of PLGA nanoparticles in human cervical mucus, Molecular Pharmaceutics. 6 (2008) 173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lai SK, Wang YY, Hida K, Cone R, Hanes J, Nanoparticles reveal that human cervicovaginal mucus is riddled with pores larger than viruses, Proceedings of the National Academy of Sciences. 107 (2010) 598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hatakeyama H, Akita H, Harashima H, The Polyethyleneglycol Dilemma: Advantage and Disadvantage of PEGylation of Liposomes for Systemic Genes and Nucleic Acids Delivery to Tumors, Biological and Pharmaceutical Bulletin. 36 (2013) 892–899. 10.1248/bpb.b13-00059. [DOI] [PubMed] [Google Scholar]
- [19].Walkey CD, Olsen JB, Guo H, Emili A, Chan WCW, Nanoparticle Size and Surface Chemistry Determine Serum Protein Adsorption and Macrophage Uptake, J. Am. Chem. Soc. 134 (2012) 2139–2147. 10.1021/ja2084338. [DOI] [PubMed] [Google Scholar]
- [20].MISHRA S, PEGylation significantly affects cellular uptake and intracellular trafficking of non-viral gene delivery particles, European Journal of Cell Biology. 83 (2004) 97–111. [DOI] [PubMed] [Google Scholar]
- [21].Ishida T, Maeda R, Ichihara M, Irimura K, Kiwada H, Accelerated clearance of PEGylated liposomes in rats after repeated injections, Journal of Controlled Release. 88 (2003) 35–42. 10.1016/S0168-3659(02)00462-5. [DOI] [PubMed] [Google Scholar]
- [22].Ichihara M, Shimizu T, Imoto A, Hashiguchi Y, Uehara Y, Ishida T, Kiwada H, Anti-PEG IgM Response against PEGylated Liposomes in Mice and Rats, Pharmaceutics. 3 (2011) 1–11. 10.3390/pharmaceutics3010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mao S, Neu M, Germershaus O, Merkel O, Sitterberg J, Bakowsky U, Kissel T, Influence of Polyethylene Glycol Chain Length on the Physicochemical and Biological Properties of Poly(ethylene imine)-graft-Poly(ethylene glycol) Block Copolymer/SiRNA Polyplexes, Bioconjugate Chem. 17 (2006) 1209–1218. 10.1021/bc060129j. [DOI] [PubMed] [Google Scholar]
- [24].Fitzsimmons REB, Uludag H, Specific effects of PEGylation on gene delivery efficacy of polyethylenimine: Interplay between PEG substitution and N/P ratio, Acta Biomaterialia. 8 (2012) 3941–3955. 10.1016/j.actbio.2012.07.015. [DOI] [PubMed] [Google Scholar]
- [25].Malek A, Czubayko F, Aigner A, PEG grafting of polyethylenimine (PEI) exerts different effects on DNA transfection and siRNA-induced gene targeting efficacy, Journal of Drug Targeting. 16 (2008) 124–139. 10.1080/10611860701849058. [DOI] [PubMed] [Google Scholar]
- [26].Zhang X, Pan SR, Hu HM, Wu GF, Feng M, Zhang W, Luo X, Poly(ethylene glycol)-block-polyethylenimine copolymers as carriers for gene delivery: Effects of PEG molecular weight and PEGylation degree, Journal of Biomedical Materials Research Part A. 84A (2008) 795–804. 10.1002/jbm.a.31343. [DOI] [PubMed] [Google Scholar]
- [27].Zhong Z, Feijen J, Lok MC, Hennink WE, Christensen LV, Yockman JW, Kim YH, Kim SW, Low Molecular Weight Linear Polyethylenimine-b-poly(ethylene glycol)-b-polyethylenimine Triblock Copolymers: Synthesis, Characterization, and in Vitro Gene Transfer Properties, Biomacromolecules. 6 (2005) 3440–3448. 10.1021/bm050505n. [DOI] [PubMed] [Google Scholar]
- [28].Mastorakos P, Zhang C, Berry S, Oh Y, Lee S, Eberhart CG, Woodworth GF, Suk JS, Hanes J, Highly PEGylated DNA Nanoparticles Provide Uniform and Widespread Gene Transfer in the Brain, Adv. Healthcare Mater. 4 (2015) 1023–1033. 10.1002/adhm.201400800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Williford J-M, Archang MM, Minn I, Ren Y, Wo M, Vandermark J, Fisher PB, Pomper MG, Mao HQ, Critical Length of PEG Grafts on lPEI/DNA Nanoparticles for Efficient in Vivo Delivery, ACS Biomater. Sci. Eng. 2 (2016) 567–578. 10.1021/acsbiomaterials.5b00551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Fang Y, Xue J, Gao S, Lu A, Yang D, Jiang H, He Y, Shi K, Cleavable PEGylation: a strategy for overcoming the “PEG dilemma” in efficient drug delivery, Drug Delivery. 24 (2017) 22–32. 10.1080/10717544.2017.1388451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Majzoub RN, Chan CL, Ewert KK, Silva BFB, Liang KS, Jacovetty EL, Carragher B, Potter CS, Safinya CR, Uptake and transfection efficiency of PEGylated cationic liposome–DNA complexes with and without RGD-tagging, Biomaterials. 35 (2014) 4996–5005. 10.1016/j.biomaterials.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Millili PG, Selekman JA, Blocker KM, Johnson DA, Naik UP, Sullivan MO, Structural and functional consequences of poly(ethylene glycol) inclusion on DNA condensation for gene delivery, Microscopy Research and Technique. 73 (2010) 866–877. 10.1002/jemt.20839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Liao WY, Li HJ, Chang MY, Tang ACL, Hoffman AS, Hsieh PCH, Comprehensive characterizations of nanoparticle biodistribution following systemic injection in mice, Nanoscale. 5 (2013) 11079–11086. 10.1039/C3NR03954D. [DOI] [PubMed] [Google Scholar]
- [34].De Jong WH, Hagens WI, Krystek P, Burger MC, Sips AJAM, Geertsma RE, Particle size-dependent organ distribution of gold nanoparticles after intravenous administration, Biomaterials. 29 (2008) 1912–1919. 10.1016/j.biomaterials.2007.12.037. [DOI] [PubMed] [Google Scholar]
- [35].Mandl HK, Quijano E, Suh HW, Sparago E, Oeck S, Grun M, Glazer PM, Saltzman WM, Optimizing biodegradable nanoparticle size for tissue-specific delivery, Journal of Controlled Release. 314 (2019) 92–101. 10.1016/j.jconrel.2019.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Griesenbach U, Alton EWFW, Moving forward: cystic fibrosis gene therapy, Hum Mol Genet. 22 (2013) R52–R58. 10.1093/hmg/ddt372. [DOI] [PubMed] [Google Scholar]
- [37].Du L, Zhao G, Lin Y, Sui H, Chan C, Ma S, He Y, Jiang S, Wu C, Yuen KY, Jin DY, Zhou Y, Zheng BJ, Intranasal Vaccination of Recombinant Adeno-Associated Virus Encoding Receptor-Binding Domain of Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) Spike Protein Induces Strong Mucosal Immune Responses and Provides Long-Term Protection against SARS-CoV Infection, The Journal of Immunology. 180 (2008) 948–956. 10.4049/jimmunol.180.2.948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Jindal AB, Nanocarriers for spleen targeting: anatomo-physiological considerations, formulation strategies and therapeutic potential, Drug Deliv. and Transl. Res. 6 (2016) 473–485. 10.1007/s13346-016-0304-0. [DOI] [PubMed] [Google Scholar]
- [39].Tietjen GT, Hosgood SA, DiRito J, Cui J, Deep D, Song E, Kraehling JR, Piotrowski-Daspit AS, Kirkiles-Smith NC, Al-Lamki R, Thiru S, Bradley JA, Saeb-Parsy K, Bradley JR, Nicholson ML, Saltzman WM, Pober JS, Nanoparticle targeting to the endothelium during normothermic machine perfusion of human kidneys, Science Translational Medicine. 9 (2017). 10.1126/scitranslmed.aam6764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Jiang Y, Gaudin A, Zhang J, Agarwal T, Song E, Kauffman AC, Tietjen GT, Wang Y, Jiang Z, Cheng CJ, Saltzman WM, A “top-down” approach to actuate poly(amine-coester) terpolymers for potent and safe mRNA delivery, Biomaterials. 176 (2018) 122–130. 10.1016/j.biomaterials.2018.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw/processed data required to reproduce these findings cannot be shared at this time as the data also forms part of an ongoing study.