

Abstract
A disintegrin and metalloprotease 10 (ADAM10) is the α-secretase for Amyloid precursor protein (APP). ADAM10 cleaves APP to generate neuroprotective soluble APPα (sAPPα), which precludes the generation of Aβ, a defining feature of Alzheimer’s disease (AD) pathophysiology. Reduced ADAM10 activity is implicated in AD but the mechanisms mediating ADAM10 modulation are unclear. We find that the plasma membrane enzyme Glycerophosphodiester phosphodiesterase 2 (GDE2) stimulates ADAM10 APP cleavage by shedding and inactivating Reversion-inducing cysteine-rich protein with Kazal motifs (RECK), a glycosylphosphatidylinositol (GPI)-anchored inhibitor of ADAM10. In AD, membrane-tethered RECK is highly elevated and GDE2 is abnormally sequestered inside neurons. Genetic ablation of GDE2 phenocopies increased membrane RECK in AD, which is causal for reduced sAPPα, increased Aβ, and synaptic protein loss. RECK reduction restores the balance of APP processing and rescues synaptic protein deficits. These studies identify GDE2 control of RECK surface activity as essential for ADAM10 α-secretase function and physiological APP processing. Moreover, our results suggest the involvement of the GDE2-RECK-ADAM10 pathway in AD pathophysiology and highlight RECK as a potential target for therapeutic development.
One-Sentence Summary:
GDE2 controls surface amounts of the metalloprotease inhibitor RECK to promote ADAM10 α-secretase activity and prevent Aβ production and synapse loss.
Introduction:
APP plays a central role in AD. APP is processed in the pathologic “amyloidogenic” pathway through sequential cleavage by β- and γ-secretases to produce Aβ peptides that can oligomerize and form toxic insoluble Aβ species (1, 2). APP is also processed via the competing “non-amyloidogenic” pathway by α-secretase (3, 4), which cleaves APP proximal to the β-secretase site and releases sAPPα, a soluble, neuroprotective N-terminal fragment (3, 5). Cleavage by α-secretase precludes β-cleavage of APP, minimizing sAPPβ generation and subsequent Aβ production. In addition, sAPPα interacts with β-secretase to inhibit its cleavage of APP, to further stimulate non-amyloidogenic APP processing (6). Several members of the ADAM family of metalloproteases are reported to have α-secretase function (7–9); however, genetic ablation and knockdown studies identify ADAM10 as the major α-secretase that cleaves APP in neurons (3, 4). Mutations of ADAM10 that reduce its activity and enhance amyloidogenic APP processing and Aβ deposition have recently been linked to late-onset AD (LOAD) (10, 11), establishing ADAM10 as a susceptibility gene for AD. Accordingly, stimulation of ADAM10 expression and activity has garnered interest as a potential treatment for AD (5, 12). Studies in the last decade have indicated that ADAM10 is tightly regulated through transcription, translation and post-translational mechanisms (5). ADAM10 transcript expression is regulated by retinoic acid (13) and its translation can be blocked through 5’ regulatory sequences, although the mechanisms underlying this control are not well understood (14). Post-translationally, ADAM10 α-cleavage activity is influenced by growth factors, cytokines and neurotransmitters and emerging studies indicate contributions of surface trafficking pathways that are mediated in part by tetraspanin proteins (15–17). Nevertheless, deeper understanding of the specific molecular mechanisms that control and promote ADAM10 activity in the adult brain and their roles in AD are still needed to inform and facilitate therapeutic development.
In the developing nervous system, ADAM10 activity is regulated by the GDE2-RECK pathway. RECK is a GPI-anchored protein that binds and inhibits metalloproteases that include ADAM10 (18–21). RECK directly binds ADAM10, inhibits ADAM10 activity in a dose-dependent manner and blocks recombinant ADAM10 cleavage activity with a Ki <15 nM (21). These observations indicate that RECK is a potent physiological inhibitor of ADAM10 activity; however, the precise mechanism of how RECK interaction with ADAM10 inhibits its activity is still to be determined. GDE2 (also known as GDPD5) is a six-transmembrane protein that contains an external enzymatic domain homologous to bacterial glycerophosphodiester phosphodiesterases. GDE2 is one of three known GPI-anchor cleaving enzymes in vertebrates that function at the cell surface, and the only one expressed in neurons (22–26). During embryonic neurogenesis, GDE2 promotes ADAM10 activity by cleaving RECK at the GPI-anchor and removing RECK from neuronal surfaces (26). RECK inactivation leads to ADAM10 shedding of the Notch ligand Delta-like 1 (Dll1), downregulation of Notch signaling in adjacent progenitors and induction of neuronal differentiation (Figure S1A) (26). Thus, GDE2 functions as an activator of ADAM10 cleavage function by regulating RECK expression on the plasma membrane. Whether GDE2 regulates ADAM10 surface activity in the adult is not known. Here, we examine if the GDE2-RECK pathway is an important determinant of ADAM10 α-secretase cleavage of APP and physiologic APP processing in the adult brain.
Results:
GDE2 distribution is disrupted in brain from patients with AD
Unlike most developmental pathways, GDE2 and RECK are not downregulated postnatally but continue to be expressed in adult brain (Figure 1A–D, Figure S2, Figure S3). To determine if the GDE2-RECK pathway acts to regulate ADAM10 α-secretase cleavage of APP in the adult brain and could be relevant to AD (Figure S1B), we examined GDE2 expression in brain sections from patients with AD and healthy controls using two separate validated antibodies (Figure S2A–D, Table S1 (27)). Rabbit rαhGDE2 and chicken cSS1 are polyclonal antibodies that detect human (h) GDE2 expressed in HEK293FT cells, and identify a band corresponding to the expected molecular weight for hGDE2 in human U2-OS osteosarcoma cell lines (Figure S2A–B). Treatment of U2-OS cells with 3 separate siRNAs against hGDE2 mRNA ablated hGDE2 expression, whereas incubation with a control siRNA of unrelated sequence did not (Figure S2B). In addition, Western blot of extracts prepared from frozen human pre-frontal cortex postmortem tissue detected a single band corresponding to the expected molecular weight of hGDE2 (Figure S2C). These experiments verify that both rαhGDE2 and cSS1 antibodies specifically detect hGDE2 protein. In inferior parietal cortex (Brodmann Area (BA) 40) of control individuals, hGDE2 localizes to the neuropil and occasionally, at low amounts within the cell soma (Figure 1A–C, Figure S3, Table S1). In BA40 cortex of late-stage Braak V/VI patients with AD (28), hGDE2 shows distinctive intracellular accumulations within the soma that are external to the nucleus (Figure 1A–C, Figure S3, Table S1). Further verifying that hGDE2 accumulates in AD, no signal is detected when primary antibodies are omitted (Figure S2D). Co-staining with antibodies for NeuN confirms hGDE2 accumulates in neurons (Figure 1B). Quantitation of neurons with low (GDE2low) or high (GDE2high) somatic hGDE2 accumulation revealed that GDE2low neurons were markedly increased in patients with AD compared with controls, and GDE2high neurons were specific to patients with AD (Figure 1C, Datafile S1, see Methods for criteria for assignment of GDE2low and GDE2high). In patients with Braak III/IV AD where degeneration of neocortex is minimal, GDE2high neurons are evident and neurons with more intense hGDE2 somatic immunoreactivity are detected (Figure 1D, Figure S4, Table S1, Datafile S1). Thus, hGDE2 neuronal accumulation precedes profound cortical degeneration in AD and appears to increase with disease severity (Figure S4, Datafile S1). Patients with Huntington’s Disease (HD) exhibit cortical and striatal neurodegeneration and loss. hGDE2 does not accumulate in neurons of BA40 cortex from age and gender-matched patients with HD and hGDE2 expression appears equivalent to controls, suggesting that GDE2 accumulation is not broadly observed across neurodegenerative diseases and shows some specificity to AD (Figure 1D, Figure S4, Table S1, Datafile S1). Because GDE2 function requires expression on the cell surface (26, 29), these observations suggest that GDE2 trafficking and function are disrupted in neurons of patients with AD.
Fig. 1. GDE2 distribution is impaired in AD.
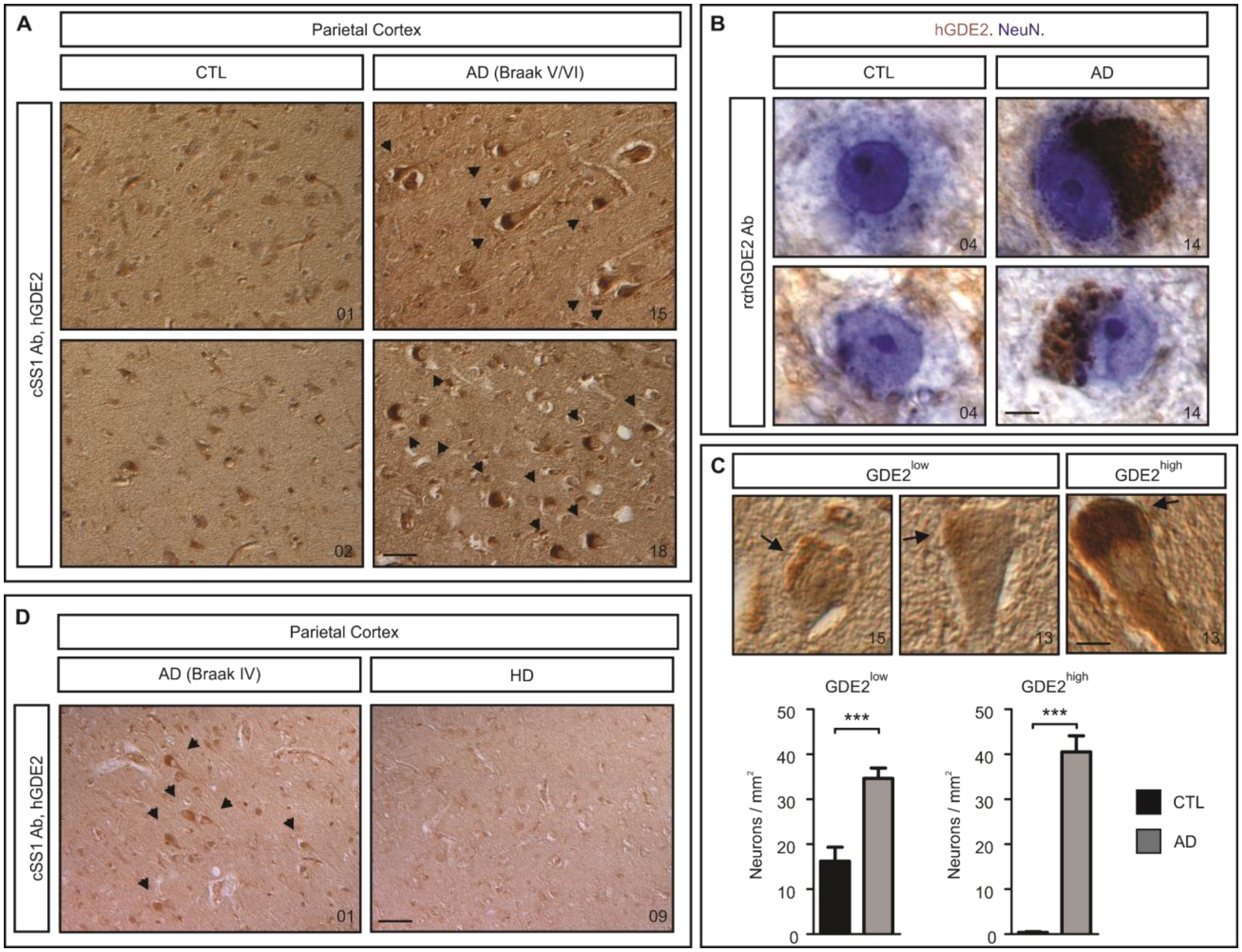
A-D, Immunohistochemical staining of human parietal (BA40) cortex sections of control individuals (CTL), and patients with AD and HD. Arrows highlight cells with GDE2 accumulations. B, NeuN (blue) marks neurons, GDE2 is labeled in brown. C, Representative images of CTL and AD neurons. Arrows highlight neurons with low and high GDE2 accumulation (GDE2low, GDE2high). Quantification of the number of GDE2low (***p=0.0002) and GDE2high (***p=1.49E-09) neurons in CTL (n=10) and AD (n=10). Total number of neurons counted: CTL = GDE2low (262), GDE2high (6) and AD = GDE2low (670), GDE2high (796). Mean ± SEM, two-tailed unpaired Student’s t-test. D, Arrows mark GDE2 accumulations. Autopsy numbers are at the bottom right of each panel. Ab = antibody. Scale bars: A, D, 3μm; B, 5μm; C, 10μm.
RECK membrane amounts are elevated in the brain of patients with AD
GDE2 releases RECK from the cell surface (Figure S5A) and ablation of GDE2 in mice (Gde2−/−) results in increased amounts of membrane-associated RECK and reduced amounts of released RECK, with no change in total RECK expression (Figure S5B, Datafile S1). To determine if RECK distribution is compromised in AD, we fractionated fresh-frozen BA40 cortex from patients with Braak V/VI-stage AD (28) and control individuals (Table S1) with Triton X-114 detergent. Membrane-tethered GPI-anchored proteins are enriched in detergent-rich fractions (DT-R), whereas released GPI-anchored proteins partition to detergent-poor fractions (DT-P) (26). Quantification of the amount of DT-R RECK normalized to total RECK expression or to the membrane protein Na+/K+ ATPase by Western blot shows a robust increase in membrane-tethered RECK in brain extracts of patients with AD compared with controls (Figure 2A, Datafile S1). Conversely, analysis of DT-P RECK normalized to total RECK or actin showed an approximately 50% reduction of released RECK in brain extracts of patients with AD (Figure 2A, Datafile S1). No change in total amounts of RECK was detected between patient groups (Figure 2A, Datafile S1). Similar observations were found for BA9 prefrontal cortex comparing patients with Braak V/VI-stage AD and controls (Figure 2B, Datafile S1) and for Braak III/IV prefrontal cortex, a stage when minimal neurodegeneration is present (Figure 2C, Datafile S1). No changes in membrane-tethered RECK, released RECK or total RECK were detected in BA40 brain extracts from patients with HD (Figure 2D, Datafile S1). Further, membrane-bound, released forms and total amounts of Contactin-1 (CNTN1), a GPI-anchored protein that is not a substrate of GDE2, were not altered between patients with AD, HD and control individuals (Figure S6A–D, Datafile S1), arguing against a general disruption of GPI-anchored protein biogenesis and trafficking in AD. Thus, the amount of membrane-tethered RECK is increased in AD coincident with GDE2 intracellular accumulation, reinforcing the notion that GDE2 activity and RECK release is disrupted in AD.
Fig. 2. Membrane RECK is increased in brain of patients with AD.
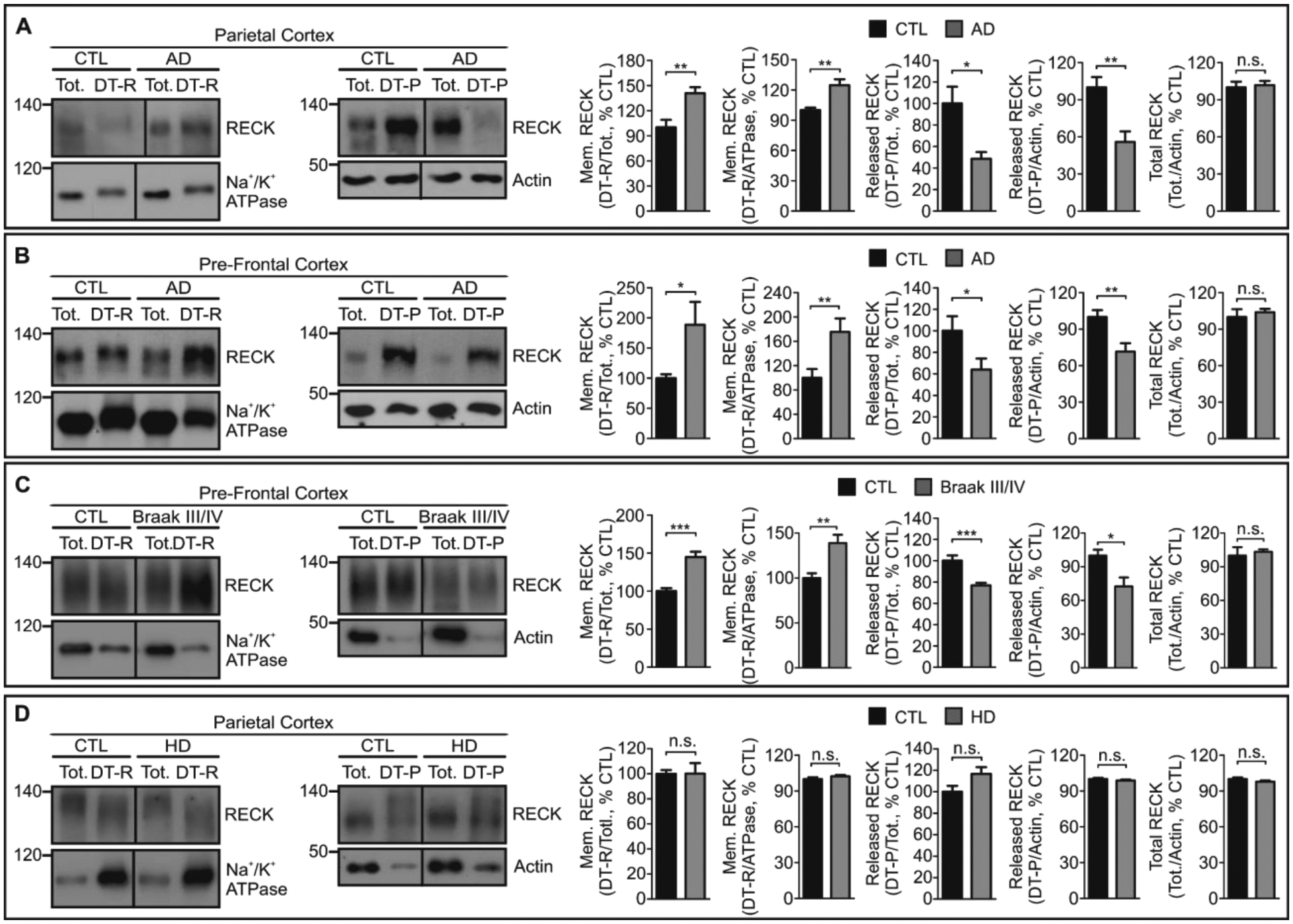
A-D, Representative Western blots of Triton X-114 fractionated parietal (BA40) and pre-frontal (BA9) cortical extracts prepared from postmortem brain from CTL individuals, and patients with AD and HD. Detergent-rich fractions (DT-R), detergent-poor fractions (DT-P), total unfractionated extract (Tot). Na+/K+ ATPase and Actin are loading controls and confirm separation of membrane and non-membrane fractions (compare between individual total fractions; between individual DT-R fractions; between individual DT-P fractions). A, Braak V/VI parietal cortex: Mem. RECK/total RECK **p=0.0088, Mem. RECK/Na+/K+ ATPase **p=0.0096; released RECK/total RECK *p=0.0123, released RECK/Actin **p=0.0084; total RECK/Actin n.s. p=0.7508. n=4 CTL, n=5 AD. B, Braak V/VI pre-frontal cortex: Mem. RECK/total RECK *p=0.0251, Mem. RECK/Na+/K+ ATPase **p=0.0083; released RECK/total RECK *p=0.0486, released RECK/Actin **p=0.0039; total RECK/Actin n.s. p=0.5837. n=13 CTL, n=12 AD. C, Braak III/IV AD pre-frontal cortex: Mem. RECK/total RECK ***p=2.55E-05, Mem. RECK/Na+/K+ ATPase **p=0.0017; released RECK/total RECK ***p=0.0005, released RECK/Actin *p=0.0102; total RECK/Actin n.s. p=0.6641. n=10 CTL, n=10 AD. D, HD parietal cortex: Mem. RECK/total RECK n.s. p=0.9840, Mem. RECK/Na+/K+ ATPase n.s. p=0.2460; released RECK/total RECK n.s. p=0.0597, released RECK/Actin n.s. p=0.3916; total RECK/Actin n.s. p=0.2244. n=10 CTL, n=10 HD. All graphs: Mean ± SEM, two-tailed unpaired Student’s t-test.
GDE2 ablation disrupts α-secretase APP cleavage and promotes β-secretase APP processing in mice
Gde2−/− mice phenocopy GDE2-RECK abnormalities in the brain of patients with AD in that they lack GDE2 activity and exhibit increased amounts of membrane-tethered surface RECK (Figure S5B–C, Datafile S1). Accordingly, Gde2−/− animals are a useful tool to examine how these cellular changes affect α-secretase function and APP processing. We focused our studies in aged mice because age is known to be a major risk factor in AD. Western blot of protein extracts prepared from aged 19-month cortices show a 16% reduction in sAPPα and a concomitant 121% rise in sAPPβ in Gde2−/− mice compared with wildtype (WT), resulting in a 64% reduction in sAPPα:sAPPβ ratios (Figure 3A, Datafile S1). Thus, loss of GDE2 results in reduced α-secretase cleavage of APP and a marked shift towards β-secretase APP processing. To examine the requirement for GDE2 in APP processing in neurons, we cultured dissociated cortical neurons from WT and Gde2−/− postnatal pups for 14 days in vitro (DIV). Western blots of extracts prepared from cultured Gde2−/− cortical neurons showed a dramatic 63% reduction in sAPPα, a 96% increase in sAPPβ, and a resulting decrease in sAPPα:sAPPβ ratios (Figure 3B, Datafile S1). Total amounts of ADAM10, BACE and APP were equivalent between WT and Gde2−/− cortical and neuronal lysates, indicating that changes in APP processing are not due to altered expression of ADAM10, BACE or APP (Figure 3A–B, Figure S7A–B, Datafile S1). These observations suggest that GDE2 functions in neurons to promote α-secretase activity and to maintain the physiological balance of APP processing. Because ADAM10 is the physiological α-secretase and principal generator of endogenous sAPPα in neurons (3, 4, 30), these observations support the model that GDE2 is required to control neuronal ADAM10 α-secretase activity.
Fig. 3. GDE2 ablation reduces α-secretase and increases β-secretase APP cleavage.
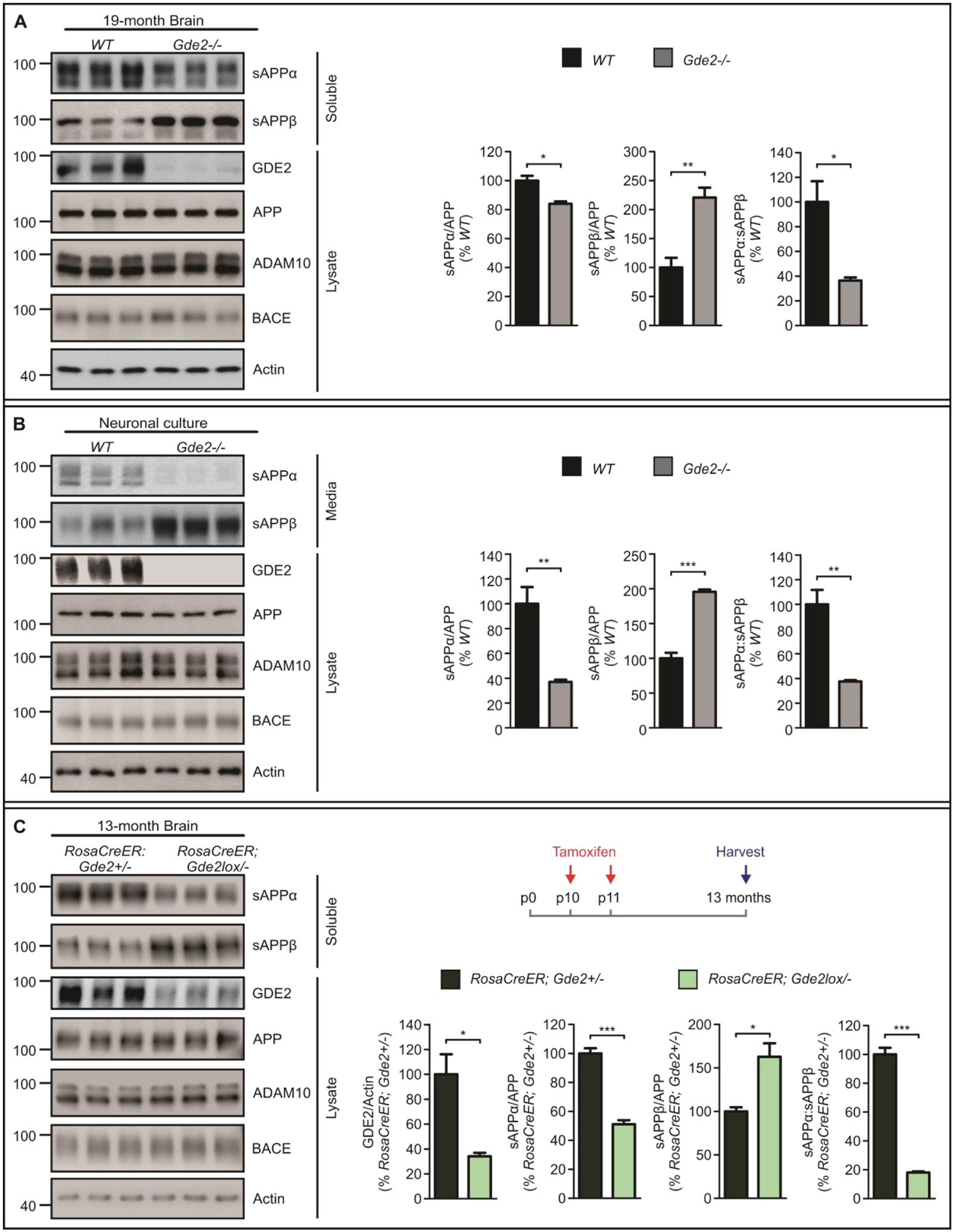
A, Western blots and graphs quantifying sAPPα (sAPPα/APP *p=0.0122), sAPPβ (sAPPβ/APP **p=0.0071) and sAPPα:sAPPβ (*p=0.0201) in 19-month Gde2−/− cortices compared with WT. (n=3). B, Western blots and graphs quantifying sAPPα (sAPPα/APP **p=0.0097), sAPPβ (sAPPβ/APP ***p=0.0004) and sAPPα:sAPPβ (**p=0.0060) in DIV14 Gde2−/− cortical neurons compared with WT. (n=3). C, Western blots and graphs quantifying GDE2 expression (GDE2/Actin *p=0.0158), sAPPα (sAPPα/APP ***p=0.0004), sAPPβ (sAPPβ/APP *p=0.0176), and sAPPα:sAPPβ ratio (***p=6.69E-05) in 13-month RosaCreER;Gde2lox/− cortical lysates (n=3). Tamoxifen was injected on postnatal (p) days 10 and 11 in RosaCreER;Gde2+/− and RosaCreER;Gde2lox/− mice, and animals were harvested at 13-months of age. All graphs: Mean ± SEM, two-tailed unpaired Student’s t-test.
GDE2 is expressed embryonically and maintained throughout life. We ablated postnatal GDE2 expression by treating RosaCreER;Gde2lox/− and RosaCreER;Gde2+/− mice with tamoxifen at postnatal day 10 (P10) and P11. This manipulation specifically disrupts GDE2 postnatal activity and retains GDE2 function during embryogenesis. Analysis at 13 months showed that GDE2 expression was reduced by 75% in tamoxifen-treated RosaCreER;Gde2lox/− animals compared with tamoxifen-treated RosaCreER;Gde2+/− controls (Figure 3C, Datafile S1). sAPPα was reduced by 50% in treated RosaCreER;Gde2lox/− mice, with a 60% increase in sAPPβ, and a corresponding 80% reduction in sAPPα:sAPPβ ratios (Figure 3C, Datafile S1). ADAM10, BACE and APP expression were equivalent between genotypes (Figure 3C, Figure S7C, Datafile S1). These observations indicate that GDE2 control of ADAM10 α-secretase activity and APP processing is separate from its developmental functions in embryogenesis.
Taken together, this set of experiments suggests that GDE2 is required for neuronal ADAM10 α-secretase activity, and that loss of GDE2 results in a switch from non-amyloidogenic APP processing to increased β-secretase cleavage of APP in mice.
GDE2 disruption accelerates Aβ pathologies in mice
The increase in sAPPβ in Gde2−/− brain (Figure 3A–C) suggested that γ-secretase APP cleavage products may be elevated in absence of GDE2. To examine this possibility, we examined Aβ40 and Aβ42 amounts in Gde2−/− animals. Enzyme-linked immunosorbent assays (ELISA) showed a marked 20% increase in Aβ42, a modest 4% increase in Aβ40 and reduced Aβ40/42 ratios in 19-month Gde2−/− cortical extracts compared to WT (Figure 4A, Datafile S1). These changes in Aβ40 and Aβ42 were recapitulated in primary cortical neuronal cultures prepared from WT and Gde2−/− cortices (Figure 4A, Datafile S1). Thus, GDE2 loss leads to an increase in Aβ peptides, in particular, the longer Aβ42 peptide associated with pathogenic Aβ species (31).
Fig. 4. GDE2 ablation promotes amyloidogenic APP processing.
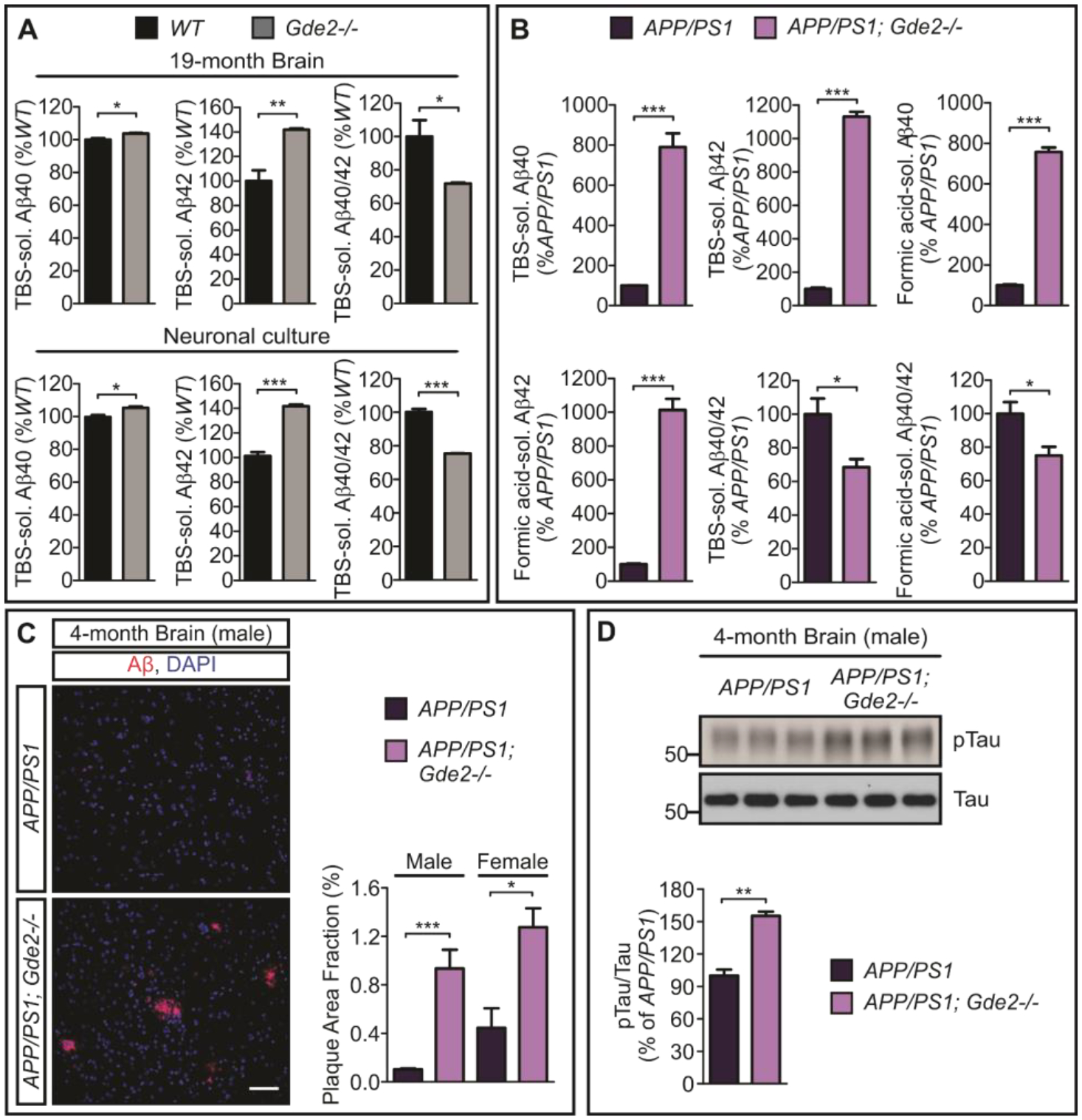
A, ELISA quantifications of TBS-soluble Aβ40 (*p=0.0252), Aβ42 (**p=0.0088) and Aβ40/42 ratios (*p=0.0461) in Gde2−/− 19-month cortical extracts (n=3); and TBS-soluble Aβ40 (*p=0.0206), Aβ42 (***p=0.0003) and Aβ40/42 ratios (***p=0.0002) in Gde2−/− DIV14 cortical neuronal cultures (n=3). B, Quantification of ELISA in APP/PS1 and APP/PS1 Gde2−/− 4-month cortical extracts for soluble Aβ peptides (TBS-soluble Aβ40 ***p=0.0006; Aβ42 ***p=4.62E-06, n=3) and Aβ aggregates (Formic acid-soluble Aβ40 ***p=7.0E-06; Aβ42 ***p=0.0001, n=3), with associated Aβ40/42 ratios (TBS-soluble Aβ40/42 ratios *p=0.0399; Formic acid-soluble Aβ40/42 ratios *p=0.0444). C, Immunostaining of 4-month APP/PS1 and APP/PS1;Gde2−/− cortical sections with corresponding quantification of amyloid load [area fraction of 6E10-positive plaque (red)], male (***p=0.0007) and female (*p=0.0208). D, Western blots and quantification of phosphorylated tau (Ser202 and Thr205 pTau/Tau, **p=0.00127) in 4-month male APP/PS1 and APP/PS1;Gde2−/− cortical extracts (n=3). All graphs: Mean ± SEM, two-tailed unpaired Student’s t-test.
APPswe;Psen1de9 (APP/PS1) mice represent a humanized mouse model of amyloidosis that contains human APP and Presenilin (γ-secretase) harboring AD-associated mutations that promote Aβ production (32). These animals accumulate Aβ peptides and develop plaque deposits by approximately 6 months of age (32). To examine the consequences of GDE2 deletion on Aβ and plaque generation, we generated APP/PS1 mice with disrupted GDE2 function (APP/PS1;Gde2−/−). Aβ peptides and Aβ aggregates can be solubilized in Tris buffer solution (TBS) and Formic acid respectively. APP/PS1;Gde2−/− animals showed a robust 8–10 fold increase in the TBS-soluble and Formic acid-soluble forms of Aβ40 and Aβ42, resulting in a marked reduction of Aβ40/42 ratios (Figure 4B, Datafile S1). In line with these changes, we observed the appearance of accelerated β-amyloid plaque deposition in APP/PS1;Gde2−/− male and female mice at 4 months of age, 2 months earlier than when Aβ pathologies are normally observed in APP/PS1 animals (32) (Figure 4C, Datafile S1). In addition, Western blot of protein extracts prepared from 4 month cortices probed with antibodies that detect phosphorylated tau at Serine 202 and Threonine 205, showed increased tau phosphorylation in APP/PS1;Gde2−/− mice compared to APP/PS1 littermates, indicating changes in tau phosphorylation dynamics in absence of GDE2 (Figure 4D, Datafile S1) (33). Taken together, these observations suggest that the loss of GDE2 leads to the increased production of pathological Aβ isoforms and that the GDE2 pathway is an important physiological “brake” on Aβ pathogenesis.
Loss of GDE2 leads to reductions in synaptic proteins and activity
Increased amyloidogenic processing of APP is associated with synaptic protein loss and impaired synaptic function (34–37). To determine if Gde2−/− animals exhibit changes in synaptic proteins, we prepared cortical extracts from 19-month Gde2−/− animals and examined presynaptic and postsynaptic protein expression by Western blot. Gde2−/− animals show robust decrease in presynaptic synaptophysin, and postsynaptic PSD95 and Homer proteins compared with controls (Figure 5A, Datafile S1). Similar reductions of pre- and postsynaptic proteins are observed in cultured Gde2−/− cortical neurons and in tamoxifen-treated RosaCreER;Gde2lox/− mice analyzed at 13 months (Figure 5B–C, Datafile S1). Treatment of cultured Gde2−/− cortical neurons with the BACE inhibitor C3 reduced Aβ40 and Aβ42 in Gde2−/− neurons by 40% and 60% respectively and increased Aβ40/42 peptides by 56% (Figure S8A, Datafile S1). However, amounts of pre- and postsynaptic proteins in Gde2−/− neurons were not restored by this amount of Aβ peptide reduction (Figure S8B, Datafile S1), suggesting the possibility that GDE2 disruption could also mediate synaptic protein loss through Aβ-independent mechanisms. We next determined if the reduction in synaptic proteins in Gde2−/− animals corresponded to a loss of synapses and synaptic activity. We visualized synapses in sections of 19-month cortices by coexpression of synaptophysin and PSD95. Gde2−/− mice showed an approximately 20% reduction in the number of puncta that expressed synaptophysin and PSD95 compared with WT (Figure 5D, Datafile S1). Consistent with the observed decrease in synapse numbers, optical recording of intracellular calcium showed reduced spike frequency in cultured Gde2−/− cortical neurons compared with WT controls (Figure S9, Datafile S1). Taken together, these observations suggest that the loss of GDE2 results in the reduction of synaptic proteins, synapse number and neuronal activity.
Fig. 5. Loss of GDE2 results in synaptic protein reduction.
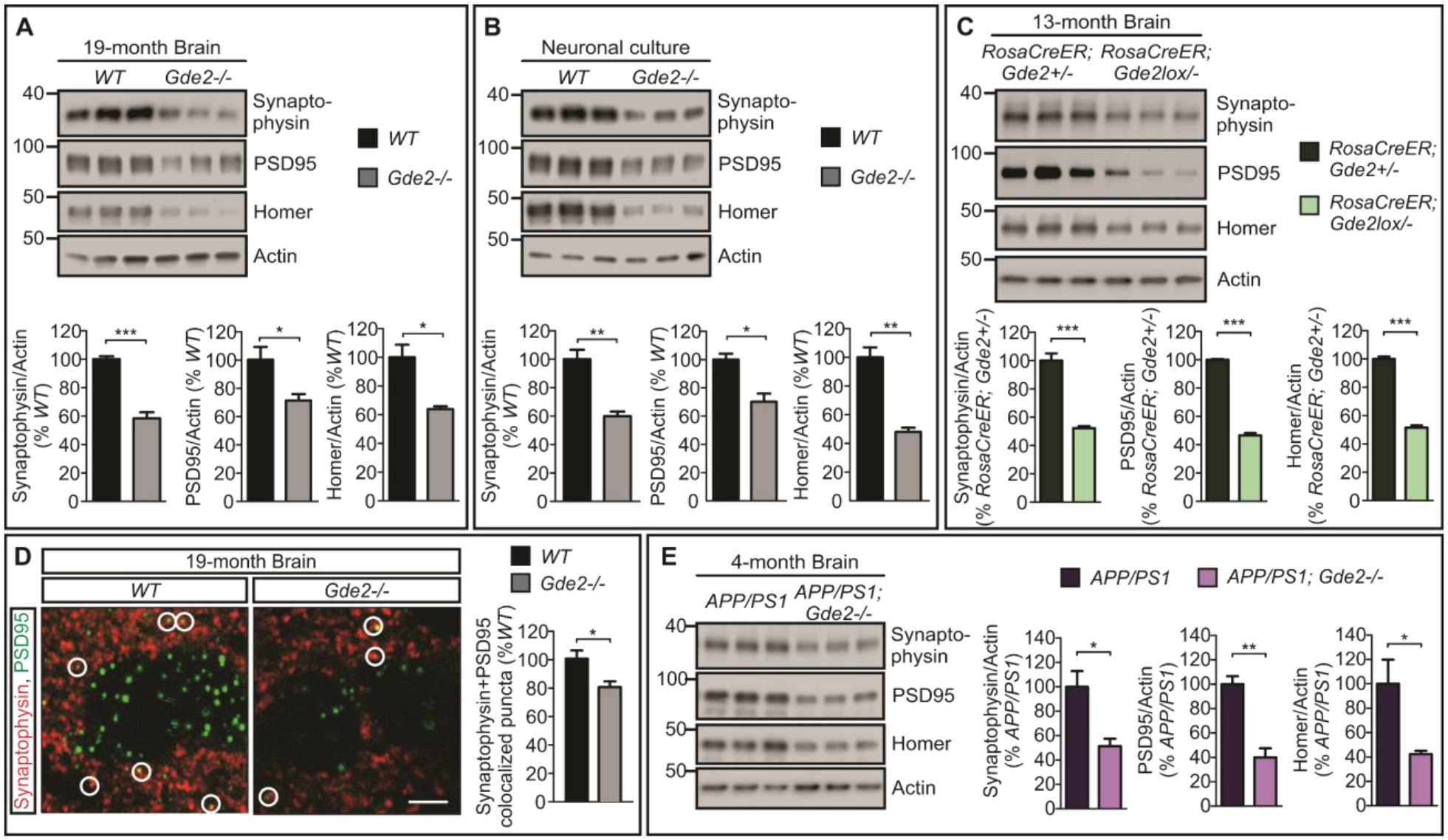
A, Western blots and quantification of Synaptophysin/Actin (***p=0.0009), PSD95/Actin (*p=0.0455) and Homer/Actin (*p=0.0160), (n=3) in 19-month WT and Gde2−/− cortical extracts. B, Western blots and quantification of Synaptophysin/Actin (**p=0.0056), PSD95/Actin (*p=0.0135) and Homer/Actin (**p=0.0023), (n=3) in extracts of DIV14 WT and Gde2−/− cortical neuronal cultures. C, Western blots and quantification of Synaptophysin/Actin (***p=0.0008), PSD95/Actin (***p=6.68E-06), and Homer/Actin (***p=3.24E-05) in cortical extracts from13month tamoxifen-treated RosaCreER;Gde2+/− and RosaCreER;Gde2lox/− animals (n=3). D, Representative images of sections from 19-month WT and Gde2−/− mouse cortex. Circles mark synapses visualized by colocalization of Synaptophysin and PSD95. Graph quantifying synapse numbers (*p=0.0475, n=3). E, Western blots and quantification of Synaptophysin/Actin (*p=0.0272), PSD95/Actin (**p=0.0038) and Homer/Actin (*p=0.0444) (n=3) in cortical extracts from brains of 4-month old APP/PS1 and APP/PS1;Gde2−/− male mice. Actin is used as a loading control. All graphs: Mean ± SEM, two-tailed unpaired Student’s t-test.
The disruption of GDE2 function increases the production of toxic Aβ peptides and accelerates plaque deposition in APP/PS1 mouse models of amyloidosis (Figure 4A–C). Analysis of synaptic proteins by Western blot of cortical extracts shows that APP/PS1;Gde2−/− mice at 4 months exhibit a marked decrease of synaptophysin, PSD95 and Homer compared with age- and gender-matched APP/PS1 animals (Figure 5E, Datafile S1), a time when synaptic changes are not normally observed in this AD mouse model. Thus, the disruption of GDE2 function in the APP/PS1 AD mouse model results in the accelerated loss of synaptic proteins in conjunction with more rapid increase in Aβ peptides and plaque generation.
Elevated RECK expression inhibits ADAM10 α-secretase activity and promotes amyloidogenic APP processing
The amount of membrane-tethered RECK is elevated when GDE2 function is disrupted in AD and in Gde2−/− mice (Figure 2A–C, Figure S5B). Surface biotinylation studies in cultured cortical neurons verify that there is a robust increase of plasma membrane RECK in absence of GDE2 (Figure S5C, Datafile S1). To examine the consequences of increased RECK expression on endogenous ADAM10 α-secretase activity and APP processing, we generated a version of RECK that replaces the GPI-anchor with the transmembrane domain of CD2 (Cluster of differentiation 2, CD2RECK; Figure S10A) (26). CD2RECK is expressed at the correct molecular weight, traffics to neuronal plasma membrane and fails to be released by GDE2 (Figure 6A, Figure S10B–C). Neuronal cultures expressing CD2RECK show a robust reduction of sAPPα, marked increase in sAPPβ, and consequent decrease in sAPPα:sAPPβ ratios (Figure 6A, Datafile S1). No change in APP, ADAM10 and BACE amounts was detected between GFP and CD2RECK conditions (Figure 6A, Figure S10D, Datafile S1). These observations suggest that increased surface RECK expression in neurons is sufficient to inhibit ADAM10 α-secretase function and promote β-secretase cleavage of APP. To test the ability of RECK to directly inhibit ADAM10-dependent APP processing, we reconstituted this pathway in heterologous cells. Co-transfection of APP and ADAM10 in COS-7 cells resulted in robust production of sAPPα, and this increase was completely abrogated in the presence of RECK (Figure S11, Datafile S1). This is consistent with known roles for RECK as a potent inhibitor of metalloproteases that include ADAM10 (19–21, 26).
Fig. 6. Surface RECK inhibits ADAM10 α-secretase activity and causes reduction in synaptic proteins.
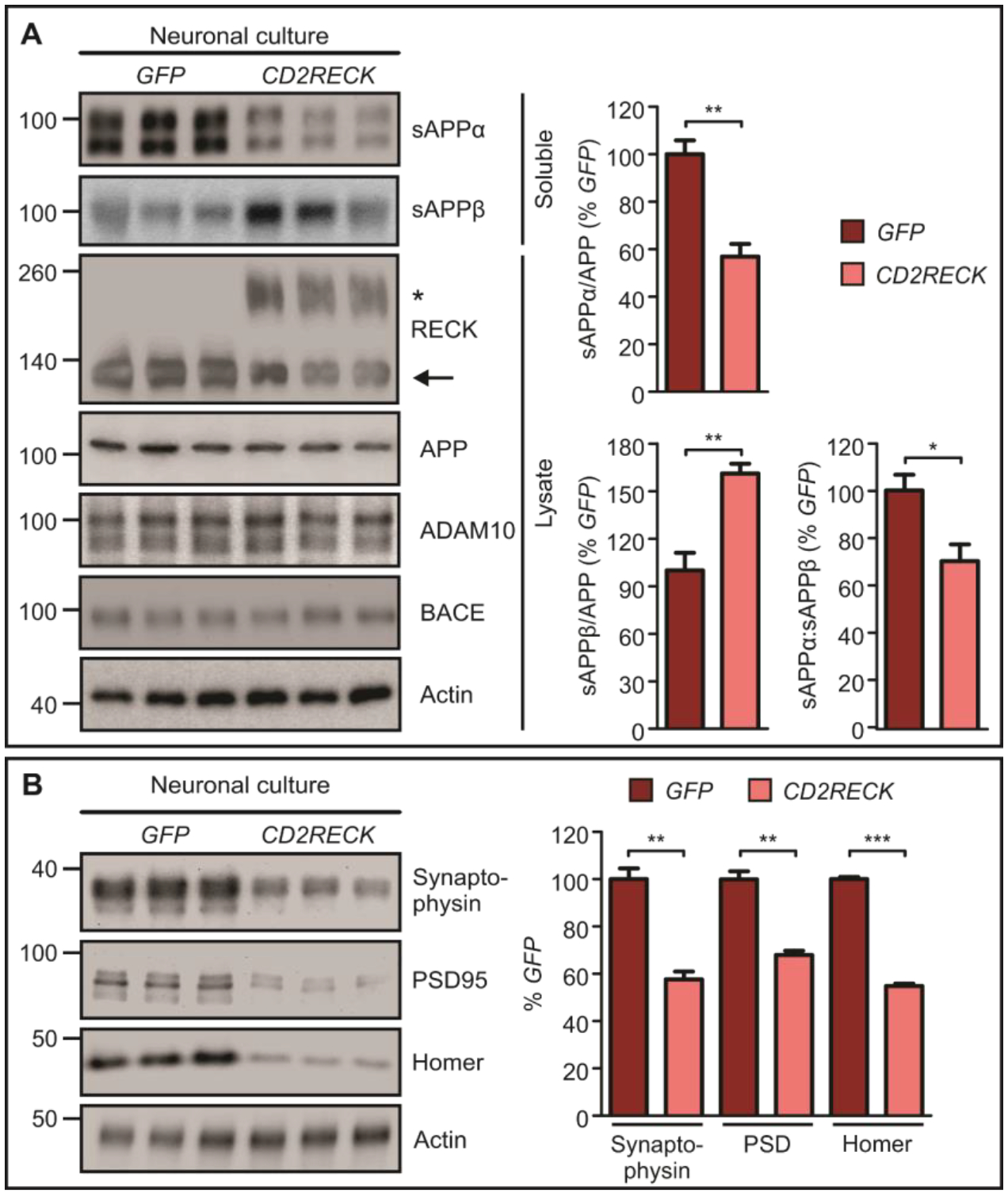
A, Western blots and quantification of sAPPα (**p=0.0056), sAPPβ (**p=0.0084), and sAPPα:sAPPβ (*p=0.0372) in GFP- and CD2RECK-transduced DIV14 cortical neuronal cultures (n=3). Asterisk marks CD2RECK expression, arrow marks endogenous RECK. B, Western blots and quantification of Synaptophysin/Actin (**p=0.0017), PSD95/Actin (**p=0.0011), and Homer/Actin (***p=5.02E-06) (n=3) in GFP- and CD2RECK-transduced DIV14 cortical neuronal cultures. Actin is used as a loading control. All graphs: Mean ± SEM, two-tailed unpaired Student’s t-test.
We next examined if CD2RECK expression was also sufficient to reduce the amount of pre- and postsynaptic proteins. Western blot analysis of primary cortical neurons infected with control or CD2RECK lentivirus showed that CD2RECK expression resulted in an approximately 40% reduction in synaptophysin, PSD95 and Homer (Figure 6B, Datafile S1). Thus, elevated expression of membrane-tethered surface RECK in neurons is sufficient to switch APP processing from the non-amyloidogenic to the amyloidogenic pathway and to induce synaptic protein loss. These studies are consistent with the model that the increase in membrane-tethered RECK in AD contributes to disease pathophysiology (Figure 2A–C).
RECK mediates GDE2-dependent regulation of APP processing
We hypothesize that GDE2 regulates RECK surface expression to promote ADAM10 α-secretase cleavage of APP and that increased RECK surface expression mediates the amyloidogenic shift in APP processing when GDE2 is disrupted. To test this hypothesis, we took advantage of our ability to reconstitute RECK regulation of ADAM10 α-secretase activity in heterologous cells. We cotransfected GDE2 with RECK, ADAM10 and APP into COS-7 cells and examined APP processing by Western blot. GDE2 expression in the presence of RECK was sufficient to restore sAPPα to the same amount generated by coexpression of APP and ADAM10 alone (Figure S11, Datafile S1). This observation supports our model that GDE2 functions at the cell surface in neurons to stimulate ADAM10 α-secretase activity by inactivating RECK (Figure S1B).
If RECK mediates GDE2-dependent effects of APP processing in vivo, then reducing RECK expression in the context of GDE2 disruption should restore the physiological balance of APP processing (Figure S12A). Reck−/− animals are embryonic lethal (20); accordingly, to test this hypothesis, we generated Gde2−/− mice that are heterozygous for Reck (Gde2−/−;Reck+/−). We confirmed by Western blot that 19-month Gde2−/−;Reck+/− animals express 62% lower amounts of RECK than Gde2−/− littermates (Figure 7A, Datafile S1). Analysis of APP processing reveals that Gde2−/−;Reck+/− mice show a marked 58% increase in sAPPα production compared with Gde2−/− siblings, consistent with a recovery of α-secretase activity (Figure 7A, Datafile S1). sAPPβ amounts are reduced by 30% and sAPPα:sAPPβ ratios are increased by 72%, thereby restoring the balance of APP processing to WT (Figure 7A, Datafile S1). In addition, pre- and post-synaptic protein amounts are markedly increased in Gde2−/−;Reck+/− mice compared with Gde2−/− animals (Figure 7B, Datafile S1). Total amounts of ADAM10, BACE and APP are unchanged between genotypes (Figure 7A, Figure S12B, Datafile S1). To determine if reducing RECK expression is sufficient to lower Aβ peptides in context of GDE2 disruption, we utilized siRNAs against Reck to knockdown RECK protein in cultured cortical neurons (Figure S12C). Compared to control unrelated siRNAs, addition of siRNA targeting Reck resulted in an approximately 75% reduction of RECK protein expression by Western blot (Figure S12D, Datafile S1). Analysis of Aβ peptides by sandwich ELISA showed that RECK knockdown resulted in a modest 10% reduction of Aβ40 peptides and a more robust 35% reduction of Aβ42 peptides in Gde2−/− neurons, leading to an increase in Aβ40/42 ratios (Figure 7C, Datafile S1). Therefore, reducing RECK expression is sufficient to increase sAPPα, restore the physiological balance of APP processing and mitigate the loss of synaptic proteins elicited by GDE2 disruption.
Fig. 7. GDE2 promotes ADAM10 α-secretase activity by regulating RECK.
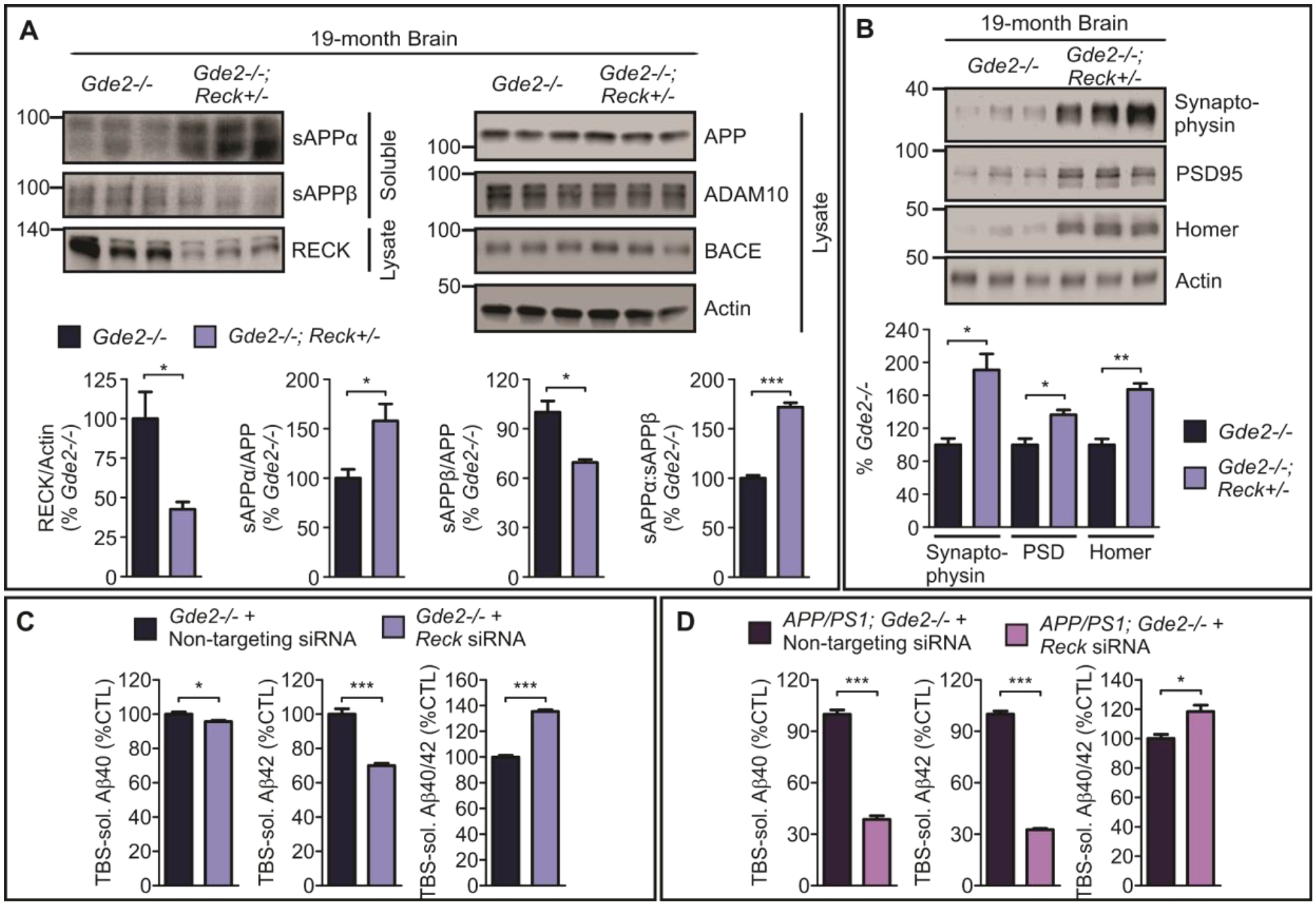
A, Western blots and quantification of RECK (*p=0.0495), sAPPα (*p=0.0403), sAPPβ (p*=0.0125), and sAPPα:sAPPβ (***p=0.0002) in extracts from 19-month Gde2−/− and Gde2−/−;Reck+/− cortices (n=3). B, Western blots and quantification of Synaptophysin/Actin (*p=0.0119), PSD95/Actin (*p=0.0184), and Homer/Actin (**p=0.0027) in extracts from 19-month Gde2−/− and Gde2−/−;Reck+/− cortices (n=3). Actin is used as a loading control. C, Quantification of ELISA for TBS-soluble Aβ40 (*p=0.0435) and Aβ42 (***p=0.0009), with associated Aβ40/42 ratio (***p=4.32E-05) (n=3) in Gde2−/− DIV14 cortical neuronal cultures treated with control or Reck siRNAs. D, ELISA analysis of APP/PS1 Gde2−/− DIV14 cortical neuronal cultures treated with control or Reck siRNAs; TBS-soluble Aβ40 ***p=4.47E-05; TBS-soluble Aβ42 ***p=4.80E-06; TBS-soluble Aβ40/42 ratios *p=0.02434, (n= 3). All graphs: Mean ± SEM, two-tailed unpaired Student’s t-test.
GDE2 loss of function in the APP/PS1 mouse model of amyloidosis results in a robust 8–10 fold increase of Aβ40 and Aβ42 indicating that the GDE2 pathway is an important negative regulator of Aβ generation (Figure 4B). To determine the consequences of RECK reduction on GDE2-dependent control of Aβ generation in APP/PS1 mice, we performed RECK knockdown experiments in cultured cortical neurons prepared from APP/PS1;Gde2−/− animals. Treatment of APP/PS1;Gde2−/− cortical neurons with Reck siRNAs resulted in an approximately 50% knockdown of RECK protein expression (Figure S12E, Datafile S1). This degree of RECK knockdown led to a robust 70% reduction in Aβ40 and Aβ42 as assayed by sandwich ELISA and an increase in Aβ40/42 ratios (Figure 7D, Datafile S1). Thus, reducing RECK expression in the context of GDE2 disruption in the APP/PS1 mouse model is an effective approach to lower amounts of Aβ40 and Aβ42 peptides and to increase Aβ40/42 ratios.
Discussion:
Our studies revealed that GDE2 regulation of RECK surface expression is an essential determinant of ADAM10 α-secretase activity in the adult brain. GDE2 disruption increased RECK surface expression, which is causal for the inhibition of ADAM10 α-secretase function, a dramatic shift to sAPPβ generation, and increased production of pathogenic Aβ peptides. These changes are accompanied by a reduction in synaptic proteins, synapse numbers and associated decreases in neuronal activity. Ablating GDE2 in the APP/PS1 model of amyloidosis accelerated the production of Aβ peptides, plaque deposition and reduced the expression of synaptic proteins, and reduction of RECK in this model robustly diminished the production of Aβ. We found that GDE2 abnormally accumulated in intracellular compartments in the brain of patients with AD and consistent with disrupted GDE2 activity, amounts of membrane-tethered RECK were specifically elevated. These observations, viewed through the lens of our mechanistic studies, identified GDE2 as an important regulator of RECK surface expression, and propose that increased plasma membrane RECK and the inhibition of ADAM10 α-secretase function are plausible mechanisms that contribute to AD pathogenesis.
Our in vivo studies combined with functional studies in cultured primary cortical neurons suggest that GDE2 promotes ADAM10 α-secretase function by regulating the surface expression of RECK in neurons. Timed ablation of GDE2 indicates that this function is distinct from its roles in the developing nervous system and underscores the importance of the GDE2 pathway in promoting ADAM10 α-secretase function in adult and aged animals. Gde2−/− animals exhibited a reduction in sAPPα production, and this decrease was particularly evident in cultured cortical neurons, suggesting that this is the prime site for GDE2 control of ADAM10 α-secretase function. This observation is in line with surface biotinylation studies, which showed a robust increase of plasma membrane RECK in neurons lacking GDE2. Further support for our model is provided by our studies in cultured neurons and in COS-7 cells, which showed that increased plasma membrane RECK expression was sufficient to inhibit ADAM10 α-secretase activity and that GDE2 effectively suppressed RECK inhibitory activity to promote ADAM10 function. Of note, the decrease in sAPPα amounts in Gde2−/− animals was concomitant with a robust increase in sAPPβ. It is possible that RECK-dependent inhibition of ADAM10 could increase the availability of APP for BACE cleavage, resulting in elevated production of sAPPβ. In addition, previous studies have shown that sAPPα can bind BACE and inhibit BACE activity (6). Accordingly, lowered amounts of sAPPα in the absence of GDE2 could further augment BACE activity and increase sAPPβ production in this context. Other potential mechanisms that could underlie the increase in sAPPβ elicited by GDE2 disruption could include changes in the dynamics of APP and BACE endosomal trafficking, the primary site of BACE-dependent APP processing in neurons, and altered BACE proteolytic activity (38, 39). Our studies showed that BACE expression was not affected by GDE2 and RECK expression. GDE2 and RECK function on the plasma membrane and not endosomes (21, 29, 40–42), and putative activation functions of RECK on aspartyl proteases such as BACE have not been reported. Accordingly, potential effects of GDE2-RECK on BACE are likely to be indirect. Deeper insight into how sAPPβ amounts are increased when the GDE2-RECK regulatory pathway is disrupted will help clarify the mechanisms underlying Aβ generation.
The disruption of GDE2 activity led to the elevation of Aβ peptides, which is in line with the observed increase of sAPPβ and the shift of APP processing towards the amyloidogenic pathway. Interestingly, the increase in Aβ40 was modest, whereas the increase in Aβ42 peptides was more robust, and this pattern was observed in Gde2−/− cortical extracts and cultured cortical neurons. Aβ peptides are generated by γ-secretase, which sequentially cleaves the membrane-embedded C-terminal fragment (CTF-β) generated by BACE to generate Aβ peptides of different lengths (1, 2). It is widely accepted that longer Aβ peptides (≥Aβ42) are more prone to aggregation of toxic Aβ-species (43, 44). The mechanism that underlies the production of longer Aβ peptides is not well understood but recent studies implicate enzyme-substrate complex stability in this process (31). How the loss of GDE2 skewed the production of Aβ peptides towards Aβ42 is unclear and further investigation into this question could deepen understanding of how longer Aβ peptides are generated. It is notable that in APP/PS1 mice, loss of GDE2 led to a robust increase of Aβ42 and Aβ40, albeit to a lesser extent of the latter. In this context, the consequences of GDE2 ablation on the timing and the amount of Aβ40 and Aβ42 peptide production is likely influenced by the dynamics of APP processing effected by the AD-associated mutations in human APP and PSEN1 (γ-secretase) proteins expressed by APP/PS1 animals (32).
Our studies of human postmortem tissue revealed that GDE2 accumulated abnormally in intracellular compartments in neurons in the brain of patients with AD, and this accumulation paralleled increases in membrane-associated RECK. Interestingly, these cellular and biochemical changes were not observed in the brain of patients with HD, arguing against a general disruption of the GDE2/RECK pathway in degenerating neurons. Our studies thus suggest that GDE2 function is disrupted in neurons of patients with AD and raises the possibility that the dysregulation of RECK surface expression is a contributing factor to AD pathophysiology. How GDE2 distribution and activity are disrupted in AD is unclear. GDE2 trafficking and activity at the cell surface are controlled by thiol-redox states that are regulated by peroxiredoxins (PRDX), which act as sensors of cellular redox. GDE2-PRDX1 thiol reduction stimulates GDE2 activity at the cell surface (45). In contrast, PRDX4 thiol oxidation of GDE2 in the ER-trans Golgi network prevents trafficking of mature GDE2 to the plasma membrane and leads to GDE2 intracellular sequestration (46). Brains of patients with AD exhibit prominent hallmarks of oxidative damage and decreased PRDX1 acetylation and thiol-reductive activity (47, 48) that could collectively impair GDE2 trafficking and function. Further studies will shed light on the mechanism and progression of GDE2 dysfunction and their contributions to AD pathogenesis.
Our studies provide evidence that increased expression of RECK on the plasma membrane inhibits ADAM10 α-secretase cleavage of APP, stimulates the production of sAPPβ and Aβ peptides and causes a loss in synaptic proteins. Genetic and siRNA reduction of Reck expression in the context of disrupted GDE2 function was sufficient to shift APP cleavage from the pathogenic amyloidogenic pathway to the protective non-amyloidogenic pathway and mitigate synaptic protein loss. Importantly, siRNA knockdown of Reck by approximately 50% in APP/PS1; Gde2−/− neurons resulted in robust reduction of Aβ peptides and improved Aβ40/42 ratios. These observations highlight RECK as a potential therapeutic target for AD and suggest that approaches to downregulate RECK expression or activity may constitute viable treatment options to mitigate AD pathophysiologies. Antisense oligonucleotides (ASOs) have proved effective in treating diseases such as spinal muscular atrophy (SMA) in human patients (49, 50); accordingly, the development of ASOs targeting RECK could be one possible intervention to mitigate pathologies associated with amyloidogenic APP processing (2, 51). Alternatively, small molecules that target RECK transcriptional regulation could be utilized. We note that reducing RECK expression by approximately 50–60% was sufficient to restore ADAM10 α-secretase function and synaptic protein expression in Gde2−/− animals, and that this amount of RECK reduction in APP/PS1;Gde2−/− neurons effectively reduced amounts of Aβ peptides and improved Aβ40/42 ratios. It is thus possible that putative therapies may not need to completely ablate RECK expression to effectively mitigate AD associated pathologies.
Although our study identifies the GDE2/RECK pathway as a mechanism that regulates ADAM10 α-secretase activity and highlights RECK as a potential therapeutic target for AD, a limitation is that our mechanistic studies are performed primarily in mouse models. Our observations in AD postmortem tissue strongly suggests that this pathway is conserved in humans; however, in-depth functional studies in human neurons are needed to solidify links to human disease. This is particularly important given that studies in model organisms have sometimes failed to translate to the human condition. GDE2 is the only known GPI-anchor cleaving enzyme in vertebrates that functions at the cell surface in neurons to release GPI-anchored proteins into the extracellular space (26), and GPI-anchored proteins can partition to the cerebrospinal fluid (CSF) (52–54). Accordingly, determining if GPI-anchored protein release is impaired in the biofluids of patients with AD could validate the relevance of GDE2 dysfunction to AD, and additionally hold promise as diagnostic biomarkers for AD.
Materials and Methods:
Study Design
The overall objectives of this study were to determine if GDE2 regulation of RECK surface expression is involved in the regulation of ADAM10 α-secretase cleavage of APP, and to determine if manipulation of this pathway can mitigate amyloidogenic APP processing. For studies using patient postmortem tissue, immunohistochemical analysis was performed on ten controls and ten patients for each condition (control, AD Braak V/VI, AD Braak III/IV, HD); regions of interest (ROIs) for each section were randomly selected for analysis and quantification. For biochemical analyses, the number of patient samples differed according to group as noted. Extensive validation of antibodies against human GDE2 was performed to ensure specificity, and two different antibodies were used to ensure reproducibility. Functional studies were performed using a combination of mouse genetic models, primary neuronal cultures and heterologous cells. Final numbers of samples analyzed for each experiment were determined by power analysis. Neuronal and cell culture experiments were typically performed with three biological replicates and three technical replicates per condition. All experiments were blinded to investigators where possible. All data points were included in the analyses and no outliers were excluded.
Mouse lines
Mice were bred and maintained in accordance with approved Johns Hopkins University Institutional Animal Care and Use Committee protocols. Gde2−/− mice, APP/PS1 mice, Reck+/− mice and ROSACreER;Gde2lox/− mice were generated, genotyped, and maintained as previously described (21, 32, 55). Tamoxifen (Sigma Aldrich) was emulsified in sunflower oil (Sigma Aldrich) at a concentration of 10 mg/ml and delivered to mice as a single intraperitoneal injection (100mg/kg) at post-natal days 10 and 11.
Tissue preparation
One hemisphere of the dissected mouse cortex was snap-frozen on dry ice and stored at −80°C for biochemistry. The other hemisphere was immersion-fixed in 4% paraformaldehyde (PFA, MP Biomedicals) in 0.1 M phosphate buffer (PB) overnight at 4°C for histological analyses. Tissues were post-fixed for 24 hours and prepared for paraffin embedding as previously described (55).
Cortical neuronal culture
1.0 × 106 dissociated cells from post-natal day 0 or 1 mouse cortices were plated on poly-L-lysine (PLL, Sigma Aldrich)-coated wells of a 6-well plate and cultured in Neurobasal medium (Thermo Fisher Scientific) supplemented with 2% B27 (Invitrogen), 5% horse serum (Life Technologies), and 2% GlutaMAX (Gibco). Cytosine arabinoside (AraC, Sigma Aldrich) was added on DIV3 to inhibit glial proliferation. From DIV4, cultures were fed every 3 days with glial-conditioned Neurobasal medium supplemented with 2% B27, 1% horse serum, and 2% GlutaMAX. Cultures were maintained at 37°C until harvest at DIV14. For viral experiments, neuronal cultures were infected with lentivirus the day of plating. For inhibitor experiments, neuronal cultures were treated with 2μM BACE inhibitor C3 on DIV19 and collected at DIV21. For knockdown experiments, siRNAs were added at DIV9. See supplementary materials for details of siRNA sequences.
Human specimens
Parietal cortex paraffin sections and fresh-frozen pre-frontal and parietal cortical samples of patients with AD and non-demented controls were from the Johns Hopkins Brain Resource Center. Samples from patients with HD were obtained from the Huntington’s Disease Center (NS 16375). For all human samples used in this study, procedures were performed under protocols approved by the Institutional Review Board at Johns Hopkins University.
Lysate preparation
Triton X-114 fractionation: Fresh-frozen mouse brain and human cortex samples were lysed and fractionated into total, DT-R, and DT-P fractions as previously described (26). The same amount of protein was loaded for each separate treatment. Human sample cohorts were batch processed at the same time to minimize variability. All extracts prepared from human tissue were surveyed by Western blot to ensure minimal protein degradation.
Surface biotinylation: Membrane proteins of cultured neurons were extracted via surface biotinylation, as previously described (26). Culture lysates were sonicated and centrifuged, and an aliquot of the supernatant was saved as the input fraction. The rest was incubated with NeutrAvidin Agarose resin (Thermo Fisher Scientific) overnight at 4°C, the beads were washed with RIPA buffer and the proteins eluted.
GDE2 Antibody Generation
Rabbit rαhGDE2 polyclonal antibody was described previously (27). The chicken cSS1 polyclonal antibody (Covance) was generated by immunizing chickens using standard protocols with immunizing peptide (GGSHTKTLIERSGR) conjugated to keyhole limpet hemocynin (KLH), and affinity purified. Rabbit anti-mouse GDE2 was previously described (24, 55).
Western Blotting
Proteins were separated under denaturing conditions using SDS-PAGE, using 7.5% or 10% Criterion TGX Precast Gels (Bio-Rad Laboratories) in Tris/Glycine buffer, wet-transferred onto PVDF membranes and blocked with 5% non-fat dry milk for 1 hour at room temperature (RT). Blocked membranes were incubated with primary antibodies (see supplementary materials) diluted in TBST overnight at 4°C. HRP-conjugated secondary antibodies (Jackson ImmunoResearch) were diluted in 5% non-fat dry milk, and membranes were incubated for 1 hour at RT. Membranes were exposed to X-ray film or developed using the KwikQuant Imager (Kindle Biosciences) following incubation with enhanced chemiluminescent (ECL) substrate (Thermo Fisher Scientific, Kindle Biosciences). Densitometry measurements of imaged blots were made with ImageJ software (National Institutes of Health).
Immunostaining
Fluorescent staining of mouse tissue sections: Mouse brain paraffin sections were cut coronally, deparaffinized in xylene, and rehydrated in an ethanol series as previously described (55). Sections were permeabilized in PBS + 0.3% Triton X-100 (PBST), followed by microwave antigen retrieval in 10 mM sodium citrate, pH 6.0 for 10 minutes. Sections were blocked with 5% bovine serum albumin (Sigma Aldrich) and incubated with primary antibodies overnight at 4°C (see supplementary materials). Sections were washed with PBS and incubated with fluorescently-conjugated secondary antibodies (Jackson ImmunoResearch). Confocal images were acquired on a Zeiss LSM 700 microscope using the same settings for all images acquired within the same experiment.
Chromogenic staining: Sections were treated as described above. After incubation with primary antibodies overnight, sections were incubated with 0.3% H2O2 in TBS to block endogenous peroxidase activity, washed, incubated in HRP-conjugated secondary antibodies for 1 hour, and rinsed. For visualization, 3,3’-diaminobenzidine (Sigma Aldrich) was applied to the sections, and the sections were washed and mounted. For co-staining, sections were incubated in a second primary antibody overnight at 4°C, washed, incubated with alkaline phosphatase-conjugated secondary antibody for 1 hour, washed, developed with Vector Blue (Vector Labs), rinsed, and mounted. Images were acquired on a Zeiss Axio Imager microscope using the same settings for all images acquired within the same experiment. All image contrast and brightness adjustments were made equally between experimental groups. Area fractions of plaque were calculated using ImageJ software as area covered by their respective markers (6E10) divided by the area of the region of interest.
Lentivirus production
EGFP or CD2RECK sequences were cloned into the lentiviral vector FUGW and sequence-verified, and lentivirus was generated using standard protocols (see supplementary materials).
Cell Culture and Transfection
HEK293FT and COS-7 cells were cultured and transfected as previously described (26). See supplementary materials.
Human bone osteosarcoma cell line U-2 OS was cultured in McCoy’s 5a Medium Modified medium (ATCC) with 10% fetal bovine serum (Sigma Aldrich). U-2 OS cells were seeded (60,000 cells per well) in 24-well plates pre-coated with 25 μg/mL PEI. The next day, cells were transfected using Lipofectamine RNAiMAX (ThermoFisher), and the medium and lysates were collected 72 hours post-transfection. See supplementary materials for details of siRNA sequences.
Human GDE2 accumulation measurement
Neurons with GDE2 accumulation were manually quantified from paraffin sections of CTL individuals (n=10) or patients with AD or HD (n=10) parietal cortex. GDE2-immunoreactive neurons were rated as low accumulation (those with accumulation less than the volume of its nucleus) or high accumulation (those with accumulation similar or higher than the volume of its nucleus). Neurons with somatic GDE2 accumulation were quantified in a 0.55 × 0.42 mm square region of interest per section (0.23mm2) that was obtained at 200x final magnification using a Zeiss Axio Imager. Neurons with GDE2 accumulations were averaged from a minimum of 7 sections per patient to ensure adequate sampling.
Sandwich ELISA
Aβ peptide amounts were assessed by sandwich ELISA using Aβ40 and Aβ42 high sensitivity kits (Wako) according to the manufacturer’s protocol (see supplementary materials for details).
Calcium imaging and analysis
Calcium flux was measured in DIV12 cortical neuronal cultures using the synthetic calcium indicator Fluo-4, AM (Thermo Scientific) using standard protocols (see supplementary materials for details).
Statistics
All experiments were performed with at least 3 biological replicates per condition, with cell culture experiments having 3 technical replicates per biological replicate. All graphs represent mean ± SEM. Statistical significance was determined using a two-tailed, unpaired Student’s t-test, with a 95% normal-based confidence interval. Significance is determined by a value of p< 0.05. Statistical analysis was performed on the raw data. GraphPad Prism (Version 5) was used for statistical analyses and the generation of graphs.
Supplementary Material
Data File S1. Individual Subject-level data (Excel file).
Extended Materials and Methods
Fig. S1. Model for GDE2 function in neurogenesis and APP processing.
Fig. S2. Validation of antibodies detecting human GDE2.
Fig. S3. Aberrant GDE2 accumulation in parietal cortex of patients with AD.
Fig. S4. GDE2 accumulates in brain of patients with Braak III/IV AD but not in brain of patients with HD.
Fig. S5. GDE2 regulates RECK surface expression.
Fig. S6. CNTN1 is not a GDE2 substrate and is unchanged in brain of patients with AD and patients with HD.
Fig.S7. APP, ADAM10 and BACE amounts are not changed by GDE2 ablation in mice.
Fig. S8. Synaptic loss in Gde2−/− neurons is not rescued by Aβ peptide reduction.
Fig. S9. GDE2 ablation reduces spike frequency in neurons.
Fig. S10. Membrane CD2RECK is not released by GDE2 and does not affect APP, ADAM10 and BACE amounts.
Fig. S11. GDE2 regulates RECK to inhibit ADAM10 α-secretase activity.
Fig. S12. siRNAs targeting Reck reduce RECK protein expression in cultured neurons.
Table S1. Basic demographic information of patients included in the study.
Acknowledgments:
We thank C. Wladyka for technical expertise; O. Pletnikova for human tissue samples; C. Ross for HD tissue samples (NS16375, HD Center); J. Nathans for RosaCreER mice; P. Worley and the Sockanathan laboratory for discussions; and the Multiphoton Imaging Core of the Johns Hopkins P30 Center for Neuroscience Research (NS050274, NIH).
Funding:
This work was funded by the following: 5T32GM08752 (NIH) for M.N.; 90068104 (AFAR) to M.N.; F31AG056088 (NIH) to M.N.; Johns Hopkins ADRC Pilot Project Award to S.S.; Johns Hopkins Innovation Award to S.S.; Johns Hopkins President’s Frontier Finalist Award to S.S.; RO1AG062671 (NIH) to S.S.; and the JHU Alzheimer’s Research Center NIA P30AG066507 for support of J.T..
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability: All data is available in the main text or supplementary materials. Reck+/− mice are available from Dr Takahashi under a Materials Transfer Agreement with Kanazawa University. All reagents generated in this study are freely available upon request.
References and Notes:
- 1.Hardy J, Selkoe DJ, The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356 (2002). [DOI] [PubMed] [Google Scholar]
- 2.De Strooper B, Karran E, The Cellular Phase of Alzheimer’s Disease. Cell 164, 603–615 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Kuhn PH, Wang H, Dislich B, Colombo A, Zeitschel U, Ellwart JW, Kremmer E, Rossner S, Lichtenthaler SF, ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J 29, 3020–3032 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jorissen E, Prox J, Bernreuther C, Weber S, Schwanbeck R, Serneels L, Snellinx A, Craessaerts K, Thathiah A, Tesseur I, Bartsch U, Weskamp G, Blobel CP, Glatzel M, De Strooper B, Saftig P, The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J Neurosci 30, 4833–4844 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lichtenthaler SF, Alpha-secretase cleavage of the amyloid precursor protein: proteolysis regulated by signaling pathways and protein trafficking. Curr Alzheimer Res 9, 165–177 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Obregon D, Hou H, Deng J, Giunta B, Tian J, Darlington D, Shahaduzzaman M, Zhu Y, Mori T, Mattson MP, Tan J, Soluble amyloid precursor protein-alpha modulates beta-secretase activity and amyloid-beta generation. Nat Commun 3, 777 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koike H, Tomioka S, Sorimachi H, Saido TC, Maruyama K, Okuyama A, Fujisawa-Sehara A, Ohno S, Suzuki K, Ishiura S, Membrane-anchored metalloprotease MDC9 has an alpha-secretase activity responsible for processing the amyloid precursor protein. The Biochemical journal 343 Pt 2, 371–375 (1999). [PMC free article] [PubMed] [Google Scholar]
- 8.Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F, Constitutive and regulated alpha-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proceedings of the National Academy of Sciences of the United States of America 96, 3922–3927 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slack BE, Ma LK, Seah CC, Constitutive shedding of the amyloid precursor protein ectodomain is up-regulated by tumour necrosis factor-alpha converting enzyme. The Biochemical journal 357, 787–794 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim M, Suh J, Romano D, Truong MH, Mullin K, Hooli B, Norton D, Tesco G, Elliott K, Wagner SL, Moir RD, Becker KD, Tanzi RE, Potential late-onset Alzheimer’s disease-associated mutations in the ADAM10 gene attenuate {alpha}-secretase activity. Hum Mol Genet 18, 3987–3996 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suh J, Choi SH, Romano DM, Gannon MA, Lesinski AN, Kim DY, Tanzi RE, ADAM10 missense mutations potentiate beta-amyloid accumulation by impairing prodomain chaperone function. Neuron 80, 385–401 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mockett BG, Richter M, Abraham WC, Muller UC, Therapeutic Potential of Secreted Amyloid Precursor Protein APPsalpha. Front Mol Neurosci 10, 30 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prinzen C, Muller U, Endres K, Fahrenholz F, Postina R, Genomic structure and functional characterization of the human ADAM10 promoter. FASEB J 19, 1522–1524 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Lammich S, Buell D, Zilow S, Ludwig AK, Nuscher B, Lichtenthaler SF, Prinzen C, Fahrenholz F, Haass C, Expression of the anti-amyloidogenic secretase ADAM10 is suppressed by its 5’-untranslated region. J Biol Chem 285, 15753–15760 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu D, Sharma C, Hemler ME, Tetraspanin12 regulates ADAM10-dependent cleavage of amyloid precursor protein. FASEB J 23, 3674–3681 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wakabayashi T, Craessaerts K, Bammens L, Bentahir M, Borgions F, Herdewijn P, Staes A, Timmerman E, Vandekerckhove J, Rubinstein E, Boucheix C, Gevaert K, De Strooper B, Analysis of the gamma-secretase interactome and validation of its association with tetraspanin-enriched microdomains. Nat Cell Biol 11, 1340–1346 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Jouannet S, Saint-Pol J, Fernandez L, Nguyen V, Charrin S, Boucheix C, Brou C, Milhiet PE, Rubinstein E, TspanC8 tetraspanins differentially regulate the cleavage of ADAM10 substrates, Notch activation and ADAM10 membrane compartmentalization. Cell Mol Life Sci 73, 1895–1915 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alexius-Lindgren M, Andersson E, Lindstedt I, Engstrom W, The RECK gene and biological malignancy--its significance in angiogenesis and inhibition of matrix metalloproteinases. Anticancer Res 34, 3867–3873 (2014). [PubMed] [Google Scholar]
- 19.Rhee JS, Coussens LM, RECKing MMP function: implications for cancer development. Trends Cell Biol 12, 209–211 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Oh J, Takahashi R, Kondo S, Mizoguchi A, Adachi E, Sasahara RM, Nishimura S, Imamura Y, Kitayama H, Alexander DB, Ide C, Horan TP, Arakawa T, Yoshida H, Nishikawa S, Itoh Y, Seiki M, Itohara S, Takahashi C, Noda M, The membrane-anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis. Cell 107, 789–800 (2001). [DOI] [PubMed] [Google Scholar]
- 21.Muraguchi T, Takegami Y, Ohtsuka T, Kitajima S, Chandana EP, Omura A, Miki T, Takahashi R, Matsumoto N, Ludwig A, Noda M, Takahashi C, RECK modulates Notch signaling during cortical neurogenesis by regulating ADAM10 activity. Nat Neurosci 10, 838–845 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Yanaka N, Mammalian glycerophosphodiester phosphodiesterases. Biosci Biotechnol Biochem 71, 1811–1818 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Rao M, Sockanathan S, Transmembrane protein GDE2 induces motor neuron differentiation in vivo. Science 309, 2212–2215 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Sabharwal P, Lee C, Park S, Rao M, Sockanathan S, GDE2 regulates subtype-specific motor neuron generation through inhibition of Notch signaling. Neuron 71, 1058–1070 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodriguez M, Choi J, Park S, Sockanathan S, Gde2 regulates cortical neuronal identity by controlling the timing of cortical progenitor differentiation. Development 139, 3870–3879 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Park S, Lee C, Sabharwal P, Zhang M, Meyers CL, Sockanathan S, GDE2 promotes neurogenesis by glycosylphosphatidylinositol-anchor cleavage of RECK. Science 339, 324–328 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matas-Rico E, van Veen M, Leyton-Puig D, van den Berg J, Koster J, Kedziora KM, Molenaar B, Weerts MJA, de Rink I, Medema RH, Giepmans BNG, Perrakis A, Jalink K, Versteeg R, Moolenaar WH, Glycerophosphodiesterase GDE2 Promotes Neuroblastoma Differentiation through Glypican Release and Is a Marker of Clinical Outcome. Cancer Cell 30, 986 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K, Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112, 389–404 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salgado-Polo F, van Veen M, van den Broek B, Jalink K, Leyton-Puig D, Perrakis A, Moolenaar WH, Matas-Rico E, Sequence-dependent trafficking and activity of GDE2, a GPI-specific phospholipase promoting neuronal differentiation. J Cell Sci 133, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Postina R, Schroeder A, Dewachter I, Bohl J, Schmitt U, Kojro E, Prinzen C, Endres K, Hiemke C, Blessing M, Flamez P, Dequenne A, Godaux E, van Leuven F, Fahrenholz F, A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. The Journal of clinical investigation 113, 1456–1464 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szaruga M, Munteanu B, Lismont S, Veugelen S, Horre K, Mercken M, Saido TC, Ryan NS, De Vos T, Savvides SN, Gallardo R, Schymkowitz J, Rousseau F, Fox NC, Hopf C, De Strooper B, Chavez-Gutierrez L, Alzheimer’s-Causing Mutations Shift Abeta Length by Destabilizing gamma-Secretase-Abetan Interactions. Cell 170, 443–456 e414 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR, Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet 13, 159–170 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Sanchez-Varo R, Trujillo-Estrada L, Sanchez-Mejias E, Torres M, Baglietto-Vargas D, Moreno-Gonzalez I, De Castro V, Jimenez S, Ruano D, Vizuete M, Davila JC, Garcia-Verdugo JM, Jimenez AJ, Vitorica J, Gutierrez A, Abnormal accumulation of autophagic vesicles correlates with axonal and synaptic pathology in young Alzheimer’s mice hippocampus. Acta Neuropathol 123, 53–70 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R, Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci 26, 10129–10140 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mucke L, Selkoe DJ, Neurotoxicity of amyloid beta-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med 2, a006338 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kamphuis W, Mamber C, Moeton M, Kooijman L, Sluijs JA, Jansen AH, Verveer M, de Groot LR, Smith VD, Rangarajan S, Rodriguez JJ, Orre M, Hol EM, GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. PLoS One 7, e42823 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eimer WA, Vassar R, Neuron loss in the 5XFAD mouse model of Alzheimer’s disease correlates with intraneuronal Abeta42 accumulation and Caspase-3 activation. Mol Neurodegener 8, 2 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haass C, Kaether C, Thinakaran G, Sisodia S, Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med 2, a006270 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Das U, Wang L, Ganguly A, Saikia JM, Wagner SL, Koo EH, Roy S, Visualizing APP and BACE-1 approximation in neurons yields insight into the amyloidogenic pathway. Nat Neurosci 19, 55–64 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cho C, Smallwood PM, Nathans J, Reck and Gpr124 Are Essential Receptor Cofactors for Wnt7a/Wnt7b-Specific Signaling in Mammalian CNS Angiogenesis and Blood-Brain Barrier Regulation. Neuron 95, 1221–1225 (2017). [DOI] [PubMed] [Google Scholar]
- 41.de Almeida GM, Yamamoto M, Morioka Y, Ogawa S, Matsuzaki T, Noda M, Critical roles for murine Reck in the regulation of vascular patterning and stabilization. Sci Rep 5, 17860 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bostaille N, Gauquier A, Twyffels L, Vanhollebeke B, Molecular insights into Adgra2/Gpr124 and Reck intracellular trafficking. Biol Open 5, 1874–1881 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haass C, Selkoe DJ, Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol 8, 101–112 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Benilova I, Karran E, De Strooper B, The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci 15, 349–357 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Yan Y, Sabharwal P, Rao M, Sockanathan S, The antioxidant enzyme Prdx1 controls neuronal differentiation by thiol-redox-dependent activation of GDE2. Cell 138, 1209–1221 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yan Y, Wladyka C, Fujii J, Sockanathan S , Prdx4 is a compartment-specific H2O2 sensor that regulates neurogenesis by controlling surface expression of GDE2. Nat Commun 6, 7006 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Butterfield DA, Drake J, Pocernich C, Castegna A, Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid beta-peptide. Trends Mol Med 7, 548–554 (2001). [DOI] [PubMed] [Google Scholar]
- 48.Choi H, Kim HJ, Kim J, Kim S, Yang J, Lee W, Park Y, Hyeon SJ, Lee DS, Ryu H, Chung J, Mook-Jung I, Increased acetylation of Peroxiredoxin1 by HDAC6 inhibition leads to recovery of Abeta-induced impaired axonal transport. Mol Neurodegener 12, 23 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, Chiriboga CA, Saito K, Servais L, Tizzano E, Topaloglu H, Tulinius M, Montes J, Glanzman AM, Bishop K, Zhong ZJ, Gheuens S, Bennett CF, Schneider E, Farwell W, De Vivo DC, Group ES, Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med 377, 1723–1732 (2017). [DOI] [PubMed] [Google Scholar]
- 50.Mercuri E, Darras BT, Chiriboga CA, Day JW, Campbell C, Connolly AM, Iannaccone ST, Kirschner J, Kuntz NL, Saito K, Shieh PB, Tulinius M, Mazzone ES, Montes J, Bishop KM, Yang Q, Foster R, Gheuens S, Bennett CF, Farwell W, Schneider E, De Vivo DC, Finkel RS, Group CS, Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N Engl J Med 378, 625–635 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Kwart D, Gregg A, Scheckel C, Murphy EA, Paquet D, Duffield M, Fak J, Olsen O, Darnell RB, Tessier-Lavigne M, A Large Panel of Isogenic APP and PSEN1 Mutant Human iPSC Neurons Reveals Shared Endosomal Abnormalities Mediated by APP beta-CTFs, Not Abeta. Neuron 104, 256–270 e255 (2019). [DOI] [PubMed] [Google Scholar]
- 52.Ayala-Sarmiento AE, Estudillo E, Perez-Sanchez G, Sierra-Sanchez A, Gonzalez-Mariscal L, Martinez-Fong D, Segovia J, GAS1 is present in the cerebrospinal fluid and is expressed in the choroid plexus of the adult rat. Histochemistry and cell biology 146, 325–336 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Castagna A, Campostrini N, Farinazzo A, Zanusso G, Monaco S, Righetti PG, Comparative two-dimensional mapping of prion protein isoforms in human cerebrospinal fluid and central nervous system. Electrophoresis 23, 339–346 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Lazzaro M, Bettegazzi B, Barbariga M, Codazzi F, Zacchetti D, Alessio M, Ceruloplasmin potentiates nitric oxide synthase activity and cytokine secretion in activated microglia. Journal of neuroinflammation 11, 164 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cave C, Park S, Rodriguez M, Nakamura M, Hoke A, Pletnikov M, Sockanathan S, GDE2 is essential for neuronal survival in the postnatal mammalian spinal cord. Mol Neurodegener 12, 8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data File S1. Individual Subject-level data (Excel file).
Extended Materials and Methods
Fig. S1. Model for GDE2 function in neurogenesis and APP processing.
Fig. S2. Validation of antibodies detecting human GDE2.
Fig. S3. Aberrant GDE2 accumulation in parietal cortex of patients with AD.
Fig. S4. GDE2 accumulates in brain of patients with Braak III/IV AD but not in brain of patients with HD.
Fig. S5. GDE2 regulates RECK surface expression.
Fig. S6. CNTN1 is not a GDE2 substrate and is unchanged in brain of patients with AD and patients with HD.
Fig.S7. APP, ADAM10 and BACE amounts are not changed by GDE2 ablation in mice.
Fig. S8. Synaptic loss in Gde2−/− neurons is not rescued by Aβ peptide reduction.
Fig. S9. GDE2 ablation reduces spike frequency in neurons.
Fig. S10. Membrane CD2RECK is not released by GDE2 and does not affect APP, ADAM10 and BACE amounts.
Fig. S11. GDE2 regulates RECK to inhibit ADAM10 α-secretase activity.
Fig. S12. siRNAs targeting Reck reduce RECK protein expression in cultured neurons.
Table S1. Basic demographic information of patients included in the study.