

Abstract
Autophagy plays an important role in the survival of cancer cells under stressful conditions, such as nutrient or oxygen deficiency. Therefore, autophagy inhibition is being considered as a novel therapeutic strategy for cancer. Decursin is a natural compound derived from Angelica gigas; it has been used in the treatment of various diseases, including cancer. However, the mechanism by which decursin regulates autophagy in gastric cancer and other carcinomas remains unclear. Here, we demonstrated that decursin reduced the growth and induced cell cycle arrest in gastric cancer cells in vitro. Decursin blocked autophagic flux by reducing the expression of lysosomal protein cathepsin C (CTSC) and attenuating its activity, thereby causing autophagic dysregulation (i.e., accumulation of LC3 and SQSTM1). Decursin also inhibited cell proliferation and cell cycle progression by inhibiting CTSC and E2F3, both of which were linked to gastric cancer aggressiveness. The antitumor effects of decursin were confirmed in vivo. We established spheroid and patient-derived organoid models and found that decursin decreased the growth of spheroids and patient-derived gastric organoids, as well as modulated the expression of CTSC and autophagy-related proteins. Hence, our findings uncovered a previously unknown mechanism by which decursin regulates cell growth and autophagy and suggests that decursin may act as a potential therapeutic agent that simultaneously inhibits cell growth and autophagy.
Keywords: Autophagy, cell cycle, cathepsin C, E2F3, decursin, gastric cancer, patient-derived gastric organoid
Introduction
Gastric cancer (GC) is the third most lethal cancer. Despite the advances in surgery and pharmacological interventions, the prognosis of GC remains poor, especially in patients with recurrent tumors [1]. In a recent clinical trial, the overall survival rate of metastatic GC patients treated with targeted therapies was found to be less than one year [2,3]. Therefore, extensive research is needed on the molecular mechanisms of GC progression to facilitate the development of new therapies and improve patient prognosis.
Autophagy is a key system involved in the degradation of intracellular components. Autophagy regulates various cellular processes, including cell growth and differentiation, organismal development, and inflammation [4]. Although autophagy’s role in cancer has been ambivalent, many recent studies have indicated autophagy as an essential energy support mechanism via the digestion of subcellular components [5]. Moreover, autophagy has been shown to inhibit apoptosis and promote angiogenesis and epithelial-to-mesenchymal transition (EMT) [4-6]. Therefore, various autophagy inhibitors are currently being tested in clinical trials, and preliminary data have confirmed their potent antitumor effects. Like in many other cancers, autophagy has dual effects on GC development. Mounting evidence suggests that autophagy promotes cancer progression in several ways. Notably, Helicobacter pylori infection, one of the main causes of GC, promotes autophagy through the metabolic regulation of host cells [7,8].
Decursin is a coumarin extracted from the plant Angelica gigas and has been shown to possess anticancer, antibacterial, and antioxidant effects [9-11]. In many cancers, including glioma, hepatocellular carcinoma, and GC, decursin has been shown to exert potent anticancer effects [12-14]. Some compounds of the coumarin family have been reported to regulate autophagy; however, the effects of decursin on autophagy remain elusive [15,16].
The aim of this study was to identify the relevance of autophagy and cell cycle regulation in the antitumor effects of decursin in GC, as well as determine whether decursin can act as an anticancer agent.
Materials and methods
Preparation of decursin
Decursin was prepared as described previously [12]. The concentration of dimethyl sulfoxide (DMSO) was 0.1% in all experiments.
Cell culture
The cancer cell lines used in this study were previously described [12]. HT29 (colon cancer), HeLa (cervical cancer), MCF-7 (breast cancer), and NCI-H460 (lung cancer) cell lines were purchased from the American Type Culture Collection (Gaithersburg, MD, USA); they were then cultured in Dulbecco’s modified Eagle medium (DMEM; Welgene) and incubated at 37°C in a humidified atmosphere with 5% CO2.
Cell proliferation assay
CCK-8 and clonogenic assays were conducted as previously described [12]. After decursin treatment, CCK-8 solution was added to the culture medium, and the absorbance at 450 nm was measured. For the clonogenic assay, cells treated with decursin for 24 h were washed with phosphate-buffered saline (PBS), and colony formation was evaluated after fixing cells with 10% formalin and staining them with 0.1% crystal violet.
Small interfering RNA (siRNA)
We designed siRNAs targeting CTSC and E2F3, as well as a non-targeting control siRNA (Table 1). Cells were transfected with siRNAs using Lipofectamine RNAiMax (Invitrogen, Carlsbad, CA, USA).
Table 1.
Sequences of siRNAs
| Gene | siRNA sequence |
|---|---|
| CTSC | 5’-GGAGAAAGUUCAUGGUAUCA-3’ |
| E2F3 | 5’-CGUCCAAUGGAUGGGCUGC-3’ |
Tumor spheroid formation
Cells were seeded in ultra-low attachment 6-well plates (Corning, Inc., Corning, NY, USA) as previously described [17]. For spheroid culture, advanced DMEM/F12 medium containing 1x B27, 1x N2, 20 ng/mL fibroblast growth factor (FGF)-basic, 20 ng/mL epidermal growth factor (EGF), 10 mM HEPES, and 1× primocin.
Transmission electron microscopy (TEM)
Cells were treated with DMSO or 50 μM decursin for 24 h. Subsequently, cells were washed three times in PBS and fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.3) overnight at 4°C. After cells had been fixed, they were post-fixed with 1% osmium tetroxide on ice for 1 h, dehydrated in an ethanol and propylene oxide series, and embedded in Epon812. Polymerization was induced using pure resin at 70°C for two days. Ultrathin sections (70 nm) were obtained using an ultramicrotome (UltraCut-UCT, Leica, Wetzlar, Germany) and collected on 150-mesh copper grids. After sections had been stained with 2% uranyl acetate for 15 min and lead citrate for 5 min, they were examined by TEM (Tecnai G2 Spirit TWIN, FEI Company, Hillsboro, OR, USA) at 120 kV.
Western blotting
Western blotting was performed as previously described [12]. Briefly, cells were washed in PBS and lysed with RIPA buffer supplemented with 1× protease and phosphatase inhibitor cocktail (GenDEPOT, Katy, TX, USA). Protein samples were separated using SDS-PAGE, transferred to a polyvinylidene difluoride membrane, and incubated with primary antibodies at 4°C overnight. After membranes had been washed, they were incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (goat anti-mouse and goat anti-rabbit IgG-HRP; Santa Cruz Biotechnology, Dallas, TX, USA).
Reverse-transcription polymerase chain reaction (RT-PCR)
The RNA preparation method was previously described [12]. Complementary DNA (cDNA) was synthesized using a qPCR RT Master Mix (TOYOBO, Osaka, Japan). RT-PCR was performed using EmeraldAmp Master Mix (TaKaRa Bio, Shiga, Japan). The primers used in this study are listed in Supplementary Table 1.
Cell cycle analysis
GC cells were treated with decursin for 24 h. Subsequently, cells were fixed in 70% ice-cold ethanol overnight at -20°C, washed with PBS, and incubated with FxCycle™ propidium iodide/RNase Staining Solution (Invitrogen) for 30 min at room temperature. Fluorescence was analyzed using a MoFlo cell sorter (Beckman Coulter, Brea, CA, USA) and the Kaluza analysis program (ver. 1.2; Beckman Coulter).
Immunocytochemistry
The main aspects of the immunocytochemistry method were previously described [12]. 5-Ethynyl-2-deoxyuridine (EdU) analysis was performed as previously described [18]. Briefly, decursin-treated cells were incubated at 37°C for 6 h in the presence of 20 μM EdU (Thermo Fisher Scientific) and then incubated with EdU antibody and Hoechst 33342 for 30 min at room temperature. pEGFP-LC3 and mRFP-GFP-LC3 plasmids were purchased from Addgene (Watertown, MA, USA). GC cells were transfected with pEGFP-LC3 or mRFP-GFP-LC3 for 24 h. After cells had been transfected, they were treated with decursin at the indicated concentration for 24 h and observed under a fluorescence microscope.
Immunohistochemistry
Immunohistochemistry was conducted as previously described [12]. Sections of 4 micrometer-thick paraffin-embedded tissues were prepared for staining. Slides were deparaffinized in xylene and rehydrated using a graded alcohol series. Sections were performed by microwaving the sections in Antigen Retrieval Buffer (pH 9.0; Dako, Carpinteria, CA, USA). Endogenous peroxidase activity was inactivated by incubation in 3% hydrogen peroxide buffer for 15 min. Slides were blocked for 20 min with protein block buffer (Dako). Primary antibodies diluted in blocking solution were added to samples and incubated overnight at 4°C. The next day, slides were incubated with the appropriate secondary antibody for 30 min and counterstained with hematoxylin.
Proteolytic activity assay
Recombinant human cathepsin C and L (rhCTSC and rhCTSL, respectively) were obtained from R&D Systems (Minneapolis, MN, USA) and activated in accordance with the manufacturer’s instructions. Briefly, rhCTSC and rhCTSL were diluted in Activation buffer to final concentrations of 20 μg/mL and 100 μg/mL, respectively; they were then incubated for 1 h at room temperature. Activated rhCTSC was diluted to 0.5 ng/μL in Assay buffer; the substrate (Gly-Arg-AMC; Bachem Americas, Inc., Torrance, CA, USA) was diluted to 20 μM in Assay buffer. The reaction was initiated by loading 50 μL of 0.5 ng/μL rhCTSC into black well plates and adding 50 μL of the substrate (20 μM). The decursin-treated group was incubated for 1 h with decursin and rhCTSC, and then reacted with the substrate.
Human tissue samples
Human GC and paired adjacent non-tumor tissues were collected from Chungnam National University Hospital. All protocols using human materials were approved by the institutional review board of Chungnam National University Hospital (Approval No.: CNUH 2018-08-015). Written informed consent was obtained from all patients involved in this study.
Establishment of patient-derived gastric organoids
Gastric tumor organoids were established as previously described [19]. Human tissues were washed 20-30 times with ice-cold PBS until they were free of impurities. Biopsies were then dissociated to single cells using a Human Tumor Dissociation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) for 1 h at 37°C. Dissociated cells were filtered through a 70-μm cell strainer (BD Biosciences, San Jose, CA, USA) into a new 50-mL tube. To remove red blood cells, the filtered cells were incubated with ACK lysis buffer for 5 min and washed with basal media (advanced DMEM/F12, 10 mM HEPES, 1× GlutaMAX, 1× penicillin/streptomycin). The cells were resuspended in 30 μL of growth factor-reduced Matrigel (Corning, Inc.) and seeded in 4-well dishes. Matrigel was then solidified by a 15-min incubation at 37°C and overlaid with 500 μL of complete organoid media. Complete media were refreshed at 2-3 day intervals; passaging of patient-derived gastric organoids (PDGOs) was performed using a cell recovery solution (Corning, Inc.) once every two weeks with a split ratio of 1:4.
Tumor formation assay in nude mice
All animal experiments were approved by the Animal Experimental Ethics Committee of Chungnam National University and were performed in accordance with the guidelines. NCI-N87 cells were washed three times with cold PBS, and 1×107 cells were subcutaneously injected into both flanks of each mouse with a 1:1 ratio of Matrigel. Tumor volume and body weight were measured twice per week for four weeks. After eight days, when tumor volumes reached approximately 100 mm3, decursin in 100 μL PBS was injected into the tumors of each mouse twice per week. DMSO in 100 μL PBS was injected as a negative control.
Bioinformatics
The online database Gene Expression Profiling Interactive Analysis (GEPIA) was used to obtain RNA sequencing data from The Cancer Genome Atlas and Genotype-Tissue Expression projects.
Statistical analysis
Data are expressed as means ± standard errors of the mean. Differences between groups were analyzed using the Student’s t-test. P-values < 0.05 were considered statistically significant. Data are representative of at least three independent experiments.
Results
Decursin inhibits cell growth and induces cell cycle arrest in GC cells
As shown in Figure 1A, decursin significantly reduced cell viability in a dose- and time-dependent manner. We also observed a reduction in cell growth through live-cell imaging and colony formation assays (Figure 1B, 1C). EdU staining was performed to further investigate the effect of decursin on cell proliferation. EdU labeling revealed that decursin strongly suppressed DNA synthesis in both GC cell lines (Figure 1D). Propidium iodide staining revealed that, after treatment with decursin, the percentage of cells in the G0/G1 phase was significantly elevated, while the percentage of cells in the S phase was reduced, indicating that decursin induced G0/G1 phase arrest (Figure 1E). Cyclin-dependent kinase (CDK) 4/6 levels were significantly reduced by decursin in a dose-dependent manner; in contrast, the expression of Cyclin D1, which interacts with CDK4/6 to promote cell cycle progression, did not change (Figure 1F). The expression of cyclin A regulated by the CDK4/6-cyclin D1 complex was also reduced. These results indicate that decursin inhibits GC growth and induces cell cycle arrest in vitro.
Figure 1.

Decursin inhibits GC cell proliferation. A. SNU216 and NCI-N87 cells were treated with indicated concentrations of decursin for indicated times. B, C. After decursin treatment for 24 h, cell growth was reduced, as demonstrated by bright field imaging and clonogenic assay (×100). Scale bar: 50 μm. D. Incorporated EdU was stained with anti-EdU, and cell nuclei were counterstained with Hoechst 33342 (×400). Scale bar: 50 μm. E. Representative results of cell cycle analysis by flow cytometry. F. Confirmation of expression of several cell cycle-related proteins. Full-length blots are presented in Supplementary Figure 6. n.s.: not significant; *P < 0.05; **P < 0.01; ***P < 0.001.
Decursin interferes with autophagic flux in GC cells
Next, we investigated the effect of decursin on autophagy. Decursin treatment markedly increased LC3-II levels in GC cells in a dose-dependent manner (Figure 2A, 2B). However, decursin-treated cells also showed SQSTM1 accumulation. The expression level of SQSTM1 is a useful indicator of autophagic flux [20], and SQSTM1 accumulation indicates inhibition of the autophagic flux. To further determine the effect of decursin on autophagic flux, we transiently expressed green fluorescent protein (GFP)-LC3, a canonical autophagosome marker, in SNU216 and NCI-N87 cells. Decursin treatment significantly increased the number of LC3 puncta compared with the control (Figure 2C). The autophagosome-specific Cyto-ID tracer dye was detected and quantified by FACS. As previously shown, decursin treatment caused autophagosome accumulation (Figure 2D). TEM was performed to visualize the autophagic phenotype in decursin-treated cells. As shown in Figure 2E, control cells displayed normal mitochondria and other organelles, while decursin-treated cells exhibited autophagic vacuoles and abnormal organelles. These results highlight the relationship between decursin and autophagy. However, SQSTM1 expression levels were gradually increased with increasing decursin concentrations (Figure 2A), suggesting that decursin promoted SQSTM1 and LC3 accumulation, impacting autophagic flux.
Figure 2.

Decursin induces autophagic alteration in GC cells. A, B. Decursin induced autophagosome formation in SNU216 and NCI-N87 cells, as revealed by conversion of LC3-I to LC3-II determined by western blotting. Full-length blots are presented in Supplementary Figure 6. C. Both cell lines were transfected with GFP-LC3 plasmid, and cells were treated with decursin for 24 h (×1000). Scale bar: 2 μm. D. Representative FACS data, showing accumulation of Cyto-ID after treatment with decursin. E. Effect of decursin on GC cell morphology. Representative TEM images are shown (×3200). Scale bar: 2 μm. The right panel shows the magnification of the boxed region. *P < 0.05; **P < 0.01; ***P < 0.001.
Decursin interferes with autophagic flux in a pH-independent manner
Next, we analyzed the expression levels of LC3 and SQSTM1 after decursin treatment. Elevated LC3 levels were observed after 12 h of treatment, and SQSTM1 levels increased in a time-dependent manner (Figure 3A-C), suggesting that autophagic flux was impaired by decursin. Decursin-mediated inhibition of autophagic flux was observed in GC, colon, cervical, breast, and lung cancer cells (Supplementary Figure 1). These findings were confirmed by treatment with decursin and bafilomycin A, a well-known inhibitor of autophagy (Figure 3D). Cells were transfected with the mRFP-GFP-LC3 plasmid to determine the mechanism involved in autophagic flux inhibition by decursin. The RFP-GFP sensor was used to distinguish the different pH values of acidic autolysosomes and neutral autophagosomes. GFP was quenched at acidic pH, while RFP exhibited relatively stable fluorescence at low pH. Therefore, green fluorescence was maintained when autophagic flux was blocked by changes in lysosomal pH. Interestingly, decursin reduced green fluorescence in a dose-dependent manner (Figure 3E). This finding suggests that autophagic flux suppression due to decursin treatment is not dependent on lysosomal pH.
Figure 3.

Decursin interferes with autophagic flux in GC cells. A. Decursin concentration was fixed at 50 μM, and treatment time was adjusted. Full-length blots are presented in Supplementary Figure 6. B, C. Expression of LC3-II and SQSTM1. D. SNU216 and NCI-N87 cells were exposed to decursin (50 μM), bafilomycin (100 nM), or a combination of both drugs for 24 h. Full-length blots are presented in Supplementary Figure 6. E. Both GC cell lines were transfected with mRFP-GFP-LC3 and treated with decursin for another 24 h, then analyzed by fluorescence microscopy (×1000). Scale bar: 2 μm.
Decursin inhibits autophagic flux by reducing CTSC levels
As decursin’s role in autophagic flux was not mediated by pH alterations, we investigated the effects of decursin on lysosomes. We confirmed that the levels of the lysosomal marker LAMP2 did not change after decursin treatment (Figure 4A and Supplementary Figure 2), indicating that decursin did not directly affect lysosomes. Subsequently, the expression of several lysosomal cathepsins was examined in decursin-treated GC cells (data not shown). CTSC expression levels were significantly reduced after decursin treatment (Figure 4A). CTSC silencing promoted autophagy dysregulation, whereas rhCTSC expression activated autophagy (Figure 4B, 4C). The loss of CTSC increased the number of LC3 puncta (Figure 4D). Moreover, the elevated level of autophagic flux caused by rhCTSC was restored to a baseline level by decursin treatment (Figure 4E). To assess the effect of decursin on CTSC activity, we performed a proteolytic analysis using Gly-Arg-AMC, a CTSC-specific substrate. Decursin reduced CTSC expression and CTSC proteolytic activity in a concentration-dependent manner (Figure 4F).
Figure 4.

Decursin reduces cathepsin C expression and activity. A. CTSC expression was confirmed in decursin-treated cells, and lysosome marker LAMP2 was identified to confirm effects on lysosomes. Full-length blots are presented in Supplementary Figure 6. B, C. Autophagic flux was evaluated by western blotting after treatment with siCTSC and rhCTSC. Full-length blots are presented in Supplementary Figure 6. D. Following co-transfection of siCTSC and GFP-LC3 plasmid, puncta were observed using immunocytochemistry (×1000). Scale bar: 2 μm. E. SNU216 and NCI-N87 cells were treated with decursin and rhCTSC, then cultured for 24 h. Full-length blots are presented in Supplementary Figure 6. F. To measure inhibition of CTSC by decursin, fluorescence values generated by CTSC cleavage were measured using Gly-Arg-AMC, a CTSC specific substrate. n.s.: not significant; *P < 0.05; **P < 0.01; ***P < 0.001.
CTSC inhibits cell cycle through E2F3
Decursin was found to induce autophagic flux disturbance through CTSC inhibition (Figure 4). Degradation of cellular components by autophagy can increase the nutrient pool required for cancer cell growth [5]. Therefore, the relationship between CTSC and cell growth was investigated. siRNA-mediated loss of CTSC significantly reduced the proliferation of GC cells and induced G0/G1 cell cycle arrest (Figure 5A, 5B). Importantly, we found that siCTSC reduced E2F3 levels (Figure 5C). In association with E2F1 and E2F2, E2F3 promotes cell growth; however, only E2F3 was reduced by siCTSC (Supplementary Figure 3A). E2F3 was also downregulated in decursin-treated cells (Figure 5D). These results indicate that decursin inhibits cancer cell growth and can be used as an anti-GC drug. E2F typically forms a complex with Rb; Rb phosphorylation releases E2F from the Rb-E2F complex, increasing transcriptional activity [21]. Therefore, the change in Rb phosphorylation status by CTSC was assessed. siCTSC did not alter Rb phosphorylation levels (Supplementary Figure 3B). Therefore, we concluded that the CTSC-dependent change in E2F3 is independent of Rb. Next, the effect of E2F3 on CTSC was evaluated using siE2F3. Unlike the decrease in E2F3 levels mediated by siCTSC, E2F3 knockdown did not affect CTSC levels (Figure 5E). However, siE2F3 promoted cell proliferation inhibition and cell cycle arrest (Figure 5F, 5G). These results suggest that E2F3 is downstream of CTSC and that decursin affects cell growth via the CTSC-E2F3 axis.
Figure 5.

Cathepsin C regulates cell cycle through E2F3. A, B. Cell proliferation and cell cycle assay after treatment with siCTSC. Both cell lines were transfected with siRNA targeting CTSC. After 24 h, cells were dissociated and seeded into 96-well plates. Proliferation was measured by CCK-8 assay at indicated times. Cell cycle analysis by FACS was performed 48 h after transfection. C. E2F3 mRNA and protein levels after treatment with siCTSC were confirmed by RT-PCR and western blotting. Full-length blots are presented in Supplementary Figure 6. D. Treatment with decursin induced reduction of E2F3 expression in a dose-dependent manner. Full-length blots are presented in Supplementary Figure 6. E. Following transfection of siE2F3, expression levels of each protein were confirmed by western blotting. Full-length blots are presented in Supplementary Figure 6. F. After 24 h of siE2F3 transfection, cells were dissociated and re-seeded in 96-well plates; CCK-8 assays were then performed at indicated times. G. E2F3 knockdown induced G0/G1 cell cycle arrest. n.s.: not significant; *P < 0.05; **P < 0.01; ***P < 0.001.
Decursin inhibits the growth of tumor spheroids and PDGOs
Many drugs show limited success in humans because in vitro experiments are based on two-dimensional (2D) cultures, which are not an accurate representation of the patient setting [22]. Thus, 3D culture models have emerged as a promising tool to test drug effectiveness. Here, we established two 3D culture models: tumor spheroids and PDGOs. NCI-N87 cells successfully formed spheroids and could be passaged (Supplementary Figures 4 and 5A). Cell stemness was enhanced in spheroid cultures and maintained for 72 h when the cells were dissociated into a monolayer (Supplementary Figure 5B). Cells with enhanced stemness were treated with decursin, and cell proliferation was reduced in a manner similar to that of decursin treatment in 2D culture (Supplementary Figure 5C, 5D). To further support these results, gastric organoids were established using tissues from three patients with GC who underwent surgery (Supplementary Table 2); the findings confirmed the effects of decursin. Histological evaluation revealed significant differences between the biopsies of non-tumor and tumor tissues (Figure 6A). Importantly, decursin reduced the size and number of organoids (Figure 6B). Consistent with previous results, decursin reduced CTSC levels and inhibited autophagic flux in PDGOs (Figure 6C). Hence, the effects of decursin were confirmed in tumor spheroids and PDGOs, enhancing the clinical relevance of our in vitro findings.
Figure 6.

Decursin reduces the growth of patient-derived gastric cancer organoids (PDGOs) and inhibits autophagic flux. A. Hematoxylin and eosin staining of adjacent non-tumor and tumor tissues (×200). Scale bar: 100 μm. B. Representative images of organoids treated with decursin (×100). Scale bar: 100 μm. C. CTSC expression and autophagic flux were confirmed by western blotting in decursin-treated organoids. Full-length blots are presented in Supplementary Figure 6.
Elevated CTSC and E2F3 levels are associated with aggressive behavior in GC
We found that the expression levels of CTSC and E2F3 were significantly upregulated in GC (Figure 7A) and that CTSC expression levels were correlated with those of E2F3 (Figure 7B). Kaplan-Meier analyses indicated a significant association between CTSC and E2F3 levels and poor survival outcomes in GC patients (Figure 7C, 7D). Western blotting in seven pairs of tumor and adjacent non-malignant tissues confirmed the elevated CTSC and E2F3 protein levels in GC tissues compared with matched non- malignant tissues (Figure 7E). A subcutaneous mouse model was established to confirm the effects of decursin on tumor growth in vivo. In contrast to control mice, decursin-treated mice showed retarded tumor growth. Body weight was not affected by the treatment (Figure 7F). Tumors were smaller in decursin-treated mice than in control mice, and histological analyses of mouse tumors revealed that control tumors were more compact than decursin-treated tumors (Figure 7G). In addition, lower levels of CTSC and E2F3 were observed in decursin-treated tumors, consistent with our in vitro results (Figure 7H). Overall, these findings suggest that decursin downregulates CTSC and suppresses tumor growth by disrupting autophagy and inhibiting CTSC and E2F3. These results also indicate that decursin suppresses GC progression by simultaneously inhibiting CTSC-mediated autophagic flux and cell cycle progression (Figure 8).
Figure 7.

Cathepsin C and E2F3 are upregulated and correlated with aggressiveness in GC. A. CTSC and E2F3 are highly expressed in GC tumors (GEPIA: http://gepia.cancer-pku.cn/). B. Correlation analysis of CTSC and E2F3 expression levels in GC. C, D. Survival analysis by Kaplan-Meier plot. Survival is lower in patients with high expression of CTSC and E2F3 than in patients with low expression of CTSC and E2F3. E. CTSC and E2F3 expression levels in tissues from patients with GC. Western blotting of protein expression levels in GC tumor and matched adjacent non-tumor tissues. Full-length blots are presented in Supplementary Figure 6. F. NCI-N87 cells were subcutaneously injected into nude mice. Tumor growth, tumor weight, and body weight were measured twice per week. Tumor volume was calculated as follows: (length × width2)/2. G. Dissected tumor tissues from BALB/c-nude mice. Tumors derived from decursin-injected mice were smaller than tumors derived from control mice. Representative hematoxylin and eosin staining indicated tumor histology (×200). Scale bar: 100 μm. H. Representative immunohistochemistry staining of CTSC and E2F3. N, non-tumor tissue; T, tumor tissue (×200). Scale bar: 100 μm.
Figure 8.
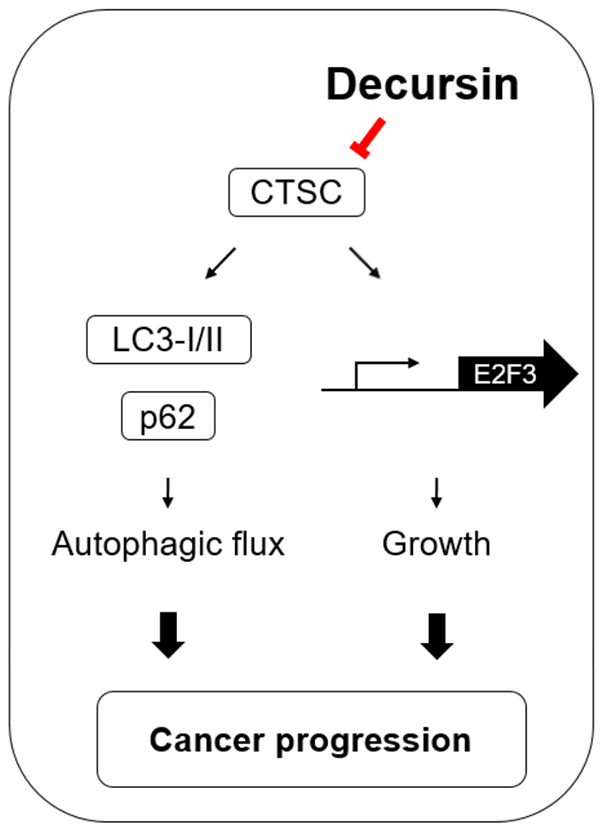
Schematic illustration showing the mechanism by which decursin inhibits the growth of GC cells through regulation of cell cycle and autophagy. Schematic representation of CTSC-mediated signaling and its biological effects in GC, implying that decursin exhibits anti-tumor activity by downregulating CTSC expression.
Discussion
In this study, we demonstrated the role of CTSC-induced autophagic flux and the cell cycle in GC progression; we also found a novel compound that inhibits CTSC. Decursin induced cell cycle arrest via CTSC-mediated inhibition of E2F3, a cell cycle regulatory transcription factor. Decursin also inhibited autophagic flux through inhibition of CTSC. Furthermore, public mRNA data for patients with GC showed that CTSC and E2F3 were highly expressed in GC and significantly associated with each other, indicating that decursin could be used as an anti-GC agent through its actions as an inhibitor of cancer cell proliferation and autophagic flux via CTSC inhibition.
Autophagy plays a complex role in cancer, where it functions as a tumor suppressor or tumor promoter [5,23]. Autophagy was initially presumed to inhibit cancer via degradation of cancer cells but has since been shown to induce cancer survival and proliferation in conditions of stress (e.g., lack of nutrients or oxygen) [24]. This biological phenomenon is regulated by multiple autophagy-related genes in various tumor types. Thus, despite possible confusion regarding the roles of autophagy, many in vitro and clinical studies have focused on its suppression.
Well-known autophagy inhibitors include chloroquine (CQ), hydroxychloroquine (HCQ), and bafilomycin A. CQ and its derivative HCQ are FDA-approved drugs currently in clinical trials for cancer treatment via inhibition of autophagy [25]. CQ and HCQ inhibit autophagy by elevating lysosomal pH and suppressing autophagosome and lysosome fusion [26]. Meanwhile, Bafilomycin A, inhibits the vacuolar-type H+-ATPase (V-ATPase) enzyme in lysosomes, thereby inhibiting lysosomal acidification [27]. 3-methyladenine inhibits autophagy by blocking autophagosome formation by inhibiting phosphatidylinositol-3-kinases [28].
Another inhibitory mechanism of autophagy involves targeting proteolytic enzymes in the lysosome. There are various types of lysosomal proteases [29,30]. Cathepsin is a lysosomal protease that plays an important role in the progression of autophagy; cathepsins can be divided into serine, cysteine, and aspartyl proteases [29]. Cathepsin levels are frequently elevated in tumors and are significantly associated with the prognosis of various cancers, including lung cancer, colorectal cancer, and breast cancer [31-33]. Cysteine cathepsin has been shown to exhibit elevated expression or activity in the tumor microenvironment; this enzyme causes proliferation, invasion, and metastasis of cancer cells [29]. Cathepsin B is a well-known cysteine cathepsin; numerous studies have shown that overexpression of cathepsin B is involved in cancer invasion and metastasis [34,35]. CTSC, also known as dipeptidyl peptidase I, is classified as a cysteine protease; it has been associated with various diseases such as cancer, sepsis, arthritis, and inflammatory disorders [36,37]. CTSC is upregulated in a variety of malignancies, including pancreatic cancer, hepatocellular carcinoma, and breast cancer [38-40]. Low CTSC expression in the gastric mucosa of Helicobacter pylori-infected patients has been reported [41], but little is known regarding the role of CTSC in GC. In the present study, decursin was found to reduce CTSC expression regardless of pH, thus leading to autophagy dysregulation.
Our results imply that CTSC may be an appropriate therapeutic target. Moreover, some studies have proposed the treatment of multiple diseases through CTSC inhibition. Consistent with our data, herbal medicine-derived timosaponin inhibits renal cell carcinoma metastasis by reducing CTSC expression through the AKT/miR-129-5p axis; moreover, olfactomedin has been shown to inhibit CTSC-mediated protease activity [42,43]. Because most studies have been focused on its structure, the mechanism of action by which CTSC contributes to cancer progression is poorly understood. Therefore, this mechanism should be identified.
Many cathepsins have been reported to affect cell growth, as well as invasion or metastasis. The cell cycle consumes a large amount of energy; autophagic degradation can provide a useful source of energy. Cathepsin B enhances the proliferation of colorectal cancer cells by cleaving p27, a cell cycle inhibitor; cathepsin S-mediated BRCA degradation results in enhanced growth of breast cancer cells [44,45]. A role for CTSC in cancer cell proliferation has not yet been reported. A notable finding in this study was that decursin-mediated CTSC reduction inhibited E2F3 expression and caused a reduction in the proliferation of GC cells. E2F3 plays an important role in the proliferation of normal cells but is frequently overexpressed in cancer, causing abnormal cell proliferation [46-48]. Moreover, autophagy inhibitors have been reported to produce synergistic effects when combined with cell cycle inhibitors or conventional chemotherapy agents [49,50]. Therefore, it is notable that decursin treatment alone can cause both autophagy and cell cycle dysregulation by suppressing CTSC expression.
Enhanced cancer stemness is presumed to be the main cause of cancer treatment failure due to drug resistance, which is a substantial challenge in drug development [51]. Thus, consideration of drug resistance using a 3D culture model with enhanced stemness may be an attractive approach. Spheroids are an easily reproducible experimental model that allows in vitro screening for drug resistance; organoids, another 3D culture model, mimic the structures and properties of the organs from which they are derived [52,53]. In addition, patient-derived organoids carry a specific patient’s genetic characteristics, which can help determine the drug response of that patient; this may be a useful model for personalized therapy [19]. Decursin induced a reduction in autophagic flux and cell growth in spheroids and organoids, implying that it may be able to overcome drug resistance.
Conclusions
In conclusion, this study revealed the importance of a new mechanism involving autophagy and cell growth through decursin-induced CTSC reduction. Decursin is a potential therapeutic candidate for the treatment of GC; determining its role and safety of the compound as a specific and selective inhibitor should be a priority.
Acknowledgements
This study was supported by grants from the Chungnam National University, the National Research Foundation of Korea (NRF-2017R1D1A1B04034638, NRF-2017R1A5A2015385, NRF-2018M3A9H3023077) and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HR20C0025).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Jim MA, Pinheiro PS, Carreira H, Espey DK, Wiggins CL, Weir HK. Stomach cancer survival in the United States by race and stage (2001-2009): findings from the CONCORD-2 study. Cancer. 2017;123(Suppl 24):4994–5013. doi: 10.1002/cncr.30881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T, Aprile G, Kulikov E, Hill J, Lehle M, Ruschoff J, Kang YK ToGA Trial Investigators. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–697. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- 3.Fuchs CS, Tomasek J, Yong CJ, Dumitru F, Passalacqua R, Goswami C, Safran H, Dos Santos LV, Aprile G, Ferry DR, Melichar B, Tehfe M, Topuzov E, Zalcberg JR, Chau I, Campbell W, Sivanandan C, Pikiel J, Koshiji M, Hsu Y, Liepa AM, Gao L, Schwartz JD, Tabernero J REGARD Trial Investigators. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): an international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet. 2014;383:31–39. doi: 10.1016/S0140-6736(13)61719-5. [DOI] [PubMed] [Google Scholar]
- 4.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 5.Kimmelman AC, White E. Autophagy and tumor metabolism. Cell Metab. 2017;25:1037–1043. doi: 10.1016/j.cmet.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mowers EE, Sharifi MN, Macleod KF. Autophagy in cancer metastasis. Oncogene. 2017;36:1619–1630. doi: 10.1038/onc.2016.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Terebiznik MR, Raju D, Vazquez CL, Torbricki K, Kulkarni R, Blanke SR, Yoshimori T, Colombo MI, Jones NL. Effect of Helicobacter pylori’s vacuolating cytotoxin on the autophagy pathway in gastric epithelial cells. Autophagy. 2009;5:370–379. doi: 10.4161/auto.5.3.7663. [DOI] [PubMed] [Google Scholar]
- 8.Chu YT, Wang YH, Wu JJ, Lei HY. Invasion and multiplication of Helicobacter pylori in gastric epithelial cells and implications for antibiotic resistance. Infect Immun. 2010;78:4157–4165. doi: 10.1128/IAI.00524-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee S, Shin DS, Kim JS, Oh KB, Kang SS. Antibacterial coumarins from Angelica gigas roots. Arch Pharm Res. 2003;26:449–452. doi: 10.1007/BF02976860. [DOI] [PubMed] [Google Scholar]
- 10.Zhang J, Li L, Jiang C, Xing C, Kim SH, Lu J. Anti-cancer and other bioactivities of Korean Angelica gigas Nakai (AGN) and its major pyranocoumarin compounds. Anticancer Agents Med Chem. 2012;12:1239–1254. doi: 10.2174/187152012803833071. [DOI] [PubMed] [Google Scholar]
- 11.Li L, Li W, Jung SW, Lee YW, Kim YH. Protective effects of decursin and decursinol angelate against amyloid beta-protein-induced oxidative stress in the PC12 cell line: the role of Nrf2 and antioxidant enzymes. Biosci Biotechnol Biochem. 2011;75:434–442. doi: 10.1271/bbb.100606. [DOI] [PubMed] [Google Scholar]
- 12.Kim S, Kim JE, Kim N, Joo M, Lee MW, Jeon HJ, Ryu H, Song IC, Song GY, Lee HJ. Decursin inhibits tumor growth, migration, and invasion in gastric cancer by down-regulating CXCR7 expression. Am J Cancer Res. 2019;9:2007–2018. [PMC free article] [PubMed] [Google Scholar]
- 13.Oh ST, Lee S, Hua C, Koo BS, Pak SC, Kim DI, Jeon S, Shin BA. Decursin induces apoptosis in glioblastoma cells, but not in glial cells via a mitochondria-related caspase pathway. Korean J Physiol Pharmacol. 2019;23:29–35. doi: 10.4196/kjpp.2019.23.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li J, Wang H, Wang L, Tan R, Zhu M, Zhong X, Zhang Y, Chen B, Wang L. Decursin inhibits the growth of HepG2 hepatocellular carcinoma cells via Hippo/YAP signaling pathway. Phytother Res. 2018;32:2456–2465. doi: 10.1002/ptr.6184. [DOI] [PubMed] [Google Scholar]
- 15.Xu H, Laraia L, Schneider L, Louven K, Strohmann C, Antonchick AP, Waldmann H. Highly enantioselective catalytic vinylogous propargylation of coumarins yields a class of autophagy inhibitors. Angew Chem Int Ed Engl. 2017;56:11232–11236. doi: 10.1002/anie.201706005. [DOI] [PubMed] [Google Scholar]
- 16.Zhang X, Dong Y, Zeng X, Liang X, Li X, Tao W, Chen H, Jiang Y, Mei L, Feng SS. The effect of autophagy inhibitors on drug delivery using biodegradable polymer nanoparticles in cancer treatment. Biomaterials. 2014;35:1932–1943. doi: 10.1016/j.biomaterials.2013.10.034. [DOI] [PubMed] [Google Scholar]
- 17.Lee JW, Sung JS, Park YS, Chung S, Kim YH. Isolation of spheroid-forming single cells from gastric cancer cell lines: enrichment of cancer stem-like cells. Biotechniques. 2018;65:197–203. doi: 10.2144/btn-2018-0046. [DOI] [PubMed] [Google Scholar]
- 18.Park J, Son Y, Lee NG, Lee K, Lee DG, Song J, Lee J, Kim S, Cho MJ, Jang JH, Lee J, Park JG, Kim YG, Kim JS, Lee J, Cho YS, Park YJ, Han BS, Bae KH, Han S, Kang B, Haam S, Lee SH, Lee SC, Min JK. DSG2 is a functional cell surface marker for identification and isolation of human pluripotent stem cells. Stem Cell Reports. 2018;11:115–127. doi: 10.1016/j.stemcr.2018.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan HHN, Siu HC, Law S, Ho SL, Yue SSK, Tsui WY, Chan D, Chan AS, Ma S, Lam KO, Bartfeld S, Man AHY, Lee BCH, Chan ASY, Wong JWH, Cheng PSW, Chan AKW, Zhang J, Shi J, Fan X, Kwong DLW, Mak TW, Yuen ST, Clevers H, Leung SY. A comprehensive human gastric cancer organoid biobank captures tumor subtype heterogeneity and enables therapeutic screening. Cell Stem Cell. 2018;23:882–897. e811. doi: 10.1016/j.stem.2018.09.016. [DOI] [PubMed] [Google Scholar]
- 20.Bjorkoy G, Lamark T, Pankiv S, Overvatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181–197. doi: 10.1016/S0076-6879(08)03612-4. [DOI] [PubMed] [Google Scholar]
- 21.Harbour JW, Dean DC. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev. 2000;14:2393–2409. doi: 10.1101/gad.813200. [DOI] [PubMed] [Google Scholar]
- 22.Edwards AM, Arrowsmith CH, Bountra C, Bunnage ME, Feldmann M, Knight JC, Patel DD, Prinos P, Taylor MD, Sundstrom M SGC Open Source Target-Discovery Partnership. Preclinical target validation using patient-derived cells. Nat Rev Drug Discov. 2015;14:149–150. doi: 10.1038/nrd4565. [DOI] [PubMed] [Google Scholar]
- 23.Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17:528–542. doi: 10.1038/nrc.2017.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roukos DH, Kappas AM. Perspectives in the treatment of gastric cancer. Nat Clin Pract Oncol. 2005;2:98–107. doi: 10.1038/ncponc0099. [DOI] [PubMed] [Google Scholar]
- 25.Rangwala R, Leone R, Chang YC, Fecher LA, Schuchter LM, Kramer A, Tan KS, Heitjan DF, Rodgers G, Gallagher M, Piao S, Troxel AB, Evans TL, DeMichele AM, Nathanson KL, O’Dwyer PJ, Kaiser J, Pontiggia L, Davis LE, Amaravadi RK. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy. 2014;10:1369–1379. doi: 10.4161/auto.29118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drose S, Altendorf K. Bafilomycins and concanamycins as inhibitors of V-ATPases and P-ATPases. J Exp Biol. 1997;200:1–8. doi: 10.1242/jeb.200.1.1. [DOI] [PubMed] [Google Scholar]
- 28.Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, Ong CN, Codogno P, Shen HM. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem. 2010;285:10850–10861. doi: 10.1074/jbc.M109.080796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olson OC, Joyce JA. Cysteine cathepsin proteases: regulators of cancer progression and therapeutic response. Nat Rev Cancer. 2015;15:712–729. doi: 10.1038/nrc4027. [DOI] [PubMed] [Google Scholar]
- 30.Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kayser K, Richter N, Hufnagl P, Kayser G, Kos J, Werle B. Expression, proliferation activity and clinical significance of cathepsin B and cathepsin L in operated lung cancer. Anticancer Res. 2003;23:2767–2772. [PubMed] [Google Scholar]
- 32.Sevenich L, Bowman RL, Mason SD, Quail DF, Rapaport F, Elie BT, Brogi E, Brastianos PK, Hahn WC, Holsinger LJ, Massague J, Leslie CS, Joyce JA. Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nat Cell Biol. 2014;16:876–888. doi: 10.1038/ncb3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gormley JA, Hegarty SM, O’Grady A, Stevenson MR, Burden RE, Barrett HL, Scott CJ, Johnston JA, Wilson RH, Kay EW, Johnston PG, Olwill SA. The role of cathepsin S as a marker of prognosis and predictor of chemotherapy benefit in adjuvant CRC: a pilot study. Br J Cancer. 2011;105:1487–1494. doi: 10.1038/bjc.2011.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Szpaderska AM, Frankfater A. An intracellular form of cathepsin B contributes to invasiveness in cancer. Cancer Res. 2001;61:3493–3500. [PubMed] [Google Scholar]
- 35.Podgorski I, Sloane BF. Cathepsin B and its role(s) in cancer progression. Biochem Soc Symp. 2003:263–276. doi: 10.1042/bss0700263. [DOI] [PubMed] [Google Scholar]
- 36.Guay D, Beaulieu C, Percival MD. Therapeutic utility and medicinal chemistry of cathepsin C inhibitors. Curr Top Med Chem. 2010;10:708–716. doi: 10.2174/156802610791113469. [DOI] [PubMed] [Google Scholar]
- 37.Korkmaz B, Caughey GH, Chapple I, Gauthier F, Hirschfeld J, Jenne DE, Kettritz R, Lalmanach G, Lamort AS, Lauritzen C, Legowska M, Lesner A, Marchand-Adam S, McKaig SJ, Moss C, Pedersen J, Roberts H, Schreiber A, Seren S, Thakker NS. Therapeutic targeting of cathepsin C: from pathophysiology to treatment. Pharmacol Ther. 2018;190:202–236. doi: 10.1016/j.pharmthera.2018.05.011. [DOI] [PubMed] [Google Scholar]
- 38.Zhang GP, Yue X, Li SQ. Cathepsin C interacts with TNF-alpha/p38 MAPK signaling pathway to promote proliferation and metastasis in hepatocellular carcinoma. Cancer Res Treat. 2020;52:10–23. doi: 10.4143/crt.2019.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joyce JA, Hanahan D. Multiple roles for cysteine cathepsins in cancer. Cell Cycle. 2004;3:1516–1619. doi: 10.4161/cc.3.12.1289. [DOI] [PubMed] [Google Scholar]
- 40.Tan GJ, Peng ZK, Lu JP, Tang FQ. Cathepsins mediate tumor metastasis. World J Biol Chem. 2013;4:91–101. doi: 10.4331/wjbc.v4.i4.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu YG, Teng YS, Cheng P, Kong H, Lv YP, Mao FY, Wu XL, Hao CJ, Chen W, Yang SM, Zhang JY, Peng LS, Wang TT, Han B, Ma Q, Zou QM, Zhuang Y. Abrogation of cathepsin C by Helicobacter pylori impairs neutrophil activation to promote gastric infection. FASEB J. 2019;33:5018–5033. doi: 10.1096/fj.201802016RR. [DOI] [PubMed] [Google Scholar]
- 42.Chiang KC, Lai CY, Chiou HL, Lin CL, Chen YS, Kao SH, Hsieh YH. Timosaponin AIII inhibits metastasis of renal carcinoma cells through suppressing cathepsin C expression by AKT/miR-129-5p axis. J Cell Physiol. 2019;234:13332–13341. doi: 10.1002/jcp.28010. [DOI] [PubMed] [Google Scholar]
- 43.Liu W, Yan M, Liu Y, McLeish KR, Coleman WG Jr, Rodgers GP. Olfactomedin 4 inhibits cathepsin C-mediated protease activities, thereby modulating neutrophil killing of Staphylococcus aureus and Escherichia coli in mice. J Immunol. 2012;189:2460–2467. doi: 10.4049/jimmunol.1103179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bian B, Mongrain S, Cagnol S, Langlois MJ, Boulanger J, Bernatchez G, Carrier JC, Boudreau F, Rivard N. Cathepsin B promotes colorectal tumorigenesis, cell invasion, and metastasis. Mol Carcinog. 2016;55:671–687. doi: 10.1002/mc.22312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim S, Jin H, Seo HR, Lee HJ, Lee YS. Regulating BRCA1 protein stability by cathepsin S-mediated ubiquitin degradation. Cell Death Differ. 2019;26:812–825. doi: 10.1038/s41418-018-0153-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Humbert PO, Verona R, Trimarchi JM, Rogers C, Dandapani S, Lees JA. E2f3 is critical for normal cellular proliferation. Genes Dev. 2000;14:690–703. [PMC free article] [PubMed] [Google Scholar]
- 47.Gao Y, Li H, Ma X, Fan Y, Ni D, Zhang Y, Huang Q, Liu K, Li X, Wang L, Yao Y, Ai Q, Zhang X. E2F3 upregulation promotes tumor malignancy through the transcriptional activation of HIF-2alpha in clear cell renal cell carcinoma. Oncotarget. 2017;8:54021–54036. doi: 10.18632/oncotarget.10568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Foster CS, Falconer A, Dodson AR, Norman AR, Dennis N, Fletcher A, Southgate C, Dowe A, Dearnaley D, Jhavar S, Eeles R, Feber A, Cooper CS. Transcription factor E2F3 overexpressed in prostate cancer independently predicts clinical outcome. Oncogene. 2004;23:5871–5879. doi: 10.1038/sj.onc.1207800. [DOI] [PubMed] [Google Scholar]
- 49.Vijayaraghavan S, Karakas C, Doostan I, Chen X, Bui T, Yi M, Raghavendra AS, Zhao Y, Bashour SI, Ibrahim NK, Karuturi M, Wang J, Winkler JD, Amaravadi RK, Hunt KK, Tripathy D, Keyomarsi K. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat Commun. 2017;8:15916. doi: 10.1038/ncomms15916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhan Y, Wang K, Li Q, Zou Y, Chen B, Gong Q, Ho HI, Yin T, Zhang F, Lu Y, Wu W, Zhang Y, Tan Y, Du B, Liu X, Xiao J. The novel autophagy inhibitor alpha-hederin promoted paclitaxel cytotoxicity by increasing reactive oxygen species accumulation in non-small cell lung cancer cells. Int J Mol Sci. 2018;19:3221. doi: 10.3390/ijms19103221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–4751. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lv D, Hu Z, Lu L, Lu H, Xu X. Three-dimensional cell culture: a powerful tool in tumor research and drug discovery. Oncol Lett. 2017;14:6999–7010. doi: 10.3892/ol.2017.7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, van Houdt W, van Gorp J, Taylor-Weiner A, Kester L, McLaren-Douglas A, Blokker J, Jaksani S, Bartfeld S, Volckman R, van Sluis P, Li VS, Seepo S, Sekhar Pedamallu C, Cibulskis K, Carter SL, McKenna A, Lawrence MS, Lichtenstein L, Stewart C, Koster J, Versteeg R, van Oudenaarden A, Saez-Rodriguez J, Vries RG, Getz G, Wessels L, Stratton MR, McDermott U, Meyerson M, Garnett MJ, Clevers H. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015;161:933–945. doi: 10.1016/j.cell.2015.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.