

Abstract
This study aimed to investigate the interaction of A-kinase-interacting protein 1 (AKIP1) with C-X-C motif chemokine ligand (CXCL)1, CXCL2, CXCL8, and their effects on regulating glioblastoma multiforme (GBM) malignant behaviors. AKIP1 expression was modified by pcDNA and pGPH1 vectors in U-87 MG and U-251 MG cells. Subsequently, multiple compensative experiments were conducted via adding CXCL1, CXCL2 and CXCL8 in the pGPH1-AKIP1 (AKIP1 knockdown) transfected U-87 MG and U-251 MG cells, respectively. Furthermore, AKIP1, CXCL1/2/8 expressions in 10 GBM and 10 low-grade glioma (LGG) tumor samples were detected. AKIP1 was elevated in various GBM cell lines compared to normal human astrocytes. AKIP1 overexpression promoted U-87 MG and U-251 MG cell proliferation and invasion while inhibited apoptosis; and it enhanced chemoresistance to temozolomide (but not cisplatin) and radiation resistance; then AKIP1 knockdown showed the opposite effects. Meanwhile, AKIP1 positively regulated CXCL1/2/8, NF-κB pathway, AKT pathway and PD-L1 expression. Further multiple compensative experiments uncovered that CXCL1 and CXCL8 promoted proliferation, invasion, chemoradiation resistance, NF-κB pathway, AKT pathway and PD-L1 expression in U-87 MG and U-251 MG cells, also in pGPH1-AKIP1 (AKIP1 knockdown) transfected U-87 MG and U-251 MG cells; although CXCL2 exhibited similar treads, but its effect was much weaker. Besides, NF-κB pathway inhibitor and AKT pathway inhibitor attenuated the effect of CXCL1&CXCL8 on promoting GBM cell malignant behaviors. Clinically AKIP1 and CXCL1/8 were elevated in GBM compared to LGG tumor samples, and they were inter-correlated. AKIP1 promotes GBM viability, mobility and chemoradiation resistance via regulating CXCL1 and CXCL8 mediated NF-κB and AKT pathways.
Keywords: A-kinase-interacting protein 1, C-X-C motif chemokine ligand, glioblastoma multiforme, chemoradiation resistance, NF-κB and AKT pathways
Introduction
Glioblastoma multiforme (GBM) is the most challenging malignant primary brain tumor to treat, accounting for approximately 57% of all gliomas and 48% of all primary malignant central nervous system (CNS) tumors [1]. Based on the latest registry data report, GBM occurs in 4.23 cases per 100,000 populations every year, ranging from 2.00 per 100,000 populations in Asian/Pacific race to 4.71 per 100,000 populations in non-Hispanic white race [2]. The common treatment strategy for GBM is surgical resection followed by chemoradiotherapy; meanwhile, with the advancements of biological and medical technology, targeted therapy and immune therapy have been introduced recently [3,4]. Despite of these achievements, the prognosis of GBM is still not obviously improved with 1-year survival rate of 41.4% and 5-year survival rate of 5.4% [2]. The heterogeneity, rapid progress and treatment resistance are key factors affecting the GBM prognosis, bring to the front that the importance of the deep understanding regarding GBM progression and treatment resistance is essential.
A-kinase-interacting protein 1 (AKIP1), initially observed in breast cancer and named as breast cancer-associated protein 3 (BCA3), is previously observed to be a key factor regulating NF-κB via multiple ways such as: binding to p65 and the cyclin D1 promoter in a neddylation-dependent way, as well as interaction with cAMP-dependent protein kinase (PKA) signaling pathway [5-7]. Subsequently, AKIP1 is further reported to be a tumor promoter and treatment resistance factor in cancers via multiple ways such as regulating AKT/GSK-3β/Snail pathway, VEGF pathway, Wnt/β-catenin/CBP pathway [8-11]. Importantly, AKIP1 is recently discovered to regulate the excretion of CXCL families such as C-X-C motif chemokine ligand (CXCL)1, CXCL2, CXCL8 to further promote cancer angiogenesis and growth; meanwhile, their interaction correlates with worse disease features and unsatisfied prognosis in both solid tumors and hematological malignancies [12-14]. Besides, NF-κB pathway which is directly modified by AKIP1, plays a critical role in PD-1/PD-L1 interaction through several ways such as its involvement in TCR-CD28 signaling [15-17]. Interestingly, CXCL members that are regulated by AKIP1, also closely interacts with NF-κB pathway, AKT pathway and are implicated in the regulation of immune checkpoint expressions [18-28]. Considering the above-mentioned potential interaction among AKIP1, CXCL1/2/8, NF-κB pathway, AKT pathway and PD-1/PD-L1 linkage, as well as their role in carcinogenesis, we hypothesized AKIP1, CXCL1, CXCL2 and CXCL8 might be involved in the GBM progression and treatment resistance. However, no related research has clarified this issue.
Therefore, this study aimed to investigate the effect of AKIP1, CXCL1, CXCL2 and CXCL8 on regulating GBM cell viability, mobility and chemoradiotherapy sensitivity, and their modifications on NF-κB pathway, AKT pathway and PD-L1 expression.
Methods
Cell culture
Human GBM cell lines U-251 MG (ECACC, UK), T98G (ATCC, USA) and U-87 MG (ATCC, USA) were cultured in Eagle’s Minimum Essential Medium (Gibco, USA) containing 10% fetal bovine serum (FBS) (Gibco, USA). Human GBM cell line LN-229 (ATCC, USA) was cultured in Dulbecco’s Modified Eagle Medium (Gibco, USA) containing 10% FBS (Gibco, USA). Normal human astrocytes (NHA) (Lonza, Switzerland) were cultured in AGMTM Astrocyte Growth Medium Bullet KitTM (Lonza, Switzerland). All cell lines were contained in a 37°C, 5% CO2 incubator. The expression of AKIP1 in cells (NHA as control) was determined by reverse transcription quantitative polymerase chain reaction (RT-qPCR) and western blot.
Transfection detection
For overexpression, AKIP1 and negative control (NC) DNA fragment were cloned into pcDNA3.1 vector (Genepharma, China) to construct the pcDNA-AKIP1 plasmid and pcDNA-NC plasmid. For knock-down, Short hairpin RNA (ShRNA) targeting AKIP1 and NC shRNA were designed and cloned into pGPH1 vector (Genepharma, China) to construct the pGPH1-AKIP1 plasmid and pGPH1-NC plasmid. The pcDNA-AKIP1, pcDNA-NC, pGPH1-AKIP1 and pGPH1-NC plasmids were transfected into U-87 MG and U-251 MG cells with the application of Lipofectamine 3000 (Invitrogen, USA), respectively. After transfection, the cells were divided into pcDNA-AKIP1, pcDNA-NC, pGPH1-AKIP1 and pGPH1-NC groups accordingly.
Cell proliferation, apoptosis and invasion detection
At 24 hour (h) after transfection, the expression of AKIP1 in pcDNA-AKIP1, pcDNA-NC, pGPH1-AKIP1 and pGPH1-NC group cells was evaluated by RT-qPCR and western blot. At 0 h, 24 h, 48 h and 72 h post transfection, cell counting kit-8 (Dojindo, Japan) was applied to detect cell proliferation following the kit’s instruction. At 48 h after transfection, Annexin V Apoptosis Detection Kit FITC (Invitrogen, USA) was used to assess cell apoptosis following the manufacturer’s instruction. After transfection, Matrigel Basement Membrane Matrix (BD, USA) coated transwell chamber was used to detect cell invasion according to the methods described in the previous study [29].
Chemosensitivity detection
After transfection, cells in pcDNA-AKIP1, pcDNA-NC, pGPH1-AKIP1 and pGPH1-NC groups were treated with different concentrations of temozolomide (TMZ) (Sigma, USA) or cisplatin (Sigma, USA) for 72 h. The concentration ranges of TMZ (Sigma, USA) and cisplatin (Sigma, USA) were 0-320 μM and 0-80 μM, respectively. The cell viability was evaluated by cell counting kit-8 (Dojindo, Japan) referring to the kit’s protocol and the relative cell viability was calculated according to the methods described in the previous study (the viability of cells without treatment was set as 100%) [30]. Meanwhile, after 80 μM TMZ or 40 μM cisplatin treatment for 72 h, cell apoptosis detection was performed with Annexin V Apoptosis Detection Kit FITC (Invitrogen, USA) according to the kit’s instruction.
Irradiation sensitivity detection
After transfection, cell irradiation was performed at room temperature by using Faxitron Cabinet X-ray System (Faxitron, IL), with a dose rate of 0.36 Gy/min at various doses (0, 2, 4, 6, and 8 Gy). Then cell viability was detected by cell counting kit-8 (Dojindo, Japan) and relative cell viability was calculated by setting the viability of cells without treatment as 100%. At the same time, cell apoptosis was detected with Annexin V Apoptosis Detection Kit FITC (Invitrogen, USA) according to the kit’s instruction after cells being treated with 6 Gy irradiation.
CXCL1/2/8 expression, PD-L1 expression, NF-κB and AKT pathways detection
AKIP1 is reported to elevate the CXCL1, CXCL2, and CXCL8 expressions in the promotion of tumor growth in cancers other than GBM [12-14]. Meanwhile, the NF-κB pathway, AKT pathway and PD-L1 play important roles in the regulation of metastasis, chemosensitivity and radiation sensitivity by AKIP1 [7,8,31-34]. To evaluate whether CXCL1/2/8, PD-L1, NF-κB pathway, AKT pathway were regulated by AKIP1 in GBM, the protein expressions of CXC L1, CXCL2 and CXCL8 in cells were evaluated by Enzyme-linked immunosorbent assay (ELISA) 24 h after transfection, and the expressions of NF-κB p60, phoso-NF-κB p60 (p-NF-κB p60), AKT, phoso-AKT (p-AKT) and PD-L1 were assessed by western blot.
ELISA
At 24 h after transfection, cell supernatant was collected. Then, the CXCL1, CXCL2 and CXCL8 in supernatant was measured by ELISA. The Human CXCL1 ELISA Kit (Invitrogen, USA), Human CXCL2 ELSA Kit (R&D, USA) and Human CXCL8 ELSIA Kit (R&D, USA) were applied to perform ELISA. The detection was carried out following the kit’s procedure.
Multiple compensative experiments
The pGPH1-NC and pGPH1-AKIP1 cells were constructed with the methods mentioned in “Transfection” subsection. Then, the pGPH1-NC and pGPH1-AKIP1 cells were incubated with 250 pg/ml CXCL1 (R&D, USA), 350 pg/ml CXCL2 (R&D, USA) and 400 pg/ml CXCL8 (R&D, USA), respectively. The cells were then divided into pGPH1-NC, pGPH1-AKIP1, pGPH1-NC&CXCL1, pGPH1-AKIP1&CXCL1, pGPH1-NC&CXCL2, pGPH1-AKIP1&CXCL2, pGPH1-NC&CXCL8, pGPH1-AKIP1&CXCL8 groups for analyses.
Furthermore, the pGPH1-NC and pGPH1-AKIP1 cells were incubated with 250 pg/ml CXCL1 (R&D, USA) plus 400 pg/ml CXCL8 (R&D, USA) simultaneously. And the cells were divided into pGPH1-NC, pGPH1-AKIP1, pGPH1-NC&CXCL1&CXCL8, pGPH1-AKIP1&CXCL1&CXCL8 groups for analyses.
Cell proliferation, cell apoptosis and cell invasion, chemosensitivity, radiation sensitivity were determined with methods mentioned above. At 48 h after incubation, the expressions of NF-κB p65, p-NF-κB p65, AKT, p-AKT and PD-L1 were detected by western blot.
LY and CAPE treatment
250 pg/ml CXCL1 (R&D, USA) plus 400 pg/ml CXCL8 (R&D, USA) was added to culture normal U-87 MG and U-251 MG cells. Then they were treated by 10 μM AKT pathway inhibitor (LY294002 (LY)) (Sigma, USA) and 50 μM NF-κB pathway inhibitor (caffeic acid phenethyl ester (CAPE)) (Sigma, USA), respectively, followed by detection of cell proliferation, apoptosis and invasion with methods mentioned above.
RT-qPCR
After the extraction of total RNA with TRIzol™ Reagent (Invitrogen, USA), the reverse transcription of cDNA was performed by the application of ReverTra Ace® qPCR RT Kit (Toyobo, Japan). Then the qPCR was carried out using ReverTra Ace® qPCR RT Kit (Toyobo, Japan). The relative expression of AKIP1 was calculated using 2-ΔΔCt with GAPDH serving as the internal reference. The primers sequences were listed as follows: AKIP1, forward primer: 5’ AGAACATCTCTAAGGACCTCTACAT 3’; reverse primer: 5’ TCCAGAATCAACTGCTACCACAT 3’; GAPDH, forward primer: 5’ GACCACAGTCCATGCCATCAC 3’; reverse primer: 5’ ACGCCTGCTTCACCACCTT 3’.
Western blot
The total protein was extracted with RIPA Buffer (Sigma, USA). Protein concentration was assessed using a Pierce™ BCA Protein Assay Kit (Thermo, USA). After thermal denaturation, 20 μg protein was separated by TruPAGE™ Precast Gels (Sigma, USA) and transferred onto polyvinylidene fluoride membrane (PALL, USA). After blocking with 5% BSA (Sigma, USA), the membrane was incubated with primary antibodies at 4°C overnight. Subsequently, secondary antibody was applied to incubate with the membrane at 37°C for 1 h. Finally, a Novex™ECL Chemiluminescent Substrate Reagent Kit (Invitrogen, USA) was used to detect the immunoreactive bands. The antibodies applied in western blot were listed in Table 1.
Table 1.
Antibodies used in western blot
| Antibody | Company | Dilution |
|---|---|---|
| Primary Antibody | ||
| Rabbit polyclonal to AKIP1 | Abcam (UK) | 1:500 |
| Rabbit monoclonal to NF-kB p65 | Abcam (UK) | 1:2000 |
| Rabbit monoclonal to p-NF-kB p65 | Abcam (UK) | 1:1000 |
| Rabbit polyclonal to AKT | Abcam (UK) | 1:500 |
| Rabbit polyclonal to p-AKT | Abcam (UK) | 1:500 |
| Rabbit monoclonal to PD-L1 | Abcam (UK) | 1:1000 |
| Rabbit polyclonal to GAPDH | CST (USA) | 1:1000 |
| Secondary Antibody | ||
| Goat Anti-Rabbit IgG-HRP | CST (USA) | 1:3000 |
Clinical samples and detections
After the ethics approval by our institution and informed consents obtained from patients or their families, 10 tumor samples from GBM patients and 10 tumor samples from age/gender matched low-grade glioma (LGG) patients were retrieved from Specimen Room. Then AKIP1 CXCL1, CXCL2 and CXCL8 expressions in tumor tissues were detected by immunohistochemistry (IHC), then scored by a semi-quantative method according to a previous study [14]. The antibodies information were as follows: Rabbit AKIP1 Polyclonal Antibody (1:50 dilution, Invitrogen, USA); Rabbit CXCL1 Polyclonal Antibody (1:100 dilution, Invitrogen, USA); Rabbit CXCL2 Recombinant Monoclonal Antibody (1:50 dilution, Invitrogen, USA); Rabbit CXCL8 Polyclonal Antibody (1:50 dilution, Invitrogen, USA); horseradish peroxidase-conjugated Goat anti-Rabbit IgG (H+L) secondary antibody (1:10000 dilution, Invitrogen, USA).
Statistical analysis
Data was described as mean with standard deviation (SD). GraphPad Prism Software version 7.02 (GraphPad Software Inc., USA) was used for data analyses and graph plotting. One-way ANOVA followed by Dunnett’s multiple comparisons test was used to determine comparisons between control and other groups. One-way ANOVA followed by Tukey’s multiple comparisons test was used to determine multiple comparisons among groups. Unpaired t test was used to compare data between GBM and LGG patients. Pearson correlation test was used to analyze the correlation among indexes in GBM patients. P value <0.05 was considered as significant. P value >0.05, <0.05, <0.01, and <0.001 were respectively marked as no significance (NS), *, **, and ***.
Results
The effect of AKIP1 modification on GBM proliferation, apoptosis and invasion
AKIP1 expression was elevated in GBM cell lines compare to control cell line (NHA) (Supplementary Figure 1A, 1B). After transfection of pcDNA and pGPH1 vectors, AKIP1 was increased in pcDNA-AKIP1 group compared to pcDNA-NC group, while decreased in pGPH1-AKIP1 group compared to pGPH1-NC group in both U-87 MG cells (Figure 1A, 1B) and U-251 MG cells (Figure 1H, 1I).
Figure 1.

Cell proliferation, apoptosis and invasion after transfection. AKIP1 mRNA expression (A) and protein expression (B) among groups in U-87 MG cells; Cell proliferation (C), cell apoptosis rate (D, E), invasive cell count (F, G) among groups in U-87 MG cells. AKIP1 mRNA expression (H) and protein expression (I) among groups in U-251 MG cells; Cell proliferation (J), cell apoptosis rate (K, L), invasive cell count (M, N) among groups in U-251 MG cells. AKIP1, A-kinase-interacting protein 1; NC, negative control; AV, Annexin V; PI, prodium Iodide.
Importantly, U-87 MG cell proliferation (Figure 1C), cell invasion (Figure 1F, 1G) were enhanced, while cell apoptosis rate (Figure 1D, 1E) was reduced in pcDNA-AKIP1 group compared to pcDNA-NC group; oppositely, U-87 MG cell proliferation (Figure 1C), cell invasion (Figure 1F, 1G) were attenuated, while cell apoptosis rate (Figure 1D, 1E) was promoted in pGPH1-AKIP1 group compared to pGPH1-NC group. These indexes among groups showed similar treads in U251 MG cells as well (Figure 1J-N), except for no difference of cell invasion between pcDNA-AKIP1 group and pcDNA-NC group. These data indicated that AKIP1 promoted GBM proliferation and invasion but inhibit its apoptosis.
The effect of AKIP1 modification on GBM chemosensitivity to TMZ and cisplatin
The relative viability was enhanced in pcDNA-AKIP1 group compared to pcDNA-NC group, while reduced in pGPH1-AKIP1 group compared to pGPH1-NC group in U-87 MG cells under 40-320 μM TMZ treatment (Figure 2A). However, no difference of relative viability was observed among these four groups in U-87 MG cells under various concentrations of cisplatin treatment (Figure 2B), except for that relative viability was enhanced in pcDNA-AKIP1 group compared to pcDNA-NC group of U-87 MG cells under 40 μM cisplatin treatment. In addition, the above findings were also validated in another GBM cell line (U-251 MG cells) (Figure 2C, 2D).
Figure 2.
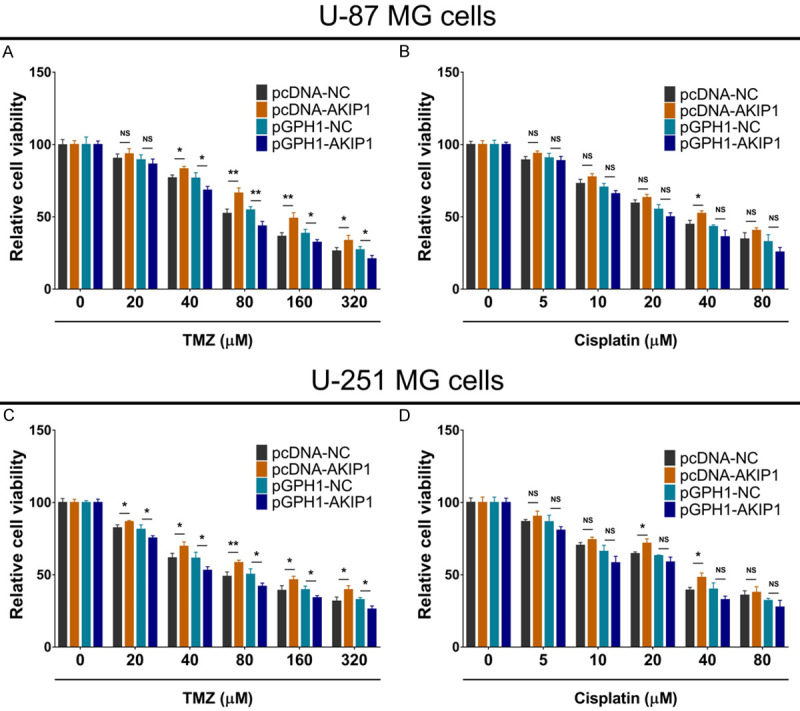
Relative cell viability under chemotherapy after transfection. Relative cell viability under 0-320 μM TMZ treatment (A) or 0-80 μM cisplatin treatment (B) among groups in U-87 MG cells. Relative cell viability under 0-320 μM TMZ treatment (C) or 0-80 μM cisplatin treatment (D) among groups in U-251 MG cells. AKIP1, A-kinase-interacting protein 1; NC, negative control; TMZ, temozolomide.
Subsequently, we detected cell apoptosis rate in each group after TMZ and cisplatin treatment with IC50 concentration, respectively. Then we found that under 80 μM TMZ treatment, both U-87 MG cell and U-251 MG cell apoptosis rates were inhibited in pcDNA-AKIP1 group compared to pcDNA-NC group, while promoted in pGPH1-AKIP1 group compared to pGPH1-NC group (Figure 3A, 3B, 3E and 3F). However, under 40 μM cisplatin treatment, both U-87 MG cell and U-251 MG cell apoptosis rates were of no difference between pcDNA-AKIP1 group and pcDNA-NC group, but was elevated in pGPH1-AKIP1 group compared to pGPH1-NC group (Figure 3C, 3D, 3G and 3H).
Figure 3.

Cell apoptosis rate under chemotherapy after transfection. Cell apoptosis rate under 80 μM TMZ treatment (A, B) or 40 μM cisplatin treatment (C, D) among groups in U-87 MG cells. Cell apoptosis rate under 80 μM TMZ treatment (E, F) or 40 μM cisplatin treatment (G, H) among groups in U-251 MG cells. AKIP1, A-kinase-interacting protein 1; NC, negative control; TMZ, temozolomide; AV, Annexin V; PI, prodium Iodide.
These data suggested that AKIP1 enhanced GBM resistance to TMZ but not cisplatin.
The effect of AKIP1 modification on GBM radiation sensitivity
The relative viability was enhanced in pcDNA-AKIP1 group compared to pcDNA-NC group, while reduced in pGPH1-AKIP1 group compared to pGPH1-NC group, in U-87 MG cells underwent 4-10 Gy radiation treatment and in U-251 MG cells underwent 2-10 Gy radiation treatment (Figure 4A, 4D). Furthermore, under 6 Gy (IC50 dose) radiation treatment, cell apoptosis was repressed in pcDNA-AKIP1 group compared to pcDNA-NC group, while enhanced in pGPH1-AKIP1 group compared to pGPH1-NC group (Figure 4B, 4C, 4E and 4F). These data implied that AKIP1 promoted GBM resistance to radiation treatment.
Figure 4.
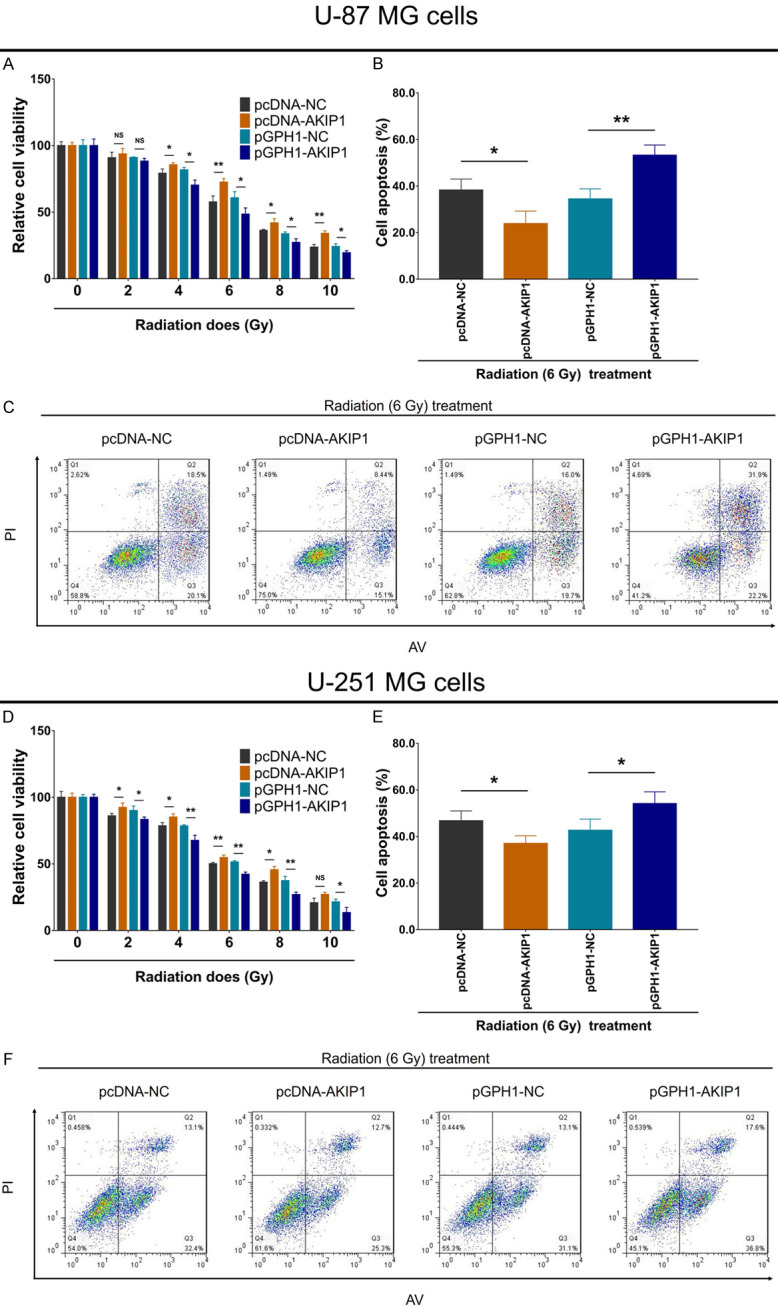
Relative cell viability and cell apoptosis rate under radiation therapy after transfection. Relative cell viability under 0-10 Gy radiation treatment (A), cell apoptosis rate under 6 Gy radiation treatment (B, C), among groups in U-87 MG cells. Relative cell viability under 0-10 Gy radiation treatment (D), cell apoptosis rate under 6 Gy radiation treatment (E, F), among groups in U-251 MG cells. AKIP1, A-kinase-interacting protein 1; NC, negative control; TMZ, temozolomide.
The effect of AKIP1 modification on CXCL1/2/8, NF-κB, AKT and PD-L1 expressions in GBM
In U-87 MG and U-251 MG cells, CXCL1 and CXCL8 were both elevated in pcDNA-AKIP1 group compared to pcDNA-NC group, while were reduced in pGPH1-AKIP1 group compared to pGPH1-NC group (Figure 5A, 5C, 5E and 5G). However, CXCL2 was increased in cDNA-AKIP1 group compared to pcDNA-NC group, while was decreased in pGPH1-AKIP1 group compared to pGPH1-NC group in U-251 MG cells (Figure 5F) but not in U-87 MG cells (Figure 5B). These indicated that AKIP1 positively regulated CXCL1, CXCL2 (weak effect) and CXCL8 in GBM.
Figure 5.

CXCL1/2/8, NF-κB, AKT and PD-L1 expressions after transfection. Supernatant CXCL1 (A), CXCL2 (B), CXCL8 (C) levels, and cell NF-κB p65, p-NF-κB p65, AKT, p-AKT, PD-L1 (D) expressions among groups in U-87 MG cells. Supernatant CXCL1 (E), CXCL2 (F), CXCL8 (G) levels, and cell NF-κB p65, p-NF-κB p65, AKT, p-AKT, PD-L1 (H) expressions among groups in U-251 MG cells. AKIP1, A-kinase-interacting protein 1; NC, negative control; CXCL, C-X-C motif chemokine ligand.
Furthermore, in U-87 MG (Figure 5D) and U-251 MG (Figure 5H) cells, p-NF-κB p65, p-AKT and PD-L1 expressions were all raised in pcDNA-AKIP1 group compared to pcDNA-NC group, but reduced in pGPH1-AKIP1 group compared to pGPH1-NC group. These indicated that AKIP1 activated NF-κB and AKT pathways, meanwhile increased PD-L1 expression.
Interaction of CXCL1/2/8 with AKIP1 knockdown on GBM proliferation, apoptosis and invasion
In order to further evaluate the interaction of AKIP1 with CXCL1/2/8 and their effects on regulating GBM malignant behaviors as well as chemoradiation sensitivity, multiple compensative experiments were conducted.
In both U-87 MG (Figure 6A-C) and U-251 MG (Figure 6D-F) cells, CXCL1 and CXCL8 enhanced cell proliferation, inhibited apoptosis, while CXCL2 did not affect them. Furthermore, in pGPH1-AKIP1 treated U-87 MG (Figure 6A-C) and U-251 MG (Figure 6D-F) cells, CXCL1, CXCL2 and CXCL8 all promoted cell proliferation, while CXCL1 and CXCL8 but not CXCL2 reduced cell apoptosis.
Figure 6.
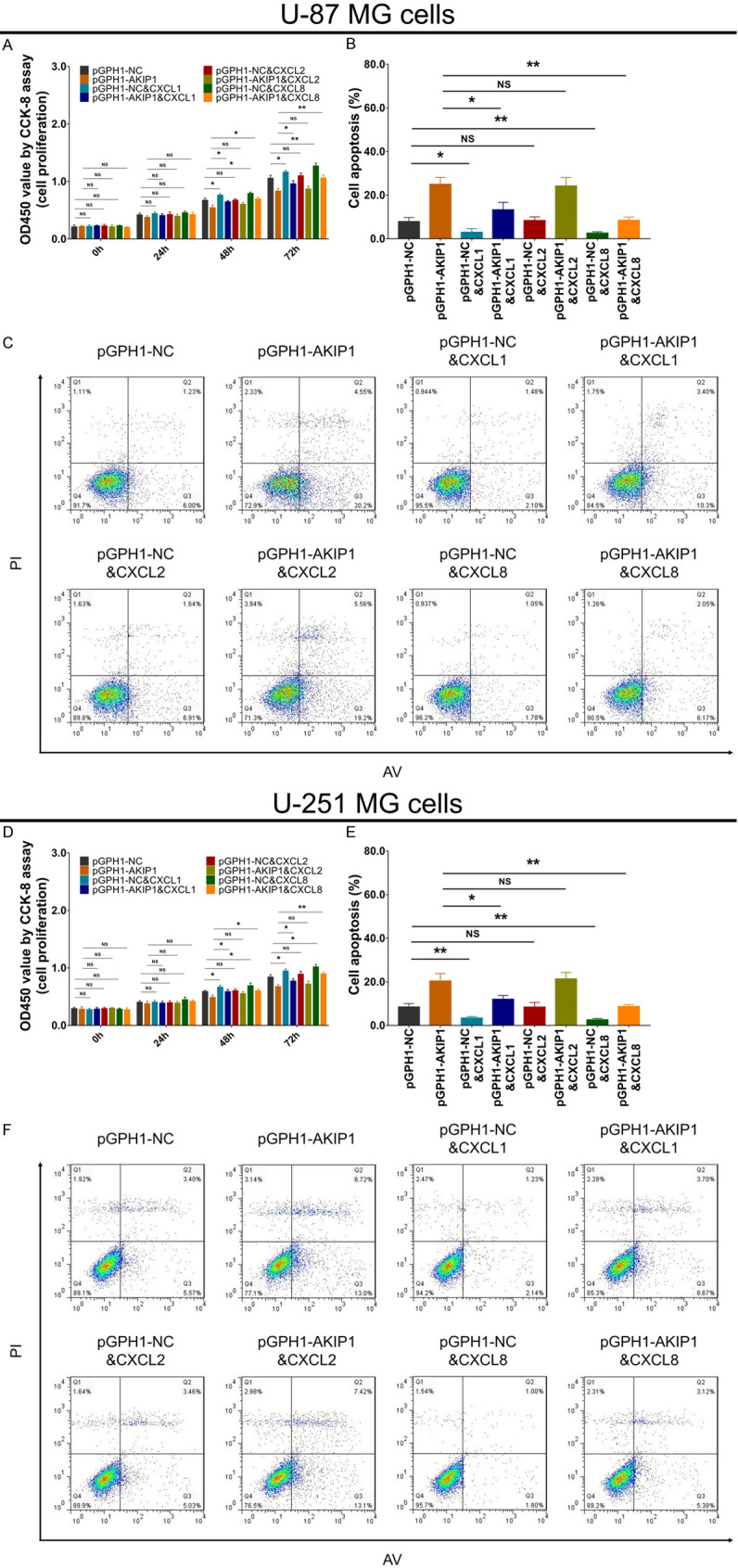
Cell proliferation and apoptosis in multiple compensative experiments. Cell proliferation (A) and cell apoptosis rate (B, C) among groups in U-87 MG cells. Cell proliferation (D) and cell apoptosis rate (E, F) among groups in U-251 MG cells. AKIP1, A-kinase-interacting protein 1; NC, negative control; CXCL, C-X-C motif chemokine ligand; AV, Annexin V; PI, prodium Iodide.
As to invasion ability, in both U-87 MG (Figure 7A, 7B) and U-251 MG (Figure 7C, 7D) cells, CXCL1 and CXCL8 promoted cell invasion, while CXCL2 did not work. In addition, in pGPH1-AKIP1 treated U-87 MG (Figure 7A, 7B) and U-251 MG (Figure 7C, 7D) cells, CXCL1 and CXCL8 also accelerated cell invasion, but CXCL2 could not.
Figure 7.
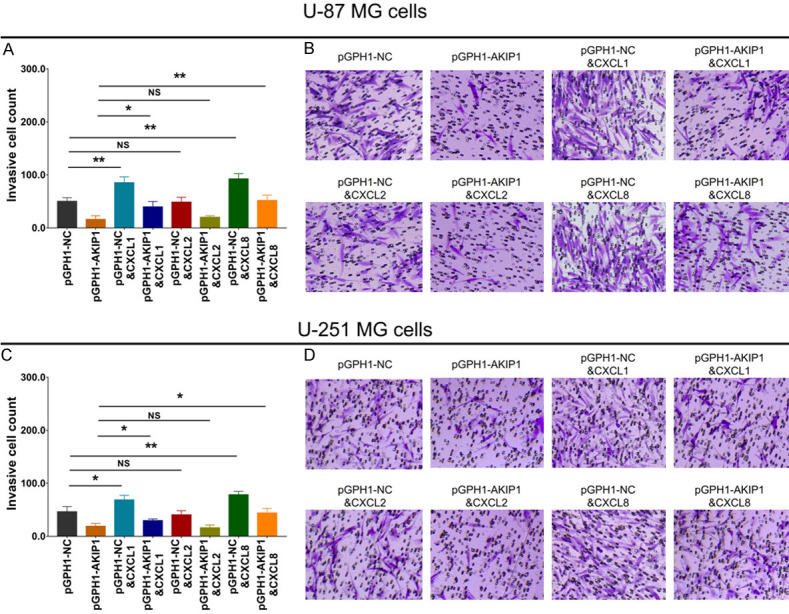
Cell invasion in multiple compensative experiments. Invasive cell count (A, B) among groups in U-87 MG cells. Invasive cell count (C, D) among groups in U-251 MG cells. AKIP1, A-kinase-interacting protein 1; NC, negative control; CXCL, C-X-C motif chemokine ligand.
Findings from compensative experiments indicated that AKIP1 regulated GBM proliferation, apoptosis and invasion via stimulating CXCL1 and CXCL8.
Interaction of CXCL1/2/8 with AKIP1 knockdown on GBM chemoradiation sensitivity
In U-87 MG cells (Figure 8A-C), CXCL1 and CXCL8 increased relative cell viability under 80 μM TMZ treatment, 40 μM cisplatin treatment, or 6 Gy radiation treatment; while CXCL2 only increased relative cell viability under 80 μM TMZ treatment, but not 40 μM cisplatin treatment, or 6 Gy radiation treatment. In pGPH1-AKIP1 treated U-87 MG cells (Figure 8A-C), CXCL1 only increased relative cell viability under 80 μM TMZ treatment, but not 40 μM cisplatin treatment or 6 Gy radiation treatment; CXCL2 did not affect the relative cell viability under 80 μM TMZ treatment, 40 μM cisplatin treatment or 6 Gy radiation treatment; notably, CXCL8 increased relative cell viability under 80 μM TMZ treatment, 40 μM cisplatin treatment, and 6 Gy radiation treatment. As to in U-251 MG cells, the related findings were similar as in U-87 MG cells (Figure 8D-F). At the same time, the related findings in pGPH1-AKIP1 treated U-251 MG cells were similar as in pGPH1-AKIP1 treated U-87 MG cells (Figure 8D-F); however, the effects of CXCL1, CXCL2 and CXCL8 were less.
Figure 8.
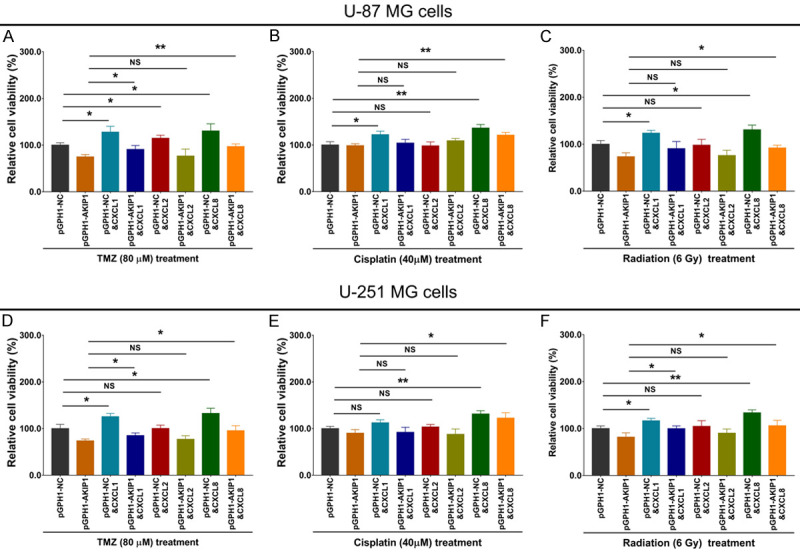
Chemoradiation sensitivity in multiple compensative experiments. Relative cell viability under 80 μM TMZ treatment (A), or 40 μM cisplatin treatment (B), or 6 Gy radiation treatment (C) among groups in U-87 MG cells. Relative cell viability under 80 μM TMZ treatment (D), or 40 μM cisplatin treatment (E), or 6 Gy radiation treatment (F) among groups in U-251 MG cells. AKIP1, A-kinase-interacting protein 1; NC, negative control; CXCL, C-X-C motif chemokine ligand; TMZ, temozolomide.
Interaction of CXCL1/2/8 with AKIP1 knockdown on NF-κB, AKT and PD-L1 expressions in GBM
In both U-87 MG (Figure 9A) and U-251 MG (Figure 9B) cells, CXCL1 and CXCL8 increased p-NF-κB p65, p-AKT and PD-L1 expressions, while CXCL2 could not. Furthermore, in pGPH1-AKIP1 treated U-87 MG (Figure 9A) and U-251 MG (Figure 9B) cells, CXCL1 and CXCL8 also elevated p-NF-κB p65, p-AKT and PD-L1 expressions, but CXCL2 lacked effect. As for the detailed molecule mechanism between NF-κB/AKT and PD-L1 expression, it has been disclosed by Kyoto Encyclopedia of Genes and Genomes (KEGG) database (https://www.kegg.jp/) that NF-κB directly regulates PD-L1 expression, while AKT indirectly regulates PD-L1 expression via activating IKK and subsequent NF-κB (Supplementary Figure 2). These data of compensative experiments suggested that AKIP1 regulated NF-κB pathway, AKT pathway and PD-L1 expression via stimulating CXCL1 and CXCL8.
Figure 9.
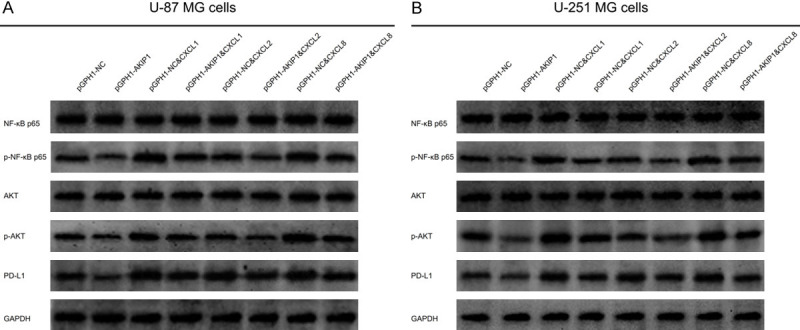
NF-κB, AKT and PD-L1 expressions in multiple compensative experiments. NF-κB p65, p-NF-κB p65, AKT, p-AKT, PD-L1 expressions among groups in U-87 MG cells (A) and U-251 MG cells (B). AKIP1, A-kinase-interacting protein 1; NC, negative control; CXCL, C-X-C motif chemokine ligand.
Validation of CXCL1 plus CXCL8 attenuating AKIP1 knockdown effect on GBM activities
CXCL1 plus CXCL8 simultaneous treatment greatly promoted GBM cell proliferation and invasion, as well as NF-κB and AKT pathways, while reduced cell apoptosis in pGPH1-AKIP1 treated U-87 MG (Supplementary Figure 3A-F) and U-251 MG (Supplementary Figure 3G-L) cells. Meanwhile, CXCL1 plus CXCL8 simultaneous treatment also increased relative cell viability under 80 μM TMZ, 40 μM cisplatin or 6 Gy radiation treatment in both pGPH1-AKIP1 treated U-87 MG (Supplementary Figure 4A-C) and U-251 MG (Supplementary Figure 4D-F) cells. Notably, pGPH1-AKIP1 did not affect relative GBM cell viability under cisplatin treatment (Supplementary Figure 4B, 4E). These data verified that AKIP1 regulated GBM viability, mobility and chemoradiation resistance via regulating CXCL1 and CXCL8.
LY and CAPE treatment crippled the effect of CXCL1&CXCL8 on GBM malignant behaviors
Both AKT pathway inhibitor (LY) and NF-κB pathway inhibitor (CAPE) reduced proliferation and invasion while enhanced apoptosis in CXCL1&CXCL8 treated U-87 MG cells (Supplementary Figure 5A-E) and U-87 MG cells (Supplementary Figure 5F-J). Combining the above all data, it was speculated that AKIP1 promoted GBM malignant behaviors and chemoradiation resistance via regulating CXCL1 and CXCL8 mediated NF-κB and AKT pathways.
Correlation of AKIP1 with CXCL1/2/8 in GBM clinical tumor tissues
In order to further validate the interaction of AKIP1 with CXCL1/2/8, we further detected them in the GBM tumor samples and LGG tumor samples with the IHC examples presented in Figure 10A. Then we found that AKIP1, CXCL1 and CXCL8 were elevated in GBM tumor samples compared with LGG tumor samples; while CXCL2 showed a trend to be higher in GBM tumor samples compared with LGG tumor samples but without statistical significance (Figure 10B). Furthermore, AKIP1 was positively correlated with CXCL1 and CXCL8 in GBM tumor tissues, while only showed a trend to be correlated with CXCL2 but without statistical significance (Figure 10C).
Figure 10.
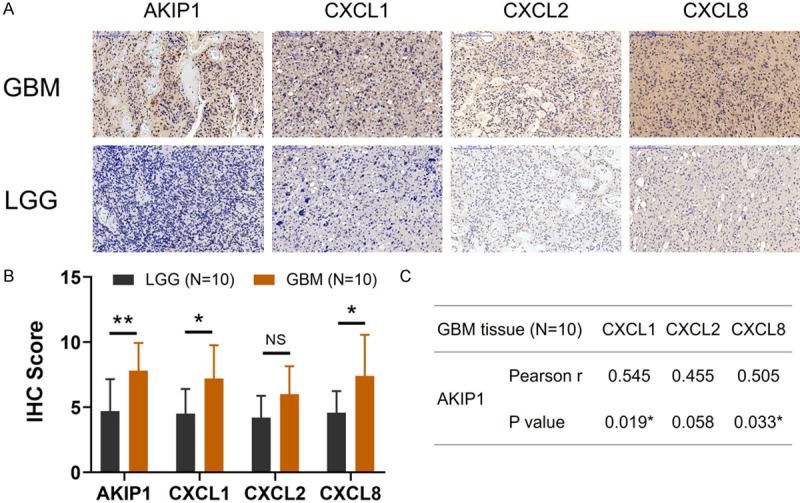
AKIP1 and CXCL1/2/8 expressions in GBM and LGG clinical tumor samples. Examples of IHC images about AKIP1, CXCL1, CXCL2, CXCL8 expressions in GBM and LGG clinical tumor samples (A). Comparison of AKIP1, CXCL1, CXCL2, CXCL8 expressions between GBM and LGG clinical tumor samples (B). Correlation of AKIP1 with CXCL1, CXCL2 and CXCL8 in GBM tumor samples (C). AKIP1, A-kinase-interacting protein 1; CXCL, C-X-C motif chemokine ligand; GBM, glioblastoma multiforme; LGG, low-grade glioma; IHC, immunohistochemistry.
In addition, we referred to GEPIA database (http://gepia.cancer-pku.cn/) to validate our findings, which was a newly developed interactive web server for analyzing the RNA sequencing expression data from the TCGA and the GTEx projects, using a standard processing pipeline. The findings were shown in Supplementary Figure 6, which observed that AKIP1, CXCL1, CXCL8 (but not CXCL2) were elevated in tumor tissue compared to non-tumor tissue of various cancers including GBM (Supplementary Figure 6A-D); furthermore, tumor AKIP1 positively correlated with CXCL1 and CXCL8 but not CXCL2 in GBM (Supplementary Figure 6E-G). These data further validated our study findings.
Discussion
GBM is the most deteriorative tumor among all primary brain tumors with an extremely pejorative prognosis, whose median survival time is only 12-15 months, and the 5-years survival rate only approximately 5% [1,2,35]. So as to improve the prognosis of GBM, several novel drugs have been introduced recently and now under clinical trials, such as cilengitide, bevacizumab, cediranib, enzastaurin, rindopepimut, etc. [36-40]. Encouragingly, PD-1/PD-L1 immune-checkpoint inhibitors are also proposed to treat GBM which shows some improvements on patients’ survival [41-43]. However, the prognosis of GBM is still bad mainly due the quick progress, treatment resistance and recurrence [1,2,44]. In order to shed light on the novel mechanisms and treatment targets of GBM, we performed the current study to investigate the effect of AKIP1 on GBM cell malignant behaviors and chemoradiotherapy sensitivity as well as CXCL1/2/8; meanwhile, to further explore their regulation on NF-κB pathway, AKT pathway and PD-L1 expression.
AKIP1 is firstly discovered as a factor interacting with the N-terminal 30 residues of PKA [45]; then it’s recognized as an important modifier of NF-κB signaling through its relationship with p65 and PKA [7,46]. Furthermore, due to its multiple phosphorylation sites and functional domains, AKIP1 interacts with multiple proteins, through which it functions in various biological processes and disease development/progression [47]. Recently, AKIP1 is also reported to be a key carcinogenic factor not only in solid tumors but also in hematological malignancies. For instance, AKIP1 enhances cervical cancer epithelial-mesenchymal transition and metastasis through activating PI3K/AKT pathway [48]. Meanwhile, AKIP1 activates Wnt/β-catenin/cyclic AMP response element-binding protein (CBP) pathway then accelerates hepatocellular carcinoma relapse [10]. Furthermore, AKIP1 overexpression could stimulate AKT/GSK-3β/Snail signaling and facilitates breast cancer metastasis [8]. Interestingly, AKIP1 also correlates with induction therapy response and disease progression in acute myeloid leukemia [49].
In our present study, AKIP1 overexpression was observed to promote GBM proliferation, invasion and inhibit apoptosis, while AKIP1 knockdown showed opposite trends. These could explained by: (1) AKIP could directly interacts with p65 and PKA to stimulate NF-κB pathway to exasperate these GBM malignant behaviors [7,12,45], which was further validated by our subsequent experiment data that AKIP1 activated NF-κB pathway in GBM; (2) AKIP1 might modify the AKT pathway to stimulate GBM viability and mobility [48], which was then validated by our subsequent experiments that AKIP1 activated AKT pathway in GBM; (3) AKIP1 would bind with multiple cancer-related proteins via its massive phosphorylation sites and functional domains, then enhanced the GBM malignant behaviors. Whereas, this hypothesis needed future works for validation [47]. Furthermore, our study also found that AKIP1 overexpression repressed GBM sensitivity to TMZ treatment and radiation treatment, while AKIP1 knockdown presented the reversed trends. The possible explanations might be that AKIP1 modified several treatment-resistance pathways such as NF-κB, AKT, β-catenin signaling pathways to inhibit GBM sensitivity to chemoradiation treatment [10,45,48].
As to the potential downstream factors of AKIP1, CXCL families have been introduced recently. For example, AKIP1 expedites cervical cancer viability and mobility in vitro, enhances angiogenesis and tumor growth in vivo via interaction with CXCL1, CXCL2, CXCL8 and NF-κB pathway [11]; meanwhile, AKIP1 inter-correlate with CXCL1, CXCL2, and is implicated in prostate cancer treatment outcomes [14]; Furthermore, AKIP1 also relates to CXCL1, CXCL2, which exhibits potential to be predictor for disease progression and survival [13]. More encouragingly, CXCL families are well-known for their tumor promoting roles in numerous cancers including GBM. For instance, CXCL1 promotes GBM proliferation and radiation resistance via activating NF-κB signaling [21]; CXCL2 upregulation is related to unfavorable outcomes in GBM; CXCL8 interacts with NF-κB pathway then involves in the treatment response and recurrence of GBM [22-24].
Considering the regulation network among AKIP1 and CXCL families (especially CXCL1, CXCL2 and CXCL8), as well as their involvements in carcinogenic pathways such as NF-κB, AKT, β-catenin, etc. [7,10-14,22-24,45,47,48], we further analyzed their interactions in GBM and effects on GBM malignant behaviors. Interestingly, we observed that AKIP1 positively regulated CXCL1, CXCL2 and CXCL8 expressions in GBM, while the latter three did not affect AKIP1 expression. These might due to the regulation network of AKIP1 on pathways such as NF-κB, AKT and so on, and CXCL1, CXCL2 as well as CXCL8 are closely related to these pathways, thus, AKIP1 positively regulated these three chemokines [7,10-14,22-24,45,47,48]. Furthermore, we observed the addition of CXCL1, CXCL2 (the effect was weak) and CXCL8 respectively reversed the effect of AKIP1 knockdown on GBM cell behaviors to some extents by compensative experiments, which suggested that AKIP1 modified GBM malignant behaviors via promoting CXCL1, CXCL2 and CXCL8. These findings might benefit from (1) The inter-correlation of AKIP1 with CXCL1, CXCL2 and CXCL8 in cancers; (2) The modification of CXCL1, CXCL2 and CXCL8 on several critical oncogenic pathways including NF-κB, AKT, etc. [21-28]. In addition, we also discovered that AKIP1 modified GBM chemoradiation sensitivity via promoting CXCL1, CXCL2 and CXCL8, which might due to their inter-regulation relationship and the effects of CXCL families on cancer treatment sensitivities [25,27,28]. These findings were subsequently validated by our clinical explorations with GBM patients, which exhibited that AKIP1 expression positively correlated with CXCL1, CXCL2 (weak correlation), CXCL8 in GBM tumor tissues, and they were all elevated in GBM tissue samples compared to LGG tissue samples.
In addition to the above findings, we also tried to investigate the regulated pathways (that involved in the tumor progression and treatment regulation) by AKIP1, CXCL1, CXCL2 and CXCL8, which showed that AKIP1 overexpression activated NF-κB pathway and AKT pathway, while its knockdown inactivated these pathways; besides, CXCL1, CXCL2 (the effect was weak) and CXCL8 treatments also stimulated these two pathways, then their addition would attenuate the regulation of AKIP1 knockdown on these two pathways as well. These findings were not surprising which were in accordant with previous studies about AKIP1, CXCL1, CXCL2 and CXCL8 on regulating NF-κB and AKT pathways in other cancers apart from GBM [7,12,21-28,45,48]. Besides, due to the implication of CXCL families with PD-1/PD-L1 linkage [20], as well as the involvement of pathways regulated by AKIP1 and CXCL families in PD-1/PD-L1 linkage [15-17], we also detected whether GBM PD-L1 expression was regulated by AKIP1, CXCL1, CXCL2 and CXCL8. We observed that AKIP1 overexpression increased while AKIP1 knockdown decreased PD-L1 expression in GBM, and CXCL1, CXCL2 (Weak effect) and CXCL8 additions could elevated PD-L1 expression in both treatment-naïve GBM and AKIP1 knockdown treated GBM. These findings might provide more evidence for AKIP1 as a potential treatment target in GBM.
In conclusion, AKIP1 promotes GBM viability, mobility and chemoradiation resistance via regulating CXCL1 and CXCL8 mediated NF-κB and AKT pathways.
Acknowledgements
This work was supported by the Natural Science Foundation of Heilongjiang Province (LH2020H044), the First Clinical College of Harbin Medical University Foundation (2013Lx03).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2011-2015. Neuro Oncol. 2018;20:iv1–iv86. doi: 10.1093/neuonc/noy131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ostrom QT, Cote DJ, Ascha M, Kruchko C, Barnholtz-Sloan JS. Adult glioma incidence and survival by race or ethnicity in the united states from 2000 to 2014. JAMA Oncol. 2018;4:1254–1262. doi: 10.1001/jamaoncol.2018.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kanderi T, Gupta V. Glioblastoma Multiforme. Treasure Island (FL): StatPearls; 2020. [PubMed] [Google Scholar]
- 4.Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol. 2018;15:422–442. doi: 10.1038/s41571-018-0003-5. [DOI] [PubMed] [Google Scholar]
- 5.Gao F, Cheng J, Shi T, Yeh ET. Neddylation of a breast cancer-associated protein recruits a class III histone deacetylase that represses NFkappaB-dependent transcription. Nat Cell Biol. 2006;8:1171–1177. doi: 10.1038/ncb1483. [DOI] [PubMed] [Google Scholar]
- 6.Yu KP, Itokawa T, Zhu ML, Syam S, Seth A, Insogna K. Breast cancer-associated gene 3 (BCA3) is a novel Rac1-interacting protein. J Bone Miner Res. 2007;22:628–637. doi: 10.1359/jbmr.070105. [DOI] [PubMed] [Google Scholar]
- 7.Gao N, Hibi Y, Cueno M, Asamitsu K, Okamoto T. A-kinase-interacting protein 1 (AKIP1) acts as a molecular determinant of PKA in NF-kappaB signaling. J Biol Chem. 2010;285:28097–28104. doi: 10.1074/jbc.M110.116566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mo D, Li X, Li C, Liang J, Zeng T, Su N, Jiang Q, Huang J. Overexpression of AKIP1 predicts poor prognosis of patients with breast carcinoma and promotes cancer metastasis through Akt/GSK-3beta/Snail pathway. Am J Transl Res. 2016;8:4951–4959. [PMC free article] [PubMed] [Google Scholar]
- 9.Lin C, Song L, Liu A, Gong H, Lin X, Wu J, Li M, Li J. Overexpression of AKIP1 promotes angiogenesis and lymphangiogenesis in human esophageal squamous cell carcinoma. Oncogene. 2015;34:384–393. doi: 10.1038/onc.2013.559. [DOI] [PubMed] [Google Scholar]
- 10.Cui Y, Wu X, Lin C, Zhang X, Ye L, Ren L, Chen M, Yang M, Li Y, Li M, Li J, Guan J, Song L. AKIP1 promotes early recurrence of hepatocellular carcinoma through activating the Wnt/beta-catenin/CBP signaling pathway. Oncogene. 2019;38:5516–5529. doi: 10.1038/s41388-019-0807-5. [DOI] [PubMed] [Google Scholar]
- 11.Leung TH, Ngan HY. Interaction of TAp73 and breast cancer-associated gene 3 enhances the sensitivity of cervical cancer cells in response to irradiation-induced apoptosis. Cancer Res. 2010;70:6486–6496. doi: 10.1158/0008-5472.CAN-10-0688. [DOI] [PubMed] [Google Scholar]
- 12.Zhang W, Wu Q, Wang C, Yang L, Liu P, Ma C. AKIP1 promotes angiogenesis and tumor growth by upregulating CXC-chemokines in cervical cancer cells. Mol Cell Biochem. 2018;448:311–320. doi: 10.1007/s11010-018-3335-7. [DOI] [PubMed] [Google Scholar]
- 13.Hao X, Gu M, Sun J, Cong L. A-kinase interacting protein 1 might serve as a novel biomarker for worse prognosis through the interaction of chemokine (C-X-C motif) ligand 1/chemokine (C-X-C motif) ligand 2 in acute myeloid leukemia. J Clin Lab Anal. 2020;34:e23052. doi: 10.1002/jcla.23052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang D, Luo Y, Guo Y, Li G, Li F. A-kinase interacting protein 1, a potential biomarker associated with advanced tumor features and CXCL1/2 in prostate cancer. Int J Biol Markers. 2020;35:74–81. doi: 10.1177/1724600820914944. [DOI] [PubMed] [Google Scholar]
- 15.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schildberg FA, Klein SR, Freeman GJ, Sharpe AH. Coinhibitory pathways in the B7-CD28 ligand-receptor family. Immunity. 2016;44:955–972. doi: 10.1016/j.immuni.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Betzler AC, Theodoraki MN, Schuler PJ, Doscher J, Laban S, Hoffmann TK, Brunner C. NF-kappaB and its role in checkpoint control. Int J Mol Sci. 2020;21:3949. doi: 10.3390/ijms21113949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee EY, Lee ZH, Song YW. The interaction between CXCL10 and cytokines in chronic inflammatory arthritis. Autoimmun Rev. 2013;12:554–557. doi: 10.1016/j.autrev.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 19.Takeyama Y, Kato M, Tamada S, Azuma Y, Shimizu Y, Iguchi T, Yamasaki T, Gi M, Wanibuchi H, Nakatani T. Myeloid-derived suppressor cells are essential partners for immune checkpoint inhibitors in the treatment of cisplatin-resistant bladder cancer. Cancer Lett. 2020;479:89–99. doi: 10.1016/j.canlet.2020.03.013. [DOI] [PubMed] [Google Scholar]
- 20.Inoue C, Miki Y, Saito R, Hata S, Abe J, Sato I, Okada Y, Sasano H. PD-L1 induction by cancer-associated fibroblast-derived factors in lung adenocarcinoma cells. Cancers (Basel) 2019;11:1257. doi: 10.3390/cancers11091257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alafate W, Li X, Zuo J, Zhang H, Xiang J, Wu W, Xie W, Bai X, Wang M, Wang J. Elevation of CXCL1 indicates poor prognosis and radioresistance by inducing mesenchymal transition in glioblastoma. CNS Neurosci Ther. 2020;26:475–485. doi: 10.1111/cns.13297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gabellini C, Castellini L, Trisciuoglio D, Kracht M, Zupi G, Del Bufalo D. Involvement of nuclear factor-kappa B in bcl-xL-induced interleukin 8 expression in glioblastoma. J Neurochem. 2008;107:871–882. doi: 10.1111/j.1471-4159.2008.05661.x. [DOI] [PubMed] [Google Scholar]
- 23.Wipfler K, Cornish AS, Guda C. Comparative molecular characterization of typical and exceptional responders in glioblastoma. Oncotarget. 2018;9:28421–28433. doi: 10.18632/oncotarget.25420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo X, Xu S, Zhong Y, Tu T, Xu Y, Li X, Wang B, Yang F. High gene expression levels of VEGFA and CXCL8 in the peritumoral brain zone are associated with the recurrence of glioblastoma: a bioinformatics analysis. Oncol Lett. 2019;18:6171–6179. doi: 10.3892/ol.2019.10988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wen Z, Liu Q, Wu J, Xu B, Wang J, Liang L, Guo Y, Peng M, Zhao Y, Liao Q. Fibroblast activation protein alpha-positive pancreatic stellate cells promote the migration and invasion of pancreatic cancer by CXCL1-mediated Akt phosphorylation. Ann Transl Med. 2019;7:532. doi: 10.21037/atm.2019.09.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu J, Zhu MD, Zhang X, Tian H, Zhang JH, Wu XB, Gao YJ. NFkappaB-mediated CXCL1 production in spinal cord astrocytes contributes to the maintenance of bone cancer pain in mice. J Neuroinflammation. 2014;11:38. doi: 10.1186/1742-2094-11-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilson C, Maxwell PJ, Longley DB, Wilson RH, Johnston PG, Waugh DJ. Constitutive and treatment-induced CXCL8-signalling selectively modulates the efficacy of anti-metabolite therapeutics in metastatic prostate cancer. PLoS One. 2012;7:e36545. doi: 10.1371/journal.pone.0036545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palomeras S, Diaz-Lagares A, Vinas G, Setien F, Ferreira HJ, Oliveras G, Crujeiras AB, Hernandez A, Lum DH, Welm AL, Esteller M, Puig T. Epigenetic silencing of TGFBI confers resistance to trastuzumab in human breast cancer. Breast Cancer Res. 2019;21:79. doi: 10.1186/s13058-019-1160-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu B, Wang H, Zhang L, Sun C, Li H, Jiang C, Liu X. High expression of RAD18 in glioma induces radiotherapy resistance via down-regulating P53 expression. Biomed Pharmacother. 2019;112:108555. doi: 10.1016/j.biopha.2019.01.016. [DOI] [PubMed] [Google Scholar]
- 30.Ye LY, Hu S, Xu HE, Xu RR, Kong H, Zeng XN, Xie WP, Wang H. The effect of tetrandrine combined with cisplatin on proliferation and apoptosis of A549/DDP cells and A549 cells. Cancer Cell Int. 2017;17:40. doi: 10.1186/s12935-017-0410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tilborghs S, Corthouts J, Verhoeven Y, Arias D, Rolfo C, Trinh XB, van Dam PA. The role of nuclear factor-kappa B signaling in human cervical cancer. Crit Rev Oncol Hematol. 2017;120:141–150. doi: 10.1016/j.critrevonc.2017.11.001. [DOI] [PubMed] [Google Scholar]
- 32.Tsubaki M, Takeda T, Noguchi M, Jinushi M, Seki S, Morii Y, Shimomura K, Imano M, Satou T, Nishida S. Overactivation of akt contributes to MEK inhibitor primary and acquired resistance in colorectal cancer cells. Cancers (Basel) 2019;11:1866. doi: 10.3390/cancers11121866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Searle EJ, Telfer BA, Mukherjee D, Forster DM, Davies BR, Williams KJ, Stratford IJ, Illidge TM. Akt inhibition improves long-term tumour control following radiotherapy by altering the microenvironment. EMBO Mol Med. 2017;9:1646–1659. doi: 10.15252/emmm.201707767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jin X, Ding D, Yan Y, Li H, Wang B, Ma L, Ye Z, Ma T, Wu Q, Rodrigues DN, Kohli M, Jimenez R, Wang L, Goodrich DW, de Bono J, Dong H, Wu H, Zhu R, Huang H. Phosphorylated RB promotes cancer immunity by inhibiting NF-kappaB activation and PD-L1 expression. Mol Cell. 2019;73:22–35. e26. doi: 10.1016/j.molcel.2018.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thakkar JP, Dolecek TA, Horbinski C, Ostrom QT, Lightner DD, Barnholtz-Sloan JS, Villano JL. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol Biomarkers Prev. 2014;23:1985–1996. doi: 10.1158/1055-9965.EPI-14-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nabors LB, Fink KL, Mikkelsen T, Grujicic D, Tarnawski R, Nam DH, Mazurkiewicz M, Salacz M, Ashby L, Zagonel V, Depenni R, Perry JR, Hicking C, Picard M, Hegi ME, Lhermitte B, Reardon DA. Two cilengitide regimens in combination with standard treatment for patients with newly diagnosed glioblastoma and unmethylated MGMT gene promoter: results of the open-label, controlled, randomized phase II CORE study. Neuro Oncol. 2015;17:708–717. doi: 10.1093/neuonc/nou356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taphoorn MJ, Henriksson R, Bottomley A, Cloughesy T, Wick W, Mason WP, Saran F, Nishikawa R, Hilton M, Theodore-Oklota C, Ravelo A, Chinot OL. Health-related quality of life in a randomized phase III study of bevacizumab, temozolomide, and radiotherapy in newly diagnosed glioblastoma. J. Clin. Oncol. 2015;33:2166–2175. doi: 10.1200/JCO.2014.60.3217. [DOI] [PubMed] [Google Scholar]
- 38.Batchelor TT, Sorensen AG, di Tomaso E, Zhang WT, Duda DG, Cohen KS, Kozak KR, Cahill DP, Chen PJ, Zhu M, Ancukiewicz M, Mrugala MM, Plotkin S, Drappatz J, Louis DN, Ivy P, Scadden DT, Benner T, Loeffler JS, Wen PY, Jain RK. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11:83–95. doi: 10.1016/j.ccr.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wick W, Puduvalli VK, Chamberlain MC, van den Bent MJ, Carpentier AF, Cher LM, Mason W, Weller M, Hong S, Musib L, Liepa AM, Thornton DE, Fine HA. Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J. Clin. Oncol. 2010;28:1168–1174. doi: 10.1200/JCO.2009.23.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, Ashby L, Mechtler L, Goldlust SA, Iwamoto F, Drappatz J, O’Rourke DM, Wong M, Hamilton MG, Finocchiaro G, Perry J, Wick W, Green J, He Y, Turner CD, Yellin MJ, Keler T, Davis TA, Stupp R, Sampson JH ACT IV trial investigators. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017;18:1373–1385. doi: 10.1016/S1470-2045(17)30517-X. [DOI] [PubMed] [Google Scholar]
- 41.Caccese M, Indraccolo S, Zagonel V, Lombardi G. PD-1/PD-L1 immune-checkpoint inhibitors in glioblastoma: a concise review. Crit Rev Oncol Hematol. 2019;135:128–134. doi: 10.1016/j.critrevonc.2018.12.002. [DOI] [PubMed] [Google Scholar]
- 42.Litak J, Mazurek M, Grochowski C, Kamieniak P, Rolinski J. PD-L1/PD-1 axis in glioblastoma multiforme. Int J Mol Sci. 2019;20:5347. doi: 10.3390/ijms20215347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, Wang AC, Ellingson BM, Rytlewski JA, Sanders CM, Kawaguchi ES, Du L, Li G, Yong WH, Gaffey SC, Cohen AL, Mellinghoff IK, Lee EQ, Reardon DA, O’Brien BJ, Butowski NA, Nghiemphu PL, Clarke JL, Arrillaga-Romany IC, Colman H, Kaley TJ, de Groot JF, Liau LM, Wen PY, Prins RM. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25:477–486. doi: 10.1038/s41591-018-0337-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Osuka S, Van Meir EG. Overcoming therapeutic resistance in glioblastoma: the way forward. J Clin Invest. 2017;127:415–426. doi: 10.1172/JCI89587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sastri M, Barraclough DM, Carmichael PT, Taylor SS. A-kinase-interacting protein localizes protein kinase A in the nucleus. Proc Natl Acad Sci U S A. 2005;102:349–354. doi: 10.1073/pnas.0408608102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gao N, Asamitsu K, Hibi Y, Ueno T, Okamoto T. AKIP1 enhances NF-kappaB-dependent gene expression by promoting the nuclear retention and phosphorylation of p65. J Biol Chem. 2008;283:7834–7843. doi: 10.1074/jbc.M710285200. [DOI] [PubMed] [Google Scholar]
- 47.Yao C, Yu KP, Philbrick W, Sun BH, Simpson C, Zhang C, Insogna K. Breast cancer-associated gene 3 interacts with Rac1 and augments NF-kappaB signaling in vitro, but has no effect on RANKL-induced bone resorption in vivo. Int J Mol Med. 2017;40:1067–1077. doi: 10.3892/ijmm.2017.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang X, Liu S, Zhu Y. A-kinase-interacting protein 1 promotes EMT and metastasis via PI3K/Akt/IKKbeta pathway in cervical cancer. Cell Biochem Funct. 2020;38:782–791. doi: 10.1002/cbf.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yan Y, Li X, Gao J. Identification of A-kinase interacting protein 1 as a potential biomarker for risk and prognosis of acute myeloid leukemia. J Clin Lab Anal. 2020;34:e23055. doi: 10.1002/jcla.23055. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.