

Abstract
Systemic inflammation-related etiologic pathways via inflammatory cytokines in the development of colorectal cancer (CRC) have not been convincingly determined and may be confounded by lifestyle factors or reverse causality. We investigated the genetically predicted C-reactive protein (CRP) phenotype in the potential causal pathway of primary CRC risk in postmenopausal women in a Mendelian randomization (MR) framework. We employed individual-level data of the Women’s Health Initiative Database for Genotypes and Phenotypes Study, which consists of 5 genome-wide association (GWA) studies, including 10,142 women, 737 of whom developed primary CRC. We examined 61 GWA single-nucleotide polymorphisms (SNPs) associated with CRP by using weighted/penalized MR weighted-medians and MR gene-environment interactions that allow some relaxation of the strict variable requirements and attenuate the heterogeneous estimates of outlying SNPs. In lifestyle-stratification analyses, genetically determined CRP exhibited its effects on the decreased CRC risk in non-viscerally obese and high-fat diet subgroups. In contrast, genetically driven CRP was associated with an increased risk for CRC in women who smoked ≥ 15 cigarettes/day, with significant interaction of the gene-smoking relationship. Further, a substantially increased risk of CRC induced by CRP was observed in relatively short-term users (< 5 years) of estrogen (E)-only and also longer-term users (5 to > 10 years) of E plus progestin. Our findings may provide novel evidence on immune-related etiologic pathways connected to CRC risk and suggest the possible use of CRP as a CRC-predictive biomarker in women with particular behaviors and CRP marker-informed interventions to reduce CRC risk.
Keywords: Genetically driven C-reactive protein, mendelian randomization, abdominal obesity, saturated fatty acids, smoking, exogenous estrogen, colorectal cancer
Introduction
Colorectal cancer (CRC) in postmenopausal women ages 50 years and older accounts for the majority (approximately 90%) of newly diagnosed female CRC cases and related deaths [1], thus mainly contributing to CRC’s third ranking in cancer incidence and mortality in women of the United States and Westernized countries [2,3]. Of the established risk factors, the inflammatory process plays a critical role in promoting CRC development [4]. In particular, inflammation-induced colorectal carcinogenesis can be driven by local inflammation of the colorectal mucosa owing to inflammatory bowel disease [5-7]. Further, as supported by evidence that reduction of systemic inflammation has the tissue-specific effect of reduced inflammation in the colorectal mucosa [8], systemic inflammation is considered one of the driving mechanisms behind CRC development [9,10]. However, whether systemic inflammation has a similar carcinogenic effect via inflammatory cytokines produced in an innate immunity response to the inflammation is less certain. In particular, C-reactive protein (CRP), a major acute-phase reactant synthesized in response to acute and chronic low-grade inflammation, is postulated as a key pro-inflammatory biomarker that has been implicated in colorectal carcinogenesis [11-15]. The molecular carcinogenic mechanism by which an elevated circulating level of CRP influences the risk for CRC is only partially understood. For example, CRP acts as a lectin-like oxidized low-density lipoprotein receptor-I (LOX-I) ligand, elevating the mRNA expression of the LOX-I gene that is important to CRC growth in CRC cell lines [12]. In addition, CRP directly interacts with higher levels of lipopolysaccharide (LPS) immunoglobulin and LPS-binding protein (LBP), biomarkers of gut hyperpermeability, inducing increased systemic exposure to intestinal bacterial products [16,17]; CRP then alters the gut microbiome, gradually forming a microenvironment that is an important step in colorectal carcinogenesis [13-15].
Higher levels of CRP due to local inflammation have been associated with a high risk of CRC [5,18]. However, findings from previous observational and genetic epidemiologic studies of elevated circulating CRP concentrations not directly related to local inflammation, and CRC risk are mixed: some positive associations with CRC [19-21] and advanced colorectal adenoma [22-24], null results [10,25,26], and inverse relationships [27-29]. Of note, few studies [10,21] of the association between the plasma level of CRP and CRC risk found significant relationships among only subjects with a short period of follow-up (< 2 and 5 years), suggesting the potential existence of reverse causality between CRP and CRC risk. Overall, the conflict between those findings may be partly explained by selection bias, confounding and/or interaction effects of lifestyle factors, relatively short-time exposure to the biomarker, and reverse causation; it thus calls for in-depth research, for example, Mendelian randomization (MR) study, which can address those challenges and potentially provide improved causal inference of the immune-related etiologic pathways connected to CRC risk.
MR examines the exposure (e.g., CRP) on an outcome (e.g., CRC risk) using genetic variants as an “instrumental” variable [30]. This approach may establish a relatively unbiased causal inference between CRP and CRC risk by first reducing potential biases and confounding given random assignment of exposure owing to randomly assorted genetic alleles at the time of gamete formation; the overall observed allelic effects are thus less likely to be influenced by environmental factors. Second, MR can address short-term exposures to inflammatory biomarkers by examining the associated alleles as a proxy for lifelong exposure [30,31]. Third, MR may also reduce the potential of reverse causation because alleles precede the phenotype and relevant clinical outcomes [31]; thus, in an MR framework, the elevated level of CRP is less likely to result from the immune response caused by premalignant or preclinical tumor growth. Finally, an MR study can be comparable to randomized clinical trials in inferring a causal relationship when the genetic instruments are fulfilled the conditions required for genomic validity in MR analysis [30]. To date, few MR studies have evaluated genetically determined CRP in relation to CRC risk, with inconsistent reports: 1 null result [32] and 2 positive relationships [21,33].
In the study detailed here, we conducted MR analysis with a focus on postmenopausal women, a vulnerable population with a high incidence of inflammation [34] and CRC. We incorporated a large number of genome-wide single-nucleotide polymorphisms (SNPs) obtained from our earlier and other genome-wide association studies (GWASs) [35-38]. As in previous MR studies, we used a traditional MR estimate from the inverse-variance weighed (IVW) method. Further, we performed more recently developed analyses, including weighted median (WM) and penalized weighted median (PWM) estimates [39,40], and also estimated corrected MR that accounts for gene-environment (G×E) interactions [41]. These approaches allow some relaxation of the strict rules for MR instrumental variables by down-weighting the contribution of heterogeneity of genetic variants to analysis and by incorporating interactions between genes and lifestyles into our MR analysis. We ultimately tested the hypothesis that genetically predicted CRP that interacts with lifestyle factors is causally associated with CRC risk.
Materials and methods
Study population
The data of our study population were obtained from the Women’s Health Initiative (WHI) Database for Genotypes and Phenotypes (dbGaP), which encompasses the 2 WHI study arms, Clinical Trials and Observation Studies, representing one of the largest studies to date on postmenopausal women in the U.S. In particular, we used the biggest substudies of the WHI dbGaP, the Harmonized and Imputed GWASs, which contribute to a joint genomic effort across the 5 WHI GWASs (Table S1; AS264, GARNET, GECCO, HIPFX, and WHIMS). Details on the study designs and rationale are available elsewhere [42]. Briefly, the WHI is a long-term prospective cohort study focusing on strategies for preventing diseases among postmenopausal women. Healthy women were enrolled in the WHI study between 1993 and 1998 at more than 40 clinical centers across the U.S. if they were 50-79 years old, postmenopausal, expected to stay near the clinical centers for at least 3 years after enrollment, and able to provide written informed consent. Participants were eligible for the WHI dbGaP study if they had met the eligibility requirements for data submission to dbGaP and had provided DNA samples. Of 16,088 women in total who reported their race/ethnicity as non-Hispanic white, we applied multilevel exclusion criteria: in the GWAS for CRP, genomic data quality control (QC), resulting in 10,798 women (Table S1); and in the associations between GWAS CRP-SNPs and CRC risk, < 1 year follow-up for cancer outcomes and a diagnosis of any cancer type at screening, finally leading to a total of 10,142 women. They had received follow-ups through August 29, 2014, with a mean follow-up of 16 years, and 737 (7% of the eligible 10,142 women) had developed primary CRC. The studies were approved by the institutional review boards of each WHI clinical center and the University of California, Los Angeles.
Lifestyle variables and CRC outcome
Participants had completed self-administered questionnaires at screening, providing demographic and lifestyle information, and data collection was periodically monitored by the WHI coordinating clinical centers for data QC. With 41 variables initially selected from literature review [11,43-61] for their association with inflammation and CRC, we conducted preliminary analyses such as univariate and multivariate/stepwise regressions and a multicollinearity test, resulting in a final selection of the following 16 variables for our analysis: demographic and socioeconomic factors (age, education, and family income); a family history of CRC; comorbidity (cardiovascular disease ever and high cholesterol requiring medication ever); behaviors (exercise and cigarette smoking); dietary factors (daily alcohol and saturated fatty acids [SFA] intake); and reproductive history (age at menopause, durations of past oral contraceptive [OC] use and of exogenous estrogen [E]-only and E plus progestin [E+P] use). Trained staff had measured anthropometric characteristics, including height, weight, and waist and hip circumferences.
For our lifestyle-stratified analyses for the G×E tests, the following variables as potential effect modifiers on the basis of previous studies [12,22-24,29,57,62-70] and relevant cutoff values were used: 30 kg/m2 body mass index (BMI), 88 cm waist circumference, 0.85 waist-to-hip ratio (WHR), 10 hours/week metabolic equivalents (METs), and 9% calories/day from SFA on the basis of obesity and relevant lifestyle guidelines [71-73]; at least 15 cigarettes/day; 14 g alcohol intake/day (1 drink standard for women) [74]; a 5-year interval of E-only/E+P use ranging from never used to used 15 years or longer; and a 7-year median of OC use.
CRC outcomes were determined by a committee of physicians’ review of medical records, pathology and cytology reports, and tumor registry abstracts, and finally coded into the central WHI database according to the National Cancer Institute’s Surveillance, Epidemiology, and End-Results guidelines [75]. The time from enrollment to CRC development, censoring, or study end point was calculated and presented in years for the purpose of analysis.
Genotyping and instrumental variables
Genotyped data were derived from the WHI GWASs database in dbGaP, which had been normalized to the Genome Reference Consortium Human Build 37 and imputed using the 1000 Genomes reference panel [76]. Details of the data-cleaning process have been discussed previously [35,42,77]. In brief, DNA was obtained from blood samples at baseline and genotyped using several GWAS platforms [42]. The SNPs’ harmonization was checked via pairwise concordance among all samples across the GWASs. Through the first and second data QC steps, SNPs were filtered on a missing call rate of < 2%, a Hardy-Weinberg equilibrium of P ≥ 1E-04, and Ȓ 2 ≥ 0.6 imputation quality [78]. Individuals with unexpected duplicates, first- and second-degree relatives, and outliers on the basis of genetic principal components (PCs) were excluded.
We selected GWA CRP-SNPs from 4 GWASs. Our earlier GWAS [35] used the WHI Harmonized and Imputed GWASs data for the association with CRP as a binary outcome (> 3.0 mg/L, reflecting chronic low-grade inflammation status [79,80]). We used results from this earlier GWAS; of 82 SNPs, 5 index/independent SNPs not in linkage disequilibrium (i.e., LD < 0.3) were selected (Table S1). Analyzing the same population for genes/SNPs in association with CRP as well as CRC risk can reduce bias derived from different population structures between phenotype and outcomes, one of the biases which have frequently been issued in MR analysis. In addition, we included the 3 outside GWASs [36-38] that recently reported GWA CRP-SNPs as a naturally log-transformed continuous variable (mg/L). They used different genotype and analytic strategies: HapMap-based 1000 Genomes imputed data analysis [36], exome-wide common and low/rare coding variants search [37], and genome-wide analysis of discovery panel combined with replication panel [38]. Of 89 SNPs obtained from those 3 studies, 16 SNPs overlapped, so findings from the most recent study were selected. In total, 61 independent SNPs (5 from our study plus 56 from others; LD < 0.3) were finally included in our analysis (Table S1). For all SNPs, the allele associated with a higher CRP level was assigned as an alternative (risk) allele, with the other as a reference allele.
Statistical analysis
The effect of each SNP on CRC risk was calculated in our study population via Cox regression with the adjustment in the first stage by age and 10 genetic PCs and in the second stage by lifestyle covariates in addition to age and 10 genetic PCs. The proportional hazard assumption was checked via a Schoenfeld residual plot and ρ evaluation. The Cox results from each of the 5 GWASs were combined via fixed-effect meta-analysis, by which the potential winner’s bias can be reduced [81].
We first checked MR assumptions to see whether our data met the conditions required for a valid causal inference. Three basic assumptions are essential for our genetic instruments to be valid for MR analysis: the genetic variants are (i) solidly predictive of the phenotype, (ii) independent of confounding factors that connect the phenotype-outcomes, and (iii) independent of the outcomes, conditioning on the phenotype and the confounding factors linked to phenotype-outcome relations (i.e., no pleiotropic pathways other than the phenotype) [82]. The first assumption was initially addressed with the selection of CRP SNPs at genome-wide significance. The inter-individual variability explained by the selected SNPs was approximately 6% [35,37,38], and the F-statistic was 107.80 on the basis of our sample size and number of instrumental variables [83]; with the traditional threshold of the 10 F-statistic [84], the strength of our genetic instruments are considered sufficient. Unlike the first assumption, the second and third may not be fully empirically tested because they rely on all the confounders, both measured and unmeasured [40]. For the horizontal pleiotropic effect on the association between CRP and CRC risk, we excluded pleiotropic SNPs (Table S2) that were associated with obesity (BMI and WHR), diabetic syndromes and diabetes (fasting glucose and insulin, post 2-hour glucose, and type 2 diabetes [T2DM]), and dyslipidemia (low-/high-density lipoproteins, total cholesterol, and triglycerides) [30,85]. While our 5 GWA CRP-SNPs did not overlap, 4 SNPs (with BMI, post 2-hour glucose, T2DM, and dyslipidemia) of the other 56 GWA CRP-SNPs were excluded. Also, we accounted for the covariates (listed above in the Lifestyle variables subsection) in the analyses of SNPs and CRC risk to remove the potential confounding effect. Further, we performed an MR-Egger analysis for assessing vertical directional pleiotropy (i.e., the third assumption) and checked whether the pleiotropic effects were skewed in one direction rather than being balanced [86].
Having demonstrated that our genetic instruments met the basic MR conditions, we conducted the MR analysis separately by analytic type of CRP phenotype - binary or continuous. In addition to the IVW method [87], we employed recent MR methods, including WM/PWM estimates [39,40] and MR G×E interactions [41]. Of note, the WM/PWM estimates allow up to 50% of genetic variants’ invalidity. Specifically, the WM method assigns a weight to the ordered estimate and establishes linearity between neighboring estimates, yielding a more stable estimate than the IVW does if the precision of the individual estimates varies substantially [40]. When the estimates from invalid instruments are not balanced about the true effect, use of the WM is inappropriate, but the PWM method can minimize this issue by down-weighting outlying genetic variants with heterogeneous estimates [40]. The PWM can also give better results if there is directional pleiotropy. In each of the separate MR analyses (i.e., CRP as binary or continuous), we conducted hypothesis-driven lifestyle-stratified analyses, and using the MR G×E method [41], calculated a corrected MR estimate that takes into consideration the interactions between CRP SNPs and selected lifestyles (BMI, waist, WHR, MET, % calories from SFA, family history of CRC, smoking, alcohol, and hormone therapy, such as OC, E-only, and E+P use). For this G×E analysis, we created a weighted genetic score (GS) using a polygenic additive model [88] with our 5 and the 56 outside GWA CRP-SNPs. Next, we rescaled the GS to the unit of CRP by conducting a linear regression; using β0 (slope) and β1 (intercept), we computed the scaled CRP-GS (= β0 + β1×GS), where 2 GSs were perfectly correlated (r = 1.0) [39,88].
In the MR analysis of our 5 GWA SNPs, we further adjusted for a correlation between CRP phenotype and CRC in which exposure and outcome were evaluated within the same population. For parameters necessary for the MR analysis, the change in CRP (> 3.0 vs. ≤ 3.0 mg/L) in log-odds and the mean change in log-transformed CRP per allele were obtained from our WHI GWAS and the 3 other earlier ones, respectively. The final MR results were reported as risk ratios (i.e., hazard ratios [HRs] and 95% confidence intervals [CIs]) for the change in CRC risk per unit increase in log-odds or log-transformed CRP.
The heterogeneity among MR estimates, reflecting additional pleiotropic evidence, was estimated via Cochran’s Q test. A 2-tailed P < 0.05 was statistically significant. R3.6.3 with survival, metafor, forestplot, ggplot2, and ggthemes packages was used.
Results
The associations between each of the 61 GWA CRP-SNPs (5 for binary, reflecting chronic inflammation, and 56 for the naturally log-transformed continuous CRP variable) and CRC risk from the meta-analyses of the 5 WHI GWASs are shown in Table 1; the analytic results from the 2-stage adjustment are presented, including the first stage of adjustment for age and 10 genetic PCs and the second stage of adjustment for lifestyle covariates in addition to age and 10 genetic PCs. The pooled analysis for the genetic instruments combining 5 SNPs (Figure 1A) and 56 SNPs (Figure 1B) each did not yield statistically significant associations (the 5 SNPs’ PWM-HR2nd-stage = 1.20, 95% CI: 0.85-1.68; the 56 SNPs’ PWM-HR2nd-stage = 1.29, 95% CI: 0.83-1.99). Removing pleiotropic SNPs did not apparently change the pooled estimate.
Table 1.
Two-stage multiple Cox regressions of genome-wide SNPs associated with CRP predicting colorectal cancer risk
| SNP | Chr | Position¥ | Gene | Allele | Stage 1 | Stage 2 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Adjustment for age and 10 PCs | Adjustment for covariates* | ||||||||||||
| in addition to age and 10 PCs | |||||||||||||
|
|
|
|
|||||||||||
| Ref | Alt | HR | (95% CI) | p | p-het† | HR | (95% CI) | p | p-het† | ||||
| GWAS examining CRP as a binary outcome reflecting high immune response and chronic inflammation (CRP > 3.0 mg/L) | |||||||||||||
| rs2794520 | 1 | 159678816 | CRPP1/CRP | C | T | 0.97 | (0.87-1.08) | 0.555 | 0.271 | 0.95 | (0.86-1.06) | 0.361 | 0.563 |
| rs2243458 | 12 | 121424490 | HNF1A | C | T | 1.02 | (0.91-1.13) | 0.792 | 0.047 | 1.06 | (0.95-1.19) | 0.276 | 0.047 |
| rs1169311 | 12 | 121440731 | C12orf43 | C | T | 1.02 | (0.91-1.13) | 0.775 | 0.168 | 1.05 | (0.94-1.17) | 0.398 | 0.098 |
| rs429358 | 19 | 45411941 | APOE | T | C | 0.95 | (0.80-1.13) | 0.554 | 0.745 | 0.95 | (0.80-1.13) | 0.568 | 0.511 |
| rs5117 | 19 | 45418790 | APOC1 | T | C | 1.02 | (0.72-1.45) | 0.893 | 0.216 | 1.05 | (0.74-1.49) | 0.793 | 0.157 |
| GWASs analyzing CRP as a continuous variable that was naturally log-transformed (mg/L) | |||||||||||||
| rs75460349 | 1 | 27180088 | ZDHHC18 | C | A | 1.16 | (0.78-1.73) | 0.459 | 0.970 | 1.16 | (0.78-1.74) | 0.469 | 0.877 |
| rs2293476 | 1 | 40036847 | PABPC4 | G | C | 1.04 | (0.92-1.18) | 0.525 | 0.954 | 1.05 | (0.92-1.19) | 0.490 | 0.904 |
| rs1805096 | 1 | 66102257 | LEPR | A | G | 1.03 | (0.92-1.15) | 0.608 | 0.298 | 1.03 | (0.92-1.15) | 0.619 | 0.376 |
| rs469772 | 1 | 91530305 | ZNF644 | T | C | 1.12 | (0.99-1.28) | 0.081 | 0.239 | 1.08 | (0.95-1.23) | 0.257 | 0.257 |
| rs4129267 | 1 | 154426264 | IL6R | T | C | 0.92 | (0.83-1.02) | 0.122 | 0.467 | 0.90 | (0.81-1.00) | 0.051 | 0.587 |
| rs2794520 | 1 | 159678816 | CRPP1/CRP | T | C | 1.03 | (0.93-1.15) | 0.555 | 0.271 | 1.05 | (0.95-1.17) | 0.361 | 0.563 |
| rs1800947 | 1 | 159683438 | CRP | G | C | 1.30 | (0.88-1.92) | 0.180 | 0.422 | 1.37 | (0.92-2.05) | 0.125 | 0.401 |
| rs1417938 | 1 | 159684186 | CRP | T | C | 1.02 | (0.91-1.14) | 0.694 | 0.576 | 1.03 | (0.92-1.15) | 0.657 | 0.539 |
| rs10925027 | 1 | 247612562 | NLRP3 | C | T | 1.15 | (0.97-1.36) | 0.117 | 0.725 | 1.21 | (1.01-1.46) | 0.043 | 0.576 |
| rs12995480 | 2 | 629881 | TMEM18 | T | C | 1.07 | (0.94-1.22) | 0.328 | 0.897 | 1.01 | (0.88-1.16) | 0.888 | 0.969 |
| rs1260326 | 2 | 27730940 | GCKR | C | T | 0.97 | (0.87-1.09) | 0.636 | 0.785 | 1.00 | (0.89-1.12) | 0.999 | 0.765 |
| rs4246598 | 2 | 88438050 | FABP1 | C | A | 0.97 | (0.87-1.07) | 0.529 | 0.526 | 0.98 | (0.88-1.09) | 0.757 | 0.597 |
| rs9284725 | 2 | 102744854 | IL1R1 | A | C | 1.04 | (0.92-1.18) | 0.525 | 0.778 | 1.04 | (0.91-1.18) | 0.576 | 0.779 |
| rs13409371 | 2 | 113838145 | IL1F10 | G | A | 1.03 | (0.93-1.15) | 0.559 | 0.834 | 1.02 | (0.92-1.14) | 0.691 | 0.782 |
| rs1441169 | 2 | 214033530 | IKZF2 | G | A | 1.05 | (0.95-1.16) | 0.363 | 0.399 | 1.07 | (0.96-1.19) | 0.215 | 0.741 |
| rs2352975 | 3 | 49891885 | TRAIP | T | C | 1.16 | (0.95-1.41) | 0.144 | 0.842 | 1.17 | (0.95-1.43) | 0.130 | 0.966 |
| rs1514895 | 3 | 170705693 | EIF5A2 | A | G | 1.06 | (0.94-1.18) | 0.354 | 0.934 | 1.08 | (0.96-1.21) | 0.212 | 0.934 |
| rs4705952 | 5 | 131839618 | IRF1 | A | G | 1.07 | (0.93-1.23) | 0.335 | 0.216 | 1.03 | (0.89-1.18) | 0.709 | 0.083 |
| rs17658229 | 5 | 172191052 | DUSP1 | T | C | 0.97 | (0.72-1.32) | 0.868 | 0.055 | 0.93 | (0.68-1.27) | 0.652 | 0.166 |
| rs9271608 | 6 | 32591588 | HLA-DQA1 | A | G | 1.13 | (0.71-1.80) | 0.606 | 0.699 | 1.20 | (0.74-1.94) | 0.453 | 0.567 |
| rs12202641 | 6 | 116314634 | FRK | T | C | 0.91 | (0.82-1.01) | 0.087 | 0.167 | 0.90 | (0.80-1.00) | 0.050 | 0.124 |
| rs6901250 | 6 | 117114025 | GPRC6A | G | A | 1.02 | (0.91-1.15) | 0.679 | 0.716 | 1.06 | (0.95-1.20) | 0.292 | 0.656 |
| rs1490384 | 6 | 126851160 | CENPW | T | C | 0.94 | (0.85-1.05) | 0.295 | 0.507 | 0.96 | (0.86-1.07) | 0.420 | 0.596 |
| rs9385532 | 6 | 130371227 | L3MBTL3 | T | C | 0.96 | (0.86-1.07) | 0.485 | 0.614 | 1.00 | (0.90-1.12) | 0.974 | 0.860 |
| rs1880241 | 7 | 22759469 | IL6 | G | A | 0.94 | (0.85-1.04) | 0.234 | 0.016 | 0.99 | (0.89-1.10) | 0.830 | 0.013 |
| rs2710804 | 7 | 36084529 | EEPD1 | T | C | 0.93 | (0.83-1.04) | 0.184 | 0.468 | 0.93 | (0.83-1.04) | 0.194 | 0.279 |
| rs13233571 | 7 | 72971231 | BCL7B | T | C | 1.01 | (0.87-1.19) | 0.857 | 1.000 | 0.94 | (0.80-1.11) | 0.478 | 0.993 |
| rs4841132 | 8 | 9183596 | PPP1R3B | A | G | 0.85 | (0.70-1.03) | 0.098 | 0.824 | 0.88 | (0.72-1.06) | 0.175 | 0.598 |
| rs2064009 | 8 | 117007850 | TRPS1 | C | T | 0.96 | (0.86-1.07) | 0.444 | 0.550 | 0.95 | (0.85-1.05) | 0.317 | 0.526 |
| rs2891677 | 8 | 126344208 | NSMCE2 | C | T | 1.00 | (0.90-1.11) | 0.975 | 0.843 | 1.03 | (0.93-1.14) | 0.582 | 0.992 |
| rs643434 | 9 | 136142355 | ABO | G | A | 0.93 | (0.84-1.04) | 0.201 | 0.541 | 0.96 | (0.86-1.07) | 0.449 | 0.489 |
| rs1051338 | 10 | 91007360 | LIPA | T | G | 1.02 | (0.90-1.14) | 0.802 | 0.581 | 1.02 | (0.91-1.15) | 0.731 | 0.452 |
| rs10832027 | 11 | 13357183 | ARNTL | G | A | 0.96 | (0.85-1.07) | 0.427 | 0.540 | 0.95 | (0.85-1.07) | 0.416 | 0.835 |
| rs10838687 | 11 | 47312892 | MADD | G | T | 0.99 | (0.87-1.13) | 0.894 | 0.811 | 0.98 | (0.86-1.12) | 0.807 | 0.891 |
| rs1582763 | 11 | 60021948 | MS4A4A | A | G | 1.04 | (0.93-1.16) | 0.490 | 0.952 | 1.00 | (0.89-1.11) | 0.955 | 0.955 |
| rs7121935 | 11 | 72496148 | STARD10 | A | G | 0.99 | (0.88-1.11) | 0.836 | 0.330 | 1.02 | (0.90-1.14) | 0.803 | 0.184 |
| rs11108056 | 12 | 95855385 | METAP2 | G | C | 0.99 | (0.83-1.19) | 0.951 | 0.131 | 1.02 | (0.84-1.23) | 0.842 | 0.199 |
| rs10778215 | 12 | 103537266 | C12orf42 | A | T | 0.96 | (0.87-1.06) | 0.430 | 0.922 | 0.99 | (0.90-1.10) | 0.904 | 0.740 |
| rs7310409 | 12 | 121424861 | HNF1A | A | G | 0.99 | (0.89-1.11) | 0.910 | 0.159 | 0.98 | (0.88-1.09) | 0.653 | 0.103 |
| rs2239222 | 14 | 73011885 | RGS6 | A | G | 1.03 | (0.91-1.18) | 0.603 | 0.835 | 1.05 | (0.92-1.19) | 0.476 | 0.526 |
| rs112635299 | 14 | 94838142 | SERPINA1/SERPINA2P | T | G | 0.88 | (0.57-1.36) | 0.570 | 0.199 | 0.95 | (0.62-1.48) | 0.828 | 0.364 |
| rs4774590 | 15 | 51745277 | DMXL2 | A | G | 1.17 | (1.05-1.30) | 0.004 | 0.139 | 1.17 | (1.05-1.30) | 0.004 | 0.229 |
| rs1189402 | 15 | 53728154 | WDR72 | G | A | 0.93 | (0.83-1.05) | 0.241 | 0.831 | 0.95 | (0.84-1.07) | 0.372 | 0.822 |
| rs340005 | 15 | 60878030 | RORA | G | A | 1.02 | (0.92-1.14) | 0.703 | 0.342 | 0.96 | (0.86-1.07) | 0.464 | 0.634 |
| rs10521222 | 16 | 51158710 | SALL1 | T | C | 1.41 | (0.86-2.30) | 0.172 | 0.510 | 1.16 | (0.68-1.98) | 0.587 | 0.559 |
| rs1558902 | 16 | 53803574 | FTO | T | A | 1.01 | (0.91-1.13) | 0.834 | 0.791 | 1.02 | (0.91-1.14) | 0.732 | 0.851 |
| rs178810 | 17 | 16097430 | NCOR1 | C | T | 0.95 | (0.86-1.06) | 0.388 | 0.457 | 0.97 | (0.87-1.08) | 0.546 | 0.357 |
| rs10512597 | 17 | 72699833 | CD300LF/RAB37 | T | C | 0.95 | (0.82-1.09) | 0.443 | 0.097 | 0.97 | (0.84-1.12) | 0.682 | 0.142 |
| rs2852151 | 18 | 12841176 | PTPN2 | G | A | 0.96 | (0.87-1.07) | 0.491 | 0.276 | 0.95 | (0.85-1.06) | 0.358 | 0.430 |
| rs4092465 | 18 | 55080437 | ONECUT2 | A | G | 0.95 | (0.75-1.19) | 0.646 | 0.915 | 0.93 | (0.74-1.18) | 0.564 | 0.957 |
| rs12960928 | 18 | 57897803 | MC4R | T | C | 0.99 | (0.88-1.12) | 0.915 | 0.814 | 0.99 | (0.88-1.11) | 0.818 | 0.875 |
| rs4420638 | 19 | 45422946 | APOC1 | G | A | 1.03 | (0.70-1.50) | 0.892 | 0.766 | 0.93 | (0.63-1.38) | 0.731 | 0.669 |
| rs1800961 | 20 | 43042364 | HNF4A | T | C | 0.90 | (0.53-1.52) | 0.687 | 0.372 | 0.89 | (0.51-1.55) | 0.681 | 0.392 |
| rs2315008 | 20 | 62343956 | ZGPAT | T | G | 1.08 | (0.96-1.20) | 0.195 | 0.620 | 1.10 | (0.98-1.22) | 0.096 | 0.961 |
| rs2836878 | 21 | 40465534 | PSMG1 | A | G | 1.11 | (0.99-1.24) | 0.078 | 0.288 | 1.11 | (0.99-1.24) | 0.080 | 0.319 |
| rs6001193 | 22 | 39074737 | TOMM22 | G | A | 1.01 | (0.91-1.13) | 0.846 | 0.320 | 1.04 | (0.93-1.17) | 0.445 | 0.606 |
Alt, alternative; Chr, chromosome; CI, confidence interval; CRP, C-reactive protein; GWAS, genome-wide association study; HR, hazard ratio; PCs, principal components; Ref, reference; SFA, saturated fatty acids; SNP, single-nucleotide polymorphism; WHI, Women’s Health Initiative studies.
Covariates adjusted in the analyses include education; annual family income; family history of colorectal cancer; cardiovascular disease ever; high cholesterol requiring medication ever; body mass index; waist-to-hip ratio; physical activity; % calories from SFA/day; dietary alcohol in g/day; number of cigarettes/day; age at menopause; duration of oral contraceptive use; and durations of exogenous estrogen (E)-only and E plus progestin use.
GRCh 37 coordinated.
Heterogeneity in estimates among the 5 WHI sub-GWASs was evaluated by Cochran’s Q test with fixed effects.
Figure 1.
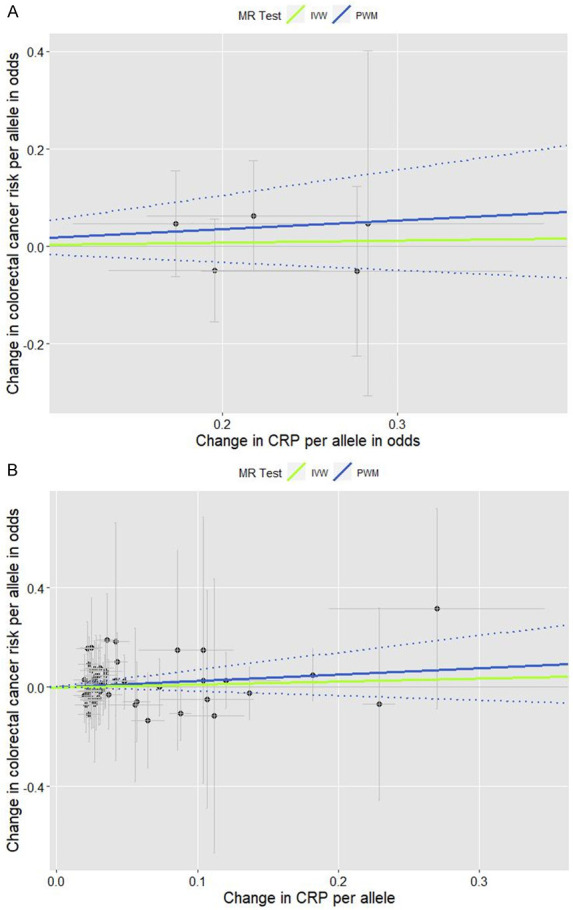
Scatter plots for the effects of individual CRP-genetic instrumental variables on colorectal cancer risk. Each black dot reflects a genome-wide CRP-raising genetic variant. The blue lines indicate penalized weighted median estimates and 95% CIs. (CI, confidence interval; CRP, C-reactive protein; HR, hazard ratio; IVW, inverse-variance weighted; MR, Mendelian randomization; PWM, penalized weighted median; SNP, single-nucleotide polymorphism). A. Five genome-wide SNPs associated with high immune response and chronic inflammation (CRP > 3.0 mg/L) (PWM HR2nd-stage = 1.20, 95% CI: 0.85-1.68; MR-Egger intercept p value = 0.673). B. Fifty-six genome-wide SNPs associated with CRP phenotype that was naturally log-transformed (mg/L) (PWM HR2nd-stage = 1.29, 95% CI: 0.83-1.99; MR-Egger intercept p value = 0.824).
Obesity-related lifestyle factors and smoking
We further performed lifestyle-specific stratification analyses. In the SFA-stratified analysis of our 5 CRP-SNPs (Table 2), a 1-unit increase in the genetically predicted chronic inflammation (defined as > 3.0 mg/L of CRP) was associated with approximately 3 times higher risk for CRC in the low fat-diet subgroup (SFA < 9%), particularly in the MR stage 2 of adjustment for multiple lifestyle variables (WM-HR2nd-stage = 2.88, 95% CI: 1.24-6.68; PWM-HR2nd-stage = 3.06, 95% CI: 1.21-7.72). In contrast, the inverse relationship between genetically determined chronic inflammation and CRC risk was observed in the high fat-diet subgroup (SFA ≥ 9%), specifically in the first MR stage (IVW-HR1st-stage = 0.79, 95% CI: 0.64-0.96).
Table 2.
Mendelian randomization analysis: the effect of genetically predicted chronic inflammation status on colorectal cancer risk by dietary fat intake
| GWAS examining CRP as a binary outcome reflecting high immune response and chronic inflammation (CRP > 3.0 mg/L) | ||||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Analytic method | Stage 1 | Stage 2 | ||||||
| Adjustment for age and 10 PCs | Adjustment for covariates* | |||||||
| in addition to age and 10 PCs | ||||||||
|
|
|
|||||||
| HR¶ | (95% CI) | p | p-het† | HR¶ | (95% CI) | p | p-het† | |
| <% calories from SFA < 9.0%> | ||||||||
| Inverse-variance weighted | 1.53 | (0.78-2.98) | 0.156 | 0.009 | 1.92 | (0.47-7.89) | 0.270 | 0.102 |
| Weighted median | 1.69 | (0.80-3.53) | 0.168 | 0.089 | 2.88 | (1.24-6.68) | 0.014 | 0.003 |
| Penalized weighted median | 1.68 | (0.82-3.47) | 0.158 | 0.083 | 3.06 | (1.21-7.72) | 0.018 | 0.004 |
| MR-Egger: slope | 2.96 | (0.01-762.22) | 0.578 | 14.75 | (0.01-1.12e+06) | 0.502 | ||
| intercept | 0.87 | (0.27-2.76) | 0.726 | 0.65 | (0.06-6.77) | 0.600 | ||
| <% calories from SFA ≥ 9.0%> | ||||||||
| Inverse-variance weighted | 0.79 | (0.64-0.96) | 0.029 | 0.80 | (0.52-1.24) | 0.235 | ||
| Weighted median | 0.82 | (0.57-1.18) | 0.292 | 0.70 | (0.47-1.03) | 0.067 | ||
| Penalized weighted median | 0.82 | (0.57-1.18) | 0.290 | 0.70 | (0.47-1.03) | 0.072 | ||
| MR-Egger: slope | 0.38 | (0.14-1.06) | 0.057 | 0.25 | (0.01-4.96) | 0.238 | ||
| intercept | 1.17 | (0.94-1.44) | 0.106 | 1.28 | (0.69-2.38) | 0.301 | ||
CI, confidence interval; CRP, C-reactive protein; GWAS, genome-wide association study; HR, hazard ratio; MR, Mendelian randomization; PCs, principal components; SFA, saturated fatty acid; SNP, single-nucleotide polymorphism. Numbers in bold face are statistically significant.
Covariates that were adjusted for in analyses of the association between GWA CRP-SNPs and colorectal cancer risk include education; annual family income; family history of colorectal cancer; cardiovascular disease ever; high cholesterol requiring medication ever; body mass index; waist-to-hip ratio; physical activity; % calories from SFA/day; dietary alcohol in g/day; number of cigarettes/day; age at menopause; duration of oral contraceptive use; and durations of exogenous estrogen (E)-only and E plus progestin use. Variable tested for stratification was not included as a covariate in the stage 2 analysis.
The MR estimate (except weighted/penalized weighted medians) was adjusted for a correlation between CRP phenotype and colorectal cancer within the same population.
p value for heterogeneity test in MR estimates across strata (< 9.0% from SFA vs. ≥ 9.0% from SFA) was evaluated with Cochran’s Q test.
Different patterns were observed in the waist circumference-stratified analysis (Figure 2A). Genetically determined chronic inflammation was associated with 20% reduced risk for CRC among the non-viscerally obese group, whereas a proportionate relationship (15% increased CRC risk) was observed with limited statistical power among the viscerally obese group.
Figure 2.
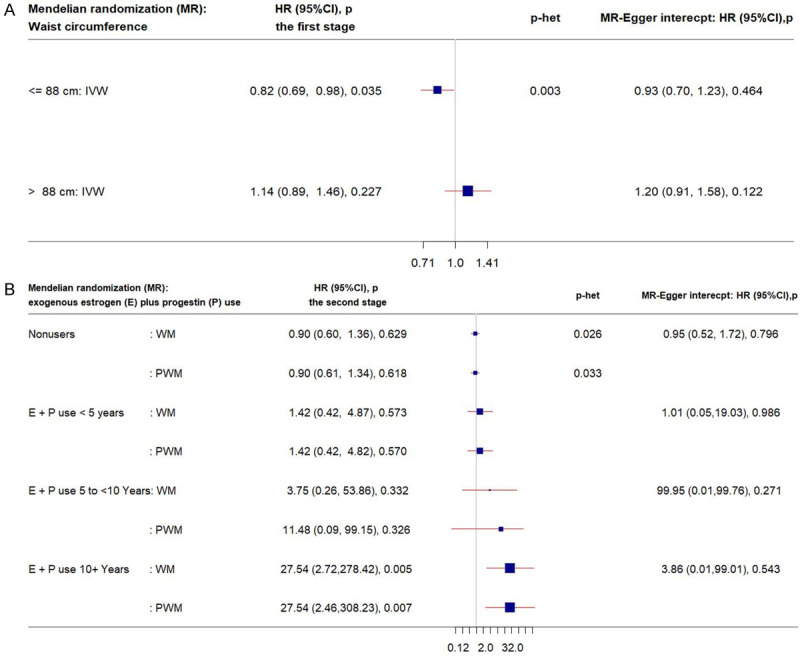
Forest plots of MR estimates. The plots show the effects of genetically predicted chronic inflammation status (CRP > 3.0 mg/L) on colorectal cancer risk in obesity/hormone use-specific strata, presented as 95% CIs (red lines) of the estimates and the inverse-variance weights and the weighted and penalized weighted medians (percentages proportional to the size of the blue squares). The MR estimates were calculated in the 2 stages on the SNP-colorectal cancer association: in the first stage, only age and 10 PCs and in the second stage, lifestyle covariates in addition to age and 10 PCs were adjusted. p-het is from the heterogeneity test in MR estimates across strata. (CI, confidence interval; CRP, C-reactive protein; E, exogeneous estrogen; HR, hazard ratio; IVW, inverse-variance weight; MR, Mendelian randomization; SNP, single-nucleotide polymorphism; P, progestin; PC, principal component; PWM, penalized weighted median; WM, weighted median). A. IVW MR estimates by waist circumference. B. WM/PWM MR estimates by E plus P use.
In the smoking-stratified analysis of our 5 and the outside 56 GWA CRP-SNPs (Table 3), a 1-unit increase in the log-transformed genetically predicted CRP was associated with 50% or greater risk of CRC among never smokers, but only in the first MR stage. The elevated effect on CRC risk was more profound in the first stage of MR analysis after excluding pleiotropic SNPs. Similarly, among smokers with 15 or more cigarettes/day, genetically driven chronic inflammation was associated with 60% increased risk for CRC in the first MR stage. By taking into account our 5 GWA CRP-SNPs’ interactions with cigarette smoking, the corrected MR estimate indicated a greater risk for CRC in relation to genetically elevated chronic inflammation (MR G×E HR = 2.59, 95% CI: 1.13-5.91). The MR-Egger tests showed no significant evidence of directional pleiotropy across the tested associations.
Table 3.
Mendelian randomization analysis: the effect of genetically predicted CRP (as log-transformed or categorized) on colorectal cancer risk by number of cigarettes per day
| Analytic method | All SNPs | After exclusion of pleiotropic SNPs | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||||||||||||
| Stage 1 | Stage 2 | Stage 1 | Stage 2 | ||||||||||||||||
| Adjustment for age and 10 PCs | Adjustment for covariates* | Adjustment for age and 10 PCs | Adjustment for covariates* | ||||||||||||||||
| in addition to age and 10 PCs | in addition to age and 10 PCs | ||||||||||||||||||
|
|
|
|
|
||||||||||||||||
| HR | (95% CI) | p | p-het† | HR | (95% CI) | p | p-het† | HR | (95% CI) | p | p-het† | HR | (95% CI) | p | p-het† | ||||
| GWASs analyzing CRP as a continuous variable that was naturally log-transformed (mg/L) | |||||||||||||||||||
| <Never smokers> | |||||||||||||||||||
| Inverse-variance weighted | 1.48 | (1.01-2.18) | 0.045 | 0.795 | 1.51 | (0.97-2.37) | 0.069 | 0.309 | 1.53 | (1.02-2.30) | 0.041 | 0.742 | 1.57 | (0.97-2.54) | 0.063 | 0.235 | |||
| Weighted median | 1.82 | (1.02-3.26) | 0.044 | 1.59 | (0.85-2.96) | 0.146 | 1.82 | (0.97-3.44) | 0.064 | 1.75 | (0.91-3.34) | 0.092 | |||||||
| Penalized weighted median | 1.82 | (1.02-3.25) | 0.044 | 1.65 | (0.87-3.12) | 0.125 | 1.82 | (1.004-3.31) | 0.049 | 1.79 | (0.94-3.42) | 0.079 | |||||||
| MR-Egger: slope | 1.58 | (0.88-2.84) | 0.122 | 1.44 | (0.73-2.86) | 0.286 | 1.59 | (0.85-2.95) | 0.140 | 1.46 | (0.71-3.01) | 0.300 | |||||||
| intercept | 1.00 | (0.96-1.03) | 0.766 | 1.004 | (0.97-1.04) | 0.853 | 1.00 | (0.96-1.03) | 0.876 | 1.01 | (0.97-1.05) | 0.780 | |||||||
| GWAS examining CRP as a binary outcome reflecting high immune response and chronic inflammation (CRP > 3.0 mg/L) | |||||||||||||||||||
| <≥ 15 cigarettes/d> | |||||||||||||||||||
| Inverse-variance weighted¶ | 1.58 | (1.03-2.41) | 0.041 | 0.908 | 1.63 | (0.91-2.93) | 0.083 | 0.788 | |||||||||||
| Weighted median | 1.52 | (0.74-3.12) | 0.256 | 1.62 | (0.76-3.46) | 0.210 | |||||||||||||
| Penalized weighted median | 1.52 | (0.72-3.19) | 0.269 | 1.62 | (0.77-3.41) | 0.201 | |||||||||||||
| MR-Egger: slope | 0.36 | (0.04-3.26) | 0.236 | 0.29 | (0.01-10.57) | 0.351 | |||||||||||||
| intercept | 1.37 | (0.86-2.18) | 0.119 | 1.44 | (0.68-3.06) | 0.219 | |||||||||||||
| <MR G×E interaction: scaled GWA CRP weighted-genetic score€ × number of cigarettes/d> | |||||||||||||||||||
| G×E univariate slope§ | 2.59 | 1.13-5.91 | 0.032 | ||||||||||||||||
CI, confidence interval; CRP, C-reactive protein; GWAS, genome-wide association study; HR, hazard ratio; MR, Mendelian randomization; PCs, principal components; SFA, saturated fatty acid; SNP, single-nucleotide polymorphism. Numbers in bold face are statistically significant.
Covariates that were adjusted for in analyses of the association between GWA CRP-SNPs and colorectal cancer risk include education; annual family income; family history of colorectal cancer; cardiovascular disease ever; high cholesterol requiring medication ever; body mass index; waist-to-hip ratio; physical activity; % calories from SFA/day; dietary alcohol in g/day; number of cigarettes/day; age at menopause; duration of oral contraceptive use; and durations of exogenous estrogen (E)-only and E plus progestin use. Variable tested for stratification was not included as a covariate in the stage 2 analysis.
p value for heterogeneity test in estimates among GWA CRP-SNPs was evaluated with Cochran’s Q test with fixed effects.
The MR estimate was adjusted for a correlation between CRP phenotype and colorectal cancer within the same population.
The MR G×E slope reflects an MR estimate from univariate analysis that was corrected for G×E interaction with a modeled (cigarette) variable.
The scaled weighted-genetic score was estimated with 5 CRP SNPs whose effect size reflects chronic inflammation (CRP > 3.0 mg/L) per allele.
Hormone therapy
Whereas genetically predicted chronic inflammation was not associated with CRC risk among E+P nonusers and relatively short-term users (i.e., < 5 years), long-term E+P users showed strongly positive relationships (Figure 2B). For example, among long-term E+P users (10 years or longer), women with genetically elevated chronic inflammation had 27 times higher risk for CRC. Similar null associations with CRC risk were observed among E+P nonusers and short-term users in the MR analysis for genetically elevated CRP phenotype (Table S3). Further, among long-term E+P users (5 to < 10 years), a 1-unit increase in the log-transformed genetically determined CRP was associated with 20 times or higher risk for CRC (PWM-HR1st-stage = 28.60, 95% CI: 1.63-501.89; PWM-HR1st-stage after excluding pleiotropy = 21.24, 95% CI: 1.30-346.88).
Similar patterns were observed in the E-only subgroups (Figure 3). Among nonusers, no association between genetically elevated CRP and CRC risk was observed. Notably different from the E+P subgroups, genetically elevated CRP with increased CRC risk was found only among relatively shortterm users (< 5 years). No directional pleiotropic effects across the analyses were observed.
Figure 3.
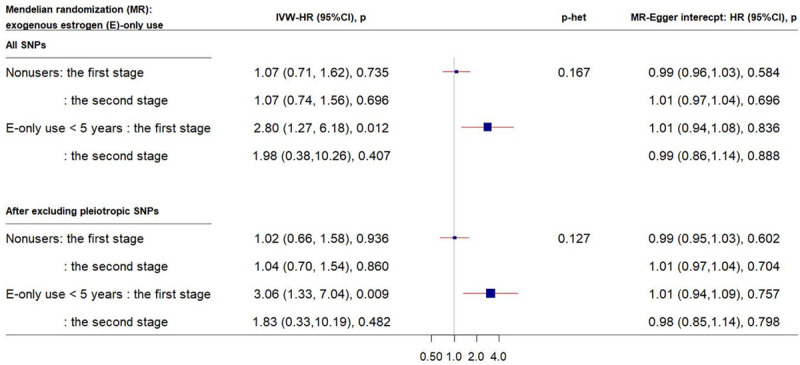
Forest plot of MR estimates by E-only use. The plot shows the effects of genetically predicted CRP phenotype on colorectal cancer risk in E-only subgroups, presented as 95% CIs (red lines) of the estimates and the inverse-variance weights (percentages proportional to the size of the blue squares). The MR estimates were calculated in the 2 stages on the SNP-colorectal cancer association: in the first stage, only age and 10 PCs and in the second stage, lifestyle covariates in addition to age and 10 PCs were adjusted. p-het is from the heterogeneity test in MR estimates across strata. (CI, confidence interval; CRP, C-reactive protein; E, exogeneous estrogen; HR, hazard ratio; IVW, inverse-variance weight; MR, Mendelian randomization; OC, oral contraceptive; SNP, single-nucleotide polymorphism; PC, principal component).
No other subgroups presented a statistically significant increased risk for CRC in association with genetically determined chronic inflammation or CRP levels (Tables S3 and S4). Further, in the MR G×E interactions, MR estimates corrected for cigarette smoking, breast feeding, and depressive symptoms reached statistical significance, and no pleiotropic effect of the estimates was detected (Table S5).
Discussion
We examined the genetically predicted effect of CRP levels (a natural log-transformed 1 mg/L increase or > 3 mg/L vs. ≤ 3.0 mg/L, indicating chronic low-grade inflammation) on postmenopausal CRC risk by performing MR analysis and found that genetically determined CRP levels and chronic inflammation status were associated with increased risk for CRC in subgroups of women with particular lifestyle behaviors. The MR findings, if the modeled genetic instruments are not affected by vertical and horizontal pleiotropic effects, can be comparable with those from randomized clinical trials, thus providing a fairly robust causal inference [30,87]. Our MR analysis removed pleiotropic SNPs in relation to obesity and glucose and lipid metabolism, which may confound the CRP-cancer risk association, and further identified a wide range of confounding factors that were accounted for in the genetic analysis with CRC outcomes. Our MR estimates, such as WM and PWM, allow some relaxation of the rigid conditions for MR instruments, thus yielding a more robust estimate of the causal effect than a traditional MR estimate. In addition, the MR approach can examine the lifelong effect of exposure (CRP) on CRC risk, and it is less susceptible to reverse causality. Thus, although our MR study was not designed to directly evaluate biologic mechanisms, our findings indicate that the long-standing effect of CRP lies in the causal pathway of CRC development. To the best of our knowledge, this study is the first to report the causal relationship between genetically determined CRP phenotype and CRC risk within lifestyle-specific subgroups in an MR framework.
In the stratification analyses by obesity, we detected a reduced risk of CRC in relation to CRP in non-viscerally obese women, whereas increased CRP-associated CRC risk was observed even with a lack of statistical power in viscerally obese women. In line with results from previous studies [62-65], our finding indicates that larger waist circumference, a biomarker of visceral fat accumulation, is more important than overall obesity (as indicated by a high BMI) in CRC risk in women. Our results further support the hypothesis [66,67] that chronic inflammation is one of the interconnected pathways that mediate the adiposity-CRC association, but additional studies on larger populations are warranted to confirm our results.
In obesity-related lifestyle-specific analyses, particularly in the dietary-SFA stratification, CRP-increased risk for CRC in the low fat-intake subgroup was observed, whereas CRP-reduced risk for CRC in the high fat-intake subgroup. Previous studies [89-91] showed that intake of n-3 polyunsaturated fatty acids improved intestinal barriers, leading to reduced exposure to intestinal bacterial products, thus contributing to less inflammatory potential and inhibition of production of pro-inflammatory cytokines such as CRP. Also, in contrast to the results from the aforementioned studies, one study [12] demonstrated that CRP together with oxidized low-density lipoprotein interact with LOX-I receptors, elevating the mRNA expression of genes (e.g., LOX-I, CEA, and MMP1/2) important in colorectal tumorigenesis. However, no published studies to date have examined the effect of SFA intake on CRP levels in relation to colorectal carcinogenesis. Our finding of the protective effect of CRP on CRC risk when the intake of SFAs is high suggests that different fat types of lipid metabolism can be implicated in the immune-related CRC pathways and calls for further biologic-mechanism studies.
Cigarette smoking is an established risk factor for CRC, accounting for 15-20% of CRC cases [92,93] with a dose-response relationship of daily cigarette consumption [94]. Tobacco-derived carcinogens reach the colorectal mucosa through the digestive tract and the circulatory system, causing the potential carcinogenesis in this target organ [95]. Circulating levels of inflammatory cytokines such as CRP have been directly associated with smoking in CRC case-control studies [29,67]. Our data, with an average 15-year induction period from onset of smoking to cancer formation, despite relatively short-term exposure [96,97], showed strong risk for CRC in relation to CRP in women who smoked ≥ 15 cigarettes/day. Moreover, consistent with other studies that reported an interaction of smoking with the CRP-CRC risk association [22-24], our study indicates the presence of a significant CRP-induced causal pathway for CRC development, which was corrected for the gene-smoking interaction. Altogether, the inflammatory pathways that interact with smoking can be postulated to be involved in colorectal carcinogenesis. Interestingly, our MR analysis of the CRP-increased CRC risk was also observed in never-smokers to a slightly lesser extent; this may be the result from unmeasured confounders or the existence of other epigenetic molecular pathways, thus further studies addressing these issues with a more comprehensive molecular dataset are required.
In the analyses stratified by hormone therapy, a substantially increased risk of CRC in relation to CRP was found in the E+P users. In particular, CRP-induced high risk for CRC was found in longer-term E+P users (≥ 5 years) while no association of the CRP-CRC risk was found in nonusers and relatively short-term E+P users (< 5 years). Lifetime cumulative exposure to estrogen, particularly to the opposed (E+P) hormone use, has been considered a protective factor for CRC risk [62,68,69], as shown in in vitro and case-control studies revealing that introduction of an E-receptor gene resulted in marked growth suppression in CRC cells and was associated with low risk for microsatellite instability-positive CRC [98,99]. However, this protective effect was observed only in relatively short-term E+P users (< 5 years) [68,69]. Further, unlike natural progesterone, the synthetic progestin contained in the E+P combination has an affinity for androgen and mineralocorticoid receptors, leading to cell proliferation and anti-apoptosis, which in turn may contribute to colorectal carcinogenesis [100]. Taken together, that evidence and our findings suggest that longer-term cumulative exposure to the opposed estrogen may be implicated in the colorectal tumorigenic pathway that is presumably mediated by inflammation. In contrast, only the relatively short-term E-only users (< 5 years) in our MR analysis had an increased risk of CRC in relation to CRP, a finding supported by another WHI study that had a 7-year follow-up, [70]; this further warrants studies on a larger population with longer-term follow-up of biologic-function investigation for more conclusive results.
We tested for MR directional pleiotropy and reduced any potential pleiotropic effect by using multifaceted approaches: removal of pleiotropic SNPs, adjustment of a wide range of confounding factors, corrections of MR estimates for gene-lifestyle interactions, and use of WM/PWM estimates. Nonetheless, our findings could be biased due to residual and unmeasured confounding factors. Also, potential nonlinearity in the SNP-exposure and exposure-outcome relationships would tend toward a null result, so generating a spurious association is less likely [101]. Our results could have inflated false-positive rates due to multiple comparisons. Finally, our findings should not be extrapolated to other races or ethnicity, in whom the interrelationships between genetic instruments, CRP, and CRC risk may be different.
In summary, we attempted to improve the causal inference between genetically elevated CRP and CRC risk in postmenopausal women by quantifying the association in an MR framework. We obtained evidence of the potential causal association between lifelong exposure to CRP and CRC risk, particularly in women with low intake of SFAs, regular smokers, and relatively short-term users of unopposed and long-term users of opposed estrogen. Further biologic-mechanism studies may help to elucidate the inflammatory-induced colorectal carcinogenesis pathways that interact with fatty acids, smoking, and hormone therapy. Our findings may yield novel evidence on immune-related etiologic pathways connected to CRC risk, and suggest the possible use of CRP as a CRC-predictive biomarker in women with particular behaviors and CRP marker-informed interventions to reduce CRC risk.
Acknowledgements
This study was supported by the National Institute of Nursing Research of the National Institutes of Health under Award Number K01NR017852. Part of the data for this project was provided by the WHI program, which is funded by the National Heart, Lung, and Blood Institute, the National Institutes of Health, and the U.S. Department of Health and Human Services through contracts HHSN268201100046C, HHSN268201100001C, HHSN268201100002C, HHSN268201100003C, HHSN268201100004C, and HHSN271201100004C. The datasets used for the analyses described in this manuscript were obtained from dbGaP at http://www.ncbi.nlm.nih.gov/sites/entrez?db=gap through dbGaP accession (phs000200.v11.p3).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.American Cancer Society. Colorectal cancer facts & figures 2017-2019. Atlanta: American Cancer Society, Inc.; 2017. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/colorectal-cancer-facts-and-figures/colorectal-cancer-facts-and-figures-2017-2019.pdf. [Google Scholar]
- 2.American Cancer Society. Cancer fact and figures, 2020. Atlanta: American Cancer Society, Inc.; https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2020/cancer-facts-and-figures-2020.pdf. [Google Scholar]
- 3.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 4.Groblewska M, Mroczko B, Wereszczynska-Siemiatkowska U, Kedra B, Lukaszewicz M, Baniukiewicz A, Szmitkowski M. Serum interleukin 6 (IL-6) and C-reactive protein (CRP) levels in colorectal adenoma and cancer patients. Clin Chem Lab Med. 2008;46:1423–1428. doi: 10.1515/CCLM.2008.278. [DOI] [PubMed] [Google Scholar]
- 5.Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer. 2013;13:759–771. doi: 10.1038/nrc3611. [DOI] [PubMed] [Google Scholar]
- 6.Jess T, Rungoe C, Peyrin-Biroulet L. Risk of colorectal cancer in patients with ulcerative colitis: a meta-analysis of population-based cohort studies. Clin Gastroenterol Hepatol. 2012;10:639–645. doi: 10.1016/j.cgh.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 7.Laukoetter MG, Mennigen R, Hannig CM, Osada N, Rijcken E, Vowinkel T, Krieglstein CF, Senninger N, Anthoni C, Bruewer M. Intestinal cancer risk in Crohn’s disease: a meta-analysis. J Gastrointest Surg. 2011;15:576–583. doi: 10.1007/s11605-010-1402-9. [DOI] [PubMed] [Google Scholar]
- 8.Pendyala S, Neff LM, Suarez-Farinas M, Holt PR. Diet-induced weight loss reduces colorectal inflammation: implications for colorectal carcinogenesis. Am J Clin Nutr. 2011;93:234–242. doi: 10.3945/ajcn.110.002683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ulrich CM, Himbert C, Holowatyj AN, Hursting SD. Energy balance and gastrointestinal cancer: risk, interventions, outcomes and mechanisms. Nat Rev Gastroenterol Hepatol. 2018;15:683–698. doi: 10.1038/s41575-018-0053-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boden S, Myte R, Harbs J, Sundkvist A, Zingmark C, Lofgren Burstrom A, Palmqvist R, Harlid S, Van Guelpen B. C-reactive protein and future risk of clinical and molecular subtypes of colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2020;29:1482–1491. doi: 10.1158/1055-9965.EPI-19-1339. [DOI] [PubMed] [Google Scholar]
- 11.Disis ML. Immune regulation of cancer. J. Clin. Oncol. 2010;28:4531–4538. doi: 10.1200/JCO.2009.27.2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghazi-Khanloosani M, Bandegi AR, Kokhaei P, Barati M, Pakdel A. CRP and LOX-1: a mechanism for increasing the tumorigenic potential of colorectal cancer carcinoma cell line. Pathol Oncol Res. 2019;25:1467–1475. doi: 10.1007/s12253-018-0507-4. [DOI] [PubMed] [Google Scholar]
- 13.Liu L, Tabung FK, Zhang X, Nowak JA, Qian ZR, Hamada T, Nevo D, Bullman S, Mima K, Kosumi K, da Silva A, Song M, Cao Y, Twombly TS, Shi Y, Liu H, Gu M, Koh H, Li W, Du C, Chen Y, Li C, Li W, Mehta RS, Wu K, Wang M, Kostic AD, Giannakis M, Garrett WS, Hutthenhower C, Chan AT, Fuchs CS, Nishihara R, Ogino S, Giovannucci EL. Diets that promote colon inflammation associate with risk of colorectal carcinomas that contain fusobacterium nucleatum. Clin Gastroenterol Hepatol. 2018;16:1622–1631. e1623. doi: 10.1016/j.cgh.2018.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, Yu X, Yu E, Wang N, Cai Q, Shuai Q, Yan F, Jiang L, Wang H, Liu J, Chen Y, Li Z, Jiang Q. Changes in gut microbiota and plasma inflammatory factors across the stages of colorectal tumorigenesis: a case-control study. BMC Microbiol. 2018;18:92. doi: 10.1186/s12866-018-1232-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Proenca MA, Biselli JM, Succi M, Severino FE, Berardinelli GN, Caetano A, Reis RM, Hughes DJ, Silva AE. Relationship between Fusobacterium nucleatum, inflammatory mediators and microRNAs in colorectal carcinogenesis. World J Gastroenterol. 2018;24:5351–5365. doi: 10.3748/wjg.v24.i47.5351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang B, Bostick RM, Tran HQ, Gewirtz AT, Campbell PT, Fedirko V. Circulating biomarkers of gut barrier function: correlates and nonresponse to calcium supplementation among colon adenoma patients. Cancer Epidemiol Biomarkers Prev. 2016;25:318–326. doi: 10.1158/1055-9965.EPI-15-0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Umoh FI, Kato I, Ren J, Wachowiak PL, Ruffin MT 4th, Turgeon DK, Sen A, Brenner DE, Djuric Z. Markers of systemic exposures to products of intestinal bacteria in a dietary intervention study. Eur J Nutr. 2016;55:793–798. doi: 10.1007/s00394-015-0900-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ananthakrishnan AN, Cheng SC, Cai T, Cagan A, Gainer VS, Szolovits P, Shaw SY, Churchill S, Karlson EW, Murphy SN, Kohane I, Liao KP. Serum inflammatory markers and risk of colorectal cancer in patients with inflammatory bowel diseases. Clin Gastroenterol Hepatol. 2014;12:1342–1348. e1341. doi: 10.1016/j.cgh.2013.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Erlinger TP, Platz EA, Rifai N, Helzlsouer KJ. C-reactive protein and the risk of incident colorectal cancer. JAMA. 2004;291:585–590. doi: 10.1001/jama.291.5.585. [DOI] [PubMed] [Google Scholar]
- 20.Slattery ML, Curtin K, Poole EM, Duggan DJ, Samowitz WS, Peters U, Caan BJ, Potter JD, Ulrich CM. Genetic variation in C-reactive protein in relation to colon and rectal cancer risk and survival. Int J Cancer. 2011;128:2726–2734. doi: 10.1002/ijc.25721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prizment AE, Folsom AR, Dreyfus J, Anderson KE, Visvanathan K, Joshu CE, Platz EA, Pankow JS. Plasma C-reactive protein, genetic risk score, and risk of common cancers in the atherosclerosis risk in communities study. Cancer Causes Control. 2013;24:2077–2087. doi: 10.1007/s10552-013-0285-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Godos J, Biondi A, Galvano F, Basile F, Sciacca S, Giovannucci EL, Grosso G. Markers of systemic inflammation and colorectal adenoma risk: meta-analysis of observational studies. World J Gastroenterol. 2017;23:1909–1919. doi: 10.3748/wjg.v23.i10.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davenport JR, Cai Q, Ness RM, Milne G, Zhao Z, Smalley WE, Zheng W, Shrubsole MJ. Evaluation of pro-inflammatory markers plasma C-reactive protein and urinary prostaglandin-E2 metabolite in colorectal adenoma risk. Mol Carcinog. 2016;55:1251–1261. doi: 10.1002/mc.22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Otake T, Uezono K, Takahashi R, Fukumoto J, Tabata S, Abe H, Tajima O, Mizoue T, Ohnaka K, Kono S. C-reactive protein and colorectal adenomas: self defense forces health study. Cancer Sci. 2009;100:709–714. doi: 10.1111/j.1349-7006.2009.01107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim C, Zhang X, Chan AT, Sesso HD, Rifai N, Stampfer MJ, Ma J. Inflammatory biomarkers, aspirin, and risk of colorectal cancer: findings from the physicians’ health study. Cancer Epidemiol. 2016;44:65–70. doi: 10.1016/j.canep.2016.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang G, Cao LY, Chen SH, Xie SH, Feng XS, Lyu ZY, Guo LW, Li F, Su K, Chang S, Ren JS, Dai M, Li N, Wu SL, He J. A prospective follow-up study on the association between serum level of C-reactive protein and risk of digestive system cancers in Chinese women. Zhonghua Zhong Liu Za Zhi. 2016;38:876–880. doi: 10.3760/cma.j.issn.0253-3766.2016.11.015. [DOI] [PubMed] [Google Scholar]
- 27.Zhang SM, Buring JE, Lee IM, Cook NR, Ridker PM. C-reactive protein levels are not associated with increased risk for colorectal cancer in women. Ann Intern Med. 2005;142:425–432. doi: 10.7326/0003-4819-142-6-200503150-00008. [DOI] [PubMed] [Google Scholar]
- 28.Gunter MJ, Cross AJ, Huang WY, Stanczyk FZ, Purdue M, Xue X, Schoen R, Limburg PJ, Schatzkin A, Sinha R, Hayes RB. A prospective evaluation of C-reactive protein levels and colorectal adenoma development. Cancer Epidemiol Biomarkers Prev. 2011;20:537–544. doi: 10.1158/1055-9965.EPI-10-1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ognjanovic S, Yamamoto J, Saltzman B, Franke A, Ognjanovic M, Yokochi L, Vogt T, Decker R, Le Marchand L. Serum CRP and IL-6, genetic variants and risk of colorectal adenoma in a multiethnic population. Cancer Causes Control. 2010;21:1131–1138. doi: 10.1007/s10552-010-9540-7. [DOI] [PubMed] [Google Scholar]
- 30.Shu X, Wu L, Khankari NK, Shu XO, Wang TJ, Michailidou K, Bolla MK, Wang Q, Dennis J, Milne RL, Schmidt MK, Pharoah PDP, Andrulis IL, Hunter DJ, Simard J, Easton DF, Zheng W Breast Cancer Association Consortium. Associations of obesity and circulating insulin and glucose with breast cancer risk: a mendelian randomization analysis. Int J Epidemiol. 2019;48:795–806. doi: 10.1093/ije/dyy201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Merino J, Leong A, Posner DC, Porneala B, Masana L, Dupuis J, Florez JC. Genetically driven hyperglycemia increases risk of coronary artery disease separately from type 2 diabetes. Diabetes Care. 2017;40:687–693. doi: 10.2337/dc16-2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang X, Dai JY, Albanes D, Arndt V, Berndt SI, Bezieau S, Brenner H, Buchanan DD, Butterbach K, Caan B, Casey G, Campbell PT, Chan AT, Chen Z, Chang-Claude J, Cotterchio M, Easton DF, Giles GG, Giovannucci E, Grady WM, Hoffmeister M, Hopper JL, Hsu L, Jenkins MA, Joshi AD, Lampe JW, Larsson SC, Lejbkowicz F, Li L, Lindblom A, Le Marchand L, Martin V, Milne RL, Moreno V, Newcomb PA, Offitt K, Ogino S, Pharoah PDP, Pinchev M, Potter JD, Rennert HS, Rennert G, Saliba W, Schafmayer C, Schoen RE, Schrotz-King P, Slattery ML, Song M, Stegmaier C, Weinstein SJ, Wolk A, Woods MO, Wu AH, Gruber SB, Peters U, White E. Mendelian randomization analysis of C-reactive protein on colorectal cancer risk. Int J Epidemiol. 2019;48:767–780. doi: 10.1093/ije/dyy244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nimptsch K, Aleksandrova K, Boeing H, Janke J, Lee YA, Jenab M, Bueno-de-Mesquita HB, Jansen EH, Tsilidis KK, Trichopoulou A, Weiderpass E, Wu C, Overvad K, Tjonneland A, Boutron-Ruault MC, Dossus L, Racine A, Kaaks R, Canzian F, Lagiou P, Trichopoulos D, Palli D, Agnoli C, Tumino R, Vineis P, Panico S, Johansson A, Van Guelpen B, Khaw KT, Wareham N, Peeters PH, Quiros JR, Vencesla Garcia A, Molina-Montes E, Dorronsoro M, Chirlaque MD, Barricarte Gurrea A, Key TJ, Duarte-Salles T, Stepien M, Gunter MJ, Riboli E, Pischon T. Association of CRP genetic variants with blood concentrations of C-reactive protein and colorectal cancer risk. Int J Cancer. 2015;136:1181–1192. doi: 10.1002/ijc.29086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ellis J, Lange EM, Li J, Dupuis J, Baumert J, Walston JD, Keating BJ, Durda P, Fox ER, Palmer CD, Meng YA, Young T, Farlow DN, Schnabel RB, Marzi CS, Larkin E, Martin LW, Bis JC, Auer P, Ramachandran VS, Gabriel SB, Willis MS, Pankow JS, Papanicolaou GJ, Rotter JI, Ballantyne CM, Gross MD, Lettre G, Wilson JG, Peters U, Koenig W, Tracy RP, Redline S, Reiner AP, Benjamin EJ, Lange LA. Large multiethnic candidate gene study for C-reactive protein levels: identification of a novel association at CD36 in African Americans. Hum Genet. 2014;133:985–995. doi: 10.1007/s00439-014-1439-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jung SY, Scott P, Papp J, Sobel E, Pellegrini M, Yu H, Han S, Zhang ZF. Genome-wide association analysis of pro-inflammatory cytokines and gene-lifestyle interaction for invasive breast cancer risk: the WHI dbGaP Study. Cancer Prev Res (Phila) 2020 doi: 10.1158/1940-6207.CAPR-20-0256. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ligthart S, Vaez A, Võsa U, Stathopoulou MG, de Vries PS, Prins BP, Van der Most PJ, Tanaka T, Naderi E, Rose LM, Wu Y, Karlsson R, Barbalic M, Lin H, Pool R, Zhu G, Macé A, Sidore C, Trompet S, Mangino M, Sabater-Lleal M, Kemp JP, Abbasi A, Kacprowski T, Verweij N, Smith AV, Huang T, Marzi C, Feitosa MF, Lohman KK, Kleber ME, Milaneschi Y, Mueller C, Huq M, Vlachopoulou E, Lyytikäinen LP, Oldmeadow C, Deelen J, Perola M, Zhao JH, Feenstra B LifeLines Cohort Study; Amini M CHARGE Inflammation Working Group. Lahti J, Schraut KE, Fornage M, Suktitipat B, Chen WM, Li X, Nutile T, Malerba G, Luan J, Bak T, Schork N, Del Greco M F, Thiering E, Mahajan A, Marioni RE, Mihailov E, Eriksson J, Ozel AB, Zhang W, Nethander M, Cheng YC, Aslibekyan S, Ang W, Gandin I, Yengo L, Portas L, Kooperberg C, Hofer E, Rajan KB, Schurmann C, den Hollander W, Ahluwalia TS, Zhao J, Draisma HHM, Ford I, Timpson N, Teumer A, Huang H, Wahl S, Liu Y, Huang J, Uh HW, Geller F, Joshi PK, Yanek LR, Trabetti E, Lehne B, Vozzi D, Verbanck M, Biino G, Saba Y, Meulenbelt I, O’Connell JR, Laakso M, Giulianini F, Magnusson PKE, Ballantyne CM, Hottenga JJ, Montgomery GW, Rivadineira F, Rueedi R, Steri M, Herzig KH, Stott DJ, Menni C, Frånberg M, St Pourcain B, Felix SB, Pers TH, Bakker SJL, Kraft P, Peters A, Vaidya D, Delgado G, Smit JH, Großmann V, Sinisalo J, Seppälä I, Williams SR, Holliday EG, Moed M, Langenberg C, Räikkönen K, Ding J, Campbell H, Sale MM, Chen YI, James AL, Ruggiero D, Soranzo N, Hartman CA, Smith EN, Berenson GS, Fuchsberger C, Hernandez D, Tiesler CMT, Giedraitis V, Liewald D, Fischer K, Mellström D, Larsson A, Wang Y, Scott WR, Lorentzon M, Beilby J, Ryan KA, Pennell CE, Vuckovic D, Balkau B, Concas MP, Schmidt R, Mendes de Leon CF, Bottinger EP, Kloppenburg M, Paternoster L, Boehnke M, Musk AW, Willemsen G, Evans DM, Madden PAF, Kähönen M, Kutalik Z, Zoledziewska M, Karhunen V, Kritchevsky SB, Sattar N, Lachance G, Clarke R, Harris TB, Raitakari OT, Attia JR, van Heemst D, Kajantie E, Sorice R, Gambaro G, Scott RA, Hicks AA, Ferrucci L, Standl M, Lindgren CM, Starr JM, Karlsson M, Lind L, Li JZ, Chambers JC, Mori TA, de Geus EJCN, Heath AC, Martin NG, Auvinen J, Buckley BM, de Craen AJM, Waldenberger M, Strauch K, Meitinger T, Scott RJ, McEvoy M, Beekman M, Bombieri C, Ridker PM, Mohlke KL, Pedersen NL, Morrison AC, Boomsma DI, Whitfield JB, Strachan DP, Hofman A, Vollenweider P, Cucca F, Jarvelin MR, Jukema JW, Spector TD, Hamsten A, Zeller T, Uitterlinden AG, Nauck M, Gudnason V, Qi L, Grallert H, Borecki IB, Rotter JI, März W, Wild PS, Lokki ML, Boyle M, Salomaa V, Melbye M, Eriksson JG, Wilson JF, Penninx BWJH, Becker DM, Worrall BB, Gibson G, Krauss RM, Ciullo M, Zaza G, Wareham NJ, Oldehinkel AJ, Palmer LJ, Murray SS, Pramstaller PP, Bandinelli S, Heinrich J, Ingelsson E, Deary IJ, Mägi R, Vandenput L, van der Harst P, Desch KC, Kooner JS, Ohlsson C, Hayward C, Lehtimäki T, Shuldiner AR, Arnett DK, Beilin LJ, Robino A, Froguel P, Pirastu M, Jess T, Koenig W, Loos RJF, Evans DA, Schmidt H, Smith GD, Slagboom PE, Eiriksdottir G, Morris AP, Psaty BM, Tracy RP, Nolte IM, Boerwinkle E, Visvikis-Siest S, Reiner AP, Gross M, Bis JC, Franke L, Franco OH, Benjamin EJ, Chasman DI, Dupuis J, Snieder H, Dehghan A, Alizadeh BZ. Genome analyses of >200,000 individuals identify 58 loci for chronic inflammation and highlight pathways that link inflammation and complex disorders. Am J Hum Genet. 2018;103:691–706. doi: 10.1016/j.ajhg.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schick UM, Auer PL, Bis JC, Lin H, Wei P, Pankratz N, Lange LA, Brody J, Stitziel NO, Kim DS, Carlson CS, Fornage M, Haessler J, Hsu L, Jackson RD, Kooperberg C, Leal SM, Psaty BM, Boerwinkle E, Tracy R, Ardissino D, Shah S, Willer C, Loos R, Melander O, McPherson R, Hovingh K, Reilly M, Watkins H, Girelli D, Fontanillas P, Chasman DI, Gabriel SB, Gibbs R, Nickerson DA, Kathiresan S, Peters U, Dupuis J, Wilson JG, Rich SS, Morrison AC, Benjamin EJ, Gross MD, Reiner AP. Association of exome sequences with plasma C-reactive protein levels in >9000 participants. Hum Mol Genet. 2015;24:559–571. doi: 10.1093/hmg/ddu450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dehghan A, Dupuis J, Barbalic M, Bis JC, Eiriksdottir G, Lu C, Pellikka N, Wallaschofski H, Kettunen J, Henneman P, Baumert J, Strachan DP, Fuchsberger C, Vitart V, Wilson JF, Paré G, Naitza S, Rudock ME, Surakka I, de Geus EJ, Alizadeh BZ, Guralnik J, Shuldiner A, Tanaka T, Zee RY, Schnabel RB, Nambi V, Kavousi M, Ripatti S, Nauck M, Smith NL, Smith AV, Sundvall J, Scheet P, Liu Y, Ruokonen A, Rose LM, Larson MG, Hoogeveen RC, Freimer NB, Teumer A, Tracy RP, Launer LJ, Buring JE, Yamamoto JF, Folsom AR, Sijbrands EJ, Pankow J, Elliott P, Keaney JF, Sun W, Sarin AP, Fontes JD, Badola S, Astor BC, Hofman A, Pouta A, Werdan K, Greiser KH, Kuss O, Meyer zu Schwabedissen HE, Thiery J, Jamshidi Y, Nolte IM, Soranzo N, Spector TD, Völzke H, Parker AN, Aspelund T, Bates D, Young L, Tsui K, Siscovick DS, Guo X, Rotter JI, Uda M, Schlessinger D, Rudan I, Hicks AA, Penninx BW, Thorand B, Gieger C, Coresh J, Willemsen G, Harris TB, Uitterlinden AG, Järvelin MR, Rice K, Radke D, Salomaa V, Willems van Dijk K, Boerwinkle E, Vasan RS, Ferrucci L, Gibson QD, Bandinelli S, Snieder H, Boomsma DI, Xiao X, Campbell H, Hayward C, Pramstaller PP, van Duijn CM, Peltonen L, Psaty BM, Gudnason V, Ridker PM, Homuth G, Koenig W, Ballantyne CM, Witteman JC, Benjamin EJ, Perola M, Chasman DI. Meta-analysis of genome-wide association studies in >80 000 subjects identifies multiple loci for C-reactive protein levels. Circulation. 2011;123:731–738. doi: 10.1161/CIRCULATIONAHA.110.948570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qian F, Wang S, Mitchell J, McGuffog L, Barrowdale D, Leslie G, Oosterwijk JC, Chung WK, Evans DG, Engel C, Kast K, Aalfs CM, Adank MA, Adlard J, Agnarsson BA, Aittomäki K, Alducci E, Andrulis IL, Arun BK, Ausems MGEM, Azzollini J, Barouk-Simonet E, Barwell J, Belotti M, Benitez J, Berger A, Borg A, Bradbury AR, Brunet J, Buys SS, Caldes T, Caligo MA, Campbell I, Caputo SM, Chiquette J, Claes KBM, Margriet Collée J, Couch FJ, Coupier I, Daly MB, Davidson R, Diez O, Domchek SM, Donaldson A, Dorfling CM, Eeles R, Feliubadaló L, Foretova L, Fowler J, Friedman E, Frost D, Ganz PA, Garber J, Garcia-Barberan V, Glendon G, Godwin AK, Gómez Garcia EB, Gronwald J, Hahnen E, Hamann U, Henderson A, Hendricks CB, Hopper JL, Hulick PJ, Imyanitov EN, Isaacs C, Izatt L, Izquierdo Á, Jakubowska A, Kaczmarek K, Kang E, Karlan BY, Kets CM, Kim SW, Kim Z, Kwong A, Laitman Y, Lasset C, Hyuk Lee M, Won Lee J, Lee J, Lester J, Lesueur F, Loud JT, Lubinski J, Mebirouk N, Meijers-Heijboer HEJ, Meindl A, Miller A, Montagna M, Mooij TM, Morrison PJ, Mouret-Fourme E, Nathanson KL, Neuhausen SL, Nevanlinna H, Niederacher D, Nielsen FC, Nussbaum RL, Offit K, Olah E, Ong KR, Ottini L, Park SK, Peterlongo P, Pfeiler G, Phelan CM, Poppe B, Pradhan N, Radice P, Ramus SJ, Rantala J, Robson M, Rodriguez GC, Schmutzler RK, Hutten Selkirk CG, Shah PD, Simard J, Singer CF, Sokolowska J, Stoppa-Lyonnet D, Sutter C, Yen Tan Y, Teixeira RM, Teo SH, Terry MB, Thomassen M, Tischkowitz M, Toland AE, Tucker KM, Tung N, van Asperen CJ, van Engelen K, van Rensburg EJ, Wang-Gohrke S, Wappenschmidt B, Weitzel JN, Yannoukakos D GEMO Study Collaborators; HEBON; EMBRACE. Greene MH, Rookus MA, Easton DF, Chenevix-Trench G, Antoniou AC, Goldgar DE, Olopade OI, Rebbeck TR, Huo D. Height and body mass index as modifiers of breast cancer risk in BRCA1/2 mutation carriers: a mendelian randomization study. J Natl Cancer Inst. 2019;111:350–364. doi: 10.1093/jnci/djy132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40:304–314. doi: 10.1002/gepi.21965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spiller W, Slichter D, Bowden J, Davey Smith G. Detecting and correcting for bias in Mendelian randomization analyses using Gene-by-Environment interactions. Int J Epidemiol. 2019;48:702–712. doi: 10.1093/ije/dyy204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.NCBI: WHI harmonized and imputed GWAS data. A sub-study of Women’s Health Initiative. 2019. https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000746.v3.p3.
- 43.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Castellano-Castillo D, Morcillo S, Clemente-Postigo M, Crujeiras AB, Fernandez-Garcia JC, Torres E, Tinahones FJ, Macias-Gonzalez M. Adipose tissue inflammation and VDR expression and methylation in colorectal cancer. Clin Epigenetics. 2018;10:60. doi: 10.1186/s13148-018-0493-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Himbert C, Ose J, Nattenmuller J, Warby CA, Holowatyj AN, Bohm J, Lin T, Haffa M, Gigic B, Hardikar S, Scherer D, Zielske L, Schrotz-King P, Kolsch T, Siegel EM, Shibata D, Ulrich A, Schneider M, Hursting SD, Kauczor HU, Ulrich CM. Body fatness, adipose tissue compartments, and biomarkers of inflammation and angiogenesis in colorectal cancer: the ColoCare study. Cancer Epidemiol Biomarkers Prev. 2019;28:76–82. doi: 10.1158/1055-9965.EPI-18-0654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pfalzer AC, Leung K, Crott JW, Kim SJ, Tai AK, Parnell LD, Kamanu FK, Liu Z, Rogers G, Shea MK, Garcia PE, Mason JB. Incremental elevations in TNFalpha and IL6 in the human colon and procancerous changes in the mucosal transcriptome accompany adiposity. Cancer Epidemiol Biomarkers Prev. 2018;27:1416–1423. doi: 10.1158/1055-9965.EPI-18-0121. [DOI] [PubMed] [Google Scholar]
- 47.Wu S, Hsu LA, Teng MS, Lin JF, Chou HH, Lee MC, Wu YM, Su CW, Ko YL. Interactive effects of C-reactive protein levels on the association between APOE variants and triglyceride levels in a Taiwanese population. Lipids Health Dis. 2016;15:94. doi: 10.1186/s12944-016-0262-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu M, Lee MH, Mak VW, Tomlinson B. Effect of central obesity, low high-density lipoprotein cholesterol and C-reactive protein polymorphisms on C-reactive protein levels during treatment with Rosuvastatin (10 mg Daily) Am J Cardiol. 2010;106:1588–1593. doi: 10.1016/j.amjcard.2010.07.044. [DOI] [PubMed] [Google Scholar]
- 49.Amaral WZ, Krueger RF, Ryff CD, Coe CL. Genetic and environmental determinants of population variation in interleukin-6, its soluble receptor and C-reactive protein: insights from identical and fraternal twins. Brain Behav Immun. 2015;49:171–181. doi: 10.1016/j.bbi.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fraser A, May M, Lowe G, Rumley A, Smith GD, Ebrahim S, Lawlor DA. Interleukin-6 and incident coronary heart disease: results from the British Women’s Heart and Health Study. Atherosclerosis. 2009;202:567–572. doi: 10.1016/j.atherosclerosis.2008.04.048. [DOI] [PubMed] [Google Scholar]
- 51.Winters-Stone KM, Wood LJ, Stoyles S, Dieckmann NF. The effects of resistance exercise on biomarkers of breast cancer prognosis: a pooled analysis of three randomized trials. Cancer Epidemiol Biomarkers Prev. 2018;27:146–153. doi: 10.1158/1055-9965.EPI-17-0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lynch BM, Friedenreich CM, Winkler EA, Healy GN, Vallance JK, Eakin EG, Owen N. Associations of objectively assessed physical activity and sedentary time with biomarkers of breast cancer risk in postmenopausal women: findings from NHANES (2003-2006) Breast Cancer Res Treat. 2011;130:183–194. doi: 10.1007/s10549-011-1559-2. [DOI] [PubMed] [Google Scholar]
- 53.van Gemert WA, May AM, Schuit AJ, Oosterhof BY, Peeters PH, Monninkhof EM. Effect of weight loss with or without exercise on inflammatory markers and adipokines in postmenopausal women: the SHAPE-2 trial, a randomized controlled trial. Cancer Epidemiol Biomarkers Prev. 2016;25:799–806. doi: 10.1158/1055-9965.EPI-15-1065. [DOI] [PubMed] [Google Scholar]
- 54.Rojo-Martinez G, Soriguer F, Colomo N, Calle A, Goday A, Bordiu E, Delgado E, Menendez E, Ortega E, Urrutia I, Girbes J, Castano L, Catala M, Gaztambide S, Valdes S. Factors determining high-sensitivity C-reactive protein values in the Spanish population. Di@bet.es study. Eur J Clin Invest. 2013;43:1–10. doi: 10.1111/eci.12002. [DOI] [PubMed] [Google Scholar]
- 55.Dias JA, Wirfalt E, Drake I, Gullberg B, Hedblad B, Persson M, Engstrom G, Nilsson J, Schiopu A, Fredrikson GN, Bjorkbacka H. A high quality diet is associated with reduced systemic inflammation in middle-aged individuals. Atherosclerosis. 2015;238:38–44. doi: 10.1016/j.atherosclerosis.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 56.Bermudez EA, Rifai N, Buring JE, Manson JE, Ridker PM. Relation between markers of systemic vascular inflammation and smoking in women. Am J Cardiol. 2002;89:1117–1119. doi: 10.1016/s0002-9149(02)02284-1. [DOI] [PubMed] [Google Scholar]
- 57.Stewart SH, Mainous AG 3rd, Gilbert G. Relation between alcohol consumption and C-reactive protein levels in the adult US population. J Am Board Fam Pract. 2002;15:437–442. [PubMed] [Google Scholar]
- 58.Golkhalkhali B, Rajandram R, Paliany AS, Ho GF, Wan Ishak WZ, Johari CS, Chin KF. Strain-specific probiotic (microbial cell preparation) and omega-3 fatty acid in modulating quality of life and inflammatory markers in colorectal cancer patients: a randomized controlled trial. Asia Pac J Clin Oncol. 2018;14:179–191. doi: 10.1111/ajco.12758. [DOI] [PubMed] [Google Scholar]
- 59.Miranda DO, Anatriello E, Azevedo LR, Cordeiro JFC, Peria FM, Floria-Santos M, Pereira-da-Silva G. Elevated serum levels of proinflammatory cytokines potentially correlate with depression and anxiety in colorectal cancer patients in different stages of the antitumor therapy. Cytokine. 2018;104:72–77. doi: 10.1016/j.cyto.2017.09.030. [DOI] [PubMed] [Google Scholar]
- 60.Nielsen FH. Dietary magnesium and chronic disease. Adv Chronic Kidney Dis. 2018;25:230–235. doi: 10.1053/j.ackd.2017.11.005. [DOI] [PubMed] [Google Scholar]
- 61.Cho YA, Lee J, Oh JH, Chang HJ, Sohn DK, Shin A, Kim J. Inflammatory dietary pattern, IL-17F genetic variant, and the risk of colorectal cancer. Nutrients. 2018;10:724. doi: 10.3390/nu10060724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pischon T, Lahmann PH, Boeing H, Friedenreich C, Norat T, Tjonneland A, Halkjaer J, Overvad K, Clavel-Chapelon F, Boutron-Ruault MC, Guernec G, Bergmann MM, Linseisen J, Becker N, Trichopoulou A, Trichopoulos D, Sieri S, Palli D, Tumino R, Vineis P, Panico S, Peeters PH, Bueno-de-Mesquita HB, Boshuizen HC, Van Guelpen B, Palmqvist R, Berglund G, Gonzalez CA, Dorronsoro M, Barricarte A, Navarro C, Martinez C, Quiros JR, Roddam A, Allen N, Bingham S, Khaw KT, Ferrari P, Kaaks R, Slimani N, Riboli E. Body size and risk of colon and rectal cancer in the European prospective investigation into cancer and nutrition (EPIC) J Natl Cancer Inst. 2006;98:920–931. doi: 10.1093/jnci/djj246. [DOI] [PubMed] [Google Scholar]
- 63.Dashti SG, Viallon V, Simpson JA, Karahalios A, Moreno-Betancur M, English DR, Gunter MJ, Murphy N. Explaining the link between adiposity and colorectal cancer risk in men and postmenopausal women in the UK Biobank: a sequential causal mediation analysis. Int J Cancer. 2020;147:1881–1894. doi: 10.1002/ijc.32980. [DOI] [PubMed] [Google Scholar]
- 64.Aleksandrova K, Drogan D, Boeing H, Jenab M, Bas Bueno-de-Mesquita H, Jansen E, van Duijnhoven FJ, Rinaldi S, Fedirko V, Romieu I, Kaaks R, Riboli E, Gunter MJ, Romaguera D, Westhpal S, Overvad K, Tjonneland A, Halkjaer J, Boutron-Ruault MC, Clavel-Chapelon F, Lukanova A, Trichopoulou A, Trichopoulos D, Vidalis P, Panico S, Agnoli C, Palli D, Tumino R, Vineis P, Buckland G, Sanchez-Cruz JJ, Dorronsoro M, Diaz MJ, Barricarte A, Ramon Quiros J, Peeters PH, May AM, Hallmans G, Palmqvist R, Crowe FL, Khaw KT, Wareham N, Pischon T. Adiposity, mediating biomarkers and risk of colon cancer in the European prospective investigation into cancer and nutrition study. Int J Cancer. 2014;134:612–621. doi: 10.1002/ijc.28368. [DOI] [PubMed] [Google Scholar]
- 65.Aleksandrova K, Schlesinger S, Fedirko V, Jenab M, Bueno-de-Mesquita B, Freisling H, Romieu I, Pischon T, Kaaks R, Gunter MJ, Dahm CC, Overvad K, Rostgaard-Hansen AL, Tjonneland A, Trichopoulou A, Bamia C, Lagiou P, Agnoli C, Mattiello A, Bradbury K, Khaw KT, Riboli E, Boeing H. Metabolic mediators of the association between adult weight gain and colorectal cancer: data from the European prospective investigation into cancer and nutrition (EPIC) cohort. Am J Epidemiol. 2017;185:751–764. doi: 10.1093/aje/kww194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Murphy N, Jenab M, Gunter MJ. Adiposity and gastrointestinal cancers: epidemiology, mechanisms and future directions. Nat Rev Gastroenterol Hepatol. 2018;15:659–670. doi: 10.1038/s41575-018-0038-1. [DOI] [PubMed] [Google Scholar]
- 67.Kim S, Keku TO, Martin C, Galanko J, Woosley JT, Schroeder JC, Satia JA, Halabi S, Sandler RS. Circulating levels of inflammatory cytokines and risk of colorectal adenomas. Cancer Res. 2008;68:323–328. doi: 10.1158/0008-5472.CAN-07-2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288:321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- 69.Chlebowski RT, Wactawski-Wende J, Ritenbaugh C, Hubbell FA, Ascensao J, Rodabough RJ, Rosenberg CA, Taylor VM, Harris R, Chen C, Adams-Campbell LL, White E. Estrogen plus progestin and colorectal cancer in postmenopausal women. N Engl J Med. 2004;350:991–1004. doi: 10.1056/NEJMoa032071. [DOI] [PubMed] [Google Scholar]
- 70.Anderson GL, Limacher M, Assaf AR, Bassford T, Beresford SA, Black H, Bonds D, Brunner R, Brzyski R, Caan B, Chlebowski R, Curb D, Gass M, Hays J, Heiss G, Hendrix S, Howard BV, Hsia J, Hubbell A, Jackson R, Johnson KC, Judd H, Kotchen JM, Kuller L, LaCroix AZ, Lane D, Langer RD, Lasser N, Lewis CE, Manson J, Margolis K, Ockene J, O’Sullivan MJ, Phillips L, Prentice RL, Ritenbaugh C, Robbins J, Rossouw JE, Sarto G, Stefanick ML, Van Horn L, Wactawski-Wende J, Wallace R, Wassertheil-Smoller S. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA. 2004;291:1701–1712. doi: 10.1001/jama.291.14.1701. [DOI] [PubMed] [Google Scholar]
- 71.Center for disease control and prevention. Overweight and obesity. Defining adult overweight and obesity. https://www.cdc.gov/obesity/adult/defining.html.
- 72.Haskell WL, Lee IM, Pate RR, Powell KE, Blair SN, Franklin BA, Macera CA, Heath GW, Thompson PD, Bauman A. Physical activity and public health: updated recommendation for adults from the American College of Sports Medicine and the American Heart Association. Med Sci Sports Exerc. 2007;39:1423–1434. doi: 10.1249/mss.0b013e3180616b27. [DOI] [PubMed] [Google Scholar]
- 73.Van Horn L, Carson JA, Appel LJ, Burke LE, Economos C, Karmally W, Lancaster K, Lichtenstein AH, Johnson RK, Thomas RJ, Vos M, Wylie-Rosett J, Kris-Etherton P. Recommended dietary pattern to achieve adherence to the American Heart Association/American College of Cardiology (AHA/ACC) guidelines: a scientific statement from the American Heart Association. Circulation. 2016;134:e505–e529. doi: 10.1161/CIR.0000000000000462. [DOI] [PubMed] [Google Scholar]
- 74.2015-2020 dietary guidelines for Americans. 8th Edition. 2015. http://health.gov/sites/default/files/2019-09/2015-2020_Dietary_Guidelines.pdf.
- 75.National Cancer Institute. SEER program: comparative staging guide for cancer. June 1993: https://seer.cancer.gov/archive/manuals/historic/comp_stage1.1.pdf.
- 76.NCBI: WHI harmonized and imputed GWAS data. A sub-study of Women’s Health Initiative. http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000746.v1.p3.
- 77.Jung SY, Mancuso N, Yu H, Papp J, Sobel E, Zhang ZF. Genome-wide meta-analysis of gene-environmental interaction for insulin resistance phenotypes and breast cancer risk in postmenopausal women. Cancer Prev Res (Phila) 2019;12:31–42. doi: 10.1158/1940-6207.CAPR-18-0180. [DOI] [PubMed] [Google Scholar]
- 78.Schumacher FR, Al Olama AA, Berndt SI, Benlloch S, Ahmed M, Saunders EJ, Dadaev T, Leongamornlert D, Anokian E, Cieza-Borrella C, Goh C, Brook MN, Sheng X, Fachal L, Dennis J, Tyrer J, Muir K, Lophatananon A, Stevens VL, Gapstur SM, Carter BD, Tangen CM, Goodman PJ, Thompson IM Jr, Batra J, Chambers S, Moya L, Clements J, Horvath L, Tilley W, Risbridger GP, Gronberg H, Aly M, Nordström T, Pharoah P, Pashayan N, Schleutker J, Tammela TLJ, Sipeky C, Auvinen A, Albanes D, Weinstein S, Wolk A, Håkansson N, West CML, Dunning AM, Burnet N, Mucci LA, Giovannucci E, Andriole GL, Cussenot O, Cancel-Tassin G, Koutros S, Beane Freeman LE, Sorensen KD, Orntoft TF, Borre M, Maehle L, Grindedal EM, Neal DE, Donovan JL, Hamdy FC, Martin RM, Travis RC, Key TJ, Hamilton RJ, Fleshner NE, Finelli A, Ingles SA, Stern MC, Rosenstein BS, Kerns SL, Ostrer H, Lu YJ, Zhang HW, Feng N, Mao X, Guo X, Wang G, Sun Z, Giles GG, Southey MC, MacInnis RJ, FitzGerald LM, Kibel AS, Drake BF, Vega A, Gómez-Caamaño A, Szulkin R, Eklund M, Kogevinas M, Llorca J, Castaño-Vinyals G, Penney KL, Stampfer M, Park JY, Sellers TA, Lin HY, Stanford JL, Cybulski C, Wokolorczyk D, Lubinski J, Ostrander EA, Geybels MS, Nordestgaard BG, Nielsen SF, Weischer M, Bisbjerg R, Røder MA, Iversen P, Brenner H, Cuk K, Holleczek B, Maier C, Luedeke M, Schnoeller T, Kim J, Logothetis CJ, John EM, Teixeira MR, Paulo P, Cardoso M, Neuhausen SL, Steele L, Ding YC, De Ruyck K, De Meerleer G, Ost P, Razack A, Lim J, Teo SH, Lin DW, Newcomb LF, Lessel D, Gamulin M, Kulis T, Kaneva R, Usmani N, Singhal S, Slavov C, Mitev V, Parliament M, Claessens F, Joniau S, Van den Broeck T, Larkin S, Townsend PA, Aukim-Hastie C, Gago-Dominguez M, Castelao JE, Martinez ME, Roobol MJ, Jenster G, van Schaik RHN, Menegaux F, Truong T, Koudou YA, Xu J, Khaw KT, Cannon-Albright L, Pandha H, Michael A, Thibodeau SN, McDonnell SK, Schaid DJ, Lindstrom S, Turman C, Ma J, Hunter DJ, Riboli E, Siddiq A, Canzian F, Kolonel LN, Le Marchand L, Hoover RN, Machiela MJ, Cui Z, Kraft P, Amos CI, Conti DV, Easton DF, Wiklund F, Chanock SJ, Henderson BE, Kote-Jarai Z, Haiman CA, Eeles RA Profile Study; Australian Prostate Cancer BioResource (APCB); IMPACT Study; Canary PASS Investigators; Breast and Prostate Cancer Cohort Consortium (BPC3); PRACTICAL (Prostate Cancer Association Group to Investigate Cancer-Associated Alterations in the Genome) Consortium; Cancer of the Prostate in Sweden (CAPS); Prostate Cancer Genome-wide Association Study of Uncommon Susceptibility Loci (PEGASUS); Genetic Associations and Mechanisms in Oncology (GAME-ON)/Elucidating Loci Involved in Prostate Cancer Susceptibility (ELLIPSE) Consortium. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat Genet. 2018;50:928–936. doi: 10.1038/s41588-018-0142-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tao Q, Ang TFA, DeCarli C, Auerbach SH, Devine S, Stein TD, Zhang X, Massaro J, Au R, Qiu WQ. Association of chronic low-grade inflammation with risk of Alzheimer disease in ApoE4 carriers. JAMA Netw Open. 2018;1:e183597. doi: 10.1001/jamanetworkopen.2018.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Buckley DI, Fu R, Freeman M, Rogers K, Helfand M. C-reactive protein as a risk factor for coronary heart disease: a systematic review and meta-analyses for the U.S. Preventive Services Task Force. Ann Intern Med. 2009;151:483–495. doi: 10.7326/0003-4819-151-7-200910060-00009. [DOI] [PubMed] [Google Scholar]
- 81.Bowden J, Dudbridge F. Unbiased estimation of odds ratios: combining genomewide association scans with replication studies. Genet Epidemiol. 2009;33:406–418. doi: 10.1002/gepi.20394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.VanderWeele TJ, Tchetgen Tchetgen EJ, Cornelis M, Kraft P. Methodological challenges in mendelian randomization. Epidemiology. 2014;25:427–435. doi: 10.1097/EDE.0000000000000081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Burgess S, Thompson SG. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol. 2011;40:755–764. doi: 10.1093/ije/dyr036. [DOI] [PubMed] [Google Scholar]
- 84.Pierce BL, Ahsan H, Vanderweele TJ. Power and instrument strength requirements for Mendelian randomization studies using multiple genetic variants. Int J Epidemiol. 2011;40:740–752. doi: 10.1093/ije/dyq151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Carreras-Torres R, Johansson M, Gaborieau V, Haycock PC, Wade KH, Relton CL, Martin RM, Davey Smith G, Brennan P. The role of obesity, type 2 diabetes, and metabolic factors in pancreatic cancer: a mendelian randomization study. J Natl Cancer Inst. 2017;109:djx012. doi: 10.1093/jnci/djx012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44:512–525. doi: 10.1093/ije/dyv080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37:658–665. doi: 10.1002/gepi.21758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Guo Y, Warren Andersen S, Shu XO, Michailidou K, Bolla MK, Wang Q, Garcia-Closas M, Milne RL, Schmidt MK, Chang-Claude J, Dunning A, Bojesen SE, Ahsan H, Aittomäki K, Andrulis IL, Anton-Culver H, Arndt V, Beckmann MW, Beeghly-Fadiel A, Benitez J, Bogdanova NV, Bonanni B, Børresen-Dale AL, Brand J, Brauch H, Brenner H, Brüning T, Burwinkel B, Casey G, Chenevix-Trench G, Couch FJ, Cox A, Cross SS, Czene K, Devilee P, Dörk T, Dumont M, Fasching PA, Figueroa J, Flesch-Janys D, Fletcher O, Flyger H, Fostira F, Gammon M, Giles GG, Guénel P, Haiman CA, Hamann U, Hooning MJ, Hopper JL, Jakubowska A, Jasmine F, Jenkins M, John EM, Johnson N, Jones ME, Kabisch M, Kibriya M, Knight JA, Koppert LB, Kosma VM, Kristensen V, Le Marchand L, Lee E, Li J, Lindblom A, Luben R, Lubinski J, Malone KE, Mannermaa A, Margolin S, Marme F, McLean C, Meijers-Heijboer H, Meindl A, Neuhausen SL, Nevanlinna H, Neven P, Olson JE, Perez JI, Perkins B, Peterlongo P, Phillips KA, Pylkäs K, Rudolph A, Santella R, Sawyer EJ, Schmutzler RK, Seynaeve C, Shah M, Shrubsole MJ, Southey MC, Swerdlow AJ, Toland AE, Tomlinson I, Torres D, Truong T, Ursin G, Van Der Luijt RB, Verhoef S, Whittemore AS, Winqvist R, Zhao H, Zhao S, Hall P, Simard J, Kraft P, Pharoah P, Hunter D, Easton DF, Zheng W. Genetically predicted body mass index and breast cancer risk: mendelian randomization analyses of data from 145,000 women of european descent. PLoS Med. 2016;13:e1002105. doi: 10.1371/journal.pmed.1002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hassanali Z, Ametaj BN, Field CJ, Proctor SD, Vine DF. Dietary supplementation of n-3 PUFA reduces weight gain and improves postprandial lipaemia and the associated inflammatory response in the obese JCR:LA-cp rat. Diabetes Obes Metab. 2010;12:139–147. doi: 10.1111/j.1463-1326.2009.01130.x. [DOI] [PubMed] [Google Scholar]
- 90.Laugerette F, Furet JP, Debard C, Daira P, Loizon E, Geloen A, Soulage CO, Simonet C, Lefils-Lacourtablaise J, Bernoud-Hubac N, Bodennec J, Peretti N, Vidal H, Michalski MC. Oil composition of high-fat diet affects metabolic inflammation differently in connection with endotoxin receptors in mice. Am J Physiol Endocrinol Metab. 2012;302:E374–386. doi: 10.1152/ajpendo.00314.2011. [DOI] [PubMed] [Google Scholar]
- 91.Mocellin MC, Pastore e Silva Jde A, Camargo Cde Q, Fabre ME, Gevaerd S, Naliwaiko K, Moreno YM, Nunes EA, Trindade EB. Fish oil decreases C-reactive protein/albumin ratio improving nutritional prognosis and plasma fatty acid profile in colorectal cancer patients. Lipids. 2013;48:879–888. doi: 10.1007/s11745-013-3816-0. [DOI] [PubMed] [Google Scholar]
- 92.Giovannucci E, Martinez ME. Tobacco, colorectal cancer, and adenomas: a review of the evidence. J Natl Cancer Inst. 1996;88:1717–1730. doi: 10.1093/jnci/88.23.1717. [DOI] [PubMed] [Google Scholar]
- 93.Chan AT, Giovannucci EL. Primary prevention of colorectal cancer. Gastroenterology. 2010;138:2029–2043. e2010. doi: 10.1053/j.gastro.2010.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hartz A, He T, Ross JJ. Risk factors for colon cancer in 150,912 postmenopausal women. Cancer Causes Control. 2012;23:1599–1605. doi: 10.1007/s10552-012-0037-4. [DOI] [PubMed] [Google Scholar]
- 95.Yamasaki E, Ames BN. Concentration of mutagens from urine by absorption with the nonpolar resin XAD-2: cigarette smokers have mutagenic urine. Proc Natl Acad Sci U S A. 1977;74:3555–3559. doi: 10.1073/pnas.74.8.3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Giovannucci E. An updated review of the epidemiological evidence that cigarette smoking increases risk of colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2001;10:725–731. [PubMed] [Google Scholar]
- 97.Giovannucci E, Colditz GA, Stampfer MJ, Hunter D, Rosner BA, Willett WC, Speizer FE. A prospective study of cigarette smoking and risk of colorectal adenoma and colorectal cancer in U.S. women. J Natl Cancer Inst. 1994;86:192–199. doi: 10.1093/jnci/86.3.192. [DOI] [PubMed] [Google Scholar]
- 98.Issa JP, Ottaviano YL, Celano P, Hamilton SR, Davidson NE, Baylin SB. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet. 1994;7:536–540. doi: 10.1038/ng0894-536. [DOI] [PubMed] [Google Scholar]
- 99.Slattery ML, Potter JD, Curtin K, Edwards S, Ma KN, Anderson K, Schaffer D, Samowitz WS. Estrogens reduce and withdrawal of estrogens increase risk of microsatellite instability-positive colon cancer. Cancer Res. 2001;61:126–130. [PubMed] [Google Scholar]
- 100.Cogliano V, Grosse Y, Baan R, Straif K, Secretan B, El Ghissassi F. Carcinogenicity of combined oestrogen-progestagen contraceptives and menopausal treatment. Lancet Oncol. 2005;6:552–553. doi: 10.1016/s1470-2045(05)70273-4. [DOI] [PubMed] [Google Scholar]
- 101.Smith GD, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32:1–22. doi: 10.1093/ije/dyg070. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.