

Abstract
Tumor recurrence is a common clinical dilemma in diffuse gliomas. We aimed to identify a recurrence-related signature to predict the prognosis for glioma patients. In the public Chinese Glioma Genome Atlas dataset, we enrolled multi-omics data including genome, epigenome and transcriptome across primary and recurrent gliomas. We included RNA sequencing data from the batch 1 patients (325 patients) as the training set, while RNA sequencing data from the batch 2 patients (693 patients) were selected as the validation set. The R language was used for subsequent analysis. Compared with primary gliomas, more somatic mutations and copy number alterations were revealed in recurrent gliomas. In recurrent gliomas, we identified 113 genes whose methylation levels were significantly different from those of the primary glioma. Through differential expression analysis between primary and recurrent gliomas, we screened 121 recurrence-related genes. Based on these 121 gene expression profiles, consensus clustering of 325 patients yielded two robust groups with different molecular and prognostic features. We developed a recurrence-related risk signature with the lasso regression algorithm. High-risk group had shorter survival and earlier tumor recurrence than the low-risk group. Compared with traditional indicators, the signature showed better prognostic value. In addition, we constructed a nomogram model to predict glioma survival. Functional characteristics analysis found that the signature was associated with cell division and cell cycle. Immune analysis suggested that immunosuppressive status and macrophages might promote glioma recurrence. We demonstrated a novel 18-gene signature that could effectively predict recurrence and prognosis for glioma patients.
Keywords: Diffuse glioma, recurrence, prognosis, multi-omics, Chinese glioma genome atlas
Introduction
Gliomas are one of the most refractory diseases in the world, accounting for the highest proportion of primary malignancies in the central nervous system. Compared with lower grade glioma (LGG, WHO II-III), glioblastoma (GBM, WHO Grade IV) has the highest fatality rate [1]. The current maximum surgical resection combined with standard chemoradiotherapy only prolongs the median survival of patients with GBM to 14.6 months [2,3]. In 2016, the concept of molecular classification was included for the first time in WHO tumor classification of central nervous system [4]. Combined traditional histopathology with the alteration of IDH and chromosome 1p/19q, diffuse gliomas were classified into 5 subtypes: LGG with IDH-mutant and 1p/19q-codeleted, LGG with IDH-mutant and 1p/19q-intact, LGG with IDH-wildtype, GBM with IDH-mutant, GBM with IDH-wildtype. Each subtype shows specific clinical and prognostic characteristics [5].
Due to tumor invasion, heterogeneity and treatment resistance, tumor recurrence has become the ultimate dilemma for glioma patients. And most LGG generally progress to aggressive GBM within 10 years, leading to treatment failure and poor prognosis [4,6]. Genomic alterations that drive glioma recurrence are different from those in the initial tumor and increasing studies have focused on the evolving genetic landscape of glioma from primary to recurrence. With whole exome sequencing, Johnson et al. detected that more than half of the mutations in the initial tumor disappeared when the 43% of tumor recurred [7]. Through genetic analysis of matched primary and recurrent GBM, Kim et al. observed that recurrent glioblastoma had more genetic variation in the core driver pathway than primary glioblastoma [8]. Meanwhile, changes in gene expression levels are also observed in recurrent gliomas compared to primary tumors. Through immunohistochemical analysis in recurrent GBM, Stark et al. revealed that MLH1 expression was significantly down-regulated [9]. Moreover, recurrent glioblastomas were characterized by downregulation of p53 and MSH2 [10]. Increasing studies have begun to identify key genes as recurrence biomarkers. Deng et al. identified 10 key genes and underlying molecular mechanisms in recurrent LGGs [11]. Jun et al. found that WWP2 could potentially predict recurrence in glioma patients [12]. Genomic variation is closely related to glioma recurrence, but recurrence-related genes have not been systematically analyzed.
Our study integrated multi-omics data including genome, epigenome and transcriptome across primary and recurrent gliomas. And we screened differentially expressed genes between primary and recurrent gliomas and found that these genes could cluster glioma patients into groups with distinct clinical and molecular characteristics. Next, we built a recurrence-related signature with the CGGA_325 dataset and used CGGA_693 as the validation dataset. Then we verified recurrence-related signature as an independent prognostic indicator with good predictive power. An individualized nomogram model, integrating signature, grade and 1p/19q codeletion, was constructed to predict 1-year, 3-year and 5-year OS survival for glioma patients. By analyzing the biological functions with DAVID and GSEA method, we found that the signature was closely related to the malignancy of cancer, covering cell division and cycle. Finally, immune microenvironment analysis suggested that immunosuppressive status and macrophages might promote glioma recurrence. To summarize, our research might contribute to the understanding of glioma recurrence and personalized precision treatment.
Methods
Data collection
We downloaded RNA sequencing data and clinicopathological information of two batches of glioma samples from the public Chinese Glioma Genome Atlas (CGGA) dataset (http://www.cgga.org.cn) [13,14]. The batch 1 (CGGA_325) included 325 gliomas as the training set, and the batch 2 (CGGA_693) included 693 gliomas as the validation set. Whole exome sequencing (WEseq) data of 286 glioma samples were collected from the CGGA dataset, including 180 primary gliomas and 106 recurrent gliomas. SAVI2 software was used to identify somatic mutations as previously mentioned [6] and CNVkit software was used to call copy number alterations [15]. Genome-wide DNA methylation microarray (methyl-array) data of 151 glioma samples from CGGA dataset were enrolled in our study, including 133 primary gliomas and 18 recurrent gliomas. Two neuropathologists jointly confirmed and issued the pathological reports according to the WHO classification of CNS tumors in 2016. Overall survival (OS) was defined from surgery to death or last follow-up. Progression-free survival (PFS) was calculated from surgery to tumor recurrence or last follow-up date on which the patient was known to be progression-free [16]. The CGGA dataset was approved by the Beijing Tiantan Hospital Capital Medical University Institutional Review Board (IRB KY2013-017-01), and all patients signed the informed consent [17].
IDH1/2 mutations
IDH1/2 mutations of patients from CGGA datasets were detected by pyrosequencing technique or whole exome sequencing technique (WES) [18,19].
Screening of differential expressed genes and consensus clustering
Using RNA sequencing data from CGGA_325 dataset, we screened 121 differentially expressed genes (fold change > 2 or < 0.5 and p value of Student’s t test < 0.05) between primary and recurrent gliomas. These genes are used for subsequent consensus clustering with R package “ConsensusClusterPlus”. The optimal number of unsupervised classes in 325 patients from the CGGA_325 dataset was determined by cumulative distribution function (CDF) and consensus matrices [20].
Signature development
We performed univariate Cox regression analysis with 121 genes and screened 108 genes related to prognosis (P < 0.05). Our signature was developed from the Least Absolute Shrinkage and Selection Operator (LASSO) regression algorithm [21]. Based on 10-fold cross-validation within the CGGA_325 dataset, the penalty parameter λ was chosen to produce the minimum mean cross-validated error for the Cox model. Thus we obtained 18 genes with corresponding regression coefficients. The risk score of each patient in CGGA_325 validation set was calculated using a linear combination of signature gene expression weighted by their regression coefficients. Risk score = (exprgene1 × coefficientgene1) + (exprgene2 × coefficientgene2) + … + (exprgene18 × coefficientgene18). In the CGGA_693 validation set, we used regression coefficients from CGGA_325 dataset to calculate the risk score. We divided patients into high- and low-risk groups using the median risk score.
DAVID functional annotation and gene set enrichment analysis (GSEA)
We filtered genes that were significantly associated with risk scores (Pearson |R| > 0.5, P < 0.05) in CGGA_325 dataset. And then we performed Gene ontology (GO) analysis and KEGG pathway analysis with positively or negatively associated genes in DAVID (https://david.ncifcrf.gov/) [22]. We performed Gene set enrichment analysis (GSEA) to find enriched functions with GSEA 4.0.1 software (http://www.broadinstitute.org/gsea/index.jsp) [23]. Gene sets ranged from 15 to 500 in size and the number of permutations was 1000. Generally, the enrichment is significant when |NES| > 1 and NOM p-value < 0.05.
Single-cell RNA sequencing (scRNA-seq)
Two IDH-wild glioblastoma samples were collected from Beijing Tiantan Hospital, Capital Medical University and fresh tumor samples were digested with trypsin to obtain cell suspension. The cell viability reached 80% and the cell number reached 5 × 105 for the next step of library construction. The Chromium Single Cell 3’ Library and Gel Bead kit v2 (120267, 10 × Genomics) were used to construct cDNA libraries. Single-cell RNA sequencing was performed on Illumina HiSeq platform and data output was processed with Cell Ranger (v3.0.2, 10 × Genomics). UMAP (Uniform Manifold Approximation and Projection) was used for dimension reduction and cell subtypes were identified by cell-specific markers. InferCNV was performed to access chromosomal copy number variation with single-cell RNA expression data by exploring the intensity of gene expression in the tumor genome compared to reference “normal” cells.
Statistical analysis
R software (version 3.6.1, https://www.r-project.org/) was mainly used for statistical analysis. We evaluated the prognostic significance with Kaplan-Meier survival curve and two-sided log-rank test. Clinicopathological differences were accessed by Student’s t-test or Chi-square test. We used coxph function in “survival” package for univariate and multivariate cox regression analysis. Time-dependent ROC curve (timeROC) was drawn with an R package “timeROC” to predict one-, three- and five-year survival time of patients [24,25]. The nomogram model we constructed was obtained by integrating signature and pathological indicators using R package “rms”. CIBERSORT [26] was performed to assess the abundance of cell types and “macrophages” were the sum of M0, M1 and M2 macrophages. Other figures were drawn with R packages, including ComplexHeatmap, gglpot2, pheatmap, Hmisc and circlize. Values of P < 0.05 were considered significant.
Results
Different patterns of genomic alterations between primary and recurrent gliomas
To investigate the genetic heterogeneity between primary and recurrent gliomas, we analyzed somatic mutations and copy number alterations from CGGA dataset. By comparing the frequency of mutations in primary and recurrent gliomas, more somatic mutations were found in recurrent gliomas (primary vs. recurrent, 33 vs. 92 mutations per person). In Figure 1A, we showed the genes which was most commonly mutated in gliomas, like IDH1, TP53, ATRX, CIC, NOTCH1, EGFR, FUBP1, PDGFRA, PTEN, PIK3CA, PIK3CG, PIK3R1, NF1, MET, LTBP4, CDKN2A, RB1 and PTPN11. And most of these genes show an increased tendency of mutation in recurrent gliomas. Mutations in EGFR were significantly enriched in recurrent gliomas (Chi-square test, p value < 0.05). EGFR was a transmembrane protein, which could transmit important extracellular growth factor signals into cells. Mutation or amplification in EGFR promoted the development of tumors, especially in lung cancer and gliomas [27,28].
Figure 1.

Genomic alterations and methylation differences in primary and recurrent gliomas. A. Somatic mutations and copy number alterations analysis in primary and recurrent gliomas of CGGA dataset. Chi-square test, *P < 0.05. B. Genes in recurrent gliomas with different DNA methylation compared to primary gliomas. C. RNA expression of 5 genes was significantly negatively correlated with methylation level by the Pearson correlation coefficient.
In addition to somatic mutations, we also analyzed copy number alterations in primary and recurrent gliomas. More copy number alterations were revealed in recurrent gliomas (primary vs. recurrent, 2025 vs. 2135 CNAs per person). In the bottom half of Figure 1, we showed the most common copy number alterations in gliomas. Deleted or amplified regions were the most frequently identified in recurrent gliomas, such as CDKN2A, CDKN2B, MLLT3, RB1, LCP1, CACNA1D and RAD21 (Chi-square test, P < 0.05).
Differences in DNA methylation between primary and recurrent gliomas
To investigate the changes of DNA methylation in recurrent gliomas, we analyzed gene methylation profiles. In recurrent gliomas, we screened 88 genes whose methylation levels were down-regulated (Fold change < 1, P < 0.01) and 25 genes whose methylation levels were up-regulated (Fold change > 1, P < 0.01, Figure 1B). Among them, the RNA expression and methylation level of 5 genes were significantly negatively correlated (Figure 1C), including AHR (Cor = -0.28, P = 0.002), BAZ1A (Cor = -0.26, P = 0.003), DDA1 (Cor = -0.24, P = 0.008), IFI16 (Cor = -0.24, P = 0.007), CD48 (Cor = -0.21, P = 0.024).
Gliomas were stratified based on a set of recurrence-related genes
To understand the RNA expression level in recurrent gliomas, we collected RNA sequencing data of 325 gliomas from CGGA dataset (CGGA_325, batch 1). CGGA_325 dataset included 229 primary gliomas and 92 recurrent gliomas (4 unknown). Based on the threshold of fold change > 2 or < 0.5 and p value of Student’s t test < 0.05, 121 differentially expressed genes between primary and recurrent gliomas were screened, including 70 down-regulated genes and 51 up-regulated genes (Figure 2A). Between k = 2 and k = 10, consensus clustering of 325 glioma patients identified two stable subgroups (Figures 2B-D and S1). Figure 2E showed a heatmap of two subgroups defined by 121 differentially expressed genes. The survival curve revealed that Group 2 lived significantly longer than Group 1 (P < 0.0001, Figure 2F). We also observed significant clinicopathological differences in two groups of glioma patients (Table 1). More patients in Group 1 were linked with recurrent status, older age at diagnosis, mesenchymal or classical subtypes, GBM, IDH wild-type and 1p/19q non-codeletion (P < 0.001), while more patients in Group 2 were primary, younger, proneural or neural subtypes, lower grade, IDH mutant, 1p/19q co-deleted (P < 0.001). These results suggested a correlation between recurrence-related genes expression and prognosis, as well as clinicopathology of glioma patients.
Figure 2.
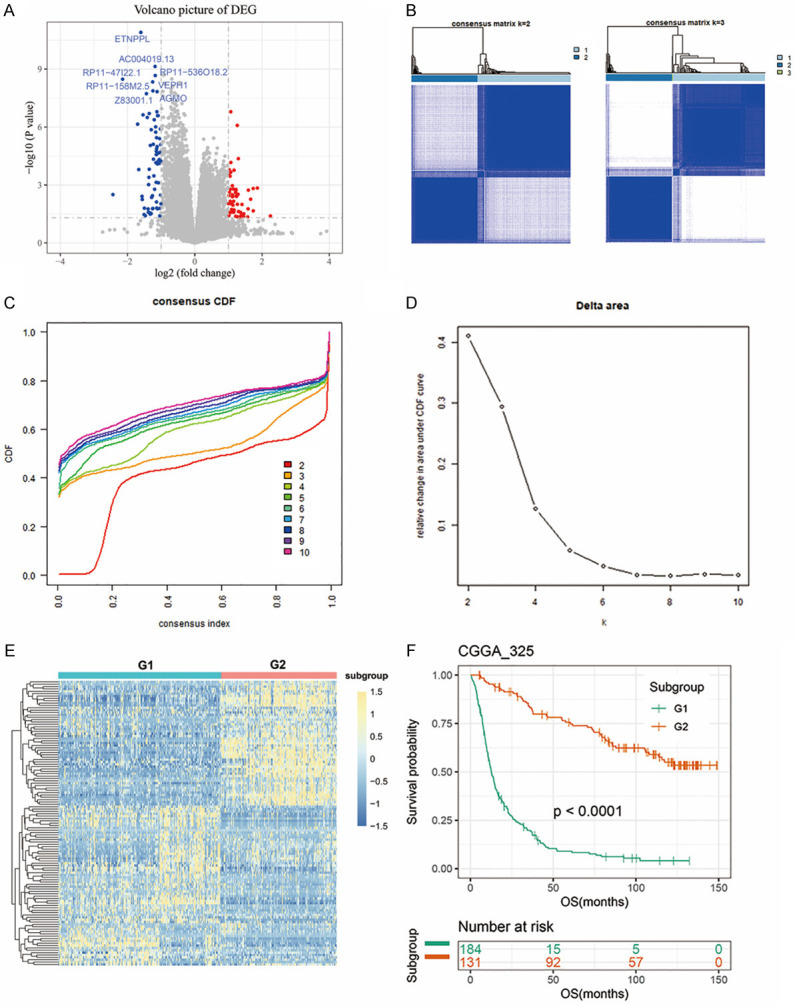
Recurrence-related genes classified glioma patients with distinct prognostic and clinical characteristics. A. The volcano plot showed the differentially expressed genes between primary and recurrent gliomas in CGGA_325 dataset (fold change > 2 or < 0.5 and p value of Student’s t test < 0.05). Red dots, up-regulated genes; blue dots, down-regulated genes. B. Consensus clustering matrix of samples from CGGA_325 dataset for k = 2 and 3. C. CDF value of consensus clustering for k from 2 to 10. D. The relative change in the area under CDF curve for k from 2 to 10. E. Heatmap of the two subgroups defined by 121 recurrence-related genes. F. Kaplan-Meier survival curve of patients from Group 1 and Group 2.
Table 1.
Characteristics of patients in group 1 and group 2 in CGGA_325 dataset
| Characteristics | n | G1 | G2 | P-value |
|---|---|---|---|---|
| Total Cases | 325 | 190 | 135 | |
| PR_type | ||||
| Primary | 229 | 116 | 113 | < 0.001 |
| Recurrent | 92 | 70 | 22 | |
| NA | 4 | 4 | 0 | |
| Age | ||||
| Mean (range) | 43 (8-79) | 45 (8-79) | 40 (17-61) | < 0.001 |
| Gender | ||||
| Female | 122 | 65 | 57 | 0.176 |
| Male | 203 | 125 | 78 | |
| TCGA subtype | ||||
| Proneural | 102 | 41 | 61 | < 0.001 |
| Neural | 81 | 15 | 66 | |
| Classical | 74 | 68 | 6 | |
| Mesenchymal | 68 | 66 | 2 | |
| Grade | ||||
| II | 103 | 13 | 90 | < 0.001 |
| III | 79 | 48 | 31 | |
| IV | 139 | 125 | 14 | |
| NA | 4 | 4 | 0 | |
| IDH status | ||||
| Mutant | 175 | 59 | 116 | < 0.001 |
| Wildtype | 149 | 131 | 18 | |
| NA | 1 | 0 | 1 | |
| 1p/19q status | ||||
| Codel | 67 | 4 | 63 | < 0.001 |
| Noncodel | 250 | 180 | 70 | |
| NA | 8 | 6 | 2 | |
| Radio_status | ||||
| Yes | 258 | 143 | 115 | 0.035 |
| No | 51 | 37 | 14 | |
| NA | 16 | 10 | 6 | |
| Chemo_status | ||||
| Yes | 178 | 119 | 59 | 0.001 |
| No | 124 | 59 | 65 | |
| NA | 23 | 12 | 11 |
Identification of a recurrence-related prognostic signature in gliomas
Since recurrence-related genes expression were closely associated with patient prognosis, we intended to construct a recurrence-related signature for prognosis prediction. First, we screened 108 prognostic genes out of the differential genes with univariate regression analysis in the CGGA_325 dataset (P < 0.05). Then, we screened 18 genes as active covariables through LASSO algorithm (Figure 3A) and regression coefficients of 18 genes were shown in Figure 3B. We then built an 18-gene risk signature with gene expression level and regression coefficients. The algorithm was as follows: signature risk score = (HOXA10 * 0.092) + (HOXD9 * 0.075) + (OR2I1P * 0.072) + (RP11-277P 12.20 * 0.056) + (ZNF560 * 0.053) + (FOXM1 * 0.030) + (TRH * 0.027) + (RP11-93B14.5 * 0.019) + (SNORD3B-2 * 0.018) + (AC062021.1 * -0.039) + (KCNJ11 * -0.042) + (AC053503.11 * -0.044) + (GS1-18A18.1 * -0.071) + (RP11-47I22.1 * -0.094) + (PRLHR * -0.125) + (CD8BP * -0.136) + (KSR2 * -0.206) + (INHBA-AS1 * -0.314). Then we divided patients into high- and low-risk groups according to the median risk score, and found significant differences in clinicopathological features (Figure 3C and Table 2). Compared to the low-risk group (16%), more recurrent patients were found in high-risk group (42%, P < 0.001). Patients were older in high-risk group (mean age = 45) than in low-risk group (mean age = 41, P = 0.006). Compared with low-risk group (17%), a large number of patients with GBM (WHO IV) were found in the high-risk group (71%, P < 0.001). Furthermore, classical and mesenchymal subtypes were found in 72% of high-risk patients and 15% of low-risk patients (P < 0.001). Moreover, IDH mutation appeared in 32% of high-risk patients and 76% of low-risk patients (P < 0.001), while 1p/19q codeletion appeared in 2% and 39% of high-risk and low-risk patients (P < 0.001).
Figure 3.
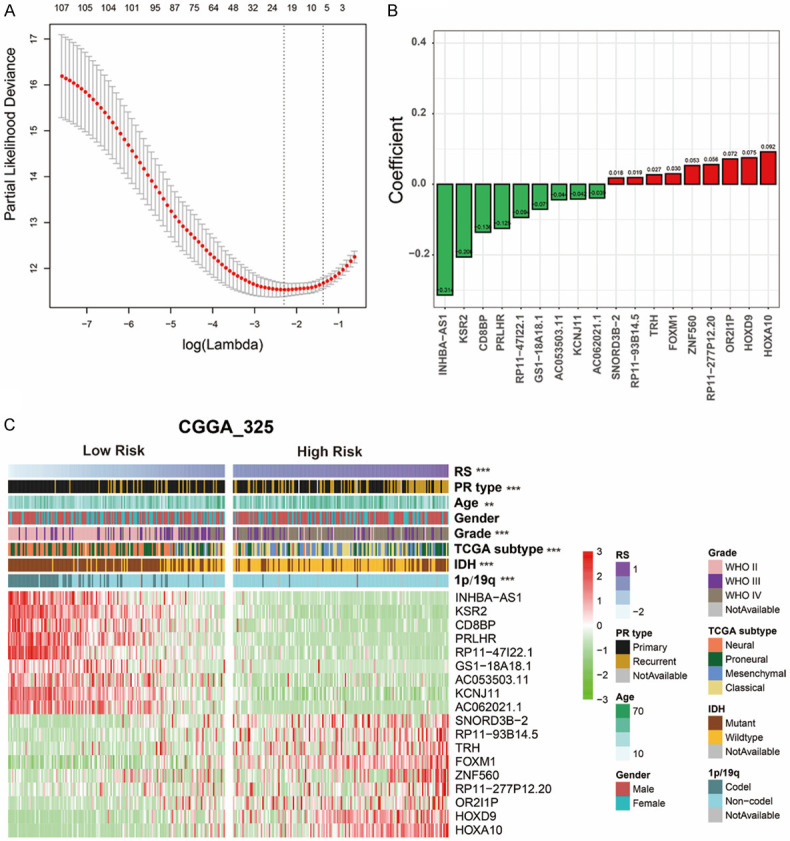
Development of a recurrence-related signature by LASSO analysis. A. Partial likelihood deviance as function of regularization parameter λ in the CGGA_325 training dataset. B. 18 genes screened by LASSO and corresponding regression coefficients. C. Heatmap and clinicopathological features of patients grouped by signature. **P < 0.01, ***P < 0.001, ****P < 0.0001. Abbreviations: RS, Risk Score; IDH, isocitrate dehydrogenase.
Table 2.
Correlation between 18-gene-based risk scores and clinicopathological factors of glioma patients in the two cohorts
| Characteristics | Training set CGGA_325 cohort | Validation set CGGA_693 cohort | ||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| Low-risk group (n = 163) | High-risk group (n = 162) | P-Value | Low-risk group (n = 347) | High-risk group (n = 346) | P-Value | |
| PR_type | ||||||
| Primary | 137 | 92 | < 0.001 | 240 | 182 | < 0.001 |
| Recurrent | 26 | 66 | 107 | 164 | ||
| NA | 0 | 4 | 0 | 0 | ||
| Age | ||||||
| Mean (range) | 41 (17-74) | 45 (8-79) | 0.006 | 41 (11-69) | 45 (13-76) | < 0.001 |
| Gender | ||||||
| Female | 66 | 56 | 0.323 | 156 | 139 | 0.232 |
| Male | 97 | 106 | 191 | 207 | ||
| TCGA subtype | ||||||
| Proneural | 65 | 37 | < 0.001 | NA | NA | |
| Neural | 73 | 8 | NA | NA | ||
| Classical | 16 | 58 | NA | NA | ||
| Mesenchymal | 9 | 59 | NA | NA | ||
| Grade | ||||||
| II | 93 | 10 | < 0.001 | 150 | 38 | < 0.001 |
| III | 43 | 36 | 150 | 105 | ||
| IV | 27 | 112 | 46 | 203 | ||
| NA | 0 | 4 | 1 | NA | ||
| IDH status | ||||||
| Mutant | 123 | 52 | < 0.001 | 247 | 109 | < 0.001 |
| Wildtype | 39 | 110 | 61 | 225 | ||
| NA | 1 | 0 | 39 | 12 | ||
| 1p/19q status | ||||||
| Codel | 64 | 3 | < 0.001 | 129 | 16 | < 0.001 |
| Noncodel | 96 | 154 | 195 | 283 | ||
| NA | 3 | 5 | 23 | 47 | ||
Correlation between recurrence-related signature and pathological features in gliomas
Next, we compared the risk scores of patients with distinct pathological features (Figure 4A). As the WHO grade increased, the risk score increased significantly (P < 0.0001). Moreover, risk score was higher in patients with wild IDH or 1p/19q non-codeletion significantly (P < 0.0001). For CGGA_693 validation dataset, we also built the risk score for each patient with 18-gene regression coefficients from the training set. The results of the validation set were consistent with the above results (Figure S2, Table 2 and Figure 4B). Then, ROC curve was used to analyze the specificity and sensitivity of recurrence-related signature to predict pathological features in two datasets (Figure 4C and 4D). Our signature risk score could well predict glioma grade (AUC 0.801 or 0.807 in CGGA_325 or CGGA_693), IDH mutation status (AUC 0.750 or 0.763) and 1p/19q co-deletion status (AUC 0.930 or 0.841), which was better than age and gender. Briefly, these findings showed that the recurrence-related signature was closely associated with pathological features in gliomas.
Figure 4.
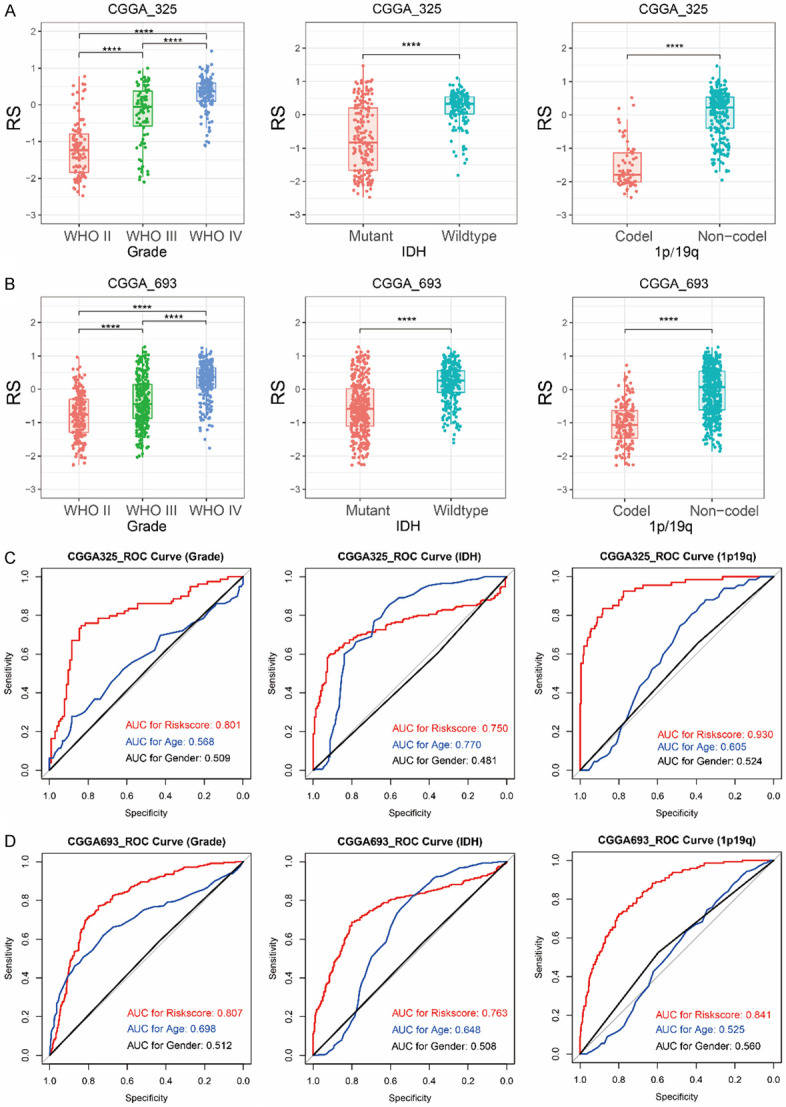
Association between risk score (RS) and pathological characteristics (Grade, IDH mutation and 1p/19q codeletion). Differences in the distribution of risk score (RS) among patients with different pathological characteristics in CGGA_325 dataset (A) and CGGA_693 dataset (B). ROC curves revealed the predictive value of risk score, age and gender for pathological characteristics (Grade, IDH mutation and 1p/19q codeletion) in CGGA_325 dataset (C) and CGGA_693 dataset (D). ****P < 0.0001.
Prognostic analysis of the recurrence-associated signature
Considering the correlation between signature and pathological features, we further evaluated its prognostic value. Survival curve showed that the low-risk patients had significantly longer overall survival than high-risk patients (P < 0.0001, Figure 5A). Then we observed that the survival difference was also significant in lower grade glioma (LGG) or GBM (P < 0.001). In 2016, the latest classification of the CNS tumor combined traditional histopathology with the status of IDH and 1p/19q, gliomas were divided into five subtypes [4]. Due to different clinical outcome of five glioma subtypes, we further evaluated in the five various populations and found excellent performance of our signature (P < 0.05). Similarly, we evaluated prognostic value of signature in the CGGA_693 validation group, and the survival curve showed consistent results (P < 0.05, Figure S3A).
Figure 5.

Survival prediction of the recurrence-related signature in CGGA_325 dataset. A. Overall survival prediction of the signature in all grade gliomas, lower grade gliomas (LGG, grade II and III), glioblastoma (GBM, grade IV), LGG with IDH-mutant and 1p/19q-codeleted, LGG with IDH-mutant and 1p/19q-intact, LGG with IDH-wildtype, GBM with IDH-mutant, GBM with IDH-wildtype. B. Progression free survival prediction of the signature in all grade gliomas, lower grade gliomas (LGG, grade II and III), glioblastoma (GBM, grade IV), LGG with IDH-mutant and 1p/19q-codeleted, LGG with IDH-mutant and 1p/19q-intact, LGG with IDH-wildtype, GBM with IDH-mutant, GBM with IDH-wildtype. Survival difference was determined by a log-rank test.
Next, we further evaluated the predictive value of signature for glioma recurrence. We collected the progression-free survival (PFS) of patients and the Kaplan-Meier curve indicated that the PFS of low-risk patients was significantly longer than that of high-risk patients (P < 0.001, Figure 5B). Dividing patients into LGG or GBM, or subdividing them into five subtypes, we still got consistent results. Meanwhile, we validated in the CGGA_693 dataset (Figure S3B).
Subsequently, univariate and multivariate analyses showed that recurrence-related signature could serve as a prognostic indicator, independent of age, gender, WHO grade, IDH mutation and 1p/19q codeletion in both CGGA_325 and CGGA_693 datasets (P < 0.0001, Table 3).
Table 3.
Variables related to OS in gliomas: univariate and multivariate analysis
| CGGA325 | Univariate Cox Regression | Multivariate Cox Regression | ||||
|
|
|
|||||
| HR | 95% CI | p Value | HR | 95% CI | p Value | |
|
| ||||||
| Gender (male vs. female) | 0.930 | 0.707-1.223 | 0.602 | |||
| Age (≥45 vs. < 45) | 1.990 | 1.518-2.610 | 6.39e-07* | 1.316 | 0.971-1.784 | 0.076 |
| Grade (GBM vs. LGG) | 4.821 | 3.586-6.481 | < 2e-16* | 1.987 | 1.402-2.816 | 0.0001* |
| IDH (wild vs. mutant type) | 2.848 | 2.155-3.765 | 1.89e-13* | 0.8233 | 0.581-1.168 | 0.275 |
| 1p/19q (non-codel vs. codel) | 5.978 | 3.663-9.755 | 8.28e-13* | 3.0347 | 1.776-5.186 | 4.91e-05* |
| Risk score (high vs. low) | 6.159 | 4.538-8.357 | < 2e-16* | 3.1822 | 2.179-4.647 | 2.09e-09* |
|
| ||||||
| CGGA693 | Univariate Cox Regression | Multivariate Cox Regression | ||||
|
|
|
|||||
| HR | 95% CI | p Value | HR | 95% CI | p Value | |
|
| ||||||
| Gender (male vs. female) | 1.012 | 0.826-1.241 | 0.907 | |||
| Age (≥45 vs. < 45) | 1.808 | 1.477-2.213 | 9.19e-09* | 1.314 | 1.044-1.652 | 0.020* |
| Grade (GBM vs. LGG) | 3.979 | 3.227-4.906 | < 2e-16* | 1.996 | 1.506-2.646 | 1.55e-06* |
| IDH (wild vs. mutant type) | 3.238 | 2.616-4.009 | < 2e-16* | 1.326 | 1.006-1.747 | 0.045* |
| 1p/19q (non-codel vs. codel) | 3.806 | 2.713-5.339 | 1.02e-14* | 1.742 | 1.180-2.572 | 0.005* |
| Risk score (high vs. low) | 4.375 | 3.503-5.464 | < 2e-16* | 2.443 | 1.839-3.245 | 6.97e-10* |
HR, hazard ratio; CI, confidence interval;
Significant.
A survival prediction model based on recurrence-related signature
Furthermore, we investigated the power of signature in predicting 1-year, 3-year and 5-year survival of glioma patients with the “timeROC” algorithm. We also included traditional indicators for comparison. The 1-year, 3-year and 5-year AUC values of recurrence-related signature were 0.8389, 0.8964 and 0.9234 (Figure 6A), respectively, superior to age (0.6019, 0.6551 and 0.6627) and grade (0.7648, 0.8477 and 0.8557). In the CGGA_693 validation group, the 1-year, 3-year and 5-year AUC values of recurrence-related signature were 0.7481, 0.8284 and 0.8401, superior to age (0.6102, 0.6277 and 0.5900) and grade (0.7466, 0.7766 and 0.7679) (Figure S4A). These results illustrated the power of our signature to predict prognosis of glioma patients.
Figure 6.
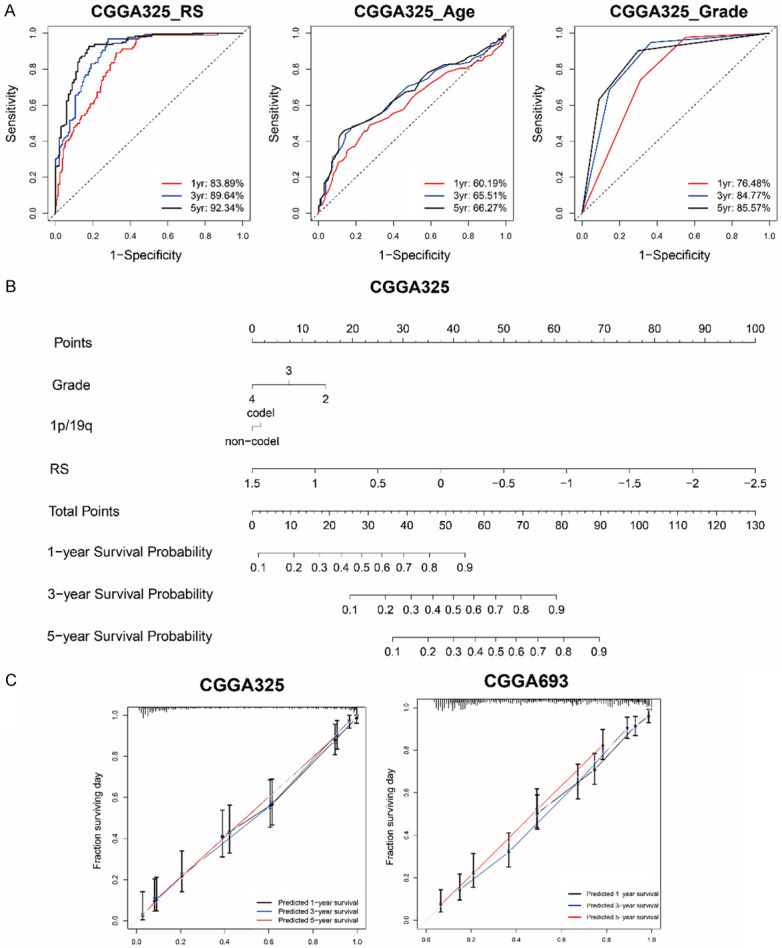
A survival prediction model for glioma patients based on recurrence-related signature. A. 1-year, 3-year and 5-year ROC curves indicated the sensitivity and specificity of signature risk score, age and grade in CGGA_325 dataset. B. A nomogram prediction model was developed by integrating the signature RS with the pathologic features in the CGGA_325 dataset. C. Calibration curves of nomogram for predicting overall survival at 1-year (black line), 3-year (blue line) and 5-year (red line) in the CGGA_325 and CGGA_693 dataset.
Then a nomogram model was constructed using independent prognostic indicators (risk score, grade, 1p/19p) in Cox regression analysis (Figures 6B and S4B). In CGGA_325 and CGGA_693 dataset, the C-indices of the model were 0.804 and 0.772 respectively. Moreover, the calibration curve for probability of survival also revealed satisfactory consistency with the predictions for 1-year, 3-year and 5-year OS in two datasets (Figure 6C).
Functional annotation of recurrence-related signature
To reveal underlying functional characteristics of signature, we performed Gene Ontology (GO) analysis in DAVID with CGGA_325 dataset. First, we performed Pearson correlation and screened genes related to signature (1875 positively and 951 negatively related genes, Pearson |R| > 0.5, P < 0.05). Positively related genes were linked to biological processes of tumor cell proliferation, covering “cell division”, “mitotic nuclear division”, “DNA replication”, “sister chromatid cohesion”, “G1/S transition of mitotic cell cycle” and so on (Figure 7A). And negatively related genes were enriched in the biological processes of normal neural function, such as “chemical synaptic transmission”, “neurotransmitter secretion”, “ion transmembrane transport”, “learning”, and “glutamate secretion” (Figure 7C).
Figure 7.

Functional annotation of recurrence-related signature in CGGA_325 dataset. With Gene Ontology (GO) analysis in DAVID, we analyzed biological processes of signature positively related genes (A) and negatively related genes (C). With KEGG pathway analysis in DAVID, we also revealed the enrichment pathways of genes that were positively (B) or negatively (D) associated with signature. Go terms enriched in high-risk patients (E) or low-risk patients (F) were revealed through Gene set enrichment analysis (GSEA).
In addition, DAVID also showed the enrichment of KEGG pathway. We noticed positively related genes were mainly enriched in the KEGG pathway including “cell cycle”, “protein processing in endoplasmic reticulum”, “focal adhesion”, and “ECM-receptor interaction” (Figure 7B). And negatively related genes were involved in “neuroactive ligand-receptor interaction”, “retrograde endocannabinoid signaling”, “GABAergic synapse”, and “glutamatergic synapse” (Figure 7D). We further verified the above results in CGGA_693 dataset (Figure S5).
Finally, GSEA analysis was performed to verify functional differences among different patients. GO terms of cell cycle were found to be enriched in the high-risk group, including “mitotic cell cycle checkpoint” (NES = 1.958, Nominal P < 0.001), “regulation of cell cycle phase transition” (NES = 1.938, Nominal P < 0.001), and “regulation of cell cycle G1 S phase transition” (NES = 1.933, Nominal P < 0.001, Figure 7E). Whereas in the low-risk group, GO terms including “regulation of postsynaptic membrane potential” (NES = -1.888, Nominal P = 0.002), “ligand gated ion channel activity” (NES = -1.825, Nominal P < 0.001), and “glutamate receptor signaling pathway” (NES = -1.843, Nominal P < 0.001) were significantly enriched (Figure 7F).
Recurrence-related signature was associated with the tumor immune microenvironment
Considering the association between tumor recurrence and immune microenvironment [29,30], we included immune checkpoint genes for analysis. In Figures 8A and S6A, circus plots showed recurrence-related signature was positively associated with immune checkpoint genes (LAG3, CTLA4, PD-L1, B7-H3, PD1, IDO1, CD80 and TIM-3) in two datasets, revealing immunosuppressive status in high-risk group. Next, we explored which immune cell types may play an important mechanism. By CIBERSORT analysis, we found that macrophages were significantly upregulated in the high-risk group (P < 0.05, Figures 8B and S6B). We then performed single-cell RNA sequencing (scRNA-seq) on two IDH-wildtype glioblastoma samples (SC1 was RS-high and SC2 was RS-low) and all the cells were clustered into three subgroups: tumor cells, macrophages and lymphocytes (Figure 8C). By comparing normal cells, inferCNV showed significant copy number variation in malignant cells (Figure 8D). Separately, the proportion of macrophages in SC1 (45.7%) was significantly higher than that in SC2 (19.4%) (P < 2.2e-16, Figure 8E and 8F).
Figure 8.

Recurrence-related signature and immune microenvironment. A. Pearson correlation of eight immune checkpoint genes and signature in all grade gliomas, lower grade gliomas (LGG) and glioblastoma multiforme (GBM). B. Tumor-associated macrophages in high-risk patients compared to low-risk patients by CIBERSORT. C. Two single-cell RNA-sequencing samples (SC1 was RS-high and SC2 was RS-low) were integrated and cells from each sample were differently colored. All cells from two samples were clustered into three groups. D. The inferCNV heatmap included reference normal cells (upper part) and malignant tumor cells (lower part). Red and blue represented chromosome amplification and deletion respectively. E. SC1 (left) and SC2 (right) in the UMAP plot. F. Macrophage quantitative results of single cell analysis (P < 2.2e-16 by chi-squared test).
Discussion
Currently, most glioma researches have focused on primary newly diagnosed tumors, while the clinical and biological characteristics of recurrent gliomas are still poorly understood. This is due to some unavoidable reasons. A minority of patients with recurrent gliomas are eligible for surgical treatment, resulting in the slow collection of these samples and hindering the establishment of large sample banks [31]. Recurrent gliomas also contain large areas of necrotic tissue with a small percentage of tumor cells [32]. The low quality of the samples makes subsequent testing and analysis difficult and biased. Although the Cancer Genome Atlas (TCGA) database, now widely used in the world, contains a large amount of glioma with RNA-seq data (699 gliomas), the recurrent gliomas are rare (only 30 gliomas). In our study, 1,018 samples with RNA-seq and clinical data have been collected from CGGA dataset, including 363 recurrent gliomas (92 gliomas from CGGA RNA-seq batch 1 and 271 gliomas from CGGA RNA-seq batch 2). This certainly helps us to delve into the differences between primary and recurrent gliomas, especially at the molecular level.
There are some possible mechanisms for the recurrence and progression of gliomas. First, the characteristic of invasive growth of glioma restricts the complete resection of tumor by surgery [33]. In addition, postoperative adjuvant therapy (radiotherapy, chemotherapy, etc.) cannot kill all tumor cells [34]. Meanwhile, chemotherapy and radiotherapy might catalyze and accelerate tumor cell cloning revolution [35]. Finally, tumor cells adapt to the tumor microenvironment (TME), including resident cells, the blood-brain barrier and complex immunosuppressive environment [36]. To further understand the mechanism of glioma recurrence, we collected multi-omics data of glioma across primary and recurrent patients. Genomic analysis revealed more somatic mutations in recurrent gliomas and recurrent EGFR mutations were significantly enriched (Figure 1A). Meanwhile, deleted or amplified regions were the most frequently identified in recurrent gliomas, such as CDKN2A, CDKN2B, MLLT3, RB1, LCP1, CACNA1D and RAD21. With methylation microarray data, we identified 113 genes whose methylation levels were significantly different between primary and recurrent glioma (Figure 1B). Among 113 genes, RNA expression and methylation level of 5 genes were significantly negatively correlated (Figure 1C). Next, we analyzed gene expression profiles of primary and recurrent gliomas and screened differentially expressed genes. Based on the expression profiles of these 121 genes, consensus clustering identified two distinct clusters (k = 2) with significant differences in molecular and prognosis (Figure 2 and Table 1).
Next, we screened 108 genes related to survival with COX univariate analysis. And LASSO regression COX model was performed to obtain genes with cumulative effect of survival prediction. Finally, we constructed a recurrence-related risk signatures of 18 genes in gliomas. We noticed that the high-risk patients tended to older, recurrent, higher grade, IDH wild and 1p/19q intact (Figures 3C, S2 and Table 2), which suggested that signature might lead to poor prognosis for glioma patients.
We then analyzed the prognostic significance of the signature in gliomas, including in different grade gliomas and five subtypes of WHO 2016 classification (Figures 5A and S3A). Of note, when analyzing the progression-free survival (PFS), we found that the recurrence time of high-risk patients was significantly shorter than that of the low-risk patients (Figures 5B and S3B), indicating the ability to predict the recurrence of glioma patients. Next, we demonstrated that recurrence-related signature was an independent prognostic factor after adjusting for clinical and pathological indicators with univariate and multivariate analysis (Table 3). Meanwhile, ROC curve analysis for 1-year, 3-year and 5-year overall survival showed that the predictive power of signature (AUC 0.8389, 0.8964 and 0.9234) was stronger than traditional indicators age (AUC 0.6019, 0.6551 and 0.6627) and grade (AUC 0.7648, 0.8477 and 0.8557) (Figure 6A). By combining recurrence-related signature with pathological features, we could optimize the prediction effect of patients’ prognosis (Figures 6B, 6C and S4).
In addition, through DAVID functional annotation and Gene Set Enrichment Analysis, “cell division” and “cell cycle” were significantly correlated with the high-risk group (Figure 7). Dysregulation of cell cycle is common in tumor cells and is closely associated with tumor recurrence. Magbanua et al. found that neoadjuvant chemotherapy in breast cancer brought changes to the tumor cell cycle, which are associated with tumor recurrence [37]. El-Gendi et al. demonstrated the prognostic potential of cell cycle regulators p53, p63 and cyclinD1 in bladder cancer recurrence [38]. Kim et al. constructed a risk score model of the cell cycle profiling to predict the early recurrence of breast cancer [39]. And in gliomas, Zhang et al. found that gene mutations in cell cycle pathways were more common in recurrent glioma through genomic analysis [40]. Therefore, the in-depth research on tumor cell cycle may contribute to the understanding of the recurrence of gliomas and provide new insight for individualized treatment of gliomas.
As the tumor progressed at different stages, the components of immune invasion in the tumor also changed, which were associated with recurrence of the tumor [29]. We found that our recurrence-related signature in gliomas was positively correlated with the immune checkpoints, suggesting the immunosuppressive status of high-risk patients. Immune cells are the main component of tumor microenvironment. Takashi Takeshita et al. found that immune cells were associated with late tumor recurrence in breast cancer [41]. Matteo Fassan et al. found that compared to patients with low CD4+ T cell infiltration, all patients with high CD4+ T cell infiltration recurred [42]. Andrea Walens et al. revealed that TNFα-CCL5-macrophage axis could promote the recurrence of breast cancer by recruiting macrophages [43]. Lang Rao et al. reported that hybrid cell membrane nanovesicles blocked the polarization of tumor-associated macrophages from M1 phenotype to M2 phenotype, preventing recurrence and metastasis of malignant melanoma [44]. In our study, CIBERSORT and scRNA-seq analysis showed that macrophages were significantly upregulated in high-risk tumor, indicating the possible role of macrophages in the recurrence of glioma.
In summary, we comprehensively analyzed multi-omics data of gliomas across primary and recurrent patients and found the prognostic significance of recurrence-related genes. We built a recurrence-related signature to stratify patients with survival differences and predict the recurrence risk of glioma. However, the application of our signature to clinical diagnosis and treatment still needs further research and verification.
Acknowledgements
This study was funded by Provincial Key R&D Program, Science and Technology Department of Zhejiang Province (Grant No.2017C03018); Key Program of Administration of Traditional Chinese Medicine, Zhejiang Province (No.2018ZZ015); National Natural Science Foundation of China (NSFC)/Research Grants Council (RGC) Joint Research Scheme (81761168038); Beijing Municipal Administration of Hospitals’ Mission Plan (SML20180501); National Natural Science Foundation of China (81902337); National Natural Science Foundation of China (81902528); National Natural Science Foundation of China (82002647). QWW conceptualized the data, involved in formal analysis and methodology, investigated the study, and wrote the original draft. YJZ conceptualized the study, supervised the data, reviewed, and edited the manuscript. ZZ, ZSB and TJ investigated and supervised the data.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 2.Jiang T, Mao Y, Ma W, Mao Q, You Y, Yang X, Jiang C, Kang C, Li X, Chen L, Qiu X, Wang W, Li W, Yao Y, Li S, Li S, Wu A, Sai K, Bai H, Li G, Chen B, Yao K, Wei X, Liu X, Zhang Z, Dai Y, Lv S, Wang L, Lin Z, Dong J, Xu G, Ma X, Cai J, Zhang W, Wang H, Chen L, Zhang C, Yang P, Yan W, Liu Z, Hu H, Chen J, Liu Y, Yang Y, Wang Z, Wang Z, Wang Y, You G, Han L, Bao Z, Liu Y, Wang Y, Fan X, Liu S, Liu X, Wang Y, Wang Q. CGCG clinical practice guidelines for the management of adult diffuse gliomas. Cancer Lett. 2016;375:263–273. doi: 10.1016/j.canlet.2016.01.024. [DOI] [PubMed] [Google Scholar]
- 3.Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin. 2010;60:166–193. doi: 10.3322/caac.20069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW. The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131:803–820. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 5.Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, Rheinbay E, Miller CR, Vitucci M, Morozova O, Robertson AG, Noushmehr H, Laird PW, Cherniack AD, Akbani R, Huse JT, Ciriello G, Poisson LM, Barnholtz-Sloan JS, Berger MS, Brennan C, Colen RR, Colman H, Flanders AE, Giannini C, Grifford M, Iavarone A, Jain R, Joseph I, Kim J, Kasaian K, Mikkelsen T, Murray BA, O’Neill BP, Pachter L, Parsons DW, Sougnez C, Sulman EP, Vandenberg SR, Van Meir EG, von Deimling A, Zhang H, Crain D, Lau K, Mallery D, Morris S, Paulauskis J, Penny R, Shelton T, Sherman M, Yena P, Black A, Bowen J, Dicostanzo K, Gastier-Foster J, Leraas KM, Lichtenberg TM, Pierson CR, Ramirez NC, Taylor C, Weaver S, Wise L, Zmuda E, Davidsen T, Demchok JA, Eley G, Ferguson ML, Hutter CM, Mills Shaw KR, Ozenberger BA, Sheth M, Sofia HJ, Tarnuzzer R, Wang Z, Yang L, Zenklusen JC, Ayala B, Baboud J, Chudamani S, Jensen MA, Liu J, Pihl T, Raman R, Wan Y, Wu Y, Ally A, Auman JT, Balasundaram M, Balu S, Baylin SB, Beroukhim R, Bootwalla MS, Bowlby R, Bristow CA, Brooks D, Butterfield Y, Carlsen R, Carter S, Chin L, Chu A, Chuah E, Cibulskis K, Clarke A, Coetzee SG, Dhalla N, Fennell T, Fisher S, Gabriel S, Getz G, Gibbs R, Guin R, Hadjipanayis A, Hayes DN, Hinoue T, Hoadley K, Holt RA, Hoyle AP, Jefferys SR, Jones S, Jones CD, Kucherlapati R, Lai PH, Lander E, Lee S, Lichtenstein L, Ma Y, Maglinte DT, Mahadeshwar HS, Marra MA, Mayo M, Meng S, Meyerson ML, Mieczkowski PA, Moore RA, Mose LE, Mungall AJ, Pantazi A, Parfenov M, Park PJ, Parker JS, Perou CM, Protopopov A, Ren X, Roach J, Sabedot TS, Schein J, Schumacher SE, Seidman JG, Seth S, Shen H, Simons JV, Sipahimalani P, Soloway MG, Song X, Sun H, Tabak B, Tam A, Tan D, Tang J, Thiessen N, Triche T Jr, Van Den Berg DJ, Veluvolu U, Waring S, Weisenberger DJ, Wilkerson MD, Wong T, Wu J, Xi L, Xu AW, Yang L, Zack TI, Zhang J, Aksoy BA, Arachchi H, Benz C, Bernard B, Carlin D, Cho J, DiCara D, Frazer S, Fuller GN, Gao J, Gehlenborg N, Haussler D, Heiman DI, Iype L, Jacobsen A, Ju Z, Katzman S, Kim H, Knijnenburg T, Kreisberg RB, Lawrence MS, Lee W, Leinonen K, Lin P, Ling S, Liu W, Liu Y, Liu Y, Lu Y, Mills G, Ng S, Noble MS, Paull E, Rao A, Reynolds S, Saksena G, Sanborn Z, Sander C, Schultz N, Senbabaoglu Y, Shen R, Shmulevich I, Sinha R, Stuart J, Sumer SO, Sun Y, Tasman N, Taylor BS, Voet D, Weinhold N, Weinstein JN, Yang D, Yoshihara K, Zheng S, Zhang W, Zou L, Abel T, Sadeghi S, Cohen ML, Eschbacher J, Hattab EM, Raghunathan A, Schniederjan MJ, Aziz D, Barnett G, Barrett W, Bigner DD, Boice L, Brewer C, Calatozzolo C, Campos B, Carlotti CG Jr, Chan TA, Cuppini L, Curley E, Cuzzubbo S, Devine K, DiMeco F, Duell R, Elder JB, Fehrenbach A, Finocchiaro G, Friedman W, Fulop J, Gardner J, Hermes B, Herold-Mende C, Jungk C, Kendler A, Lehman NL, Lipp E, Liu O, Mandt R, McGraw M, McLendon R, McPherson C, Neder L, Nguyen P, Noss A, Nunziata R, Ostrom QT, Palmer C, Perin A, Pollo B, Potapov A, Potapova O, Rathmell WK, Rotin D, Scarpace L, Schilero C, Senecal K, Shimmel K, Shurkhay V, Sifri S, Singh R, Sloan AE, Smolenski K, Staugaitis SM, Steele R, Thorne L, Tirapelli DP, Unterberg A, Vallurupalli M, Wang Y, Warnick R, Williams F, Wolinsky Y, Bell S, Rosenberg M, Stewart C, Huang F, Grimsby JL, Radenbaugh AJ, Zhang J. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015;372:2481–2498. doi: 10.1056/NEJMoa1402121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu H, Mu Q, Bao Z, Chen Y, Liu Y, Chen J, Wang K, Wang Z, Nam Y, Jiang B, Sa JK, Cho HJ, Her NG, Zhang C, Zhao Z, Zhang Y, Zeng F, Wu F, Kang X, Liu Y, Qian Z, Wang Z, Huang R, Wang Q, Zhang W, Qiu X, Li W, Nam DH, Fan X, Wang J, Jiang T. Mutational landscape of secondary glioblastoma guides MET-targeted trial in brain tumor. Cell. 2018;175:1665–1678. e1618. doi: 10.1016/j.cell.2018.09.038. [DOI] [PubMed] [Google Scholar]
- 7.Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, Fouse SD, Yamamoto S, Ueda H, Tatsuno K, Asthana S, Jalbert LE, Nelson SJ, Bollen AW, Gustafson WC, Charron E, Weiss WA, Smirnov IV, Song JS, Olshen AB, Cha S, Zhao Y, Moore RA, Mungall AJ, Jones SJM, Hirst M, Marra MA, Saito N, Aburatani H, Mukasa A, Berger MS, Chang SM, Taylor BS, Costello JF. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014;343:189–193. doi: 10.1126/science.1239947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim J, Lee IH, Cho HJ, Park CK, Jung YS, Kim Y, Nam SH, Kim BS, Johnson MD, Kong DS, Seol HJ, Lee JI, Joo KM, Yoon Y, Park WY, Lee J, Park PJ, Nam DH. Spatiotemporal evolution of the primary glioblastoma genome. Cancer Cell. 2015;28:318–328. doi: 10.1016/j.ccell.2015.07.013. [DOI] [PubMed] [Google Scholar]
- 9.Stark AM, Doukas A, Hugo HH, Mehdorn HM. The expression of mismatch repair proteins MLH1, MSH2 and MSH6 correlates with the Ki67 proliferation index and survival in patients with recurrent glioblastoma. Neurol Res. 2010;32:816–820. doi: 10.1179/016164110X12645013515052. [DOI] [PubMed] [Google Scholar]
- 10.Stark AM, Witzel P, Strege RJ, Hugo HH, Mehdorn HM. p53, mdm2, EGFR, and msh2 expression in paired initial and recurrent glioblastoma multiforme. J Neurol Neurosurg Psychiatry. 2003;74:779–783. doi: 10.1136/jnnp.74.6.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deng T, Gong YZ, Wang XK, Liao XW, Huang KT, Zhu GZ, Chen HN, Guo FZ, Mo LG, Li LQ. Use of genome-scale integrated analysis to identify key genes and potential molecular mechanisms in recurrence of lower-grade brain glioma. Med Sci Monit. 2019;25:3716–3727. doi: 10.12659/MSM.913602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liang J, Qi WF, Xie S, Wang WF, Zhang XL, Zhou XP, Hu JX, Yu RT. Expression of WW domain-containing protein 2 is correlated with pathological grade and recurrence of glioma. J Cancer Res Ther. 2017;13:1032–1037. doi: 10.4103/0973-1482.176176. [DOI] [PubMed] [Google Scholar]
- 13.Zhao Z, Meng F, Wang W, Wang Z, Zhang C, Jiang T. Comprehensive RNA-seq transcriptomic profiling in the malignant progression of gliomas. Sci Data. 2017;4:170024. doi: 10.1038/sdata.2017.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bao ZS, Chen HM, Yang MY, Zhang CB, Yu K, Ye WL, Hu BQ, Yan W, Zhang W, Akers J, Ramakrishnan V, Li J, Carter B, Liu YW, Hu HM, Wang Z, Li MY, Yao K, Qiu XG, Kang CS, You YP, Fan XL, Song WS, Li RQ, Su XD, Chen CC, Jiang T. RNA-seq of 272 gliomas revealed a novel, recurrent PTPRZ1-MET fusion transcript in secondary glioblastomas. Genome Res. 2014;24:1765–1773. doi: 10.1101/gr.165126.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol. 2016;12:e1004873. doi: 10.1371/journal.pcbi.1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang QW, Wang YW, Wang ZL, Bao ZS. Clinical and molecular characterization of incidentally discovered lower-grade gliomas with enrichment of aerobic respiration. Onco Targets Ther. 2020;13:9533–9542. doi: 10.2147/OTT.S248623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang QW, Liu HJ, Zhao Z, Zhang Y, Wang Z, Jiang T, Bao ZS. Prognostic correlation of autophagy-related gene expression-based risk signature in patients with glioblastoma. Onco Targets Ther. 2020;13:95–107. doi: 10.2147/OTT.S238332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan W, Zhang W, You G, Bao Z, Wang Y, Liu Y, Kang C, You Y, Wang L, Jiang T. Correlation of IDH1 mutation with clinicopathologic factors and prognosis in primary glioblastoma: a report of 118 patients from China. PLoS One. 2012;7:e30339. doi: 10.1371/journal.pone.0030339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Q, Wang Z, Bao Z, Zhang C, Wang Z, Jiang T. PABPC1 relevant bioinformatic profiling and prognostic value in gliomas. Future Oncol. 2020;16:4279–4288. doi: 10.2217/fon-2019-0268. [DOI] [PubMed] [Google Scholar]
- 20.Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26:1572–1573. doi: 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao J, Kwan PW, Shi D. Sparse kernel learning with LASSO and bayesian inference algorithm. Neural Netw. 2010;23:257–264. doi: 10.1016/j.neunet.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 22.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 23.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blanche P, Dartigues JF, Jacqmin-Gadda H. Estimating and comparing time-dependent areas under receiver operating characteristic curves for censored event times with competing risks. Stat Med. 2013;32:5381–5397. doi: 10.1002/sim.5958. [DOI] [PubMed] [Google Scholar]
- 25.Wang Q, Wang Z, Li G, Zhang C, Bao Z, Wang Z, You G, Jiang T. Identification of IDH-mutant gliomas by a prognostic signature according to gene expression profiling. Aging (Albany NY) 2018;10:1977–1988. doi: 10.18632/aging.101521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.da Cunha Santos G, Shepherd FA, Tsao MS. EGFR mutations and lung cancer. Annu Rev Pathol. 2011;6:49–69. doi: 10.1146/annurev-pathol-011110-130206. [DOI] [PubMed] [Google Scholar]
- 28.Sigismund S, Avanzato D, Lanzetti L. Emerging functions of the EGFR in cancer. Mol Oncol. 2018;12:3–20. doi: 10.1002/1878-0261.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, Bruneval P, Fridman WH, Becker C, Pagès F, Speicher MR, Trajanoski Z, Galon J. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39:782–795. doi: 10.1016/j.immuni.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 30.Yan X, Jiao SC, Zhang GQ, Guan Y, Wang JL. Tumor-associated immune factors are associated with recurrence and metastasis in non-small cell lung cancer. Cancer Gene Ther. 2017;24:57–63. doi: 10.1038/cgt.2016.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weller M, Cloughesy T, Perry JR, Wick W. Standards of care for treatment of recurrent glioblastoma--are we there yet? Neuro Oncol. 2013;15:4–27. doi: 10.1093/neuonc/nos273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marucci G, Fabbri PV, Morandi L, De Biase D, Di Oto E, Tallini G, Sturiale C, Franceschi E, Frezza GP, Foschini MP. Pathological spectrum in recurrences of glioblastoma multiforme. Pathologica. 2015;107:1–8. [PubMed] [Google Scholar]
- 33.Chaichana KL, Jusue-Torres I, Navarro-Ramirez R, Raza SM, Pascual-Gallego M, Ibrahim A, Hernandez-Hermann M, Gomez L, Ye X, Weingart JD, Olivi A, Blakeley J, Gallia GL, Lim M, Brem H, Quinones-Hinojosa A. Establishing percent resection and residual volume thresholds affecting survival and recurrence for patients with newly diagnosed intracranial glioblastoma. Neuro Oncol. 2014;16:113–122. doi: 10.1093/neuonc/not137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frosina G. DNA repair and resistance of gliomas to chemotherapy and radiotherapy. Mol Cancer Res. 2009;7:989–999. doi: 10.1158/1541-7786.MCR-09-0030. [DOI] [PubMed] [Google Scholar]
- 35.Barthel FP, Johnson KC, Varn FS, Moskalik AD, Tanner G, Kocakavuk E, Anderson KJ, Abiola O, Aldape K, Alfaro KD, Alpar D, Amin SB, Ashley DM, Bandopadhayay P, Barnholtz-Sloan JS, Beroukhim R, Bock C, Brastianos PK, Brat DJ, Brodbelt AR, Bruns AF, Bulsara KR, Chakrabarty A, Chakravarti A, Chuang JH, Claus EB, Cochran EJ, Connelly J, Costello JF, Finocchiaro G, Fletcher MN, French PJ, Gan HK, Gilbert MR, Gould PV, Grimmer MR, Iavarone A, Ismail A, Jenkinson MD, Khasraw M, Kim H, Kouwenhoven MCM, LaViolette PS, Li M, Lichter P, Ligon KL, Lowman AK, Malta TM, Mazor T, McDonald KL, Molinaro AM, Nam DH, Nayyar N, Ng HK, Ngan CY, Niclou SP, Niers JM, Noushmehr H, Noorbakhsh J, Ormond DR, Park CK, Poisson LM, Rabadan R, Radlwimmer B, Rao G, Reifenberger G, Sa JK, Schuster M, Shaw BL, Short SC, Smitt PAS, Sloan AE, Smits M, Suzuki H, Tabatabai G, Van Meir EG, Watts C, Weller M, Wesseling P, Westerman BA, Widhalm G, Woehrer A, Yung WKA, Zadeh G, Huse JT, De Groot JF, Stead LF, Verhaak RGW GLASS Consortium. Longitudinal molecular trajectories of diffuse glioma in adults. Nature. 2019;576:112–120. doi: 10.1038/s41586-019-1775-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quail DF, Joyce JA. The microenvironmental landscape of brain tumors. Cancer Cell. 2017;31:326–341. doi: 10.1016/j.ccell.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Magbanua MJ, Wolf DM, Yau C, Davis SE, Crothers J, Au A, Haqq CM, Livasy C, Rugo HS, Esserman L, Park JW, van’t Veer LJ. Serial expression analysis of breast tumors during neoadjuvant chemotherapy reveals changes in cell cycle and immune pathways associated with recurrence and response. Breast Cancer Res. 2015;17:73. doi: 10.1186/s13058-015-0582-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.El-Gendi S, Abu-Sheasha G. Ki-67 and cell cycle regulators p53, p63 and cyclinD1 as prognostic markers for recurrence/progression of bladder urothelial carcinoma. Pathol Oncol Res. 2018;24:309–322. doi: 10.1007/s12253-017-0250-2. [DOI] [PubMed] [Google Scholar]
- 39.Kim SJ, Masuda N, Tsukamoto F, Inaji H, Akiyama F, Sonoo H, Kurebayashi J, Yoshidome K, Tsujimoto M, Takei H, Masuda S, Nakamura S, Noguchi S. The cell cycle profiling-risk score based on CDK1 and 2 predicts early recurrence in node-negative, hormone receptor-positive breast cancer treated with endocrine therapy. Cancer Lett. 2014;355:217–223. doi: 10.1016/j.canlet.2014.08.042. [DOI] [PubMed] [Google Scholar]
- 40.Zhang L, Liu Z, Li J, Huang T, Wang Y, Chang L, Zheng W, Ma Y, Chen F, Gong X, Yuan Q, Teaw S, Fang X, Song T, Huo L, Li X, Xia X, Liu Z, Wu J. Genomic analysis of primary and recurrent gliomas reveals clinical outcome related molecular features. Sci Rep. 2019;9:16058. doi: 10.1038/s41598-019-52515-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takeshita T, Yan L, Asaoka M, Rashid O, Takabe K. Late recurrence of breast cancer is associated with pro-cancerous immune microenvironment in the primary tum or. Sci Rep. 2019;9:16942. doi: 10.1038/s41598-019-53482-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fassan M, Cavallin F, Guzzardo V, Kotsafti A, Scarpa M, Cagol M, Chiarion-Sileni V, Maria Saadeh L, Alfieri R, Castagliuolo I, Rugge M, Castoro C, Scarpa M. PD-L1 expression, CD8+ and CD4+ lymphocyte rate are predictive of pathological complete response after neoadjuvant chemoradiotherapy for squamous cell cancer of the thoracic esophagus. Cancer Med. 2019;8:6036–6048. doi: 10.1002/cam4.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walens A, DiMarco AV, Lupo R, Kroger BR, Damrauer JS, Alvarez JV. CCL5 promotes breast cancer recurrence through macrophage recruitment in residual tumors. Elife. 2019;8:e43653. doi: 10.7554/eLife.43653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rao L, Wu L, Liu Z, Tian R, Yu G, Zhou Z, Yang K, Xiong HG, Zhang A, Yu GT, Sun W, Xu H, Guo J, Li A, Chen H, Sun ZJ, Fu YX, Chen X. Hybrid cellular membrane nanovesicles amplify macrophage immune responses against cancer recurrence and metastasis. Nat Commun. 2020;11:4909. doi: 10.1038/s41467-020-18626-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.