

Abstract
The overexpression of ATP-binding cassette transporters subfamily B member 1 (ABCB1) is known to be the primary trigger of multidrug resistance (MDR) in colorectal cancer (CRC), leading to chemotherapy failure. However, factors that regulate chemoresistance in CRC cells are largely unknown. To identify proteins involved in MDR in CRC, we used proteomics and transcriptomics approaches to analyze HCT8/T cells and parental HCT8 cells. Results showed that the expression of insulin-like growth factor-2 mRNA-binding protein 3 (IGF2BP3) was upregulated in HCT8/T cells, and siIGF2BP3 remarkably elevated the sensitivity of HCT8/T cells to DOX. Overexpression of IGF2BP3 promoted ABCB1 expression, and reduced the sensitivity to ABCB1 substrates. Conversely, knockdown of IGF2BP3 reduced ABCB1 expression, and increased the sensitivity to ABCB1 substrates in vitro and in vivo. This phenomeon was further confirmed by the strong association of IGF2BP3 and ABCB1 expression with DOX sensitivity. Mechanistically, IGF2BP3, as a N6-methyladenosine (m6A) reader, directly bound to the m6A-modified region of ABCB1 mRNA, thereby promoting the stability and expression of ABCB1 mRNA. Overall, the results showed that IGF2BP3 bound to the m6A modification region of ABCB1 mRNA, and conferred chemoresistance in CRC cells via upregulation of ABCB1. These findings suggest that IGF2BP3 might be a potential biomarker for predicting the development of MDR in CRC. Targeting IGF2BP3 might be an important chemotherapeutic strategy for preventing MDR development in CRC.
Keywords: Colorectal cancer, chemoresistance, IGF2BP3, ABCB1, m6A
Introduction
Colorectal cancer (CRC) is the third most diagnosed malignant tumor globally [1]. Though considerable advancements have been made in the treatment of CRC, acquired resistance of cancer cells to chemotherapeutic drugs remains a major obstacle [2]. Almost 90% of patients develop a some degree of multidrug resistance (MDR) and die of cancer [3]. Several mechanisms drive the development of MDR, such as increased drug efflux, cell cycle arrest, altered drug targets and dysregulated apoptosis [4]. However, our current understanding of these and other unidentified molecular mechanisms leading to MDR development is not complete.
Overexpression of the ATP-binding cassette (ABC) transporters has been shown to increase efflux of anti-cancer drugs there decrease drug efficacy [5]. So far, 49 ABC transporter family members have been identified, including ABCB1, ABCG2, and ABCC1. ABCB1, encoding P-glycoprotein (P-gp), is one of the best studied molecules in chemoresistance [6]. It renders cancer cells resistant to many chemotherapeutic agents, e.g., paclitaxel (PTX), doxorubicin (DOX), vincristine (VCR), and vinorelbine (NVB) [7]. However, upstream regulatory factors that lead to deregulation of ABCB1 in chemoresistant CRC are largely unknown.
Epigenetic regulatory mechanisms, such as post-transcriptional modifications of RNA, are considered key regulators of various biological processes [8]. Among the more than 100 mRNA modifications, the N6-methyladenosine (m6A) modification is the most ubiquitous in eukaryotic mRNAs. Thousands of RNA transcripts possess m6A modifications with distinct distribution patterns [9]. m6A modification is reversible and controls the fate of RNA. m6A modifications is primarily regulated by writers, erasers and readers [10]. Methytransferase-like 3 (METTL3) and METTL14 have been proven to act as m6A writers in mammalian cells [11]. Previous studies demonstrate that FTO (fat-mass and obesity-associated protein), one of RNA demethylases, functions as an m6A eraser and modulates leukemia cells transformation [12]. m6A-binding proteins, including YTHDF1, YTHDF2, YTHDC1 and eIF3, have been recognized as m6A readers, which modulate the fate of target transcripts by specifically recognizing m6A [10,13].
In previous report, we reported that ABCB1 was remarkably higher in MDR cells in contrast to parental cells [14]. ABCB1 also conferred MDR cells resistance to numerous anticancer agents, such as PTX, VCR and NVB. Advances in high-throughput research technologies have revealed that a panel of molecules, rather than an individual molecule, contribute to the chemoresistant phenotype [15]. In this study, to identify additional key factors, we conducted large-scale and unbiased analytical tools of comparative omics to explore the characteristics of MDR in CRC cell lines HCT8 and its chemoresistant counterpart HCT8/T. Herein, we found that IGF2BP3 was markedly upregulated in MDR HCT8/T cells. The mammalian IGF2BP (insulin-like growth factor-2 mRNA-binding protein) family consists of three RNA-binding proteins, IGF2BP1, IGF2BP2, and IGF2BP3 [16]. They are composed of 6 conserved RNA-binding domains with 2 N-terminal RRM (RNA recognition motifs) and 4 C-terminal hnRNPK homology (KH) domains [17]. It has been reported that IGF2BPs, as a novel family of m6A readers, preferentially recognize m6A-modified mRNAs and promote the stability of numerous mRNA targets in an m6A-dependent manner, thus globally enhancing gene expression output [18]. Moreover, IGF2BPs enhance mRNA storage or inhibit mRNA degradation under stress conditions, and facilitate its translation [19]. These reports reveal that m6A-reading process influences mRNA stability and translation, and highlight the functional significance of IGF2BPs as m6A readers involved in post-transcriptional gene modulation [18,19].
Our study investigated the effect of IGF2BP3 on sensitivity of ABCB1 substrates in CRC cells, and the mechanism driving m6A-dependent regulation of ABCB1 expression.
Materials and methods
Cell culture
Human CRC cells (HCT8, HCT15, HCT116, SW1463, SW480, SW620, HT29 and DLD1) were bought from the American Type Culture Collection (ATCC, Manassas, USA). The paclitaxel resistant cell HCT8/T was a generous gift from Prof. Jian Ding of the Shanghai Institute of Materia Medica. Cells were inoculated and grown as per the manufacturer provided protocols and regularly authenticated through morphologic assessments. They were evaluated for Mycoplasma contaminations.
Drugs and reagents
Paclitaxel (PTX), Oxaliplatin (OXA) and Topotecan (TPT) were purchased from Sigma-Aldrich (St. Louis, USA); Vincristine (VCR), Doxorubicin (DOX), Vinorelbine (NVB) and Actinomycin D (Act D) were purchased from MCE (Shanghai, China) while Puromycin dihydrochloride (Puro) and Blasticidin S (BSD) were obtained from Beyotime (Shanghai, China). For the in vitro assays, all the drugs were dissolved in dimethyl sulfoxide (DMSO), aliquoted, stored at -20°C. Prior to each experiment, the solutions were diluted to the desired levels in normal saline. For the in vivo studies, PTX injection TESU was obtained from Haikou Pharmaceutical Factory (Haikou, China). The concentration of DMSO was ≤ 0.1%.
Proteomics by MRM analysis
Proteins were extracted from 3×107 HCT8 as well as HCT8/T cells and stored at -80°C until processing, as previously described [20]. Briefly, the samples were detected using MRM mass spectrometry (n = 2). The HCT8 cell ratios were used for the normalization of protein expression levels. Normalized proteomics analysis was performed at Majorbio lnc (Shanghai, China). The screening conditions were that the mean relative fold change of differentially expressed genes (DEGs) was ≥ 1.33 or ≤ 0.75, P < 0.05, and that the DEGs were associated with MDR.
RNA-seq
RNA isolation from 6×107 HCT8 and HCT8/T cells was performed using TRIzol and phenol-chloroform, followed by isopropanol precipitation. The purified mRNA was treated with DNase I and sequenced using the Illumina mRNA-sequencing protocol (n = 3), as previously described [20]. The HCT8 cell ratios were used to normalize mRNA expression levels. Transcriptomics analysis was performed at Majorbio lnc (Shanghai, China). The screening condition was that the mean relative fold change of DEGs is ≥ 2.0 or ≤ 0.5, and P < 0.05.
Western blot assessment
Western blot assay was used to determine cellular concentrations of the specified proteins, as previously described [21]. Densitometry with Image J was used to quantify the corresponding bands. Antibodies against GAPDH and ABCB1 were obtained from Santa Cruz Biotechnology (1:1000, Santa Cruz, CA, USA), IGF2BP3, ALDH3A1, AKR1C1, and ABCG2 were obtained from Cell Signaling Technology (1:1000, Danvers, MA, USA), ABCC1 was obtained from Abcam (1:1000, Cambridge, UK), while METTL3 and METTL14 were obtained from Proteintech (1:1000, Wuhan, China). HRP-labelled goat anti-mouse and anti-rabbit secondary antibodies were obtained from Multi Sciences Lianke Biotech (1:5000, Hangzhou, China).
Proliferation suppression assays
Cells were inoculated into 96-well plates and incubated overnight. They were treated with graded levels of specified agents for 72 h. Then, the Cell Counting Kit 8 (CCK8; Dojindo, Japan) was used to evaluate cell proliferation, as previously described [21]. The suppression rate (%) was computed as [1-(A450treated/A450vehicle)] ×100%. The average IC50 values, expressed as mean ± SD, were determined by the Logit approach from three independent assays.
RNA interference
Cells were inoculated into 6-well plates and incubated overnight. Then, specific siRNAs (100 nM) against genes of interest or a scrambled siRNA (the negative control) (RayBiotech, Guangzhou, China) were transfected for 48 h using the RNAiMAX Transfection System (Invitrogen, CA, USA) as per the manufacturer’s instructions.
qRT-PCR
The TRIzol reagent (Life Technologies, CA, USA) was used for RNA isolation. The M-MLV reverse transcriptase (Promega, WI, USA) was used to convert RNA (500 ng) into cDNA, followed by amplification using the SYBR Premix EX TaqII Kit (TaKaRa, Tokyo, Japan) on the CFX96 Touch Deep Well Real-Time PCR System (Bio-rad, CA, USA). Primers were synthesized at Genewiz (Suzhou, China) and their sequences are follows: 5’-CTGCACCACCAACTGCTTAG-3’ (forward) and 5’-TTCAGCTCAGGGATGACCTT-3’ (reverse) for GAPDH, 5’-CAGGTGGAGGCAAATCTTCGT-3’ (forward) and 5’-ACCCTGTTAATCCGTTCGTTTT-3’ (reverse) for ABCG2, 5’-CTCTATCTCTCCCGACATGACC-3’ (forward) and 5’-AGCAGACGATCCACAGCAAAA-3’ (reverse) for ABCC1, 5’-TATATCGGAAACCTCAGCGAGA-3’ (forward) and 5’-GGACCGAGTGCTCAACTTCT-3’ (reverse) for IGF2BP3, 5’-CTGCACCACCAACTGCTTAG-3’ (forward) and 5’-GGGCTTGAGGACCACTGAG-3’ (reverse) for ALDH3A1, 5’-TTCATGCCTGTCCTGGGATTT-3’ (forward) and 5’-CTGGCTTTACAGACACTGGAAAA-3’ (reverse) for AKR1C1, 5’-AGCACGGCTCCATATACATACC-3’ (forward) and 5’-TGGACCACTAAAGGAGAAAGGT-3’ (reverse) for ESR2, 5’-GTGGCATCGTTGAGGAGTG-3’ (forward) and 5’-CACGTCCCTCTCGGACTTG-3’ (reverse) for IGF2.
Stable IGF2BP3 knockdown and overexpression
The short hairpin RNA (shRNA)-mediated silencing of IGF2BP3 was performed to interfere with the expression of IGF2BP3. The shIGF2BP3 sequences were as follows: shIGF2BP3-1, 5’-CGGTGAATGAACTTCAGAATT-3’ and shIGF2BP3-2, 5’-GGTGAAACTTGAAGCTCATAT-3’. The forward and reverse sequences of each shRNA were annealed and sub-cloned into a BSD-resistant lentiviral vector (pLV.I4-BSD). A shNC vector was used as a negative control. These procedures were performed at Huiyuanyuan (Guangzhou, China).
The IGF2BP3 coding sequence was cloned through PCR amplification and inserted into a Puro-resistant lentiviral vector (pLVX-EF1a-IRES-Puro) by homologous recombination to develop a plasmid overexpressing IGF2BP3. An empty vector was used as a control.
Cells were infected with lentivirus particles and selected by BSD or Puro to establish stable cell lines for further experiments. The IGF2BP3 overexpressing and control HCT8 cells were named as HCT8+IGF2BP3 and HCT8+Vector, respectively while the IGF2BP3 knockdown and control HCT8/T cells were named as HCT8/T shIGF2BP3 and HCT8/T shNC, respectively.
m6A RIP-qPCR
m6A modifications of individual genes in 6×107 HCT8/T and HTC8 cells were done using the Magna MeRIP™ m6A Kit (Millipore, MA, USA), as previously reported [22]. Briefly, the RNA extracted from HCT8/T cells were chemically fragmented into 100 nucleotides or smaller fragments. Then, magnetic immunoprecipitation was performed using a monoclonal antibody against m6A. After immunoprecipitation, the isolated RNA fragments were run through qRT-PCR. The corresponding m6A enrichment in each IP sample was calculated by normalizing to the input, as per the manufacturer’s instructions. The percent of each IP sample was measured: m6A RIP/input (%) = 2(Ct input-Log2 [dilution factor]-Ct IP). The m6A site of ABCB1 mRNA (CDS:562) was predicted through the RMBase v2.0 database (http://rna.sysu.edu.cn/rmbase/index.php) [23]. The m6A RIP-qPCR for individual genes was performed using specific primers: 5’-CGGTCTCAGAACTGTTTGTTTC-3’ (forward) and 5’-AAACCAAAGTGGTCCACAAA-3’ (reverse) for EEF1A positive, 5’-GGATGGAAAGTCACCCGTAAG-3’ (forward) and 5’-TTGTCAGTTGGACGAGTTGG-3’ (reverse) for EEF1A negative, 5’-CCAAAATTTCACGTCTTGGTG-3’ (forward) and 5’-AAGTTCTTCTTCTTTGCTCCTC-3’ (reverse) for ABCB1.
RIP-qPCR
The RNA-binding protein immunoprecipitation (RIP) was done in 3×107 HCT8 and HCT8/T cells with or without siMETTL3/14 using Magna RIP™ Kit (Millipore, MA, USA), as previously reported [22]. Briefly, magnetic beads coated with distinct antibodies against mouse IgG or RIPAb+™ IGF2BP3 (Millipore, MA, USA) were incubated with cell lysates. The RNA was washed, purified, and analyzed by qRT-PCR to establish the relative interactions between IGF2BP3 proteins and target gene transcripts. IGF2BP3 enrichment of each IP sample was normalized to the corresponding IgG, as per the manufacturer’s instructions. The mRNA fold with anti-IGF2BP3 antibody was measured: Fold enrichment = 2(Ct IgG-Ct IGF2BP3). Moreover, primers for the m6A modification region of ABCB1 mRNA were the same as those of m6A RIP-qPCR.
RNA stability assay
Plasmid or siRNA transfected HCT8/T cells were dispensed into 6-well plates. After 24 h, they were exposed to 5 μg/ml Act D and collected at specified time points. The samples were extracted and analyzed by qRT-PCR.
Nude mice xenograft tumor assay
Female BALB/c nu/nu mice (5-6 weeks old, 16-18 g) were obtained from Jihui Laboratory Animal Care Co. Ltd. (Shanghai, China). The study protocols involving mice were approved by the Institutional Animal Care and Use Committee of East China Normal University. HCT8/T xenografts derived from shNC or shIGF2BP3-1 HCT8/T cells were established through subcutaneous inoculation of cells (6×106) into nude mice. After 1-2 passages, well-established xenografts were sectioned into 1.5 mm3 segments. Then, the sections were subcutaneously transplanted into the right flank of nude mice. The xenografts were allowed to grow to a volume of 100-150 mm3 after which the mice were randomly assigned into three groups. Each group (n = 7) was administered with specified treatments: i. vehicle (q3d); ii. NVB (4 mg/kg, intraperitoneal injection/i.p., q3d); iii. PTX (20 mg/kg, intraperitoneal injection/i.p., q3d).
Xenograft sizes and mice body weights were individually determined after every two days. Measurement of the xenografts involved the maximal width (X), and the length (Y) from which the volume (V) as (X2Y)/2 was computed. The relative tumor volume (RTV) was computed as Vt/V0, with V0 and Vt signifying the volume before and after treatments, respectively. Moreover, the in vivo anticancer activity of every treatment was assessed through its T/C (%) value. T/C (%) was computed as (TRTV/CRTV) ×100%, whereby, TRTV and CRTV denoted the RTV of the treatment and vehicle groups, respectively. Tumor growth inhibition was computed as TGI% = (1-(mean tumor volume of the treatment group on the first day-mean tumor volume of the treatment group on the end day)/(mean tumor volume of the control group on the first day - mean tumor volume of the control group on the end day)) ×100%.
Statistical analyses
Data were presented as the mean ± SD. All statistical analyses were performed using the GraphPad Prism 7.0 software. Comparisons between two groups were performed using a two-tailed Student’s t-test. P < 0.05 was set as the threshold for statistical significance.
Results
IGF2BP3 was upregulated in HCT8/T cells and affected DOX sensitivity
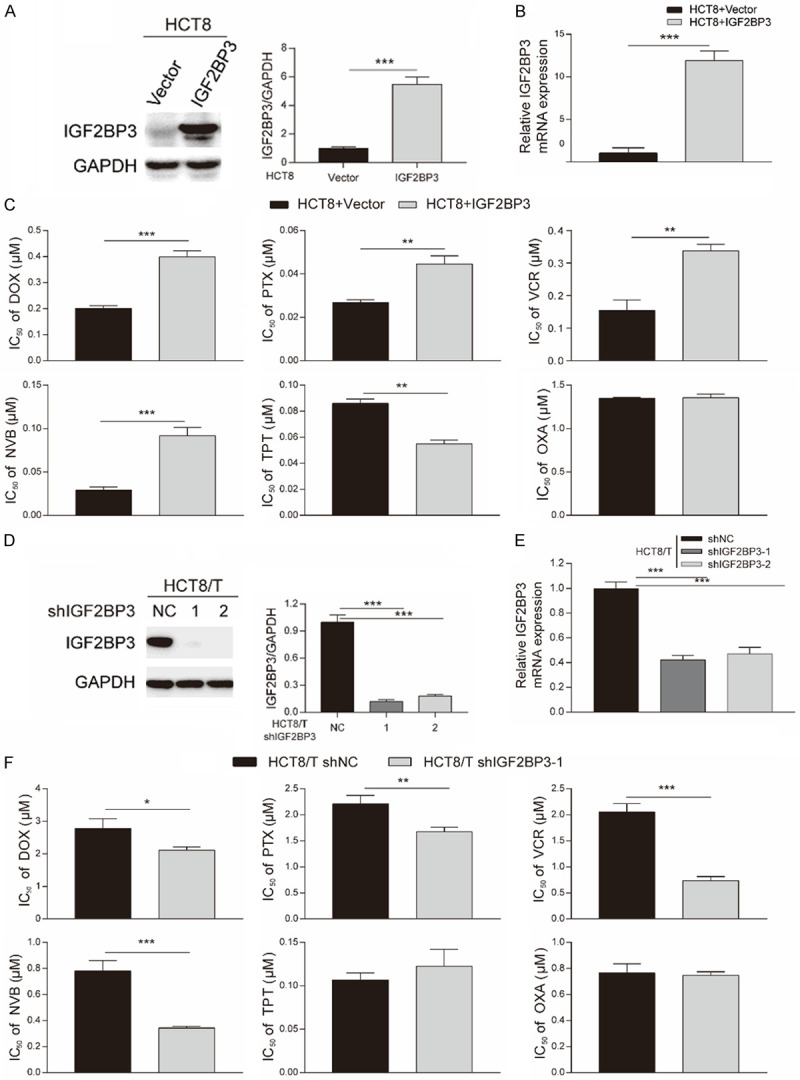
HCT8/T cells exhibited high expression of ABCB1 (Figure 1F), the most well-known marker of MDR, and extremely resistant to ABCB1 substrates (DOX, NVB, VCR, PTX), in contrast with its parental HCT8 cells (Table 1). However, sensitivity of HCT8/T cells to TPT and OXA, which are not ABCB1 substrates, was barely altered. Development of MDR is multifactorial, and may be affected by unknown biological mechanisms. To ascertain whether there are other MDR-related biomarkers, besides ABCB1, we analyzed expression profiles of this pair of cell lines using omics assays. Proteomics and transcriptomics analyses revealed 41 (Figure 1A) and 92 (Figure 1B) differentially expressed genes, respectively. Results of principal component analysis (PCA) corroborated the reproducibility of omics data without outliers (Figure 1C). Further analysis of the two groups revealed that 4 genes, namely ABCB1, IGF2BP3, ALDH3A1 and AKR1C1, were shared and dysregulated at both protein and mRNA levels (Figure 1D). Specifically, ABCB1 and IGF2BP3 were upregulated whereas ALDH3A1 and AKR1C1 were downregulated in HCT8/T cells (Figure 1A and 1B), suggesting that they may be regulating mechanisms of MDR in HCT8/T cells. Next, we validated expression patterns of the 4 shared genes using qRT-PCR (Figure 1E) and Western blot assay (Figure 1F). To identify whether other differentially expressed genes, besides ABCB1, affected cytotoxic agents sensitivity of CRC cells, we used specific siRNAs to silence IGF2BP3 in HCT8/T cells, as well as ALDH3A1 and AKR1C1 in HCT8 cells. Results revealed downregulation of IGF2BP3 (Figure 1G) in HCT8/T cells as well as ALDH3A1 and AKR1C1 in HCT8 cells (Figure 1I). siIGF2BP3 remarkably elevated the sensitivity of HCT8/T cells to DOX, by 0.76-fold (Figure 1H and Table 2), whereas siALDH3A1 and siAKR1C1 had little effect on DOX sensitivity of HCT8 cells (Figure 1J). Overall, these data suggested that among the 3 dysregulated factors, IGF2BP3 had the greatest impact on drug sensitivity of HCT8 and HCT8/T cells.
Figure 1.

IGF2BP3 was upregulated in HCT8/T cells and affected DOX sensitivity. A. A Heat map profiling of relative protein expression in HCT8/T cells in contrast with HCT8 cells (n = 2). The protein expression of parental HCT8 cells was normalized to 1. B. Heat map profiling of relative mRNA expression in HCT8/T cells in contrast with HCT8 cells (n = 3). mRNA expression of parental HCT8 cells was normalized to 1. C. Results of principal components analysis (PCA) in omics data. Points were colored according to group and ellipses were used describe the distribution of points in each group. PCA of proteomics data in HCT8/T cells and HCT8 cells (left). R2 [1] = 0.557, R2 [2] = 0.271. PCA of transcriptomics data in HCT8/T cells and HCT8 cells (right). R2 [1] = 0.437, R2 [2] = 0.228. D. Overlapping genes between proteomics data and transcriptomics data. E, F. qRT-PCR and Western blotting analysis of expression of four shared genes in HCT8 and HCT8/T cells. G. Western blotting analysis of IGF2BP3 protein expression in HCT8/T cells treated with siIGF2BP3 for 48 h. H. IC50 values of DOX in siRNA-transfected HCT8/T cells. Cells were transfected with the indicated siRNAs, and treated with DOX for 72 h and then subjected to CCK8 assays. I. Western blotting analysis of ALDH3A1 and AKR1C1 proteins in HCT8 cells treated with siRNA for 48 h. J. IC50 values of DOX in siRNA-transfected HCT8 cells. Densitometry analysis of protein bands was performed using Image J. Data are shown as mean ± SD of n = 3 independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (Student’s t-test).
Table 1.
Sensitivity of MDR HCT8/T cells to cytotoxic agents in comparison to the sensitivity of parental HCT8 cells
| Compounds | IC50 (mean ± SD) (μM) | RF | |
|---|---|---|---|
|
| |||
| HCT8 | HCT8/T | ||
| DOX | 0.1569±0.0143 | 2.8243±0.1302** | 18.00 |
| PTX | 0.0165±0.0012 | 3.0580±0.0366*** | 185.33 |
| VCR | 0.0446±0.0085 | 2.1749±0.0713*** | 48.76 |
| NVB | 0.1126±0.0045 | 2.9270±0.0520*** | 25.99 |
| TPT | 0.0829±0.0142 | 0.0711±0.0095 | 0.86 |
| OXA | 0.7637±0.0011 | 0.8735±0.1226 | 1.14 |
Cells were treated with different concentrations of agents for 72 h. Cytotoxicity of drugs in HCT8 and HCT8/T cells was assessed by CCK8 assays. The reversal-fold (RF) was calculated by dividing the IC50 value of HCT8/T cells by that of HCT8 cells. Data are shown as the mean ± SD of three independent assays.
P < 0.01 (Student’s t-test);
P < 0.001 (Student’s t-test).
Table 2.
Changes in DOX sensitivity induced by siRNAs transfection
| Cell lines | IC50 (mean ± SD) (μM) | RF |
|---|---|---|
|
| ||
| DOX | ||
| HCT8 siNC | 0.1007±0.0084 | 1.00 |
| HCT8 siALDH3A1-2 | 0.0921±0.0074 | 0.91 |
| HCT8 siAKR1C1-2 | 0.1182±0.0122 | 1.17 |
| HCT8/T siNC | 2.4575±0.0780 | 1.00 |
| HCT8/T siIGF2BP3-1 | 1.8635±0.1029** | 0.76 |
DOX cytotoxicity was detected by CCK8 assays in HCT8 and HCT8/T cells subjected to siRNAs. The reversal-fold (RF) was calculated by dividing the IC50 value of DOX for cells transfected with siRNAs by that transfected with siNC. Data are shown as the mean ± SD of three independent assays.
P < 0.01 (Student’s t-test).
Overexpression of IGF2BP3 desensitized HCT8 cells to ABCB1 substrates
We investigated the possible role of IGF2BP3 in promoting chemoresistance by overexpressing IGF2BP3 expression in parental HCT8 cells, then validated efficiency of this overexpression via Western blotting (Figure 2A) and qRT-PCR (Figure 2B) analyses. We also performed CCK8 assays to determine the changes of drug sensitivity due to IGF2BP3 overexpression (Figure 2C). Results showed that sensitivity of various ABCB1 substrates, including DOX, PTX, VCR and NVB was significantly diminished (up to 3.17-fold) in HCT8+IGF2BP3, relative to control HCT8+Vector cells (Table 3). However, IGF2BP3 overexpression did not reduce the cytotoxicity of TPT and OXA, which were not ABCB1 substrates. These results indicated that overexpressing IGF2BP3 remarkably suppressed sensitivity of HCT8 cells to chemotherapeutic agents as ABCB1 substrates.
Figure 2.
Overexpression and knockdown of IGF2BP3 affected the sensitivity of HCT8 and HCT8/T cells to ABCB1 substrates. A, B. The efficiency of IGF2BP3 overexpression in HCT8 cells by Western blotting and qRT-PCR analysis. C. IC50 values of chemotherapeutic drugs in HCT8 cells overexpressing IGF2BP3 and control cells. The cells were exposed to different concentrations of drugs for 72 h and then subjected to CCK8 assays. D, E. The efficiency of IGF2BP3 knockdown in HCT8/T cells by Western blotting and qRT-PCR analysis. F. IC50 values of chemotherapeutic drugs in HCT8/T cells with IGF2BP3 knockdown and control cells. The cells were treated with different concentrations of drugs for 72 h and then subjected to CCK8 assays. Densitometry analysis of protein bands was performed with Image J. Data are shown as mean ± SD of n = 3 independent assays. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (Student’s t-test).
Table 3.
Sensitivity of IGF2BP3-overexpressing HCT8 cells to cytotoxic agents in comparison with that of control HCT8 cells
| Compounds | IC50 (mean ± SD) (μM) | RF | |
|---|---|---|---|
|
| |||
| HCT8+Vector | HCT8+IGF2BP3 | ||
| DOX | 0.2008±0.0087 | 0.3987±0.0195*** | 1.98 |
| PTX | 0.0268±0.0011 | 0.0446±0.0031** | 1.66 |
| VCR | 0.1542±0.0264 | 0.3381±0.0161** | 2.19 |
| NVB | 0.0289±0.0031 | 0.0918±0.0077*** | 3.17 |
| TPT | 0.0859±0.0028 | 0.0549±0.0025 | 0.64 |
| OXA | 1.3482±0.0094 | 1.3562±0.0341 | 1.01 |
The experiments were conducted as described in Materials and methods. The reversal-fold (RF) was calculated by dividing the IC50 values of agents for HCT8+IGF2BP3 cells by that for HCT8+Vector cells. Data are shown as mean ± SD (n = 3).
P < 0.01 (Student’s t-test);
P < 0.001 (Student’s t-test).
Knockdown of IGF2BP3 resensitized HCT8/T cells to ABCB1 substratess
Since RNA interference did not confer complete and long-term obliteration of IGF2BP3 expression, we selected HCT8/T cells to stably silence IGF2BP3 expression using IGF2BP3-targeting shRNAs. We then validated silencing efficiency via Western blot (Figure 2D) and qRT-PCR (Figure 2E) analyses. Furthermore, we tested IGF2BP3’s potential role in MDR, control and IGF2BP3-depleted cells by analyzing their sensitivity to chemotherapeutic agents (Figure 2F). In contrast to the above results, HCT8/T shIGF2BP3-1 cells’ sensitivity to ABCB1 substrates was increased by 0.38-fold (Table 4), whereas those for TPT and OXA were not altered. Moreover, loss of IGF2BP3 resulted in lower MDR levels in HCT8/T cells. Taken together, these results indicated that IGF2BP3 contributed to chemoresistance of HCT8/T cells to ABCB1 substrates.
Table 4.
Sensitivity of IGF2BP3-knockdown HCT8/T cells to cytotoxic agents in comparison with that of the control HCT8/T cells
| Compounds | IC50 (mean ± SD) (μM) | RF | |
|---|---|---|---|
|
| |||
| HCT8/T shNC | HCT8/T shIGF2BP3-1 | ||
| DOX | 2.7807±0.2405 | 2.1127±0.0805* | 0.76 |
| PTX | 2.2103±0.1309 | 1.6718±0.0715** | 0.76 |
| VCR | 2.0528±0.1326 | 0.7846±0.0986*** | 0.38 |
| NVB | 0.7798±0.0663 | 0.3624±0.0240*** | 0.46 |
| TPT | 0.0174±0.0023 | 0.0155±0.0020 | 0.89 |
| OXA | 0.7644±0.0578 | 0.7462±0.0221 | 0.98 |
The experiments were conducted as described in Materials and methods. The reversal-fold (RF) was calculated by dividing the IC50 values of agents for HCT8/T shIGF2BP3-1 cells by that for HCT8/T shNC cells. Data are shown as mean ± SD (n = 3).
P < 0.05 (Student’s t-test);
P < 0.01 (Student’s t-test);
P < 0.001 (Student’s t-test).
IGF2BP3 regulated ABCB1 expression
ABCB1, ABCC1 and ABCG2 are the most well-studied ABC transport proteins, critical for chemoresistance of cancer cells. Notably, we found that ABCB1 was overexpressed in HCT8/T cells, in contrast with HCT8 cells, whereas ABCC1 and ABCG2 were not altered (Figure 3A), which was consistent with previous results (Figure 1F). We then correlated IGF2BP3 and ABCB1, to ascertain how IGF2BP3 affects sensitivity to ABCB1 substrates. IGF2BP3 protein expression was assessed in HCT8/T cells after siABCB1 transfection, with resulting Western blots showing that RNA interference successfully silenced ABCB1, but did not change IGF2BP3 expression (Figure 3B). Next, we analyzed expression levels of ABCB1, ABCC1 and ABCG2 in HCT8 and HCT8/T cells transfected with IGF2BP3-related plasmids. Overexpressing IGF2BP3 in HCT8 cells remarkably upregulated ABCB1 expression, at both protein (Figure 3C) and mRNA (Figure 3D) levels, but had no effect on ABCC1 and ABCG2. In contrast, depletion of IGF2BP3 in HCT8/T cells remarkably downregulated ABCB1 expression at both protein (Figure 3E) and mRNA (Figure 3F) levels, but had no effect on ABCC1 and ABCG2. These results affirmed ABCB1’s role in IGF2BP3-regulated chemoresistance of HCT8/T cells.
Figure 3.
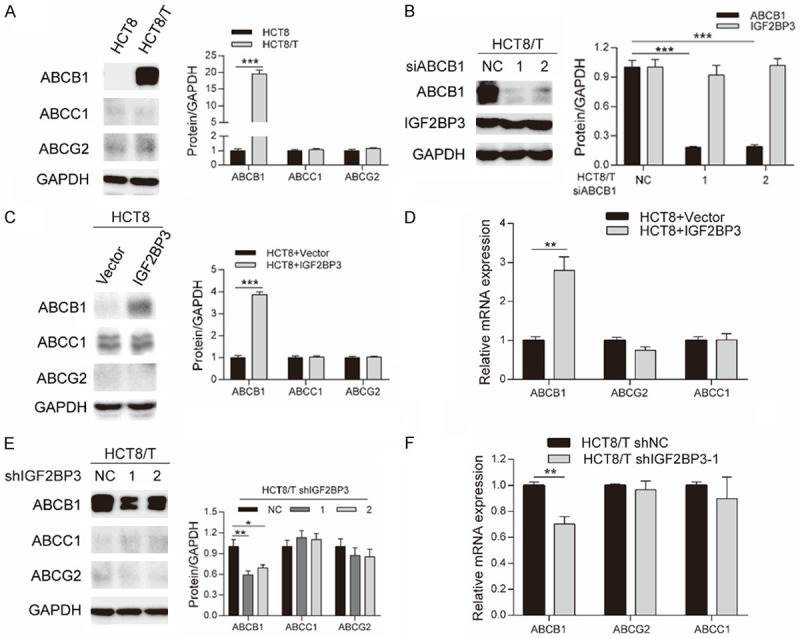
IGF2BP3 regulated ABCB1 expression. A. Results of Western blotting assay showing protein expression of ABCB1, ABCC1 and ABCG2 in HCT8 and HCT8/T cells. B. Results of Western blotting assay showing protein expression of ABCB1 and IGF2BP3 in HCT8/T cells treated with siABCB1 for 48 h. C, D. Results of Western blotting and qRT-PCR assays showing the expression level of ABCB1, ABCC1 and ABCG2 in IGF2BP3 overexpressing and control HCT8 cells. E, F. Results of Western blotting and qRT-PCR assays showing expression level of ABCB1, ABCC1 and ABCG2 in IGF2BP3 knockdown and control HCT8/T cells. Densitometry analysis for protein bands was performed using Image J. Data are shown as mean ± SD of n = 3 independent assays. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (Student’s t-test).
Binding of RNA m6A by IGF2BP3 enhanced stability of ABCB1 mRNA
We elucidated the underlying mechanism of IGF2BP3-regulated expression of ABCB1. Previous studies have suggested that members of the IGF2BP family, as m6A readers, might be playing a distinct role in modulating the fate of methylated mRNA [18]. For example, IGF2BPs promoted stability and storage of target mRNAs, and subsequently upregulated expression of target factors by binding to m6A modified mRNA regions [19]. Based on this, we hypothesized that IGF2BP3 might bind to m6A-modified ABCB1 mRNA. To determine the potential epigenetic mechanisms responsible for IGF2BP3-mediated ABCB1 upregulation, we first performed m6A RIP-qPCR to confirm the methylated site of ABCB1 mRNA. For control experiments of EEF1A mRNA, we chose stop codon and exon 5 as the positive and negative control regions, respectively. We designed ABCB1 primers of the potential m6A site for m6A RIP-qPCR, located at 562 bp in CDS region predicted by RMBase v2.0 database (Figure 4A). Results revealed successful m6A modification in the CDS region of ABCB1 mRNA in HCT8/T and HCT8 cells, and the levels of m6A modified ABCB1 mRNA in HCT8/T and HCT8 cells were not significantly different (Figure 4A).
Figure 4.
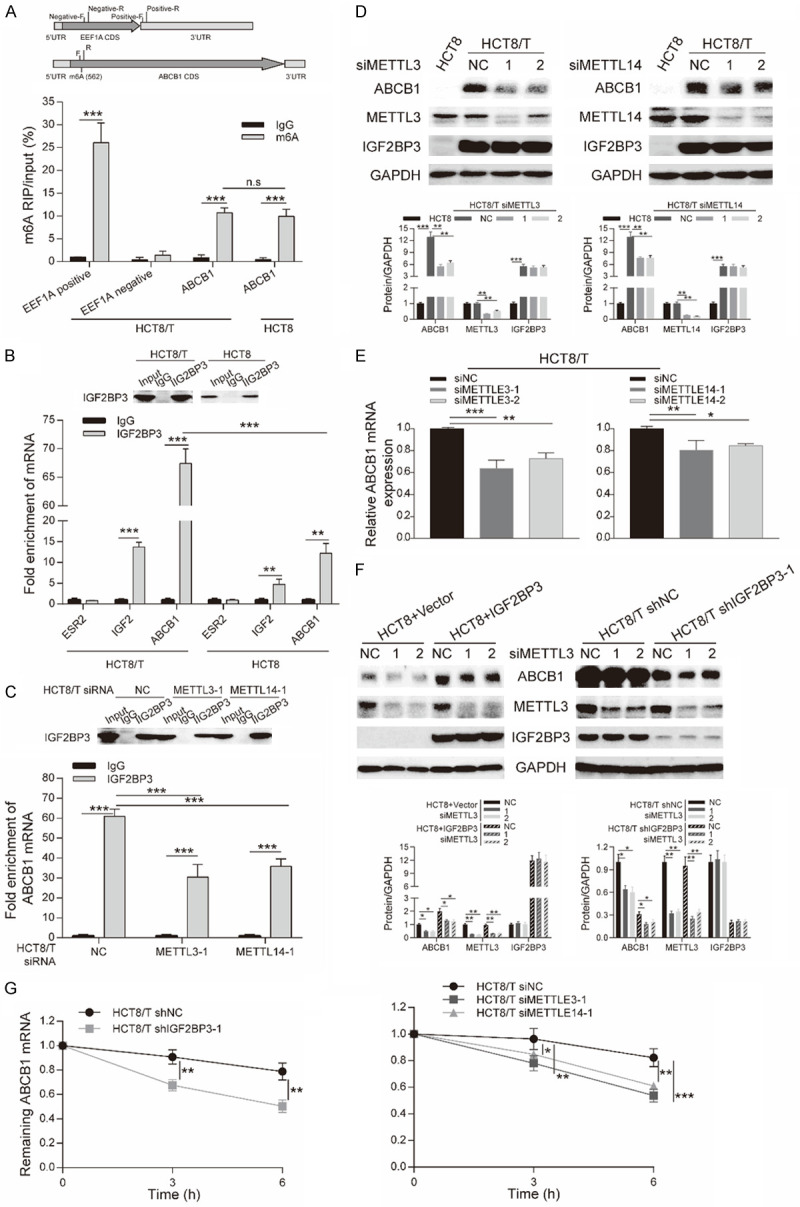
Binding of RNA m6A by IGF2BP3 enhanced stability of ABCB1 mRNA. A. Primers for m6A RIP-qPCR displayed in the structure of EEF1A mRNA and ABCB1 mRNA (upper). m6A RIP-qPCR analysis of ABCB1 mRNA in HCT8/T and HCT8 cells (lower). m6A RIP was performed using mRNAs samples extracted from HCT8/T and HCT8 cells with anti-m6A antibody, and expression level of m6A modified mRNAs was determined by qRT-PCR. Input was used for normalization. B. Western blotting (upper) and RT-qPCR (lower) analysis of RIP assays in HCT8/T and HCT8 cells. IGF2BP3-associated mRNAs were isolated from HCT8/T and HCT8 cells by IP using anti-IGF2BP3 antibody. IgG was used for normalization. Fold enrichment of ESR2, IGF2 and m6A-modified ABCB1 mRNA bound to IGF2BP3 was measured by qRT-PCR. Primers for the m6A modification region of ABCB1 mRNA is the same as that in m6A RIP-qPCR assay. C. Western blotting (upper) and RT-qPCR (lower) analysis of RIP assays in HCT8/T cells treated with siMETTL3 or siMETTL14 for 48 h. IGF2BP3-associated ABCB1 mRNAs were isolated from HCT8/T cells with or without siRNA by IP using anti-IGF2BP3 antibody. IgG was used for normalization. Fold enrichment of m6A-modified ABCB1 mRNA bound to IGF2BP3 was determined by qRT-PCR. Primers of the m6A modification region of ABCB1 mRNA were the same as those used in m6A RIP-qPCR. D, E. Western blotting and qRT-PCR analysis of ABCB1 expression in HCT8/T cells treated with siMETTL3 or siMETTL14 for 48 h. F. Western blotting analysis performed in HCT8 cells with IGF2BP3 overexpression and control HCT8 cells or IGF2BP3 knockdown and control HCT8/T cells upon METTL3 silencing. G. Remaining ABCB1 mRNA in IGF2BP3-knockdown or control HCT8/T cells treated with 5 μg/ml Act-D at 0, 3, or 6 h (left), and remaining ABCB1 mRNA in HCT8/T cells treated with siMETTL3 or siMETTL14 for 48 h, and then with 5 μg/ml Act-D at 0, 3, or 6 h (right). Densitometry analysis of protein bands was performed using Image J. Data are shown as mean ± SD of n = 3 independent assays. n.s, P >0.05, *, P <0.05; **, P < 0.01; ***, P < 0.001 (Student’s t-test).
To ascertain whether IGF2BP3 directly interacts with the m6A-modified region of ABCB1 mRNA, we performed RIP-qPCR, with IGF2 and estrogen receptor 2 (ESR2) included as positive and negative control, respectively [16,24]. The fold enrichment of ABCB1 mRNA normalized to IgG and IGF2 was 67.45±2.08 and 4.93±0.21 in HCT8/T cells respectively, while 12.20±1.91 and 2.69±0.58 in HCT8 cells (Figure 4B). Therefore, significant enrichment of ABCB1 mRNA with IGF2BP3 in HCT8/T and HCT8 cells, in contrast to IGF2 transcript. Additionally, IGF2BP3 had a significantly higher binding affinity for the m6A modification region of ABCB1 mRNA in HCT8/T cells, relative to HCT8 cells. METTL3 and METTL14 are m6A writers that represent the two most essential components of the methytransferase complex that catalyzes methylation at N6-adenosine [11]. To test the requirement of m6A modification for the binding between IGF2BP3 and ABCB1 mRNA, we performed RIP-qPCR in HCT8/T cells with or without METTL3/14 silencing, then validated the results using Western blot assay (Figure 4C, 4D). In summary, depletion of METTL3/14 remarkably impaired IGF2BP3’s interaction with ABCB1 mRNA, affirming that m6A modification of ABCB1 mRNA promoted its binding on IGF2BP3 (Figure 4C). These results suggested that IGF2BP3, reported as an m6A reader, recognized and bound to the m6A modification region of ABCB1 transcripts.
To further identify whether ABCB1 output was affected by m6A levels, we analyzed ABCB1 expression in HCT8 and HCT8/T cells with or without METTL3/14 silencing. We found no changes in METTL3/14 expression in HCT8 and HCT8/T cells (Figure 4D), hence there were no changes in m6A levels of ABCB1 mRNA between HCT8 and HCT8/T cells (Figure 4A). In addition, siMETTL3/14 did not alter IGF2BP3 expression, while ABCB1 expression in HCT8/T cells exhibited a significant downregulation at both protein (Figure 4D) and mRNA (Figure 4E) levels following METTL3/14 knockdown. Moreover, METTL3 silencing reversed upregulation of ABCB1 induced by IGF2BP3 overexpression in HCT8 cells, but intensified downregulation of ABCB1 induced by IGF2BP3 knockdown in HCT8/T cells (Figure 4F). This result indicated that m6A modification upregulated ABCB1, suggesting a positive correlation between this modification and expression. The dual regulation of ABCB1 expression by both IGF2BP3 and METTL3/14 suggested that IGF2BP3 regulated ABCB1 expression in an m6A-dependent manner. We further conducted an ABCB1 mRNA stability assay in IGF2BP3-, and METTL3/14-depleted as well as control cells, to evaluate the effect of m6A reader and writer. Results revealed that IGF2BP3 or METTL3/14 knockdown accelerated mRNA decay in ABCB1 in HCT8/T cells (Figure 4G). Moreover, depletion of IGF2BP3 or reduction of m6A levels decreased ABCB1 mRNA stability and consequently inhibited its expression. Taken together, our data demonstrated that recognition of m6A modification in ABCB1 mRNA by IGF2BP3 enhanced ABCB1 mRNA stability and expression.
ABCB1 expression and DOX sensitivity were positively associated with IGF2BP3 expression in various CRC cell lines
To further explore the function of IGF2BP3 and predict the sensitivity of ABCB1 substrates, we analyzed IGF2BP3 and ABCB1 expression in eight CRC cell lines via Western blotting (Figure 5A and Table 5). Results revealed a positive correlation between ABCB1 and IGF2BP3 expression (Figure 5B). Next, we evaluated CRC cell lines for sensitivity to DOX with CCK8 assays. IC50 values are displayed in Figure 5C and Table 5. Cells with high IC50 values of DOX had higher expression of IGF2BP3 (r = 0.8446) and ABCB1 (r = 0.9695), than those with low IC50 values (Figure 5D). These results showed that DOX sensitivity was positively associated with IGF2BP3 expression in measured CRC cell lines. Overall, these data confirmed that IGF2BP3 contributed to chemoresistance by regulating ABCB1 expression in CRC cell lines.
Figure 5.
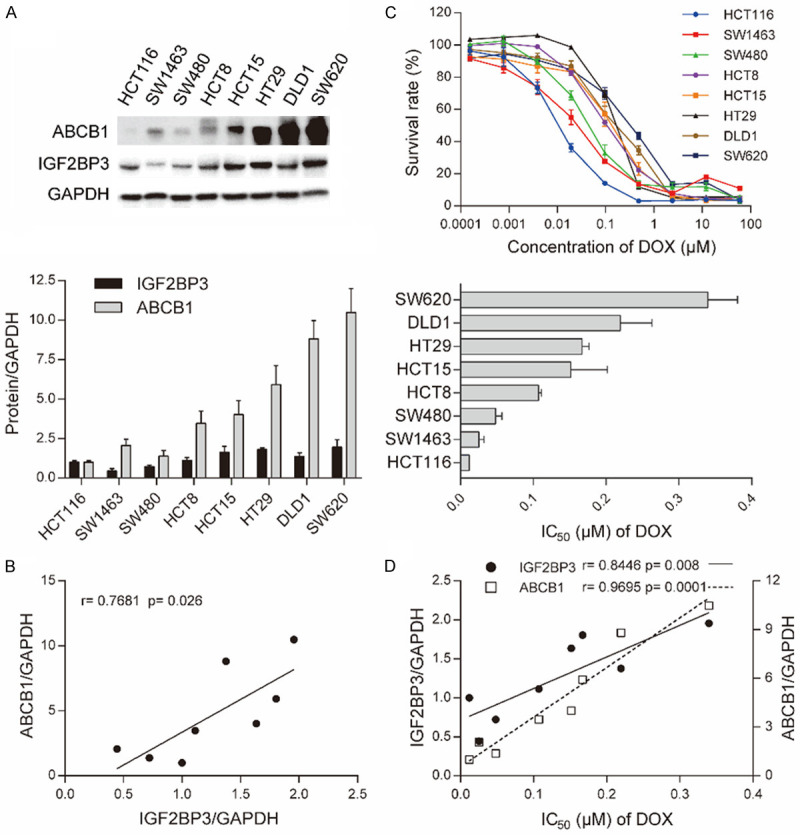
ABCB1 expression and DOX sensitivity was positively associated with IGF2BP3 in CRC cell lines. A. Western blotting analysis showing protein expression of ABCB1 and IGF2BP3 in 8 kinds of CRC cell lines (upper) and quantitative analysis of densitometry values as determined using Image J (lower). The relative expression of protein/GAPDH was normalized by HCT116 cells. B. Pearson correlation analysis showing the association between ABCB1 and IGF2BP3 expression in CRC cell lines. C. Cell survival curves for CRC cell lines treated with DOX for 72 h (upper) and IC50 values plotted (lower). D. Pearson correlation analysis showing the association of ABCB1 and IGF2BP3 expression with IC50 values of DOX. Data are shown as mean ± SD of n = 3 independent assays. *, P <0.05; **, P <0.01, ***, P < 0.001 (Pearson correlation).
Table 5.
Correlation among ABCB1 expression, DOX sensitivity and IGF2BP3 expression in CRC lines
| CRC cell lines | Relative expression of IGF2BP3 | Relative expression of ABCB1 | DOX |
|---|---|---|---|
|
| |||
| IC50 (mean ± SD) (μM) | |||
| SW620 | 1.9557±0.4754 | 10.4842±1.5194 | 0.3397±0.0335 |
| DLD1 | 1.3759±0.2247 | 8.8091±1.1620 | 0.2195±0.0354 |
| HT29 | 1.8044±0.1104 | 5.9175±1.1977 | 0.1669±0.0079 |
| HCT15 | 1.6345±0.3697 | 4.0163±0.8954 | 0.1515±0.0411 |
| HCT8 | 1.1126±0.1827 | 3.4658±0.7837 | 0.1071±0.0032 |
| SW480 | 0.7210±0.0952 | 1.3822±0.3628 | 0.0482±0.0070 |
| SW1463 | 0.4431±0.1431 | 2.0618±0.3937 | 0.0252±0.0058 |
| HCT116 | 1.0000±0.1015 | 1.0000±0.1039 | 0.0119±0.0010 |
The experiments were conducted as described in Materials and methods. Data are shown as mean ± SD (n = 3).
Knockdown of IGF2BP3 enhanced the response of HCT8/T xenografts to NVB and PTX treatment
To explore whether IGF2BP3 influences in vivo sensitivity to ABCB1 substrates, we conducted IGF2BP3 knockdown HCT8/T cells and control cells to establish xenograft models in nude mice. HCT8/T shIGF2BP3-1 cell-originated xenografts grew much faster than shNC cell- originated xenografts (Figure 6A). Intraperitoneal administration of NVB and PTX markedly repressed the growth of shIGF2BP3 tumor xenografts by 46.07% and 56.82%, respectively, but inhibited shNC tumor xenografts only by 2.18% and 15.83% (Figure 6A and Table 6). The tumor weight of shIGF2BP3 xenografts treated with NVB and PTX was remarkably lower in contrast with that treated with vehicle (Figure 6B, 6D). However, there was no remarkable difference in tumor weight among shNC xenografts treated with vehicle, NVB and PTX. Moreover, the ABCB1 substrates did not affect body weight of each group (Figure 6C).
Figure 6.
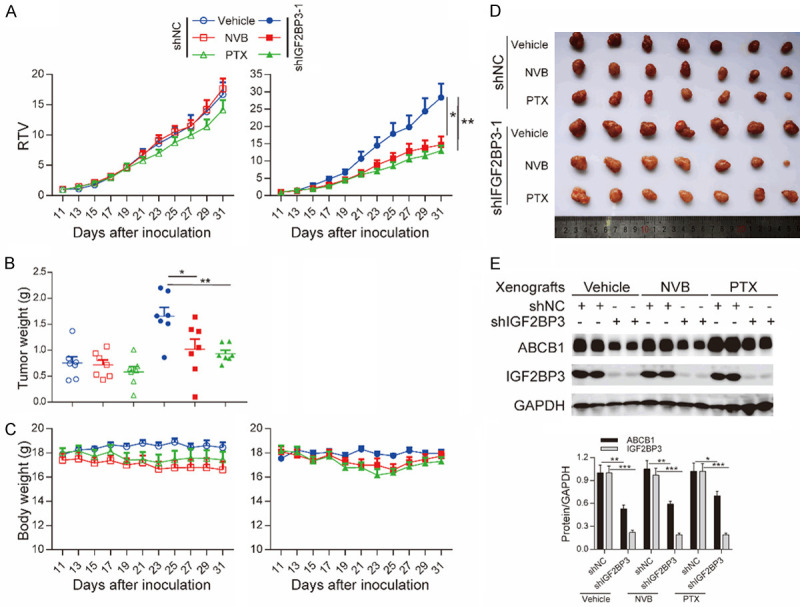
Loss of IGF2BP3 enhanced the response of HCT8/T xenografts to NVB and PTX. NVB or PTX were intraperitoneally administered to nude mice bearing xenografts derived from IGF2BP3-knockdown or control HCT8/T cells for 20 days. A-C. Changes in relative tumor volume (RTV), tumor weight and body weight along the days of inoculation. D. Images of the xenografts at the end of treatments. E. Western blotting analysis showing the expression of ABCB1 and IGF2BP3 proteins in xenograft tumors. Densitometry analysis of protein bands was performed using Image J. *, P <0.05; **, P <0.01, ***, P < 0.001 (Student’s t-test).
Table 6.
Loss of IGF2BP3 enhanced the response of HCT8/T xenografts to NVB and PTX
| Cell lines | Group | Schedule | Body weight (g) | Tumor volume (mm3) | Tumor weight (g) | RTV | T/C | TGI (%) | ||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||||
| D16 | D36 | D16 | D36 | |||||||
| HCT8/T shNC | Vehicle | / | 17.87±0.20 | 18.44±0.43 | 75.42±8.31 | 1235.14±182.40 | 0.76±0.11 | 16.72±2.00 | / | / |
| NVB | 4 mg/kg; q3d; iP | 17.40±0.53 | 16.60±0.71 | 75.80±8.34 | 1259.77±100.69 | 0.72±0.09 | 17.65±1.65 | 1.06 | -2.18 | |
| PTX | 20 mg/kg; q3d; ip | 18.03±0.36 | 17.43±0.66 | 74.54±8.36 | 1050.67±16.01 | 0.58±0.29 | 14.17±1.61 | 0.85 | 15.83 | |
| HCT8/T shIGF2BP3 | Vehicle | / | 17.53±0.20 | 17.96±0.28 | 92.11±8.31 | 2476.17±272.92 | 1.66±0.16 | 28.37±4.00 | / | / |
| NVB | 4 mg/kg; q3d; iP | 18.09±0.44 | 17.74±0.35 | 91.46±7.92 | 1377.19±247.76 | 1.02±0.18* | 14.66±2.43* | 0.52 | 46.07 | |
| PTX | 20 mg/kg; q3d; ip | 18.17±0.44 | 17.30±0.35 | 90.77±6.78 | 1120.31±108.87 | 0.93±0.06** | 12.96±1.49** | 0.46 | 56.82 | |
Changes in body weight, tumor volume, tumor weight and relative tumor volume (RTV) after HCT8/T shNC and shIGF2BP3 cells inoculation. The experiments were conducted as described in Materials and methods. Data are presented as mean ± SEM (n = 7) for each group.
P < 0.05 (Student’s t-test);
P < 0.01 (Student’s t-test).
Western blotting demonstrated that IGF2BP3 and ABCB1 were expressed in the xenografts (Figure 6E), similar to the matching cell lines (Figures 2D, 3E). These data demonstrated that loss of IGF2BP3 enhanced the response of HCT8/T xenografts to NVB and PTX treatment.
Discussion
Colorectal cancer is among the leading causes of tumor related morbidities and mortalities across the world [25,26]. In addition, chemotherapy is the standard form of treatment, although MDR remains a major setback to the successful management of CRC in clinical practice. However, additional underlying mechanisms of MDR in CRC other than the ABC transporters, remain largely unclear. Therefore, the present study analyzed the protein and mRNA profiles of MDR HCT8/T cells and their parental HCT8 cells to gain further insights on the molecular mechanisms underlying MDR and identify new biological targets suitable for predicting sensitivity to chemotherapeutic agents. Bioinformatics screening revealed that IGF2BP3, ALDH3A1 and AKR1C1 were altered and may have a role in MDR, in addition to ABCB1. However, only IGF2BP3 was shown to have a marked effect on sensitivity to DOX.
Additionally, IGF2BP3 is a member of the IGF2BP family and is considered to be an oncofetal factor [16]. It plays a central role in cell growth and migration during embryogenesis and its expression is nearly undetectable in non-neoplastic adult tissues. However, previous studies reported that IGF2BP3 is highly expressed in numerous malignant tumors and has a remarkable prognostic value in various types of cancer, including CRC [27]. Moreover, IGF2BP3 promotes cell proliferation by triggering the expression of oncogenes, IGF2 and MYC through mRNA stabilization. It also has a significant effect on cellular adhesion and infiltration by stabilizing the CD44 mRNA [28]. In addition, previous reports showed that IGF2BP3 promotes the chemoresistance of triple-negative breast cancer by regulating the expression of ABCG2 [24]. Therefore, in order to further investigate the link between IGF2BP3 and chemoresistance, the present study generated two pairs of cell lines, namely; HCT8+Vehicle vs. HCT8+IGF2BP3 and HCT8/T shNC vs. HCT8/T shIGF2BP3. The results revealed that overexpression of IGF2BP3 significantly attenuated the sensitivity of HCT8 cells to ABCB1 substrates. However, the targeted knockdown of IGF2BP3 remarkably restored the sensitivity of HCT8/T cells to ABCB1 substrates in vitro and in vivo. Furthermore, there was a strong correlation between the expression levels of IGF2BP3 and the IC50 values of DOX in a panel of CRC cell lines (r = 0.8446, P = 0.008). These results therefore suggested that the upregulation of IGF2BP3 was associated with chemoresistance in CRC cells and the cells became more sensitive when expressing less IGF2BP3.
Moreover, IGF2BP3 had no effect on the sensitivity of HCT8 and HCT8/T cells to non-ABCB1 substrates. In order to further explore the mechanisms underlying the effect of IGF2BP3 on sensitivity to ABCB1 substrates, the study assessed ABCB1 expression in two pairs of stably transfected cell lines. The results revealed that upregulation of IGF2BP3 led to an increase in the expression of ABCB1 while depletion of IGF2BP3 inhibited ABCB1 expression. Additionally, the expression levels of IGF2BP3 in a panel of CRC cell lines was positively correlated with ABCB1, further supporting the conclusion that IGF2BP3 contributed to chemoresistance by promoting the expression of ABCB1 in CRC. It is therefore possible that the positive effect of IGF2BP3 in enhancing the expression of ABCB1 might be responsible for regulating chemoresistance to ABCB1 substrates. To the best of our knowledge, this was the first study to propose a direct link between IGF2BP3, ABCB1 and chemoresistance.
On the other hand, IGF2BPs constitutes a distinct family of m6A readers that target thousands of mRNA transcripts by recognizing the m6A modified sequence [8]. According to previous reports, IGF2BPs protect target mRNAs from degradation in the P-body while promoting translation after being exported to the cytoplasm, by mobilizing mRNA stabilizers consisting of HuR and MATR3 [19]. With regard to heat shock, IGF2BP-containing complexes are translocated to stress granules for the storage of mRNA targets. Moreover, IGF2BPs promote the stability and storage of their targeted mRNAs in an m6A-dependent approach, thus affecting the output of gene expression. Therefore, it was imperative to explore accurate mechanisms underlying the effects of IGF2BP3, as an m6A reader, on the expression of ABCB1. Consistently, the results revealed that IGF2BP3 acted as an m6A reader and bound to the m6A modification region of ABCB1 mRNA. The findings also revealed a decrease in ABCB1 following the depletion of METTL3/14. The METTL3/14-IGF2BP3 axis was able to regulate the expression of ABCB1. Furthermore, accelerated mRNA decay of ABCB1 upon IGF2BP3 or METTL3/14 depletion was confirmed. In general, these results suggested that IGF2BP3 directly bound to the m6A modification region of ABCB1 mRNA then controlled the half-life of ABCB1 mRNA in an m6A-dependent manner. Therefore, IGF2BP3 promoted the stability of ABCB1 mRNA and enhanced the expression of ABCB1 in an m6A-dependent manner.
Figure 7 shows that the ABCB1 mRNA was methylated de novo by m6A writers, the methytransferase complex, including METTL3 and METTL14. In addition, the ABCB1 mRNAs with m6A modifications were recognized by the IGF2BP3 protein, as an m6A reader. Therefore, IGF2BP3 stabilized ABCB1 mRNAs and facilitated their expression. Under stress conditions, such as the presence of anti-cancer drugs, the expression of IGF2BP3 was upregulated and more methylated ABCB1 mRNAs were subsequently recognized. Furthermore, the stability and expression of mRNAs was significantly enhanced. Consequently, increasing the expression of ABCB1 promoted chemoresistance in CRC cells, leading to the progression of MDR.
Figure 7.
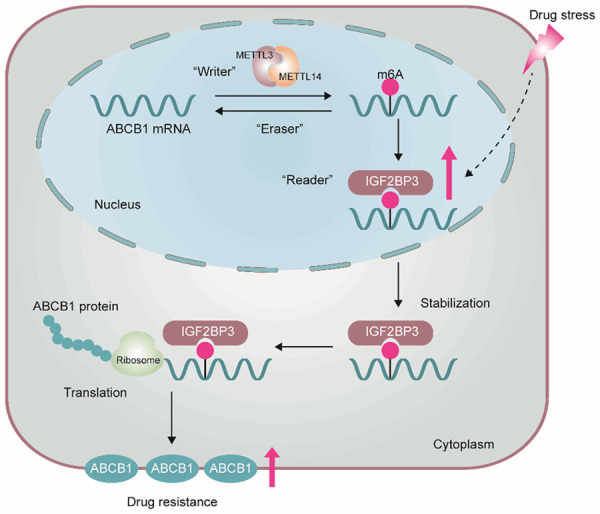
A proposed model of IGF2BP3-mediated regulation of m6A modified ABCB1 mRNA. ABCB1 mRNA was methylated by m6A writers, METTL3 and METTL14. Then, ABCB1 mRNAs with m6A modifications were recognized by IGF2BP3, an m6A reader. IGF2BP3 protected ABCB1 mRNAs from degradation. Under stress conditions, the expression of IGF2BP3 was upregulated, and more methylated ABCB1 mRNAs were subsequently recognized, which enhanced mRNAs stability and expression. Overexpression of ABCB1 promoted CRC cells chemoresistance.
In conclusion, the present study for the first time identified the key role of IGF2BP3 in mediating chemoresistance in CRC cells by upregulating ABCB1. The study also uncovered novel mechanisms through which IGF2BP3 regulated the expression of ABCB1 by binding m6A modified mRNAs. Therefore, these findings identified IGF2BP3 to be a candidate target in the clinical management of CRC. Additionally, IGF2BP3 might function as a predictive biosignature for standard chemotherapy and holds potential as a feasible target for overcoming chemoresistance.
Acknowledgements
This work was supported by funds from the National Nature Science Foundation of China (No. 81872496 and 81873056), and the Science and Technology Commission of Shanghai Municipality (20S11902200 and 16DZ2280100).
Disclosure of conflict of interest
None.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Meisenberg C, Gilbert DC, Chalmers A, Haley V, Gollins S, Ward SE, El-Khamisy SF. Clinical and cellular roles for TDP1 and TOP1 in modulating colorectal cancer response to irinotecan. Mol Cancer Ther. 2015;14:575–585. doi: 10.1158/1535-7163.MCT-14-0762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li W, Zhang H, Assaraf YG, Zhao K, Xu X, Xie J, Yang DH, Chen ZS. Overcoming ABC transporter-mediated multidrug resistance: molecular mechanisms and novel therapeutic drug strategies. Drug Resist Updat. 2016;27:14–29. doi: 10.1016/j.drup.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 4.Kumar A, Jaitak V. Natural products as multidrug resistance modulators in cancer. Eur J Med Chem. 2019;176:268–291. doi: 10.1016/j.ejmech.2019.05.027. [DOI] [PubMed] [Google Scholar]
- 5.Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 6.Zeino M, Paulsen MS, Zehl M, Urban E, Kopp B, Efferth T. Identification of new P-glycoprotein inhibitors derived from cardiotonic steroids. Biochem Pharmacol. 2015;93:11–24. doi: 10.1016/j.bcp.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 7.Longley DB, Johnston PG. Molecular mechanisms of drug resistance. J Pathol. 2005;205:275–292. doi: 10.1002/path.1706. [DOI] [PubMed] [Google Scholar]
- 8.Lan Q, Liu PY, Haase J, Bell JL, Hüttelmaier S, Liu T. The critical role of RNA m(6)A methylation in cancer. Cancer Res. 2019;79:1285–1292. doi: 10.1158/0008-5472.CAN-18-2965. [DOI] [PubMed] [Google Scholar]
- 9.Sun T, Wu R, Ming L. The role of m6A RNA methylation in cancer. Biomed Pharmacother. 2019;112:108613. doi: 10.1016/j.biopha.2019.108613. [DOI] [PubMed] [Google Scholar]
- 10.Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. 2019;20:608–624. doi: 10.1038/s41580-019-0168-5. [DOI] [PubMed] [Google Scholar]
- 11.Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, Dai Q, Chen W, He C. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93–95. doi: 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J, Zhu L, Shi Y, Liu J, Lin L, Chen X. m6A demethylase FTO promotes hepatocellular carcinoma tumorigenesis via mediating PKM2 demethylation. Am J Transl Res. 2019;11:6084–6092. [PMC free article] [PubMed] [Google Scholar]
- 13.Zhuang M, Li X, Zhu J, Zhang J, Niu F, Liang F, Chen M, Li D, Han P, Ji SJ. The m6A reader YTHDF1 regulates axon guidance through translational control of Robo3.1 expression. Nucleic Acids Res. 2019;47:4765–4777. doi: 10.1093/nar/gkz157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fang Y, Sun J, Zhong X, Hu R, Gao J, Duan G, Ji C, Chen L, Zhang W, Miao C, Aisa HA, Zhang X. ES2 enhances the efficacy of chemotherapeutic agents in ABCB1-overexpressing cancer cells in vitro and in vivo. Pharmacol Res. 2018;129:388–399. doi: 10.1016/j.phrs.2017.11.001. [DOI] [PubMed] [Google Scholar]
- 15.Li XH, Li C, Xiao ZQ. Proteomics for identifying mechanisms and biomarkers of drug resistance in cancer. J Proteomics. 2011;74:2642–2649. doi: 10.1016/j.jprot.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 16.Lederer M, Bley N, Schleifer C, Hüttelmaier S. The role of the oncofetal IGF2 mRNA-binding protein 3 (IGF2BP3) in cancer. Semin Cancer Biol. 2014;29:3–12. doi: 10.1016/j.semcancer.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 17.Bell JL, Wächter K, Mühleck B, Pazaitis N, Köhn M, Lederer M, Hüttelmaier S. Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs): post-transcriptional drivers of cancer progression? Cell Mol Life Sci. 2013;70:2657–2675. doi: 10.1007/s00018-012-1186-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deng X, Su R, Weng H, Huang H, Li Z, Chen J. RNA N(6)-methyladenosine modification in cancers: current status and perspectives. Cell Res. 2018;28:507–517. doi: 10.1038/s41422-018-0034-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, Zhao BS, Mesquita A, Liu C, Yuan CL, Hu YC, Hüttelmaier S, Skibbe JR, Su R, Deng X, Dong L, Sun M, Li C, Nachtergaele S, Wang Y, Hu C, Ferchen K, Greis KD, Jiang X, Wei M, Qu L, Guan JL, He C, Yang J, Chen J. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20:285–295. doi: 10.1038/s41556-018-0045-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang X, Yan J, Zhang M, Wang Y, Chen Y, Fu X, Wei R, Zheng XL, Liu Z, Zhang X, Yang H, Hao B, Shen YY, Su Y, Cong X, Huang M, Tan M, Ding J, Geng M. Targeting epigenetic crosstalk as a therapeutic strategy for EZH2-aberrant solid tumors. Cell. 2018;175:186–199. e119. doi: 10.1016/j.cell.2018.08.058. [DOI] [PubMed] [Google Scholar]
- 21.Li T, Hu PS, Zuo Z, Lin JF, Li X, Wu QN, Chen ZH, Zeng ZL, Wang F, Zheng J, Chen D, Li B, Kang TB, Xie D, Lin D, Ju HQ, Xu RH. METTL3 facilitates tumor progression via an m(6)A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol Cancer. 2019;18:112. doi: 10.1186/s12943-019-1038-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang ZM, Liao XM, Chen Y, Shen YY, Yang XY, Su Y, Sun YM, Gao YL, Ding J, Zhang A, He JX, Miao ZH. Combining 53BP1 with BRCA1 as a biomarker to predict the sensitivity of poly(ADP-ribose) polymerase (PARP) inhibitors. Acta Pharmacol Sin. 2017;38:1038–1047. doi: 10.1038/aps.2017.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xuan JJ, Sun WJ, Lin PH, Zhou KR, Liu S, Zheng LL, Qu LH, Yang JH. RMBase v2.0: deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res. 2018;46:D327–D334. doi: 10.1093/nar/gkx934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Samanta S, Pursell B, Mercurio AM. IMP3 protein promotes chemoresistance in breast cancer cells by regulating breast cancer resistance protein (ABCG2) expression. J Biol Chem. 2013;288:12569–12573. doi: 10.1074/jbc.C112.442319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dekker E, Tanis PJ, Vleugels JLA, Kasi PM, Wallace MB. Colorectal cancer. Lancet. 2019;394:1467–1480. doi: 10.1016/S0140-6736(19)32319-0. [DOI] [PubMed] [Google Scholar]
- 26.Arul M, Roslani AC, Cheah SH. Heterogeneity in cancer cells: variation in drug response in different primary and secondary colorectal cancer cell lines in vitro. In Vitro Cell Dev Biol Anim. 2017;53:435–447. doi: 10.1007/s11626-016-0126-x. [DOI] [PubMed] [Google Scholar]
- 27.Xu W, Sheng Y, Guo Y, Huang Z, Huang Y, Wen D, Liu CY, Cui L, Yang Y, Du P. Increased IGF2BP3 expression promotes the aggressive phenotypes of colorectal cancer cells in vitro and vivo. J Cell Physiol. 2019;234:18466–18479. doi: 10.1002/jcp.28483. [DOI] [PubMed] [Google Scholar]
- 28.Vikesaa J, Hansen TV, Jønson L, Borup R, Wewer UM, Christiansen J, Nielsen FC. RNA-binding IMPs promote cell adhesion and invadopodia formation. EMBO J. 2006;25:1456–1468. doi: 10.1038/sj.emboj.7601039. [DOI] [PMC free article] [PubMed] [Google Scholar]