

Summary
We developed a cost sensitive isotope labelling procedure using a fed‐batch fermentation method and tested its efficiency producing the 15N‐, 13C‐ and 15N/13C‐labelled variants of an amyloidogenic miniprotein (E5: EEEAVRLYIQWLKEGGPSSGRPPPS). E5 is a surface active protein, which forms amyloids in solution. Here, we confirm, using both PM‐IRRAS and AFM measurements, that the air–water interface triggers structural rearrangement and promotes the amyloid formation of E5, and thus it is a suitable test protein to work out efficient isotope labelling schemes even for such difficult sequences. E. coli cells expressing the recombinant, ubiquitin‐fused miniprotein were grown in minimal media containing either unlabelled nutrients, or 15N‐NH4Cl and/or 13C‐D‐Glc. The consumption rates of NH4Cl and D‐Glc were quantitatively monitored during fermentation and their ratio was established to be 1:5 (for NH4Cl: D‐Glc). One‐ and two‐step feeding schemes were custom‐optimized to enhance isotope incorporation expressing five different E5 miniprotein variants. With the currently optimized protocols we could achieve a 1.5‐ to 5‐fold increase of yields of several miniproteins coupled to a similar magnitude of cost reduction as compared to flask labelling protocols.
We developed a cost sensitive isotope labelling procedure using a fed‐batch fermentation method and tested its efficiency producing the 15N‐, 13C‐ and 15N/13C‐labelled variants of an amyloidogenic miniprotein E5, which is a surface active and forms amyloids in solution. With the currently optimized protocols we could achieve a 1.5‐ to 5‐fold increase of yields of several miniproteins coupled to a similar magnitude of cost reduction as compared to flask labelling protocols.

Introduction
The 5‐10 mg scale production of non‐selective isotopic labelled polypeptide hormones (e.g. insulin, incretins; Baeshen et al., 2014) and miniproteins (e.g. Trp‐cage, cyclotides, PPα‐Tyr; Neidigh, 2002; Baker et al., 2017; Gould, 2017) is crucial for structure determination, binding studies, lead optimization etc. Solution or solid‐state NMR structure determinationis practically the only experimental method to provide atomic‐level structural information concerning shorter or midsize proteins requires labelled materials for thorough investigations that reveal the structure and dynamics of such systems. Before the widespread use of recombinant expression methodologies, solid phase peptide synthesis (SPPS) was the only way to produce 15N, 13C labelled peptides but at extreme costs (Merrifield, 1964). The latter method requires N‐protected isotopic labelled amino acid residues, largely increasing the production cost, especially for longer peptides or proteins (Stráner et al., 2016). In contrast, recombinant expression is a widely used environmentally friendly (green) and economical technique. Using a suitable strain of E. coli bacteria, high growth rates and fast protein production can be achieved, expressing directly the required gene or its constructs, such as the gene cloned side‐by‐side with a selected fusion protein. Host cells can be cultured and genetically manipulated in a water‐based environment. Isotope labelling in bacteria is also cost‐effective, as only two ingredients, NH4Cl and D‐Glc, have to be 15N and 13C labelled to achieve uniform 15N‐, 13C‐incorporation.
The challenge in expressing medium‐sized (20–40 residue‐long) polypeptides or miniproteins is that their size makes them extremely vulnerable to proteolytic degradation. However, applying suitable fusion tag proteins (glutathione‐S‐transferase, GST; thioredoxin, TRX; ubiquitin; small ubiquitin‐related modifier, SUMO; ketosteroid isomerase, KSI; Williamson et al., 2000; Bogomolovas et al., 2009; Panavas et al., 2009; Zorko, 2010) has been identified as a powerful strategy to overcome this difficulty. In this work, we used ubiquitin as a fusion tag (Bommarius et al., 2010). Ubiquitin fusion system has numerous advantages: the ubiquitin tag is relatively small (11 kDa), hence the target peptide has larger contribution to the total mass of the fusion protein and the yeast ubiquitin hydrolase (YUH) enzyme can cleave the fusion protein from the polypeptide without any remaining and unwanted residue at N‐terminus (Kohno et al., 1998). One of the major drawbacks of bacterial expression systems are that in certain cases the expression levels can be too low (< 1–2 mg l−1). Eukaryotic proteins without post‐translational modifications essential for their fold, toxic peptides, peptides or proteins with aggregation tendency and proteins lacking their complex disulfide bond pattern are typical systems inefficiently synthesized by bacteria. Changing the host cell to a higher organism (yeast, insect or mammalian cell) can be a solution to this problem; however, it extremely increases the isotope labelling costs. Another option is upscaling, however large volume apparatus (e.g. 30‐50 l fermenter) are still uncommon in most academic laboratories.
During scale up, the number of cells is augmented, achieved either by a volumetric increase of the cell culture or by concentration enhancement. In an everyday biochemical environment even a moderate scale increase, for example from 1 to 10 l, introduces unexpected challenges to work with. In addition, this strategy results in the vast loss of isotope labelled ingredients. Furthermore, if instead of using a single batch, multiple flasks are in use, then the differences in metabolic rates can result in severe timing issues (e.g. induction and/or harvesting) to be matched. On the other hand, if cell concentration is increased to reach a higher yield by using a fermenter for example, then normal size laboratory equipment as well as improved and tunable biotechnology protocols can be applied (Klopp et al., 2018). However, the close monitoring of the pH, air flux and the concentration of selected metabolites requires a disciplined and automation‐oriented technology.
When conditions such as temperature, pH, nutrient concentration, oxygen concentration for aerobic strains, etc., are optimal, bacteria grow at an exponential rate. However, if conditions are inappropriate, growth becomes limited. In nutrient limited growth, the rate limiting molecule is that which the medium runs out of the first, introducing a lag‐phase, followed by a slower‐than‐exponential phase that utilizes an alternative nutrient (diauxic growth). The change caused by the petering of the limiting nutrient is called the diauxic shift (Monod, 1942). During exponential growth, the dissolved oxygen concentration steadily decreases, as the volume of the biomass increases. However, during the lag‐phase there is practically no bacterial growth, and thus the oxygen concentration increases, indicating a shortage of a limiting nutrient. Several fermentation protocols to produce recombinant proteins are known in the literature. Working with labelled nutrients, cost‐effective batch and fed‐batch fermentation techniques are usually applied along the entire process (Studts, 1999; Ross et al., 2004). Cai et al. (1998) used a fed‐batch fermentation process for 15N‐ and 13C/15N‐labelling, in which the cells were grown in the presence of unlabelled isotopes in the initial phase. In a comprehensive study, several fermentation protocols were implemented with auto‐ or IPTG‐inducing on minimal or rich media (Klopp et al., 2018).
Here we present an economical fermentation protocol to produce isotope labelled polypeptides and miniproteins. During the development of our protocol we used a self‐aggregating sequence, E5 (EEEAVRLYIQWLKEGGPSSGRPPPS; Rovó et al., 2013) derived from Exendin‐4 miniprotein as a test system, which forms amyloid near the physiological conditions (80 µM < c prot. < 800 µM, 10 mM < c salt < 100 mM, 4 < pH < 5, T = 37 °C) after a few hours (Taricska et al., 2020) and thus disturbs and hinders high yield protein expression. We examined the structural rearrangement of E5 at air–water surface and its dependence on storage time. By using both PM‐IRRAS and AFM measurements early amyloid formation triggered by the air–water interface could be effectively monitored. We then successfully applied our new protocol for the expression of Exendin‐4 itself and its further variants, namely E11, E19, Tc5b‐D9N (Rovó et al., 2014; Scheme 1).
Fig. 5.
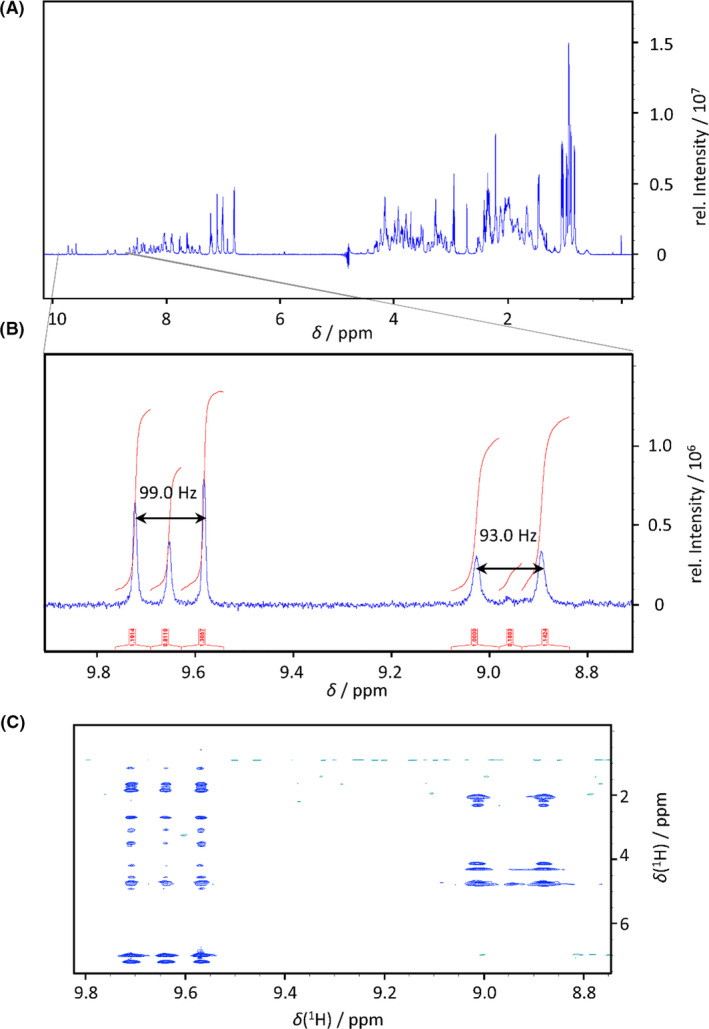
(A) 1H‐NMR spectrum of 15N‐E5 expressed in fermenter. (B) The zoom of the 1H‐NMR spectrum shows two different signal groups in the H(N) region: one is a typical resonance of backbone peptide H(N) (right) with a doublet with 1J‐coupling of 93.0 Hz (assigned to Glu3), while the other belongs to the Trp indole side chain H(N) with a 1J‐coupling of 99.0 Hz (left). (C) The zoom of the homonuclear 1H,1H NOESY spectrum proves that the three signals of Trp side chain and the two signals of the backbone belong to the same H(N) due to the same crosspeak pattern.
Scheme 1.

The primary sequence of Exendin‐4, a drug used in the treatment of type II Diabetes Mellitus and its four variants: E5, E11, E19, Tc5b‐D9N.
Results and discussion
Our fermentation method for non‐selective isotope labelling (15N, 13C) is based on the fact that the isotope carrying (15N‐NH4Cl and 13C‐D‐Glc) components are added to the cell culture at the time of the diauxic shift, t 1 (Fig. 1A). Prior to this, unlabelled 14N‐NH4Cl and 12C‐D‐Glc are the exclusive nitrogen and carbon sources used in the minimal medium (batch phase, between t 0–t 1; Fig. 1A). As both the isotope source and the timing were examined thoroughly, the synchronized induction led to optimal use of isotope sources: predominantly to produce the required protein. In practice, cytosolic accumulation of both the ingredients and the metabolites could result in incomplete labelling, therefore it is common to overuse the isotope source, adding them in two consecutive steps (first feeding phase, between t 1a–t 1b and second feeding phase, between t 1b–t 2; Fig. 1B; Cai et al., 1998). As the superiority of two‐step feeding has never been unambiguously probed, we have carried out the following two experiments. Using a one‐step feeding protocol, the time of induction and isotope addition was synchronized to the oxygen peak (Fig. 1A). During the two‐step feeding approach, one quarter of the total amount of isotope is added in synchrony with to the initial oxygen peak, followed by induction and addition of the remaining three‐quarters when the second oxygen peak is detected (Fig. 1B). The extent of isotope incorporation achieved with each protocol was examined by NMR spectroscopy choosing one well separated signal. 1H‐NMR spectrum of 15N‐E5 between 8.8 ppm and 9.1 ppm reveals a doublet, with 1JHN‐coupling of ~ 93.0 Hz with a singlet at the centre (8.96 ppm), belonging to the unlabelled derivative. Based on the ratio of integrals of the doublet and the singlet signals, we determined the ratio of the concentration of the labelled and unlabelled protein. This ratio was found to be 96% if the two‐step feeding protocol was applied, suggesting a more complete isotope incorporation (Fig. 1D) than in case of the one‐step feeding protocol (Fig. 1C) when it was 91%. This might be due to presence of greater excess of cytosolic unlabelled metabolites in case of the latter. Based on these findings, we selected the two‐step feeding protocol for further use.
Fig. 1.
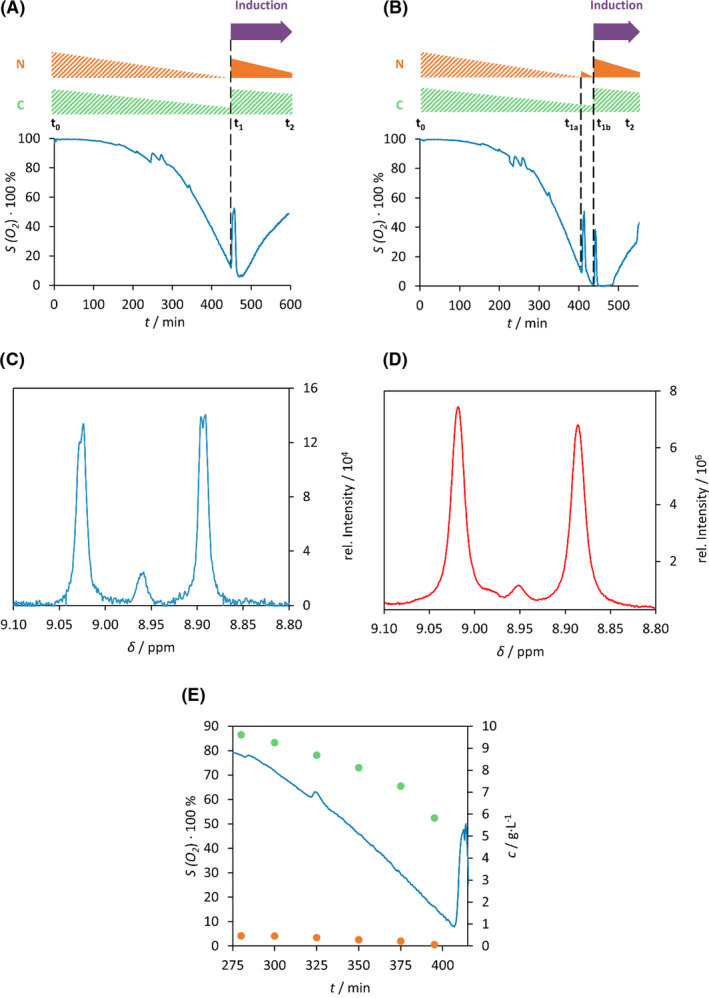
Comparison of the two batch‐fed strategy. (A) During isotope labelled expression using one‐step and (B) two‐step feeding scheme, the dissolved oxygen (%) was monitored as function of the time. The relative consumption of D‐Glc (green triangle) and NH4Cl (orange triangle) are depicted schematically above using solid colours for 13C‐ or 15N‐, while striped colours for natural abundant (12C‐ or 14N‐) ingredients. Purple arrow points to the time of induction. t 0: beginning of fermentation; t 1, time of feeding; t 1a, t 1b, time of the first or second feeding; t 2, end of the fermentation. Selected 1H‐NMR signals of backbone amid H(N) of Glu3 of 15N‐labelled E5: using either (C) the one‐step (red line) or (D) two‐step feeding (blue line) fermentation protocol. The doublet stands for the 1H‐15N one‐bond coupling non‐existent in a 1H‐14N system resulting in a singlet. (E) Monitoring both D‐Glc (green dot) and NH4Cl (orange dot) concentration (g·l−1) changes with variation of the dissolved oxygen (%) (blue line) as function of the fermentation time (min).
It is important to distinguish whether the nitrogen or carbon source is exhausted at the time of the diauxic shift. Bacteria consume NH4Cl and D‐Glc at a different rate, however the ratio of the consumption rates remains constant over time. Therefore, it is possible to determine a priori whether NH4Cl or D‐Glc will be the limiting nutrient, if the characteristic ratio of consumption rates is established. Thus, the concentration changes of the carbon and nitrogen source were measured as function of the time using initially 10 g of D‐Glc and 1 g of NH4Cl (Fig. 1E). The concentration of NH4Cl was monitored by absorption spectroscopy detecting the indophenol directly derived from the ammonium ion. D‐Glc concentration was followed by commercial blood Glc metre and by using a novel method quantifying the concentration of a D‐Glc derivative by RP‐HPLC. Concentration of D‐Glc decreased rapidly, while that of NH4Cl decreased only moderately. When NH4Cl was depleted completely, more than 5 g of D‐Glc remained in the media. Hence, the ratio of the consumption rates of D‐Glc and NH4Cl was determined to be 1:5. This constant was used as a benchmark value to develop the media recipes for the various types of labelling methods, in particular the mass ratio of carbon and nitrogen sources. For 15N‐labelling, we used 1:10 15N‐NH4Cl : unlabelled 12C‐D‐Glc mass ratio: to guarantee the continual excess of D‐Glc during the expression. 1:3 unlabelled or 15N‐NH4Cl:13C‐D‐Glc mass ratio was used for the expression of 13C‐ and 13C/15N‐miniproteins, which results in nitrogen source overabundance. To generate 15N‐isotope labelled miniproteins, 1 g of unlabelled 14N‐NH4Cl and 10 g of unlabelled 12C‐D‐Glc were loaded into the initial media, using the following protocol: after the batch phase, 0.25 g of 15N‐NH4Cl (one quarter of the total amount to be fed) was added, the second diauxic shift, the remaining 0.75 g of 15N‐NH4Cl was loaded into the culture with additional 10 g of unlabelled 12C‐D‐Glc ensuring the carbon excess. In parallel with the second feeding, the induction of the protein expression was performed. The applied protocols and labelling schemes are summarized in Table 1 and described in detail in Supplementary material (S1).
Table 1.
Summary of the initial and additional amount of D‐Glc and NH4Cl added to the samples making either 15N or 13C as well as 15N/13C labelled miniproteins.
|
Batch phase t 0 < t(min) a < t 1a |
First feeding phase t 1a < t(min) < t 1b |
Second feeding phase t 1b < t(min) < t 2 |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| N (g) b | C (g) b | N (g) | C (g) | N (g) | C (g) | ||||
| U c | L d | U | L | L | U | L | U | L | |
| 15N‐miniprotein | 1 | 0 | 10 | 0.25 | 0 | 0 | 0.75 | 10 | 0 |
| 13C‐miniprotein | 1 | 0 | 3 | 0 | 0.75 | 1 | 0 | 0 | 2.25 |
| 13C/15N‐miniprotein | 0 | 1 | 3 | 0 | 0.75 | 0 | 1 | 0 | 2.25 |
t 0, t 1a, t 1b, t 2 are the same codification as in Fig. 1A and B.
N = mass of NH4Cl; C = mass of D‐Glc.
U = natural abundance isotopic source.
L = isotopically enriched (13C and/or 15N) source.
Next, our new protocol was tested on four further miniproteins (namely: Exendin‐4, E11, E19 and Tc5b‐D9N) using our fermentation strategy (Table 2). We found that yields increased 1.5‐5‐fold with respect to flask expression protocol suggesting that our new protocol is suitable for producing difficult‐to‐express miniproteins and peptides with satisfactory yields.
Table 2.
Yield and cost comparison of the flask expression and fermentation of Exendin‐4 and four variants, namely Tc5b‐D9N, E5, E11 and E19. Ni‐IMAC chromatograms of raw materials were used to estimate the yields and the purity was checked by analytical HPLC (an example for E5 is shown in Supplementary material S2).
| Protein name and labelling type | Calculated yield a (mg l−1) | Estimated cost (EUR/10 mg) | ||
|---|---|---|---|---|
| Ferm. | Flask | Ferm. | Flask | |
| E11, 15N | 64.8 | 31.2 | 18.7 | 38.8 |
| E19, 15N | 150.2 | 33.5 | 8.1 | 36.2 |
| E5, 15N | 99.6 | 19.4 | 12.2 | 62.4 |
| Exendin‐4, 15N | 87.0 | 31.4 | 13.9 | 38.6 |
| Tc5b D9N, 15N | 19.1 | 12.1 | 63.5 | 100.1 |
Raw material yield based on the integral volume of the absorbance curve (see methods), determined after completing the first Ni‐IMAC step.
Immediate purification of the miniproteins following the expression proved to be crucial, significantly affecting the final yields. Following our first attempts, cell pellets were stored at −20 °C after fermentation. The cells which remained in frozen state at −20 °C for an extended time, did not provide reproducible yields, most likely due to aggregation processes leading to fibril formation to various extents. This is probably caused by the conformational heterogeneity of the systems that is sustained even at −20 °C (Yang et al., 2015). The formation of β amyloid fibril structure requires conformational change from the globular to elongated forms and the formation of numerous intermolecular contacts between the latter. If amyloid formation is triggered during the expression, the overall yield will decrease due to the precipitation of the fibrils.
Our isotope labelling (15N, 13C) protocol was developed using E5 known to be amyloidogenic sequence, which forms amyloid near physiological conditions after a few hours in solution (Yang et al., 2015). We followed the process by PM‐IRRAS and AFM experiments. We established that E5 suffers a structural rearrangement at air–water surface causing reduced yields during protein expression and purification.
The PM‐IRRAS technology allows the measurement of surface specific FTIR spectra of materials relying on the differences in the reflection of p‐ and s‐polarized light from interfaces. This gives information of the chemical composition, phase transition, structure and orientation of the molecules in the examined sample. Polarization modulation results in the elimination of the background signals, such as water vapour and CO2, thus it is not necessary to use protective gases or vacuum during the measurements. E5 is a highly surface active molecule, it accumulates promptly in air–water interfaces (Fig. 2A), similarly to other well‐known globular proteins like bovine serum albumin (BSA) and ovalbumin (OVA). Thus, PM‐IRRAS measurements were carried out at the air–water surface of spread E5 films (Fig. 2B). Multiple peaks could be identified in the amide I region indicating the presence of the protein molecules at the surface and allowing for the determination of the protein secondary structure (Shanmukh et al., 2005; Yang et al., 2015). The most notable feature is the presence of prominent peaks around 1630 cm−1 that can be assigned to β‐sheet structures. Since in bulk phase no β‐structure is detectable (Taricska et al., 2019), the appearance of β‐sheet and β‐turn structures and the decreased presence of random coil and α‐structures points to surface induced structural changes.
Fig. 2.
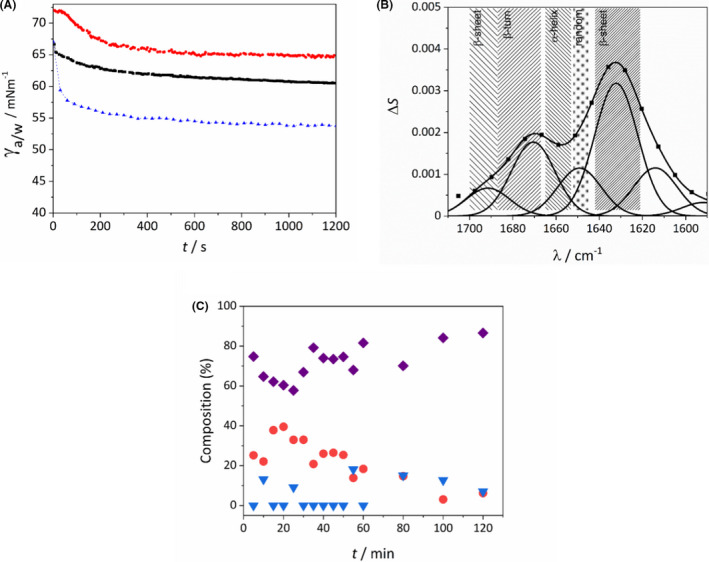
(A) Time dependence of the surface tension (γ) of ovalbumin (OVA, red dots), E5 (black rectangles) and bovine serum albumin (BSA, blue triangles) at solution/air surface, OVA and BSA are included as a reference. (B) Representative PM‐IRRAS spectrum of E5 recorded at the air–water interface at 15 °C. (C) Secondary structure composition (purple rectangles: β‐sheets; red circles: α‐helices and random coil structures; blue triangles: β‐turn motives) of E5 interfacial films at 15 °C as a function of film age.
To characterize the time dependence of this structural rearrangement, measurements were carried out at 15 °C subphase temperature as a function of film age. Already in the first measurement, started within 1 min after film deposition corresponding to a film age between 1 and 6 min, an excess in β‐structures can be seen (Fig. 2C). Over time a further time dependent reorganization could be detected with an enrichment of β‐structures. Due to the time resolution of the measurement technique the initial refolding could not be observed but finding predominantly β‐structures even in the first measurements indicate that it is a readily occurring, fast process.
We also found significant increase in the readiness of β‐structure formation in E5 solutions stored for extended time periods at −20 °C. The lowest starting β‐component (72.5%) was observed for the freshly prepared solution, while the corresponding value for the film prepared using an E5 solution stored for two weeks at 4 °C is 85%. The difference becomes smaller as the interfacial film ages, converging to 85–90% (Fig. 3A).
Fig. 3.
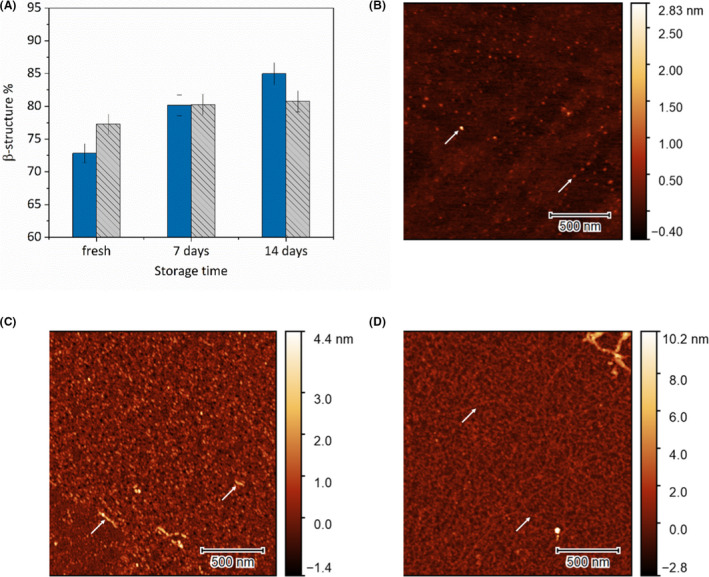
(A) Percentage of β‐sheet + β‐turn structures 5, 60 and 120 min after film formation at 15 °C as a function of solution storage time. Atomic force microscopy images of E5 interfacial films created from (B) freshly prepared solution, (C) solution stored for 7 days and (D) solution stored for 14 days. White arrows indicate amyloid fibrils.
Amyloid formation was also followed using AFM. When the E5 interfacial film was created from a freshly prepared solution, a typical, uniform molecular film could be observed with AFM. Only a small number of aggregates could be seen with typical diameters of 0.5–1.0 nm (Fig. 3B). Films created from samples stored for 7 days exhibit a significantly altered morphology. The surface is covered with small, uniformly sized aggregates in the size range of 2–3 nm (Fig. 3C). These aggregates could be identified as globular oligomers of the E5 that can be considered as the primary building blocks of amyloid fibrils (Dovidchenko et al., 2014). Already in this state, occasionally bigger fibril‐like aggregates could be observed. After 14 day of storage, while most of the surface is still covered with the globular aggregates, many of these have started to fuse together to form a network of protofibrils. These protofibrils have the same diameter of 2–3 nm as the globular aggregates indicating that they are most likely formed by the linking of these units (Fig. 3D). Our findings indicate that in solution the secondary structure of the miniprotein becomes unstable over time. When the samples come into contact with an interface, it can initialize the amyloid formation.
Because of this effect, target proteins were isolated immediately, and purified a day after expression. To achieve prompt comparison between fermentation and flask expression methods, one‐step purification was applied. After a Ni‐IMAC chromatography, the yields were calculated from the peak area that belongs to the target protein. The samples were purified using additional chromatography steps, if needed (Fig. 4).
Fig. 4.
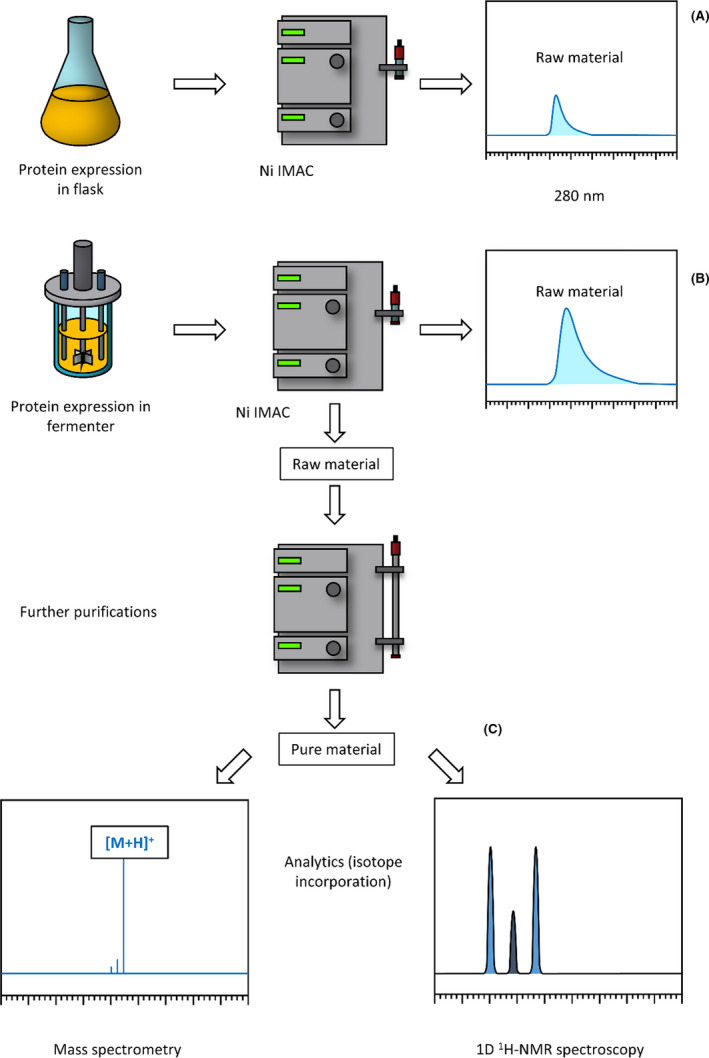
The comparative scheme of protein expression using either a flask or fermenter completed with both product purification and spectroscopy (MS and 1H‐NMR) measurements. (A) and (B) routes stand for direct raw material yield measurements obtained either by flask or fermenter. (C) route depicts raw material purification followed by quality control measurements such as isotope incorporation with NMR spectroscopy.
Compared to the isotope labelling methods in flask, 2‐ to 5‐fold increase was reached in almost every case. The estimated cost reduction followed the same pattern in each case. The lowest yield development (1.5‐fold) was detected in the case of Tc5b‐D9N, and the highest in the case of E19 (5‐fold). During the study of proper isotope incorporation into 15N‐E5, an unusual phenomenon was observed. The zoom of the 1H‐NMR spectrum between 8.7–9.9 ppm showed two different signal groups in H(N) region: one is a typical resonance of backbone peptide H(N) with a doublet with 1J‐coupling of 93.0 Hz (assigned to Glu3), while the other belongs to the Trp indole side chain H(N) with a 1J‐coupling of 99.0 Hz (Fig. 5A and B). The latter group is a doublet with a singlet in the centre, indicating only partial isotope labelling of Trp side chain. Similar effect was seen in the 1H,1H‐NOESY spectrum (Fig. 5C). In contrast, the signals belonging to backbone H(N)s clearly demonstrate a high isotope labelling ratio – in this region the intensity of the unlabelled signals in the centre are very low. The phenomenon is probably caused by the improper depletion of unlabelled indole, the metabolic precursor of Trp, which is present in increased amounts in the cytosol during the diauxic shift (Gaimster, 2015).
Conclusion
Lab‐scale fermenters ensure greater control of bacterial culture processes and provide highly economical and scaled‐up procedures to prepare isotope labelled miniproteins and peptides as compared to flask applications. Here we developed three protocols to produce 13C‐, 15N‐ and 13C/15N‐labelled miniproteins. Our strategy consists of a batch phase, when cells grow in media containing mostly unlabelled isotopes, and a feeding phase, where the labelled isotopes were added in a two‐step manner We found that two‐step feeding protocol provides greater extent of isotope incorporation than one‐step feeding, and thus, could reach a 1.5‐ to 5‐fold increase of yields and a similar magnitude of cost reduction compared to flask labelling methods. Our finding about improper isotope incorporation of the NH of the Trp indole ring suggests, that in some cases indole resonances might ‘underestimate’ the overall isotope incorporation efficacy.
The protocols are summarized here:
To produce 13C‐labelled proteins, cells are grown in minimal media containing 1 g of unlabelled 14N‐NH4Cl and 3 g of unlabelled 12C‐D‐Glc. At the first oxygen peak 0.75 g of 13C‐D‐Glc is added, and at the second oxygen peak further 2.25 g of 13C‐D‐Glc is added to the media.
For making 15N‐labelled proteins, cells are grown in minimal media containing 1 g of unlabelled 14N‐NH4Cl and 10 g of unlabelled 12C‐D‐Glc during the batch phase. At the first, oxygen peak 0.25 g of 15N‐NH4Cl is added, and at the second oxygen peak further 0.75 g of 15N‐NH4Cl is added to the media.
To produce 13C/15N‐labelled proteins, cells grow in minimal media containing 1 g of 15N‐NH4Cl and 3 g of unlabelled 12C‐D‐Glc. At the first, oxygen peak 0.75 g of 13C‐D‐Glc is added, and at the second oxygen peak further 2.25 g of 13C‐D‐Glc is added to the media.
The protocols described here are straightforward, cost effective and can be adapted to produce any recombinant protein, especially for those miniproteins which are expressed with low yields due to their tendency of aggregation and amyloid formation. This was demonstrated for the case of E5, a miniprotein that is surface active and suffers an immediate structural rearrangement at air–water surface. We showed that the PM‐IRRAS/AFM methodologies can provide detailed molecular information on aggregate formation. We propose that in case of E5 the process is initiated by the formation of metastable pre‐aggregates of the globular form of the protein. Reaching the air–water interface triggers the restructuring and rearrangement of these pre‐organized, high concentration clusters leading to β‐amyloid formation. We concluded that extended storage of such sensitive samples in solution or cell pellet form leads to extensive material loss and thus should be avoided.
Experimental procedures
Cultivation
Fermentations were carried out in BIOSTAT A 2 L benchtop bioreactor (Sartorius). The inoculation procedure and fermentation protocols are presented in detail in the Supplementary material. Culturing was typically completed at 37°C, pH 7.0, 600 r.p.m. stirring, 1500 cm3 min−1 aeration. Expression was induced with 1 mM IPTG. Following fermentation, cells were pelleted by centrifugation, followed by suspension in a Lysis buffer (50 mM NaPi, 300 mM NaCl, pH 8.0) and stored at −20°C. Flask expression controls were performed in 2YT media. For 15N‐labelling, 1 g of 15N‐NH4Cl and 4 g of unlabelled 12C‐D‐Glc was added to the media. For 13C/15N‐labelling, the media were supplied with 1 g of 15N‐NH4Cl and 2 g of 13C‐D‐Glc.
Purification of the recombinant miniproteins
After thawing the suspension, cells were disrupted by sonication and cell debris was removed by centrifugation. The supernatant containing the protein of interest was purified with one of the two following methods depending on further uses. (i) If prompt information about protein yield was required, one‐step Ni immobilized metal ion affinity chromatography (Ni‐IMAC) was applied. The supernatant was loaded onto a HisTrap HP 5 ml column, and after washing with Wash buffer (Lysis buffer containing 20 mM imidazole), the protein of interest was eluted with Elution buffer (Lysis buffer containing 250 mM imidazole). Protein yield was calculated from area of absorbance peak at 280 nm. At this point, the samples could be flash frozen and stored at −80°C or purified further according to the second method. (ii) When purified samples were needed, the second strategy, a three‐step purification protocol was applied as described (Rovó et al., 2013). Briefly, the sample was dialysed against 2 l wash buffer twice, after the first Ni‐IMAC step. The ubiquitin tag was cleaved by His‐tagged YUH followed by a 3–4 h long digestion. Sample integrity was checked by SDS PAGE. Both Ubiquitin and YUH were removed during the second Ni‐IMAC step. Finally, the peptides were purified by RP‐HPLC on a C‐18 column, using a water/acetonitrile gradient (eluent A being 0.1% TFA in water and eluent B being 0.08% TFA and 80% acetonitrile in water). Collected fractions were pooled and lyophilized, and their identities were determined by a PerkinElmer Sciex API2000 mass spectrometer equipped with an electrospray ionization source.
PM‐IRRAS experiments
Doubly distilled water, checked by its surface tension (72.0 mN m−1 at 25°C) and conductivity (<5 mS), was used in all polarization modulation infrared reflection absorption spectroscopy (PM‐IRRAS) experiments as a subphase and for the preparation of E5 aqueous solutions. Surface films of the protein were created by carefully spreading 50 μl aqueous solution of E5 at a concentration of 200 μM onto a subphase of freshly doubly distilled water (pH 6.5) in a Langmuir‐balance (Biolin Scientific KSV Nima Medium trough). When otherwise not specified, the E5 solutions were prepared with doubly distilled water immediately before spreading. Subphase temperature was controlled by a circulating thermostat in the 7–25°C range. Polarization modulation infrared reflection absorption spectra of the formed films were collected in situ at the air–water interface using a KSV Nima PM‐IRRAS instrument, composed of a Fourier transform IR spectrometer equipped with a polarization modulation unit (Hinds Instruments, PEM‐100, ZnSe photoelastic modulator) on one arm of a goniometer and a Peltier cooled detector on the other arm. The incident angle of 76° relative to the normal of the air–water interface was used during measurements to maximize signal‐to‐noise ratio. The incident IR beam was modulated by the photoelastic modulator at 1500 cm−1 frequency with a retardation of λ/2. Using the modulator, it is possible to simultaneously record two spectra, a sum or reference spectrum and a difference or surface sensitive spectrum. The resulting PM‐IRRAS signal can be given by the following equation:
where R p and R s are the parallel (p) and perpendicular (s) polarized reflectances (Blaudez et al., 1993).
Normalized spectra a given as
where S 0 is the signal measured on pure water surface at respective temperatures, while S is the signal from the protein covered surface. For each spectrum, an acquisition time of 5 min was used to record 3000 spectra in the 800–4000 cm−1. Spectra showed good reproducibility on separate films and the presented spectra are the average of at least 5 recordings. The normalized spectra were baseline corrected and the amide I band in the 1600–1700 cm−1 range was subjected to peak fitting using second‐derivative peak identification and Gaussian curve‐fitting with the fityk software.
Atomic force microscopy (AFM)
The interfacial films of the E5 miniprotein was also studied applying atomic force microscopy. Following the PM‐IRRAS measurements, the films were transferred onto a solid support using the Langmuir–Schaeffer method. Freshly cleaved mica was used as the flat substrate. Transferred films were dried in vacuum before imaging in air at room temperature. Sample morphology was studied with a Nanosurf FlexAFM atomic force microscope, operating in dynamic mode. Tap190GD‐G cantilever (BudgetSensors) with a nominal tip radius of less than 10 nm was used for the measurements. Images were recorded over 2 × 2 μm2 window areas at 10 randomly selected locations with a resolution of 512 pixels/line.
Surface tension measurement
The surface tension of aqueous protein solutions (c = 0.1 g l−1) was determined with an accuracy of 0.1 mNm−1 by the axisymmetric drop shape analysis using the OCA15 + instrument (Dataphysics, Filderstadt, Germany). Apart from the E5 miniprotein, the surface tension of bovine serum albumin (BSA) and ovalbumin (OVA) solutions were also measured as a reference. The profile of capillary surface required to determine the surface/interfacial tension is obtained by analysing the shape of the pendant drop using a CCD camera coupled to a video image profile digitizer board connected to a personal computer (Hill et al., 2008). Drop of 8 μl was formed and the surface tension was recorded as a function of the time for 20 min with a frequency of 10 frames per min.
Measurement of NH4Cl concentration
The NH4Cl concentration was measured by using the Spectroquant Ammonium Test kit (Merck, Darmstadt, Germany), using an eight‐point calibration curve in the 0.0–140.0 mg l−1 range. This assay is based on the conversion of the NH4 + to an indophenol derivative via the Berthelot reaction and the measurement of the absorption of the indophenol derivative (Searle, 1984).
Measurement of D‐Glc concentration
The D‐Glc concentration was measured using two different methods. At higher D‐Glc concentrations, a commercial blood Glc meter (e.g. Méry Plusz TD‐4255) was employed. The instrument was five‐point calibrated using standard D‐Glc solutions in the 2.0–10.0 g l−1 range. Lower D‐Glc concentrations (2.0 > c > 0.1 g l−1) were measured using a D‐Glc derivative, quantified by RP‐HPLC as follows (Iqbal, 2009). Cells were removed by centrifugation and the diluted sample was filtered through an Amicon Ultra Ultracel 3K 0.5 ml ultrafilter, hence eliminating most macromolecular content. 200 µl DNPH solution (10 mM 2,4‐dinitrophenyl hydrazine, 1 V/V% H2SO4, 4 V/V% H2O, 95 V/V% EtOH) was added to 200 µl filtrate and the mixture was incubated at 65°C for 1 h. DNPH and D‐Glc reacted in a quantitative manner and resulted in the UV active 2,4‐dinitrophenyl hydrazone derivative. After derivatization, the sample was separated by RP‐HPLC on a Phenomenex Gemini C18 5 µm column using the same water/acetonitrile gradient as mentioned above. The DNPH derivative’s peak area (t R ≈ 12 min) was calculated and compared to D‐Glc concentrations: correlation was determined using a five‐point calibration standard of D‐Glc from the 0.1–0.4 g l−1 concentration range.
NMR spectroscopy
15N‐labelled miniproteins were dissolved in water (c ~ 1 mM), and 10% D2O with 1% DSS standard was added and pH was adjusted to 7.0. The NMR measurements were carried out at T = 25 °C on a Bruker Avance III 700 MHz spectrometer equipped with a 5 mm Prodigy TCI H&F–C/ND, z‐gradient probehead operating at 700.05 MHz for 1H nucleus. Temperature calibration was performed using a standard methanol solution. All chemical shifts were referenced taking account the internal 1H‐resonance of DSS standard. 1H‐NMR and 1H,1H‐NOESY (mixing time: 150 ms) spectra were collected. All spectra were processed with the Bruker Topspin software.
Conflict of interest
None declared.
Supporting information
Supplementary material S1. Fermentation protocols.
Supplementary material S2. chromatogram and HPLC profile of 15 N‐E5 miniprotein.
Acknowledgements
NMR spectrometer measurement time (700 MHz Bruker) was courtesy of MedInProt Grant Facilitating Access to Instruments from the Hungarian Academy of Sciences. This research project was supported by the European Union and the State of Hungary and financed by the European Regional Development Fund (VEKOP‐2.3.2‐16‐2017‐00014) grant of the NKFIH of the Hungarian Academy of Sciences. The authors gratefully acknowledge Tünde Tóth and Dániel Fülöp for surface tension and PM‐IRRAS measurements and Dóra Karancsiné Menyhárd for the fruitful discussions regarding the wording. This work was completed in the ELTE Thematic Excellence Programme supported by the Hungarian Ministry for Innovation and Technology. Project no. 2018‐1.2.1‐NKP‐2018‐00005 has been implemented with the support provided from the National Research, Development and Innovation Fund of Hungary, financed under the 2018‐1.2.1‐NKP funding scheme.
Microbial Biotechnology (2021) 14(3), 1107–1119
Funding information
This research project was supported by the European Union and the State of Hungary and financed by the European Regional Development Fund (VEKOP‐2.3.2‐16‐2017‐00014) grant of the NKFIH of the Hungarian Academy of Sciences.
References
- Baeshen, N.A. , Baeshen, M.N. , Sheikh, A. , Bora, R.S. , Ahmed, M.M.M. , Ramadan, H.A.I. , et al. (2014) Cell factories for insulin production. Microb Cell Factories 13: 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, E.G. , Bartlett, G.J. , Porter Goff, K.L. , and Woolfson, D.N. (2017) Miniprotein design: past, present, and prospects. Acc Chem Res 50: 2085–2092. [DOI] [PubMed] [Google Scholar]
- Blaudez, D. , Buffeteau, T. , Cornut, J.C. , Desbat, B. , Escafre, N. , Pezolet, M. , and Turlet, J.M. (1993) Polarization‐modulated FT‐IR spectroscopy of a spread monolayer at the air/water interface. Appl Spectrosc 47: 869–874. [Google Scholar]
- Bogomolovas, J. , Simon, B. , Sattler, M. , and Stier, G. (2009) Screening of fusion partners for high yield expression and purification of bioactive viscotoxins. Protein Expr Purif 64: 16–23. [DOI] [PubMed] [Google Scholar]
- Bommarius, B. , Jenssen, H. , Elliott, M. , Kindrachuk, J. , Pasupuleti, M. , Gieren, H. , et al. (2010) Cost‐effective expression and purification of antimicrobial and host defense peptides in Escherichia coli . Peptides 31: 1957–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, M. , Huang, Y. , Sakaguchi, K. , Clore, G.M. , Gronenborn, A.M. , and Craigie, R. (1998) An efficient and cost‐effective isotope labeling protocol for proteins expressed in Escherichia coli. J Biomol NMR 11: 97–102. [DOI] [PubMed] [Google Scholar]
- Dovidchenko, N.V. , Leonova, E.I. , and Galzitskaya, O.V. (2014) Mechanisms of amyloid fibril formation. Biochemistry 79: 1515–1527. [DOI] [PubMed] [Google Scholar]
- Gaimster, H. , and Summers, D. (2015) Regulation of indole signalling during the transition of E. coli from exponential to stationary phase. PLoS One 10: e0136691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould, A. , and Camarero, J.A. (2017) Cyclotides: overview and biotechnological applications. ChemBioChem 18: 1350–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, K. , Horváth‐Szanics, E. , Hajós, G.y. , and Kiss, É. (2008) Surface and interfacial properties of water‐soluble wheat proteins. Colloids Surf Physicochem Eng Asp 319: 180–187. [Google Scholar]
- Iqbal, M.Z. , and Novalin, S. (2009) Analysis of formose sugar and formaldehyde by high‐performance liquid chromatography. J Chromatogr A 1216: 5116–5121. [DOI] [PubMed] [Google Scholar]
- Klopp, J. , Winterhalter, A. , Gébleux, R. , Scherer‐Becker, D. , Ostermeier, C. , and Gossert, A.D. (2018) Cost‐effective large‐scale expression of proteins for NMR studies. J Biomol NMR 71: 247–262. [DOI] [PubMed] [Google Scholar]
- Kohno, T. , Kusunoki, H. , Sato, K. , and Wakamatsu, K. (1998) A new general method for the biosynthesis of stable isotope‐enriched peptides using a decahistidine‐tagged ubiquitin fusion system: An application to the production of mastoparan‐X uniformly enriched with 15N and 15N/13C. J Biomol NMR 12: 109–121. [DOI] [PubMed] [Google Scholar]
- Merrifield, R.B. (1964) Solid‐phase peptide synthesis. III. An improved synthesis of Bradykinin. Biochemistry 3: 1385–1390. [DOI] [PubMed] [Google Scholar]
- Monod, J. (1942) Recherches sur la croissance des cultures bactériennes. Paris, France: Hermann & cie. [Google Scholar]
- Neidigh, J.W. , and Andersen, N.H. (2002) Peptide conformational changes induced by tryptophan–phosphocholine interactions in a micelle. Biopolymers 65: 354–361. [DOI] [PubMed] [Google Scholar]
- Panavas, T. , Sanders, C. , and Butt, T.R. (2009) SUMO fusion technology for enhanced protein production in prokaryotic and eukaryotic expression systems. In SUMO Protocols. Methods in Molecular Biology. Ulrich, H.D. (ed). Totowa, NJ: Humana Press, pp. 303–317. [DOI] [PubMed] [Google Scholar]
- Ross, A. , Kessler, W. , Krumme, D. , Menge, U. , Wissing, J. , van den Heuvel, J. , and Flohé, L. (2004) Optimised fermentation strategy for 13C/15N recombinant protein labelling in Escherichia coli for NMR‐structure analysis. J Biotechnol 108: 31–39. [DOI] [PubMed] [Google Scholar]
- Rovó, P. , Farkas, V. , Stráner, P. , Szabó, M. , Jermendy, Á. , Hegyi, O. , et al. (2014) Rational design of α‐Helix‐stabilized Exendin‐4 analogues. Biochemistry 53: 3540–3552. [DOI] [PubMed] [Google Scholar]
- Rovó, P. , Stráner, P. , Láng, A. , Bartha, I. , Huszár, K. , Nyitray, L. , and Perczel, A. (2013) Structural insights into the Trp‐Cage folding intermediate formation. Chemistry 19: 2628–2640. [DOI] [PubMed] [Google Scholar]
- Searle, P.L. (1984) The berthelot or indophenol reaction and its use in the analytical chemistry of nitrogen. A review. Analyst 109: 549–568. [Google Scholar]
- Shanmukh, S. , Biswas, N. , Waring, A.J. , Walther, F.J. , Wang, Z. , Chang, Y. , et al. (2005) Structure and properties of phospholipid–peptide monolayers containing monomeric SP‐B1–25: II. Peptide conformation by infrared spectroscopy. Biophys Chem 113: 233–244. [DOI] [PubMed] [Google Scholar]
- Stráner, P. , Taricska, N. , Szabó, M. , Tóth, K.G. , and Perczel, A. (2016) Bacterial expression and/or solid phase peptide synthesis of 20–40 amino acid long polypeptides and miniproteins, the case study of Class B GPCR ligands. Curr Protein Pept Sci 17: 147–155. [DOI] [PubMed] [Google Scholar]
- Studts, J.M. , and Fox, B.G. (1999) Application of fed‐batch fermentation to the preparation of isotopically labeled or selenomethionyl‐labeled proteins. Protein Expr Purif 16: 109–119. [DOI] [PubMed] [Google Scholar]
- Taricska, N. , Bokor, M. , Menyhárd, D.K. , Tompa, K. , and Perczel, A. (2019) Hydration shell differentiates folded and disordered states of a Trp‐cage miniprotein, allowing characterization of structural heterogeneity by wide‐line NMR measurements. Sci Rep 9: 2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taricska, N. , Horváth, D. , Menyhárd, D.K. , Ákontz‐Kiss, H. , Noji, M. , So, M. , et al. (2020) The route from the folded to the amyloid state: exploring the potential energy surface of a drug‐like miniprotein. Chemistry 26: 1968–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson, P.T.F. , Roth, J.F. , Haddingham, T. , and Watts, A. (2000) Expression and purification of recombinant neurotensin in Escherichia coli . Protein Expr Purif 19: 271–275. [DOI] [PubMed] [Google Scholar]
- Yang, H. , Yang, S. , Kong, J. , Dong, A. , and Yu, S. (2015) Obtaining information about protein secondary structures in aqueous solution using Fourier transform IR spectroscopy. Nat Protoc 10: 382–396. [DOI] [PubMed] [Google Scholar]
- Zorko, M. , and Jerala, R. (2010) Production of recombinant antimicrobial peptides in bacteria. In Antimicrobial Peptides. Methods in Molecular Biology. Giuliani, A. , and Rinaldi, A.C. (eds). Totowa, NJ: Humana Press, pp. 61–76. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material S1. Fermentation protocols.
Supplementary material S2. chromatogram and HPLC profile of 15 N‐E5 miniprotein.