

Summary
Aquatic environments are the recipients of many sources of environmental stress that trigger both local and global changes. To evaluate the associated risks to organisms and ecosystems more sensitive and accurate strategies are required. The analysis of the microbiome is one of the most promising candidates for environmental diagnosis of aquatic systems. Culture‐independent interconnected meta‐omic approaches are being increasing used to fill the gaps that classical microbial approaches cannot resolve. Here, we provide a prospective view of the increasing application of these high‐throughput molecular technologies to evaluate the structure and functional activity of microbial communities in response to changes and disturbances in the environment, mostly of anthropogenic origin. Some relevant topics are reviewed, such as: (i) the use of microorganisms for water quality assessment, highlighting the incidence of antimicrobial resistance as an increasingly serious threat to global public health; (ii) the crucial role of microorganisms and their complex relationships with the ongoing climate change, and other stress threats; (iii) the responses of the environmental microbiome to extreme pollution conditions, such as acid mine drainage or oil spills. Moreover, protists and viruses, due to their huge impacts on the structure of microbial communities, are emerging candidates for the assessment of aquatic environmental health.
Aquatic environments are the recipients of many sources of environmental stress that trigger both local and global changes. We provide a prospective view of the increasing application of high‐throughput molecular technologies to evaluate the structure and functional activity of microbial communities in response to changes and disturbances in the environment.

Introduction
Freshwater and marine ecosystems are subjected to many drivers of environmental changes. In most cases, they are associated with human activities, which also result in global changes, such as an increase in temperature, acidification, salinity or reduction of the oxygen content. In other cases, the changes at local or regional scales are a consequence of the human population growth and the increasing use of littoral areas. Impacting human activities have risen especially in coastal and shelf areas, which are the most productive areas of the seas (Ramírez et al., 2018). One of the main consequences of the human activities is the release to the ocean and coastal areas of chemical stressors such as persistent organic compounds (including PAHs, PCBs and pesticides), metals and emerging pollutants (e.g. pharmaceuticals ‐antibiotics, anti‐inflammatory, anti‐depressives‐, personal care products, nanomaterials, micro‐/nano‐plastics, marine biotoxins; Sauvé and Desrosiers, 2014; Geissen et al., 2015). Due to lack of sanitation or to inefficient treatments, a high proportion of pollutants (e.g. antibiotics) from urban or industrial wastewater treatment plants (WWTP) are directly discharged into aquatic systems (Geissen et al., 2015).
Given the thousands of chemicals released into the aquatic environment, major efforts are necessary to evaluate the associated risks to organisms and ecosystems (Fernández‐Cisnal et al., 2017). Additionally, in the natural environment, pollutants are not found individually, and they occur as complex mixtures of hazardous interacting substances. The action of these compounds should thus be assessed considering their cumulative effects and interactions (European Marine Board, 2019). Besides chemical stress, multi‐stress related to other environmental issues, many of them as result of climate change (e.g. acidification, increase in temperature and UV), can jointly affect the resiliency and health of aquatic ecosystems (Holmstrup et al., 2010; Bejaoui et al., 2020). To assess the impact of pollution and other sources of environmental stress, a relation between the occurrence and the effect should be measured. Aquatic ecotoxicological studies need to find appropriate model organisms (used as biomonitors) that respond sensitively to complex environmental threats (Zhou et al., 2008; Gago‐Tinoco et al., 2014; Parmar et al., 2016; Fernández‐Cisnal et al., 2017). Invertebrates, fish, mammals and plants have been usually used as bioindicator organisms of pollution (Burger, 2006; Zhou et al., 2008; Li et al., 2019).
The analysis of the microbiome is one of the most promising candidates for environmental diagnosis of aquatic ecosystems due to: (i) the ubiquitous presence of microbial organisms in any environmental compartment, (ii) their direct and immediate contact with the pollutants and other stressors, (iii) their sensitive response to environmental stresses expressed by changes in their structure, diversity and functional activity, which affects matter and flow energy in the ecosystems, and (iv) the ongoing increasing availability of high‐throughput techniques to elucidate those changes in both the microbial community structure and its metabolic patterns (Caruso, 2013; Bouchez et al., 2016; Wang et al., 2016).
Use of microbial parameters to monitor environmental pollution started in the 80s, mainly to analyse the quality of drinking water, as most aquatic pollution was produced by unprocessed faecal discharges or uncontrolled effluents from WWTP (Bae and Park, 2014). In fact, the quality of drinking water is routinely monitored by the determination of enterobacteria (Caruso, 2013; Jang et al., 2017; Gorski et al., 2019). Traditionally, bacteria levels in such waters have been quantified by the cultivation of samples under laboratory conditions or even microscopic counting, but these methodologies, although sensitive, are relative slow and do not allow for a quick detection and, thus, an immediate response to remediate the pollution events (Storey et al., 2011; Caruso, 2013; Gorski et al., 2019). Several bioassays to measure pollution have been developed based on the changes in physicochemical parameters triggered by one or multiple toxic chemicals. Common determinations include O2 consumption/CO2 production, bioluminescence, activity of microbial enzymes, or the transformation of elements such as carbon, sulphur, or nitrogen (Tothill and Turner, 1996; Bae and Park, 2014; Hassan et al., 2016). Whole‐cell biosensors devices have been designed to detect the presence of biological analytes, including pollutants, using living organisms that respond in some measurable and predictable way. Although these devices are being used widely to determine aquatic pollution, they are not pollutant‐specific, and their determinations can be biased by the presence of growth inhibitory chemicals in the samples or by the microbial biosensor profile (OECD, 2010; Hassan et al., 2016; Gorski et al., 2019). Recent methodological innovations to enumerate enteric pathogens in water include sensitive nucleic acid‐based methods, being quantitative PCR (qPCR) the most often used. Nevertheless, these methodologies also have their drawbacks, the main one being the detection of false positives due to the persistence of dead cells or naked DNA in the water samples. Finally, metagenomics has significant potential for bacterial quantification, although the application of this technique for water testing is still under development (Gorski et al., 2019).
One of the main issues in ecotoxicology is the development and use of much more sensitive tools for the in‐field monitoring of the impact and sublethal effects of pollutants and other sources of environmental stress. Advances in this area are undoubtedly related to the technical progress of specific methodologies. Next, we will review the use of advanced molecular methodologies, particularly those designed to fill the gaps that classical microbial approaches cannot resolve, to assess the main drivers and pressures on aquatic ecosystems.
Application of high‐throughput molecular microbiological tools
Considering that uncultured microorganisms represent the great majority of the planet´s biodiversity, culture‐independent methods are essential to understand the genetic diversity, population structure, metabolic activities, and environmental roles of microbes (Riesenfeld et al., 2004; Lloyd et al., 2018). In this sense, the emergence and increasingly extensive use of new high‐throughput sequencing (HTS) technologies, together with the rapidly growing of metagenomic sequence data from different environments, and the application of new user‐friendly bioinformatics and statistical tools, are facilitating our understanding of environmental microbial communities (Riesenfeld et al., 2004; Glöckner and Joint, 2010; Heidelberg et al., 2010; Delmont et al., 2011; Barberán et al., 2012; Neelakanta and Sultana, 2013; Coutinho et al., 2015; Zuñiga et al., 2017; Coutinho et al., 2018; Tangherlini et al., 2018). For instance, when comparing the diversity and distribution of 77 metagenomes, the microbial profiles from different ecosystems (e.g. oceans, coral atolls, deep oceans, Antarctic aquatic environments, Arctic snows, soils, hypersaline sediments, sludge, microbial cell biofilms, acid mine biofilms, polluted air and animal microbial populations) were found to be clearly different (Delmont et al., 2011). Thus, environmental responses can be evaluated by comparing the metagenomes from different conditions, for example their involvement in, and response to, climate change and environmental stress, their response to pollutants and biodegradation capacities, as well as the risks they represent to human health.
The integration of culture‐independent, interconnected omic approaches can provide detailed information concerning: how microbial communities assemble and interact, their predominant metabolic activities, their progress over time, and their responses to environmental perturbations (Fig. 1; Ram et al., 2005; Simon and Daniel, 2011; Hettich et al., 2012; Zarraonaindia et al., 2013; Franzosa et al., 2015; Tan et al., 2015). While metagenomics identifies the potential function of a microbial community in the environment, metatranscriptomics determines which microbes are active and which genes are transcribed, while metaproteomics informs of which proteins are actually expressed; finally, metabolomics may be used to evaluate changes in metabolic fluxes and metabolite levels which are the final result of complex interactions (Wilmes and Bond, 2009; Siggins et al., 2012; Zarraonaindia et al., 2013; Wang et al., 2014; Aguiar‐Pulido et al., 2016; Herbst et al., 2016; Wang et al., 2016; Zuñiga et al., 2017).
Fig. 1.
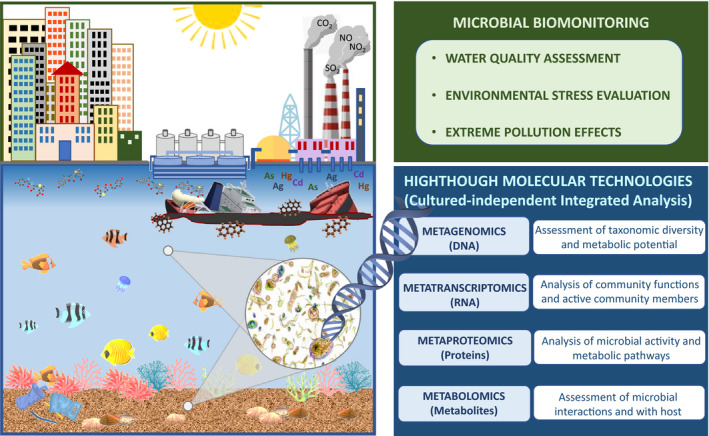
Application of high‐throughput molecular technologies for the microbial biomonitoring of aquatic environments.
Below we provide a prospective view of the increasing application of high‐throughput molecular technologies to evaluate the structure and activity of microbial communities in response to changes and disturbances in the environment, mostly of anthropogenic origin (Fig. 1). In relation to this, recently, the Microbiome Stress Project, an open access database of environmental and host‐associated 16S rRNA, has collected cross‐study analyses of microbial community responses to stressors (Rocca et al., 2019). Here, we address the application of HTS technologies to microbial water quality assessment, highlighting the incidence of antimicrobial resistance as an increasingly serious threat to global public health. Next, we review approaches trying to evaluate the crucial role of microorganisms and their complex relationships with the ongoing climate change, and other environmental stress conditions. Finally, we will assess how the environmental microbiome responds to extreme pollution conditions, such as acid mine drainage or oil spills. The unprecedented advances in these areas represent a valuable source of information that is increasingly being incorporated into the improvement of bioprocesses and environmental engineering applications (e.g. wastewater treatment, bioremediation) to achieve a more sustainable environment (Czaplicki and Gunsch, 2016; Techtmann and Hazen, 2016). Table 1 includes a bibliographic compilation of the application of HTS technologies to microbial water quality assessment.
Table 1.
Most relevant examples of the application of high‐throughput molecular technologies to microbial water quality assessment.
| Quality assessment tasks | HTS technologies used | Main goals | References |
|---|---|---|---|
| Antimicrobial resistance | Metagenomic (Illumina sequencing) analysis | The trend of the total ARG abundances in different environments matched well with the levels of anthropogenic impacts | Li et al. (2015) |
| WWTP effluents substantially influence on the dispersal of microorganisms and ARGs in receiving aquatic sediments | Chu et al. (2018) | ||
| In WWTP, removal efficiency of ARGs in anaerobic sludge treatment is much less efficient than the sewage treatment | Yang et al. (2014) | ||
| 16S rRNA, metagenomic & metatranscriptomic (Illumina sequencing) analyses | Conventional treatment process within WWTP strongly influences resistance genes and their transcriptional activities | Ju et al. (2019) | |
| Metagenomic (Oxford Nanopore & Illumina sequencing) analyses | Most ARGs detected in all compartments of WWTP are carried by plasmids, which have a significant role in facilitating the survival and persistence of multidrug resistant bacteria | Che et al. (2019) | |
| Relationships with climate change and environmental stress | Metagenomic (Sanger shotgun sequencing) and metaproteomic (MS/MS) analyses | Metabolic components (nitrification, anaerobic ammonium oxidation, denitrification, and inorganic C fixation) were differentially expressed in ocean OMZ, and covaried with ubiquitous microbes | Hawley et al. (2014) |
| 16S rRNA & metagenomic (Illumina sequencing) analyses | Free‐living microbial communities in coral reefs have a high potential to infer environmental parameters due to their environmental sensitivity and predictability | Glasl et al. (2019) | |
| Microbiome flexibility facilitates rapid responses of corals to environmental changes (e.g. high temperature) and increases stress tolerance | Ziegler et al. (2017) | ||
| Corals microbiota plays a critical role in the defence of their host against pathogens | Grottoli et al. (2018) | ||
| Metagenomic (FLX Titanium bar‐coded pyrosequencing) analysis | Reduced pH and oil contamination can adversely affect the structure and functioning of sediment benthic communities, including bacteria | Coelho et al. (2015, 2016) | |
| qPCR, 16S rRNA & metagenomic (Illumina sequencing) analyses | Marine benthic microorganisms are susceptible to changes in ocean carbonate chemistry and sweater temperature | Currie et al. (2017) | |
| 16S rRNA & metagenomic (FLX pyrosequencing) analyses | Exposure to oil‐contaminated sediments led to distinct shifts in commensal bacterial population structures in the gill and intestine of the southern flounder fish | Brown‐Peterson et al. (2015) | |
| Metagenomic (Illumina sequencing) analysis | The endogenous xenobiotic metabolism of the Manila clam hepatopancreas is impacted by its microbiome, which is a key component in the host response to chemical stress | Milan et al. (2018) | |
| 16S rRNA & metagenomic (Ion Torrent sequencing) analyses | TiO2 nanoparticles shift the haemolymph microbiome composition of the bivalve Mytilus galloprovincialis, which may affect its immune system | Auguste et al. (2019) | |
| Responses to extremely polluted environments | Bar‐coded 16S rRNA & metagenomic (GS‐FLX Titanium pyrosequencing) analyses | Solution pH is the major factor explaining microbial community differences in AMD environments. Microbial communities develop a critical strategy of stress adaptation to highly acidic/metal‐rich conditions, by increasing the expression of genes involved in many key functions | Kuang et al. (2013, 2016) |
| Metagenomic & metatranscriptomic (sequencing) analyses | Gene transcriptional profiles of microorganisms, as response and adaptation mechanisms, are closely related to the physicochemical characteristics of AMD sites | Chen et al. (2015) | |
| Bar‐coded 16S rRNA, metagenomic & metatranscriptomic (sequencing) analyses | In prokaryotes, multiple strategies for resource acquisition and energy generation, and mechanisms of adaptation and response to AMD environmental stress, are revealed. | Hua et al. (2015) | |
| 16S rRNA, metagenomics & metatranscriptomic (454 GS‐FLX Titanium sequencing) analyses | The identity of metabolically active microbes from DWH oil spill and their roles in petroleum consumption are revealed | Rivers et al. (2013) | |
| 16S rRNA & metagenomic (Illumina sequencing) analyses | Surface sediment‐associated microbes can account for rapid depletion of aliphatic and simple aromatics of the DWH oil spill, while PAHs are more recalcitrant to degradation | Mason et al. (2014) | |
| After DWH oil spill, early microbial responders which degrade AHs were replaced by populations capable of PAHs decomposition, and at longer times a typical beach community was re‐established | Rodriguez‐R et al. (2015) |
Used abbreviations: AHs, aliphatic hydrocarbons; AMD, acid mine drainage; ARGs, antibiotic resistance genes; DWH, Deepwater Horizon; HTS; high‐throughput sequencing; OMZ, oxygen minimum zones; PAHs, polycyclic aromatic hydrocarbons; WWTP, wastewater treatment plants.
Water quality assessment: antimicrobial resistance as a global threat to public health
The prevalence and dissemination of antimicrobial resistance (AMR) is an increasingly serious threat to global public health (WHO, 2015; Qiao et al., 2018; Thomas et al., 2020), that has turned into an emergency in low‐income countries (Bastaraud et al., 2020). Consequently, a significant reduction in the effectiveness of antibiotics and other antimicrobial agents (antifungals, antivirals, antimalarials, and anthelmintics) threatens our ability to treat common infectious diseases, resulting in prolonged illness, disability, and death (Wright, 2010; Almakki et al., 2019). Over the past two decades, the misuse and overuse of antimicrobials have accelerated this process (Almakki et al., 2019). Antibiotics are poorly metabolized by humans and animals and are readily excreted, entering the environment through wastewater and manure (Qiao et al., 2018). Several studies have reported on the co‐selection of antibiotic resistant bacteria (ARB) and antibiotic resistance genes (ARGs) in contaminated environments (Almakki et al., 2019; Chen et al., 2019; Thomas et al., 2020). Antibiotic production plants, livestock farms and aquaculture, and discharges from wastewater treatment plants (WWTP) contribute to the increase of ARBs and ARGs reported in aquatic environments (Qiao et al., 2018). Thus, ARGs have been detected in natural water bodies and drinking water, soils and even deep ocean sediments (Yang et al., 2014), and their increased prevalence as a result of human activities has led to their characterization as an emerging environmental contaminant of growing concern (Pruden et al., 2006; Yang et al., 2014). Table 2 contains a compilation of the ARGs detected in WWTP, showing that genes encoding resistance to all classes of antibiotics are widespread in influent, effluent and activated sludge of WWTP. A detailed review of available literature on the methods of analysis of the different genes is included in Pazda et al., 2019. The state of the art of the environmental antibiotic resistance in China has also been recently reviewed (Qiao et al., 2018). DNA‐based techniques are being increasingly used to detect and quantify resistant genes in environmental samples. Compared to the former, amplification‐based approaches (PCR, qPCR), which target a limited number of primers‐available ARGs, HTS‐based analysis surveys a more complete metagenomic profiling of ARGs in environmental samples (Qiao et al., 2018; Sukhum et al., 2019). The development of annotated and manually curated databases of ARGs (e.g. ARBD, CARD, SNC‐ARBD) has allowed for the rapid screening of metagenomic data sets for resistance (Yang et al., 2014; Li et al., 2015; Sukhum et al., 2019). Thus, comparative metagenomics has shown the spread of ARGs in river environments because of the selective pressure resulting from antibiotic use (Yang et al., 2014; Chen et al., 2019). Anthropogenic activities contributed > 93% for sulphonamide, beta‐lactam, and aminoglycoside ARGs (Chen et al., 2019). Shotgun metagenomics also showed a higher abundance of ARGs in the Lake Michigan sediments identical to those present in the effluents of two different WWTP that discharge into the lake; most abundant genes (i.e., strA, dfrE, acrB, adeJ, mexB and semeE) were attributed to organisms belonging to Helicobacteraceae (Helicobacter), Legionellaceae (Legionella), Moraxellaceae (Acinetobacter and Moraxella), and Neisseriaceae (Neisseria) (Chu et al., 2018). An HTS‐based metagenomic approach was also used to investigate the wide‐spectrum profiles of ARGs in 50 samples from 10 environments. Network analysis showed that the trend of total ARGs abundances matched well with the levels of anthropogenic impacts on the different environments. Based on the co‐occurrence pattern revealed by network analysis, tetM and aminoglycoside resistance protein were proposed as indicators to evaluate the quantity of other 23 co‐occurring ARGs in multiple environmental samples. Five bacterial genera (Blautia, Clostridium, Enterococcus, Bacteroides and Escherichia) and one archaea (Methanobrevibacter) were speculated as the possible ARGs host (Li et al., 2015).
Table 2.
Antibiotic resistance genes detected in wastewater treatment plants (Compiled from Rizzo et al., 2013; Pazda et al., 2019).
| Antibiotic class | Mechanism of action | Mechanism of bacterial resistance | Gene name a |
|---|---|---|---|
| Aminoglycosides | Inhibiting protein synthesis |
Drug modification by adenylation Drug modification by phosphorylation |
aad[A1, A2, A13, B] aph[A, A‐3, A‐6, 2]; str[A, B]; strB |
| Beta‐lactams | Interfering with the bacterial cell wall biosynthesis by inactivation of penicillin‐binding proteins |
Penicillin‐binding protein Inactivation by beta‐lactamases (Drug degradation) |
mecA amp[C, R]; bla[CIT, CMY, CTX, FOX, GES, IMP, NPS, OXA, PER, PSE, SHV, TEM, TLA, VEB, VIM]; cfx[A, A3] |
| Glycopeptides | Interfering with cell wall biosynthesis | Produces a modified peptidoglycan pentapeptide | vanA |
| Macrolides | Inhibiting early stages of protein synthesis (binding to the 50S bacterial ribosome subunit) |
Methylation of 23S rRNA target side Cleavage of the drug lactone ring Inactivation of macrolide phosphotransferases Active pumping of the drug |
erm[A, B, C, F, O] ere[A, A2, B] mph[(A), (B), BM] mef[A, E]; mel; msrA |
| Quinolones | Inhibiting bacterial DNA replication enzymes (DNA gyrase & topoisomerase IV) |
Modification of target enzymes DNA gyrase and topoisomerase IV protection Modification by acetylation |
gyr[A, B]; parC qnr[A, A3, B, B1, B2, B4, B5, D, S, S1, S2, VC] aacA6‐ib‐cr |
| Sulphonamides | Inhibiting the dihydropteroate synthase, enzyme involved in the folic acid synthesis | Competitive inhibitors of dihydropteroate synthase, enzyme involved in folic acid synthesis | sul[1, 2, 3, A] |
| Tetracyclines | Inhibiting bacterial protein synthesis (binding to the 30S ribosomal subunit and preventing the aminoacyl tRNA association) |
Ribosome protection Drug export by membrane associated proteins Enzymatic inactivation of the drug |
ortA, tet[B(P), M, O, Q, S, T, W, X] tet[A, A(C), A(P), B, C, D, E, G, H, J, K, L, V, Y, Z, 31, 35, 36, 39] tetX |
| Trimethoprim | Inhibiting Dihydrofolate reductase (DHFR) | Increased synthesis of DHFR | dfr[II, V, XIII, 13, 16, 17, A19, B2, A20, A3, A12, A13, A21, A22, A33, B1, B5, B6, B8, D, E]; dhfr[V, VII, VIII, IX, XII, XV, A1, A14] |
| Multidrug | Multidrug resistance | Accumulation of many genes encoding resistance to a single drug and multidrug efflux pumps operation | acr[B, D]; ade[A, J]; amrB; mdt[F, G, H, N, O]; mex[B, D, F, I, W, Y]; norM; orf11; qac[B, EΔ1, EΔ1‐01, F, G2, H]; sedY; sme[B, E] |
For simplicity, gene variations are presented within square brackets.
Metagenomic sequencing has shown that mobile genetic elements‐associated ARGs dominated the resistome in WWTP (Chu et al., 2018; Che et al., 2019; Ju et al., 2019; Pazda et al., 2019). Thus, by combining Oxford Nanopore and Illumina sequencing it was shown that most of the ARGs detected in WWTP were carried by plasmids, and integrative and conjugative elements (ICEs). Particularly, tetA and sul1 genes carried by plasmids, and cfxA, mefA/E, tetQ and tetM genes carried by ICEs, had a persistent prevalence. Remarkably, four potential antimicrobial‐resistant pathogens (i.e. Enterococcus faecium, Klebsiella pneumoniae, Acinetobacter baumannii, and Pseudomonas aeruginosa), harbouring a high diversity of ARGs, were identified (Che et al., 2019). Conventional biological treatment processes, such as activate sludge and anaerobic digestion, do not only no efficiently reduce ARGs, but these can even proliferate throughout the biological treatment stages and their abundance increase in the effluent (Rizzo et al., 2013; Yang et al., 2014; Mao et al., 2015; Rafraf et al., 2016; Ju et al., 2019; Pazda et al., 2019; Yin et al., 2019). WWTP are thus recognized as hotspots of vertical and horizontal gene transfer of ARGs (Che et al., 2019; Pazda et al., 2019). The amount of ARBs and ARGs discharged by WWTP into the environment depends on the bacterial biomass remaining in the final effluent (Ju et al., 2019). Thus, urgent measures are necessary to limit the use of antimicrobials in medicine and animal production, and to increase the efficiency of wastewater treatments (Rizzo et al., 2013; Pazda et al., 2019). In that regard, more advanced technologies such as Advanced Oxidation Processes (AOPs) have been developed to effectively degrade antibiotics and improve the removal of ARBs and ARGs in WWTP (Alexander et al., 2016; Pazda et al., 2019; Rodriguez‐Chueca et al., 2019; Shen et al., 2019; Wang and Zhuan, 2020).
Several studies have shown that heavy metal contamination can play an important role in the proliferation of antibiotic resistance by inducing ARGs, and metal‐resistant genes (MRGs), via co‐selection mechanisms (Seiler and Berendonk, 2012; Mao et al., 2015; Li et al., 2017; Ju et al., 2019; Thomas et al., 2020). Thus, metagenomic analysis, using 16S rRNA gene amplicon sequencing, showed significant differences in the relative abundance and diversity of certain ARGs (and MRGs) in metal/radionuclide contaminated soils (Thomas et al., 2020). Recently, flow cytometry and 16S rRNA sequencing have shown that silver nanoparticles (AgNPs) clearly affect the microbial community structure and suggest a potential negative impact on WWTP functions (Guo et al., 2019). Finally, given the increased presence of microplastics as aquatic emerging water pollutants, their role as potential vectors for harmful microbes and propagation of ARBs and ARGs is a matter of growing concern (Bastaraud et al., 2020; Song et al., 2020; Zhang et al., 2020).
Microbiome relationships with global climate change and environmental stress
Global climate change due to human‐induced stressors (e.g. increased sea surface temperature, ocean acidification and reduced oxygen content, increased ultraviolet radiation, reduced salinity) has the potential to affect marine ecosystems adversely (Coelho et al., 2013; FAQ, 2017; Cavicchioli et al., 2019). Different microbial components respond with different sensitivity to changes in ocean temperature, oxygen concentration, pH and food supply, providing evidence that global change will affect different groups of microorganisms differentially (Danovaro et al., 2016; Currie et al., 2017; Danovaro et al., 2017). Comparative analysis of bacteria and archaea based on qPCR allowed to conclude that climate change will primarily affect deep‐sea benthic archaea, with important consequences concerning global biogeochemical cycles (Danovaro et al., 2016).
Climate‐induced warming of the upper ocean is contributing to the reduction of the oxygen content of the global ocean and to the expansion of oxygen minimum zones (OMZ) (Ulloa et al., 2012; Hawley et al., 2014; Khan et al., 2018). Multi‐omic molecular approaches have shown that diverse microbial communities are contributing there to major losses of fixed nitrogen and the production of climate active greenhouse gases (Ulloa et al., 2012; Hawley et al., 2014; Hawley et al., 2017). The combined application of metagenomics and metaproteomics showed that the expression of metabolic pathway components for nitrification, anaerobic ammonium oxidation (anammox), denitrification, and inorganic carbon fixation was differentially expressed in these areas, and covaried with ubiquitous OMZ microbes (e.g. Thaumarchaeota, Nitrospina, Nitrospira, Planctomycetes; Hawley et al., 2014).
In a climate change scenario, increased sea surface temperatures and ocean acidification also contribute significantly to the ongoing decline of coral reef ecosystems. Increasingly serious consequences are predicted, given that coral reefs are among the most biologically diverse and economically important ecosystems (Hoegh‐Guldberg et al., 2007; Sharp and Ritchie, 2012). Within corals, microbes play a critical role, ultimately governing the overall health and resilience of these reef systems and influencing their response to environmental changes (Sharp and Ritchie, 2012; Chiarello et al., 2020). HTS methods have allowed the study of the composition of coral microbiomes, its spatial‐temporal variability, plus its role in corals´ health and in response to environmental changes, in a broad range of coral specimens (Sharp and Ritchie, 2012; Hernandez‐Agreda et al., 2017). Thus, recently, a comprehensive microbial database of multiple coral reef microbiomes was generated by 16S rRNA gene sequencing. When this database was coupled to environmental parameters, seawater microbiomes were shown to be good indicators since they respond in a very sensitive and predictable way to the environmental perturbations (Glasl et al., 2019). Network analysis of metagenomic data has shown that microbial species commonly found in corals play potential roles in host nutrient metabolism, carbon, nitrogen and sulphur cycles, and host detoxification (Wegley et al., 2007; Sharp and Ritchie, 2012; Cai et al., 2018). The response to environmental changes differs according to the composition of the microbial community and suggests a key role of the microbiome in the acclimatization/adaptive responses of the coral host to the stress conditions (Ziegler et al., 2017; Voolstra and Ziegler, 2020). It is no wonder that stress‐tolerant corals have a more stable and diverse microbiome and are also more physiologically resilient to ocean warming and acidification (Grottoli et al., 2018). Microbiota that inhabits corals also play a critical role in the defence of their host against pathogens. Thus, the presence of a high number of genes involved in antibacterial compound biosynthesis has been detected in the metagenomes from corals (Thurber et al., 2009). Additionally, high resolution mass spectrometry imaging (MALDI‐MSI) has allowed the detection of the metabolites responsible for the antifungal properties of coral microbiota (Fig. 2; Moree et al., 2013; Moree et al., 2014).
Fig. 2.
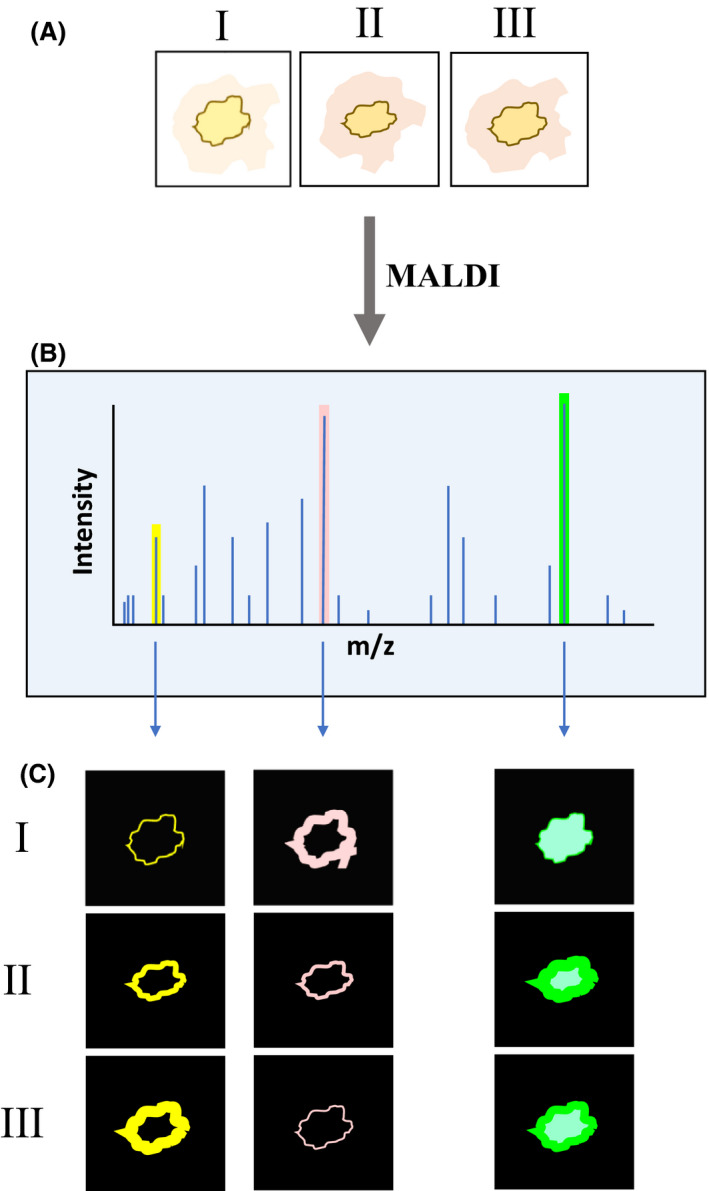
Overview of MALDI‐MSI applied to the study of metabolic exchange interactions of microorganisms.
A. Schematic representation of microorganisms (e.g. bacterial, and fungal species isolated from a coral specimen) grown under different conditions (I–III).
B. The average signal from the m/z of all the spectra obtained in the imaging runs.
C. Separate signals of three specific m/z ions obtained in each of the studied environmental conditions. The signals correspond to the different metabolites produced by the bacterial and fungal species at the site of interaction. Metabolites with antifungal properties, antibiotic compounds, and signalling molecules can be identified.
Mass spectrometry imaging (MSI) has been proposed as a general tool to study the metabolic exchange patterns of microorganisms, which may have important implications based on the hypothesis that microbes could have an impact on their environmental niches or receive the impact of environmental changes (Gonzalez et al., 2012; Maloof et al., 2020). In the same way, HTS technologies have been used to study the impacts of different stresses on different tissues´ microbial communities and its associated consequences on the host’s metabolism and health, for example the effects of oil‐contaminated sediments in the gill and intestine of the juvenile southern flounder (Brown‐Peterson et al., 2015), hypoxia in the digestive gland of the eastern oyster (Khan et al., 2018), chemical contamination and seasonal fluctuations in the hepatopancreas of the Manila clam (Milan et al., 2018), and nTiO2 in the haemolymph of a marine bivalve (Auguste et al., 2019). In this sense, it has been shown that host´s microbial abundance/diversity and/or its metabolic activity, are significantly affected by stressful conditions and may contribute to their toxicity (Brown‐Peterson et al., 2015; Khan et al., 2018; Milan et al., 2018), thus compromising host´s health status and susceptibility to diseases (Auguste et al., 2019). Then, to understand how animals respond to chemical stress a key component of such response, the microbiota, must be taken into account (Milan et al., 2018).
Climate change conditions (ocean warming and acidification, increased UV radiation) can alter the speciation, solubility and bioavailability of various pollutants, and, when coexist, they exert combined effects on the functions and services of marine ecosystems (Coelho et al., 2013; Louvado et al., 2015; Zeng et al., 2015; Coelho et al., 2016). For instance, on the one hand acidification increases the biotoxicity of heavy metals and reduces the degradation of organic pollutants, on the other hand heavy metals and oils could decrease the photosynthesis rate and increase the respiration of marine organisms, both leading to ocean acidification (Zeng et al., 2015). Metagenomic analysis showed that the interaction between reduced seawater pH and oil hydrocarbon contamination significantly altered the microbial estuarine community, greatly reducing the relative abundance of oil‐degrading sulphate‐reducing bacteria (order Desulfobacterales, and Desulfosarcina/Desulfococcus clade) (Coelho et al., 2015; Coelho et al., 2016). A reduction in specific archaeal groups and an increase of several hydrocarbonoclastic fungi was also found. All together this points to an impairment of the ability of the ecosystems to recover from acute oil contamination events in future acidified marine environments (Coelho et al., 2016). Significantly, despite its potentially harmful effects, UV‐B radiation appears to minimize the synergistic effects of seawater acidification and oil pollution (Coelho et al., 2015).
Microbial responses to extremely polluted environments
Microorganisms living under extreme conditions have attracted considerable attention because of their peculiar physiology and ecology. In response to extreme pollution conditions, microbes can undergo significant structural and metabolic adaptations to survive (Desai et al., 2010; Bouhajja et al., 2016; Malla et al., 2018).
Acid mine drainage (AMD), the wastewater released by the mining‐industry, represents an extremely harsh environment and a major environmental challenge worldwide. It is characterized by a low pH and high concentrations of metals and sulphates (Méndez‐García et al., 2015; Chen et al., 2016). In the last decade, HTS technologies have extensively increased our knowledge of microbial diversity in AMD ecosystems (Kuang et al., 2013; Chen et al., 2016). Phylogenetic differentiation among microbial communities from diverse AMD sites across Southeast China has been widely studied by metagenomics and metatranscriptomics (Kuang et al., 2013; Chen et al., 2015; Kuang et al., 2016). Unexpectedly, a high microbial diversity was found in these extremely acidophilic conditions, highlighting the high transcriptional activities exhibited by the abundant taxa of Acidithiobacillus, Leptospirillum and Acidiphilium (Chen et al., 2015). Studies identified pH as a strong predictor of relative lineage abundance. Thus, Betaproteobacteria (dominated by the genus Ferrovum) were predominant under moderate pH conditions, whereas Alphaproteobacteria, Euryarchaeota, Gammaproteobacteria and Nitrospira were better adapted to acidic environments (Kuang et al., 2013). Metatranscriptomic analyses have shown that natural acidophilic microbial communities develop elaborate adaptation mechanisms to extreme conditions by regulating the expression of genes involved in many essential key functions. That includes multiple strategies for resource acquisition and energy generation (carbon, nitrogen and phosphate utilization, iron and sulphur oxidation for energy conservation), and responses to environmental stresses (low pH adaptation, resistance to heavy metals and to oxidative stress) (Chen et al., 2015; Hua et al., 2015; Méndez‐García et al., 2015; Kuang et al., 2016). Combining genomic analysis with mass spectrometry‐based proteomic methods, essential activities and metabolic functions in a natural AMD microbial biofilm community were studied. A total of 2,033 proteins were identified, 48% of which are from the dominant biofilm organism, Leptospirillum group II. Significantly, key proteins for the survival in these extreme environments were highly expressed; for example, proteins involved in protein refolding, as chaperones, and in the response to oxidative stress, such as thioredoxins and peroxiredoxins (Ram et al., 2005). Metagenome sequencing and functional gene annotations revealed that heavy metal polluted sediments from the Yellow River (Gansu Province, China) contain a larger number of genes related to DNA recombination and repair, and heavy metal resistance. Additionally, metal polluted sediments had a higher viral abundance, suggesting virus‐mediated heavy metal resistance gene transfer as an adaptation mechanism (Chen et al., 2018). Other studies highlight the increased proportion of metal‐resistant genes and the presence of mobile genetic elements at the metal polluted sites (Gillan et al., 2015; Jacquiod et al., 2018). The remarkable insights into the composition of microorganisms, and their metabolic adaptations, in AMD extreme ecosystems provide significant clues for biotechnological applications (Chen et al., 2016; Villegas‐Plazas et al., 2019), with iron and sulphur‐oxidizers being used in biomining, and the activities of sulphate reducers which are essential in the AMD bioremediation systems (Chen et al., 2016).
Increasing global petroleum demand has brought a dramatic increase in oil spills and leakage accidents all over the world, with devastating consequences for the local marine environment, given the high toxicity of petroleum compounds and its derivatives (Xue et al., 2015). This has promoted intensive research to understand how microbial communities respond to hydrocarbon pollution, with the final goal of providing new biotechnological strategies for the recovery of the affected areas (Atlas and Bragg, 2009; Acosta‐González et al., 2015; Acosta‐González and Marqués, 2016; Bouhajja et al., 2016). The magnitude of accidental spills, such as the Deepwater Horizon oil spill (April 20, 2010; northern Gulf of Mexico), encouraged studies with a high level of resolution using meta‐omic based molecular analyses, as previously reviewed (King et al., 2015; Bouhajja et al., 2016). For instance, after the spill, the responses in the sediments were evaluated, using a combination of 16S rRNA sequencing and screening of shotgun metagenomic data, to characterize the microbial community and to explore the nitrogen and hydrocarbon metabolic potentials, respectively. Uncultured Gammaproteobacterium and Colwellia species were shown to be dominant in the most heavily oil‐impacted seawater and sediments, where pathways of denitrification and degradation of aliphatic and simple aromatic compounds were also abundant (Rivers et al., 2013; Mason et al., 2014). Hydrocarbon degraders were dominated by Gammaproteobacteria, Oceanospirillales in the deep‐sea plume and the Alcanivorax genus in coastal systems and in sea surface oil, while fungi increased in oil salt marshes and beach sands (King et al., 2015). Complex successional taxonomic and functional patterns in coastal sands persisted when evolution was followed for over one year after the spill. Thus, the microbial community first shifted towards generalist populations of oil‐degrading bacteria and then, a year after the disturbance, to a typical beach community, although significantly different from the original one before the spill, with little or no oil hydrocarbon degradation ability and enriched by xenobiotic‐sensitive archaeal taxa (Rodriguez‐R et al., 2015).
New prospects for development: microbes are more than bacteria
Although all microorganisms play key roles in aquatic ecosystems, meta‐omic studies focused on non‐bacterial microbes are scarce due to taxonomic limitations (Pawlowski et al., 2016). Protists can have huge impacts on the structure of microbial communities as they are the most common predators in aquatic environments (Sherr and Sherr, 2002). Furthermore, they are components of the phytoplankton communities that accounts for over half of the global primary production, and, thus, play a vital role in aquatic food webs (Chavez et al., 2011). Additionally, they are generally broadly distributed even within extreme habitats, have short life spans, and respond sensitively to environmental variables (Sherr and Sherr, 2002; Heidelberg et al., 2010). Several non‐HTS‐based studies have proposed protists for pollution biomonitoring. Thus, significant disturbances were successfully linked to salt, trace metals or organic pollution. Significantly, the presence of pollutants produces a decrease in protists global richness (Shannon Index) and abundance (Roe et al., 2010; Desrosiers et al., 2013; Roe and Patterson, 2014). However, the methodology used in these studies was usually time consuming as the different species were manually identified by microscopy (Roe et al., 2010; Desrosiers et al., 2013; Roe and Patterson, 2014). Some specific taxons have been proposed as suitable bioindicators of certain stresses, for example Difflugia oblonga of eutrophization (Roe et al., 2010). Of particular interest is the fact that protists have proved to be excellent bioindicators to monitor the quality of wastewater effluents (Foissner, 2016). Recently, a combination of HTS and conventional methods (e.g. accessory pigment analysis) has been used to characterize the major phytoplankton groups in an estuarine area (North Caroline, USA). 18S rRNA sequencing showed that nutrients´ concentration and salinity influenced the phytoplankton taxonomic composition, being Trebouxiophyceae the most dominant family (Gong et al., 2020).
As already mentioned, faecal bacteria have been traditionally used to monitor wastewater pollution in aquatic environments, where they are also determined for health check as infectious agents (Jang et al., 2017). Some viruses (e.g. waterborne enteric or human polyomaviruses) are highly resilient to wastewater treatments what makes them excellent biomonitors of faecal pollution (Rachmadi et al., 2016; Farkas et al., 2020). Actually, enteric‐ and polyoma viruses have proven to be more persistent in the environment than initially thought, even resisting UV, chlorination, pH changes or salty environments (Fong and Lipp, 2005; Rachmadi et al., 2016; Farkas et al., 2020). In contrast, enveloped corona‐, Ebola‐ or influenza‐viruses are more susceptible due to their labile outer lipid layer (Polo et al., 2020). In the case of molecular viral determinations, PCR‐based methods are being widely used as they are not only quite sensitive but also highly specific. However, nucleic acids detection do not provide information about viral infectivity, as the loss of the cover in enveloped viruses leads to a reduced infection capacity in aqueous systems (Fong and Lipp, 2005; Polo et al., 2020). Thus, polyomavirus can be detected in almost all types of aquatic environments from coastal to tap waters, but there is no proof so far that these can be a source of infection (Rachmadi et al., 2016). Viruses are also major players in the ocean ecosystems where they are estimated to be at least ten times more abundant than the microbiome, and they kill approximately 20% of their microbial biomass daily, which the subsequent impact on nutrient and energy cycles (Suttle, 2007; Dávila‐Ramos et al., 2019). There are several methodological bottlenecks linked to the widespread use of viruses as biomonitors, such as the need for: (i) an initial concentration phase, since their concentration is usually low in abiotic matrices because they cannot regrow outside their hosts; (ii) complex methods to extract nucleic acids from matrices that can contain inhibitors of the enzymes used for molecular biology analysis; (iii) good viral databases for analysing massive sequencing results. Nevertheless, several recent studies are focusing on the study of the ocean virome (Hayes et al., 2017; Thurber et al., 2017; Dávila‐Ramos et al., 2019), that, although not directly related to environmental stress responses, are providing new sequences from an enormous diversity of unknown viruses that are greatly improving available databases, for example the Global Ocean Virome dataset. Although viral communities vary according to the depth, season, and distance from the littoral, top levels of richness functional capacity were found in the coastal areas (Hayes et al., 2017), otherwise the most exposed to pollution. Nevertheless, to‐date we are not aware of any studies relating virome alterations to pollution. Finally, in the current Sars‐CoV2 pandemic situation, the importance of viruses as infective agents and the search for potential viral biomarkers to monitor the quality of waters has become even more urgent. Thus, levels of Sars‐CoV2 in wastewaters have proven to be an excellent bioindicator for the COVID infection detection of the global population and to model and monitor the pandemic (Kumar et al., 2020; Peccia et al., 2020; Polo et al., 2020).
Final remarks
Microorganisms are the most diverse forms of life on Earth, where they play essential roles (e.g. biogeochemical cycles, marine food web, global change) and inhabit all possible environments, including the most extreme and hostile ones. Microbial structure and activity are highly sensitive to natural and anthropogenic stressors, with consequences for global environmental health, and, thus, perfect potential sensors of environmental disturbances. However, most microbial lineages numerically dominant in all major environments on Earth, known as the ‘microbial dark matter’, have never been characterized in laboratory cultures, and represent a bulk of undiscovered physiologies that may be crucial to the ecosystem (Riesenfeld et al., 2004; Lloyd et al., 2018). Thus, HTS techniques are emerging as a valuable source of information to assess the worrying effects of anthropogenic activity on the environmental microbiota. To achieve this, further improvements in the extraction of biomolecules from complex matrices (e.g. wastewaters, biofilms, sediments, soils), together with greater developments in high‐throughput molecular technologies, more consistent annotated databases, and more powerful bioinformatics software for big data analysis, are required. Altogether, these developments will allow us to evaluate the structure and functions of microbial communities better, and their consequences, in response to changes and aggressions on the environment, especially in an evolving climate change scenario.
In addition to the natural environmental studies described in this review, microcosm/mesocosm experiments have been proposed to study the interactive effects of global change on organisms from different trophic levels, including microorganisms. This approach makes it possible to simulate the exposure of estuarine benthic communities under controlled laboratory conditions to directly associate changes with the source of environmental disturbance or stress (Coelho et al., 2015; Coelho et al., 2016; Louvado et al., 2019). Also, an emergent field of study is the evaluation of how bacterial interactions and their symbiotic relationships with higher‐level organisms are affected in response to environmental perturbations, mostly considering the vital importance of microbial communities in the hosts’ development and health. Thus, further development and more extensive application of MALDI‐MSI to image host‐microbiome symbioses and their metabolic interactions look quite promising. In this sense, a spatial metabolomics pipeline (metaFISH) has been recently presented that combines fluorescence in situ hybridization (FISH) microscopy and atmospheric pressure MALDI‐MSI to image host‐microbiome symbioses and their metabolic interactions (Geier et al., 2020). Another area of future development and application, that is not without important technological problems to solve, is the mass spectral identification of microbes in complex environmental matrices and the evaluation of changes in response to stressful conditions, to be used as fingerprints of the environmental aggression (Welker and Moore, 2011; Maloof et al., 2020).
Nowadays, in a global change scenario with the increase of anthropogenic activities, to get a sustainable development is an essential objective for our societies. The improvement in the management of natural resources and the reduction of the number and quantity of substances released to the environment (e.g. from wastewater, industrial activities, agriculture, etc.) are priority actions. To support these objectives, the ongoing application of molecular technologies can shed light on the factors governing the growth, dynamics, and metabolism of indigenous microbial communities. This knowledge will provide useful tools for a better risk assessment, management, and design of more efficient bio‐engineering applications in fields as wastewater treatment or bioremediation.
Conflict of interest
The authors have no conflict of interest to declare.
Acknowledgements
This study was supported by the Spanish Ministry of Economy and Competitiveness (CTM2016‐75908‐R), by the Spanish Ministry of Science and Innovation (PID2019‐110049RB‐I00), by the European Regional Development Fund (UCO‐FEDER‐1262384‐R), and by the Chelonia Association (Mares Circulares project).
Microbial Biotechnology (2021) 14(3), 870–885
Funding information
This study was supported by the Spanish Ministry of Economy and Competitiveness (CTM2016‐75908‐R), by the Spanish Ministry of Science and Innovation (PID2019‐110049RB‐I00), by the European Regional Development Fund (UCO‐FEDER‐1262384‐R), and by the Chelonia Association (Mares Circulares project).
References
- Acosta‐González, A. , and Marqués, S. (2016) Bacterial diversity in oil‐polluted marine coastal sediments. Curr Opin Biotechnol 38: 24–32. [DOI] [PubMed] [Google Scholar]
- Acosta‐González, A. , Martirani‐von Abercron, S.M. , Rosselló‐Móra, R. , Wittich, R.M. , and Marqués, S. (2015) The effect of oil spills on the bacterial diversity and catabolic function in coastal sediments: a case study on the Prestige oil spill. Environ Sci Pollut Res Int 22: 15200–15214. [DOI] [PubMed] [Google Scholar]
- Aguiar‐Pulido, V. , Huang, W. , Suarez‐Ulloa, V. , Cickovski, T. , Mathee, K. , and Narasimhan, G. (2016) Metagenomics, metatranscriptomics, and metabolomics approaches for microbiome analysis. Evol Bioinform Online 12: 5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, J. , Knopp, G. , Dotsch, A. , Wieland, A. , and Schwartz, T. (2016) Ozone treatment of conditioned wastewater selects antibiotic resistance genes, opportunistic bacteria, and induce strong population shifts. Sci Total Environ 559: 103–112. [DOI] [PubMed] [Google Scholar]
- Almakki, A. , Jumas‐Bilak, E. , Marchandin, H. , and Licznar‐Fajardo, P. (2019) Antibiotic resistance in urban runoff. Sci Total Environ 667: 64–76. [DOI] [PubMed] [Google Scholar]
- Atlas, R. , and Bragg, J. (2009) Bioremediation of marine oil spills: when and when not ‐ the Exxon Valdez experience. Microb Biotechnol 2: 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auguste, M. , Lasa, A. , Pallavicini, A. , Gualdi, S. , Vezzulli, L. , and Canesi, L. (2019) Exposure to TiO2 nanoparticles induces shifts in the microbiota composition of Mytilus galloprovincialis hemolymph. Sci Total Environ 670: 129–137. [DOI] [PubMed] [Google Scholar]
- Bae, M.J. , and Park, Y.S. (2014) Biological early warning system based on the responses of aquatic organisms to disturbances: a review. Sci Total Environ 466–467: 635–649. [DOI] [PubMed] [Google Scholar]
- Barberán, A. , Fernández‐Guerra, A. , Bohannan, B.J. , and Casamayor, E.O. (2012) Exploration of community traits as ecological markers in microbial metagenomes. Mol Ecol 21: 1909–1917. [DOI] [PubMed] [Google Scholar]
- Bastaraud, A. , Cecchi, P. , Handschumacher, P. , Altmann, M. , and Jambou, R. (2020) Urbanization and waterborne pathogen emergence in low‐income countries: where and how to conduct surveys? Int J Environ Res Public Health 17: 480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejaoui, S. , Michán, C. , Telahigue, K. , Nechi, S. , Cafsi, M.E. , Soudani, N. , et al. (2020) Metal body burden and tissue oxidative status in the bivalve Venerupis decussata from Tunisian coastal lagoons. Mar Environ Res 159: 105000. [DOI] [PubMed] [Google Scholar]
- Bouchez, T. , Blieux, A.L. , Dequiedt, S. , Domaizon, I. , Dufresne, A. , Ferreira, S. , et al. (2016) Molecular microbiology methods for environmental diganosis. Environ Chem Lett 14: 423–441. [Google Scholar]
- Bouhajja, E. , Agathos, S.N. , and George, I.F. (2016) Metagenomics: probing pollutant fate in natural and engineered ecosystems. Biotechnol Adv 34: 1413–1426. [DOI] [PubMed] [Google Scholar]
- Brown‐Peterson, N.J. , Krasnec, M. , Takeshita, R. , Ryan, C.N. , Griffitt, K.J. , Lay, C. , et al. (2015) A multiple endpoint analysis of the effects of chronic exposure to sediment contaminated with Deepwater Horizon oil on juvenile Southern flounder and their associated microbiomes. Aquat Toxicol 165: 197–209. [DOI] [PubMed] [Google Scholar]
- Burger, J. (2006) Bioindicators: a review of their use in the environmental literature 1970–2005. Environ Bioindic 1: 136–144. [Google Scholar]
- Cai, L. , Tian, R.M. , Zhou, G. , Tong, H. , Wong, Y.H. , Zhang, W. , et al. (2018) Exploring coral microbiome assemblages in the South China Sea. Sci Rep 8: 2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruso, G. (2013) Microbes and their use as indicators of pollution. J Pollut Eff Cont 1: 1000e1102. [Google Scholar]
- Cavicchioli, R. , Ripple, W.J. , Timmis, K.N. , Azam, F. , Bakken, L.R. , Baylis, M. , et al. (2019) Scientists' warning to humanity: microorganisms and climate change. Nat Rev Microbiol 17: 569–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez, F.P. , Messie, M. , and Pennington, J.T. (2011) Marine primary production in relation to climate variability and change. Ann Rev Mar Sci 3: 227–260. [DOI] [PubMed] [Google Scholar]
- Che, Y. , Xia, Y. , Liu, L. , Li, A.D. , Yang, Y. , and Zhang, T. (2019) Mobile antibiotic resistome in wastewater treatment plants revealed by Nanopore metagenomic sequencing. Microbiome 7: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H. , Bai, X. , Jing, L. , Chen, R. , and Teng, Y. (2019) Characterization of antibiotic resistance genes in the sediments of an urban river revealed by comparative metagenomics analysis. Sci Total Environ 653: 1513–1521. [DOI] [PubMed] [Google Scholar]
- Chen, L.X. , Hu, M. , Huang, L.N. , Hua, Z.S. , Kuang, J.L. , Li, S.J. , and Shu, W.S. (2015) Comparative metagenomic and metatranscriptomic analyses of microbial communities in acid mine drainage. ISME J 9: 1579–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, L.X. , Huang, L.N. , Méndez‐García, C. , Kuang, J.L. , Hua, Z.S. , Liu, J. , and Shu, W.S. (2016) Microbial communities, processes and functions in acid mine drainage ecosystems. Curr Opin Biotechnol 38: 150–158. [DOI] [PubMed] [Google Scholar]
- Chen, Y. , Jiang, Y. , Huang, H. , Mou, L. , Ru, J. , Zhao, J. , and Xiao, S. (2018) Long‐term and high‐concentration heavy‐metal contamination strongly influences the microbiome and functional genes in Yellow River sediments. Sci Total Environ 637–638: 1400–1412. [DOI] [PubMed] [Google Scholar]
- Chiarello, M. , Auguet, J.C. , Graham, N.A.J. , Claverie, T. , Sucre, E. , Bouvier, C. , et al. (2020) Exceptional but vulnerable microbial diversity in coral reef animal surface microbiomes. Proc Biol Sci 287: 20200642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu, B.T.T. , Petrovich, M.L. , Chaudhary, A. , Wright, D. , Murphy, B. , Wells, G. , and Poretsky, R. (2018) Metagenomics reveals the impact of wastewater treatment plants on the dispersal of microorganisms and genes in aquatic sediments. Appl Environ Microbiol 84: e02168‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho, F.J. , Cleary, D.F. , Rocha, R.J. , Calado, R. , Castanheira, J.M. , Rocha, S.M. , et al. (2015) Unraveling the interactive effects of climate change and oil contamination on laboratory‐simulated estuarine benthic communities. Glob Chang Biol 21: 1871–1886. [DOI] [PubMed] [Google Scholar]
- Coelho, F.J. , Cleary, D.F. , Costa, R. , Ferreira, M. , Polonia, A.R. , Silva, A.M. , et al. (2016) Multitaxon activity profiling reveals differential microbial response to reduced seawater pH and oil pollution. Mol Ecol 25: 4645–4659. [DOI] [PubMed] [Google Scholar]
- Coelho, F.J. , Santos, A.L. , Coimbra, J. , Almeida, A. , Cunha, A. , Cleary, D.F. , et al. (2013) Interactive effects of global climate change and pollution on marine microbes: the way ahead. Ecol Evol 3: 1808–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho, F.H. , Gregoracci, G.B. , Walter, J.M. , Thompson, C.C. , and Thompson, F.L. (2018) Metagenomics sheds light on the ecology of marine microbes and their viruses. Trends Microbiol 26: 955–965. [DOI] [PubMed] [Google Scholar]
- Coutinho, F.H. , Meirelles, P.M. , Moreira, A.P. , Paranhos, R.P. , Dutilh, B.E. , and Thompson, F.L. (2015) Niche distribution and influence of environmental parameters in marine microbial communities: a systematic review. PeerJ 3: e1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currie, A.R. , Tait, K. , Parry, H. , de Francisco‐Mora, B. , Hicks, N. , Osborn, A.M. , et al. (2017) Marine microbial gene abundance and community composition in response to ocean acidification and elevated temperature in two contrasting coastal marine sediments. Front Microbiol 8: 1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czaplicki, L.M. , and Gunsch, C.K. (2016) Reflection on molecular approaches influencing state‐of‐the‐art bioremediation design: culturing to microbial community fingerprinting to omics. J Environ Eng 142: 03116002. 10.1061/(ASCE)EE.1943-7870.0001141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danovaro, R. , Corinaldesi, C. , Dell'Anno, A. , and Rastelli, E. (2017) Potential impact of global climate change on benthic deep‐sea microbes. FEMS Microbiol Lett 364: fnx214. [DOI] [PubMed] [Google Scholar]
- Danovaro, R. , Molari, M. , Corinaldesi, C. , and Dell'Anno, A. (2016) Macroecological drivers of archaea and bacteria in benthic deep‐sea ecosystems. Sci Adv 2: e1500961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dávila‐Ramos, S. , Castelán‐Sánchez, H.G. , Martínez‐Ávila, L. , Sánchez‐Carbente, M.D.R. , Peralta, R. , Hernández‐Mendoza, A. , et al. (2019) A review on viral metagenomics in extreme environments. Front Microbiol 10: 2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmont, T.O. , Malandain, C. , Prestat, E. , Larose, C. , Monier, J.M. , Simonet, P. , and Vogel, T.M. (2011) Metagenomic mining for microbiologists. ISME J 5: 1837–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai, C. , Pathak, H. , and Madamwar, D. (2010) Advances in molecular and "‐omics" technologies to gauge microbial communities and bioremediation at xenobiotic/anthropogen contaminated sites. Bioresour Technol 101: 1558–1569. [DOI] [PubMed] [Google Scholar]
- Desrosiers, C. , Leflaive, J. , Eulin, A. , and Ten‐Hage, L. (2013) Bioindicators in marine waters: benthic diatoms as a tool to assess water quality from eutrophic to oligotrophic coastal ecosystems. Ecol Indic 32: 25–34. [Google Scholar]
- European Marine Board . (2019) Navigating the future V. Position Paper 24: Marine Science for a Sustainable Future. [Google Scholar]
- FAQ . (2017) Microbes and climate change. Report on an American Academy of Microbiology and American Geophysical Union Colloquium held in Washington, DC, in March 2016. [PubMed] [Google Scholar]
- Farkas, K. , Walker, D.I. , Adriaenssens, E.M. , McDonald, J.E. , Hillary, L.S. , Malham, S.K. , and Jones, D.L. (2020) Viral indicators for tracking domestic wastewater contamination in the aquatic environment. Water Res 181: 115926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández‐Cisnal, R. , García‐Sevillano, M.A. , Gómez‐Ariza, J.L. , Pueyo, C. , López‐Barea, J. , and Abril, N. (2017) 2D‐DIGE as a proteomic biomarker discovery tool in environmental studies with Procambarus clarkii . Sci Total Environ 584–585: 813–827. [DOI] [PubMed] [Google Scholar]
- Foissner, W. (2016) Protists as bioindicators in activated sludge: Identification, ecology and future needs. Eur J Protistol 55: 75–94. [DOI] [PubMed] [Google Scholar]
- Fong, T.T. , and Lipp, E.K. (2005) Enteric viruses of humans and animals in aquatic environments: health risks, detection, and potential water quality assessment tools. Microbiol Mol Biol Rev 69: 357–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzosa, E.A. , Hsu, T. , Sirota‐Madi, A. , Shafquat, A. , Abu‐Ali, G. , Morgan, X.C. , and Huttenhower, C. (2015) Sequencing and beyond: integrating molecular 'omics' for microbial community profiling. Nat Rev Microbiol 13: 360–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gago‐Tinoco, A. , González‐Domínguez, R. , García‐Barrera, T. , Blasco‐Moreno, J. , Bebianno, M.J. , and Gómez‐Ariza, J.L. (2014) Metabolic signatures associated with environmental pollution by metals in Donana National Park using P. clarkii as bioindicator. Environ Sci Pollut Res Int 21: 13315–13323. [DOI] [PubMed] [Google Scholar]
- Geier, B. , Sogin, E.M. , Michellod, D. , Janda, M. , Kompauer, M. , Spengler, B. , et al. (2020) Spatial metabolomics of in situ host‐microbe interactions at the micrometre scale. Nat Microbiol 5: 498–510. [DOI] [PubMed] [Google Scholar]
- Geissen, V. , Mol, H. , Klumpp, E. , Umlauf, G. , Nadal, M. , van der Ploeg, M. , et al. (2015) Emerging pollutants in the environment: a challenge for water resource management. Int Soil Water Conse 3: 57–65. [Google Scholar]
- Gillan, D.C. , Roosa, S. , Kunath, B. , Billon, G. , and Wattiez, R. (2015) The long‐term adaptation of bacterial communities in metal‐contaminated sediments: a metaproteogenomic study. Environ Microbiol 17: 1991–2005. [DOI] [PubMed] [Google Scholar]
- Glasl, B. , Bourne, D.G. , Frade, P.R. , Thomas, T. , Schaffelke, B. , and Webster, N.S. (2019) Microbial indicators of environmental perturbations in coral reef ecosystems. Microbiome 7: 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glöckner, F.O. , and Joint, I. (2010) Marine microbial genomics in Europe: current status and perspectives. Microb Biotechnol 3: 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong, W. , Hall, N. , Paerl, H. , and Marchetti, A. (2020) Phytoplankton composition in a eutrophic estuary: Comparison of multiple taxonomic approaches and influence of environmental factors. Environ Microbiol 22: 4718–4731. [DOI] [PubMed] [Google Scholar]
- Gonzalez, D.J. , Xu, Y. , Yang, Y.L. , Esquenazi, E. , Liu, W.T. , Edlund, A. , et al. (2012) Observing the invisible through imaging mass spectrometry, a window into the metabolic exchange patterns of microbes. J Proteomics 75: 5069–5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski, L. , Rivadeneira, P. , and Cooley, M.B. (2019) New strategies for the enumeration of enteric pathogens in water. Environ Microbiol Rep 11: 765–776. [DOI] [PubMed] [Google Scholar]
- Grottoli, A.G. , Dalcin Martins, P. , Wilkins, M.J. , Johnston, M.D. , Warner, M.E. , Cai, W.J. , et al. (2018) Coral physiology and microbiome dynamics under combined warming and ocean acidification. PLoS One 13: e0191156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, Y. , Cichocki, N. , Schattenberg, F. , Geffers, R. , Harms, H. , and Muller, S. (2019) AgNPs change microbial community structures of wastewater. Front Microbiol 9: 3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan, S.H. , Van Ginkel, S.W. , Hussein, M.A. , Abskharon, R. , and Oh, S.E. (2016) Toxicity assessment using different bioassays and microbial biosensors. Environ Int 92–93: 106–118. [DOI] [PubMed] [Google Scholar]
- Hawley, A.K. , Brewer, H.M. , Norbeck, A.D. , Pasa‐Tolic, L. , and Hallam, S.J. (2014) Metaproteomics reveals differential modes of metabolic coupling among ubiquitous oxygen minimum zone microbes. Proc Natl Acad Sci USA 111: 11395–11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley, A.K. , Torres‐Beltran, M. , Zaikova, E. , Walsh, D.A. , Mueller, A. , Scofield, M. , et al. (2017) A compendium of multi‐omic sequence information from the Saanich Inlet water column. Sci Data 4: 170160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes, S. , Mahony, J. , Nauta, A. , and van Sinderen, D. (2017) Metagenomic approaches to assess bacteriophages in various environmental niches. Viruses 9: 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberg, K.B. , Gilbert, J.A. , and Joint, I. (2010) Marine genomics: at the interface of marine microbial ecology and biodiscovery. Microb Biotechnol 3: 531–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst, F.A. , Lunsmann, V. , Kjeldal, H. , Jehmlich, N. , Tholey, A. , von Bergen, M. , et al. (2016) Enhancing metaproteomics–the value of models and defined environmental microbial systems. Proteomics 16: 783–798. [DOI] [PubMed] [Google Scholar]
- Hernandez‐Agreda, A. , Gates, R.D. , and Ainsworth, T.D. (2017) Defining the core microbiome in corals' microbial soup. Trends Microbiol 25: 125–140. [DOI] [PubMed] [Google Scholar]
- Hettich, R.L. , Sharma, R. , Chourey, K. , and Giannone, R.J. (2012) Microbial metaproteomics: identifying the repertoire of proteins that microorganisms use to compete and cooperate in complex environmental communities. Curr Opin Microbiol 15: 373–380. [DOI] [PubMed] [Google Scholar]
- Hoegh‐Guldberg, O. , Mumby, P.J. , Hooten, A.J. , Steneck, R.S. , Greenfield, P. , Gomez, E. , et al. (2007) Coral reefs under rapid climate change and ocean acidification. Science 318: 1737–1742. [DOI] [PubMed] [Google Scholar]
- Holmstrup, M. , Bindesbol, A.M. , Oostingh, G.J. , Duschl, A. , Scheil, V. , Kohler, H.R. , et al. (2010) Interactions between effects of environmental chemicals and natural stressors: a review. Sci Total Environ 408: 3746–3762. [DOI] [PubMed] [Google Scholar]
- Hua, Z.S. , Han, Y.J. , Chen, L.X. , Liu, J. , Hu, M. , Li, S.J. , et al. (2015) Ecological roles of dominant and rare prokaryotes in acid mine drainage revealed by metagenomics and metatranscriptomics. ISME J 9: 1280–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquiod, S. , Cyriaque, V. , Riber, L. , Al‐Soud, W.A. , Gillan, D.C. , Wattiez, R. , and Sorensen, S.J. (2018) Long‐term industrial metal contamination unexpectedly shaped diversity and activity response of sediment microbiome. J Hazard Mater 344: 299–307. [DOI] [PubMed] [Google Scholar]
- Jang, J. , Hur, H.G. , Sadowsky, M.J. , Byappanahalli, M.N. , Yan, T. , and Ishii, S. (2017) Environmental Escherichia coli: ecology and public health implications‐a review. J Appl Microbiol 123: 570–581. [DOI] [PubMed] [Google Scholar]
- Ju, F. , Beck, K. , Yin, X. , Maccagnan, A. , McArdell, C.S. , Singer, H.P. , et al. (2019) Wastewater treatment plant resistomes are shaped by bacterial composition, genetic exchange, and upregulated expression in the effluent microbiomes. ISME J 13: 346–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, B. , Clinton, S.M. , Hamp, T.J. , Oliver, J.D. , and Ringwood, A.H. (2018) Potential impacts of hypoxia and a warming ocean on oyster microbiomes. Mar Environ Res 139: 27–34. [DOI] [PubMed] [Google Scholar]
- King, G.M. , Kostka, J.E. , Hazen, T.C. , and Sobecky, P.A. (2015) Microbial responses to the Deepwater Horizon oil spill: from coastal wetlands to the deep sea. Ann Rev Mar Sci 7: 377–401. [DOI] [PubMed] [Google Scholar]
- Kuang, J.L. , Huang, L.N. , Chen, L.X. , Hua, Z.S. , Li, S.J. , Hu, M. , et al. (2013) Contemporary environmental variation determines microbial diversity patterns in acid mine drainage. ISME J 7: 1038–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang, J. , Huang, L. , He, Z. , Chen, L. , Hua, Z. , Jia, P. , et al. (2016) Predicting taxonomic and functional structure of microbial communities in acid mine drainage. ISME J 10: 1527–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, M. , Mohapatra, S. , Mazumder, P. , Singh, A. , Honda, R. , Lin, C. , et al. (2020) Making waves perspectives of modelling and monitoring of SARS‐CoV‐2 in aquatic environment for COVID‐19 pandemic. Curr Pollut Rep 6: 468–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, B. , Yang, Y. , Ma, L. , Ju, F. , Guo, F. , Tiedje, J.M. , and Zhang, T. (2015) Metagenomic and network analysis reveal wide distribution and co‐occurrence of environmental antibiotic resistance genes. ISME J 9: 2490–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Lusher, A.L. , Rotchell, J.M. , Deudero, S. , Turra, A. , Brate, I.L.N. , et al. (2019) Using mussel as a global bioindicator of coastal microplastic pollution. Environ Pollut 244: 522–533. [DOI] [PubMed] [Google Scholar]
- Li, L.G. , Xia, Y. , and Zhang, T. (2017) Co‐occurrence of antibiotic and metal resistance genes revealed in complete genome collection. ISME J 11: 651–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd, K.G. , Steen, A.D. , Ladau, J. , Yin, J. , and Crosby, L. (2018) Phylogenetically novel uncultured microbial cells dominate earth microbiomes. mSystems 3: e00055‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louvado, A. , Coelho, F. , Oliveira, V. , Gomes, H. , Cleary, D.F.R. , Simoes, M.M.Q. , et al. (2019) Microcosm evaluation of the impact of oil contamination and chemical dispersant addition on bacterial communities and sediment remediation of an estuarine port environment. J Appl Microbiol 127: 134–149. [DOI] [PubMed] [Google Scholar]
- Louvado, A. , Gomes, N.C. , Simoes, M.M. , Almeida, A. , Cleary, D.F. , and Cunha, A. (2015) Polycyclic aromatic hydrocarbons in deep sea sediments: microbe‐pollutant interactions in a remote environment. Sci Total Environ 526: 312–328. [DOI] [PubMed] [Google Scholar]
- Malla, M.A. , Dubey, A. , Yadav, S. , Kumar, A. , Hashem, A. , and Abd Allah, E.F. (2018) Understanding and designing the strategies for the microbe‐mediated remediation of environmental contaminants using omics approaches. Front Microbiol 9: 1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloof, K.A. , Reinders, A.N. , and Tucker, K.R. (2020) Applications of mass spectrometry imaging in the environmental sciences. Curr Opin Environ Sci Health 18: 54–62. [Google Scholar]
- Mao, D. , Yu, S. , Rysz, M. , Luo, Y. , Yang, F. , Li, F. , et al. (2015) Prevalence and proliferation of antibiotic resistance genes in two municipal wastewater treatment plants. Water Res 85: 458–466. [DOI] [PubMed] [Google Scholar]
- Mason, O.U. , Scott, N.M. , Gonzalez, A. , Robbins‐Pianka, A. , Baelum, J. , Kimbrel, J. , et al. (2014) Metagenomics reveals sediment microbial community response to Deepwater Horizon oil spill. ISME J 8: 1464–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méndez‐García, C. , Peláez, A.I. , Mesa, V. , Sánchez, J. , Golyshina, O.V. , and Ferrer, M. (2015) Microbial diversity and metabolic networks in acid mine drainage habitats. Front Microbiol 6: 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milan, M. , Carraro, L. , Fariselli, P. , Martino, M.E. , Cavalieri, D. , Vitali, F. , et al. (2018) Microbiota and environmental stress: how pollution affects microbial communities in Manila clams. Aquat Toxicol 194: 195–207. [DOI] [PubMed] [Google Scholar]
- Moree, W.J. , McConnell, O.J. , Nguyen, D.D. , Sanchez, L.M. , Yang, Y.L. , Zhao, X. , et al. (2014) Microbiota of healthy corals are active against fungi in a light‐dependent manner. ACS Chem Biol 9: 2300–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moree, W.J. , Yang, J.Y. , Zhao, X. , Liu, W.T. , Aparicio, M. , Atencio, L. , et al. (2013) Imaging mass spectrometry of a coral microbe interaction with fungi. J Chem Ecol 39: 1045–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelakanta, G. , and Sultana, H. (2013) The use of metagenomic approaches to analyze changes in microbial communities. Microbiol Insights 6: 37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OECD . (2010) Test No. 209: Activated Sludge, Respiration Inhibition Test (Carbon and Ammonium Oxidation), OECD Guidelines for the Testing of Chemicals, Section 2. OECD Publishing, Paris. 10.1787/9789264070080-en [DOI] [Google Scholar]
- Parmar, T.K. , Rawtani, D. , and Agrawal, Y.K. (2016) Bioindicators: the natural indicator of environmental pollution. Front Life Sci 9: 110–118. [Google Scholar]
- Pawlowski, J. , Lejzerowicz, F. , Apotheloz‐Perret‐Gentil, L. , Visco, J. , and Esling, P. (2016) Protist metabarcoding and environmental biomonitoring: Time for change. Eur J Protistol 55: 12–25. [DOI] [PubMed] [Google Scholar]
- Pazda, M. , Kumirska, J. , Stepnowski, P. , and Mulkiewicz, E. (2019) Antibiotic resistance genes identified in wastewater treatment plant systems – a review. Sci Total Environ 697: 134023. [DOI] [PubMed] [Google Scholar]
- Peccia, J. , Zulli, A. , Brackney, D.E. , Grubaugh, N.D. , Kaplan, E.H. , Casanovas‐Massana, A. , et al. (2020) Measurement of SARS‐CoV‐2 RNA in wastewater tracks community infection dynamics. Nat Biotechnol 38: 1164–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo, D. , Quintela‐Baluja, M. , Corbishley, A. , Jones, D.L. , Singer, A.C. , Graham, D.W. , and Romalde, J.L. (2020) Making waves: wastewater‐based epidemiology for COVID‐19 – approaches and challenges for surveillance and prediction. Water Res 186: 116404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruden, A. , Pei, R. , Storteboom, H. , and Carlson, K.H. (2006) Antibiotic resistance genes as emerging contaminants: studies in northern Colorado. Environ Sci Technol 40: 7445–7450. [DOI] [PubMed] [Google Scholar]
- Qiao, M. , Ying, G.G. , Singer, A.C. , and Zhu, Y.G. (2018) Review of antibiotic resistance in China and its environment. Environ Int 110: 160–172. [DOI] [PubMed] [Google Scholar]
- Rachmadi, A.T. , Torrey, J.R. , and Kitajima, M. (2016) Human polyomavirus: advantages and limitations as a human‐specific viral marker in aquatic environments. Water Res 105: 456–469. [DOI] [PubMed] [Google Scholar]
- Rafraf, I.D. , Lekunberri, I. , Sànchez‐Melsió, A. , Aouni, M. , Borrego, C.M. , and Balcázar, J.L. (2016) Abundance of antibiotic resistance genes in five municipal wastewater treatment plants in the Monastir Governorate, Tunisia. Environ Pollut 219: 353–358. [DOI] [PubMed] [Google Scholar]
- Ram, R.J. , Verberkmoes, N.C. , Thelen, M.P. , Tyson, G.W. , Baker, B.J. , Blake, R.C. 2nd , et al. (2005) Community proteomics of a natural microbial biofilm. Science 308: 1915–1920. [PubMed] [Google Scholar]
- Ramírez, F. , Coll, M. , Navarro, J. , Bustamante, J. , and Green, A.J. (2018) Spatial congruence between multiple stressors in the Mediterranean Sea may reduce its resilience to climate impacts. Sci Rep 8: 14871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riesenfeld, C.S. , Schloss, P.D. , and Handelsman, J. (2004) Metagenomics: genomic analysis of microbial communities. Annu Rev Genet 38: 525–552. [DOI] [PubMed] [Google Scholar]
- Rivers, A.R. , Sharma, S. , Tringe, S.G. , Martin, J. , Joye, S. , and Moran, A.M. (2013) Transcriptional response of bathypelagic marine bacterioplankton to the Deepwater Horizon oil spill. ISME J 7: 2315–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo, L. , Manaia, C. , Merlin, C. , Schwartz, T. , Dagot, C. , Ploy, M.C. , et al. (2013) Urban wastewater treatment plants as hotspots for antibiotic resistant bacteria and genes spread into the environment: a review. Sci Total Environ 447: 345–360. [DOI] [PubMed] [Google Scholar]
- Rocca, J.D. , Simonin, M. , Blaszczak, J.R. , Ernakovich, J.G. , Gibbons, S.M. , Midani, F.S. , and Washburne, A.D. (2019) The microbiome stress project: toward a global meta‐analysis of environmental stressors and their effects on microbial communities. Front Microbiol 9: 3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez‐Chueca, J. , Varella della Giustina, S. , Rocha, J. , Fernandes, T. , Pablos, C. , Encinas, Á. , et al. (2019) Assessment of full‐scale tertiary wastewater treatment by UV‐C based‐AOPs: Removal or persistence of antibiotics and antibiotic resistance genes? Sci Total Environ 652: 1051–1061. [DOI] [PubMed] [Google Scholar]
- Rodriguez‐R, L.M. , Overholt, W.A. , Hagan, C. , Huettel, M. , Kostka, J.E. , and Konstantinidis, K.T. (2015) Microbial community successional patterns in beach sands impacted by the Deepwater Horizon oil spill. ISME J 9: 1928–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe, H.M. , Patterson, R.T. , and Swindles, G.T. (2010) Controls on the contemporary distribution of lake thecamoebians (testate amoebae) within the Greater Toronto Area and their potential as water quality indicators. J Paleolimnol 43: 955–975. [Google Scholar]
- Roe, H.M. , and Patterson, R.T. (2014) Arcellacea (Testate amoebae) as bio‐indicators of road salt contamination in lakes. Microb Ecol 68: 299–313. [DOI] [PubMed] [Google Scholar]
- Sauvé, S. , and Desrosiers, M. (2014) A review of what is an emerging contaminant. Chem Cent J 8: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler, C. , and Berendonk, T.U. (2012) Heavy metal driven co‐selection of antibiotic resistance in soil and water bodies impacted by agriculture and aquaculture. Front Microbiol 3: 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp, K.H. , and Ritchie, K.B. (2012) Multi‐partner interations in corals in the face of climate change. Biol Bull 223: 66–77. [DOI] [PubMed] [Google Scholar]
- Shen, Y. , Chu, L. , Zhuan, R. , Xiang, X. , Sun, H. , and Wang, J. (2019) Degradation of antibiotics and antibiotic resistance genes in fermentation residues by ionizing radiation: a new insight into a sustainable management of antibiotic fermentative residuals. J Environ Manage 232: 171–178. [DOI] [PubMed] [Google Scholar]
- Sherr, E.B. , and Sherr, B.F. (2002) Significance of predation by protists in aquatic microbial food webs. Antonie Van Leeuwenhoek 81: 293–308. [DOI] [PubMed] [Google Scholar]
- Siggins, A. , Gunnigle, E. , and Abram, F. (2012) Exploring mixed microbial community functioning: recent advances in metaproteomics. FEMS Microbiol Ecol 80: 265–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon, C. , and Daniel, R. (2011) Metagenomic analyses: past and future trends. Appl Environ Microbiol 77: 1153–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, J. , Jongmans‐Hochschulz, E. , Mauder, N. , Imirzalioglu, C. , Wichels, A. , and Gerdts, G. (2020) The Travelling Particles: Investigating microplastics as possible transport vectors for multidrug resistant E. coli in the Weser estuary (Germany). Sci Total Environ 720: 137603. [DOI] [PubMed] [Google Scholar]
- Storey, M.V. , van der Gaag, B. , and Burns, B.P. (2011) Advances in on‐line drinking water quality monitoring and early warning systems. Water Res 45: 741–747. [DOI] [PubMed] [Google Scholar]
- Sukhum, K.V. , Diorio‐Toth, L. , and Dantas, G. (2019) Genomic and metagenomic approaches for predictive surveillance of emerging pathogens and antibiotic resistance. Clin Pharmacol Ther 106: 512–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttle, C.A. (2007) Marine viruses‐major players in the global ecosystem. Nat Rev Microbiol 5: 801–812. [DOI] [PubMed] [Google Scholar]
- Tan, B. , Ng, C. , Nshimyimana, J.P. , Loh, L.L. , Gin, K.Y. , and Thompson, J.R. (2015) Next‐generation sequencing (NGS) for assessment of microbial water quality: current progress, challenges, and future opportunities. Front Microbiol 6: 1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tangherlini, M. , Miralto, M. , Colantuono, C. , Sangiovanni, M. , Dell' Anno, A. , Corinaldesi, C. , et al. (2018) GLOSSary: the GLobal Ocean 16S subunit web accessible resource. BMC Bioinformatics 19: 443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Techtmann, S.M. , and Hazen, T.C. (2016) Metagenomic applications in environmental monitoring and bioremediation. J Ind Microbiol Biotechnol 43: 1345–1354. [DOI] [PubMed] [Google Scholar]
- Thomas, J.C. , Oladeinde, A. , Kieran, T.J. , Finger, J.W. , Bayona‐Vásquez, N.J. , Cartee, J.C. , et al. (2020) Co‐occurrence of antibiotic, biocide, and heavy metal resistance genes in bacteria from metal and radionuclide contaminated soils at the Savannah River Site. Microb Biotechnol 13: 1179–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurber, R.V. , Payet, J.P. , Thurber, A.R. , and Correa, A.M. (2017) Virus‐host interactions and their roles in coral reef health and disease. Nat Rev Microbiol 15: 205–216. [DOI] [PubMed] [Google Scholar]
- Thurber, R.V. , Willner‐Hall, D. , Rodriguez‐Mueller, B. , Desnues, C. , Edwards, R.A. , Angly, F. , et al. (2009) Metagenomic analysis of stressed coral holobions. Environ Microbiol 11: 2148–2163. [DOI] [PubMed] [Google Scholar]
- Tothill, I.E. , and Turner, A.P.F. (1996) Developments in bioassay methods for toxicity testing in water treatment. Trends Analyt Chem 15: 178–188. [Google Scholar]
- Ulloa, O. , Canfield, D.E. , DeLong, E.F. , Letelier, R.M. , and Stewart, F.J. (2012) Microbial oceanography of anoxic oxygen minimum zones. Proc Natl Acad Sci USA 109: 15996–16003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villegas‐Plazas, M. , Sanabria, J. , and Junca, H. (2019) A composite taxonomical and functional framework of microbiomes under acid mine drainage bioremediation systems. J Environ Manage 251: 109581. [DOI] [PubMed] [Google Scholar]
- Voolstra, C.R. , and Ziegler, M. (2020) Adapting with microbial help: microbiome flexibility facilitates rapid responses to environmental change. BioEssays 42: e2000004. [DOI] [PubMed] [Google Scholar]
- Wang, D.Z. , Kong, L.F. , Li, Y.Y. , and Xie, Z.X. (2016) Environmental microbial community proteomics: status, challenges and perspectives. Int J Mol Sci 17: 1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, D.Z. , Xie, Z.X. , and Zhang, S.F. (2014) Marine metaproteomics: current status and future directions. J Proteomics 97: 27–35. [DOI] [PubMed] [Google Scholar]
- Wang, J. , and Zhuan, R. (2020) Degradation of antibiotics by advanced oxidation processes: an overview. Sci Total Environ 701: 135023. [DOI] [PubMed] [Google Scholar]
- Wegley, L. , Edwards, R. , Rodriguez‐Brito, B. , Liu, H. , and Rohwer, F. (2007) Metagenomic analysis of the microbial community associated with the coral Porites astreoides . Environ Microbiol 9: 2707–2719. [DOI] [PubMed] [Google Scholar]
- Welker, M. , and Moore, E.R. (2011) Applications of whole‐cell matrix‐assisted laser‐desorption/ionization time‐of‐flight mass spectrometry in systematic microbiology. Syst Appl Microbiol 34: 2–11. [DOI] [PubMed] [Google Scholar]
- WHO . (2015) Global Action Plan on Antimicrobial Resistance. World Health Organization. [DOI] [PubMed] [Google Scholar]
- Wilmes, P. , and Bond, P.L. (2009) Microbial community proteomics: elucidating the catalysts and metabolic mechanisms that drive the Earth's biogeochemical cycles. Curr Opin Microbiol 12: 310–317. [DOI] [PubMed] [Google Scholar]
- Wright, G.D. (2010) Antibiotic resistance in the environment: a link to the clinic? Curr Opin Microbiol 13: 589–594. [DOI] [PubMed] [Google Scholar]
- Xue, J. , Yu, Y. , Bai, Y. , Wang, L. , and Wu, Y. (2015) Marine oil‐degrading microorganisms and biodegradation process of petroleum hydrocarbon in marine environments: a review. Curr Microbiol 71: 220–228. [DOI] [PubMed] [Google Scholar]
- Yang, Y. , Li, B. , Zou, S. , Fang, H.H. , and Zhang, T. (2014) Fate of antibiotic resistance genes in sewage treatment plant revealed by metagenomic approach. Water Res 62: 97–106. [DOI] [PubMed] [Google Scholar]
- Yin, X. , Deng, Y. , Ma, L. , Wang, Y. , Chan, L.Y.L. , and Zhang, T. (2019) Exploration of the antibiotic resistome in a wastewater treatment plant by a nine‐year longitudinal metagenomic study. Environ Int 133: 105270. [DOI] [PubMed] [Google Scholar]
- Zarraonaindia, I. , Smith, D.P. , and Gilbert, J.A. (2013) Beyond the genome: community‐level analysis of the microbial world. Biol Philos 28: 261–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, X. , Chen, X. , and Zhuang, J. (2015) The positive relationship between ocean acidification and pollution. Mar Pollut Bull 91: 14–21. [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Lu, J. , Wu, J. , Wang, J. , and Luo, Y. (2020) Potential risks of microplastics combined with superbugs: Enrichment of antibiotic resistant bacteria on the surface of microplastics in mariculture system. Ecotoxicol Environ Saf 187: 109852. [DOI] [PubMed] [Google Scholar]
- Zhou, Q. , Zhang, J. , Fu, J. , Shi, J. , and Jiang, G. (2008) Biomonitoring: an appealing tool for assessment of metal pollution in the aquatic ecosystem. Anal Chim Acta 606: 135–150. [DOI] [PubMed] [Google Scholar]
- Ziegler, M. , Seneca, F.O. , Yum, L.K. , Palumbi, S.R. , and Voolstra, C.R. (2017) Bacterial community dynamics are linked to patterns of coral heat tolerance. Nat Commun 8: 14213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuñiga, C. , Zaramela, L. , and Zengler, K. (2017) Elucidation of complexity and prediction of interactions in microbial communities. Microb Biotechnol 10: 1500–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]