

Abstract
Background
In high grade serous ovarian cancer (HGSOC), there is a spectrum of sensitivity to first line platinum‐based chemotherapy. This study molecularly characterizes HGSOC patients from two distinct groups of chemotherapy responders (good vs. poor).
Methods
Following primary debulking surgery and intravenous carboplatin/paclitaxel, women with stage III–IV HGSOC were grouped by response. Patients in the good response (GR) and poor response (PR) groups respectively had a progression‐free intervals (PFI) of ≥12 and ≤6 months. Analysis of surgical specimens interrogated genomic and immunologic features using whole exome sequencing. RNA‐sequencing detected gene expression outliers and inference of immune infiltrate, with validation by targeted NanoString arrays. PD‐L1 expression was scored by immunohistochemistry (IHC).
Results
A total of 39 patient samples were analyzed (GR = 20; PR = 19). Median PFI for GR and PR patient cohorts was 32 and 3 months, respectively. GR tumors were enriched for loss‐of‐function BRCA2 mutations and had a significantly higher nonsynonymous mutation rate compared to PR tumors (p = 0.001). Samples from the PR cohort were characterized by mutations in MGA and RAD51B and trended towards a greater rate of amplification of PIK3CA, MECOM, and ATR in comparison to GR tumors. Gene expression analysis by NanoString correlated increased PARP4 with PR and increased PD‐L1 and EMSY with GR. There was greater tumor immune cell infiltration and higher immune cell PD‐L1 protein expression in the GR group.
Conclusions
Our research demonstrates that tumors from HGSOC patients responding poorly to first line chemotherapy have a distinct molecular profile characterized by actionable drug targets including PARP4.
Keywords: genomic profiling, high grade serous, immune profiling, ovarian carcinoma, platinum resistance
In patients with suboptimally‐debulked, advanced high grade serous ovarian cancer (HGSOC), the spectrum of responses to first line platinum‐based chemotherapy (PtC) vary from durable to non‐existent, highlighting an unmet medical need for those with poor responses. Our study molecularly characterizes a cohort of HGSOC patients with distinct responses, either good or poor, to PtC. The good and poor responders have median progression‐free intervals of 32 and 3 months, respectively, and molecular analysis of tumors identifies differences in DNA damage response pathways and immunity, namely, tumors from poor responders to first line PtC have a distinct molecular profile characterized by actionable drug targets including PARP4.

1. INTRODUCTION
Ovarian cancer is the leading cause of gynecological cancer mortality in North America. 1 The primary treatment for high grade serous ovarian cancer (HGSOC), the most common subtype of this disease, consists of surgical resection and combination platinum‐based chemotherapy. Although advanced stage, poor performance status, and residual disease (RD) >1 cm after debulking surgery are strong predictors of an adverse prognosis, sensitivity to first‐line carboplatin and paclitaxel chemotherapy is a critical factor in overall outcome. 2 However, not all patients who share adverse prognostic factors at diagnosis do poorly, suggesting there are underlying biological differences between tumors from patients at the extremes of the outcome spectrum. Patients with primary platinum‐resistance (disease‐free interval [DFI] < 6 months) have the worst prognosis with a median survival of only 9–12 months, and fewer than 15% of these patients respond to subsequent chemotherapy. 3 Given that the biology of platinum resistance is not fully understood and the tumor characteristics predicating a poor response to treatment are largely unknown, the development of new treatment strategies has been limited, representing an impediment to new and better treatments.
Many mechanisms have been proposed to explain the variability in treatment responses to platinum‐based chemotherapy, including active efflux, enzymatic inactivation of platinum agents, and increased DNA repair capacity. 4 Patients with platinum‐sensitive HGSOC (disease‐free interval ≥ 6 months) have seen an improvement in outcome largely attributed to the study of the DNA damage response (DDR) mechanisms as mediators of chemosensitivity. 5 Specifically, the exploitation of homologous recombination deficiency (HRD) has led to the development and approval of PARP inhibitors in platinum‐sensitive patients with germline and somatic BRCA1 and BRCA2 mutations and patients with tumors displaying HRD. 6 , 7 The characterization of HRD beyond BRCA1 and BRCA2 mutation status is an active area of research and includes the study of loss of heterozygosity, epigenetic silencing of the BRCA1 promotor, EMSY amplification, and the deletion of core HRD genes. 8 Furthermore, higher PD‐L1 expression has been noted in patients with defective DDR genes including BRCA1, 9 suggesting an interplay between DDR deficiency and the tumor/stromal immunophenotype, but does little to explain the >25% of patients with tumors that are resistant or refractory to platinum chemotherapy. 10
No biomarker/signature predictive of platinum‐resistance in HGSOC has yielded clinically translatable benefit 11 despite encouraging developments in gene expression profiling. This may be due to high levels of DNA instability in HGSOC leading to inconsistencies in reports. For instance, amplification of CCNE1, the gene encoding cell‐cycle checkpoint regulator Cyclin E1, has been identified as a potential marker of platinum‐resistant HGSOC 10 , 11 although conflicting survival data exists. 12 , 13 Therefore, the definition of tumor molecular characteristics associated with poor chemotherapy response is a crucial area of study with the potential to lead to early initiation of additional therapies, reduction in morbidity and cost associated with futile therapies, and informs the development of therapeutic alternatives to chemotherapy. We hypothesize that genomic and immunophenotypic features of HGSOC will characterize differential response to first‐line chemotherapy. In this study, we sought to explore these two aspects through analysis of tumors from primary debulking surgery using whole exome sequencing, whole transcriptome sequencing, targeted immuno‐ and DDR‐specific codeset profiling by NanoString gene expression panels, and immunohistochemistry (IHC).
2. MATERIALS AND METHODS
2.1. Study design and patient population
A retrospective chart review of HGSOC patients from 2004 to 2017 was conducted at The Ottawa Hospital Cancer Centre (TOHCC), Ottawa, Canada (Figure S1). Patients had advanced stage III or IV ovarian cancer, histologically confirmed HGSOC, suboptimal primary debulking with ≥1 cm RD remaining after surgery, receipt of at least four adjuvant cycles of first‐line chemotherapy with carboplatin/paclitaxel or cisplatin/paclitaxel doublet, and adequate archival tumor sample for molecular analysis. From 39 patients meeting eligibility criteria, patients were classified as having either a good response (GR, n = 20) or poor response (PR, n = 19), defined as progression‐free interval (PFI) ≥12 and ≤6 months. 12 PFI was defined as the time between last chemotherapy treatment and disease recurrence. Assessment of response to chemotherapy and disease progression was based on RECIST v 1.1 criteria or CA‐125 progression criteria as defined by the GCIG. 13 The study was approved by the Ottawa Health Science Network (#20150500‐01H) and University of Toronto Research Ethics Board (#34640). Consent was obtained for living patients in accordance with the declaration of Helsinki, a waiver of consent was obtained from the research ethics board for deceased patients. For enrolled patients, clinicopathologic data including PFI and overall survival (OS) were collected from the electronic medical record up to March 2018.
2.2. Sample preparation
Formalin‐fixed paraffin embedded (FFPE) tumor blocks were utilized for molecular analysis following quality review of a hematoxylin and eosin (H&E) slide. A reference pathologist (H.S.) identified specific tumor‐rich areas which were then macrodissected from 10‐µm tissue sections using a sterile razor blade followed by deparaffinization in 1 ml of xylene. Genomic DNA and total RNA were isolated using the Qiagen AllPrep FFPE Tissue Kit (QIAGEN) followed by fluorometric quantification using a Qubit 3.0 instrument (Life Technologies). RNA integrity was assessed using the Agilent TapeStation on a High Sensitivity RNA ScreenTape.
2.3. Whole exome and transcriptome sequencing
Transcriptome sequencing libraries were constructed from 200 ng of total RNA using the Illumina TruSeq Stranded Total RNA Library Prep Gold kit from which >69 million paired‐end sequencing reads were generated on the Illumina NextSeq550 platform using V2 chemistry and reagents. Exome libraries were made from 100 ng of genomic DNA using the Kapa HyperPrep Kit which were enriched using Agilent SureSelect XT Human All Exon V6+ COSMIC reagents. Exome libraries were sequenced to a mean exon coverage of 100X on the Illumina HiSeq2500 platform using V4 chemistry and reagents. All reads were processed following the GATK Best Practices framework including read alignment against the hg19 human reference using BwaMem v 0.7.12, 14 called somatic mutations using MuTect2 v 1.1 15 and annotated variants using Variant Effect Predictor v92. 16 As matched normal material was lacking, likely germline variants were removed with GnomAD population frequency >0.01% in any population (r2.0.1). 17 To control for potential FFPE‐induced sequencing artifacts, variants with allele fractions <10% were removed but retained OncoKB variants in the 5%–10% range. To assess allele‐specific copy number profiles, loss of heterozygosity, and estimates of purity and ploidy, we used CNVKIT v0.9.1 18 using a pooled reference set of 60 peripheral blood samples from individuals unrelated to the study. To identify candidate high level amplifications and homozygous deletions, we only considered copy number variations (CNVs) with log2R > 0.7 (high level gain) and <−0.7 (deep deletions) as used by convention in cBioPortal. 19
2.4. Transcriptome informatics
Total RNA data quality was assessed using FastQC and ReSeOC v 2.6.4 20 prior to read alignment with STAR aligner v2.6.0c. 21 BAM files were preprocessed similar to exome methods, except an additional trimming of soft‐clipped reads was performed prior to in/del realignment and base recalibration. RNA abundance was quantified with RSEM v1.3.0 22 to generate an expression matrix. RODIC 23 was used to identify expression outliers, ESTIMATE 24 for immunological gene signatures/infiltrates, and ssGSEA 25 for pathway analysis. HaplotypeCaller 26 was used to generate variant call files (VCF) prior to Variant Effect Predictor v92 16 analysis of annotated mutations. 27
2.5. Total RNA preparation and NanoString gene expression profiling
An H&E stain was performed on one tumor section per patient, and a blinded pathologist reviewed the sections for tumor content (M.R.). The tumor component was maccrodissected from FFPE, and RNA was extracted using RNeasy FFPE Extraction kit (Qiagen) following manufacturer's instructions. RNA was quantified using Qubit 2.0 Flourometer (Life Technologies) and hybridized 150 ng of each sample with the codeset at 65°C for 21 h and kept at 4°C < 1 h before preparing cartridges. Patient samples were randomly distributed in four cartridges and included tumor reference and background controls (water). NanoString assays were performed by following the standard protocol “Setting up 12 nCounter Assays (MAN‐C0003‐03, 2008–2013)”. AZ‐designed DDR‐max codeset was used in this project. Cartridges were read immediately after being prepared, on the AZ GEN2 Digital Analyzer station with high resolution selected (3 h enhanced, 555 fields of view captured). In addition to 27 housekeeping genes, the AZ‐custom designed DDR‐max codeset includes 753 genes from the core DDR pathways and (HRD, nonhomologous end‐joining [NHEJ], mismatch repair [MMR], base excision repair [BER], nucleotide excision repair [NER], and replication stress [RS]) and previously published gene signatures predicting for response to DNA damaging chemotherapy. 28 , 29 , 30 , 31 NanoString‐designed probes were verified to ensure recognition of the canonical gene transcripts and normalized each sample with the reference samples using nSolver Analysis Software version 4.0. We then performed a housekeeping genes normalization step (assuming constant gene expression across test samples).
2.6. Histology and immunohistochemistry
FFPE tissues were cut into 4‐µm serial sections and mounted on charged slides. The H&E was assessed by a blinded pathologist (M.R.) for the percentage of tumor surface containing immune cells (ICs) (lymphocytes, plasma cells, and macrophages). PD‐L1 staining was conducted using Ventana PD‐L1 (SP263) IHC assay, investigational use only, according to the manufacturer's instructions on the Benchmark Ultra stainer (Ventana). PD‐L1 IHC stained slides were blindly assessed for tumor cell (TCs) PD‐L1 expression and for percentage of PD‐L1 positive ICs (over total ICs). Ataxia telangiectasia mutated (ATM) IHC staining was performed using the ab32420 antibody as previously described, 32 and the percentage of ATM positive tumor cell nuclei was scored blindly.
2.7. Statistical analysis
For analyses where responder status was the predictor or independent variable, Fisher's exact test for categorical outcomes and an independent samples t test or a Wilcoxon rank‐sum test for continuous outcomes, significance cut‐off p < 0.05, were used. Associations of protein expression with clinical, pathological and molecular features, and chemotherapy response were evaluated with Fisher's exact and Wilcoxon–Mann Whitney tests, as required. To find genes associated with PFI, PFI was treated as the outcome variable in a survival model and gene expression values, as measured by the NanoString technology, were used as predictor variables in univariate analyses. PFI was modeled, adjusting for censoring, as a lognormal distribution and therefore patients with PFI values equal to zero had 0.5 added to the PFI value (all values must be >0).
3. RESULTS
3.1. Extreme response cohorts characterized by significant differences in PFI and OS
Chart review identified 39 patients that met study inclusion/exclusion criteria (Figure S1). Baseline characteristics were similar in the two extreme response groups (Table 1). There were no statistically significant differences in age, stage, BRCA1 and BRCA2 carrier status, residual disease, vascular invasion, or number of lines of therapy between the two groups. There was a higher median CA125 in the PR group compared to the GR group (1678 vs. 599, p = 0.04). At data cut‐off, the median PFI for GR and PR cohorts was significantly different at 32 and 3 months (p < 0.001), respectively. The median OS was 65.5 months in the GR group and 23 months in the PR group (p < 0.001), in support of the two groups having extremes in overall outcomes. Patients with pathogenic germline BRCA1 and BRCA2 mutations trended towards both a higher PFI and OS than the germline BRCA1 and BRCA2 negative patients.
TABLE 1.
Study population demographic characteristics
| Total (n = 39) | Good responders (n = 20) b | Poor responders (n = 19) b | p value | |
|---|---|---|---|---|
| Age at diagnosis, years | ||||
| Mean age at diagnosis (range) [CI] | 59.4 (39−82) [56.3−62.5] | 59.2 [54.0−64.4] | 59.7 [55.8−63.5] | 0.88 |
| Stage | ||||
| 3 | 36 | 20 | 16 | 0.11 |
| 4 | 3 | 0 | 3 | |
| Ca125 | ||||
| Value at diagnosis (mean) | 2254 | 1526 | 2982 | 0.04 |
| Value at diagnosis (median) | 1085 | 599 | 1678 | |
| BRCA germline status a | ||||
| Positive (Carrier) | 5 | 4 | 1 | 0.18 |
| Negative (WT) | 31 | 14 | 17 | |
| Unknown | 3 | 2 | 1 | |
| Residual disease | ||||
| Mean (in cm) [CI] | 5.4 [4.3−6.4] | 4.4 [3.1−5.8] | 6.4 [4.7−8.1] | 0.069 |
| Vascular invasion | ||||
| Positive | 18 | 11 | 7 | 0.73 |
| Negative | 16 | 8 | 8 | |
| Not documented | 5 | 1 | 4 | |
| Total lines of chemotherapy | ||||
| Mean [CI] | 2.8 [2.3−3.3] | 2.4 [1.6−3.1] | 3.2 [2.6−3.8] | 0.09 |
| Progression‐free interval | ||||
| Median (months) | 10 | 32 | 3 | <0.001 |
| gBRCA+ | 11 | 50.5 (4) | 0 (1) | |
| gBRCA− | 6 | 32.5 (14) | 4 (17) | |
| Unknown | 17 | 25.5 (2) | 3 (1) | |
| Overall survival | ||||
| Median (months) | 41 | 65.5 | 23 | <0.001 |
| gBRCA+ | 60 | 76.5 (4) | 37(1) | |
| gBRCA− | 41 | 71.5 (14) | 23 (17) | |
| Unknown | 29 | 33.5 (2) | 16 (1) | |
Abbreviations: CI, 95% confidence interval; WT, wild‐type.
BRCA1 and BRCA2 germline mutation status as assessed by local testing.
Sample size shown within parentheses.
3.2. Mutations of BRCA2 but not BRCA1 distinguish GR and PR cohorts
Exome evaluation of somatic mutations in all 39 tumors revealed previously identified genes of relevance in HGSOC including BRCA1, BRCA2, and TP53, as well as additional oncogenic mutations in genes associated with PI3K/AKT/mTOR signaling, DDR and epigenetic regulation (Figure 1; Table S1). Comparison of tumor mutation burden found that GR tumors had significantly higher non‐synonymous mutation rate compared to the PR cohort (median 5.52 vs. 4.47 nonsynonymous mutations per callable Mb, p = 0.001). The most frequently mutated genes with known relevance to cancer were TP53 (39/39 cases, 100%), BRCA1 and BRCA2 (15/39 including five germline, 38%), DDR genes MLH3, MSH2, MSH6, and RAD51B (8/39, 21%, none hypermutant), PIK3CA (5/39 all missense hotspot mutations, 13%), MGA (4/39, 10%), CSMD3 (3/39, 8%), NF1 (3/39, 2 frameshift, 8%), AURKA (2/39, 5%), and CNTRL (2/39, 5%). Of the eight somatic BRCA2 mutations, seven were found in GR tumors (p = 0.044) and included an in‐frame deletion, two nonsense mutations, two frameshift insertions, and two missense mutations (one located in the helical domain). The single BRCA2 mutation in a PR tumor was a p. Arg2108Lys missense mutation outside of a functional domain. Whereas BRCA2 nonsense, insertion/deletion, and splice‐site loss‐of‐function (LOF) mutations were found almost exclusively in the GR group (p = 0.047, Fisher's exact test), LOF BRCA1 mutations were found in both GR and PR tumors (5/20 vs. 3/19, p = 0.45, Fisher's exact test). LOF mutations in MGA (2/39) and RAD51B (2/39) mutations were only found in PR tumors.
FIGURE 1.
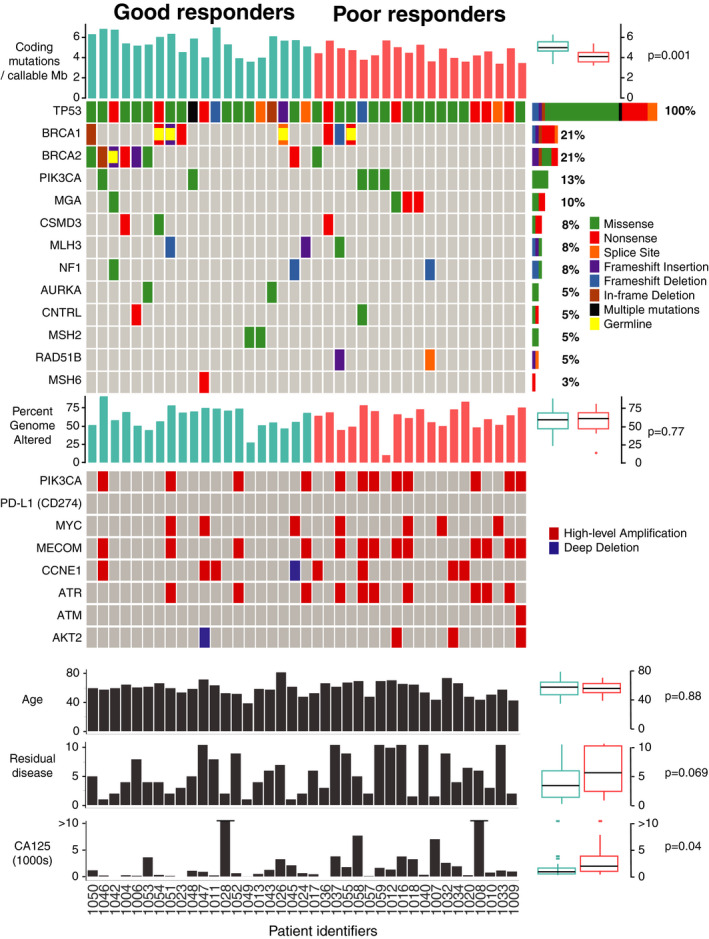
Summary of genetic alterations in the good and poor response cohorts to first line platinum doublet chemotherapy. Each column represents one patient's tumor sample. Tumor mutation burden (TMB) expressed as the number of nonsynonymous mutations per number of callable coding bases (Mb) in each sample. The value for each case is represented by a bar graph, with a summary of results to the right of the panel, expressed as a boxplot with the median bracketed by first and third quartiles and whiskers showing points within 1.5 times the interquartile range and outliers shown as individual points. p = 0.001 by Wilcoxon rank‐sum test. OncoPrint summary of genetic alterations (as described in adjacent legend) in the most frequently altered genes for each tumor sample. Overall frequency of alterations for each gene are listed as a percentage to the right of the panel. BRCA1 and BRCA2 are marked as germline mutations if germline status was reported in the clinical chart. Percent genome altered (including amplification and deletions) is shown, per subject, in a bar graph and summarized to the right of the panel as a boxplot, with p = 0.77 by Wilcoxon rank‐sum test. Amplifications (defined as CNVs with log2R > 0.7) and deletions (defined as CNVs with log2R < −0.7) are visually represented in an oncoprint diagram for each case. Clinical characteristics (age, residual disease, and CA125) are expressed for each patient in a bar graph, with the boxplot summary to the right of the panel, p values were calculated using the Wilcoxon rank‐sum test
Tumors were characterized by considerable aneuploidy consistent with HGSOC in both GR and PR patients, yet the percentage of genome altered by a copy number amplification or deletion was not significantly different between the two groups (median 61.32% vs. 62.32%, p = 0.77) (Figure 1; Table S2). An analysis of candidate genes associated with DDR and PI3K/AKT/mTOR signaling showed a trend towards greater copy number amplifications in the PR group. Specifically, PR tumors had greater number of high‐level copy number amplifications affecting PIK3CA (GR 4/20 vs. PR 8/19), MECOM (4/20 vs. 9/19), and ATR (3/20 vs. 7/19). CCNE1 and MYC were each amplified in 18% of all cases (7/39), and this number was nearly equally distributed among PR and GR groups. In the GR group, one deep deletion was observed in CCNE1.
3.3. Differentially expressed DDR genes are correlated with chemotherapy response patterns
To test whether there was correlation between clinical response and expression of DDR genes, a specific DDR panel codeset for NanoString analysis was applied, specifically suited to provide high‐quality results in RNA extracted from FFPE tissue. The analysis by NanoString nCounter of 753 DDR genes of the 39 HGSOC tissues (20 GR, 19 PR) showed that 127 genes were expressed at significantly higher levels in GR patients, whereas 13 genes were significantly more expressed in PR patients. TFIIH, POLR2B, CCNC, LIG3, POLD3, RFC2, POLE3 genes were amongst the most highly expressed in GR samples. In contrast, PR tumors had elevated levels of PARP4, POLK, CDK7, RAD17 and TP53BP1 (Figure 2A; Table S3). Analysis of protein association networks carried out for the 127 genes or selected subsets (top 20% of genes with highest correlation to PFI (Figure 2C; Figure S2A), or top 20% of genes with the lowest p value) showed that GR tumors were enriched for expression of genes involved in DNA replication/cell cycle, BER, NER, and HRD pathways (measured by lowest false discovery rate [FDR]) (Figure S2C). In contrast, the PR group showed an enrichment of genes associated with negative regulation of cell cycle (i.e., PTEN, APC, and VASH1). The PR tumors also showed an enrichment of genes involved in NER (Figure 2B; Figure S2B).
FIGURE 2.
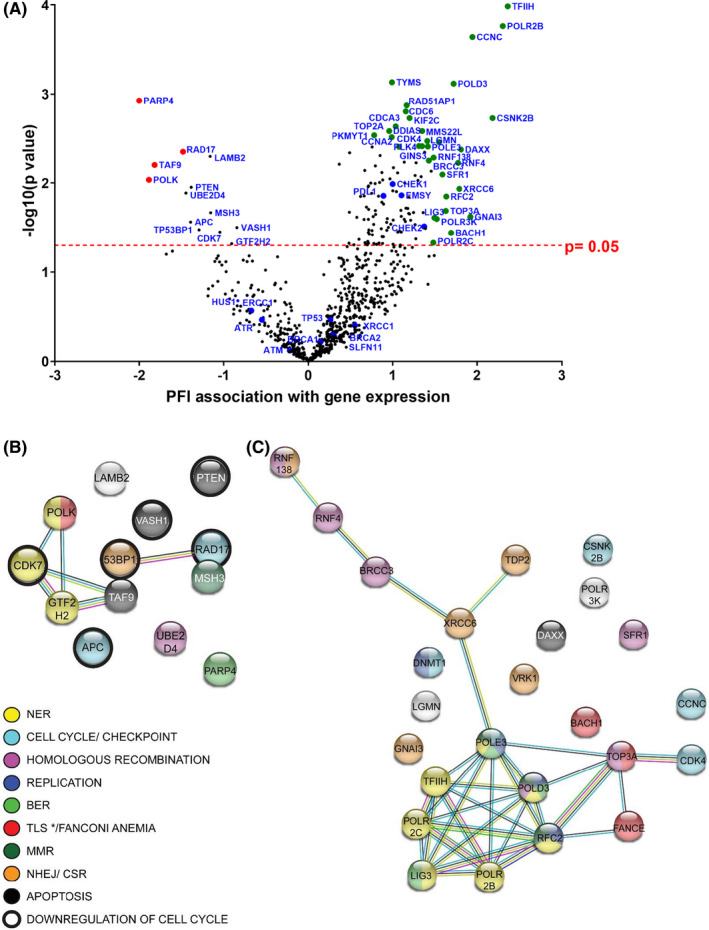
Transcriptomic analysis of tumor samples from good (GR) and poor (PR) responders using NanoString DDRmax codeset. (A) Volcano plot showing correlation of gene expression with patient PFI (X axis, greater value correlates with longer PFI). Y axis represents the level of confidence of the statistical analysis for each gene (p value 0.05 is depicted by a dashed line and was used as a cut‐off value). Green and red dots: genes best correlated to long and short PFI, and with lowest p value (list expanded in Table S3), blue dots: DDR genes closely examined. Protein association networks of top genes the expression of which correlates best with shorter PFI (B) and longest PFI (C) (left and right genes on volcano plot, respectively), with a p value < 0.05. Color code refers to curated KEGG pathways, where * denotes that TLS is specific to the short PFI network. Pathways were generated with String software. Red color refers to Translesion synthesis in panel (B) and Fanconi Anemia pathway in panel (C). BER, base excision repair; MMR, mismatch repair; NER, nucleotide excision repair; NHEJ/CSR, nonhomologous end joining/class‐switch recombination; TLS, translesion synthesis
The expression levels of selected genes known to be relevant in DDR pathways were examined by NanoString nCounter (Figure 3). Expression of two genes implicated in the response to DNA damaging chemotherapy were significantly higher in the GR group, EMSY (p = 0.008), a repressor of BRCA2 transactivation; and programmed death ligand 1 (PD‐L1) (CD274) (p = 0.014), an immune inhibitory receptor ligand frequently used as a biomarker for immune checkpoint inhibition (Figure 3). There were no amplifications in EMSY or CD274 in any case tested (CD274 shown in Figure 1). Although the ATR gene was more frequently amplified in the PR group (Figure 1), this did not translate to a significant difference in ATR mRNA expression between GR and PR groups when pooled (Figure 3). Furthermore, although ATR gene expression (mRNA z‐Scores) from RNA‐seq trended towards correlation with gene copy number, NanoString analysis showed no statistically significant difference in mRNA log2 gene expression when amplified cases were examined compared to all others, independently of platinum‐response (Figure S3A,B). ATM expression was also evaluated and although all samples were ATM‐expressing, neither RNA expression nor protein expression (IHC) was differential between response groups (Figure 3; Figure S4A). In addition, no notable DNA alterations to ATM were found (data not shown). Furthermore, no significant differences in mRNA expression were found between the GR and PR groups for the DDR genes: BRCA1, BRCA2, ERCC1, SLFN11, TP53, and XRCC1 (Figure 3).
FIGURE 3.
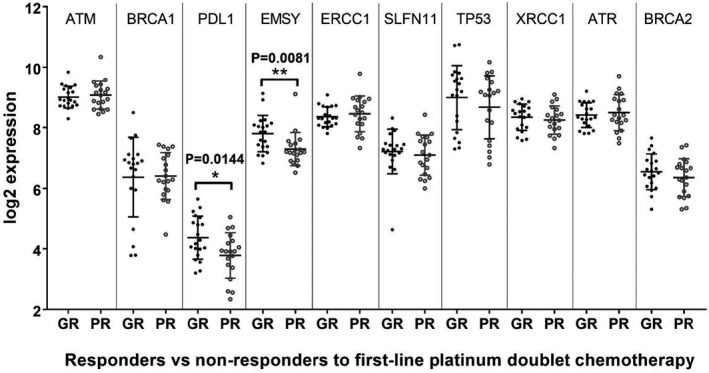
Transcriptomic analysis of tumor samples from good (GR) and poor (PR) responders showing selected NanoString gene expression data. Gene expression (log2 expression) values for selected genes shown with mean ± SEM for each response group, where GR, good response; PR, poor response. Data analyzed with R‐limma package (version 3.30.13), multiple hypotheses not tested
Increased PD‐L1 mRNA levels in the GR group correlated with BRCA2 mutation status (p = 0.013) but not with BRCA1 mutations (Figure 4A) and PD‐L1 protein expression in tumor immune cells was significantly higher in those patients with mutations in BRCA2 (p = 0.029) (Figure 4B). EMSY expression did not significantly correlate with BRCA status (Figure S4B).
FIGURE 4.
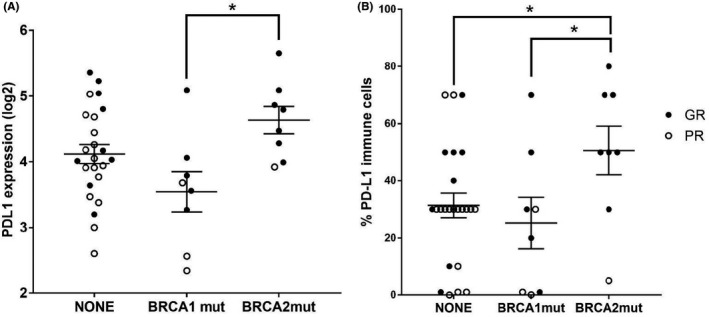
Transcriptomic and immunohistochemical (IHC) analysis of tumor samples from good (GR) and poor (PR) responders establishes a link between BRCA mutation status and PD‐L1 expression in tumors and tumor associated immune cells. (A) PD‐L1 mRNA expression in tumors from both response groups classified by BRCA1 and BRCA2 mutation status. Data shown as mean ± SEM (* indicates p = 0.013). (B) Percentage of PD‐L1 protein expression in tumor immune cells, assessed by IHC, within tumors from both response groups, based on BRCA1 and BRCA2 mutation status. Data shown as mean ± SEM (* and ** indicate p = 0.029 and p = 0.01, respectively)
3.4. Increased intratumoral inflammation and immune cell PD‐L1 expression correlates with chemotherapy response
Based on the positive association of PD‐L1 gene expression with favorable chemotherapy response, PD‐L1 IHC was conducted to confirm increased protein expression and delineate tissue expression patterns. Intratumoral inflammation estimated by H&E was significantly higher in the GR group at 24% (±16% SD) than in the PR group at 8% (±7% SD) (Figure 5A,D). In addition, the mean percentage of PD‐L1 expressing immune cells by IHC was higher in GR tumors than in PR tumors at 42% (±23% SD) and 24% (±21% SD), respectively (p < 0.001 Mann–Whitney) (Figure 5B,E,F), based on the number of positive cells. PD‐L1 protein expression was low in tumor cells, and there was no difference in PD‐L1 expression between the two groups (Figure 5C). Immune cell infiltration was either patchy in the stroma (Figure 5E), observed at the invasive margin (Figure S5A) or diffuse in between tumor cells (Figure S5B).
FIGURE 5.
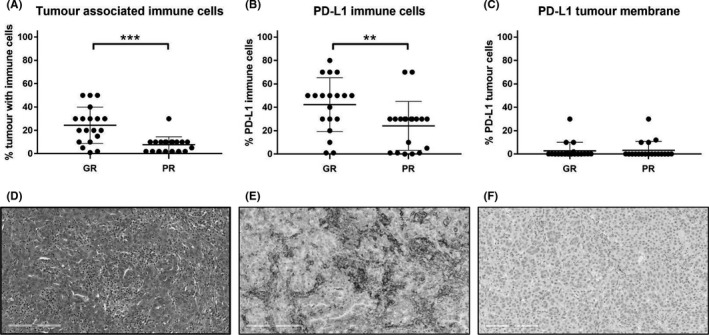
Immunohistochemical evaluation of patient tumor specimens: immune cell infiltration and PD‐L1 expression on the surface of tumor cells (TC) and immune cells (IC). (A) Percentage of tumor associated immune cells in responders compared to nonresponders to first‐line platinum doublet chemotherapy, using H&E ***p < 0.001. (B) Amongst immune cells (ICs), percentage of PD‐L1 positive ICs were significantly higher in responders as compared to nonresponders. (PD‐L1 positivity defined as 25% membrane positivity in the absence of an established definition for HGSOC) **p < 0.01. (C) Percentage of PD‐L1 positive tumor cells in both response cohorts. No difference between GR and PR groups. (D) Representative H&E of a good responder tumor with dense inflammation (immune cells are morphologically lymphocytes, plasma cells and macrophages), 20×. (E) Same tumor as in (D) stained with SP263 showing patchy infiltration of tumor stroma by PD‐L1 positive immune cells. PD‐L1 (SP263) 20×. (F) Poor responder tumortumor with absence of positive PD‐L1 tumor associated immune cells. PD‐L1 (SP263) 20× (PD‐L1 antibody: SP263, IUO Ventana Benchmark assay)
To assess whether these patterns may be associated with specific immune infiltrates, RNA‐seq of 29 tumors (Table S5) was performed to assess total infiltration (ESTIMATE algorithm) and infiltration of specific immune cell types (CIBERSORT). Overall, we did not find a significant difference in aggregate immune or stromal scores between the GR and PR groups (Figure S6), nor were there significant differences using a focused analysis of 22 immune cell signatures (Figure S7). Ultimately, larger cohorts or high‐resolution single cell expression analysis may yield insight into these patterns.
4. DISCUSSION
4.1. Patients with similar clinical and demographic characteristics exhibit distinct survival outcomes in HGSOC
Platinum resistance poses a great clinical challenge in the treatment of HGSOC as up to 30% of patients develop recurrent disease within 6 months of first line treatment 10 and virtually all patients eventually fail platinum therapy. In the treatment of patients with suboptimal debulking, the addition of bevacizumab to systemic chemotherapy is a therapeutic option; however this strategy lacks a predictive biomarker and has no overall survival benefit when used in the front‐line setting. 33 The options for treatment of poor prognosis patients in the first‐line setting are limited, stressing the need to explore novel therapies. In this study, we compare the molecular profile of tumors from patients undergoing primary debulking surgery, who have had good and poor responses to first‐line platinum‐based chemotherapy.
The GR and PR cohorts in the present study are noteworthy for their extreme differences in median PFI and OS (32 months vs. 3 months, and 65.5 months vs. 23 months, respectively). In suboptimally debulked stage III, HGSOC patients treated with platinum doublet chemotherapy, the previously reported PFI and OS were 14.1–16.9 and 35–45.1 months, respectively, 2 suggesting that our cohort truly represents two distinct outcome groups. Given our limited cohort of patient specimens with variable responses, it is noteworthy that our molecular findings corroborate results from large‐scale genomic characterisation efforts in stage II–IV HGSOC tumors. Thus, suggesting that many molecular features are in fact characteristics of the disease, rather than markers of response. An example of this is the ubiquitous mutation rate of TP53, which is 100% in our tumor cohort. 34 , 35
4.2. Alterations in genes responsible for DNA repair mechanisms characterize GR and PR groups
The BRCA1and BRCA2 mutation rate of 38% (15/39) in the present study is higher than the The Cancer Genome Atlas (TCGA) reported rate of 22%, 35 although the selection of good responders likely enriches our BRCA mutant cohort. Our finding that BRCA2 mutations were significantly more frequent in the GR group is in accordance with evidence that mutations in BRCA1 and especially BRCA2, confer chemo‐sensitivity in ovarian cancer. 5 Retrospective studies have correlated BRCA1 and BRCA2 mutations, higher tumor mutation burden and infiltration by T cells with improved survival in HGSOC, 36 , 37 and BRCA2 mutations were found to be enriched among long‐term responders to PARP inhibition. 38
Aside from expected mutations in BRCA1 and BRCA2 and TP53, mutated tumor suppressor genes are uncommon in HGSOC. 35 Rather, HGSOC is characterized by marked genomic instability, with frequent DNA gains and losses and up to 50% of tumors demonstrating HR deficiency. 35 Terms like BRCAness and HRD characterize the myriad genes that can produce a defective DNA repair phenotype and influence sensitivity to agents such as chemotherapy and PARPi. One gene belonging to this group is EMSY, a repressor of BRCA2 transactivation that localizes to sites of DNA repair and the amplification of which has been associated with an adverse prognosis. 39 Amplification of EMSY, and consequent suppression of BRCA2 transcriptional activity, has been proposed as an additional mechanism of defective DNA double‐strand break (DSB) repair in HGSOC. 40 TCGA data suggest that the impact of BRCAness on the chemo‐responsiveness of a tumor is dependent on the mechanism by which BRCA is silenced (e.g., mutation vs. epigenetic silencing do not carry the same predictive value). 35 In this study, there were no amplifications or mutations in EMSY at the DNA level, but EMSY mRNA levels were significantly increased within the GR group, supporting a role for EMSY upregulation as a marker of chemosensitivity.
Ataxia telangiectasia mutated (ATM) and Rad3‐related protein (ATR) are core players in the DDR pathway, with central roles coordinating response to DSBs and replication stress (RS). 41 RS represents the initial insult from stalled or collapsed DNA replication forks during oncogenesis, which places ATR in a pivotal position to confront growing genomic instability within a developing cancer. Therefore, this provides rationale for ATR's role in inherent chemotherapy resistance, as it stabilizes stalled replication forks and allows DSB repair. 42 Although ATR was not mutated in our cohort and its expression was not significantly different in GR and PR samples, we report ATR amplification, as determined by exome sequencing, at a higher frequency (26%, 10/39 cases) than in the TCGA datasets (8%). 19 , 43 When correlated with response, the divide grew, with ATR amplification noted in 15% of GR and in 37% of PR, but this was not statistically significant in this relatively small cohort.
4.3. Differential genetic alterations in MYC/PI3K pathways between response groups
Amplification of Chromosome 3q (Chr3q) has been frequently described in up to 50% of HGSOCs. 44 , 45 Our study noted amplifications in several genes of interest located on Chr3q including ATR, MECOM, and PIK3CA. That PIK3CA, which is commonly amplified in HGSOC (18%), 35 was amplified in our study predominantly in the PR group (42% of cases), is noteworthy. The amplification rate of MECOM and CCNE1 in our study cohort was consistent with TCGA data. 35 Our study also found various genetic alterations detected in MGA that were concentrated in the PR group. MGA, a transcription factor which interacts with c‐MYC, is a putative tumor suppressor gene. Inactivating mutations in MGA may contribute to solid tumor development and have been detected in colorectal cancer, adenocarcinomas of the lung and small‐cell lung cancers. 46 , 47
4.4. NanoString gene expression analysis highlights differential expression of DDR gene between response groups
The broad screen of mRNA expression data using the NanoString DDRmax codeset showed association between longer survival and high expression of genes involved in DNA replication, NER, BER, and HR repair pathways.
The expression of genes such as TFIIH, POLR2B, POLR2C, CCNC, RFC2, LIG3, and POLD3, involved in NER and transcription, correlated with PFI (Figure 2A). We also observed an enrichment of NER genes in the PR group, which is in agreement with previous data relating high expression of NER genes to platinum resistance in ovarian carcinoma, as platinum‐DNA adducts are resolved by NER mechanisms. 48 These data are also consistent with other reports showing that low expression of POLD3 is a marker of poor prognosis. 49 Select HR genes were also overexpressed in patients with long PFI, as it is the case of MMS22L, RAD51AP1, RAD54B, and RAD54L. Noticeably, the overexpression of genes involved in DNA replication (CDC6, CDT1, MCM2, MCM3, MCM5, MCM10, CDK2, CCNA2) in patients with long PFI links enhanced replication potential, on which platinum relies to exert DNA damage, with GR (Figure 2A; Figure S2A; Table S3). Conversely, genes highly expressed in tumors from individuals with short PFI were enriched in processes that negatively regulate cell cycle progression. This is in agreement with previous reports 50 suggesting that a slower proliferation rate would enable cells to repair damaged DNA. In the PR group, genes PARP4 and RAD17 were more highly expressed (Figure 2A). RAD17 recruits the 9‐1‐1 complex in response to RS to activate ATR, hence its higher expression in patients with lower PFI could contribute to their resistance to platinum therapy. ATR and HUS1 (which encodes one of the components of the 9‐1‐1 complex) exhibited the same trend but lacked significance (below cut‐off in Figure 2A). Therefore, more efforts into the study of DNA replication genes as potential biomarkers predicting the efficiency of platinum‐based therapy are warranted.
Several reports link PARP4 overexpression with multidrug resistance genes such as MVP, and, in particular, it was shown by IHC methods that higher levels of PARP4 correlate with higher grade ovarian cancer. 51 , 52 Furthermore, high expression of PARP4 has been reported in breast cancer with poor outcomes. 53 Several PARP inhibitors have already been approved for cancer treatment and, in light of our results and those of previous reports, PARP4 emerges as a candidate actionable target for platinum‐resistant HGSOC.
4.5. Higher immune context in the GR cohort linked to BRCA2 mutations
Our finding that patients with good chemotherapy response have a higher tumor mutation burden is consistent with other studies. 37 , 54 , 55 Evidence supports that the predicted neo‐antigen load is higher in BRCA1‐ and BRCA2‐mutant and HR‐deficient cancers, 54 owing to the theory that impaired DDR leads to genetic alterations and putative neo‐antigens, thus favoring recognition by the immune system.
The connection between BRCA1 and BRCA2 gene mutations and PD‐L1 expression has been previously described in HGSOC, with some discordance in the literature. This study makes the important observation that tumors from patients with BRCA2 mutations had increased PD‐L1 mRNA expression and a higher percentage of PD‐L1 protein expression in tumor‐associated ICs compared to patients with BRCA1 mutations. This is in agreement with two studies reporting that PD‐L1 expression correlated with BRCA1 and BRCA2 mutation status, 54 , 56 but in contrast to another, which reported no association with BRCA1 or BRCA2 mutation, or somatic mutation load, using TGCA data. 57 PD‐L1 expression and CD8+ TILs are associated with favorable prognosis in HGSOC and this was found regardless of the extent of RD following cytoreduction, receipt of standard treatment and germline BRCA1 status. 57 , 58 , 59 Taken together, our results, support the existing body of literature characterizing BRCA1 and BRCA2 mutations as predictive of chemo‐response and potentially associated with high tumor immune cell infiltration and PD‐L1 IHC expression. However, this observation does not necessarily correlate with long‐term survival benefit. 56
In primary ovarian tumors, PD‐L1 expression has been predominantly reported on immune cells, rather than on tumor cells 57 , 60 and our results corroborate a low tumor cell expression level of PD‐L1. Although our analyses examined all immune cells, increased PD‐L1 IC expression has been primarily reported in CD65+ tumor‐associated macrophages. Expression of PD‐L1 in HGSOC has been described as focal and patchy in one large study and the frequency of CD8+ TIL positivity (Grade 2/3 and Stage I–III) is reported to be around 95%, with 57.4% of cases being PD‐L1 positive and 37.4% being PD‐L1 negative. 57 This high positivity rate is greater than we observed but the discrepancy may be due to methodology differences and the immune cell types examined. Agents that recruit CD8+ T cells to the tumor milieu such as immunostimulants (i.e., cytokines) and DNA damaging agents that promote immunogenic cell death (such as chemotherapy), may alter the tumor microenvironment to favorably influence therapeutic responses and therefore new combination therapies are an area of clinical interest.
4.6. Future directions
In this analysis, as no matched normal control samples were available, it was not possible to definitively identify BRCA1 and BRCA2 mutations as somatic or germline in all patients. Results of germline testing were not available on all patients at the time of data collection. As we did not have matched normal controls, all genomic findings underwent stringent filtering to ensure false positive signals were sufficiently reduced. The differential amplification in PIK3CA, MECOM, and ATR while interesting signals require validation using an orthogonal method and a larger cohort given the high degree of aneuploidy found in ovarian carcinoma.
Despite these limitations, our gene expression data supports a clear role for DNA repair genes in both the favorable and poor response groups. Our work suggests interplay between DDR pathways and the tumor immune microenvironment in shaping platinum response. Durable remissions were associated with a high tumor mutation burden, evidence of DNA repair disruption (via BRCA2 mutations and EMSY overexpression), and tumor infiltration of PD‐L1 expressing immune cells. Platinum resistant tumors were characterized by increased expression of PARP4, NER pathway genes, and lower expression of DNA replication genes as compared to platinum sensitive tumors. These findings may be applied to larger studies to advance therapeutic options for patients experiencing inherent platinum resistance.
CONFLICT OF INTEREST
GG has acted as principal investigator on an AstraZeneca‐sponsored clinical trial, outside the submitted work. GG and JW have participated in AstraZeneca advisory boards, outside the submitted work. GG has received travel grants from AstraZeneca, outside the submitted work. GG and JW have received honoraria from AstraZeneca, outside the submitted work. MR, GNJ, SEL, PMC, DH, and NL are/were employees of AstraZeneca, and MR, GNJ, PMC, DH, and NL own AstraZeneca stock. The other authors declare no potential conflicts of interest relevant to this publication.
AUTHOR CONTRIBUTIONS
Johanne I Weberpals: study conception, study design, data acquisition, data analysis, data interpretation, manuscript writing, manuscript review, and approval; Trevor J. Pugh: study design, data acquisition, data analysis, data interpretation, manuscript writing, manuscript review, and approval; Paola Marco‐Casanova: study design, data acquisition, data analysis, data interpretation, manuscript writing, manuscript review, and approval; Glenwood D. Goss: study conception, study design, data analysis, data interpretation, manuscript review, and approval; Natalie Andrews Wright: data acquisition, data analysis, data interpretation, manuscript writing, manuscript review, and approval; Prisni Rath: data acquisition, data analysis, data interpretation, manuscript review, and approval; Jonathon Torchia: data acquisition, data analysis, data interpretation, manuscript review, and approval; Alexander Fortuna: data acquisition, data analysis, data interpretation, manuscript review, and approval; Gemma N Jones: data acquisition, data analysis, data interpretation, manuscript review, and approval; Martine P Roudier: data acquisition, data analysis, data interpretation, manuscript review, and approval; Laurence Bernard: data analysis, data interpretation, manuscript review, and approval; Bryan Lo: data analysis, data interpretation, manuscript review, and approval; Dax Torti: data acquisition, data analysis, data interpretation, manuscript review, and approval; Alberto Leon: data acquisition, data analysis, data interpretation, manuscript review, and approval; Kayla Marsh: data acquisition, manuscript review, and approval; Darren Hodgson: study conception, study design, data interpretation, manuscript review, and approval; Marc Duciaume: data acquisition, data analysis, manuscript review, and approval; William J Howat: data acquisition, manuscript review, and approval; Natalia Lukashchuk: data analysis, data interpretation, manuscript review, and approval; Stanley E. Lazic: data analysis, data interpretation, manuscript review, and approval; Doreen Whelan: data acquisition, data analysis, manuscript review, and approval; Harmanjatinder S. Sekhon: study design, data acquisition, data analysis, data interpretation, manuscript review, and approval.
Supporting information
Fig S1‐S7
Table S1
Table S2
Table S3
ACKNOWLEDGEMENTS
The authors wish to thank the patients included in this study for their participation, as well as Dr. Tim Ramsay (statistician, OHRI) for his consultation on this manuscript.
Funding information
This research was conducted with support from AstraZeneca Canada Inc. and with the support of the Ontario Institute for Cancer Research through funding provided by the Government of Ontario, Ministry of Research, Innovation and Science and the Princess Margaret Cancer Foundation. TJP is supported by the Canada Research Chairs program.
DATA AVAILABILITY STATEMENT
Some of the data that supports the findings of this study are available in the supporting information of this article (i.e., deidentified data), and the remainder of the data that support the findings of this study are available on request from the corresponding author, these are not publicly available due to privacy or ethical restrictions (i.e., associations of clinical data and molecular data).
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7‐34. [DOI] [PubMed] [Google Scholar]
- 2. Winter WE, Maxwell GL, Tian C, et al.; Gynecologic Oncology Group Study . Prognostic factors for stage III epithelial ovarian cancer: a gynecologic oncology group study. J Clin Oncol. 2007;25(24):3621‐3627. [DOI] [PubMed] [Google Scholar]
- 3. Davis A, Tinker AV, Friedlander M. “Platinum resistant” ovarian cancer: what is it, who to treat and how to measure benefit? Gynecol Oncol. 2014;133(3):624‐631. [DOI] [PubMed] [Google Scholar]
- 4. Galluzzi L, Senovilla L, Vitale I, et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31(15):1869‐1883. [DOI] [PubMed] [Google Scholar]
- 5. Alsop K, Fereday S, Meldrum C, et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation‐positive women with ovarian cancer: a report from the Australian ovarian cancer study group. J Clin Oncol. 2012;30(21):2654‐2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in platinum‐sensitive relapsed ovarian cancer. N Engl J Med. 2012;366(15):1382‐1392. [DOI] [PubMed] [Google Scholar]
- 7. Ledermann JA. PARP inhibitors in ovarian cancer. Ann Oncol. 2016;27(September):i40‐i44. [DOI] [PubMed] [Google Scholar]
- 8. Zhang S, Yuan Y, Hao D. A genomic instability score in discriminating nonequivalent outcomes of BRCA1/2 mutations and in predicting outcomes of ovarian cancer treated with platinum‐based chemotherapy. PLoS One. 2014;9(12):e113169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Clarke B, Tinker AV, Lee C‐H, et al. Intraepithelial T cells and prognosis in ovarian carcinoma: novel associations with stage, tumor type, and BRCA1 loss. Mod Pathol. 2009;22(3):393‐402. [DOI] [PubMed] [Google Scholar]
- 10. Cannistra SA. Cancer of the ovary. N Engl J Med. 2004;351(24):2519‐2529. [DOI] [PubMed] [Google Scholar]
- 11. Colombo P‐E, Fabbro M, Theillet C, Bibeau F, Rouanet P, Ray‐Coquard I. Sensitivity and resistance to treatment in the primary management of epithelial ovarian cancer. Crit Rev Oncol Hematol. 2014;89(2):207‐216. [DOI] [PubMed] [Google Scholar]
- 12. Stuart GCE, Kitchener H, Bacon M, et al. 2010 Gynecologic Cancer InterGroup (GCIG) consensus statement on clinical trials in ovarian cancer: report from the fourth ovarian cancer consensus conference. Int J Gynecol Cancer. 2011;21(4):750‐755. [DOI] [PubMed] [Google Scholar]
- 13. Rustin GJS, Vergote I, Eisenhauer E, et al.; Gynecological Cancer Intergroup . Definitions for response and progression in ovarian cancer clinical trials incorporating RECIST 1.1 and CA 125 agreed by the Gynecological Cancer Intergroup (GCIG). Int J Gynecol Cancer. 2011;21(2):419‐423. [DOI] [PubMed] [Google Scholar]
- 14. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics. 2009;25(14):1754‐1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McLaren W, Gil L, Hunt SE, et al. The ensembl variant effect predictor. Genome Biol. 2016;17(1):122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: Genome‐wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol. 2016;12(4):e1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data: figure 1. Cancer Discov. 2012;2(5):401‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Andrews S. FastQC: a quality control tool for high throughput sequence data [Internet]. 2010. Available from http://www.bioinformatics.babraham.ac.uk/projects/fastqc
- 21. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics. 2013;29(1):15‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA‐Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Torchia J. RNA Seq Outlier Detection (RODIC). 2018.
- 24. Yoshihara K, Shahmoradgoli M, Martínez E, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545‐15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Poplin R, Ruano‐Rubio V, DePristo MA, et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv. 2017. [Google Scholar]
- 27. McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 2010;20(9):1297‐1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ihnen M, zu Eulenburg C, Kolarova T, et al. Therapeutic potential of the poly(ADP‐ribose) polymerase inhibitor rucaparib for the treatment of sporadic human ovarian cancer. Mol Cancer Ther. 2013;12(6):1002‐1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Konstantinopoulos PA, Spentzos D, Karlan BY, et al. Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. J Clin Oncol. 2010;28(22):3555‐3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mulligan JM, Hill LA, Deharo S, et al. Identification and validation of an anthracycline/cyclophosphamide–based chemotherapy response assay in breast cancer. J Natl Cancer Inst. 2014;106(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Peng G, Chun‐Jen Lin C, Mo W, et al. Genome‐wide transcriptome profiling of homologous recombination DNA repair. Nat Commun. 2014;5:3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Villaruz LC, Jones H, Dacic S, et al. ATM protein is deficient in over 40% of lung adenocarcinomas. Oncotarget. 2016;7(36):57714‐57725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Burger RA, Enserro D, Tewari KS, Brady MF, Bookman MA, Fleming GF. Final overall survival (OS) analysis of an international randomized trial evaluating bevacizumab (BEV) in the primary treatment of advanced ovarian cancer: a NRG oncology/Gynecologic Oncology Group (GOG) study. J Clin Oncol. 2018;36(15_suppl):5517. [Google Scholar]
- 34. Ahmed AA, Etemadmoghadam D, Temple J, et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J Pathol. 2010;221(1):49‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cancer Genome Atlas Research Network . Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huang Y‐W. Association of BRCA1/2 mutations with ovarian cancer prognosis: an updated meta‐analysis. Medicine. 2018;97(2):e9380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang SYC, Lheureux S, Karakasis K, et al. Landscape of genomic alterations in high‐grade serous ovarian cancer from exceptional long‐ and short‐term survivors. Genome Med. 2018;10(1):81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lheureux S, Lai Z, Dougherty BA, et al. Long‐term responders on olaparib maintenance in high‐grade serous ovarian cancer: clinical and molecular characterization. Clin Cancer Res. 2017;23(15):4086‐4094. [DOI] [PubMed] [Google Scholar]
- 39. Hughes‐Davies L, Huntsman D, Ruas M, et al. EMSY links the BRCA2 pathway to sporadic breast and ovarian cancer. Cell. 2003;115(5):523‐535. [DOI] [PubMed] [Google Scholar]
- 40. Cousineau I, Belmaaza A. EMSY overexpression disrupts the BRCA2/RAD51 pathway in the DNA‐damage response: implications for chromosomal instability/recombination syndromes as checkpoint diseases. Mol Genet Genomics. 2011;285(4):325‐340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lecona E, Fernandez‐Capetillo O. Targeting ATR in cancer. Nat Rev Cancer. 2018;18(9):586‐595. [DOI] [PubMed] [Google Scholar]
- 42. Saldivar JC, Cortez D, Cimprich KA. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol. 2017;18(10):622‐636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gorringe KL, Jacobs S, Thompson ER, et al. High‐resolution single nucleotide polymorphism array analysis of epithelial ovarian cancer reveals numerous microdeletions and amplifications. Clin Cancer Res. 2007;13(16):4731‐4739. [DOI] [PubMed] [Google Scholar]
- 45. Iwabuchi H, Sakamoto M, Sakunaga H, et al. Genetic analysis of benign, low‐grade, and high‐grade ovarian tumors. Cancer Res. 1995;55(24):6172‐6180. [PubMed] [Google Scholar]
- 46. Jo YS, Kim MS, Yoo NJ, Lee SH. Somatic mutation of a candidate tumour suppressor MGA gene and its mutational heterogeneity in colorectal cancers. Pathology. 2016;48(5):525‐527. [DOI] [PubMed] [Google Scholar]
- 47. Cancer Genome Atlas Research Network . Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543‐550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Saldivar JS, Wu X, Follen M, Gershenson D. Nucleotide excision repair pathway review I: implications in ovarian cancer and platinum sensitivity. Gynecol Oncol. 2007;107(1 Suppl 1):S56‐S71. [DOI] [PubMed] [Google Scholar]
- 49. Willis S, Villalobos VM, Gevaert O, et al. Single gene prognostic biomarkers in ovarian cancer: a meta‐analysis. PLoS One. 2016;11(2):e0149183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Roberts D, Schick J, Conway S, et al. Identification of genes associated with platinum drug sensitivity and resistance in human ovarian cancer cells. Br J Cancer. 2005;92(6):1149‐1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wojtowicz K, Januchowski R, Nowicki M, Zabel M. vPARP adjusts MVP expression in drug‐resistant cell lines in conjunction with MDR proteins. Anticancer Res. 2017;37(6):3015‐3023. [DOI] [PubMed] [Google Scholar]
- 52. Szaflarski W, Sujka‐Kordowska P, Pula B, et al. Expression profiles of vault components MVP, TEP1 and vPARP and their correlation to other multidrug resistance proteins in ovarian cancer. Int J Oncol. 2013;43(2):513‐520. [DOI] [PubMed] [Google Scholar]
- 53. Ikeda Y, Kiyotani K, Yew PY, et al. Germline PARP4 mutations in patients with primary thyroid and breast cancers. Endocr Relat Cancer. 2016;23(3):171‐179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Strickland KC, Howitt BE, Shukla SA, et al. Association and prognostic significance of BRCA1/2‐mutation status with neoantigen load, number of tumor‐infiltrating lymphocytes and expression of PD‐1/PD‐L1 in high grade serous ovarian cancer. Oncotarget. 2016;7(12):13587‐13598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Patch A‐M, Christie EL, Etemadmoghadam D, et al. Whole‐genome characterization of chemoresistant ovarian cancer. Nature. 2015;521(7553):489‐494. [DOI] [PubMed] [Google Scholar]
- 56. Wieser V, Gaugg I, Fleischer M, et al. BRCA1/2 and TP53 mutation status associates with PD‐1 and PD‐L1 expression in ovarian cancer. Oncotarget. 2018;9(25):17501‐17511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Webb JR, Milne K, Kroeger DR, Nelson BH. PD‐L1 expression is associated with tumor‐infiltrating T cells and favorable prognosis in high‐grade serous ovarian cancer. Gynecol Oncol. 2016;141(2):293‐302. [DOI] [PubMed] [Google Scholar]
- 58. Darb‐Esfahani S, Kunze CA, Kulbe H, et al. Prognostic impact of programmed cell death‐1 (PD‐1) and PD‐ligand 1 (PD‐L1) expression in cancer cells and tumor‐infiltrating lymphocytes in ovarian high grade serous carcinoma. Oncotarget. 2016;7(2):1486‐1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Goode EL, Block MS, Kalli KR, et al. Dose‐response association of CD8+ tumor‐infiltrating lymphocytes and survival time in high‐grade serous ovarian cancer. JAMA Oncol. 2017;3(12):e173290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ojalvo LS, Thompson ED, Wang T‐L, et al. Tumor‐associated macrophages and the tumor immune microenvironment of primary and recurrent epithelial ovarian cancer. Hum Pathol. 2018;74:135‐147. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S7
Table S1
Table S2
Table S3
Data Availability Statement
Some of the data that supports the findings of this study are available in the supporting information of this article (i.e., deidentified data), and the remainder of the data that support the findings of this study are available on request from the corresponding author, these are not publicly available due to privacy or ethical restrictions (i.e., associations of clinical data and molecular data).