

Summary
Cell type transition occurs during normal development and under pathological conditions. In prostate cancer bone metastasis, prostate cancer-secreted BMP4 induces endothelial cell-to-osteoblast (EC-to-OSB) transition. Such tumor-induced stromal reprogramming supports prostate cancer progression. We delineate signaling pathways mediating EC-to-OSB transition using EC lines 2H11 and SVR. We found that BMP4-activated pSmad1-Notch-Hey1 pathway inhibits EC migration and tube formation. BMP4-activated GSK3β-βcatenin-Slug pathway stimulates Osx expression. In addition, pSmad1-regulated Dlx2 converges with the Smad1 and β-catenin pathways to stimulate osteocalcin expression. By co-expressing Osx, Dlx2, Slug and Hey1, we were able to achieve EC-to-OSB transition, leading to bone matrix mineralization in the absence of BMP4. In human prostate cancer bone metastasis specimens and MDA-PCa-118b and C4-2b-BMP4 osteogenic xenografts, immunohistochemical analysis showed that β-catenin and pSmad1 are detected in activated osteoblasts rimming the tumor-induced bone. Our results elucidated the pathways and key molecules coordinating prostate cancer-induced stromal programming and provide potential targets for therapeutic intervention.
Subject Areas: Molecular Biology, Cell Biology, Cancer
Graphical abstract
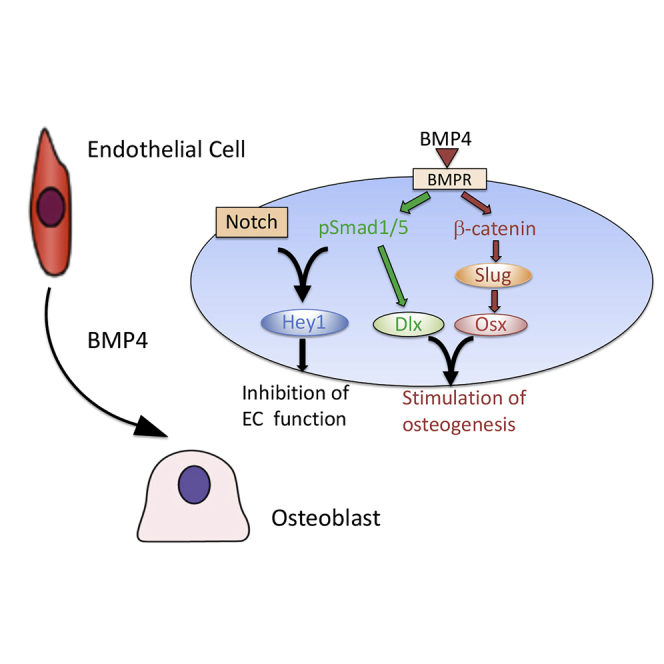
Highlights
-
•
BMP4 upregulates several pathways essential for EC-to-OSB transition
-
•
BMP4 activates pSmad1-Notch-Hey1 and GSK3β-βcatenin-Slug-OSX pathways
-
•
pSmad1-regulated Dlx2 expression connects the pSmad1 and β-catenin pathways
-
•
Coexpression of OSX, Dlx2, Slug, and Hey1 is sufficient to induce EC-to-OSB transition
Molecular Biology ; Cell Biology ; Cancer
Introduction
Bone metastases occur in the majority of men with advanced prostate cancer (PCa) as the initial site of therapy-resistant progression. Uniquely, PCa bone metastasis induces aberrant bone overgrowth, as reflected in bone scans and/or an increase in the level of serum bone alkaline phosphatase (Logothetis and Lin, 2005). PCa-induced aberrant bone formation was shown to support tumor progression in bone in animal models (Chen et al., 2007; Gordon et al., 2009; Jacob et al., 1999; Khodavirdi et al., 2006; Lee et al., 2011). Clinically, treatment with bone-targeting radiopharmaceuticals radium-223 shows life-prolonging efficacy for patients with PCa, although the survival benefit is modest (Autio et al., 2012). Thus delineating the mechanisms by which PCa induces aberrant bone formation may provide strategies to interfere with this lethal progression.
We have shown that the bone morphogenetic protein (BMP) family member BMP4 plays a role in PCa-induced osteogenic bone lesions by inducing endothelial cells (ECs) to undergo cell fate transition to osteoblasts (OSBs) (Lee et al., 2011; Lin et al., 2017). We also showed that PCa cells in human bone metastasis specimens express high levels of BMP4 (Lin et al., 2017) and that MDA-PCa-118b PDX that expressed BMP4 induced ectopic bone formation when implanted subcutaneously or intrafemorally (Li et al., 2008). By using a mouse model with endothelial-specific deletion of Osterix (OSX), the OSB cell fate determination gene, we showed that the PCa-induced aberrant bone originates from ECs that are induced to become OSBs by PCa-secreted BMP4 (Lin et al., 2017). Together, our studies revealed a mechanism, i.e., EC-to-OSB transition, which is involved in PCa-induced aberrant bone formation.
Tumor-induced stromal reprogramming is now recognized as an important mechanism by which the tumor microenvironment engages in promoting tumorigenesis. In addition to playing a role in PCa-induced bone formation, EC-to-OSB transition was also reported in fibrodysplasia ossificans progressiva (FOP) (Medici et al., 2010), a disease caused by genetic mutation in a BMP type I receptor ALK2 (R206H) that activates BMP signaling pathways, leading to the formation of ectopic bone. Thus, understanding the signaling mechanisms of EC-to-OSB transition may identify similar paradigms in other diseases and cancers and provide potential targets for therapeutic intervention. The molecular mechanisms by which BMP4 induces EC-to-OSB transition is unknown. It is likely that both inhibition of angiogenesis and activation of osteoblastogenesis may be required for EC-to-OSB transition to occur.
Regulatory networks orchestrated by key transcription factors have been shown to play a central role in cell fate determination. Precise regulation and integration of pathways that suppress or activate transcription factors that play important roles in cell fate switch are likely required for EC-to-OSB transition. In skeletal development, OSX was found as a key transcription factor for OSB cell fate determination (Sinha and Zhou, 2013) and β-catenin activation has been shown to upregulate OSX (Choi et al., 2017). In ECs, the Notch pathway was shown to play a role in suppressing EC program (Itoh et al., 2004).
In this study, we identified the molecular signaling pathways and mechanisms that converge to mediate EC-to-OSB transition in metastatic prostate cancer. We also determined the combination of transcription factors that are sufficient for EC-to-OSB transition during stromal reprogramming.
Results
Induction of EC-to-OSB transition reduces EC migration and tube formation
To decipher the mechanisms mediating BMP4-induced EC-to-OSB transition, we reasoned that pathways to reduce EC phenotypes while enhancing OSB phenotypes may be involved. We found that BMP4-treated 2H11 cells underwent a morphological transition from a flattened to a more fibroblastic phenotype (Figure 1A). This cell shape change was accompanied by a significant increase in the expression of OSX, an osteoblast cell fate determination transcription factor (Nakashima et al., 2002), and osteocalcin, a non-collagen protein marker for osteoblasts (Figure 1B). During EC-to-OSB transition, ECs need to commit to the OSB lineage followed by differentiation into osteoblasts. One of the EC characteristic is cell migration and tube formation in Matrigel (Lee et al., 2007). We observed that upon BMP4 treatment, both cell migration in vitro (Figure 1C) and tube formation on Matrigel (Figure 1D) were significantly reduced. These observations suggest that BMP4 treatment inhibits EC characteristics (angiogenesis) and enhances osteoblast phenotypes in 2H11 cells.
Figure 1.

BMP4 treatment reduces migration and tube formation in 2H11 endothelial cells and increases the expression of OSX and osteocalcin through GSK3β-β-catenin-OSX pathway
(A) Morphology of 2H11 cells in response to BMP4 (100 ng/mL) for 48 h
(B) qRT-PCR of OSX and osteocalcin mRNAs in cells treated as in (A). Signals were normalized against GAPDH and expressed as mean ± s.d.
(C) Cells treated as indicated were allowed to migrate through the membrane in a Boyden chamber for 2 h and then labeled with Calcein AM. n = 2.
(D) Cells treated as in (C) were seeded on top of Matrigel, and tube formation was analyzed for 20 h n = 2.
(E) Dose response and time course of BMP4 on β-catenin protein levels.
(F) Time course of BMP4 on β-catenin nuclear accumulation. DAPI, DNA staining in cell nucleus. Scale bars, 20 μm. Nuclear β-catenin levels were quantified using NIS-Elements and expressed as mean ± s.e.m. n, number of nuclei analyzed.
(G) qRT-PCR of BMP4-induced OSX and osteocalcin mRNAs after incubation with 0.1 or 0.2 μM PRI-724, a β-catenin inhibitor.
(H) Time course of BMP4 on phospho-GSK-3β protein levels.
(I) Inhibition of BMP4-induced GSK-3β activity by incubation with 5 μM CHIR99021. Signals were normalized against Vinculin. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, #p < 0.0001 by Student's t-test in this and subsequent experiments.
(J) Schematic summary. BMP4 signaling is mediated in part through the inhibition of GSK-3β activity (via increased GSK3β phosphorylation) that increases the level of β-catenin in the nucleus, leading to increased expression of OSX and osteogenesis-related genes in ECs.
BMP4 increases osteoblastogenesis by activating the GSK3β-β-catenin-OSX pathway
We next examined how BMP4 signaling turns on the osteoblast cell fate determinant OSX that activates the osteoblastogenesis pathway. β-Catenin has been shown to induce osteoblastic differentiation of bone marrow stromal cells through upregulation of OSX expression (Liu et al., 2015). We found that BMP4 treatment led to an increase in β-catenin levels in a dose- and time-dependent manner (Figure 1E) as well as nuclear accumulation of β-catenin (Figure 1F). Treatment of 2H11 cells with PRI-724, a β-catenin inhibitor (Tokunaga et al., 2017), significantly blocked BMP4-induced OSX and osteocalcin gene expression (Figure 1G). These results suggest that β-catenin plays a role upstream of OSX and osteocalcin during EC-to-OSB transition.
Phosphorylation of β-catenin at serine 33 and 37 by GSK-3β has been shown to lead to β-catenin degradation in the cytosol and reduction of β-catenin translocation into the nucleus (Dema et al., 2016). We found that BMP4 treatment increased the level of phosphorylated GSK-3β (Figure 1H), resulting in inactivation of GSK-3β kinase activity (Salas et al., 2003). Inhibition of GSK-3β kinase activity with 5 μM CHIR-99021 led to an increase in the level of β-catenin comparable to that in cells treated with BMP4 alone (Figure 1I). These results suggest that BMP4-induced GSK-3β phosphorylation leads to an increase of nuclear β-catenin, which is necessary for the increase in OSX and osteocalcin gene expression in ECs (Figure 1J).
BMP4 activates p38MAPK, p44/42ERK, and AKT to regulate GSK-3β phosphorylation
Next, we looked for BMP4-activated kinases that may phosphorylate GSK-3β, including p38MAPK (p38), p44/42ERK (ERK1/2), and AKT (Bikkavilli et al., 2008; Bikkavilli and Malbon, 2009). We observed that BMP4 treatment increased the phosphorylation of p38MAPK, ERK1/2, AKT, and GSK-3β after 1 h (Figure 2A). Specifically, BMP4 stimulated the phosphorylation of p38MAPK (pp38) in a dose-dependent manner (Figure 2B) and pp38 nuclear accumulation in a time-dependent manner, with elevated levels from 1 to 24 h (Figure 2C). We next treated 2H11 cells with a pan p38 inhibitor BIRB-796 (Kuma et al., 2005). We found that 2 μM BIRB-796 decreased the levels of BMP4-induced pp38 and pGSK-3β (Figure 2D), with a decrease in nuclear β-catenin levels observed by western blot (Figure 2E) and immunofluorescence (Figure 2F), and a partial inhibition of OSX (Figure 2G) and osteocalcin (Figure 2H) expression. A similar reduction in BMP4-activated pGSK-3β levels was observed when cells were treated with 1 μM U0126, an ERK1/2 inhibitor (Figure 2I), or 1 μM AKTi V, an AKT inhibitor (Figure 2J). These observations suggest that p38MAPK, ERK1/2, and AKT are among the BMP4-activated kinases that regulate the GSK3β-β-catenin axis. Our studies support that p38MAPK(ERK1/2,AKT)-GSK3β-β-catenin-OSX is one of the key BMP4-induced pathways leading to osteogenesis during EC-to-OSB transition (Figure 2K).
Figure 2.

BMP4 activates p38MAPK, ERK1/2, and AKT to phosphorylate GSK-3β
(A) 2H11 cells were treated with BMP4 (100 ng/mL) and analyzed for the protein levels of phosphorylated (p) and total (T) p38, ERK1/2, AKT, and GSK-3β. Vinculin, loading control.
(B) Dose response of BMP4 on phospho-p38MAPK (pp38). Signals were normalized against GAPDH.
(C) Time course of BMP4 on pp38 nuclear accumulation. Scale bars, 20 μm. Nuclear pp38 signals were quantified as in Figure 1F.
(D) Cells were pretreated with 2 μM BIRB-796, a pan pp38 inhibitor, for 2 h, then BMP4 was added for 48 h, and pp38 and pGSK-3β signals were normalized against Vinculin.
(E) Nuclear fractionation of 2H11 cells treated with BMP4 and/or 2 μM BIRB-796 for 48 h. Antibody against active β-catenin was used. β-Catenin signals were normalized against nuclear Lamin A/C as loading control.
(F) Cells were pretreated with 200 nM BIRB-796 for 1 h before BMP4 was added for 48 h. Scale bars, 10 μm. Nuclear β-catenin was quantified as in (C).
(G and H) qRT-PCR for (G) OSX and (H) Osteocalcin mRNAs in cells treated as in (D).
(I and J) Cells were pretreated with (I) 1 μM U0126, an ERK1/2 inhibitor, or (J) 1 μM AKTi V, an AKT inhibitor, for 1 h, before the addition of BMP4 for 1 h. pGSK-3β signals were normalized against Vinculin. ∗p < 0.05, ∗∗p < 0.01, #p < 0.0001.
(K) Schematic summary. p38, ERK1/2, and AKT are among the BMP4-activated kinases that phosphorylate GSK-3β in ECs.
BMP4 induces Dlx expression through Smad1 signaling
Although OSX plays a critical role in osteoblast cell fate determination (Nakashima et al., 2002), it is not sufficient to induce an osteogenic phenotype in 2H11 cells (Lin et al., 2017), suggesting the requirement of additional factors. The homeodomain transcription factor Dlx family proteins, including Dlx2, 3, and 5, are required for OSX function through complex formation with OSX at OSX target genes (Yang et al., 2017, 2019). We found that during EC-to-OSB transition, BMP4 induced the expression of Dlx2, Dlx3, and Dlx5 (Figures 3A, S1A, and S1C). We knocked down Dlx2, Dlx3, or Dlx5 individually in 2H11 cells by small hairpin RNA (shRNA) (Figures 3B, S1B, and S1D). In Dlx2 knockdown shDlx2#3 and shDlx2#4 clones, we found that BMP4-stimulated osteocalcin and OSX mRNA expression was significantly reduced relative to that in vector control (Figure 3B). Similar results were obtained with knockdown of Dlx3 (Figure S1B) and Dlx5 (Figure S1D). Conversely, overexpression of Dlx2 (Figure 3C), Dlx3 (Figure S1E) or Dlx5 (Figure S1F) led to the increased expression of OSX. These results indicate that Dlxs, including Dlx2, Dlx3, and Dlx5, are required for BMP4-mediated EC-to-OSB transition.
Figure 3.

BMP4 induces Dlx expression through pSmad1 signaling
(A) Dlx2 mRNA and protein level in 2H11 cells treated with BMP4 (100 ng/mL) for 48 h. Message signals were quantified against GAPDH.
(B) Dlx2 protein levels in Dlx2 knockdown clones shDlx2#3 and shDlx2#4, and the clones were treated as indicated with BMP4 and analyzed for Dlx2, osteocalcin, and OSX mRNA expression.
(C) mRNA and protein levels of Dlx2 and OSX mRNA level in Dlx2 overexpression cells.
(D) qRT-PCR of Smad1 and Smad5 mRNA levels in cells treated as indicated.
(E) Time course of BMP4 on pSmad1/5, Smad1, and Smad5 protein levels.
(F) BMP4 on nuclear localization of pSmad1/5. Scale bars, 20 μm. pSmad1 signals were quantified by NIS-Elements. n, number of nuclei analyzed.
(G) Smad1 protein levels in Smad1 knockdown clones shSmad1#1, shSmad1#5, and shSmad1#3. Signals were normalized against Vinculin.
(H) qRT-PCR of Dlx2, Dlx3, and Dlx5 mRNAs in Smad1 knockdown clones treated as indicated with BMP4. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, #p < 0.0001.
(I) Schematic summary. BMP4-induced Smad1 phosphorylation plays a role in the expression of Dlx, which regulates OSX expression in ECs.
Next, we examined the signaling pathway by which BMP4 upregulates Dlx gene expression. We first examined whether BMP4 regulates Dlxs through the p38MAPK(ERK1/2, AKT)-GSK3β-β-catenin pathway. Treatment of 2H11 with the β-catenin inhibitor, PRI-724, did not have a significant effect on BMP4-induced Dlx gene expression (Figure S2A). BMP4 is also known to activate pSmad1/5 through the canonical BMP4 signaling pathway (Yan et al., 2009). Both Smad1 and Smad5 are expressed in 2H11 cells, whereas only Smad1 mRNA and protein levels were increased upon BMP4 treatment (Figures 3D and 3E). Indeed, BMP4 stimulated a rapid increase in Smad1/5 phosphorylation (Figure 3E) and nuclear translocation of phospho-Smad1/5 (pSmad1/5) (Figure 3F) within 1–3 h. We knocked down Smad1 in 2H11 shSmad1#1, shSmad1#3, and shSmad1#5 clones (Figure 3G) and observed significant decreases in BMP4-inducible Dlx2, 3, and 5 expression relative to that in vector control (Figure 3H). These results reveal that BMP4 stimulates Dlx gene expression through Smad1 signaling during EC-to-OSB transition (Figure 3I).
BMP4-mediated Hey1 upregulation is required for inhibition of endothelial cell functions
Given that BMP4 treatment inhibits the EC characteristics of 2H11 cells (Figures 1C and 1D), we considered that additional pathways are involved in BMP4-induced EC-to-OSB transition. RNA sequencing (RNA-seq) analysis (GEO database accesion number GSE168321) of 2H11 cells treated with or without BMP4 (100 ng/mL) for 48 h detected genes whose expression is regulated by BMP4. We focused on the analysis of transcription factors that may regulate gene expression. We found that Hey1, a transcriptional repressor and mediator of Notch signaling, was the highest upregulated transcription factor, with a 13-fold increase after BMP4 treatment, followed by Dlx2 and Dlx3 (Figure 4A). Other transcription factors, including Id4, Id2, and Snai2 (Slug) were also upregulated by BMP4 during EC-to-OSB transition (Figure 4A). Upregulation of BMP4 target genes Id4 and Id2 are confirmed by qRT-PCR (Figures S2B and S2C).
Figure 4.

BMP4-mediated Hey1 upregulation is required for inhibition of endothelial cell migration and tube formation
(A) RNA-seq analysis of 2H11 cells treated with or without BMP4 (100 ng/mL) for 48 h. Transcription factors upregulated after BMP4 treatment are shown as fold increase. Data are deposited in GEO database under accession #GSE168321.
(B) qRT-PCR of Hey1 and Hey2 mRNAs in cells treated as indicated. Signals are quantified against GAPDH.
(C) Time course of BMP4 on Hey1 protein expression.
(D) qRT-PCR for mRNAs of Notch receptor isoforms in cells treated with BMP4 as indicated.
(E) Time course of BMP4 on Notch1 and Notch2 protein expression.
(F) qRT-PCR for basal (left) and BMP4-induced (right) Hey1 mRNA in Hey1 knockdown clones.
(G) Boyden chamber migration of Hey1 knockdown clones treated with BMP4 for 2 h. n = 2.
(H) Tube formation of Hey1 knockdown clones treated with or without BMP4 before seeding on top of Matrigel for 9 h. n = 2.
(I) qRT-PCR of OSX and osteocalcin mRNAs in Hey1 knockdown clones treated with or without BMP4. ∗p < 0.05, ∗∗p < 0.01.
Because Hey1 is the mediator of Notch signaling, we examined whether the Notch pathway is involved in EC-to-OSB transition. Among the two Hey family genes, Hey1 but not Hey2 mRNA levels were significantly increased by BMP4 (Figure 4B), and this is accompanied by an increase in Hey1 protein levels from 6 h up to 48 h (Figure 4C). Next, we examined whether Notch and Notch ligands are expressed in 2H11 cells. We found that the mRNAs for all four Notch isoforms, Notch1–Notch4, are present and their levels were significantly increased by BMP4 (Figure 4D). Notch1 and Notch2 protein levels were increased at 24 h and maintained at 48 h after BMP4 treatment (Figure 4E). The mRNA levels of Notch ligands Dll4 and Jagged2, but not Jagged1, were also significantly increased by BMP4 (Figures S2D–S2F). We next knocked down Hey1 in 2H11 shHey1#2 and shHey1#5 clones (Figure 4F) and observed that loss of Hey1 significantly abrogated the inhibitory effect of BMP4 on EC migration (Figure 4G) and tube formation (Figure 4H), resulting in a higher level of EC functions relative to vector control. These results suggest that Hey1 plays a role in BMP4-induced EC inhibition. Importantly, Hey1 knockdown decreased BMP4-stimulated OSX and osteocalcin gene expression relative to vector control (Figure 4I). These results suggest that the Notch pathway, through the transcription factor Hey1, is involved in the inhibition of endothelial characteristics of 2H11 cells during EC-to-OSB transition.
pSmad1 forms a complex with NICD to activate Hey1 transcription
Next, we examined whether BMP4-mediated Smad1 phosphorylation activates Hey1 gene expression. Because Smad1 is phosphorylated by BMP receptor kinase, we treated 2H11 cells with BMP4 in the presence or absence of 100 nM LDN193189, a BMP receptor kinase inhibitor. We found that LDN193189 inhibited BMP4-induced Smad1 phosphorylation (Figure 5A) and Hey1 (Figure 5B), OSX (Figure 5C), and osteocalcin (Figure 5D) gene expression. Furthermore, knockdown of Smad1 in shSmad1#1, shSmad1#3, and shSmad1#5 clones significantly decreased BMP4-induced Hey1 expression compared with that in vector control (Figure 5E). These results suggest that BMP4 stimulates Hey1 gene expression through Smad1.
Figure 5.

pSmad1 and NICD form a complex to regulate Hey1 expression
(A–D) (A) BMPR kinase inhibitor LDN193189 (100 nM) on BMP4-induced pSmad1/5 and Smad1 protein levels in 2H11 cells for 48 h. Signals were normalized against Vinculin. qRT-PCR of (B) Hey1, (C) OSX, and (D) osteocalcin mRNAs in cells treated as in (A). Signals were normalized against GAPDH.
(E) qRT-PCR of Hey1 mRNA in shSmad1#1, shSmad1#3, and shSmad1#5 clones treated with BMP4 as indicated.
(F) Time course of BMP4 on NICD and pSmad1/5 localization in the nucleus. Cells were co-immunostained for NICD (Notch1 C-terminal amino acids 2433–2467) and pSmad1/5. Nuclear signals were quantified as in Figure 1F. Scale bars, 10 μm.
(G) Cells were treated with BMP4 for 24 h and analyzed by proximity ligation assay (PLA) using anti-NICD together with anti-pSmad1/5 antibodies, and either single or no antibodies as controls. Red spots indicate PLA positivity in the nucleus. Box, enlarged. Scale bars, 10 μm. PLA spots/nucleus were quantified by NIS-Elements. n, number of nuclei. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, #p < 0.0001.
(H–J) Whole cell lysate (WCL) from control and 48-h BMP4-treated cells were immunoprecipitated (IP) with anti-Notch1, anti-Notch2, or anti-NICD antibodies followed by immunoblotting (IB) with pSmad1/5 antibody. Actin or Vinculin were used as loading controls.
(K and L) Reverse IP was performed using anti-Smad1 antibody followed with IB with anti-Notch1 or anti-Notch2 antibodies.
(M) Schematic summary. Notch pathway is involved in BMP4 inhibition of EC migration and tube formation through the formation of a pSmad1/NICD complex that regulates Hey1 gene expression.
Hey1 gene expression has been shown to be mainly activated by the Notch pathway, in which the Notch receptor is cleaved to generate a Notch intracellular domain (NICD) (Leal et al., 2012). NICD has been shown to form a complex with pSmad1 to regulate downstream Notch target genes (Beets et al., 2013; Dahlqvist et al., 2003). We found that BMP4 stimulated an increase in the level of NICD in the nucleus at 24 h (Figure 5F), where NICD signals co-stained with that of pSmad1/5 at 3 and 24 h of BMP4 stimulation (Figure 5F). To detect a potential interaction between pSmad1 and NICD in the nucleus, we performed an in situ proximity ligation assay (PLA) (Soderberg et al., 2006; Yu-Lee et al., 2018) using anti-pSmad1/5 together with anti-NICD antibodies. We found a significant increase in PLA signals, visualized as individual fluorescent spots, in the nucleus of BMP4-treated 2H11 cells (Figure 5G, see enlarged) relative to untreated control cells, indicating a close proximity between pSmad1 and NICD in the cell nucleus. No PLA signals were observed in cells incubated with only a single or no antibody as negative controls (Figure 5G). We further confirmed the association of pSmad1 with Notch by co-immunoprecipitation. A high level of pSmad1 was observed in immunoprecipitations using Notch1 (Figure 5H), Notch2 (Figure 5I), or NICD (Figure 5J) antibodies following BMP4 treatment, but there was little pSmad1 in control cells. Conversely, we found Notch1 (Figure 5K) and Notch2 (Figure 5L) in a reverse immunoprecipitation using Smad1 antibody. Together, these findings indicate that BMP4/BMPR interaction leads to Smad1 phosphorylation. pSmad1 then forms a complex with NICD to upregulate Notch target gene Hey1, which is necessary for EC-to-OSB transition (Figure 5M).
BMP4 induces Slug expression
Transcription factors, including Snail, Slug, FoxC2, and Twist, have been shown to play critical roles in cell type transition (Sipos and Galamb, 2012). Snail was shown to be critical for TGFβ-induced endothelial-to-mesenchyme transition (Medici et al., 2011). Our RNA-seq analysis showed that the transcription factor Slug was upregulated by BMP4 (Figure 4A). We confirmed that Slug mRNA levels was upregulated about 3-fold, whereas Snail was upregulated only 1.4-fold by BMP4 (Figure 6A), and Slug protein expression was upregulated in a time-dependent manner by BMP4 (Figure 6B). We knocked down Slug in 2H11 shSlug#3 and shSlug#5 clones (Figure 6C) and observed that a loss of Slug inhibited BMP4-induced upregulation of Slug mRNA and protein (Figure 6D), which resulted in a decrease in BMP4-induced osteocalcin expression (Figure 6E), suggesting that Slug expression is necessary for EC-to-OSB transition.
Figure 6.

BMP4 induces Slug expression
(A) qRT-PCR of Slug and Snail mRNAs in 48-h BMP4-treated 2H11 cells. Signals were quantified against GAPDH.
(B) Time course of BMP4 on Slug protein levels. Signals were normalized against Vinculin.
(C) Slug protein levels in Slug knockdown clones.
(D) Slug mRNA by qRT-PCR and protein levels following BMP4 treatment in Slug knockdown clones.
(E) qRT-PCR of osteocalcin mRNA in shSlug clones treated as in (D).
(F) Dose response of β-catenin inhibitor PRI-724 on BMP4-induced Slug mRNA expression in 2H11 cells.
(G) qRT-PCR of OSX mRNA in Slug knockdown clones treated as in (D).
(H) qRT-PCR of Slug and OSX mRNAs and Slug protein levels in Slug-overexpressing cells. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(I) Schematic summary. BMP4-induced β-catenin signaling regulates Slug expression. Slug then participates in the upregulation of OSX expression during EC-to-OSB transition.
Next, we examined the signaling pathway that leads to Slug upregulation. We found that knockdown of Smad1 did not affect BMP4-induced Slug upregulation (Figure S2G). Additionally, knockdown of Hey1 in 2H11 cells also did not affect BMP4-induced Slug upregulation (Figure S2H). These results indicate that Slug upregulation is not mediated through the Smad1-Notch-Hey1 pathway. In contrast, inhibition of β-catenin by PRI-724 led to a significant inhibition of BMP4-mediated Slug expression (Figure 6F). These results suggest that BMP4-induced Slug upregulation is mediated through β-catenin signaling pathway. Because Slug plays a role in cell type transition, we examined whether Slug regulates the expression of OSX, Dlx2, or Hey1, the transcription factors involved in EC-to-OSB transition. We found that knockdown of Slug decreased BMP4-induced OSX expression (Figure 6G), but did not affect that of Dlx2 (Figure S2I) or Hey1 (Figure S2J). Furthermore, overexpression of Slug in 2H11 cells significantly upregulated the expression of OSX (Figure 6H) but not that of Dlx2 (Figure S2K) or Hey1 (Figure S2L). These results suggest that Slug plays a role in EC-to-OSB transition by upregulating OSX through the β-catenin-Slug pathway (Figure 6I).
BMP4-induced pSmad1/5 and p38MAPK-β-catenin pathways occur in parallel
As BMP4 stimulates signaling through both pSmad1 and p38MPAK, we next determined whether the two signaling pathways may regulate each other's activation. 2H11 cells were treated with 2 μM BIRB-796 or 100 nM LDN193189 before the addition of BMP4. We found that blocking p38MAPK activation with BIRB inhibited BMP4-induced pp38MAPK nuclear translocation (Figure S3A, upper) but did not prevent BMP4-induced pSmad1/5 nuclear translocation (Figure S3A, lower). Conversely, blocking Smad1/5 phosphorylation with LDN193189 inhibited BMP4-induced nuclear translocation of pSmad1/5 (Figure S3A, lower), but did not affect BMP4-induced pp38 nuclear translocation (Figure S3A, upper). Furthermore, BIRB did not prevent BMP4-induced phosphorylation of Smad1 protein (Figure S3B, right). Conversely, LDN193189 did not block BMP4-induced phosphorylation of p38MAPK (Figure S3B, left). These results suggest that the BMP4-induced pSmad1/5 and p38MAPK-β-catenin pathways largely occur in parallel.
Overexpression of key transcription factors is sufficient to induce EC-to-OSB transition
Our data show that BMP4 induces the expression of Hey1, Slug, Dlx, and OSX transcription factors that are necessary for EC-to-OSB transition to occur. We next examined whether co-expression of these key factors will be sufficient for EC-to-OSB transition in the absence of BMP4. Factors were overexpressed singly or in combination, and osteocalcin gene expression was monitored as the readout. Individual overexpression of Hey1, Slug, OSX, Dlx2, Dlx3, or Dlx5 in 2H11 cells was not sufficient to induce osteocalcin expression (Figure S4A). Overexpression of various combinations of two factors showed that only the combination of OSX with Dlx2, but not Dlx3 or Dlx5, was effective in increasing osteocalcin expression, when compared with a dose-response of BMP4 induction of osteocalcin (Figure 7A). We next transiently transfected Slug, Hey1, or both into a 2H11-OSX-Dlx2 (2-factor) cell line and found that either Slug or Hey1 further increased osteocalcin levels (Figure 7B, upper), indicating that Slug or Hey1 also contributed to osteocalcin expression. Transient co-transfection of Slug and Hey1 into 2H11-OSX-Dlx2 cells did not result in further increases in osteocalcin levels, possibly due to different transfection efficiencies for Slug and Hey1 (Figures S4B and S4C). We next established a 2H11-OSX-Dlx2-Slug (3-factor) cell line and found that transient transfection of Hey1 resulted in a significant increase in osteocalcin levels compared with vector-transfected cells (Figure 7C, upper). Although overexpression of the four factors may not fully replicate the complex coordinated signal transduction that occurs upon BMP4 stimulation, our findings support that co-expression of all four factors is sufficient to achieve significant osteocalcin expression in the absence of BMP4.
Figure 7.

Overexpression of key transcription factors induces EC-to-OSB transition
(A) qRT-PCR of osteocalcin mRNA in 2H11 cells overexpressing combinations of two transcription factors. Dose response of BMP4 on osteocalcin mRNA was performed in parallel. Signals were quantified against GAPDH.
(B) 2H11-OSX-Dlx2 (2-factor) cell line was generated, transiently transfected (superscript +) with Slug, Hey1, or both, and mRNA levels for osteocalcin (upper), ALP (middle), and Col1α1 (lower) were analyzed by qRT-PCR.
(C) 2H11-OSX-Dlx2-Slug (3-factor) cell line was generated, transiently transfected with (superscript +) Hey1, and analyzed as in (B).
(D) Osteoblast mineralization using alizarin red staining on cells in (B).
(E) Mineralization analysis for cells in (C). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(F) β-Catenin and pSmad1 expression in EC-OSB cells of PCa bone metastases of clinical specimens. Activated EC-OSB cells (arrows) rimming the bone were immunopositive with β-catenin and pSmad1. (400X). B, bone.
(G) Schematic summary of BMP4-mediated EC-to-OSB transition. In tumor-derived ECs, BMP4 stimulates the canonical Smad1 pathway that upregulates Hey1 to inhibit EC properties. Smad1 signaling also upregulates Dlx that forms a complex with OSX to activate transcription of skeletal-related genes. BMP4 also stimulates a non-canonical pathway through p38MAPK(p44/42ERK,AKT)-GSK3β-βcatenin that upregulates the expression of OSX. In 2H11 cells, this non-Smad pathway also upregulates Slug that enhances OSX expression. The unique integration of the Smad1-Notch-Hey1 pathway that inhibits EC function and the p38MAPK(ERK1/2,AKT)-GSK3β-β-catenin signaling pathway that stimulates osteogenesis underlies BMP4-mediated EC-to-OSB transition.
Given that osteoblast function is characterized by the secretion of extracellular proteins, including osteocalcin, alkaline phosphatase, and type 1 collagen (Col1α1), we further determined whether the combined co-expression of all four factors could lead to the expression of osteoblast differentiation markers. 2H11-OSX-Dlx2 (2-factor) cells transfected with Slug or Hey1 or both (Figure 7B, middle and lower), and 2H11-OSX-Dlx2-Slug (3-factor) cells transfected with Hey1 (Figure 7C, middle and lower) led to a significant increase in alkaline phosphatase and type I collagen. Furthermore, when the 2H11-OSX-Dlx2 2-factor and 2H11-OSX-Dlx2-Slug 3-factor cell lines together with transiently transfected factors as indicated were cultured in osteoblast differentiation medium, they achieved mineralization as shown by alizarin red staining (Figures 7D and 7E). Together, these results indicate that co-expression of all four transcription factors is sufficient to stimulate EC-to-OSB transition as well as osteoblast mineralization in the absence of BMP4.
BMP4 induces EC-to-OSB transition in SVR cells
To examine whether EC-to-OSB transition also occurs in another EC line, we treated murine SVR ECs, derived from pancreatic islet by sequential introduction of simian virus 40 (SV40) large T (tumor) antigen and H-ras (Arbiser et al., 1997), with BMP4. We note that SVR cells do not tolerate well the serum-free condition used for BMP4 treatment. We found that BMP4 increased mRNA levels of OSX and osteocalcin and led to mineralization of SVR cells (Figure S5A). BMP4 also stimulated pSmad1/5 protein levels and nuclear translocation (Figure S5B). Blocking pSmad1 activation through inhibiting BMP receptor kinase activity with LDN193189 led to downregulation of BMP4-induced Hey1 and Dlx2 mRNA (Figure S5C, upper), downstream targets of the Smad1 pathway, resulting in a significant inhibition of OSX and osteocalcin expression (Figure S5C, lower). BMP4 also stimulated pp38MAPK and β-catenin protein levels and increased the nuclear translocation of β-catenin (Figure S5D). Inhibition of β-catenin by PRI-724 blocked the mRNA expression of OSX and osteocalcin (Figure S5E). Interestingly, Slug mRNA levels were neither upregulated by BMP4 nor altered by PRI-724 in SVR cells (Figure S5F), suggesting variations in different EC lines. These results suggest that BMP4-induced signal transduction pathways during EC-to-OSB transition in SVR cells are largely similar to those in 2H11 cells.
β-Catenin and pSmad1 are expressed in osteoblasts in prostate cancer bone metastasis specimens and MDA-PCa-118b and C4-2b-BMP4 osteogenic xenografts
Our previous studies showed that osteoblasts rimming the bone in the biopsy specimens from human bone metastasis expressed both osteocalcin and Tie2, suggesting that these cells were undergoing EC-to-OSB transition (Lin et al., 2017). We further examined whether β-catenin is expressed in the EC-OSB cells present in PCa bone metastases of clinical specimens. Specimens from bone biopsies were immunostained for β-catenin or pSmad1. EC-OSB cells in bone metastasis specimens showed staining for β-catenin and pSmad1 (Figures 7F and S6A). Similarly, in MDA-PCa-118b (Figure S6B) and C4-2b-BMP4 (Figure S6C) osteogenic xenografts, which were generated by injecting MDA-PCa-118b or C4-2b-BMP4 cells subcutaneously, immunostaining showed the expression of β-catenin and pSmad1 in EC-OSB hybrid cells that rim the tumor-induced bone. These observations suggest that β-catenin and Smad1 signaling were present in EC-to-OSB transition in PCa bone metastasis.
Together, our studies show that stromal reprograming of ECs to OSBs by BMP4 involves the integration of several pathways, including Smad1-Notch-Hey1 pathway that inhibits EC function, p38MAPK(ERK1/2,AKT)-GSK3β-βcatenin-Slug signaling pathway that stimulates osteogenesis, and Smad1-regulated Dlx2 that converges with the Smad1 and β-catenin pathways during EC-to-OSB transition (Figure 7G).
Discussion
We have delineated the molecular mechanisms underlying BMP4-induced EC-to-OSB transition. We found that stromal reprograming of ECs to OSBs by BMP4 requires the coordination of several pathways, including the Smad1-Notch-Hey1 pathway that inhibits EC function and the p38MAPK(ERK1/2,AKT)-GSK3β-βcatenin signaling pathway that stimulates osteogenesis (Figure 7G). We also showed that EC-to-OSB transition can occur by overexpressing several of the key molecules, i.e., cell fate transition factors Slug and OSX, transcriptional repressor Hey1, and transcriptional activator Dlx2, in the absence of BMP4. Our studies elucidated that these four factors are not only necessary but also sufficient for EC-to-OSB transition. Together, our findings provide a mechanism for BMP4-induced stromal reprogramming. As our previous studies showed that PCa secreted-BMP4 led to EC-to-OSB transition, understanding the complex signaling mechanisms underlying EC-to-OSB transition will provide strategies to block PCa-induced aberrant bone formation by interfering with pathways essential for EC-to-OSB transition.
Identification of the Notch-Hey1 pathway in BMP4-induced EC-to-OSB transition was unexpected. Notch signaling influences cell fate decisions, cell differentiation, tissue homeostasis, and regeneration (Liu et al., 2010); however, its involvement in EC-to-OSB transition is not known. We found that BMP4-activated Notch-Hey1 pathway inhibits EC migration and tube formation and is necessary for EC-to-OSB transition. Hey1 protein is a member of the Hey family of transcriptional repressors that function as downstream effectors of Notch signaling. Buas et al. (2010) reported that Hey1 inhibits myogenesis by repressing the expression of myogenin and Mef2C, genes critical for myogenesis. Hey1 has been shown to physically associate with other transcription factors to regulate cellular activities. For example, interactions between Hey proteins and Runx2 were shown to repress Runx2 transcriptional activity and prevent aortic calcification (Garg et al., 2005). Interaction of Hey1 with androgen receptor was thought to repress androgen receptor functions (Belandia et al., 2005). Elagib et al. (2004) showed that interaction of Hey1 with GATA1 led to inhibition of GATA1 transcriptional activity and GATA1's effect on inducing erythropoiesis. Hey family members were also shown to inhibit GATA-driven cardiac gene expression through association with GATA4/6 (Fischer et al., 2005; Kathiriya et al., 2004). A direct interaction between Hey family members and Ptf1-p48 led to inhibition of pancreatic exocrine differentiation (Ghosh and Leach, 2006). The target genes that are regulated by Hey1 and how these target genes impact EC-to-OSB transition remain to be determined.
Our studies showed that β-catenin plays a critical role in EC-to-OSB transition. Choi et al. (2017) showed that β-catenin upregulates OSX expression through direct binding to the promoter region of OSX. OSX, also known as Sp7, is a Sp family transcription factor. Although Sp family transcription factors mainly bind to GC-rich sequence, studies by Hojo et al. (2016) demonstrated that OSX does not bind to GC-rich sequence directly. Rather, OSX and Dlx form a complex that binds to AT-rich sequence (Figure 7G). Our studies showed that the expression of Dlx2, 3, and 5 is upregulated by BMP4-mediated Smad1 pathway and that knockdown of any of the three Dlx genes results in the inhibition of EC-to-OSB transition, suggesting that Dlx factors are necessary for EC-to-OSB transition. Interestingly, only Dlx2 supports osteocalcin expression when co-expressed with OSX, suggesting complex roles of Dlx family proteins in cell type transition. As Dlx expression is regulated by BMP4-mediated Smad1, whereas OSX is regulated by β-catenin pathways, these observations raise the possibility that Dlx plays a role in converging the Smad1 and β-catenin pathways during EC-to-OSB transition.
Although Snail and Slug family proteins have been shown to play important roles in cell fate transition, we found that Slug, but not Snail, expression promotes EC-to-OSB transition in 2H11 cells. In contrast, Medici et al. (2011) showed that TGFβ2 stimulates endothelial-to-mesenchyme (EndMT) transition by increasing Snail expression, suggesting that Snail and Slug play unique roles in different cellular contexts. Interestingly, we found that BMP4 does not regulate Slug expression in SVR cells. Whether Snail is involved in BMP4-induced EC-to-OSB transition in SVR cells is not clear.
Bone marrow stromal cells have been shown to be a source of osteoblasts (Hu et al., 2018). In characterizing the stem cells for bone marrow stroma, Chan et al. (2015) showed that the skeletal system contains multipotent stem cells that can differentiate into various cell lineages, including bone, cartilage, and stroma. It is possible that some of the prostate tumor-induced osteoblasts are derived from bone marrow stromal cells, which may have different signaling pathways than those in endothelial-derived osteoblasts.
A change in lineage plasticity through epithelial-to-mesenchymal transition of tumor cells is one of the established mechanisms underlying prostate tumor progression and metastasis (Aggarwal et al., 2014; Bishop et al., 2017). Our studies further elucidate that the bone microenvironment is not a static component; instead PCa interacts dynamically with the bone microenvironment. Depending on the characteristics of PCa cells, the bone microenvironment can be modified in response to signals from the PCa cells. Thus, both epithelial and stromal cell lineage reprograming are involved in PCa progression. These observations suggest that incorporating therapies that target mechanisms underlying the reprogramming of both stromal cells and tumor cells may improve therapy outcomes.
In summary, we have delineated the molecular basis of EC-to-OSB transition. Understanding how tumor cells alter stromal cell fate in the tumor microenvironment will be critical in developing strategies for cancer therapies.
Limitations of the study
We used immortalized 2H11 and SVR EC lines in our in vitro studies. As only a few specific EC lines can be induced to become osteoblasts by BMP4, this observation suggests that these cell lines may have modifications that allow EC-to-OSB transition to occur. FOP disease progression is usually initiated by local inflammation. It is likely that pathways activated by inflammatory factors are also required for EC-to-OSB transition. In the osteoblastic bone lesion of human prostate cancer bone metastasis, it is likely that tumor cells may induce an inflammatory environment to allow ectopic bone formation to take place under BMP4 stimulation. Thus, the in vitro approaches used in this study do not fully reflect the diverse tumor microenvironments encountered in vivo. Further study will be required to better characterize this process in vivo.
Resource availability
Lead contact
Information and requests for resources should be directed to and will be fulfilled by the lead contact, Sue-Hwa Lin (slin@mdanderson.org).
Materials availability
Materials (plasmids) generated in this study will be made available upon reasonable request to the lead contact. Their delivery could require a material transfer agreement.
Data and code availability
All relevant data are available from the lead contact upon request. The original mouse RNA-seq data have been deposited in the GEO database under accession number GSE168321.
Methods
All methods can be found in the accompanying transparent methods supplemental file.
Acknowledgments
This work was supported by grants from the NIH (RO1 CA174798, 5P50 CA140388, P30 CA16672), the Prostate Cancer Foundation, and Cancer Prevention and Research Institute of Texas (CPRIT RP150179, RP190252) and funds from The University of Texas MD Anderson Sister Institute Network Fund and Moonshot Program.
Author contribution
G.Y., L.-Y.Y.-L., T. Panaretakis, G.W., and S.-H.L. designed the research; G.Y., P.S., S.-C.L., Y.-C.L., J.P., X.L., J.H.S., and L.-Y.Y.-L. performed the experiments; G.Y., S.-C.L., Y.-C.L., T. Pan, J.P., C.J.L., G.E.G., L.-Y.Y.-L., and S.-H.L. analyzed the data; G.Y., L.-Y.Y.-L., G.E.G., C.J.L., T. Panaretakis, G.W., and S.-H.L. wrote the paper.
Declaration of interests
C.J.L. reports receiving commercial research grants from Janssen, Bristol-Myers Squibb, Pfizer, and ORIC Pharmaceutical, and honoraria from Bayer, Amgen, Merck, and Sharp & Dohme. He also serves as consulting/advisory for Merck, Sharp & Dohme, Bayer, and Amgen. No potential conflicts of interest were disclosed by the other authors.
Published: April 23, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102388.
Contributor Information
Li-Yuan Yu-Lee, Email: yulee@bcm.edu.
Sue-Hwa Lin, Email: slin@mdanderson.org.
Supplemental information
References
- Aggarwal R., Zhang T., Small E.J., Armstrong A.J. Neuroendocrine prostate cancer: subtypes, biology, and clinical outcomes. J. Natl. Compr. Canc. Netw. 2014;12:719–726. doi: 10.6004/jnccn.2014.0073. [DOI] [PubMed] [Google Scholar]
- Arbiser J.L., Moses M.A., Fernandez C.A., Ghiso N., Cao Y., Klauber N., Frank D., Brownlee M., Flynn E., Parangi S. Oncogenic H-ras stimulates tumor angiogenesis by two distinct pathways. Proc. Natl. Acad. Sci. U S A. 1997;94:861–866. doi: 10.1073/pnas.94.3.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autio K.A., Scher H.I., Morris M.J. Therapeutic strategies for bone metastases and their clinical sequelae in prostate cancer. Curr. Treat. Options Oncol. 2012;13:174–188. doi: 10.1007/s11864-012-0190-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beets K., Huylebroeck D., Moya I.M., Umans L., Zwijsen A. Robustness in angiogenesis: notch and BMP shaping waves. Trends Genet. 2013;29:140–149. doi: 10.1016/j.tig.2012.11.008. [DOI] [PubMed] [Google Scholar]
- Belandia B., Powell S.M., Garcia-Pedrero J.M., Walker M.M., Bevan C.L., Parker M.G. Hey1, a mediator of notch signaling, is an androgen receptor corepressor. Mol. Cell. Biol. 2005;25:1425–1436. doi: 10.1128/MCB.25.4.1425-1436.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikkavilli R.K., Feigin M.E., Malbon C.C. p38 mitogen-activated protein kinase regulates canonical Wnt-beta-catenin signaling by inactivation of GSK3beta. J. Cell Sci. 2008;121:3598–3607. doi: 10.1242/jcs.032854. [DOI] [PubMed] [Google Scholar]
- Bikkavilli R.K., Malbon C.C. Mitogen-activated protein kinases and Wnt/beta-catenin signaling: molecular conversations among signaling pathways. Commun. Integr. Biol. 2009;2:46–49. doi: 10.4161/cib.2.1.7503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop J.L., Thaper D., Vahid S., Davies A., Ketola K., Kuruma H., Jama R., Nip K.M., Angeles A., Johnson F. The master neural transcription factor BRN2 is an androgen receptor-suppressed driver of neuroendocrine differentiation in prostate cancer. Cancer Discov. 2017;7:54–71. doi: 10.1158/2159-8290.CD-15-1263. [DOI] [PubMed] [Google Scholar]
- Buas M.F., Kabak S., Kadesch T. The Notch effector Hey1 associates with myogenic target genes to repress myogenesis. J. Biol. Chem. 2010;285:1249–1258. doi: 10.1074/jbc.M109.046441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C.K., Seo E.Y., Chen J.Y., Lo D., McArdle A., Sinha R., Tevlin R., Seita J., Vincent-Tompkins J., Wearda T. Identification and specification of the mouse skeletal stem cell. Cell. 2015;160:285–298. doi: 10.1016/j.cell.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N., Ye X.C., Chu K., Navone N.M., Sage E.H., Yu-Lee L.Y., Logothetis C.J., Lin S.H. A secreted isoform of ErbB3 promotes osteonectin expression in bone and enhances the invasiveness of prostate cancer cells. Cancer Res. 2007;67:6544–6548. doi: 10.1158/0008-5472.CAN-07-1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H., Kim T.H., Yang S., Lee J.C., You H.K., Cho E.S. A reciprocal interaction between beta-catenin and osterix in cementogenesis. Sci. Rep. 2017;7:8160. doi: 10.1038/s41598-017-08607-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlqvist C., Blokzijl A., Chapman G., Falk A., Dannaeus K., Ibanez C.F., Lendahl U. Functional Notch signaling is required for BMP4-induced inhibition of myogenic differentiation. Development. 2003;130:6089–6099. doi: 10.1242/dev.00834. [DOI] [PubMed] [Google Scholar]
- Dema A., Schroter M.F., Perets E., Skroblin P., Moutty M.C., Deak V.A., Birchmeier W., Klussmann E. The A-kinase anchoring protein (AKAP) glycogen synthase kinase 3beta interaction protein (GSKIP) regulates beta-catenin through its interactions with both protein kinase A (PKA) and GSK3beta. J. Biol. Chem. 2016;291:19618–19630. doi: 10.1074/jbc.M116.738047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elagib K.E., Xiao M., Hussaini I.M., Delehanty L.L., Palmer L.A., Racke F.K., Birrer M.J., Ganapathy-Kanniappan S., McDevitt M.A., Goldfarb A.N. Jun blockade of erythropoiesis: role for repression of GATA-1 by HERP2. Mol. Cell. Biol. 2004;24:7779–7794. doi: 10.1128/MCB.24.17.7779-7794.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A., Klattig J., Kneitz B., Diez H., Maier M., Holtmann B., Englert C., Gessler M. Hey basic helix-loop-helix transcription factors are repressors of GATA4 and GATA6 and restrict expression of the GATA target gene ANF in fetal hearts. Mol. Cell. Biol. 2005;25:8960–8970. doi: 10.1128/MCB.25.20.8960-8970.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg V., Muth A.N., Ransom J.F., Schluterman M.K., Barnes R., King I.N., Grossfeld P.D., Srivastava D. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437:270–274. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- Ghosh B., Leach S.D. Interactions between hairy/enhancer of split-related proteins and the pancreatic transcription factor Ptf1-p48 modulate function of the PTF1 transcriptional complex. Biochem. J. 2006;393:679–685. doi: 10.1042/BJ20051063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon J.A., Sodek J., Hunter G.K., Goldberg H.A. Bone sialoprotein stimulates focal adhesion-related signaling pathways: role in migration and survival of breast and prostate cancer cells. J. Cell. Biochem. 2009;107:1118–1128. doi: 10.1002/jcb.22211. [DOI] [PubMed] [Google Scholar]
- Hojo H., Ohba S., He X., Lai L.P., McMahon A.P. Sp7/Osterix is restricted to bone-forming vertebrates where it acts as a Dlx Co-factor in osteoblast specification. Dev. Cell. 2016;37:238–253. doi: 10.1016/j.devcel.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L., Yin C., Zhao F., Ali A., Ma J., Qian A. Mesenchymal stem cells: cell fate decision to osteoblast or adipocyte and application in osteoporosis treatment. Int. J. Mol. Sci. 2018;19:360. doi: 10.3390/ijms19020360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh F., Itoh S., Goumans M.J., Valdimarsdottir G., Iso T., Dotto G.P., Hamamori Y., Kedes L., Kato M., ten Dijke Pt P. Synergy and antagonism between Notch and BMP receptor signaling pathways in endothelial cells. EMBO J. 2004;23:541–551. doi: 10.1038/sj.emboj.7600065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob K., Webber M., Benayahu D., Kleinman H.K. Osteonectin promotes prostate cancer cell migration and invasion: a possible mechanism for metastasis to bone. Cancer Res. 1999;59:4453–4457. [PubMed] [Google Scholar]
- Kathiriya I.S., King I.N., Murakami M., Nakagawa M., Astle J.M., Gardner K.A., Gerard R.D., Olson E.N., Srivastava D., Nakagawa O. Hairy-related transcription factors inhibit GATA-dependent cardiac gene expression through a signal-responsive mechanism. J. Biol. Chem. 2004;279:54937–54943. doi: 10.1074/jbc.M409879200. [DOI] [PubMed] [Google Scholar]
- Khodavirdi A.C., Song Z., Yang S., Zhong C., Wang S., Wu H., Pritchard C., Nelson P.S., Roy-Burman P. Increased expression of osteopontin contributes to the progression of prostate cancer. Cancer Res. 2006;66:883–888. doi: 10.1158/0008-5472.CAN-05-2816. [DOI] [PubMed] [Google Scholar]
- Kuma Y., Sabio G., Bain J., Shpiro N., Marquez R., Cuenda A. BIRB796 inhibits all p38 MAPK isoforms in vitro and in vivo. J. Biol. Chem. 2005;280:19472–19479. doi: 10.1074/jbc.M414221200. [DOI] [PubMed] [Google Scholar]
- Leal M.C., Surace E.I., Holgado M.P., Ferrari C.C., Tarelli R., Pitossi F., Wisniewski T., Castano E.M., Morelli L. Notch signaling proteins HES-1 and Hey-1 bind to insulin degrading enzyme (IDE) proximal promoter and repress its transcription and activity: implications for cellular Abeta metabolism. Biochim. Biophys. Acta. 2012;1823:227–235. doi: 10.1016/j.bbamcr.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.H., Kunz J., Lin S.H., Yu-Lee L.Y. 16-kDa prolactin inhibits endothelial cell migration by down-regulating the Ras-Tiam1-Rac1-Pak1 signaling pathway. Cancer Res. 2007;67:11045–11053. doi: 10.1158/0008-5472.CAN-07-0986. [DOI] [PubMed] [Google Scholar]
- Lee Y.C., Cheng C.J., Bilen M.A., Lu J.F., Satcher R.L., Yu-Lee L.Y., Gallick G.E., Maity S.N., Lin S.H. BMP4 promotes prostate tumor growth in bone through osteogenesis. Cancer Res. 2011;71:5194–5203. doi: 10.1158/0008-5472.CAN-10-4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.G., Mathew P., Yang J., Starbuck M.W., Zurita A.J., Liu J., Sikes C., Multani A.S., Efstathiou E., Lopez A. Androgen receptor-negative human prostate cancer cells induce osteogenesis in mice through FGF9-mediated mechanisms. J. Clin. Invest. 2008;118:2697–2710. doi: 10.1172/JCI33093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.C., Lee Y.C., Yu G., Cheng C.J., Zhou X., Chu K., Murshed M., Le N.T., Baseler L., Abe J.I. Endothelial-to-Osteoblast conversion generates osteoblastic metastasis of prostate cancer. Dev. Cell. 2017;41:467–480 e463. doi: 10.1016/j.devcel.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B., Wu S., Han L., Zhang C. beta-catenin signaling induces the osteoblastogenic differentiation of human pre-osteoblastic and bone marrow stromal cells mainly through the upregulation of osterix expression. Int. J. Mol. Med. 2015;36:1572–1582. doi: 10.3892/ijmm.2015.2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Sato C., Cerletti M., Wagers A. Notch signaling in the regulation of stem cell self-renewal and differentiation. Curr. Top. Dev. Biol. 2010;92:367–409. doi: 10.1016/S0070-2153(10)92012-7. [DOI] [PubMed] [Google Scholar]
- Logothetis C.J., Lin S.H. Osteoblasts in prostate cancer metastasis to bone. Nat. Rev. Cancer. 2005;5:21–28. doi: 10.1038/nrc1528. [DOI] [PubMed] [Google Scholar]
- Medici D., Potenta S., Kalluri R. Transforming growth factor-beta2 promotes Snail-mediated endothelial-mesenchymal transition through convergence of Smad-dependent and Smad-independent signalling. Biochem. J. 2011;437:515–520. doi: 10.1042/BJ20101500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medici D., Shore E.M., Lounev V.Y., Kaplan F.S., Kalluri R., Olsen B.R. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat. Med. 2010;16:1400–1406. doi: 10.1038/nm.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima K., Zhou X., Kunkel G., Zhang Z., Deng J.M., Behringer R.R., de Crombrugghe B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17–29. doi: 10.1016/s0092-8674(01)00622-5. [DOI] [PubMed] [Google Scholar]
- Salas T.R., Reddy S.A., Clifford J.L., Davis R.J., Kikuchi A., Lippman S.M., Menter D.G. Alleviating the suppression of glycogen synthase kinase-3beta by Akt leads to the phosphorylation of cAMP-response element-binding protein and its transactivation in intact cell nuclei. J. Biol. Chem. 2003;278:41338–41346. doi: 10.1074/jbc.M302972200. [DOI] [PubMed] [Google Scholar]
- Sinha K.M., Zhou X. Genetic and molecular control of osterix in skeletal formation. J. Cell. Biochem. 2013;114:975–984. doi: 10.1002/jcb.24439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipos F., Galamb O. Epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions in the colon. World J. Gastroenterol. 2012;18:601–608. doi: 10.3748/wjg.v18.i7.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderberg O., Gullberg M., Jarvius M., Ridderstrale K., Leuchowius K.J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L.G. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- Tokunaga Y., Osawa Y., Ohtsuki T., Hayashi Y., Yamaji K., Yamane D., Hara M., Munekata K., Tsukiyama-Kohara K., Hishima T. Selective inhibitor of Wnt/beta-catenin/CBP signaling ameliorates hepatitis C virus-induced liver fibrosis in mouse model. Sci. Rep. 2017;7:325. doi: 10.1038/s41598-017-00282-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y., Tang D., Chen M., Huang J., Xie R., Jonason J.H., Tan X., Hou W., Reynolds D., Hsu W. Axin2 controls bone remodeling through the beta-catenin-BMP signaling pathway in adult mice. J. Cell Sci. 2009;122:3566–3578. doi: 10.1242/jcs.051904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G., Yuan G., MacDougall M., Zhi C., Chen S. BMP-2 induced Dspp transcription is mediated by Dlx3/Osx signaling pathway in odontoblasts. Sci. Rep. 2017;7:10775. doi: 10.1038/s41598-017-10908-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Fan J., Cao Y., Gao R., Fan Z. Distal-less homeobox 5 promotes the osteo-/dentinogenic differentiation potential of stem cells from apical papilla by activating histone demethylase KDM4B through a positive feedback mechanism. Exp. Cell Res. 2019;374:221–230. doi: 10.1016/j.yexcr.2018.11.027. [DOI] [PubMed] [Google Scholar]
- Yu-Lee L.Y., Yu G., Lee Y.C., Lin S.C., Pan J., Pan T., Yu K.J., Liu B., Creighton C.J., Rodriguez-Canales J. Osteoblast-secreted factors mediate dormancy of metastatic prostate cancer in the bone via activation of the TGFbetaRIII-p38MAPK-pS249/T252RB pathway. Cancer Res. 2018;78:2911–2924. doi: 10.1158/0008-5472.CAN-17-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data are available from the lead contact upon request. The original mouse RNA-seq data have been deposited in the GEO database under accession number GSE168321.