

Abstract
Community-acquired pneumonia is the most common type of pneumonia and remains a leading cause of morbidity and mortality worldwide. Although many different pathogens can contribute to pneumonia, Streptococcus pneumoniae is one of the common bacterial pathogens that underlie community-acquired pneumonia. RIPK3 (receptor-interacting protein kinase 3) is widely recognized as a key modulator of inflammation and cell death. To elucidate a potential role of RIPK3 in pneumonia, we examined plasma from healthy control subjects and patients positive for streptococcal pneumonia. In human studies, RIPK3 protein concentrations were significantly elevated and were identified as a potential plasma marker of pneumococcal pneumonia. To expand these findings, we used an in vivo murine model of pneumococcal pneumonia to demonstrate that RIPK3 deficiency leads to reduced bacterial clearance, severe pathological damage, and high mortality. Our results illustrated that RIPK3 forms a complex with RIPK1, MLKL (mixed-lineage kinase domain-like protein), and MCU (mitochondrial calcium uniporter) to induce mitochondrial calcium uptake and mitochondrial reactive oxygen species(mROS) production during S. pneumoniae infection. In macrophages, RIPK3 initiated necroptosis via the mROS-mediated mitochondrial permeability transition pore opening and NLRP3 inflammasome activation via the mROS–AKT pathway to protect against S. pneumoniae. In conclusion, our study demonstrated a mechanism by which RIPK3-initiated necroptosis is essential for host defense against S. pneumoniae.
Keywords: pneumonia, Streptococcus pneumoniae, receptor-interacting protein kinase 3, mitochondrial permeability transition pore
Streptococcus pneumoniae is a gram-positive bacterium and often colonizes the upper respiratory tract, especially the nasopharynx (1). This asymptomatic colonization can progress to invasive diseases, including pneumonia, sepsis, and meningitis, depending on the host’s immune status (2). S. pneumoniae is the most common pathogen in community-acquired pneumonia and causes significant mortality, especially in children and the elderly (2, 3).
RIPK3 (receptor-interacting protein kinase 3) is a serine/threonine protein kinase that has emerged as a key modulator in cell death signaling and inflammatory pathways. Necroptosis, a form of programmed cell death, is tightly regulated by RIPK3, RIPK1, and MLKL (mixed-lineage kinase domain-like protein) (4–6). Necroptosis plays a vital role in the immune response to bacterial pathogens of the airway, such as Staphylococcus aureus, S. pneumoniae, and Serratia marcescens (4–6). Pore-forming bacterial toxins such as S. pneumoniae pneumolysin and S. marcescens ShlA toxin have been shown previously to be potent inducers of necroptosis, with heightened concentrations of MLKL detectable in nonhuman primates and mice, respectively (4–7). Recent work has illustrated that a deficiency in MLKL or RIPK3 improved survival and reduced pulmonary injury in response to pore-forming toxins secreted by S. marcescens (7). The experiments entailed in our current study are designed to build on these findings and to examine an in vivo role of necroptosis in S. pneumoniae infection.
The NLRP3 infiammasome, a multiprotein complex that contributes to the processing and secretion of proinfiammatory cytokines, can be regulated by RIPK3 under certain circumstances (8, 9). Previous work has demonstrated that NLRP3 and apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) expression are associated with pore-forming toxin–induced necroptosis in macrophages (7, 10). Specifically, NLRP3-deficient macrophages, when challenged directly with pneumolysin, were shown to be protected from necroptosis (10). As necroptosis is a major cell death pathway induced by pore-forming toxins, the experiments in our current study are designed to expand the understanding of RIPK3 on NLRP3 inflammasome activation and whether an impairment in RIPK3 expression is beneficial or detrimental to host defense against S. pneumoniae.
Mitochondria are fundamental organelles crucial for coordinating high-order cell functions, such as intracellular signal transduction, cellular metabolism, calcium homeostasis, and programmed cell death. Recent studies have shown that mitochondria are critically involved in the regulation of the innate immune system for host defense (11, 12). Calcium enters mitochondria via the MCU (mitochondrial calcium uniporter) and can enhance oxidative phosphorylation and mitochondrial reactive oxygen species (mROS) production, which is important for mediating innate antibacterial activity (13, 14). In addition, high concentrations of mROS can cause cell death via activation of the mitochondrial permeability transition pore (mPTP) (15). Our previous findings demonstrate that RIPK3 interacts with mitochondria in the pathogenesis of several lung and kidney diseases (16, 17). Multiple mechanisms, such as diminished ion homeostasis at the plasma membrane, mitochondrial damage, and generation of reactive oxygen species (ROS), have been previously demonstrated to contribute to RIPK3-mediated necroptosis (7, 10). The goal of our current study is to expand on these findings and examine whether an intrinsic mechanism between RIPK3 and mitochondria exists during S. pneumoniae infection.
In the current study, we identify elevated RIPK3 and phosphorylation of downstream effector MLKL (p-MLKL) in the plasma of patients with pneumococcal pneumonia, thereby demonstrating that an increase in RIPK3 and p-MLKL expression is associated with host response against S. pneumoniae. Using a murine model of pneumococcal pneumonia, our results demonstrate that RIPK3 deficiency leads to reduced bacterial clearance, severe pathological damage, and high lethality. Interestingly, our results illustrate that these observations were dependent on the strain of S. pneumoniae, with lethality detected in RIPK3-Adeficient mice infected with serotype 3 strain American Type Culture Collection (ATCC) 6303. Furthermore, our findings illustrate that RIPK3 forms a complex with RIPK1, MLKL, and MCU to induce mitochondrial calcium uptake and mROS production during S. pneumoniae infection. In macrophages, RIPK3 initiates necroptosis via mROS-mediated mPTP opening and NLRP3 inflammasome activation via the mROS–AKT pathway in response to S. pneumoniae. Taken together, our findings illustrate that RIPK3 is essential for host defense against S. pneumoniae.
Methods
Additional detailed methodology is included in the data supplement.
Mice
C57BL/6 mice were purchased from Jackson Laboratory. Ripk3−/−, Mlkl−/−, Ripk3fl/fl LysM-Cre, and Ripk3fl/fl Spc-Cre mice were kindly obtained from Dr. Augustine M. K. Choi. Mice were maintained in the Weill Cornell Medicine pathogen-free facility.
Human Subjects
Adults (n = 30) aged 19 years or older with streptococcal pneumonia, as diagnosed by positive airway culture (sputum, aspirates, BAL), who were admitted to Yale New Haven Hospital, Yale School of Medicine were enrolled for plasma isolation obtained from whole blood on the basis of approval by the Human Investigational Committee. Healthy age-matched control subjects (n = 30) were also recruited.
Streptococcus pneumoniae
S. pneumoniae (ATCC 6303 or D39) was grown on 10% sheep blood agar plates (BD Biosciences) overnight at 37°C (5% CO2). All mice were intranasally instilled with 1 × 107 cfu of 6303 or D39 S. pneumoniae (50 μl volume in PBS).
Cell Culture
Macrophages were prepared as previously described (18). Cells were cultured with media alone or media containing S. pneumoniae ATCC 6303 or D39 (multiplicity of infection [MOI] = 10). Akt inhibitor (MK-2206) was from Selleckchem (S1078). MitoTEMPO (SML0737) was from Millipore Sigma. Cells were pretreated with 1,2-bis(2-aminophenoxy)ethane N,N,N′,N′-tetraacetic acid acetoxymethyl ester (BAPTA-AM) (10 μM, #B6769; Thermo Fisher Scientific) in Hanks’ balanced salt solution for 20 minutes before infection with S. pneumoniae.
ELISA
Human plasma samples were analyzed for RIPK3 (CSB-EL019737HU; Cusabio) and p-MLKL (PEL-MLKL-S345–1; RayBiotech). Culture supernatants and lung homogenates were analyzed for IL-1β (#88–7013–88), IL-6 (#88–7064–88), and TNF-α (#88–7324–88) production (Thermo Fisher Scientific).
Western Blot Analysis
Lung tissues were lysed with a buffer containing tissue extraction reagent (#78510 and #87786; Thermo Fisher Scientific). Cells were lysed with a buffer containing mammalian protein extraction reagent (#78501; Thermo Fisher Scientific, #5872; Cell Signaling Technology). Immunodetection was performed using primary antibodies at 1:1,000 dilution, secondary antibodies at 1:2,000 dilution, and ECL Western Blotting Analysis System (Thermo Fisher Scientific). Images were acquired on film or by using Image Lab 5.0 software (Bio-Rad).
Coimmunoprecipitation
Coimmunoprecipitation with RIPK3 was performed using the Pierce Classic Magnetic Co-IP kit (Thermo Fisher Scientific) according to the manufacturer’s instructions using 300 μg of macrophage cell lysate and 1:100 dilution of rabbit anti-RIPK3 (#15828; Cell Signaling Technology).
Histology and Immunostaining
We used formalin-fixed, paraffin-embedded lung sections stained with hematoxylin and eosin. Immunohistochemical analysis was performed using a specific primary antibody to mouse RIPK3 (ab152130) and biotinylated secondary antibody (Vector Laboratories).
Flow Cytometry
Intracellular Ca2 + was measured with Fluo-3 (#F14218; Thermo Fisher Scientific) staining and was detected by flow cytometry at 506/526 nm. Mitochondrial Ca2 + was measured with Rhod-2 staining. The mROS was measured with MitoSOX Red mitochondrial superoxide indicator (#M36008; Thermo Fisher Scientific) staining. Mitochondrial Permeability Transition Pore Assay Kit (#K239–100; BioVision) was used to analyze the mPTP opening.
Cell Death Assessment
Cell death ratio was measured by Pierce LDH Cytotoxicity Assay Kit (#88953; Thermo Fisher Scientific) or by Real-Time Glo Viability Assay (#G9711; Promega).
Statistical Analysis
All results were expressed as the means ± SEM. All data were analyzed using GraphPad Prism software. A value of P < 0.05 was considered to be statistically significant.
Results
RIPK3 Expression Is Elevated during S. pneumoniae Infection
To explore the involvement of RIPK3 in S. pneumoniae infection, human plasma samples were collected from 30 healthy individuals and 34 patients with S. pneumoniae enrolled in the Center of Pulmonary Infection Research and Treatment at Yale University (Figure 1A). When compared with healthy control subjects, RIPK3 and p-MLKL concentrations in plasma were significantly higher in patients with S. pneumoniae (Figure 1A). To expand these findings, using a murine model of S. pneumoniae infection, we examined RIPK3 expression in the lung. Immunohistochemical analysis of RIPK3 revealed heightened expression in alveolar epithelial cells and macrophages in wild-type (WT) lung in response to S. pneumoniae infection, which was absent in Ripk3−/− lung tissue (Figure 1B). To confirm these findings, we examined RIPK3 protein expression in the lung and macrophages in response to S. pneumoniae. In WT mice, expression levels of RIPK3 and p-MLKL were increased in lung tissue at 24 hours after infection (Figures 1C and E1A in the data supplement). We next examined whether RIPK3 expression in response to S. pneumoniae was upstream of MLKL. When compared with WT mice, there was no detectable difference in RIPK3 expression in the lung in response to S. pneumoniae (Figure E1B). As macrophages play an important role in host defense against S. pneumoniae infection, we next investigated RIPK3, MLKL, and p-MLKL expression in macrophages challenged with S. pneumoniae (19). Although there was no difference in RIPK3 or MLKL expression in response to S. pneumoniae, RIPK3-dependent expression of p-MLKL was increased in WT macrophages in response to S. pneumoniae infection (Figure 1D). In the absence of RIPK3, there is diminished MLKL phosphorylation in lung (Figure 1C) and macrophages (Figure 1D) in response to S. pneumoniae. Taken together, our findings are in agreement with previously published studies and illustrate that RIPK3 expression is elevated in response to S. pneumoniae and that RIPK3-dependent MLKL phosphorylation is induced in macrophages and lung during infection (6, 7, 10).
Figure 1.

RIPK3 (receptor-interacting protein kinase 3) is upregulated both in human plasma and mouse lungs in response to Streptococcus pneumoniae (S. pne) infection. (A) Baseline characteristics and RIPK3 or phosphorylation of downstream effector MLKL (mixed-lineage kinase domain-like protein) concentrations in plasma of healthy control subjects (n = 30) and patients with S. pne (n = 34). (B and C) Wild-type (WT) or Ripk3−/− mice were intranasally instilled with 1 × 107 cfu of S. pne (ATCC 6303) and killed at 1 day after infection. PBS-treated mice were used as a control. (B) S. pne infection enhanced the local recruitment of RIPK3 in the lung as shown by immunohistochemistry. Scale bars, 20 μm. (C) Expression of RIPK3, MLKL, and p-MLKL were examined by Western blot analysis in total protein from lung homogenates. (D) Macrophages derived from WT or Ripk3−/− mice were coincubated with S. pne (MOI = 10) for indicated hours. RIPK3, MLKL, and p-MLKL expressions were determined by Western blot analysis in total protein of cell lysates. Technical and biological replicates for animal experiments, N = 5–10, and for macrophage experiments, N = 3–5. Experiments entailed in B–D were repeated at least three times, with representative findings shown. ***P < 0.001 and ****P < 0.0001. cfu = colony-forming unit; MOI = multiplicity of infection; OD = optical density; p = phosphorylation; p-MLKL = phosphorylation of downstream effector MLKL.
RIPK3 Deficiency Contributes to Increased Bacterial Burden, Lung Inflammation, and Tissue Damage in Response to S. pneumoniae
To investigate the effect of RIPK3 during pneumococcal pneumonia, WT and Ripk3-deficient (Ripk3−/−) mice were infected intranasally with 107 cfu of a serotype 3 strain of S. pneumoniae (ATCC 6303). There was a significant difference in survival between WT and Ripk3−/− mice, with WT mice exhibiting 100% survival by 10 days after infection (Figure 2A). In contrast, only 50% survival was observed in Ripk3−/− mice by 10 days after infection, with increased mortality observed by Day 5 after infection (Figure 2A). To determine the role of RIPK3 in bacterial clearance, WT and Ripk3−/− mice were infected with S. pneumoniae, and lung tissue was collected at select time points after infection. Interestingly, although WT and Ripk3−/− mice exhibited similar bacterial burdens in the lung at Day 1, there is increased bacterial clearance in WT mice Days 2 and 3 after infection (Figure 2B). In contrast, bacterial burden continues to increase in Ripk3−/−, which significantly increased lung bacterial loads detected on Days 2 and 3 after infection (Figure 2B). To assess the lung inflammation and injury, BAL fluid (BALF) was collected from WT and Ripk3−/− mice at Day 3 after infection. When compared with WT mice, Ripk3−/− mice exhibited a significant increase in total cell counts and protein content in BALF in response to S. pneumoniae (Figures 2C and 2D). We next examined the effect of RIPK3 on the lung pathology during S. pneumoniae infection. Genetic deletion of RIPK3 did not affect baseline pulmonary morphology, as PBS-instilled Ripk3−/− mice showed no visible alterations in lungs (Figure 2E). At Day 3 after infection, WT mice had subtle enhancement of lung leukocyte infiltration, whereas Ripk3−/− mice showed progressive lung inflammation with large lesions (Figure 2E). Given these findings, we next examined whether macrophage or epithelial cell expression of RIPK3 was essential for survival during S. pneumoniae infection. To this extent, we generated mice with a macrophage-specific deletion of RIPK3 (Ripk3fl/fl; LysM-Cre) as well as mice with an alveolar type II epithelial cell–specific deletion of RIPK3 (Ripk3fl/fl; SPC-Cre). In response to S. pneumoniae, mice deficient for RIPK3 in macrophages had higher mortality, whereas mice with alveolar type II epithelial cell–specific RIPK3 deficiency had similar mortality when compared with matched control animals (Figures 2F and 2G). Taken together, our results illustrate that RIPK3 expression in macrophages, but in not epithelial cells, plays a key role in the response to S. pneumoniae. Collectively, these findings demonstrate that RIPK3 deficiency leads to reduced bacterial clearance as well as severe lung inflammation and tissue damage, resulting in high mortality in Ripk3−/− mice during S. pneumoniae infection.
Figure 2.

RIPK3 deficiency leads to worse outcome during S. pne infection. Mice were intranasally instilled with S. pne (ATCC 6303) or PBS. (A) Survival study showed that Ripk3−/− mice were more susceptible to S. pne infection than WT mice (n = 10 for each group). *P < 0.05 by Mantel Cox test. (B) Pulmonary bacterial burden of WT or Ripk3−/− mice at indicated times after infection (n = 5 for each group). (C and D) Total cell counts and protein quantification in BAL fluid of WT or Ripk3−/− mice at Day 3 after infection (n = 3 PBS; n = 5 S. pne). *P < 0.05 and ***P < 0.001 by one-way or two-way ANOVA test. Data are presented as mean ± SEM. (E) At 3 days after infection, mouse lungs were harvested for histology. Representative photomicrographs (hematoxylin and eosin stained) taken from each group were shown. Scale bars, 20 μm. (F) Survival study of Ripk3fl/fl; LysM-Cre−/− or Ripk3fl/fl; LysM-Cre+/− mice (n = 10 for each group). *P < 0.05 by Mantel Cox test. (G) Survival study of Ripk3fl/fl; Spc-Cre−/− or Ripk3fl/fl; Spc-Cre+/− mice (n = 10 for each group). Technical and biological replicates for infection experiments, N = 10. Experiments were repeated at least three times, with representative findings shown. NS = no significant difference.
MLKL Deficiency Leads to Excessive Inflammation during S. pneumoniae Infection
To determine the role of MLKL during infection, we examined the response of Mlkl−/− mice after intranasal instillation with S. pneumoniae. In comparison with WT mice, there was significantly higher mortality in the Mlkl−/− mice (Figure 3A). When compared with WT mice, Mlkl−/− mice exhibited increased bacterial burden as well as higher cell counts and protein content in BALF in response to S. pneumoniae (Figures 3B–3D). Examination of lung histology illustrated that Mlkl−/− mice had large areas of consolidation and massive inflammatory cell infiltration at Day 3 after infection when compared with WT mice (Figure 3E). Together, these data suggest that MLKL can participate in the regulation of excessive inflammation and that the absence of MLKL can result in severe lung damage during S. pneumoniae infection.
Figure 3.

MLKL deficiency leads to excessive inflammation during S. pne infection. WT or Mlkl−/− mice were intranasally instilled with 107 S. pne (ATCC 6303) or PBS. (A) Survival study showed that Mlkl−/− mice were more susceptible to S. pne infection than WT mice (n = 10 for each group). ***P < 0.001 by Mantel Cox test. (B) Pulmonary bacterial burden of WT or Mlkl−/− mice at indicated times after infection (n = 5 for each group). (C and D) Total cell counts and protein quantification in BAL fluid of WT or Mlkl−/− mice at Day 3 after infection (n = 3 PBS; n = 5 S. pne). (E) At 3 days after infection, mouse lungs were harvested for histology. Representative photomicrographs (hematoxylin and eosin stained) taken from each group were shown. Scale bars, 20 μm. *P < 0.05, **P < 0.01, and ***P < 0.001 by one-way or two-way ANOVA test. Data are presented as mean ± SEM. Technical and biological replicates for infection experiments, N = 3–5 (PBS control animals) and N = 10 (S. pne). Experiments were repeated at least three times, with representative findings shown.
To examine whether increased pulmonary inflammation and decreased survival in RIPK3- and MLKL-deficient mice might be due to differences in S. pneumoniae virulence, we challenged WT, Ripk3−/−, and Mlkl−/− mice with a more virulent, mouse-adapted strain of S. pneumoniae, D39. In agreement with previously published work, challenge of WT, Ripk3−/−, and Mlkl−/− mice with a more virulent, mouse-adapted strain of S. pneumoniae, D39, resulted in reduced pulmonary injury and improved survival (Figures E2A–E2D) (5–7, 10). Similarly, although Ripk3−/− macrophages were highly susceptible to the ATCC 6303 strain of S. pneumoniae, there was a significant decrease in cell death when macrophages were infected with the D39 strain of S. pneumoniae (Figures E3A and E2B). Taken together, our results demonstrate that RIPK3 deficiency results in an S. pneumoniae strain–dependent phenotype.
RIPK3 Interacts with MCU in Mitochondria
Previous work has illustrated that the RIPK3-containing necrosome can translocate to the mitochondria, where RIPK3 can interact and modulate mitochondrial metabolism (20). To determine whether components of the RIPK3 necrosome migrate to the mitochondria, we isolated mitochondrial and cytosolic fractions from WT macrophages in response to S. pneumoniae. RIPK3 expression in mitochondria was increased in response to S. pneumoniae infection (Figure 4A). RIPK1 and MLKL, two other signaling molecules associated with RIPK3-necrosome, were also increased in mitochondria during infection (Figure 4A) (21). Recent work has illustrated a potential role for RIPK3 in mediating cell death in MCU-deficient cardiomyocytes (22). We next examined whether there is an impact of increased mitochondrial RIPK3 translocation on MCU expression during S. pneumoniae infection. In response to S. pneumoniae, there was increased MCU expression (Figure 4A). MCU functions as a mitochondrial inner membrane transport protein that regulates calcium uptake. To examine whether other mitochondrial proteins might be also be modulated by RIPK3, we next examined the expression of UCP2 (uncoupling protein 2) in response to S. pneumoniae (23). In contrast to MCU, mitochondrial expression of UCP2 in macrophages was similar at baseline and in response to S. pneumoniae (Figure 4A). We next investigated whether RIPK3 might directly interact with mitochondria. Mitochondria were isolated from WT and Ripk3−/− macrophages, and the expression of RIPK3, RIPK1, MLKL, and MCU was examined. In contrast to WT controls, there was diminished RIPK3, RIPK1, and MLKL as well as MCU upregulation in Ripk3−/− macrophages in response to S. pneumoniae (Figure 4B).
Figure 4.
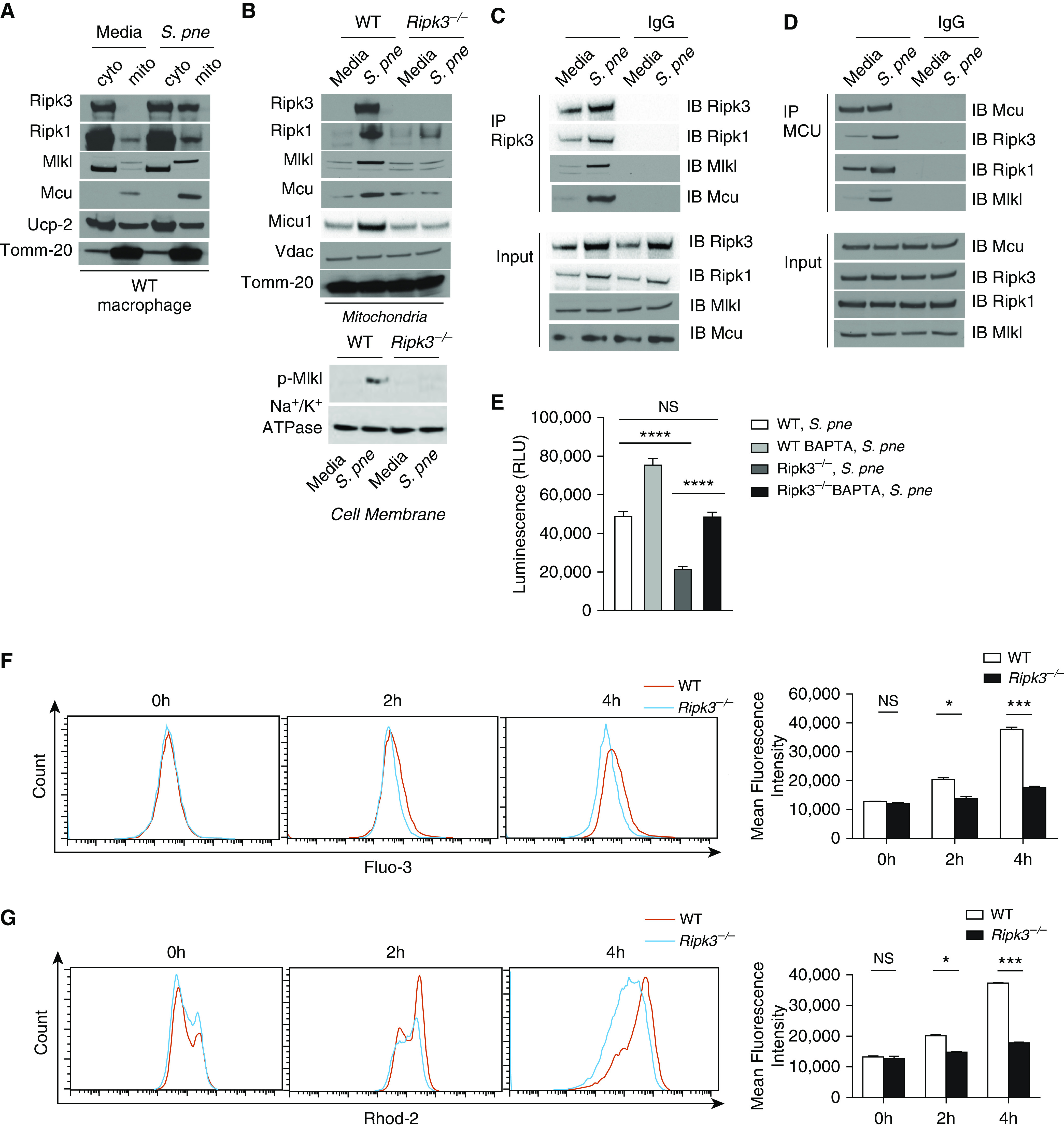
RIPK3 interacts with MCU (mitochondrial calcium uniporter) in mitochondria. Macrophages derived from WT or Ripk3−/− mice were coincubated with S. pne, strain ATCC 6303 (MOI = 10), for 4 hours. (A) Mitochondrial (mito) and cyto fractions from WT macrophages were subjected to Western blot analysis for RIPK3, RIPK1, MLKL, MCU, and UCP2 (uncoupling protein 2). (B) Mitochondria lysates from WT or Ripk3−/− macrophages were subjected to Western blot analysis for RIPK3, RIPK1, MLKL, MCU, MICU1, and VDAC (voltage-dependent anion channel). Tomm-20 was the standard. (C) Coimmunoprecipitation of RIPK3 in total protein from naive or S. pne–infected cell lysates. (D) Coimmunoprecipitation of MCU in total protein from naive or S. pne–infected cell lysates. (E) WT and Ripk3−/− macrophages were pretreated with BAPTA-AM (10 μM, 20 min) before infection with S. pne (MOI = 10) for 4 hours. Cell viability was assessed using the Real Time Glo Viability assay, with decreased luminescence being indicative of cell death. ****P < 0.0001. (F–G) Macrophages derived from WT or Ripk3−/− mice were coincubated with S. pne (MOI = 10) for indicated hours. (F) Cells were loaded with 5 μM Fluo-3, and the intracellular calcium fluorescence was detected by flow cytometric analysis. (G) Cells were loaded with 5 μM Rhod-2, and the mito calcium fluorescence was detected by flow cytometric analysis. *P < 0.05 and ***P < 0.001 by two-way ANOVA test. Data are presented as mean ± SEM. Technical and biological replicates for experiments, N = 3–5. Experiments were repeated at least three times, with representative findings shown. BAPTA = 1,2-bis(2-aminophenoxy)ethane N,N,N′,N′-tetraacetic acid; cyto = cytosolic; RLU = relative light units.
Previous studies have shown that MICU1 (mitochondrial calcium uptake 1) is a regulator of MCU (24, 25). To examine whether RIPK3-mediated changes in MCU were due to changes in MICU1 expression, we examined mitochondrial MICU1 expression in WT and Ripk3−/− macrophages at baseline and in response to S. pneumoniae. Although MICU1 expression was increased in WT mitochondria in response to S. pneumoniae, expression in Ripk3−/− mitochondria remain unchanged (Figure 4B). VDAC (voltage-dependent anion channel), located in the outer mitochondrial membrane, has been shown to control calcium permeation (26, 27). To expand our results, we examined the expression of VDAC in WT and Ripk3−/− macrophages in response to S. pneumoniae. In response to infection, there was similar expression of VDAC in mitochondria isolated from WT and Ripk3−/− macrophages (Figure 4B). As p-MLKL also targets the cell membrane, we examined whether p-MLKL concentrations were also increased in response to S. pneumoniae infection. A RIPK3-dependent increase in membrane expression of p-MLKL was observed that correlated with heightened mitochondrial expression of RIPK3 and MLKL (Figure 4B).
The defining feature of RIPK3-dependent cell death is the formation of the necrosome, a molecular complex comprising RIPK3, RIPK1 Fas-associated protein, and MLKL (28). Furthermore, it has been shown that RIPK1 can bind MCU to mediate the induction of mitochondrial calcium uptake (29). On the basis of these findings, we next performed coimmunoprecipitation assays to determine the potential interaction between RIPK3 and MCU. Our results demonstrate that RIPK3 formed a complex with RIPK1, MLKL, and MCU in mitochondria in response to S. pneumoniae infection (Figures 4C and 4D). As necroptosis results in ion dysregulation, we next examined whether calcium loss and initiation of necroptosis in response to S. pneumoniae could be modulated by pretreatment with calcium chelator BAPTA-AM. As illustrated with Figures 4E and E3, there was a significant decrease in cell viability in Ripk3−/− macrophages when compared with WT macrophages that was rescued with pretreatment with BAPTA-AM.
Because MCU is critical for Ca2 + homeostasis and MCU is regulated by RIPK3, we next measured mitochondrial and cytosolic Ca2 + in WT and Ripk3−/− macrophages. S. pneumoniae stimulation augmented the concentrations of cytosolic (Figure 4F) and mitochondrial Ca2 + (Figure 4G) in WT macrophages. Notably, mitochondrial Ca2 + uptake was blocked in the absence of RIPK3 (Figures 4F and 4G). Taken together, these results indicate that RIPK3 forms a complex with RIPK1, MLKL, and MCU to induct mitochondrial calcium uptake in response to S. pneumoniae infection.
RIPK3 Regulates mROS Production in Response to S. pneumoniae Infection
Mitochondria are a major source of ROS (30). It has been reported that mitochondrial Ca2 + uptake via MCU stimulates the Krebs cycle and oxidative phosphorylation to promote the generation of ROS (31). To determine whether RIPK3 plays a role in the regulation of mROS production, we measured mROS concentrations at baseline and in response to S. pneumoniae. In response to S. pneumoniae, there was increased mROS production in WT macrophages, which was significantly decreased in the absence of RIPK3 (Figure 5A). Treatment with MitoTEMPO, a specific scavenger of mitochondrial superoxide, suppressed mROS production in WT macrophages during S. pneumoniae infection (Figure 5B). Taken together, results of these studies indicate that RIPK3 plays a role in regulating mROS production during S. pneumoniae infection.
Figure 5.
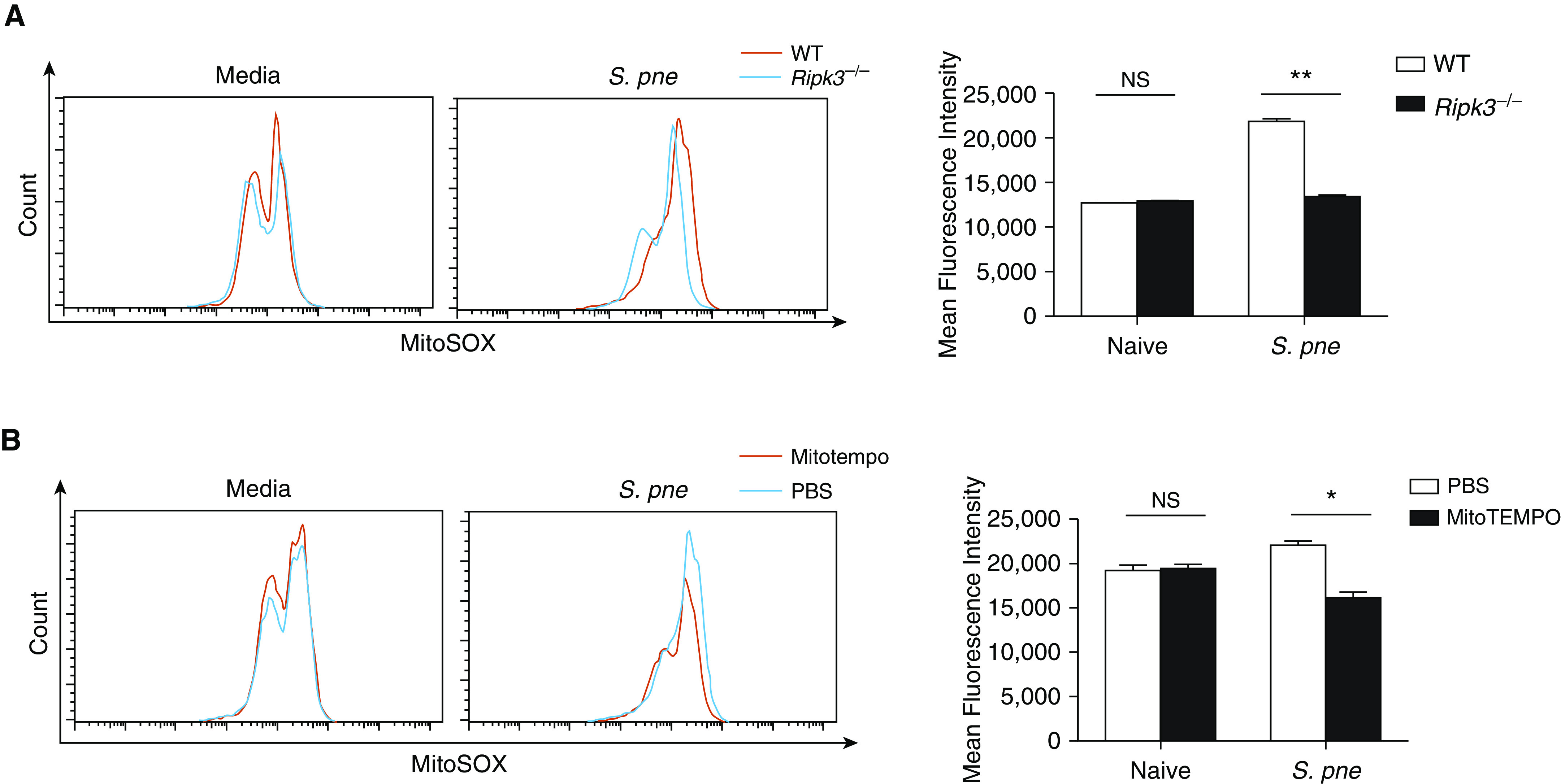
RIPK3 regulates mito reactive oxygen species (ROS) production in response to S. pne infection. Cells were loaded with 5 μM MitoSOX Red. The ROS production in mitochondria was detected by flow cytometric analysis. (A) Macrophages derived from WT or Ripk3−/− mice were coincubated with S. pne, strain ATCC 6303 (MOI = 10), for 4 hours. (B) WT macrophages were coincubated with S. pne (MOI = 10) for 4 hours with/without treatment of 20 μM MitoTEMPO. *P < 0.05 and **P < 0.01 by two-way ANOVA test. Data are presented as mean ± SEM. Technical and biological replicates for experiments, N = 3–5. Experiments were repeated at least three times, with representative findings shown.
RIPK3 Initiates Necroptosis via mROS-mediated mPTP Opening
Previous work has shown that extended or heightened amounts of ROS can trigger the formation and opening of the mPTP, a potential upstream trigger of cellular necroptosis (15, 32–34). In healthy cells, mPTP flutter between open and closed states, but during cell death, the mPTP can alter the permeability of the mitochondria. To this extent, we next examined whether decreased ROS concentrations in Ripk3−/− macrophages impacted mPTP opening in response to S. pneumoniae. In naive WT and Ripk3−/− cells, there was similar cumulative fluorescence in both the cytoplasm and mitochondria (Figures 6A and 6B). Quenching of the cytoplasmic signal by CoCl2 illustrated a significant increase in mitochondrial fluorescence in WT macrophages in response to S. pneumoniae (Figures 6A and 6C). In response to CoCl2 and subsequent treatment with ionomycin, activation of the mPTP was observed in WT macrophages, whereas Ripk3−/− treated macrophages did not exhibit a similar response (Figures 6A and 6D). As there is a detectable increase plasma membrane expression of p-MLKL, we next examined the contribution of cell membrane disruption to this phenotype. WT and Ripk3−/− macrophages were loaded with Calcein red-orange AM for 1 hour before infection with S. pneumoniae strain ATCC 6303 or D39 (Figures E3D and E3E). In response to infection, cell membrane integrity, as indicated by increased release of Calcein red-orange, begins to increase by 4 hours after infection with ATCC 6303 (Figure E3D), with similar amounts of membrane disruption detected in both WT and Ripk3−/− macrophages. In contrast, infection with D39 resulted in increased cell membrane disruption in WT macrophages (Figure E3E). Although there is detectable p-MLKL expression at the cell membrane in WT macrophages at 4 hours of infection, our results demonstrate that similar changes in cell membrane integrity occur in both WT and RIPK3-deficient macrophages in response to ATCC 6303. Taken together, our data demonstrate that RIPK3 may initiate S. pneumoniae strain–dependent cell death via mROS-mediated opening of the mPTP.
Figure 6.
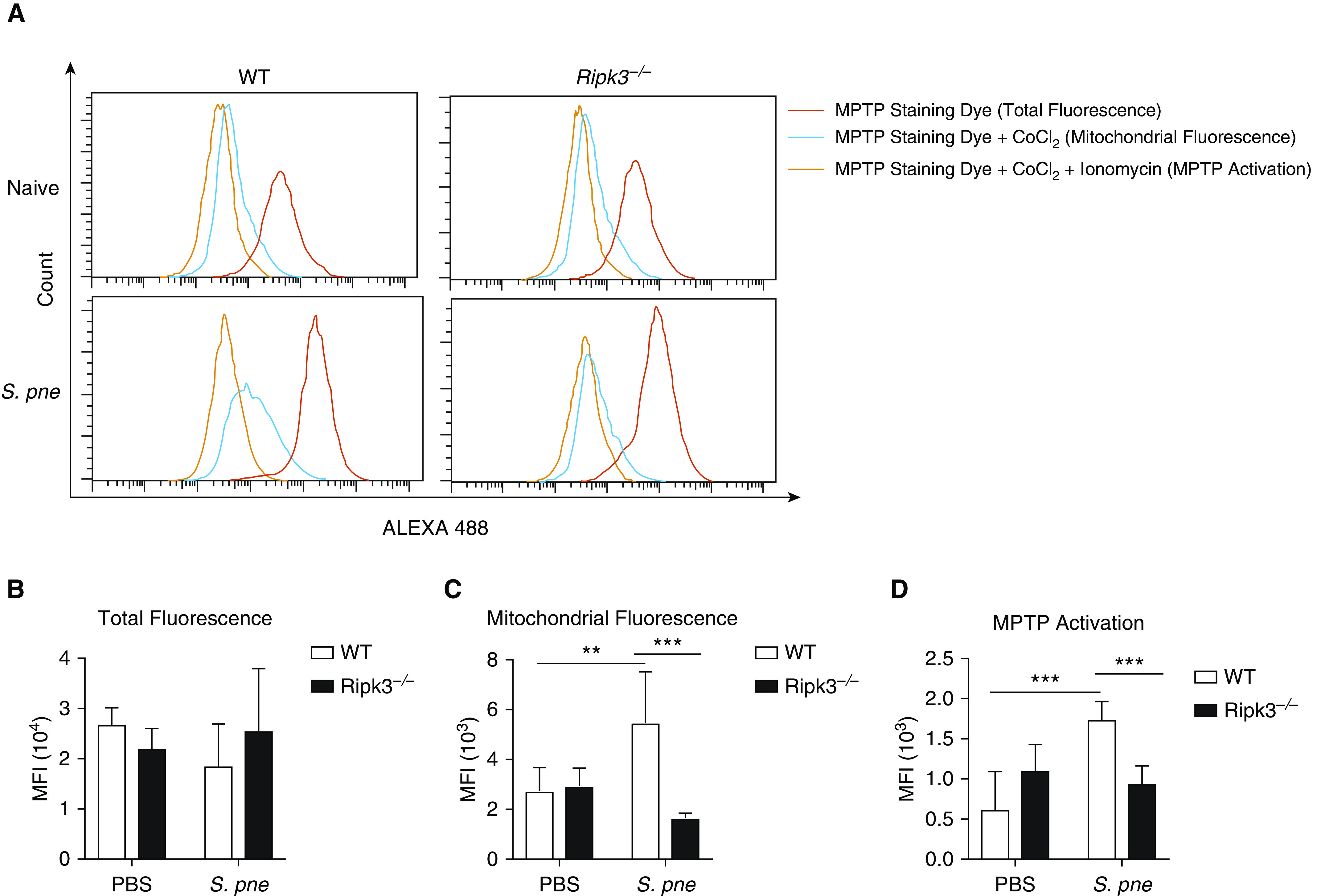
RIPK3 initiates necroptosis via mito ROS–mediated mito permeability transition pore (mPTP) opening. (A) WT macrophages, WT macrophages treated with 20 μM MitoTEMPO, and Ripk3−/− macrophages were coincubated with S. pne, strain ATCC 6303 (MOI = 10), for 4 hours. For each sample, one tube was stained with MPTP staining dye only, one tube was stained with MPTP staining dye and CoCl2, and one tube was stained with MPTP staining dye, CoCl2, and ionomycin. The mPTP opening was detected by flow cytometric analysis. (B) mPTP opening was calculated as the mean florescence intensity (MFI) of MPTP staining dye + CoCl2 minus the MFI of mPTP staining dye + CoCl2 + ionomycin. (C) WT macrophages, WT macrophages treated with 20 μM MitoTEMPO, and Ripk3−/− macrophages were coincubated with S. pne (MOI = 10) for 12 hours. (E) Cell death was measured by LDH assay. **P < 0.01 and ***P < 0.001 by two-way ANOVA test. Data are presented as mean ± SEM. Technical and biological replicates for experiments, N = 3–5. Experiments were repeated at least three times, with representative findings shown. LDH = lactate dehydrogenase; MFI = mean fluorescence intensity.
RIPK3 Initiates NLRP3 Inflammasome Activation via the mROS–AKT Pathway
Previous studies have demonstrated that RIPK3 can promote NLRP3 inflammasome activation (8, 35). We examined the impact of RIPK3 expression on cytokine production in lung homogenates after S. pneumoniae infection. Although there was an increase in cytokine production in WT lung in response to S. pneumoniae, there were significantly less IL-1β, IL-6, and TNF-α secreted in lung homogenates of Ripk3−/− mice (Figure 7A). Furthermore, Mlkl−/− mice had excessive proinflammatory cytokine production during S. pneumoniae infection (Figure E4). Specifically, as early as 1 day after infection, Mlkl−/− mice had higher IL-1β and IL-6 concentrations in lung homogenates than similarly infected WT mice (Figure E4). At Days 2 and 3 after infection, although expression of IL-1β, IL-6, and TNF-α began to decline in WT mice, there was a significant increase in cytokine production in Mlkl−/− mice (Figure E4). Given these findings, we next examined the expression of NLRP3 and IL-1β in lung lysates of WT and Ripk-knockout mice in response to S. pneumoniae. The expression of NLRP3 and IL-1β were significantly increased in WT mouse lung during S. pneumoniae infection, whereas RIPK3 deficiency blocked those effects (Figure 7B). To expand these findings, we next examined the impact of RIPK3 on cytokine production by macrophages in response to S. pneumoniae. WT macrophages produced large concentrations of IL-1β, IL-6, and TNF-α in response to S. pneumoniae. In the absence of RIPK3, there was a marked reduction in IL-1β production but no effect on IL-6 and TNF-α production (Figure 7C). We next examined whether RIPK3 deficiency in macrophages may contribute to decreased NLRP3 and IL-1β expression in response to S. pneumoniae (Figure 7D). In the absence of RIPK3, despite similar ASC expression, there was decreased NLRP3 and IL-1β production by macrophages in during S. pneumoniae infection (Figure 7D).
Figure 7.
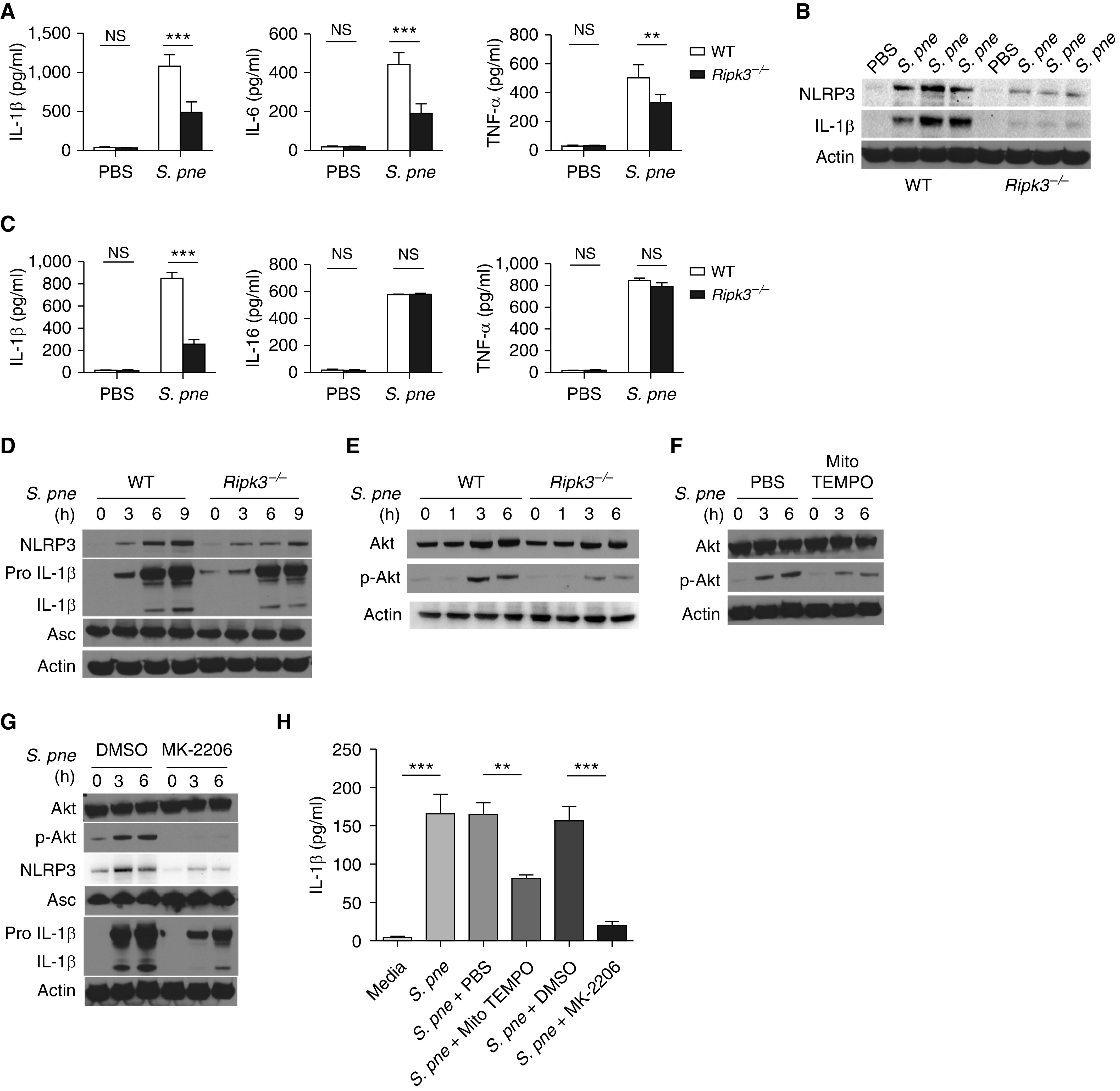
RIPK3 initiates NLRP3 inflammasome activation in response to S. pne infection. (A–B) WT or Ripk3−/− mice were intranasally instilled with 107 S. pne (ATCC 6303) and killed at 1 day after infection. (A) Cytokine concentrations were detected in the supernatants of lung tissue homogenates. (B) Pulmonary expression of NLRP3, IL-1β, and β-actin were examined by Western blot analysis in total protein from lung homogenates. (C–D) WT or Ripk3−/− macrophages were coincubated with S. pne (MOI = 10) for 18 hours. (C) Cytokine concentrations were detected in the culture supernatant. (D) NLRP3, Pro IL-1β, IL-1β, ASC, and β-actin expression were determined by Western blot analysis in total protein of cell lysis. (E–H) WT or Ripk3−/− macrophages were coincubated with S. pne (MOI = 10) for the indicated time. (E) Akt, phospho-Akt, and β-actin expression were determined by Western blot analysis in total protein of cell lysates. (F) WT macrophages were coincubated with S. pne (MOI = 10) with/without treatment of 20 μM MitoTEMPO for the indicated time. Akt, phospho-Akt, and β-actin expression were determined by Western blot analysis in total protein of cell lysates. (G) WT macrophages were coincubated with S. pne (MOI = 10) with/without treatment of 5 μM MK-2206 for the indicated time. Akt, phospho-Akt, NLRP3, Pro IL-1β, IL-1β, Asc, and β-actin expression were determined by Western blot analysis in total protein of cell lysates. (H) IL-1β concentrations were measure by ELISA in the culture supernatant of WT macrophages coincubated with S. pne (MOI = 10) for 18 hours with/without 20 μM MitoTEMPO or 5 μM MK-2206 treatment. **P < 0.01 and ***P < 0.001 by one-way ANOVA test. Data are presented as mean ± SEM. Densitometric analysis of all Western blots is included in Figure E5. Technical and biological replicates for experiments, N = 3–5. Experiments were repeated at least three times, with representative findings shown.
Previous work has demonstrated that AKT is a downstream molecule of RIPK3 (36). It has also been illustrated that mROS can modulate the AKT activation and regulate NLRP3 inflammasome activation (37, 38). On the basis of our current findings, we hypothesized that RIPK3 regulated NLRP3 inflammasome activation via the mROS–AKT pathway. Although increased AKT phosphorylation was observed in WT macrophages in response to S. pneumoniae, there was diminished expression in Ripk3−/− macrophages (Figure 7E). A reduction in mROS after treatment with MitoTEMPO reduced AKT phosphorylation in response to S. pneumoniae (Figure 7F). Treatment with MK-2206, a highly selective inhibitor of AKT, significantly inhibited AKT phosphorylation as well as NLRP3 activation (Figure 7G). Furthermore, IL-1β production in response to S. pneumoniae was reduced in response to decreased mROS (MitoTEMPO) and AKT inhibition (MK-2206 treatment) (Figure 7H). Taken together, those results demonstrate that RIPK3 regulate NLRP3 inflammasome activation via the mROS–AKT pathway.
Discussion
Necroptosis is an important part of host defense against bacterial pathogens. Previous work has demonstrated that necroptosis can promote bacterial clearance by inhibiting excessive inflammatory signaling during S. aureus infection (5). In response to additional bacterial species, such as the gram-negative bacteria S. Typhimurium, RIPK3-dependant necroptosis in macrophages is used by the pathogen to evade detection by the immune system (39). RIPK3 can play a dual role in Mycobacterial tuberculosis infection by inducing mROS production for bacteria killing as well as mediating macrophage necroptosis for extracellular release of Mycobacterium and promotion of bacteria growth (40). In our current study, we found that S. pneumoniae induced RIPK3-MLKL–dependent necroptosis in lung. Decreased expression of RIPK3 and MLKL resulted in increased bacterial loads, severe lung inflammation, and high mortality, implying that necroptosis is beneficial to host defense against S. pneumoniae. Our findings demonstrate that a deficiency in MLKL results in a significant increase in lethality during S. pneumoniae infection. Our results illustrate a critical role for MLKL association at the mitochondrial membrane and increased p-MLKL expression at the plasma membrane in response to S. pneumoniae. Therefore, in the absence of MLKL, initiation of additional molecular signaling cascades essential for host survival to S. pneumoniae may be significantly dampened. Thus, although RIPK3-mediated activation of MLKL is necessary for host survival to serotype 3 ATCC 6303, a complete deficiency of MLKL may contribute to irreversible lung injury that results in heightened pulmonary inflammation and impaired survival.
It is well established that NLRP3 inflammasome activation is required for protective immunity against S. pneumoniae infection (41, 42). IL-1β also plays a major role in resistance to pneumococcal infection (43). Our results demonstrate that S. pneumoniae can induce RIPK3-dependent NLRP3 inflammasome activation in lung. RIPK3 deficiency leads to diminished NLRP3 expression and decreased IL-1β production in lung in response to S. pneumoniae. Taken together, our results illustrate that RIPK3 regulates the balance between proinflammatory signaling for bacterial clearance and the lethal consequences of excessive inflammation. Although RIPK3 initiates NLRP3 inflammasome activation to secrete proinflammatory cytokines that guide immune cell recruitment and bacterial clearance, it can also modulate necroptosis to prevent excessive inflammation and maintain immune homeostasis by clearing dead bacteria and cell debris.
Interestingly, our results demonstrate an S. pneumoniae strain–dependent phenotype in RIPK3 deficient mice. In agreement with previously published work, challenge of WT, Ripk3−/−, and Mlkl−/− mice with a more virulent, mouse-adapted strain of S. pneumoniae, D39, resulted in reduced pulmonary injury and improved survival (5–7, 10). Similarly, although Ripk3−/− macrophages were highly susceptible to the ATCC 6303 strain of S. pneumoniae, there was a significant decrease in cell death when macrophages were infected with the D39 strain of S. pneumoniae. Our results also illustrate that there is a S. pneumoniae strain–dependent alteration in cell membrane integrity, with RIPK3-deficient macrophages being highly susceptible to ATCC 6303 and less susceptible to D39. As pore-forming toxins, such as pneumolysin, play a critical role in modulating the induction of necroptosis, future research into pore-forming toxin regulation and secretion by S. pneumoniae as well as other bacterial pathogens will need to be explored.
It has been established for decades that mitochondrial calcium uptake plays an important role in the regulation of mitochondrial function (44, 45). The delivery of Ca2 + into the mitochondrial matrix can activate mitochondrial dehydrogenases, thereby stimulating mitochondrial respiration and oxidative phosphorylation (46). However, excessive Ca2 + uptake may lead to mitochondrial damage. Ca2 + transport into mitochondria is mediated by the MCU, a 40-kD protein that, together with other proteins, forms a channel complex in the inner mitochondrial membrane (47). Previous studies have reported that deletion of the MCU completely inhibited Ca2 + uptake in heart and skeletal muscle mitochondria (48). It has also been reported that RIPK1 binds MCU to mediate mitochondrial Ca2 + uptake in colorectal cancer (29). Our results reveal a new mechanism by which RIPK3 forms a complex with RIPK1, MLKL and MCU to induct mitochondrial calcium uptake in macrophages in response to S. pneumoniae infection.
Activation and opening of the mPTP has been identified as an important step to induce necroptosis. mPTP can be activated by mitochondrial Ca2 + overload, mROS, and the components of the channel itself (49). CypD (cyclophilin D) has been regarded to play a critical role in mediating mPTP opening by activating mROS. However, most of the studies are limited to cardiac ischemia–reperfusion. Our study notably shows that RIPK3 initiates necroptosis via mROS-mediated activation of the mPTP in response to S. pneumoniae infection.
An increase in mROS induces AKT phosphorylation and downstream NLRP3 inflammasome activation to initiate proinflammatory cytokine release, thereby recruiting immune cells to clear S. pneumoniae. However, heightened accumulation of mROS can induce the activation of the mPTP and contribute to necroptosis. Previous work has illustrated the possible beneficial effect of mROS inhibitors, such as MitoTEMPO, in the treatment of diseases associated with oxidative stress, including myocardial ischemia, acute kidney injury, and age-related diseases (12, 16, 31).
Our research mainly focused on the murine model and cells to explore the function and mechanism of RIPK3. Although we have detected RIPK3 expression in plasma of patients with pneumococcal pneumonia, future work will need to be performed to measure RIPK3 expression in plasma, BALF, and urine to determine whether RIPK3 concentrations are associated with pathogen types, severity, and progression of pneumonia. Furthermore, our previous studies have demonstrated that aged mice are more susceptible to S. pneumoniae because of the impaired NLRP3 inflammasome activation and mitochondrial dysfunction in aged macrophages (11). Thus, whether RIPK3 plays a role in the immune response of the aged group is worth further exploration.
In summary, our study demonstrated that RIPK3 is essential for host defense against S. pneumoniae. RIPK3 deficiency leads to reduced bacterial clearance, severe pathological damage in the lung, and high mortality. Mechanistically, RIPK3 forms a complex with RIPK1, MLKL, and MCU to induce mitochondrial calcium uptake and mROS production during S. pneumoniae infection. In macrophages, RIPK3 initiates necroptosis via mROS-mediated mPTP opening and NLRP3 inflammasome activation via mROS-AKT pathway to protect against S. pneumoniae.
Supplementary Material
Footnotes
Supported by the U.S. National Institutes of Health R01AG052530 (H.W.S.-D.), R01AG056699 (H.W.S.-D.), American Lung Association Biomedical Grant (S.J.C.), K08HL138285 (S.J.C.), and China Scholarship Council (H.-R.H.).
Author Contributions: H.-R.H., S.J.C., C.S.D.C., J.-F.X., and H.W.S.-D. designed the experiments. H.-R.H., R.M.H., J.Y., S.B., L.S., and H.W.S.-D. performed the experiments and analyzed the data. H.-R.H., S.J.C., R.M.H., C.S.D.C., and H.W.S.-D. wrote the manuscript. All authors revised the manuscript and approved of its submission.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0312OC on February 24, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Chao Y, Marks LR, Pettigrew MM, Hakansson AP. Streptococcus pneumoniae biofilm formation and dispersion during colonization and disease. Front Cell Infect Microbiol. 2015;4:194. doi: 10.3389/fcimb.2014.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van der Poll T, Opal SM. Pathogenesis, treatment, and prevention of pneumococcal pneumonia. Lancet. 2009;374:1543–1556. doi: 10.1016/S0140-6736(09)61114-4. [DOI] [PubMed] [Google Scholar]
- 3.Sousa D, Justo I, Domínguez A, Manzur A, Izquierdo C, Ruiz L, et al. Community-acquired pneumonia in immunocompromised older patients: incidence, causative organisms and outcome. Clin Microbiol Infect. 2013;19:187–192. doi: 10.1111/j.1469-0691.2012.03765.x. [DOI] [PubMed] [Google Scholar]
- 4.Ahn D, Prince A. Participation of necroptosis in the host response to acute bacterial pneumonia. J Innate Immun. 2017;9:262–270. doi: 10.1159/000455100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kitur K, Wachtel S, Brown A, Wickersham M, Paulino F, Peñaloza HF, et al. Necroptosis promotes Staphylococcus aureus clearance by inhibiting excessive inflammatory signaling. Cell Rep. 2016;16:2219–2230. doi: 10.1016/j.celrep.2016.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kitur K, Parker D, Nieto P, Ahn DS, Cohen TS, Chung S, et al. Toxin-induced necroptosis is a major mechanism of Staphylococcus aureus lung damage. PLoS Pathog. 2015;11:e1004820. doi: 10.1371/journal.ppat.1004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.González-Juarbe N, Bradley KM, Shenoy AT, Gilley RP, Reyes LF, Hinojosa CA, et al. Pore-forming toxin-mediated ion dysregulation leads to death receptor-independent necroptosis of lung epithelial cells during bacterial pneumonia. Cell Death Differ. 2017;24:917–928. doi: 10.1038/cdd.2017.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun. 2015;6:6282. doi: 10.1038/ncomms7282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yabal M, Müller N, Adler H, Knies N, Groß CJ, Damgaard RB, et al. XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell Rep. 2014;7:1796–1808. doi: 10.1016/j.celrep.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 10.González-Juarbe N, Gilley RP, Hinojosa CA, Bradley KM, Kamei A, Gao G, et al. Pore-forming toxins induce macrophage necroptosis during acute bacterial pneumonia. PLoS Pathog. 2015;11:e1005337. doi: 10.1371/journal.ppat.1005337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Misawa T, Takahama M, Saitoh T. Mitochondria-endoplasmic reticulum contact sites mediate innate immune responses. Adv Exp Med Biol. 2017;997:187–197. doi: 10.1007/978-981-10-4567-7_14. [DOI] [PubMed] [Google Scholar]
- 12.Plataki M, Cho SJ, Harris RM, Huang HR, Yun HS, Schiffer KT, et al. Mitochondrial dysfunction in aged macrophages and lung during primary Streptococcus pneumoniae infection is improved with pirfenidone. Sci Rep. 2019;9:971. doi: 10.1038/s41598-018-37438-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Y, Zhou Z, Min W. Mitochondria, oxidative stress and innate immunity. Front Physiol. 2018;9:1487. doi: 10.3389/fphys.2018.01487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–1014. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sureshbabu A, Patino E, Ma KC, Laursen K, Finkelsztein EJ, Akchurin O, et al. RIPK3 promotes sepsis-induced acute kidney injury via mitochondrial dysfunction. JCI Insight. 2018;3:e98411. doi: 10.1172/jci.insight.98411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest. 2014;124:3987–4003. doi: 10.1172/JCI74985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol. 2008;Chapter 14:Unit 14.1. doi: 10.1002/0471142735.im1401s83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cole J, Aberdein J, Jubrail J, Dockrell DH. The role of macrophages in the innate immune response to Streptococcus pneumoniae and Staphylococcus aureus: mechanisms and contrasts. Adv Microb Physiol. 2014;65:125–202. doi: 10.1016/bs.ampbs.2014.08.004. [DOI] [PubMed] [Google Scholar]
- 20.Yang Z, Wang Y, Zhang Y, He X, Zhong CQ, Ni H, et al. RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nat Cell Biol. 2018;20:186–197. doi: 10.1038/s41556-017-0022-y. [DOI] [PubMed] [Google Scholar]
- 21.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 22.Parks RJ, Menazza S, Holmström KM, Amanakis G, Fergusson M, Ma H, et al. Cyclophilin D-mediated regulation of the permeability transition pore is altered in mice lacking the mitochondrial calcium uniporter. Cardiovasc Res. 2019;115:385–394. doi: 10.1093/cvr/cvy218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trenker M, Malli R, Fertschai I, Levak-Frank S, Graier WF. Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nat Cell Biol. 2007;9:445–452. doi: 10.1038/ncb1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xing Y, Wang M, Wang J, Nie Z, Wu G, Yang X, et al. Dimerization of MICU proteins controls Ca2+ influx through the mitochondrial Ca2+ uniporter. Cell Rep. 2019;26:1203–1212, e4. doi: 10.1016/j.celrep.2019.01.022. [DOI] [PubMed] [Google Scholar]
- 25.Phillips CB, Tsai CW, Tsai MF. The conserved aspartate ring of MCU mediates MICU1 binding and regulation in the mitochondrial calcium uniporter complex. eLife. 2019;8:e41112. doi: 10.7554/eLife.41112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gincel D, Zaid H, Shoshan-Barmatz V. Calcium binding and translocation by the voltage-dependent anion channel: a possible regulatory mechanism in mitochondrial function. Biochem J. 2001;358:147–155. doi: 10.1042/0264-6021:3580147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rapizzi E, Pinton P, Szabadkai G, Wieckowski MR, Vandecasteele G, Baird G, et al. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J Cell Biol. 2002;159:613–624. doi: 10.1083/jcb.200205091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeng F, Chen X, Cui W, Wen W, Lu F, Sun X, et al. RIPK1 binds MCU to mediate induction of mitochondrial Ca2+ uptake and promotes colorectal oncogenesis. Cancer Res. 2018;78:2876–2885. doi: 10.1158/0008-5472.CAN-17-3082. [DOI] [PubMed] [Google Scholar]
- 30.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dietl A, Maack C. Targeting mitochondrial calcium handling and reactive oxygen species in heart failure. Curr Heart Fail Rep. 2017;14:338–349. doi: 10.1007/s11897-017-0347-7. [DOI] [PubMed] [Google Scholar]
- 32.Kim JS, Jin Y, Lemasters JJ. Reactive oxygen species, but not Ca2+ overloading, trigger pH- and mitochondrial permeability transition-dependent death of adult rat myocytes after ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2006;290:H2024–H2034. doi: 10.1152/ajpheart.00683.2005. [DOI] [PubMed] [Google Scholar]
- 33.Nakayama H, Chen X, Baines CP, Klevitsky R, Zhang X, Zhang H, et al. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest. 2007;117:2431–2444. doi: 10.1172/JCI31060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 35.Kang TB, Yang SH, Toth B, Kovalenko A, Wallach D. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity. 2013;38:27–40. doi: 10.1016/j.immuni.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 36.Imamura M, Moon JS, Chung KP, Nakahira K, Muthukumar T, Shingarev R, et al. RIPK3 promotes kidney fibrosis via AKT-dependent ATP citrate lyase. JCI Insight. 2018;3:e94979. doi: 10.1172/jci.insight.94979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moon JS, Lee S, Park MA, Siempos II, Haslip M, Lee PJ, et al. UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J Clin Invest. 2015;125:665–680. doi: 10.1172/JCI78253. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38.Kim JH, Choi TG, Park S, Yun HR, Nguyen NNY, Jo YH, et al. Mitochondrial ROS-derived PTEN oxidation activates PI3K pathway for mTOR-induced myogenic autophagy. Cell Death Differ. 2018;25:1921–1937. doi: 10.1038/s41418-018-0165-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol. 2012;13:954–962. doi: 10.1038/ni.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roca FJ, Ramakrishnan L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell. 2013;153:521–534. doi: 10.1016/j.cell.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McNeela EA, Burke A, Neill DR, Baxter C, Fernandes VE, Ferreira D, et al. Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog. 2010;6:e1001191. doi: 10.1371/journal.ppat.1001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Witzenrath M, Pache F, Lorenz D, Koppe U, Gutbier B, Tabeling C, et al. The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J Immunol. 2011;187:434–440. doi: 10.4049/jimmunol.1003143. [DOI] [PubMed] [Google Scholar]
- 43.Kafka D, Ling E, Feldman G, Benharroch D, Voronov E, Givon-Lavi N, et al. Contribution of IL-1 to resistance to Streptococcus pneumoniae infection. Int Immunol. 2008;20:1139–1146. doi: 10.1093/intimm/dxn071. [DOI] [PubMed] [Google Scholar]
- 44.Deluca HF, Engstrom GW. Calcium uptake by rat kidney mitochondria. Proc Natl Acad Sci USA. 1961;47:1744–1750. doi: 10.1073/pnas.47.11.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mraz FR. Calcium and strontium uptake by rat liver and kidney mitochondria. Proc Soc Exp Biol Med. 1962;111:429–431. doi: 10.3181/00379727-111-27814. [DOI] [PubMed] [Google Scholar]
- 46.Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta. 2009;1787:1309–1316. doi: 10.1016/j.bbabio.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 47.Foskett JK, Philipson B. The mitochondrial Ca(2+) uniporter complex. J Mol Cell Cardiol. 2015;78:3–8. doi: 10.1016/j.yjmcc.2014.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15:1464–1472. doi: 10.1038/ncb2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Webster KA. Mitochondrial membrane permeabilization and cell death during myocardial infarction: roles of calcium and reactive oxygen species. Future Cardiol. 2012;8:863–884. doi: 10.2217/fca.12.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jain S, Self WH, Wunderink RG, Fakhran S, Balk R, Bramley AM, et al. CDC EPIC Study Team. Community-acquired pneumonia requiring hospitalization among U.S. Adults. N Engl J Med. 2015;373:415–427. doi: 10.1056/NEJMoa1500245. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.