

Abstract
Mitochondria have emerged as important signaling organelles where intracellular perturbations are integrated and, consequently, intracellular signaling pathways are modulated to execute appropriate cellular functions. MAVS (mitochondrial antiviral signaling protein) represents such an example that functions as a platform molecule to mediate mitochondrial innate immune signaling. Recently, multimeric aggregation of MAVS has been identified as a key molecular process for its signaling. The underlying mechanisms to regulate this, however, are still incompletely understood. We hypothesized that PINK1 (PTEN-induced kinase 1) plays an important role in the regulation of multimeric MAVS aggregation and its consequent pathobiology. To test whether PINK1 interacts with MAVS, bimolecular fluorescence complementation analysis and IP were performed. RLH (RIG-I–like helicase) and NLRP3 inflammasome signaling were evaluated by in vitro assay. In vivo functional significance of PINK1 in the regulation of MAVS signaling was evaluated from both murine modeling of influenza viral infection and bleomycin-induced experimental pulmonary fibrosis, wherein MAVS plays important roles. Multimeric MAVS aggregation was induced by mitochondria dysfunction, and, during this event, the stabilized PINK1 interacted physically with MAVS and antagonized multimeric MAVS aggregation. Accordingly, the MAVS-mediated antiviral innate immune and NLRP3 inflammasome signaling were enhanced in PINK1 deficiency. In addition, in vivo studies revealed that MAVS-mediated pulmonary antiviral innate immune responses and fibrotic responses after bleomycin injury were enhanced in PINK1 deficiency. In conclusion, these results establish a new role of PINK1 in the regulation of MAVS signaling and the consequent pulmonary pathobiology.
Keywords: PTEN‐induced kinase 1, mitochondrial antiviral signaling protein, MAVS aggregation, mitochondrial signaling, inflammasomes
Clinical Relevance
Mitochondrial dysfunction has recently emerged as a major player in a myriad of human health and diseases in which MAVS (mitochondrial antiviral signaling protein) signaling mediates critical innate immune and tissue damage responses. By demonstrating that PINK1 (PTEN-induced kinase 1), an important player of mitochondrial health, interacts with MAVS and functions to inhibit MAVS signaling and the consequent pulmonary pathobiology, including antiviral response and experimental pulmonary fibrosis, the current study provides a conceptual proof that augmenting PINK1 activity may be a novel therapeutic approach for pulmonary disorders in which PINK1–MAVS interaction plays a pathogenic role.
Mitochondria have emerged as important signaling organelles (1, 2). Mitochondrial and intracellular physiology are intricately connected, and, responding to intracellular perturbations, mitochondrial functions are influenced accordingly. The alteration of mitochondrial functions, in turn, modulates various intracellular signals to execute appropriate cellular functions (2, 3). MAVS (mitochondrial antiviral signaling protein) represents such an example and functions as a platform molecule to mediate mitochondrial antiviral or inflammasome signaling (4, 5). Upon the recognition of cytoplasmic nucleic acids, pathogen-associated molecular patterns, or danger-associated molecular patterns (DAMPs) by their cognate receptors, RLH (RIG-I–like helicase) signaling or nucleotide-binding domain, leucine-rich repeat containing, or NLRP3 (NOD-like receptor family pyrin domain containing 3) inflammasome signaling is activated, during which MAVS plays as an essential adaptor molecule to mediate these innate immune and inflammatory responses (5, 6).
Interestingly, when MAVS is activated, it aggregates together to form multimeric structures on mitochondria, and the MAVS aggregation has recently been identified as a key event for its proper functioning (5–7). Although the formation of multimeric structures is believed to be beneficial to propagate a robust host defense response, mechanisms should exist to inhibit persistent activation of MAVS-mediated signaling and keep it in homeostatic physiologic state. Otherwise, unregulated MAVS signaling would result in chronic persistent inflammatory or autoimmune disease states (7). Our understanding of the inhibitory mechanism of MAVS aggregation, however, remains inadequate.
PINK1 (PTEN-induced kinase 1) is a mitochondrially targeted serine/threonine kinase whose loss of function causes striking changes in mitochondrial structure and function (8, 9). It was identified as a gene whose mutation mediates an early-onset autosomal recessive form of parkinsonism in humans (10). Now, multiple lines of evidence suggest that PINK1 plays a key role in mitochondrial quality control, an integrated coordination of mechanisms operating to protect mitochondria against stress, monitor mitochondrial damage, and ensure the selective removal of dysfunctional mitochondrial proteins or organelles (9, 11–13). The functional role of PINK1 on the regulation of MAVS homeostasis, however, has not been reported yet.
Here, we demonstrate that PINK1 plays as a critical negative regulator of MAVS-mediated signaling by inhibiting MAVS aggregation. Responding to mitochondrial dysfunction, MAVS is massively aggregated on mitochondria, and, interestingly, the MAVS aggregation and the consequent MAVS distal signaling responses are significantly enhanced in the absence of PINK1. Our mechanistic study demonstrates that PINK1 interacts directly with MAVS and inhibits its aggregation. Furthermore, the in vivo functional significance of PINK1 that negatively regulates MAVS signaling is verified from both murine modeling of influenza viral infection and bleomycin-induced experimental pulmonary fibrosis. Taken together, the current study reveals a previously unidentified role of PINK1 that negatively regulates the function of MAVS, a crucial mitochondrial innate immune signaling adaptor molecule.
Methods
Mice
Wild-type (WT) and PINK1-null mutant (−/−) (from Dr. J. H. Shin, Harvard University), and MAVS−/− (from Dr. Z. J. Chen, University of Texas) mice were all kept on a C57BL/6J background and bred at Yale University. By breeding PINK−/− mice with MAVS−/− mice, PINK1/MAVS double-null mutant (PINK1−/−/MAVS−/−) mice were obtained. All animal experiments were approved by the Yale Animal Care and Use Committee.
Influenza Viral Infection
WT, PINK1−/−, or PINK1−/−/MAVS−/− mice were lightly anesthetized, and A/PR8/34 influenza (equivalent to 0.05 median lethal dose in C57BL/6J mice) virus was administered via nasal aspiration, as previously described by our laboratory (14). The mice were then killed at Day 5, a peak time point of type I IFN responses. Lung tissues were harvested to measure the expression of MAVS-mediated signaling.
Bleomycin-induced Pulmonary Fibrosis
WT, PINK1−/−, or PINK1−/−/MAVS−/− mice were treated with 0.4 U/kg bleomycin (NDC 61703–0332–18; Pfizer) with PBS by intratracheal administration, as previously described by our laboratory (15).
Cells
Human embryonic kidney 293 (HEK293) cells were cultured in Eagle’s minimum essential medium containing 10% FBS. WT and PINK1−/− mouse embryonic fibroblasts (MEFs) were cultured in Dulbecco’s modified Eagle medium (DMEM), supplemented with 10% FBS, 5 mg/ml penicillin, and 10 mg/ml streptomycin. Bone marrow–derived macrophages (BMDMs) were prepared following the method previously described (16).
Isolation of the Mitochondria
Mitochondria were isolated using Qproteome Mitochondria Isolation kit (QIAGEN) according to the manufacturer’s instructions.
Evaluation of the MAVS Aggregation
MEF cells were harvested for the isolation of mitochondria. Then, MAVS aggregation was evaluated by blue native PAGE (BN-PAGE), as described previously (17). Briefly, 20 μg of isolated crude mitochondrial proteins was lysed in 1% digitonin/1X NativePAGE sample loading buffer (Thermo Fisher Scientific) for 30 minutes on ice and then loaded to 3–12% BN-PAGE (Thermo Fisher Scientific) under nonreducing condition. Samples were then transferred to a polyvinylidene fluoride or polyvinylidene difluoride membrane for Western blot analysis. For the two-dimensional gel analysis, each lane of BN-PAGE was excised and loaded to two-dimensional gel (Thermo Fisher Scientific) according to the manufacturer’s instructions. For semidenaturing detergent agarose gel electrophoresis, pooled BAL cells were lysed in radioimmunoprecipitation assay buffer, and mitochondria were resuspended in 1× sample buffer (50 mM Tris/HCL, 5% glycerol, 2% SDS, and 0.0025% bromophenol blue) and loaded onto a vertical 1.5% agarose gel.
Statistical Analysis
All statistical analysis was performed with GraphPad Prism (version 6). Comparisons between two groups were performed with Student’s t test (unpaired). Values are expressed as mean ± SD. Statistical significance was defined at a level of P < 0.05.
Additional experimental methods are described in online supplement; these methods include 1) evaluation of the alteration of mitochondrial membrane potential, pH, and reactive oxygen species (ROS), 2) complex V activity, 3) plasmid construction and biomolecular fluorescence complementation assay, 4) IP and immunoblot analysis, 5) immunofluorescence staining and confocal microscopy, 6) activation of NLRP3 inflammasomes, 7) activation of type I IFN response, 8) collagen assay, and 9) TGF- β1 (transforming growth factor-β1) measurement.
Results
Mitochondrial Dysfunction Induces MAVS Aggregation
MAVS has recently been identified to aggregate together to form prion-like multimeric structures on mitochondria during its activation (5, 6). We questioned whether MAVS aggregation is incited by various mitochondrial dysfunctions. To address this, HEK293 cells were treated with diverse mitochondrial stimuli, and MAVS aggregation was evaluated by BN-PAGE, which has been extensively used for the detection of mitochondrial high-order structures (6, 17). Specifically, nigericin (a potassium ionophore promoting K+/H+ exchange), carbonyl cyanide m-chlorophenyl hydrazone (CCCP) (an uncoupler of mitochondrial oxidative phosphorylation), oligomycin A (an inhibitor of mitochondrial H+-ATP synthase), rotenone (an inhibitor of mitochondrial oxidative phosphorylation complex I and an inducer of mitochondrial ROS production), and valinomycin (a K+-specific ionophore) were used for this purpose. Intriguingly, among these agents, only nigericin, CCCP, and valinomycin induced significant aggregation of MAVS on mitochondria (Figure 1A). In contrast, the inhibition of ATP synthesis (by oligomycin A treatment), impairment of mitochondrial respiration (by rotenone treatment), or enhanced ROS production (by rotenone treatment) had only a negligible effect on the MAVS aggregation (Figure 1A). Further experiments revealed that the nature of mitochondrial dysfunction that induces MAVS aggregation might be attributed to the decrease of mitochondrial membrane potential or the proton gradient; as known, the mitochondrial membrane potential and the proton gradient contribute independently to the mitochondrial proton-motive force, a key aspect of mitochondrial function (Figures E1A–E1D) (18, 19). It is of note that the MAVS aggregation was not explained by the alteration of its total amount in whole cells (Figure 1A) or mitochondrial fractions (Figure 1A). To validate this observation further, two-dimensional native/SDS-PAGE was performed. Indeed, although no significant aggregation of MAVS was observed in control cells, multimeric aggregation of MAVS was observed after nigericin or CCCP treatment (Figure 1B). In addition, the denaturation of these MAVS aggregates revealed that ∼75 kD or ∼65 kD sizes of MAVS were major components of MAVS aggregates. To confirm the site of MAVS aggregation, we performed an immunofluorescent assay to localize the specific subcellular organelle where the MAVS aggregation takes place. Our data show that most of MAVSs are colocalized with mitochondria at baseline, and the colocalization of MAVS on mitochondria was more accentuated after nigericin stimulation compared with colocalization of MAVS with peroxisome (Figures 1C–1E). Taken together, these data demonstrate that MAVS forms multimeric aggregation on mitochondria upon various forms of mitochondrial perturbation.
Figure 1.
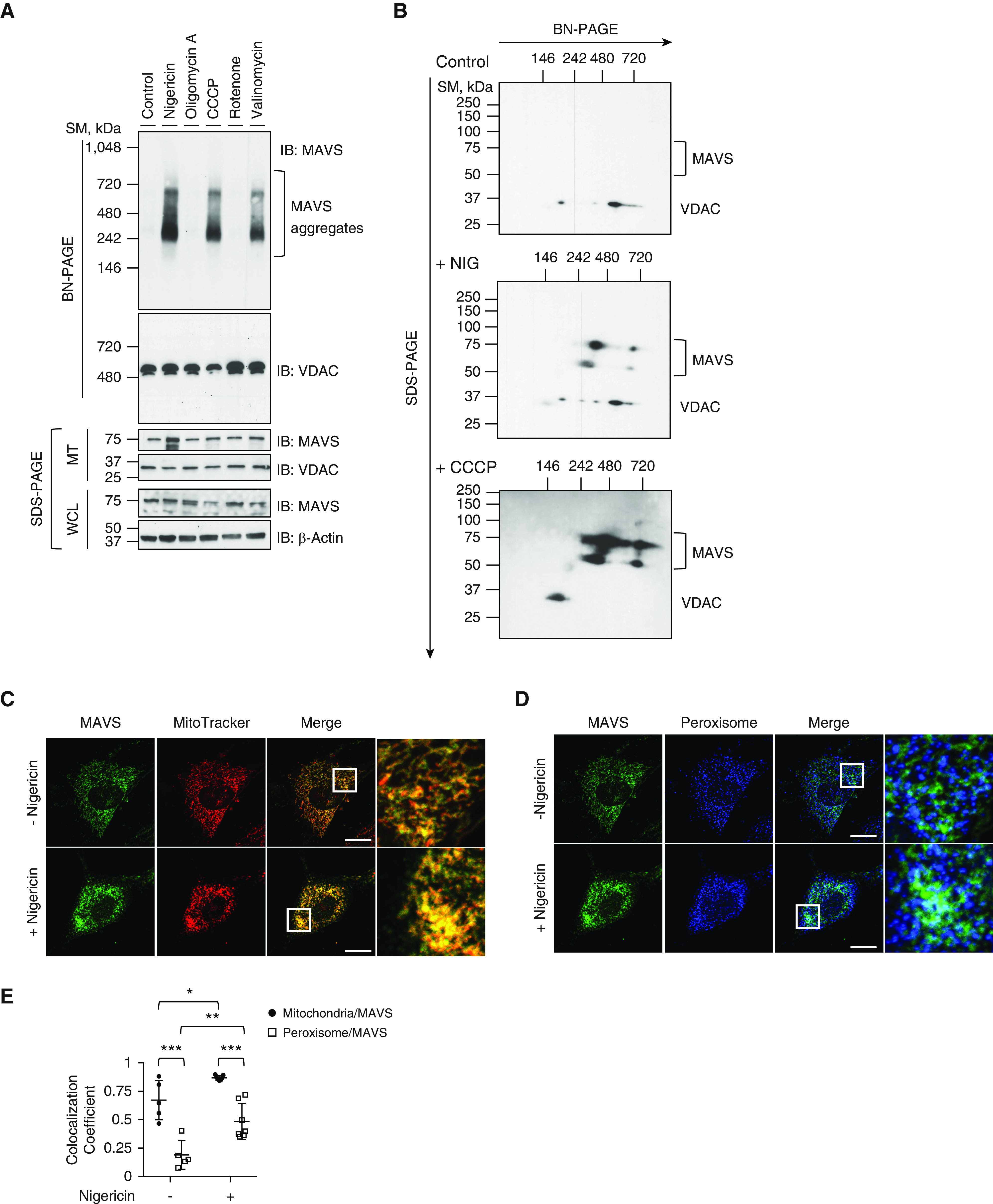
MAVS (mitochondrial antiviral signaling protein) aggregation by mitochondrial drugs. (A) Human embryonic kidney 293 (HEK293) cells were stimulated with 5 μM of nigericin, 5 μM of oligomycin A, 50 μM of carbonyl cyanide m-chlorophenyl hydrazone (CCCP), 1 μM of rotenone, or 10 μM of valinomycin for 1 hour. MTs were separated and loaded to blue native PAGE (BN-PAGE). The MAVS aggregation was analyzed by Western blot. Voltage-dependent anion channel (VDAC) was used as the loading control for mitochondrial analysis. (B) HEK293 cells were stimulated with 5 μM of nigericin or 50 μM of CCCP for 1 hour. Mitochondrial fractions were separated and loaded to BN-PAGE. For two-dimensional analysis, each lane was excised and loaded on second-dimension SDS gel. The MAVS aggregation was analyzed by Western blot. VDAC was used as the loading control. Readers may view the uncut gels for A and B in the data supplement. (C–E) Mouse embryonic fibroblasts from wild-type mice were coimmunostained with MitoTracker Orange CMTMRos (red, C) and MAVS (green, C) or with PMP70 (blue, D) and MAVS (green, D). The images for localization of MAVS and its aggregates were acquired by confocal microscopy. (E) Colocalization of MAVS to mitochondria or peroxisome was analyzed using ZEN2010 software. Scale bars, 20 μm. All experiments are repeated at least three times. Representative results are shown. Means ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001. MT = mitochondrial fraction; PINK1 = PTEN-induced kinase 1; SDS-PAGE = sodium dodecyl sulfate-PAGE; SM = size marker; WCL = whole cell lysate.
PINK1 Regulates the MAVS Aggregation
After establishing that MAVS aggregation is a conserved mechanism downstream to various mitochondrial perturbation pathways, we sought to determine the negative regulators of MAVS pathway. As PINK1 performs essential functions in mitochondrial homeostasis during stress, we hypothesized that PINK1 may play as a negative regulator of MAVS.
To test this, experiments were undertaken in which MEFs from WT and PINK1−/− mice were stimulated with nigericin. In WT MEFs, MAVS aggregation was observed after 45 minutes of stimulation of nigericin and further increased at 1 hour of nigericin stimulation. Interestingly, however, the nigericin-induced formation of the MAVS aggregates was significantly enhanced in the absence of PINK1 (Figure 2A), suggesting that PINK1 has an inhibitory role in the formation of MAVS aggregation. In addition, the experiments were undertaken to address whether PINK1 has a functional role on the removal/or resolution of MAVS aggregates. Specifically, MEFs were treated with 5 μM of nigericin for 1 hour and then washed once with serum-free DMEM and maintained with DMEM containing 10% FBS for 24 hours or 48 hours. Then, the amount of MAVS aggregates was evaluated during this period. In WT MEFs, MAVS aggregates were sustained for ∼24 hours and after 48 hours, most of MAVS aggregates had disappeared (Figure 2B). In contrast, MAVS aggregates were observed in significantly more amounts in PINK1−/− MEFs during this period and were sustained for at least 48 hours in the absence of PINK1, suggesting that PINK1 has a role on the removal/or resolution of MAVS aggregates (Figure 2B). Next, to address whether PINK1 has an inhibitory role of RLH signaling–mediated MAVS aggregation, experiments were undertaken in which MEFs from WT and PINK1−/− mice were stimulated with polyinosinic–polycytidylic acid (polyI:C), a synthetic analog double-stranded RNA that is an established stimulator of RLH signaling. Indeed, the results demonstrated that RLH signaling–mediated MAVS aggregation was significantly enhanced in the absence of PINK1 (Figure 2C). In addition, we observed that polyI:C induced a significant mitochondrial depolarization in both HEK293 cells and MEFs, respectively (Figures E2A and E2B). Overall, these data suggest that PINK1 may inhibit MAVS aggregation induced by both NLRP3 inflammasome signaling and RLH signaling.
Figure 2.
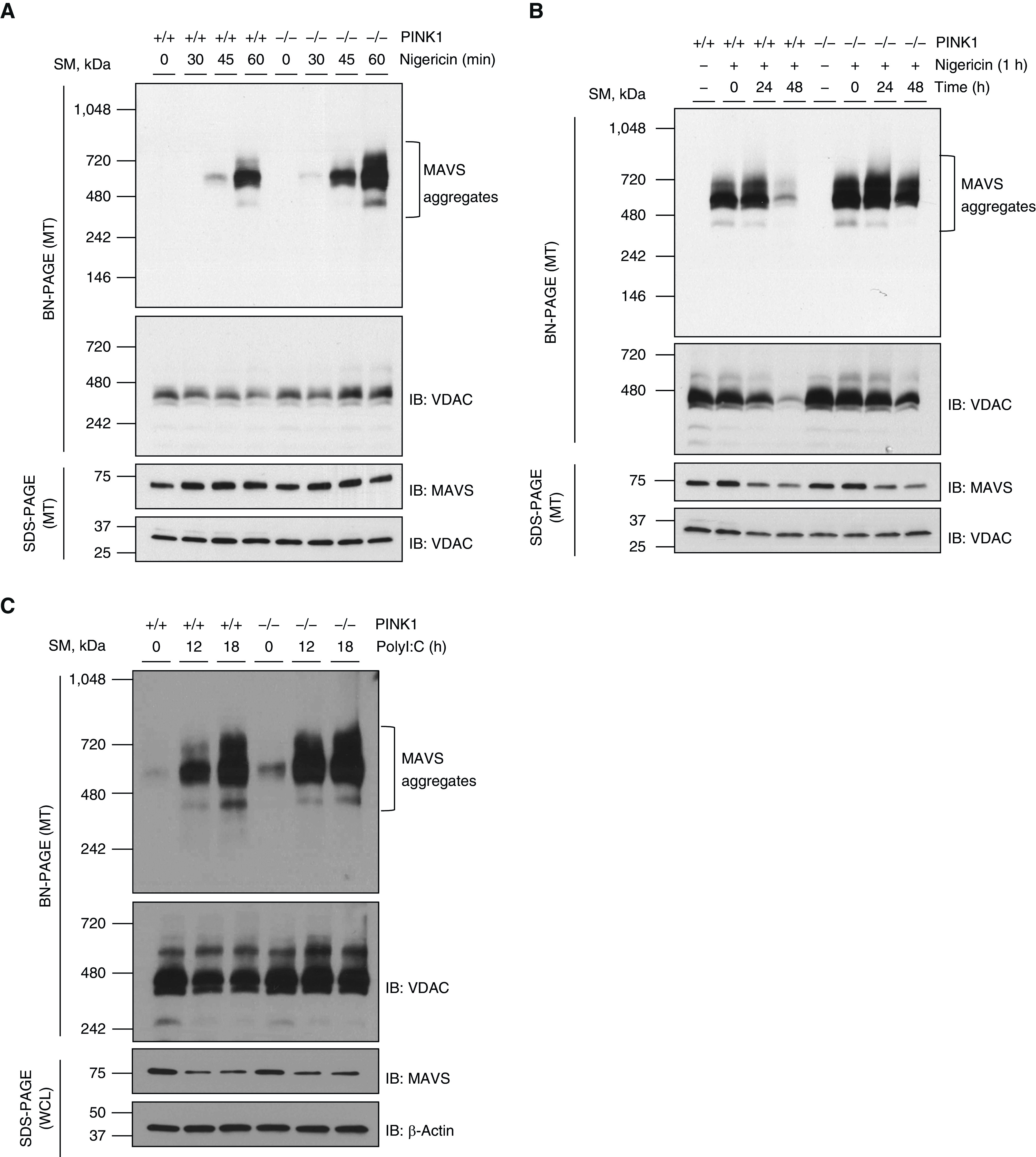
The augmentation of MAVS aggregation in the absence of PINK1. (A) Mouse embryonic fibroblasts (MEFs) from wild-type (WT) and PINK1−/− mice were stimulated with 5 μM of nigericin for indicated time. MTs were separated and loaded to BN-PAGE. The MAVS aggregation was analyzed by Western blot. (B) MEFs from WT and PINK1−/− mice were stimulated with 5 μM of nigericin for 1 hour. Then, cells were washed with serum-free Dulbecco’s modified Eagle medium (DMEM) and maintained with 10% FBS/DMEM for indicated time. MTs were separated and loaded to BN-PAGE. The MAVS aggregation was analyzed by Western blot. (C) MEFs from WT and PINK1−/− mice were stimulated with 10 μg/ml of high molecular weight polyI:C for the indicated time. MTs and WCLs were separated and loaded to BN-PAGE and SDS-PAGE, respectively. The MAVS aggregation was analyzed by Western blot. VDAC and β-actin were used as loading controls of MTs and WCLs, respectively. All experiments are repeated at least three times. Representative results are shown. PolyI:C = polyinosinic–polycytidylic acid.
PINK1 Dynamically Interacts with MAVS
Next, we sought to determine how the absence of PINK1 leads to exacerbated MAVS aggregation. Given that both PINK1 and MAVS are known to localize on mitochondria, we questioned whether both PINK1 and MAVS interact physically and thus PINK1 inhibits MAVS aggregation. To test this, we first determined whether PINK1 is recruited and interacts with MAVS on mitochondria after nigericin stimulation. Indeed, during nigericin stimulation, PINK1 was accumulated on mitochondria in both HEK293 cells and BMDMs (Figure 3A). In addition, when HEK293 cells were treated with nigericin for various time points and MAVS was immunoprecipitated, the interaction of PINK1 with MAVS was gradually increased in a time-dependent manner (Figure 3B). To further evaluate whether PINK1 interacts with MAVS, bimolecular fluorescence complementation analysis was undertaken, as described previously (20). Specifically, pcDNA3.1-mPINK1-VN and pcDNA3.1-VC-mMAVS vectors were cotransfected to MEFs, and confocal microscopy was employed to evaluate the interaction between PINK1 and MAVS. Unlike the control cells, cotransfected MEFs displayed strong green fluorescence signals when stimulated with nigericin, suggesting that PINK1 and MAVS interact dynamically with each other during the stimulation (Figure E3A). In line with the above findings, PINK1 was coimmunoprecipitated with MAVS in both HEK293 cells and MEFs after stimulation with polyI:C (Figures 3C and E3B). In addition, the bimolecular fluorescence complementation assay revealed strong green fluorescence signals from the cotransfected MEFs with polyI:C stimulation, further confirming PINK1/MAVS interaction during RLH signaling activation (Figure 3D). Taken together, these data demonstrate that MAVS is aggregated on mitochondria in the event of mitochondrial dysfunction and that, during this process, PINK1 interacts with MAVS dynamically to negatively regulate its aggregation.
Figure 3.
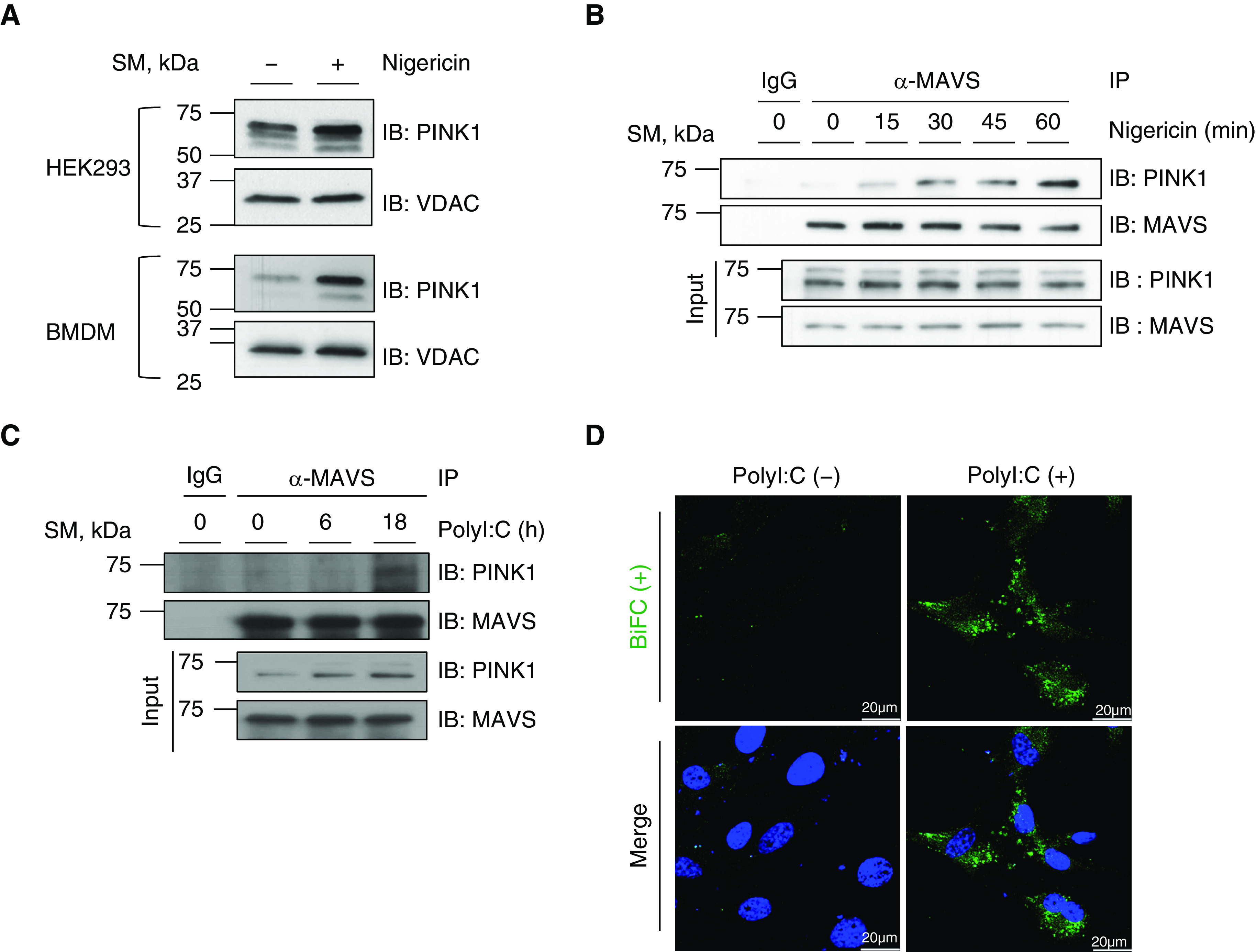
The interaction of PINK1 with MAVS. (A) HEK293 cells and BMDMs were stimulated with 5 μM of nigericin for 1 hour. MTs were separated and loaded to SDS-PAGE. The recruitment and stabilization of PINK1 on mitochondria was evaluated by Western blot. Readers may view the uncut gels for A in the data supplement. (B) HEK293 cells were stimulated with 5 μM of nigericin for the indicated time. The WCLs were immunoprecipitated with α-MAVS antibody and immunoblotted with α-PINK1 antibody. (C) HEK293 cells were stimulated with 10 μg/ml of polyI:C for the indicated time. The WCLs were immunoprecipitated with α-MAVS antibody and immunoblotted with α-PINK1 antibody. (D) MEFs were cotransfected with pcDNA3.1-mPINK1-VN and pcDNA3.1-VC-mMAVS vectors before treating with 10 μg/ml of polyI:C for 18 hours, and the interaction between PINK1 and MAVS was evaluated by confocal microscopy (green, BiFC dimer). Scale bars, 20 μM. All experiments are repeated at least three times. Representative results are shown. BiFC = bimolecular fluorescence complementation; BMDM = bone marrow–derived macrophage.
PINK1 Inhibits MAVS-mediated Antiviral Signaling
We then asked whether PINK1-mediated inhibition of MAVS aggregation leads to the attenuation of MAVS-mediated functions. Originally, MAVS has been identified to play as a key adaptor for RLH antiviral signaling (4). As such, we determined how these signals are changed in PINK1 deficiency. For this purpose, polyI:C has been extensively used to stimulate MAVS-mediated RLH signaling and induce type I IFN production in macrophages or MEFs (21). Therefore, to test whether PINK1 has a role in MAVS-mediated RLH signaling, we used BMDMs and MEFs from WT and PINK1−/− mice, respectively. As reported, polyI:C induced a significant increase of IFN-β production in both BMDMs and MEFs (Figures 4A and 4B). Interestingly, the production of IFN-β was significantly enhanced in the absence of PINK1 compared with control cells in both BMDMs and MEFs (Figures 4A and 4B). This enhancement of IFN-β production was observed at all time points with polyI:C stimulation and reached the peak after 12 hours of polyI:C stimulation (Figure 4C). To verify these responses further, the mRNA transcripts known to be responsive to RLH signaling activation were evaluated by real-time quantitative PCR analysis. Indeed, the expression levels of Ifnb, Isg56 (Ifn-stimulated gene 56), and Cxcl10 were significantly enhanced in PINK1−/− MEFs compared with those of control cells after polyI:C stimulation (Figures 4D–4F). Taken together, these data demonstrate that PINK1 plays a critical inhibitory role in the MAVS-mediated RLH signaling.
Figure 4.

Enhancement of the MAVS-mediated RLH (RIG-I–like helicase) signaling in the absence of PINK1. (A) BMDMs or (B) MEFs from WT and PINK1−/− mice were stimulated with 10 μg/ml of polyI:C for 24 hours. The supernatant was collected, and the amount of IFN-β was measured by ELISA. (C) MEFs from WT and PINK1−/− mice were stimulated with 10 μg/ml of polyI:C for the indicated time. The supernatant was collected, and the amount of IFN-β was measured by ELISA. (D–F) MEFs from WT and PINK1−/− mice were stimulated with 10 μg/ml of polyI:C. The mRNA concentration of Ifn-β (D), Isg56 (Ifn-stimulated gene 56) (E), and Cxcl10 (F) were analyzed by quantitative PCR. All experiments are repeated at least three times, and representative results are shown. Means ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001. mIFN = murine IFN.
PINK1 Inhibits MAVS-mediated NLRP3 Inflammasome Signaling
Recently, MAVS has been identified as an important adaptor for the assembly of NLRP3 inflammasome complex on mitochondria when activated with LPS and nigericin (5). Thus, as a next step to test whether PINK1 has a role in MAVS-mediated inflammasome activation, MEFs or BMDMs from WT and PINK1−/− mice were treated with LPS and nigericin. Indeed, the production of IL-1β induced by LPS and nigericin stimulation was significantly enhanced in PINK1−/− MEFs compared with WT control cells (Figure 5A). In addition, the similar enhancement of IL-1β production was observed PINK1−/− BMDMs compared with WT control cells (Figure 5B). In support of this observation, Western blot analysis revealed that the production of active IL-1β was significantly enhanced in cell supernatants from PINK1−/− BMDMs compared with WT control cells (Figure 5C). It is known that caspase-1–independent proteolytic cleavage of IL-1β can occur to produce active IL-1β (22). Hence, Western blot analysis was performed to evaluate whether the activation of caspase-1, the hallmark of NLRP3 inflammasome activation, is enhanced by LPS and nigericin stimulation in the absence of PINK1. As anticipated, the active form of caspase-1 (p20) was increased in WT BMDMs, and the activation of caspase-1 was further enhanced in the PINK1−/− BMDMs compared with WT control cells, verifying that the enhanced production of active IL-1β in the absence of PINK1 is mediated by the activation of caspase-1 (Figure 5D). As a next step, we evaluated the effects of PINK1 on the recruitment of NLRP3 on mitochondria. Indeed, after stimulation with LPS/nigericin, the amount of NLRP3 was increased in the mitochondrial fraction of BMDMs compared with those from control cells with no stimulation, and, importantly, this recruitment of NLRP3 on mitochondria was significantly enhanced in the absence of PINK1 (Figure 5E). Overall, these data demonstrate that PINK1 plays an important inhibitory role in the MAVS-mediated NLRP3 inflammasomes signaling.
Figure 5.
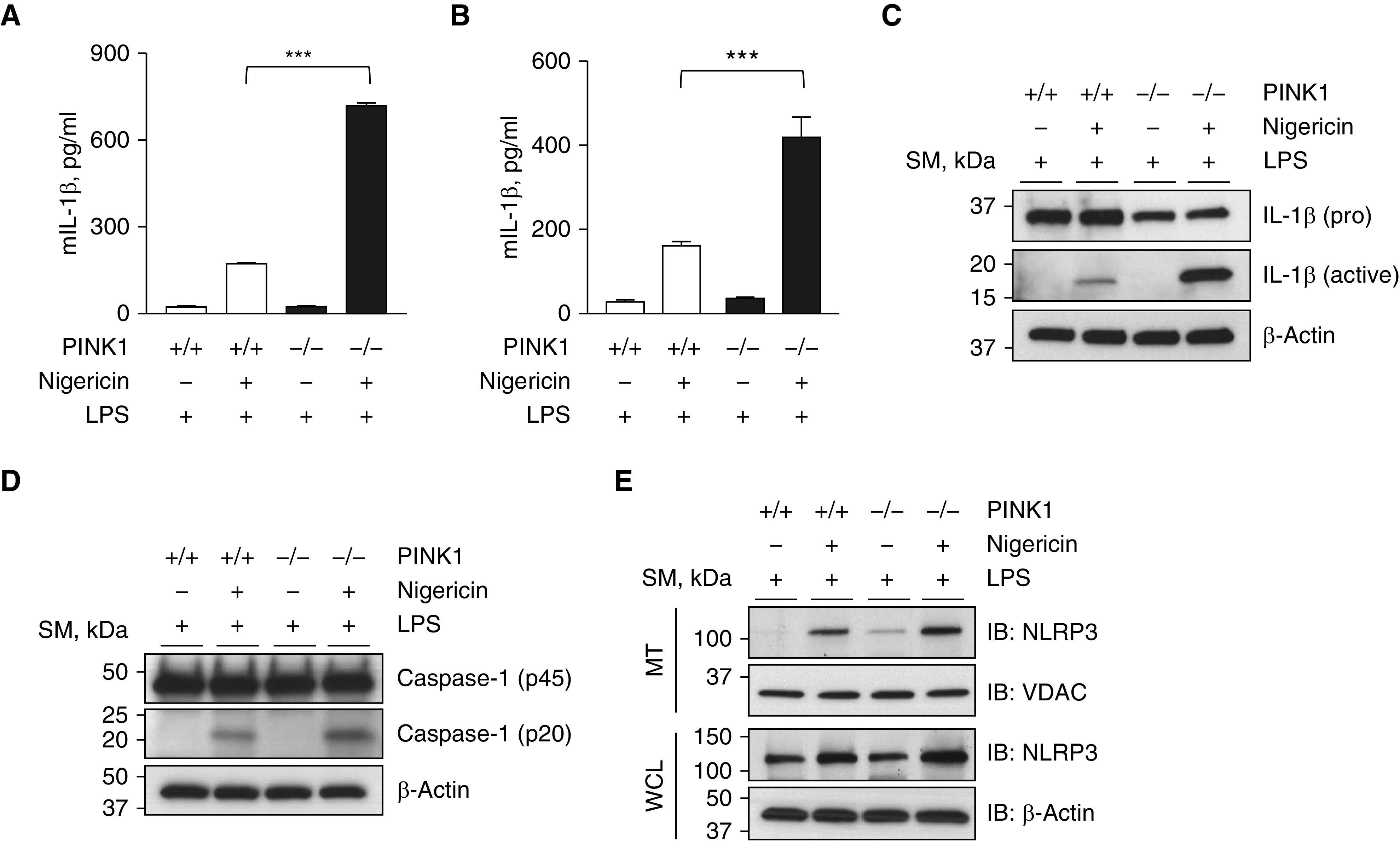
Enhancement of the MAVS-mediated inflammasome signaling in the absence of PINK1. (A) MEFs from WT and PINK1−/− mice were primed with 1 μg/ml of LPS for 4 hours and then stimulated with 5 μM of nigericin for 1 hour. The supernatant was collected, and the amount of IL-1β was measured by ELISA. (B–E) BMDMs from WT and PINK1−/− mice were primed with 1 μg/ml of LPS for 4 hours and then stimulated with 5 μM of nigericin for 1 hour. (B) The supernatant was collected, and the IL-1β amount was measured by ELISA. (C and D) The amounts of pro forms and active forms of IL-1β (C) and caspase-1 (D), respectively, in cell lysates from BMDMs were evaluated by Western blot analysis. (E) Mitochondrial fractions from BMDMs were loaded to SDS-PAGE, and the recruitment of NLRP3 inflammasome on mitochondria was evaluated by Western blot analysis. VDAC and β-actin were used as loading controls of mitochondria and WCLs, respectively. All experiments are repeated at least three times, and representative results are shown. Means ± SD. ***P < 0.001. mIL-1β = murine IL-1β; NLRP3 = NOD-like receptor family pyrin domain-containing 3.
In Vivo Functional Significance of PINK1 That Negatively Regulates MAVS-mediated Pulmonary Pathobiology
To assess the significance of PINK1 on MAVS signaling in vivo, first, we used a murine model of influenza viral infection. For influenza infection, WT or PINK1−/− mice were infected intranasally with influenza virus. To evaluate whether in vivo data are consistent to previous in vitro results, we measured the expression of type I IFN signaling in whole lungs at Day 5 after infection. Post-infection Day 5 was carefully chosen because type I IFN production reaches its peak point at Day 5 in our experimental setting. The expression levels of Ifnb, Isg56, Isg15 (ifn-stimulated gene 15), and Cxcl10 were significantly enhanced in PINK1−/− mice compared with WT mice (Figures 6B–6E), although total BAL fluid cell counts did not reach statistical significance in the case of PINK1 deficiency (Figure 6A). As expected from the literature, which demonstrated a crucial role of MAVS in the regulation of virus-induced type I IFN and its related genes (23, 24), the expression levels of Ifnb, Isg56, and Isg15 in PINK1 deficiency were significantly attenuated in PINK1−/−/MAVS−/− mice (Figure 6F and data not shown).
Figure 6.
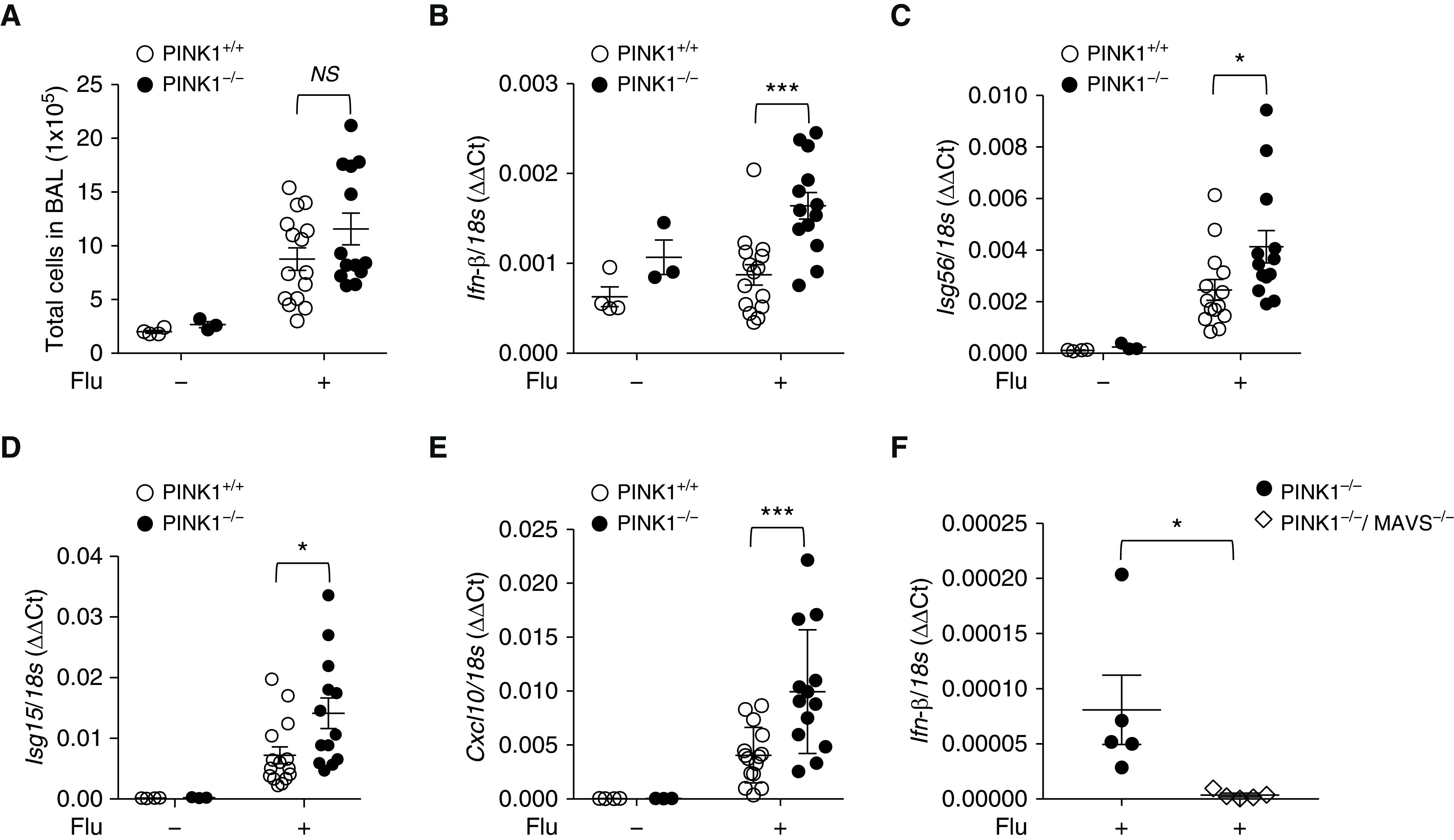
In vivo functional significance of PINK1/MAVS in influenza-infected mouse model. (A–E) 100 plaque-forming units of A/PR8/34 influenza (equivalent to 0.5 median lethal dose in C57BL/6J mice) was administered through intranasal aspiration to WT and PINK1−/− mice. Experimental groups were composed of control WT (n = 3), control PINK−/− mice (n = 4), influenza-infected WT mice (n = 15), and influenza-infected PINK1−/− mice (n = 13). The lysates of total lung tissues were obtained from the mice at Day 5 after influenza infection. (A) Total cell counts of the BAL fluid were evaluated. (B–E) The mRNA levels of Ifn-β (B), Isg56 (C), Isg15 (D), and Cxcl10 (E) were analyzed by quantitative PCR. Readers may view the uncut gels for E in the data supplement. (F) A separate in vivo experiment was undertaken in which experimental groups were composed of influenza-infected PINK1−/− mice (n = 5) and PINK1−/−/MAVS−/− mice (n = 5). The lysates of total lung tissues were obtained from the mice at Day 5 after influenza infection. The mRNA level of Ifn-β was analyzed by quantitative PCR. Means ± SEM. *P < 0.05 and ***P < 0.001. Flu = influenza virus; NS = not significant.
A recent study from our laboratory has demonstrated a crucial role of MAVS for the development of pulmonary fibrosis (15), and another group demonstrated that PINK1 deficiency promotes lung fibrosis (25). Based on these studies, to further address the regulation between PINK1 and MAVS in pulmonary fibrosis, we evaluated and compared key phenotypes from PINK1−/− mice, PINK1−/−/MAVS−/−, mice and appropriate littermate WT control mice in a bleomycin-induced pulmonary fibrosis model. Compared with PINK1−/− mice, the loss of body weight was significantly ameliorated from PINK1−/−/MAVS−/− mice at Day 8 after bleomycin injury in vivo (Figure 7A). Amounts of collagen, a key fibrotic phenotype, were significantly reduced in lungs of PINK1−/−/MAVS−/− mice compared with PINK1−/− mice after bleomycin administration (Figure 7B). In line with this, bleomycin injury–induced increase of active TGF-β, a primary factor of tissue fibrosis, was significantly attenuated in PINK1−/−/MAVS−/− mice compared with PINK1−/− mice (Figure 7C). Moreover, well-known fibrotic molecules, including phospho (p)-SMAD2, fibronectin, and α-Sma (α-smooth muscle actin), were markedly attenuated in PINK1−/−/MAVS−/− mice (Figure 7D). To address whether PINK1 has an important role on MAVS aggregation in pulmonary fibrosis model, MAVS aggregation was evaluated by semidenaturing detergent agarose gel electrophoresis. MAVS aggregation was significantly enhanced from pooled BAL cells from PINK1−/− mice compared with those from WT mice (Figure 7E), suggesting that PINK1 has an inhibitory role in the formation of MAVS aggregation in this experimental setting. Taken together, these results confirm the in vivo functional significance of PINK1 inhibiting MAVS aggregation and its signaling.
Figure 7.
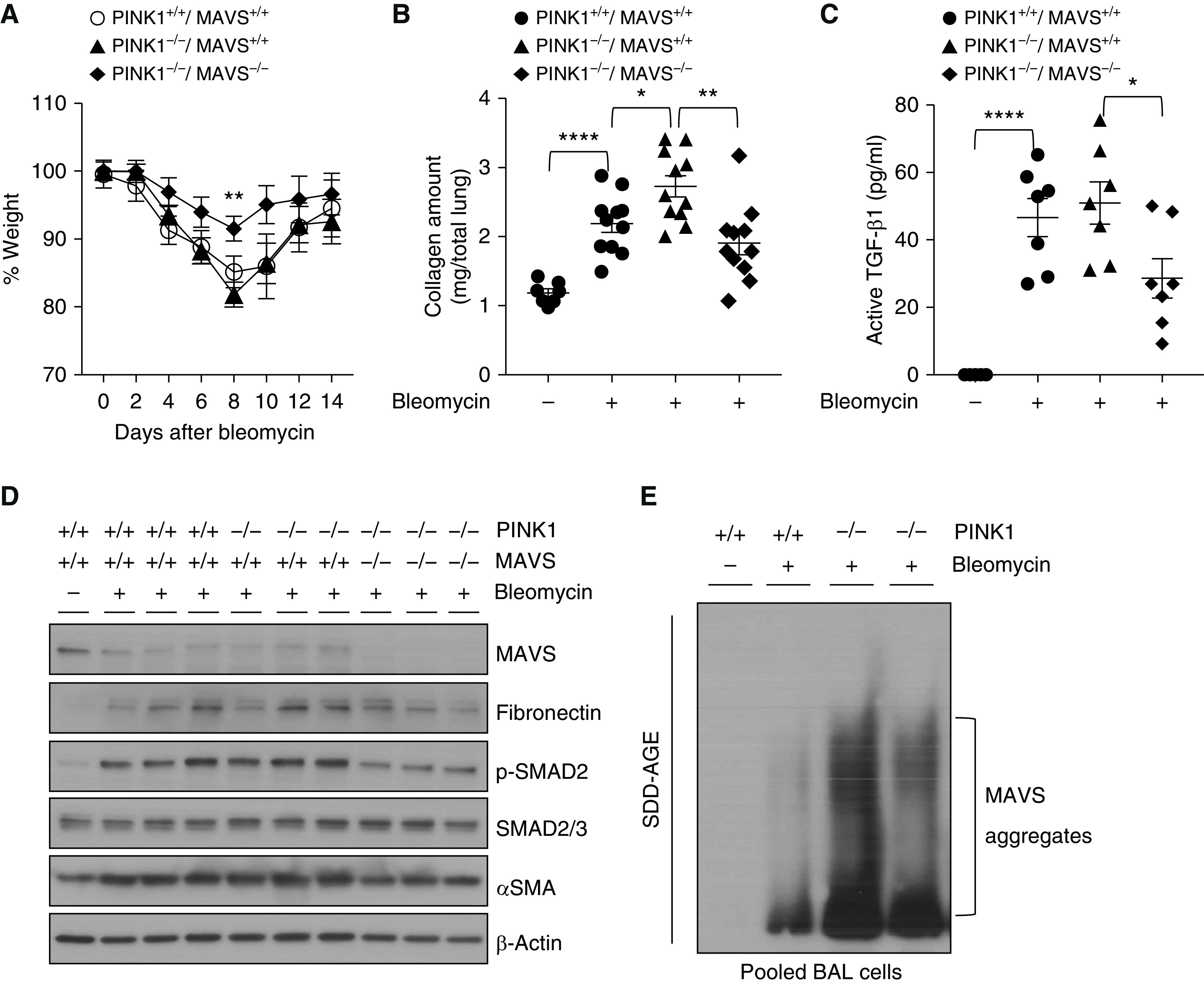
In vivo functional significance of PINK1/MAVS in bleomycin-induced pulmonary fibrosis model. (A–E) Saline (bleomycin −) or 0.4 U/kg of bleomycin (+) was administered through intratracheal route to WT, PINK1−/−, and PINK1−/−MAVS−/− mice. (A) Body weight changes were monitored during the course of the experiment. (B) Collagen contents of total lung tissues obtained from the mice killed at Day 21 after bleomycin administration were measured (n = 7 per saline-treated WT group, n = 11 per each bleomycin-treated group). (C) Active TGF-β1 (transforming growth factor-β1) concentrations at Day 14 after bleomycin administration were measured from BAL fluid by ELISA. (n = 5 per saline group; n = 7 per bleomycin-administered group). (D) Western blot analysis of MAVS, fibronectin, p-SMAD2, SMAD2/3, and α-SMA expressions in whole lung tissue lysates at Day 14 after saline or bleomycin administration. β-actin was used as a loading control. (E) The result of MAVS aggregation is presented. Pooled BAL fluid cells were separated by SDD-AGE and detected with MAVS antibody. Samples were pooled from WT mice (n = 5 per saline group and n = 3 per bleomycin-treated group) and PINK1−/− mice (n = 3 per each bleomycin-treated group), respectively. Means ± SEM. *P < 0.05, **P < 0.01, and ****P < 0.0001. SDD-AGE = semidenaturating detergent agarose gel electrophoresis.
Discussion
Our studies are novel to reveal that 1) PINK1, an important regulator of mitochondrial health, functions to inhibit MAVS-mediated innate immune signaling; 2) PINK1 interacts dynamically with MAVS during the event of mitochondrial dysfunction; and 3) PINK1 negatively regulates the prion-like multimeric formation of MAVS, a critical event of MAVS signaling. The significance of these in vitro findings is further confirmed from two murine in vivo pulmonary disease models in which MAVS has been identified to be crucial for the phenotypes.
Mitochondrial dysfunction has recently emerged as a major player in a myriad of human health and diseases (1, 26, 27). Given the role of MAVS as a pivotal adaptor in various innate immune and inflammatory signaling, it is not surprising that MAVS has a profound implication in a variety of health conditions (28–32). Indeed, the pathogenic importance of MAVS signaling in multiple human diseases has been highlighted in recent publications, including ours (15, 28, 31, 32). In this regard, our studies that demonstrate that PINK1 inhibits MAVS aggregation as well as its signaling raise an interesting question about whether PINK1 could play an important role in the pathogenesis of those human disorders. We recently reported that multimeric MAVS aggregation was significantly observed in lungs from patients with idiopathic pulmonary fibrosis (15). Mechanistic studies revealed that MAVS plays as a critical mediator of multiple DAMP signaling pathways and cellular senescence in bleomycin-induced pulmonary fibrosis in vivo (15). Combined with the observations noted in the current study, fibrogenic injury–induced DAMP signaling activation and cellular senescence response might be exaggerated in PINK1 deficiency, which is known to be associated with idiopathic pulmonary fibrosis pathogenesis (25, 33).
Currently, not much is known about the negative regulation of MAVS signaling specifically after it aggregates in response to nonviral pathogen-associated molecular pattern or DAMP stimulation. Molecular mechanisms must exist to counter-regulate the multimeric formation of the MAVS aggregation. Otherwise, uncontrolled continuation of the irreversible aggregation of MAVS would result in persistent activation of innate immune and inflammatory responses, culminating in chronic inflammatory disorders or autoimmune diseases (28, 31). Although recent studies have shed light on this important subject (34, 35), so far, most studies have been undertaken in the context of viral infection. The regulatory mechanisms of the MAVS aggregation in the context of nonviral DAMP-induced mitochondrial dysfunction have rarely been explored. In this regard, our current studies are unique in revealing that mitochondrial dysfunction can be an important trigger to induce the MAVS aggregation and that PINK1 plays an important regulatory role to inhibit the MAVS aggregation and its signaling in association with mitochondrial dysfunction.
PINK1 plays a pivotal role in the regulation of mitochondrial health by the regulation of multiple pathways, including mitochondrial dynamics, transport, biogenesis, and mitophagy (36, 37). Given the functional significance of PINK1 in mitophagic removal of dysfunctional mitochondria or mitochondrial molecules, defective mitophagy in PINK1 deficiency could play an important role in the enhancement of MAVS signaling. Regarding this, several studies indicate that, together with the MAVS signaling activation, autophagic activation is induced and negatively regulates MAVS signaling (35, 38, 39). In particular, NDP52 (nuclear domain 10 protein 52), a known mitophagy receptor recruited by PINK1 to induce mitophagy (40), has recently been identified to be involved in the autophagic inhibition of MAVS signaling (35, 39). Thus, PINK1-mediated recruitment of mitophagy receptors such as NDP52 might be related to the inhibitory function of PINK1 on MAVS aggregation and its signaling. In addition, it is of note that mitochondrial stress–induced type I IFN response and inflammation observed in PINK1 deficiency was dependent on STING (stimulator of IFN genes) (41). As such, given the existence of cross-talk between STING and MAVS (42, 43), STING might be critically involved in the enhanced MAVS signaling in PINK1 deficiency.
Several issues remain unanswered in the current study. As a kinase, the activities that PINK1 exerts require the recruitment and phosphorylation of multiple substrates (36, 44, 45). Although our data demonstrated that PINK1 interacts dynamically with MAVS on mitochondria upon mitochondrial dysfunction, currently, it is not clear whether MAVS is a novel substrate of PINK1 and whether the phosphorylation of MAVS by PINK1 is required for the resolution of aggregated MAVS. In addition, questions are raised about whether the molecular interaction of PINK1 with MAVS occurs during PINK1-mediated mitophagy, and, if so, how PINK1-mediated mitophagy operates in line with the PINK1/MAVS interaction to inhibit MAVS aggregation. From a chemiosmotic point of view, mitochondrial membrane potential and proton gradient are independent components that equally contribute to the mitochondrial proton-motive force, a key aspect of mitochondrial function (18, 19). As such, the current study might raise an intriguing question of whether PINK1 is a key regulator of mitochondrial health in the setting of a disruption of mitochondrial proton-motive force. By answering the above questions, further studies will provide a better understanding of the mechanisms that underlie the inhibition of PINK1 on MAVS function.
It will be interesting to explore whether targeting the PINK1 molecule has a potential for the regulation of MAVS signaling. Given the significance of MAVS biology, which plays a key role in multiple mitochondria-related regulation of cellular functions, including antiviral innate immunity, inflammasomes activation, and apoptosis (46, 47), the modulation of this pathway via targeting of the PINK1 function might open a new horizon for future therapeutics in medicine.
In conclusion, these studies suggest that PINK1 plays an important regulatory role in MAVS-mediated signaling via its direct interaction with MAVS, and they provide a conceptual proof that augmenting PINK1 activity may be a novel therapeutic approach for pulmonary disorders in which PINK1–MAVS interaction plays a pathogenic role.
Supplementary Material
Acknowledgments
Acknowledgment
The authors thank Susan Ardito for excellent administrative assistance.
Footnotes
Supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF-2013R1A6A3A03026988) (C.M.Y.), Veterans Affairs grant I01-BX004661 (C.S.D.C.), Department of Defense (PR181442) (C.S.D.C.), National Heart, Lung, and Blood Institute (1R01HL130283) (M.-J.K.), and National Institute on Aging (1R01AG053495) (M.-J.K.).
Author Contributions: Conceived the idea and designed the experiments: S.-H.K., H.J.S., C.M.Y., and M.-J.K. Performed experiments: S.-H.K., H.J.S., C.M.Y., and L.S. Provided important reagents/tools: S.W.L., L.S., and C.S.D.C. Provided scientific insight: S.-H.K. and M.-J.K. Analyzed data: S.-H.K., C.M.Y., L.S., and M.-J.K. Drafted the manuscript: S.-H.K., L.S., and M.-J.K.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0490OC on February 21, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Dela Cruz CS, Kang MJ. Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion. 2018;41:37–44. doi: 10.1016/j.mito.2017.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chandel NS. Evolution of mitochondria as signaling organelles. Cell Metab. 2015;22:204–206. doi: 10.1016/j.cmet.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 3.West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol. 2011;11:389–402. doi: 10.1038/nri2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 5.Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell. 2013;153:348–361. doi: 10.1016/j.cell.2013.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146:448–461. doi: 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cai X, Xu H, Chen ZJ. Prion-like polymerization in immunity and inflammation. Cold Spring Harb Perspect Biol. 2017;9:a023580. doi: 10.1101/cshperspect.a023580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas KJ, Cookson MR. The role of PTEN-induced kinase 1 in mitochondrial dysfunction and dynamics. Int J Biochem Cell Biol. 2009;41:2025–2035. doi: 10.1016/j.biocel.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scarffe LA, Stevens DA, Dawson VL, Dawson TM. Parkin and PINK1: much more than mitophagy. Trends Neurosci. 2014;37:315–324. doi: 10.1016/j.tins.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 11.Koh H, Chung J. PINK1 as a molecular checkpoint in the maintenance of mitochondrial function and integrity. Mol Cells. 2012;34:7–13. doi: 10.1007/s10059-012-0100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eiyama A, Okamoto K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr Opin Cell Biol. 2015;33:95–101. doi: 10.1016/j.ceb.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 13.Kazlauskaite A, Muqit MM. PINK1 and Parkin – mitochondrial interplay between phosphorylation and ubiquitylation in Parkinson’s disease. FEBS J. 2015;282:215–223. doi: 10.1111/febs.13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee SW, Sharma L, Kang YA, Kim SH, Chandrasekharan S, Losier A, et al. Impact of cigarette smoke exposure on the lung fibroblastic response after influenza pneumonia. Am J Respir Cell Mol Biol. 2018;59:770–781. doi: 10.1165/rcmb.2018-0004OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim SH, Lee JY, Yoon CM, Shin HJ, Lee SW, Rosas I, et al. Mitochondrial antiviral signalling protein is crucial for the development of pulmonary fibrosis. Eur Respir J. doi: 10.1183/13993003.00652-2020. [online ahead of print] 22 Oct 2020; DOI: 10.1183/13993003.00652-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472:476–480. doi: 10.1038/nature09973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wittig I, Braun HP, Schägger H. Blue native PAGE. Nat Protoc. 2006;1:418–428. doi: 10.1038/nprot.2006.62. [DOI] [PubMed] [Google Scholar]
- 18.Santo-Domingo J, Demaurex N. Perspectives on: SGP symposium on mitochondrial physiology and medicine: the renaissance of mitochondrial pH. J Gen Physiol. 2012;139:415–423. doi: 10.1085/jgp.201110767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morelli AM, Ravera S, Calzia D, Panfoli I. An update of the chemiosmotic theory as suggested by possible proton currents inside the coupling membrane. Open Biol. 2019;9:180221. doi: 10.1098/rsob.180221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seo JY, Yaneva R, Hinson ER, Cresswell P. Human cytomegalovirus directly induces the antiviral protein viperin to enhance infectivity. Science. 2011;332:1093–1097. doi: 10.1126/science.1202007. [DOI] [PubMed] [Google Scholar]
- 21.Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature. 2008;451:573–577. doi: 10.1038/nature06501. [DOI] [PubMed] [Google Scholar]
- 22.Schönbeck U, Mach F, Libby P. Generation of biologically active IL-1 beta by matrix metalloproteinases: a novel caspase-1-independent pathway of IL-1 beta processing. J Immunol. 1998;161:3340–3346. [PubMed] [Google Scholar]
- 23.Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, et al. Activation of innate immune antiviral responses by Nod2. Nat Immunol. 2009;10:1073–1080. doi: 10.1038/ni.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sen A, Pruijssers AJ, Dermody TS, García-Sastre A, Greenberg HB. The early interferon response to rotavirus is regulated by PKR and depends on MAVS/IPS-1, RIG-I, MDA-5, and IRF3. J Virol. 2011;85:3717–3732. doi: 10.1128/JVI.02634-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bueno M, Lai YC, Romero Y, Brands J, St Croix CM, Kamga C, et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest. 2015;125:521–538. doi: 10.1172/JCI74942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang MJ. Recent advances in molecular basis of lung aging and its associated diseases. Tuberc Respir Dis (Seoul) 2020;83:107–115. doi: 10.4046/trd.2020.0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kang MJ, Yoon CM, Kim BH, Lee CM, Zhou Y, Sauler M, et al. Suppression of NLRX1 in chronic obstructive pulmonary disease. J Clin Invest. 2015;125:2458–2462. doi: 10.1172/JCI71747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi CS, Qi HY, Boularan C, Huang NN, Abu-Asab M, Shelhamer JH, et al. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J Immunol. 2014;193:3080–3089. doi: 10.4049/jimmunol.1303196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hirai-Yuki A, Hensley L, McGivern DR, González-López O, Das A, Feng H, et al. MAVS-dependent host species range and pathogenicity of human hepatitis A virus. Science. 2016;353:1541–1545. doi: 10.1126/science.aaf8325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shao WH, Shu DH, Zhen Y, Hilliard B, Priest SO, Cesaroni M, et al. Prion-like aggregation of mitochondrial antiviral signaling protein in lupus patients is associated with increased levels of type I interferon. Arthritis Rheumatol. 2016;68:2697–2707. doi: 10.1002/art.39733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bellecave P, Sarasin-Filipowicz M, Donzé O, Kennel A, Gouttenoire J, Meylan E, et al. Cleavage of mitochondrial antiviral signaling protein in the liver of patients with chronic hepatitis C correlates with a reduced activation of the endogenous interferon system. Hepatology. 2010;51:1127–1136. doi: 10.1002/hep.23426. [DOI] [PubMed] [Google Scholar]
- 33.Kim SJ, Cheresh P, Jablonski RP, Rachek L, Yeldandi A, Piseaux-Aillon R, et al. Mitochondrial 8-oxoguanine DNA glycosylase mitigates alveolar epithelial cell PINK1 deficiency, mitochondrial DNA damage, apoptosis, and lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2020;318:L1084–L1096. doi: 10.1152/ajplung.00069.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoo YS, Park YY, Kim JH, Cho H, Kim SH, Lee HS, et al. The mitochondrial ubiquitin ligase MARCH5 resolves MAVS aggregates during antiviral signalling. Nat Commun. 2015;6:7910. doi: 10.1038/ncomms8910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jin S, Tian S, Luo M, Xie W, Liu T, Duan T, et al. Tetherin suppresses type I interferon signaling by targeting MAVS for NDP52-mediated selective autophagic degradation in human cells. Mol Cell. 2017;68:308–322.e4. doi: 10.1016/j.molcel.2017.09.005. [DOI] [PubMed] [Google Scholar]
- 36.Arena G, Valente EM. PINK1 in the limelight: multiple functions of an eclectic protein in human health and disease. J Pathol. 2017;241:251–263. doi: 10.1002/path.4815. [DOI] [PubMed] [Google Scholar]
- 37.McWilliams TG, Muqit MM. PINK1 and Parkin: emerging themes in mitochondrial homeostasis. Curr Opin Cell Biol. 2017;45:83–91. doi: 10.1016/j.ceb.2017.03.013. [DOI] [PubMed] [Google Scholar]
- 38.Cheng J, Liao Y, Xiao L, Wu R, Zhao S, Chen H, et al. Autophagy regulates MAVS signaling activation in a phosphorylation-dependent manner in microglia. Cell Death Differ. 2017;24:276–287. doi: 10.1038/cdd.2016.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He X, Zhu Y, Zhang Y, Geng Y, Gong J, Geng J, et al. RNF34 functions in immunity and selective mitophagy by targeting MAVS for autophagic degradation. EMBO J. 2019;38:e100978. doi: 10.15252/embj.2018100978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–314. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature. 2018;561:258–262. doi: 10.1038/s41586-018-0448-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 43.Zevini A, Olagnier D, Hiscott J. Crosstalk between cytoplasmic RIG-I and STING sensing pathways. Trends Immunol. 2017;38:194–205. doi: 10.1016/j.it.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85:257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mills EL, Kelly B, O’Neill LAJ. Mitochondria are the powerhouses of immunity. Nat Immunol. 2017;18:488–498. doi: 10.1038/ni.3704. [DOI] [PubMed] [Google Scholar]
- 47.Belgnaoui SM, Paz S, Hiscott J. Orchestrating the interferon antiviral response through the mitochondrial antiviral signaling (MAVS) adapter. Curr Opin Immunol. 2011;23:564–572. doi: 10.1016/j.coi.2011.08.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.