

Abstract
Background
Niraparib is the only poly (adenosine diphosphate-ribose)-polymerase (PARP) inhibitor available as oral monotherapy for maintenance, regardless of BRCA mutational status.
Methods
This phase I, open-label, non-randomized, dose-escalation study was conducted in Japan using a 3 + 3 design. Adults (≥20 years) with metastatic or locally advanced solid tumours were enrolled. Niraparib 200 mg (cohort 1) or 300 mg (cohort 2) was administered once daily in 21-day cycles (no drug holiday between cycles) until progressive disease (PD) or unacceptable toxicity. The primary objective was to evaluate the safety and tolerability of niraparib in Japanese patients with advanced solid tumours. The number of patients with dose-limiting toxicities in cycle 1 and number with treatment-emergent adverse events were primary endpoints. Secondary endpoints were pharmacokinetics and tumour response.
Results
There were three patients in cohort 1 and six patients in cohort 2. Only one patient, in cohort 2, developed a dose-limiting toxicity (grade 4 platelet count decreased). All patients in both cohorts developed treatment-emergent adverse events. The most common treatment-related treatment-emergent adverse events were decreased appetite (n = 2) in cohort 1, and platelet count decreased as well as aspartate aminotransferase increased (both n = 5) in cohort 2. Mean Cmax and AUC0–24 of niraparib increased dose-proportionally after multiple doses (accumulation ratio of between 1.64 and 3.65); median tmax was 3–4 h. Two patients, both in cohort 2, had a partial response to treatment.
Conclusions
Niraparib (200 or 300 mg/day) was tolerable and had a favourable pharmacokinetic profile in Japanese patients with advanced solid tumours.
Keywords: clinical trial; phase I; neoplasms; niraparib; PARP Inhibitor; pharmacokinetics, PARP
This study investigated the safety of niraparib in Japanese patients with metastatic or locally advanced solid tumours. One patient receiving 300 mg/day developed a dose-limiting toxicity (platelet count decreased)
Introduction
Poly(adenosine diphosphate [ADP]-ribose) polymerase (PARP) enzymes detect and promote DNA repair. Inhibition of PARP forces cells to rely on homologous recombination as an alternative mechanism for DNA repair (1). Some tumour-related mutations, including BRCA1 and BRCA2 mutations, result in homologous recombination deficiency (HRd); therefore, tumours with these mutations are particularly susceptible to treatment with PARP inhibitors (1).
Niraparib is a potent and highly selective oral PARP-1 and -2 inhibitor, and is approved in the USA and Europe for the maintenance treatment of platinum-sensitive recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer after a complete or partial response to platinum-based chemotherapy (2, 3). It is currently being investigated in patients with other types of solid tumours, with or without HRd-related mutations. In the phase III NOVA study, maintenance treatment with niraparib significantly prolonged progression-free survival (PFS) compared with placebo in patients with platinum-sensitive recurrent ovarian cancer, regardless of germline BRCA mutation status (4). Niraparib also significantly prolonged PFS compared with placebo, regardless of the presence or absence of HRd, in the phase III PRIMA study in patients with newly diagnosed advanced ovarian cancer that had responded to platinum-based therapy (5).
To date, niraparib is the only PARP inhibitor shown to be significantly more efficacious, statistically and clinically, than placebo when administered as monotherapy in relapsed ovarian cancer without BRCA mutations (4). Niraparib is now approved in the USA as first-line maintenance therapy for women with platinum-responsive advanced ovarian cancer (3), and is the only PARP inhibitor available as oral monotherapy for the first line maintenance treatment, regardless of BRCA mutational status (6, 7). This addresses a high previously unmet need in ovarian cancer. Niraparib is also currently the only PARP inhibitor that can be taken once daily (8), making it an attractive choice for long-term maintenance treatment.
The recommended starting dosage of niraparib in the USA and Europe is 300 mg once daily (2, 3), based on the results of phase I studies conducted in those markets showing that this was the maximum tolerated dose (MTD) (9). This was the starting dosage in the NOVA study, with a reduction to 200 or 100 mg/day allowed to manage adverse events (AEs) (4). The PRIMA protocol was amended during the trial, to allow an individualized starting dosage of 200 or 300 mg/day, depending on patients’ baseline weight and platelet count (5). The objective of the current study was to evaluate the safety and tolerability of niraparib in Japanese patients with solid tumours, as well as to investigate its anti-tumour activity and pharmacokinetics in this population.
Patients and methods
Design
This was a phase I, open-label, non-randomized, dose-escalation study conducted at a single centre in Japan between 5 April 2018 and 10 February 2020. Dose-escalation was undertaken using a 3 + 3 design (Supplementary Figure 1), in which three or six patients were successively enrolled in two cohorts at first a lower dose (cohort 1) and then a higher dose (cohort 2), with the number of patients in each cohort dependent on the development of dose-limiting toxicity (DLT). Based on this design, between six and 12 patients would be enrolled. The initial dose of niraparib was 200 mg once daily in cohort 1. If ≤1 patient in cohort 1 developed a DLT during cycle 1, then patients could be enrolled in cohort 2 and treated with niraparib 300 mg once daily.
DLTs were defined using the same criteria as those used in an overseas phase I study of niraparib in non-Japanese patients with advanced solid tumours (PN001) (9) as any treatment-related AE (regardless of severity) that led to interruption of niraparib for >14 days, as well as most treatment-related non-hematologic toxicities of grade ≥ 3, hematologic toxicity of grade ≥ 4, and febrile neutropenia grade ≥ 3 occurring during cycle 1. Complete definitions of DLTs are shown in Supplementary Table 1.
In both cohorts, niraparib was administered at the dosage they were assigned on enrolment (200 or 300 mg/day) in 21-day cycles with no drug holiday between cycles, until they experienced progressive disease (PD) or unacceptable toxicity, or withdrew for other reasons. Dosage reduction (minimum 100 mg/day) was allowed only in patients who developed DLTs during cycle 1 or any toxicity from cycle 2 onwards. Study participants were required to fast for 2 h before and after treatment administration.
This study was approved by the Ethics Review Board at the National Cancer Center Hospital in Tokyo, Japan.
Patients
Japanese adults (≥20 years) with cytologically or histologically confirmed metastatic or locally advanced solid tumours were eligible for inclusion if they had progressed on standard therapy, or if there was no standard therapy available, in the opinion of the investigator. Patients were required to have an Eastern Cooperative Oncology Group (ECOG) performance status of ≤1 and adequate organ function (bone marrow, renal, hepatic). Women of childbearing potential and men were required to practice effective contraception during treatment and until 120 days after the last dose of niraparib. Key exclusion criteria were receipt of chemotherapy, radiotherapy, hormonal or biologic therapy within 14 days before the start of cycle 1; receipt of a PARP inhibitor; treatment with any investigational agent within 28 days or 5 half-lives (whichever was longer) before study treatment; patients at high medical risk, or with a condition or treatment that might interfere with niraparib absorption or otherwise confound the results; known primary CNS tumour or CNS metastases.
Endpoints and assessments
The primary study objective was to evaluate the safety and tolerability of niraparib in Japanese patients with advanced solid tumours, with the primary endpoints being the number of patients with DLTs during cycle 1, and the number of patients with treatment-emergent AEs (TEAEs) overall. A TEAE was defined as any AE occurring after administration of the first dose of study drug and through 28 days after the last dose of study drug. TEAEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) version 22.0, which includes separate codes for thrombocytopenia and platelet count decreased. The choice of code to use for a patient with a reduction in platelet count was at the investigator’s discretion. Severity was graded using the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 4.03.
The secondary endpoint was the pharmacokinetic properties of niraparib on days 1 and 21 of cycle 1 (maximum plasma concentration [Cmax], time to Cmax [tmax] and area under the plasma concentration-time curve for 0–24 h [AUC0–24]). Minimum plasma concentration (Cmin) and accumulation ratio based on AUC0–24 and Cmax were also calculated for day 21 samples. Samples for PK analysis were serially collected for cycle 1 on day 1 at 2 h pre-dose and 1, 1.5, 2, 3, 4, 6, 8, 10 and 24 h post-dose, and on day 2 of cycle 1 at 24 h pre-dose. On days 3, 5, 8 and 15 of cycle 1, samples were collected at 2 h pre-dose and on day 21 of cycle 1 samples were collected 2 h pre-dose and at 1, 1.5, 2, 3, 4, 6, 8, 10 and 24 h post-dose. In cycle 2, plasma samples for PK analysis were collected at 2 h pre-dose on day 1.
Additional endpoints were overall response rate (ORR) according to the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 (10); laboratory safety parameters, electrocardiogram, ECOG performance status and vital signs; and pharmacokinetics of the M1 metabolite on days 1 and 21 of cycle 1. ORR was assessed at screening, cycle 3 and every three cycles thereafter by computed tomography (CT) with contrast or magnetic resonance imaging (MRI).
Statistical methods
Safety data were analysed by dose level in the safety analysis set, comprising all patients who received ≥1 dose of study drug. The number of cases and incidence of DLTs during cycle 1 were tabulated using the DLT-evaluable set, comprising all patients who had received ≥80% of planned doses of niraparib in cycle 1 (for ≥17 out of 21 days) unless interrupted by treatment-related toxicities and had sufficient follow-up data, as considered by the sponsor and investigator, to determine whether a DLT occurred. The 80% criterion was chosen as it was thought that patients who missed ≥20% or more of their dose in 21 days could not be adequately evaluated for DLT. ORR was calculated using the response-evaluable set (i.e. all patients who received ≥1 dose of study drug, had measurable disease sites at baseline, and had ≥1 post-baseline disease assessment). Pharmacokinetic parameters were summarized using descriptive statistics for each cohort and time point in the pharmacokinetic analysis set, which included all patients with sufficient dosing and pharmacokinetic data to reliably estimate ≥1 pharmacokinetic parameter. Baseline demographic and clinical characteristics were summarized using mean and standard deviation (SD) for continuous variables, and number (%) for dichotomous variables.
Results
Patients
A total of 11 patients gave informed consent, but two patients did not receive niraparib because one was experiencing AEs and the other did not meet the study entry criteria. Therefore, nine patients (five males and four females) received ≥1 dose of study drug and were included in the safety, pharmacokinetic and response-evaluable sets. These nine patients also met the criteria for the DLT-evaluable set. Patient and disease characteristics are shown in Table 1. Patients were aged a median of 67.0 (range: 41–75) years and weighed a median of 62.8 (range: 58.1–76.4) kg. Median BMI was 23.6 (range: 21.1–26.5) kg/m2. Two patients had known BRCA status; both were positive for BRCA2 and negative for BRCA1.
Table 1.
Baseline demographic and clinical characteristics
| Niraparib 200 mg/day (n = 3) | Niraparib 300 mg/day (n = 6) | Total (N = 9) | |
|---|---|---|---|
| Age, years, median (range) | 50 (41–68) | 67 (45–75) | 67 (41–75) |
| Gender, n (%) | |||
| Male | 2 (66.7) | 3 (50.0) | 5 (55.6) |
| Female | 1 (33.3) | 3 (50.0) | 4 (44.4) |
| Weight, kg, mean (SD) | 65.6 (9.0) | 64.2 (6.6) | 64.7 (6.9) |
| Range | 58.3–75.6 | 58.1–76.4 | 58.1–76.4 |
| ECOG performance status, n (%) | |||
| 0 | 2 (66.7) | 3 (50.0) | 5 (55.6) |
| 1 | 1 (33.3) | 3 (50.0) | 4 (44.4) |
| Cancer type, n (%) | |||
| Bile duct cancer | 0 | 1 (16.7) | 1 (11.1) |
| Bladder cancer | 0 | 1 (16.7) | 1 (11.1) |
| Oesophageal cancer | 1 (33.3)a | 0 | 1 (11.1) |
| Gallbladder cancer | 0 | 1 (16.7) | 1 (11.1) |
| Lung cancer | 0 | 1 (16.7)b | 1 (11.1) |
| Pancreatic cancer | 1 (33.3) | 1 (16.7) | 2 (22.2) |
| Papilla cancer | 0 | 1 (16.7) | 1 (11.1) |
| Urachal cancer | 1 (33.3) | 0 | 1 (11.1) |
| BRCA1 mutation, n (%) | |||
| Yes | 0 | 0 | 0 |
| No | 1 (33.3) | 1 (16.7) | 2 (22.2) |
| Unknown | 2 (66.7) | 5 (83.3) | 7 (77.8) |
| BRCA2 mutation, n (%) | |||
| Yes | 1 (33.3) | 1 (16.7) | 2 (22.2) |
| No | 0 | 0 | 0 |
| Unknown | 2 (66.7) | 5 (83.3) | 7 (77.8) |
| Prior surgery, n (%) | 1 (33.3) | 3 (50.0) | 4 (44.4) |
| Prior radiation, n (%) | 3 (100.0) | 1 (16.7) | 4 (44.4) |
aSquamous cell carcinoma.
bNeuroendocrine carcinoma.
BMI, body mass index; ECOG, Eastern Cooperative Oncology Group; SD, standard deviation.
Three patients participated in cohort 1 and received niraparib 200 mg/day, and six patients participated in cohort 2 and received niraparib 300 mg/day. All three patients in the 200 mg/day cohort and four patients in the 300 mg/day cohort discontinued study treatment due to PD. The remaining two patients in the 300 mg/day cohort discontinued study treatment due to TEAEs.
Exposure and safety
All patients in cohort 1 received 2 cycles of niraparib; patients in cohort 2 received between 1 and 22 cycles of niraparib (median 7.5). The median (range) number of days on treatment was 45.0 (7–462) overall, and was 41.0 (33–45) days in cohort 1 and 153.5 (7–462) days in cohort 2.
No patients in cohort 1 and one patient in cohort 2 (16.7%) developed a DLT requiring niraparib interruption. This event was a grade 4 platelet count decreased, which was seen on day 14 of cycle 1; it had resolved by day 21. The patient who developed this DLT had a baseline platelet count of 100 000 cells/μl and a body weight of 59.3 kg. Two patients in cohort 2 discontinued treatment due to a TEAE (malaise and gamma-glutamyl transferase increased). In cohort 1, no patient discontinued treatment because of a TEAE.
All patients in both cohorts developed TEAEs. In cohort 1, one patient had two grade 3 treatment-related AEs (blood alkaline phosphatase increased, white blood cell count decreased). In cohort 2, five patients had a total of 11 grade ≥ 3 treatment-related AEs. Grade 3 AEs were anaemia, which occurred in two patients, and neutropenia, febrile neutropenia, gamma-glutamyl transferase increased, and alanine aminotransferase increased, which all occurred in one patient each. Grade 4 AEs were thrombocytopenia and platelet count decreased. None of these events met the definition of a DLT as they occurred in treatment cycle 2 or later.
One patient in cohort 2 developed a serious AE—pyelonephritis leading to hospitalization—but this was considered unrelated to niraparib because the patient had comorbid nephrolithiasis.
The most common TEAEs were categorized by system organ class as gastrointestinal disorders or investigations (Table 2). The most common treatment-related TEAE in cohort 1 was decreased appetite (n = 2 [66.7%]); in cohort 2, the most common treatment-related TEAEs were platelet count decreased (n = 5 [83.3%]) and aspartate aminotransferase increased (n = 5 [83.3%]). In addition, anaemia, nausea and alkaline phosphatase increased each occurred in three patients (50.0%) in cohort 2.
Table 2.
Treatment-emergent adverse events (TEAEs) occurring in ≥2 patients in either cohort and all grade 3/4 TEAEs, by preferred term
| Patients with events, n (%) | Niraparib 200 mg/day (n = 3) | Niraparib 300 mg/day (n = 6) | ||
|---|---|---|---|---|
| Any | Grade 3/4 | Any | Grade 3/4 | |
| Any TEAE | 3 (100.0) | 1 (33.3) | 6 (100.0) | 4 (66.7) |
| Platelet count decreaseda | 1 (33.3) | 0 | 5 (83.3) | 1 (16.7) |
| AST increased | 0 | 0 | 5 (83.3) | 0 |
| Blood ALP increased | 1 (33.3) | 1 (33.3) | 4 (66.7) | 1 (16.7) |
| Nausea | 1 (33.3) | 0 | 4 (66.7) | 0 |
| Decreased appetite | 2 (66.7) | 0 | 2 (33.3) | 0 |
| Vomiting | 2 (66.7) | 0 | 2 (33.3) | 0 |
| ALT increased | 0 | 0 | 3 (50.0) | 1 (16.7) |
| Anaemia | 0 | 0 | 3 (50.0) | 1 (16.7) |
| Constipation | 1 (33.3) | 0 | 2 (33.3) | 0 |
| Fatigue | 2 (66.7) | 0 | 1 (16.7) | 0 |
| GGT increased | 0 | 0 | 3 (50.0) | 2 (33.3) |
| Malaise | 1 (33.3) | 0 | 2 (33.3) | 0 |
| Blood creatinine increased | 1 (33.3) | 0 | 1 (16.7) | 0 |
| Chest pain | 0 | 0 | 2 (33.3) | 0 |
| Diarrhoea | 0 | 0 | 2 (33.3) | 0 |
| Hypertension | 0 | 0 | 2 (33.3) | 0 |
| White blood cell count decreased | 1 (33.3) | 1 (33.3) | 1 (16.7) | 0 |
| Febrile neutropenia | 0 | 0 | 1 (16.7) | 1 (16.7) |
| Thrombocytopeniaa | 0 | 0 | 1 (16.7) | 1 (16.7) |
| Pyelonephritis | 0 | 0 | 1 (16.7) | 1 (16.7) |
| Diabetes mellitus | 0 | 0 | 1 (16.7) | 1 (16.7) |
aTEAEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) version 22.0, in which independent codes were used for thrombocytopenia and platelet count decreased. It was up to each investigator to decide which code to use when reporting a decrease in platelet levels.
ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, gamma-glutamyl transferase; TEAE, treatment-emergent adverse event.
Pharmacokinetics
Results of the single- and multiple-dose pharmacokinetic analyses of niraparib and its metabolite M1 are shown in Table 3. The plasma concentration-time profiles of niraparib after a single dose (cycle 1, day 1) and multiple doses (cycle 1, day 21) are shown in Fig. 1A and B, respectively.
Table 3.
Pharmacokinetic parameters for niraparib on day 1 and day 21 of Cycle 1
| Pharmacokinetic parametersa | Day 1 | Day 21 | ||
|---|---|---|---|---|
| Niraparib 200 mg/day (n = 3) | Niraparib 300 mg/day (n = 6) | Niraparib 200 mg/day (n = 3) | Niraparib 300 mg/day (n = 4) | |
| Niraparib | ||||
| Cmax, ng/ml | 442.9 (195.1) | 529.6 (149.2) | 729.2 (387.5) | 1167 (194.9) |
| Tmax, h | 3.2 (1.5) | 5.1 (2.8) | 3.9 (0.1) | 3.7 (1.6) |
| AUC0–24, h · ng/ml | 4931 (2905) | 6270 (2631) | 13 040 (6493) | 19 540 (3117) |
| Cmin, ng/ml | – | – | 405.8 (267.8) | 592.3 (138.2) |
| RCmax | – | – | 1.64 (0.40) | 2.39 (1.03) |
| RAUC0–24 | – | – | 2.64 (0.32) | 3.65 (1.58) |
| M1 metabolite | ||||
| Cmax, ng/ml | 390.3 (89.2) | 406.7 (154.4) | 1084 (94.0) | 1267 (211.0) |
| Tmax, h | 8.0 (3.4) | 9.6 (7.4) | 5.3 (2.4) | 8.1 (3.5) |
| AUC0–24, h · ng/ml | 6566 (1949) | 6570 (2374) | 21 300 (2888) | 24 850 (4387) |
| Cmin, ng/ml | – | – | 726.7 (134.0) | 800.7 (155.1) |
| RCmax | – | – | 2.78 (0.46) | 3.28 (1.64) |
| RAUC0–24 | – | – | 3.24 (0.62) | 3.77 (1.82) |
aAll parameters are expressed as geometric mean, (standard deviation) except Tmax which is expressed as mean, (standard deviation).
AUC0–24, area under the plasma concentration-time curve from time 0 to 24 h; Cmax, maximum observed concentration; Cmin, minimum observed concentration; RAUC0–24, accumulation ratio based on AUC0–24; RCmax, accumulation ratio based on Cmax; Tmax, time of first occurrence of Cmax.
Figure 1.
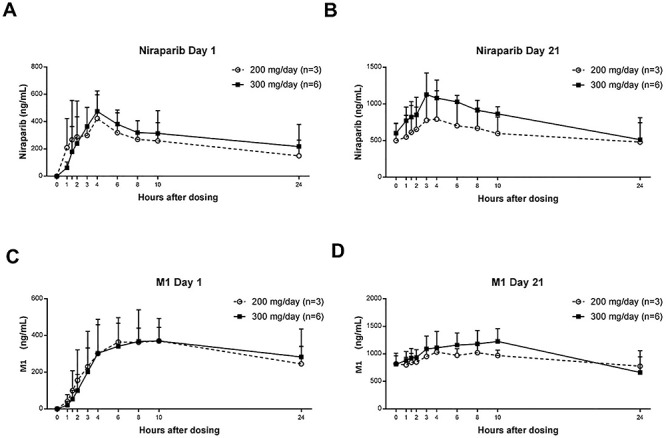
Plasma concentration of (A) niraparib after a single dose (day 1 of cycle 1), (B) niraparib after multiple doses (day 21 of cycle 1), (C) M1 after a single dose (day 1 of cycle 1), and (D) M1 after multiple doses (day 21 of cycle 1). Data are mean (standard deviation).
Following a single (cycle 1, day 1) and multiple (cycle 1, day 21) oral administration of niraparib 200 and 300 mg/day, the median tmax of niraparib was ~3–4 h. The mean Cmax and AUC0–24 of niraparib generally increased in a dose-proportional manner (Table 3). The geometric mean of accumulation ratio RCmax ranged from 1.64 to 2.39 and RAUC0–24 ranged from 2.64 to 3.65 after multiple doses.
Following a single administration (cycle 1, day 1) and multiple (cycle 1, day 21) oral administrations of niraparib 200 and 300 mg/day, the median tmax of M1 was ~4–10 h. The geometric mean of accumulation ratios RCmax and RAUC0–24 ranged from 2.78 to 3.28 and 3.24 to 3.77, respectively, on day 21 (Table 3).
Tumour response
Two patients (one with lung cancer and one with bile duct cancer; both in cohort 2) had a partial response to niraparib. Two had stable disease and four had PD. Of these four patients with PD, two had a BRCA2 mutation, one patient was receiving niraparib 200 mg/day and the other one was receiving 300 mg/day; the BRCA status of the other patients are unknown. One patient’s tumour response was not evaluable. All patients in cohort 1 had PD.
Discussion
This phase I study showed that niraparib at a dosage of 200 or 300 mg/day was generally safe and tolerable in Japanese patients with solid tumours, and that the pharmacokinetics of niraparib were characterized by rapid absorption and dose-proportional exposure over time. Only one DLT (grade 4 platelet count decreased) developed in one patient receiving niraparib 300 mg/day during cycle 1, with no DLTs in patients receiving 200 mg/day. The patient who had a DLT had a baseline platelet count <150 000 cells/μl and a body weight of <70 kg, which are both risk factors for grade ≥ 3 thrombocytopenia with niraparib 300 mg/day (11). As such, the event was considered treatment related and niraparib was interrupted; the event had resolved by day 21. Based on these results, we can conclude that niraparib at a dosage of 200 or 300 mg/day was tolerable in Japanese patients.
All patients in the 200 and 300 mg/day cohorts experienced ≥1 TEAE, with treatment-related TEAEs reported in all patients. The most frequently reported treatment-related TEAEs were platelet count decreased, aspartate aminotransferase increased, anaemia, nausea, alkaline phosphatase increased and decreased appetite. The treatment-related grade 3 or 4 TEAEs were blood alkaline phosphatase increased, white blood cell count decreased, anaemia, neutropenia, febrile neutropenia, thrombocytopenia, platelet count decreased, gamma-glutamyl transferase increased and alanine aminotransferase increased. One serious TEAE—pyelonephritis—was reported, but this was assessed to be unrelated to treatment.
In the current study, no AEs of special interest, such as myelodysplastic syndrome, acute myeloid leukaemia or hypertensive crisis, were reported. The thrombocytopenia and events coded as platelet count decreased are well known AEs with niraparib, and were reported as grade 4 TEAEs in two patients in the 300 mg/day cohort on day 14 of cycle 1 and day 8 of cycle 2, respectively. The patients recovered from these events within ~10 days. These findings in Japanese patients are consistent with previous phase II or III studies in predominantly Caucasian populations. In these studies, the most common grade 3 or 4 TEAEs with niraparib were hematologic (principally thrombocytopenia and anaemia), and the most common TEAEs of any grade were anaemia, fatigue and gastrointestinal events (e.g. nausea, vomiting and anorexia) (4, 5, 12).
A high proportion of patients in the phase III ENGOT-OV16/NOVA trial (~70%) required a dosage reduction to 200 mg once daily (11). Risk factors for grade ≥ 3 thrombocytopenia development during niraparib are bodyweight of <77 kg and baseline platelet count of <150 000 cells/μl (11, 13). However, grade 3 thrombocytopenia was most common during the first 3 months of treatment, and patients who could persist with niraparib at a dosage of 300 mg/day for 3 months rarely developed grade ≥ 3 thrombocytopenia after this time (11). With the exception of the aforementioned grade 4 events, which occurred in two patients who had a baseline platelet count of 100 000 and 185 000 cells/μl, all reported platelet count decreases were grade 1 or 2 and recovered without the need for transfusion. Overall, the safety profile was acceptable in this Japanese cohort of patients with solid tumours, and was consistent with the known safety profile of niraparib and previous clinical experience with niraparib in non-Japanese patients.
The secondary objective of this study was to evaluate the pharmacokinetics of once-daily oral niraparib in Japanese patients. Following a single dose and multiple oral administrations of niraparib 200 and 300 mg/day, the median tmax of niraparib was ~3–4 h, consistent with previous pharmacokinetic data from a predominantly Caucasian population (9). The mean Cmax and AUC0–24 after multiple doses of niraparib generally increased in a dose-proportional manner, irrespective of the DLT experienced. Geometric mean accumulation ratios (day 21/day 1) after 21 days of dosing ranged from 1.643 to 2.385 and 2.644 to 3.654 for Cmax and AUC0–24, respectively. These profiles were similar to the study results of an overseas phase I study in non-Japanese patients with advanced solid tumours (Study PN001), which showed 2- to 4-fold accumulation after multiple doses (9).
Niraparib has ~73% bioavailability after oral administration in cancer patients (14), and tumour exposure is high at steady state (15). The pharmacokinetic profile of niraparib is characterized by high volume of distribution and cell membrane permeability relative to other PARP inhibitors (15).
Among eight evaluable patients, two had a partial response and two had stable disease. Together with the tolerability data, these data suggest that both the 200 and 300 mg/day niraparib dosages can be investigated in future studies in Japanese patients, consistent with the doses used in the phase III European and US PRIMA and NOVA studies (4, 5). Furthermore, as Japanese patients generally have lower body weight compared with Caucasian patients, a niraparib starting dosage of 200 or 300 mg/day, based on the patient’s baseline body weight and platelet count, may have a better tolerability and safety profile in a Japanese patient.
The limitations of this study are consistent with those of any phase I dose-escalation evaluation, including the small number of patients enrolled and the lack of a control group.
In conclusion, niraparib was rapidly absorbed and the exposure generally increased in a dose-proportional manner. There were no safety concerns in niraparib administration to Japanese patients, demonstrating that niraparib 200 and 300 mg/day was tolerable in Japanese patients with advanced solid tumours.
Supplementary Material
{kind=link}
Contributor Information
Kan Yonemori, Department of Experimental Therapeutics, National Cancer Center Hospital, Tokyo, Japan.
Toshio Shimizu, Department of Experimental Therapeutics, National Cancer Center Hospital, Tokyo, Japan.
Shunsuke Kondo, Department of Experimental Therapeutics, National Cancer Center Hospital, Tokyo, Japan.
Satoru Iwasa, Department of Experimental Therapeutics, National Cancer Center Hospital, Tokyo, Japan.
Takafumi Koyama, Department of Experimental Therapeutics, National Cancer Center Hospital, Tokyo, Japan.
Shigehisa Kitano, Department of Experimental Therapeutics, National Cancer Center Hospital, Tokyo, Japan.
Jun Sato, Department of Experimental Therapeutics, National Cancer Center Hospital, Tokyo, Japan.
Akihiko Shimomura, Department of Experimental Therapeutics, National Cancer Center Hospital, Tokyo, Japan; Department of Breast and Medical Oncology, National Center for Global Health and Medicine, Tokyo, Japan; Department of Breast and Medical Oncology, National Cancer Center Hospital, Tokyo, Japan.
Ryota Shibaki, Department of Experimental Therapeutics, National Cancer Center Hospital, Tokyo, Japan.
Ajit Suri, Millennium Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited, Cambridge, MA, USA.
Yoichi Kase, Takeda Pharmaceutical Company Limited, Osaka, Japan.
Shuuji Sumino, Takeda Pharmaceutical Company Limited, Osaka, Japan.
Kenji Tamura, Department of Breast and Medical Oncology, National Cancer Center Hospital, Tokyo, Japan.
Noboru Yamamoto, Department of Experimental Therapeutics, National Cancer Center Hospital, Tokyo, Japan.
Data sharing statement
The datasets, including the redacted study protocol, redacted statistical analysis plan and individual participants’ data supporting the results reported in this article, will be made available within 3 months from initial request to researchers who provide a methodologically sound proposal. The data will be provided after its de-identification, in compliance with applicable privacy laws, data protection and requirements for consent and anonymization.
Author contributions
A.S., Y.K. and S.S. designed the study and reviewed drafts of the manuscript. K.Y., T.S., S.K., S.I., T.K., S.K., J.S., A.S., R.S., K.T. and N.Y. enrolled patients and reviewed drafts of the manuscript. S.S. was responsible for statistical analysis. All authors approved the final version of the manuscript for submission.
Acknowledgements
The authors thank the patients and their families and caregivers, and all clinicians for their involvement and contribution to the study. We would also like to thank TESARO, a GlaxoSmithKline company, for providing valuable advice during manuscript development; Yuka Yamamoto, medical expert of Takeda Pharmaceutical Company Limited, for providing valuable advice regarding study design and data interpretation during manuscript development; and Catherine Rees of Springer Healthcare Communications who wrote the outline and first draft of the manuscript. This medical writing assistance was funded by Takeda Pharmaceutical Company Limited.
Funding
This study was funded by Takeda Pharmaceutical Company Limited.
Declarations of interest
Kan Yonemori reports personal fees from Eisai Co., Ltd., Novartis Pharma K.K., Chugai Pharmaceutical Co., Ltd., Ono Pharmaceutical Co., Ltd., Takeda Pharmaceutical Co., Ltd. and Taiho Pharmaceutical Co., Ltd., outside the submitted work; Toshio Shimizu reports grants from Novartis Pharma K.K., Eli Lilly and Company, Bristol-Myers Squibb K.K., Daiichi-Sankyo Co., Ltd., Millenium-Takeda, 3D Medicines Co., Ltd., Chordia Therapeutics Inc., Symbio Pharmaceuticals Ltd., Five Prime Therapeutics Inc., PharmaMar, S.A., AstraZeneca K.K., Abbvie Inc., Incyte Corporation and Astellas Pharma Inc., outside the submitted work; Satoru Iwasa reports grants from Bayer Yakuhin, Ltd., outside the submitted work; Takafumi Koyama reports other funding from Chugai Pharmaceutical Co., Ltd. and Sysmex Corporation, outside the submitted work; Shigehisa Kitano reports grants and personal fees from Boehringer Ingelheim Japan, Inc., Eisai Co., Ltd., Ono pharmaceutical Co., Ltd. and Regeneron Pharmaceuticals, Inc., personal fees from AstraZeneca K.K., Chugai pharmaceutical Co., Ltd., Pfizer Japan Inc., Sanofi K.K., Nippon Kayaku Co., Ltd., Meiji Seika Pharma Co., Ltd., Taiho Pharmaceutical Co., Ltd., Novartis Pharma K.K., Daiichi-Sankyo Co., Ltd., MSD K.K., Kyowa Kirin Co., Ltd., Celgene Corporation, Sumitomo Dainippon Pharma Co., Ltd., Bristol Myers Squibb K.K., AYUMI Pharmaceutical Corporation, Rakuten Medical Inc., PMDA (Pharmaceuticals and Medical Devices Agency) and GlaxoSmithKline PLC., and grants from Astellas Pharma Inc., Gilead Sciences, Inc., AMED (Japan Agency for Medical Research and Development) and JSPS (Japan Society for the Promotion of Science), outside the submitted work; Akihiko Shimomura reports grants and personal fees from AstraZeneca K.K., Chugai Pharmaceutical Co., Ltd. and Daiichi-Sankyo Co., Ltd., personal fees from Pfizer Japan Inc., Eli Lilly and Company, Eisai Co., Ltd., Novartis Pharma K.K. and Mundipharma K.K., and grants from Taiho Pharmaceutical Co., Ltd. and Mochida Pharmaceutical Co., Ltd., outside the submitted work; Noboru Yamamoto reports personal fees from Ono Pharmaceutical Co., Ltd., Chugai Pharmaceutical Co., Ltd., AstraZeneca K.K., Pfizer Japan Inc., Eli Lilly and Company, Bristol-Myers Squibb K.K., Eisai Co., Ltd., Otsuka Pharmaceutical Co., Ltd., Takeda Pharmaceutical Co., Ltd., Boehringer Ingelheim Japan, Inc., CMIC HOLDINGS Co., Ltd. and Sysmex Corporation, and grants from Chugai Pharmaceutical Co., Ltd., Taiho Pharmaceutical Co., Ltd., Eisai Co., Ltd., Eli Lilly and Company, Quintiles Transnational Japan K.K., Astellas Pharma Inc., Bristol-Myers Squibb K.K., Novartis Pharma K.K., Daiichi-Sankyo Co., Ltd., Pfizer Japan Inc., Boehringer Ingelheim Japan, Inc., Kyowa Kirin Co., Ltd., Bayer, Ono Pharmaceutical Co., Ltd., Takeda Pharmaceutical Co., Ltd., Janssen Pharmaceutical K.K., MSD K.K., Merck & Co., Inc., GlaxoSmithKline PLC. and Sumitomo Dainippon Pharma Co., Ltd., outside the submitted work; Yoichi Kase and Shuuji Sumino are employees of Takeda Pharmaceutical Co., Ltd.; Ajit Suri is an employee of Millennium Pharmaceuticals, Inc; Shunsuke Kondo, Jun Sato, Ryota Shibaki and Kenji Tamura have nothing to disclose.
References
- 1. Lord CJ. Ashworth a. PARP inhibitors: synthetic lethality in the clinic. Science 2017;355:1152–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. European Medicines Agency . Zejula 100 mg hard capsules. Summary of product characteristics. 2017. Available from: https://www.ema.europa.eu/en/documents/product-information/zejula-epar-product-information_en.pdf (3 February 2020, date last accessed).
- 3. US Food and Drug Administration . Zejula (niraparib) capsules, for oral use. Prescribing information. 2020. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/208447s015s017lbledt.pdf (3 February 2020, date last accessed).
- 4. Mirza MR, Monk BJ, Herrstedt J, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med 2016;375:2154–64. [DOI] [PubMed] [Google Scholar]
- 5. Gonzalez-Martin A, Pothuri B, Vergote I, et al. Niraparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med 2019;381:2391–402. [DOI] [PubMed] [Google Scholar]
- 6. GlaxoSmithKline . FDA approves Zejula (niraparib) as the only once-daily PARP inhibitor in first-line monotherapy maintenance treatment for women with platinum-responsive advanced ovarian cancer regardless of biomarker status. 2020. Available from: https://www.gsk.com/en-gb/media/press-releases/fda-approves-parp-inhibitor-in-first-line-monotherapy-maintenance-treatment-for-women-with-platinum-responsive-advanced-ovarian-cancer-regardless-of-biomarker-status/ (27 July 2020, date last accessed).
- 7. Tew WP, Lacchetti C, Ellis A, et al. PARP inhibitors in the Management of Ovarian Cancer: ASCO guideline. J Clin Oncol 2020;38:3468–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. O'Cearbhaill RE. Using PARP inhibitors in advanced ovarian cancer. Oncology 2018;32:339–43. [PMC free article] [PubMed] [Google Scholar]
- 9. Sandhu SK, Schelman WR, Wilding G, et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol 2013;14:882–92. [DOI] [PubMed] [Google Scholar]
- 10. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–47. [DOI] [PubMed] [Google Scholar]
- 11. Berek JS, Matulonis UA, Peen U, et al. Safety and dose modification for patients receiving niraparib. Ann Oncol 2018;29:1784–92. [DOI] [PubMed] [Google Scholar]
- 12. Mirza MR, Avall Lundqvist E, Birrer MJ, et al. Niraparib plus bevacizumab versus niraparib alone for platinum-sensitive recurrent ovarian cancer (NSGO-AVANOVA2/ENGOT-ov24): a randomised, phase 2, superiority trial. Lancet Oncol 2019;20:1409–19. [DOI] [PubMed] [Google Scholar]
- 13. Smith JA, Le T, Martin GA, et al. Identifying the need to refine the potential patient risk factors for niraparib-induced thrombocytopenia. Gynecol Oncol 2019;152:265–9. [DOI] [PubMed] [Google Scholar]
- 14. van Andel L, Rosing H, Zhang Z, et al. Determination of the absolute oral bioavailability of niraparib by simultaneous administration of a 14C-microtracer and therapeutic dose in cancer patients. Cancer Chemother Pharmacol 2018;81:39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sun K, Mikule K, Wang Z, et al. A comparative pharmacokinetic study of PARP inhibitors demonstrates favorable properties for niraparib efficacy in preclinical tumor models. Oncotarget 2018;9:37080–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.