

Abstract
Spike (S) glycoprotein of SARS-CoV2 exists chiefly in two conformations, open and closed. Most previous structural studies on S protein have been conducted at pH 8.0, but knowledge of the conformational propensities under both physiological and endosomal pH conditions is important to inform vaccine development. Our current study employed single-particle cryoelectron microscopy to visualize multiple states of open and closed conformations of S protein at physiological pH 7.4 and near-physiological pH 6.5 and pH 8.0. Propensities of open and closed conformations were found to differ with pH changes, whereby around 68% of S protein exists in open conformation at pH 7.4. Furthermore, we noticed a continuous movement in the N-terminal domain, receptor-binding domain (RBD), S2 domain, and stalk domain of S protein conformations at various pH values. Several key residues involving RBD-neutralizing epitopes are differentially exposed in each conformation. This study will assist in developing novel therapeutic measures against SARS-CoV2.
Keywords: spike homotrimer, cryo-EM, 3D reconstruction, TEM, negative staining, pH-dependent, solvent accessibility, single particle, S-head, stalk domain
Graphical abstract
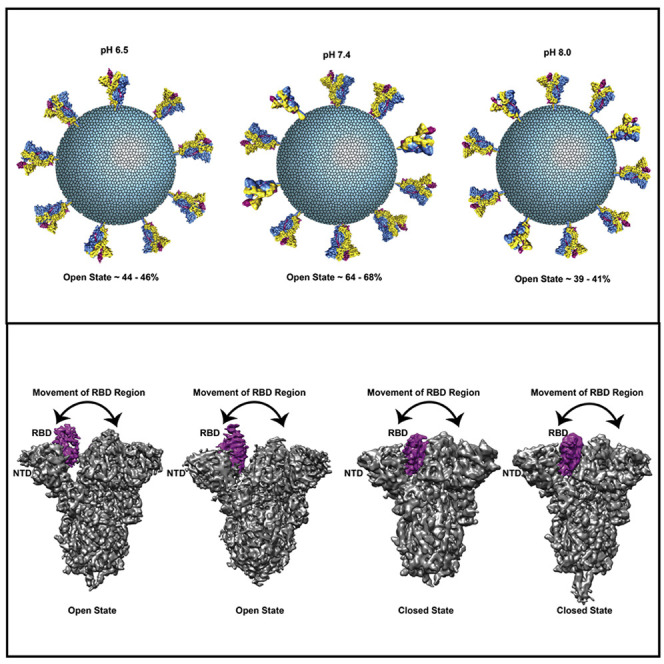
In this study, Pramanick et al. demonstrate inherent structural flexibility of NTD, RBD, and stalk domain of SARS-CoV2 Spike glycoprotein. Eleven high-resolution cryo-EM structures obtained over a range of near-physiological pH values indicate a trend of increasing open conformational state of S protein at physiological pH 7.4.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV2), belonging to the Coronaviridae family, is the third known zoonotic virus to have plagued mankind in the 21st century (Drosten et al., 2003; Huang et al., 2020; Zaki et al., 2012; Zhou et al., 2020b). This formerly unexplored coronavirus was first isolated from Hubei province, China, in December 2019 (Zhou et al., 2020a). As of November 1, 2020, the World Health Organization declared nearly 46.6 million infections and ∼1.2 million deaths worldwide (WHO Daily Report). Since the release of the complete sequence of the SARS-CoV2 genome, a plethora of studies have been performed in search of possible therapeutic and vaccine candidates. One such thoroughly investigated vaccine generation target is the heavily glycosylated homotrimeric Spike (S) protein responsible for the crown-like surface display in these so-called coronaviruses (Tortorici and Veesler, 2019). Among the three transmembrane envelope proteins, S protein is known to mediate viral entry into host cells. The S protein in its pre-fusion state has two distinct subunits called S1 and S2. S1 harbors the receptor-binding domain (RBD), N-terminal domain (NTD), and two small subunits SD1 and SD2, while the S2 subunit has three stable long helices, which tether the S1 domain as well as the S protein with the viral envelope. Early studies have shown that this recently emerged coronavirus binds tightly to human ACE2 (hACE2) receptor, thereby facilitating its transmission (Walls et al., 2020). Specifically, the RBD of the S protein exists in two prominent conformations, up (open) and down (closed) (Toelzer et al., 2020; Walls et al., 2020; Wrapp et al., 2020; Xiong et al., 2020). These surface proteins exist as dimorphic entities before and after the fusion of viral and cellular membranes. Following membrane fusion, the S protein is cleaved by host cell proteases at the boundary between the S1 and S2 subunits, transforming into an elongated post-fusion state (Kirchdoerfer et al., 2016). Thus, owing to a pivotal role of S protein in eliciting the infection cascade, it is the most well-characterized viral structural protein and is widely used to isolate neutralizing antibodies (Du et al., 2009; Elshabrawy et al., 2012; Jiang et al., 2014; Li et al., 2015; Wang et al., 2015; Ying et al., 2015; Yu et al., 2015). A large number of structural studies indicate appreciable flexibility of the RBD region and unequivocally report the presence of the distinct 1-RBD up-open and all-RBD down-closed species of S protein trimers (Ke et al., 2020; Korber et al., 2020; Melero et al., 2020; Walls et al., 2020).
Unless stabilized by mutations, purified soluble S protein trimers have not been observed to display a 2-RBD up-open or all-RBD up-open conformation that could possibly lead to a more lethal SARS-CoV2 infection (Korber et al., 2020). Cryoelectron tomography (cryo-ET) analysis of intact virions displaying S proteins affirms past knowledge and reports a minor 14% population in 2-RBD up-open state (Ke et al., 2020). The envelope proximal stalk of the S2 subunit was recently investigated in a membrane-bound state and was found to act as a hinge around which the S trimer is free to rotate (Ke et al., 2020; Turoňová et al., 2020). Extensive characterization of MERS-CoV and SARS-CoV show a moderately flexible NTD, although the most dramatic movement is restricted to the RBD region (Xiong et al., 2020). Also, recent findings have indicated the movements in RBD upon binding with ACE2 and in the apo form at endosomal acidic pH values (pH 4–5.5) (Zhou et al., 2020b). Although a wealth of available structural information has furthered understanding about RBD movement, mapping of these discrete elegantly orchestrated intermediates at physiological pH still remains elusive. Based on current understanding, we pooled the distribution of S trimers at physiological and near-physiological pH conditions. Our results suggest a milieu of myriad conformational intermediates that have unique structural shifts directly impacting the cavity lining the RBD and NTD. Interestingly, we were able to trap different conformations of the “hip,” “knee,” and “ankle” movement of the S2 stalk showing that anchoring to a lipid layer is not mandatory for rotational freedom of the S1 around the S2 in the purified S trimer at physiological pH. Moreover, we also report a predominantly receptor-accessible population of S trimer at pH 7.4 as compared with slightly acidic or alkaline pH. A thorough analysis of these diverse structural shifts will augment existing knowledge and inform vaccine development and drug design.
Results
Several high-resolution cryoelectron microscopy (cryo-EM) structures of the S protein of SARS-CoV, MERS-CoV, and SARS-CoV2 S protein have been widely characterized in the last few months (Henderson et al., 2020; Ke et al., 2020; Park et al., 2019; Toelzer et al., 2020; Walls et al., 2020; Wrapp et al., 2020; Xiong et al., 2020; Yuan et al., 2017; Zhou et al., 2020b). However, most cryo-EM structures of S protein are determined at pH 8.0, where one RBD up-open conformation and the three RBD down-closed conformations of S protein have been observed (Walls et al., 2020; Wrapp et al., 2020). Additionally, recent studies that focused on a range of endosomal acidic pH values observed variability in RBD regions in both receptor-bound and apo form, which could be attributed to pH-mediated conformational switches (Warwicker, 2020; Zhou et al., 2020b). Therefore, in this study we investigated the effect of pH on S protein, whereby we implemented our cryo-EM-based structural study at near-physiological pH, namely pH 8.0, pH 7.4, and pH 6.5. Furthermore, we inspected and classified the cryo-EM data more precisely to identify several intermediate conformations of the RBD and NTD between the up and down conformations of RBD of the S protein.
Initially, S protein was examined at three different pH conditions, as mentioned earlier, using negative-stain transmission electron microscopy (TEM) and two-dimensional (2D) classification (Figures 1A, 1B, S1A, and S1B). A room-temperature TEM study indicated no aggregation or distortion of the S protein at three different pH values. However, a low-resolution TEM study cannot adequately describe any conformational flexibility of RBD or NTD of S protein, although some 2D class averages corresponded to compact closed conformations while the rest of the 2D class averages showed dispersed conformations of the S protein (Figure 1B). Furthermore, oligomerization, homogeneity, and conformational heterogeneity of the S protein were visualized using negative-stain TEM imaging at room temperature (Figures 1A, 1B, S1A, and S1B). EM images of the S protein indicated a uniform, cone-shaped monodispersed particle population, and the particles were broadly similar at the three different pH values (Figure 1B). Therefore, we decided to further investigate the conformational changes of S protein at three different pH values using single-particle cryo-EM.
Figure 1.

Negative staining and 2D class averages of S protein at pH 8.0, 7.4, and 6.5, and 3D reconstruction at pH 8.0
(A) Representative negative-stain image of S protein.
(B) Representative reference free 2D class averages of negative-stain images at pH 8.0, 7.4, and 6.5. Class averages indicate that S protein was stable and formed a cone-shaped architecture at various pH conditions. There is no observable aggregation or distortion due to pH changes. Bottom panel shows an enlarged view of three different class averages where different orientations and the flexibility of RBD and NTD are visible (marked by red arrow).
(C) Cryo-EM micrograph and 2D class averages of S protein at pH 7.4. Cryo-EM micrographs and 2D class averages of S protein at pH 6.5 and pH 8.0 are shown in Figure S1.
(D) 3D reconstruction of 1-RBD up-open and all-RBD down-closed conformations of S protein at pH 8.0.
See also Figures S1 and S2.
Single-particle cryo-EM studies of S protein at three different pH values
In the current study, we investigated the pH-dependent morphological changes of S protein of SARS-CoV2. Viral protein conformation is highly variable, diverse, and pH dependent in many cases (Batishchev et al., 2016; Sturman et al., 1990; Yang et al., 2004; Zhou et al., 2020b). To achieve our targets, we collected data for the S protein ectodomain of SARS-CoV2 at three different pH values whereby we acquired 2,405, 4,504 and 3,166 micrographs at pH 8.0, pH 7.4, and pH 6.5, respectively, using a Talos Arctica 200kV cryo-transmission electron microscope equipped with a K2 direct electron detector. We classified and processed the cryo-EM data using the RELION 3.0 software package. Multiple three-dimensional (3D) classification steps were implemented to identify conformational heterogeneity, which enabled us to identify several previously unreported intermediate conformations of S protein (Figures S2–S4).
Cryo-EM reconstruction of S protein of SARS-CoV2 at pH 8.0 and pH 7.4
Viral S glycoprotein at the virus surface adopts a kinetically stable pre-fusion conformation that binds the host cell hACE2 receptor, initiating viral infection. The S glycoprotein of many enveloped viruses, including SARS-CoV2, undergoes a metastable pre-fusion to post-fusion conformational change during cell entry. These conformational changes of S protein depend on pH (Yang et al., 2004), proteolytic cleavage (Jaimes et al., 2020), and protein-protein interaction (Hoffmann et al., 2020). Additionally, the interaction of hACE2 receptors with the pre-fusion state of viral S glycoprotein is highly pH dependent (Zhou et al., 2020b). Therefore, in this study we performed cryo-EM studies of S protein at physiological pH (pH 7.4) and near-physiological pH (pH 8.0 and pH 6.5). There are several cryo-EM structures of S protein available at pH 8.0. Therefore, here our main intention was to characterize the S protein structure, structural variability, and conformational flexibility at pH 7.4 and pH 6.5. Recombinantly expressed S protein was well distributed and monodispersed at the three different pH conditions at cryogenic temperature (Figures 1C, S1C, and S1D). Several high-resolution cryo-EM reconstructions of S protein at three different pH conditions were obtained around 3.8- to 5.4-Å resolution (Figures 1D, 2A, 2B, 3A, 3B, and S5B–S5E). The 3D reconstructions of S protein illustrate that the membrane-proximal S2 region of S protein has 3-fold symmetry, whereas the S1 region, mainly RBD and NTD of S protein, is highly flexible and asymmetric. The S2 region of S protein is more stable than RBD and NTD. Therefore, 3D reconstructions of S protein were performed without any symmetry to identify the structural variability of the RBD domain of S protein (Figures S2–S4). We reconstructed S protein at pH 8.0, and the overall structure was in agreement with previous reports (Figure 1D) (Walls et al., 2020; Wrapp et al., 2020). The 3D classification indicated that at pH 8.0 the overall trend of variability of S protein is around 61% in the 3-RBD down-closed conformation and 39% in the 1-RBD up-open conformation (Figures 1D and 4E). As mentioned earlier, several S protein structures were already characterized by single-particle cryo-EM at pH 8.0. Therefore, in our study we concentrated on studying the conformational changes and dynamics of apo S protein RBD at physiological pH (pH 7.4). Around 1,187,595 particles from 4,504 micrographs at pH 7.4 were extracted for 2D and 3D classifications. Finally the 723,229 best particles were selected for further 3D classification, and the dataset was classified into 15 classes with C1 symmetry to visualize the structural diversity of S protein (Figure S3). Our 3D reconstructions showed 1-RBD up-open states and 3-RBD down-closed conformations of trimeric S protein, although we were also able to identify various states of open and closed conformations. Structural variability of RBD and NTD of S protein was observed at pH 7.4 (Figures 2A, 2B, and S3) and pH 6.5 (Figures 3A, 3B, and S4). Three open-state and two closed-state S protein structures were determined after 3D classification and final refinement of the pH 7.4 dataset. The dataset from the open and closed conformations could be merged separately to increase the particle number as well as the signal-to-noise ratio to obtain a higher-resolution structure. Nevertheless, our main intention was to visualize the conformational flexibility between different open or closed conformations of S protein. Therefore, the dataset from three open and two closed states of S protein were processed separately, and we investigated the flexibility and conformational dissimilarity of the RBD and NTD of all the open and closed states. Furthermore, phenix.real_space_refinement was employed to pinpoint the structural variability of RBD, and NTD in open as well as closed conformations. Remarkably, we noticed that the RBD of two closed conformations of S protein are more than 8 Å displaced from each other (Figure 2C), and a similar trend was also detected in the NTD, which was shifted toward S2 from the original conformation by more than 10 Å (Figures 2C and 2D). However, no significant movement of the S2 domain between the two different closed conformations of S protein was observed (Figure 2C), and minute displacement was observed near the boundary of the S2 and S1 regions, more precisely at amino acid T696 (0.68 Å) (Figures 2A and 2C). Similar types of displacement (3.1–10.8 Å) were observed in the recently published cryo-EM structure of S-hACE2 complex at pH 7.4 (Zhou et al., 2020b), and displacements were measured at hACE2 molecules. However, actual displacement occurs in the RBD or NTD of S1 subdomain to accommodate the hACE2 receptor, and movement is observed in hACE2 molecules. This type of inherent flexibility of RBD and NTD is prominent in the current study, which was performed at pH 7.4 without any hACE2 receptor (Figure 6). The structural analysis of complete superposition of atomic model of the S-hACE2 complex with unliganded S protein demonstrated the structural rearrangement of RBD. More precisely, conformational modification was highly significant in two hACE2-binding loop regions (L441, K444, V445, Y449, P499, T500, and Y505) to accommodate hACE2 (Figure 6). This comparison strongly supports the notion that RBD adopts various conformations at pH 7.4 to accommodate hACE2. Similarly, the structural differences between the three high-resolution cryo-EM structures of 1-RBD up-open conformation were examined, whereby three long helices at S2 region in all three open states were comparatively well aligned and almost identical (Figure 2B). However, major dissimilarity (>11 Å) was noticed in the membrane-proximal non-helical part of the S2 region of the two open states of the S protein structure (Figure 2D). Nevertheless, the RBD and NTD in the open conformations are only shifted ∼6 Å, leading to a marginal widening of the cavity lining the RBD and NTD of the open conformations (Figure 4C). Therefore, our study indicated that the S protein at physiological pH exists in a variety of conformations in accordance with previously observed open and closed conformations over a range of pH values in a recent study (Zhou et al., 2020b). Various extents of conformational diversity were also detected in the RBD, NTD, and S2 regions of S protein in up and down conformations.
Figure 2.

Cryo-EM 3D reconstruction of S protein at pH 7.4
(A and B) Solid and transparent representation of cryo-EM 3D reconstruction of S protein at pH 7.4. The transparent representation of S protein is fitted with an atomic model calculated from 3D reconstructions of S protein and PDB: 6vyb (1-RBD up) and PDB: 6zwv (3-RBD down). Yellow spheres represent T696 at the boundary of S1 and S2 regions. The RBD and NTD of the solid representation of the S protein are colored orange and blue, respectively. At pH 7.4, two 3-RBD down-closed conformations (A) and three 1-RBD up-open conformations (B) are observed. Distance between RBD and NTD is marked by black curved arrows.
(C) Comparison between two closed states, class 3 (cyan) and class 9 (purple), indicates the movement of RBD and NTD. Only one monomer of both the closed conformers are superimposed and represented here to visualize the displacement of NTD and RBD. No displacement is noticed in S2 subunit, whereas a major shift is observed in RBD (marked as red box) and NTD (marked as green box). The enlarged views of RBD and NTD are displayed alongside the full monomer. Green and red arrows show the displacement between the atomic model of class 3 (cyan) and class 9 (purple).
(D) Comparison between two 1-RBD up-open conformations, class 5 (cyan) and class 8 (purple). Difference in NTD region is shown by green arrows, RBD region by red arrows, and S2 region by orange arrows. A significant displacement is observed in S2 subunit, RBD, and NTD, marked by orange, red, and green boxes, respectively. All these three boxed out regions are shown in enlarged view beside the full monomer.
See also Figure S3 and Tables S1–S3.
Figure 3.

Cryo-EM 3D reconstruction of S protein at pH 6.5
(A and B) Solid and transparent representation of cryo-EM 3D reconstruction of S protein at pH 6.5. The transparent representation of S protein is fitted with an atomic model calculated from 3D reconstructions of S protein and PDB: 6vyb (1-RBD up) and PDB: 6zwv (3-RBD down). Yellow spheres represent T696 at the boundary of S1 and S2 region. The RBD and NTD of the solid representation of the S protein are colored orange and blue, respectively. At pH 6.5, two 3-RBD down-closed conformations (A) and two 1-RBD up-open conformations (B) are observed. Distance between RBD and NTD is marked by black curved arrows.
(C) Comparison between the atomic models of two closed (all-RBD down conformation) states, class 2 (cyan) and class 5 (purple), indicates the movement of RBD and NTD. RBD and NTD are marked by red and green boxes, respectively. Black arrows show the enlarged view of RBD and NTD, where significant displacement is observed.
(D) Comparison between atomic models of two open states, class 4 (cyan) and class 3 (purple). Difference in NTD region is shown by green arrows and RBD region by red arrows.
See also Figure S4 and Tables S1–S3.
Figure 6.

Comparison between 1-RBD up single ACE2 bound S protein with 1-RBD up apo S protein at physiological pH
Superimposition of 1-RBD up single ACE2 bound S protein (PDB: 7KNB) and 1-RBD up S protein atomic model from this study (class 5 at pH 7.4). Magnified view represents the difference in the RBD region (top right) between ACE2 bound and apo S protein. Bottom right panel illustrates good correlation between the secondary structures of S2 region of former and latter.
Figure 4.

Calculation of high-resolution symmetric closed states of S protein and measurement of cavity between closed and open conformers
(A) Solid and transparent representations of cryo-EM reconstruction of S protein at pH 7.4 with C3 symmetry.
(B) Nineteen glycan chains out of 23 predicted glycosylation sites are clearly visible in the high-resolution cryo-EM 3D reconstruction of closed-state S protein structure. All the glycan chains are colored green (transparent model). In the solid model, glycosylation residues are marked in blue.
(C) Cavity between RBD and NTD is marked by blue color in open and closed conformations at pH 7.4.
(D) Cavity between RBD and NTD is marked by blue color in open and closed conformations at pH 6.5.
(E) Bar diagram (percentage values ± standard error of mean) showing distribution of open and closed conformations of S protein at three different pH conditions (pH 8.0, 7.4, and 6.5).
See also Figures S2–S4.
Our study of the atomic models of open and closed cryo-EM maps suggested that the RBD region is significantly flexible and not ordered in the same fashion for all the conformations. Four different types of neutralizing antibodies have been reported so far: neutralizing antibody blocking hACE2 and binding only to up conformation (such as C105, B38); neutralizing antibody binding to both up and down conformations at ACE2 site (such as P2B-2F6, Fab 2-4); neutralizing antibody binding to both up and down conformations other than hACE2 interaction sites (such as S309); and neutralizing antibody binding only to up conformation without blocking hACE2-binding site (such as CR3022) (Barnes et al., 2020; Liu et al., 2020). Antibody-binding sites were recognized from previous literature and identified in the atomic models of different conformations (Figures 5A and 5B ). Relative solvent-accessible surface areas of residual side chains of atomic models obtained from the three pH studies were calculated using NACCESS (Hubbard and Thornton, 1993). Analysis of the RBD indicated varied extent of accessibility of the antibody-binding epitopes in each conformer (Figure 5C). Nearly 40 key interacting residues showed remarkable heterogeneity in solvent accessibility, which could indicate the presence of a diverse population of S proteins at neutral pH that facilitates elicitation of antibodies to epitopes that are buried in the closed conformation of the S trimer.
Figure 5.

Relative solvent-accessible surface area calculation of each residue side chain at the RBD region
(A) Amino acid residues (marked red) at RBD and NTD regions of 3-RBD down S protein, involved in interaction with neutralizing antibodies at pH 8.0, 7.4, and 6.5.
(B) Amino acid residues (marked red) at RBD and NTD regions of 1-RBD up S protein, involved in interaction with neutralizing antibodies at pH 8.0, 7.4, and 6.5.
(C) Comparison between relative solvent-accessible surface area of residue side chain at RBD region of closed and open models. Heatmap shows the change of relative solvent-accessible surface area for residue side chain (%) at the RBD region of closed and open models at pH 8.0, 7.4, and 6.5.
To obtain new structural information from our dataset, we imposed C3 symmetry on the 3D classification of particles to improve the resolution of only the closed conformations. Thereby, we refined a 3-RBD down-closed structure to a resolution of 3.8 Å (Figure 4A).
Comparison of the resultant closed structure with a glycosylated full atomic model (Woo et al., 2020) of S protein revealed that among the 23 predicted sites, our map clearly showed strong densities for 18 N-linked and 1 O-linked glycosylation sites. We were also able to visualize a previously unobserved N-linked glycosylation at His625. This also indicates that 200-kV cryo-EM is capable of achieving a high-resolution cryo-EM structure of S protein and able to resolve almost all the glycan side chains of S protein (Figure 4B). Furthermore, we noticed that approximately 68% of particles from this dataset are in 1-RBD up-open states (Figure 4E), suggesting that open S protein complexes dominate the population at physiological pH (pH 7.4). Superimposition of RBD-bound ACE2 receptor (PDB: 7A94 [Benton et al., 2020]) with closed and open atomic models obtained from our dataset reveals steric clashes with adjacent RBD in closed conformations of S homotrimers making the up conformation a prerequisite for entry of SARS-CoV2, akin to SARS-CoV (Gui et al., 2017) (Figure S6). Several past reports show that mutations stabilize the 2-RBD and 3-RBD up-open conformations, leading to majorly open forms of S homotrimer (Henderson et al., 2020; Ke et al., 2020; Korber et al., 2020). It is interesting that at pH 7.4 we are able to observe nearly 68% of S protein in a single RBD up conformation with only two proline mutations at residues 986 and 987 that are far from the RBD and unlikely to influence the aforementioned conformational distribution.
The membrane-proximal region or stalk of the S protein is also flexible and adopts multiple conformations. Based on various conformations of the stalk domain, the orientation of the S1 and S2 domain of the spike head were diverse (Figure S5A). These results support the recently published cryo-ET structure and molecular dynamics (MD) data (Ke et al., 2020; Turoňová et al., 2020). The stalk showed various lengths and orientations in all of the 3D reconstructions, affirming the flexible nature of the stalk (Ke et al., 2020; Turoňová et al., 2020). Additionally, in some reconstructions the stalk is exceptionally long (Figures S3 and S5A), altering the position of the spike head and its distance from the virion surface. However, we were unable to resolve the complete stalk domain in all of the high-resolution structures. The stalk domain was also visible in most of the high-resolution S protein structure, but the density of stalk appeared at a higher contour level. This also supported our previous observations that the stalk domain is extremely flexible and adopts multiple conformations. Recently published MD simulation results describe the “hip,” “knee,” and “ankle” of the stalk region that could be identified in all of the low-resolution single-particle reconstructions (Turoňová et al., 2020). Thus, these single-particle cryo-EM reconstructions showed the conformational flexibility of stalk domain and spike-head rotation at different directions relative to the viral membrane. Nevertheless, we were unable to find the 60° twist of the stalk domain (Ke et al., 2020), which might be difficult to identify without the viral membrane. Single-particle cryo-EM reconstructions of this study have revealed that the inherent flexibility of the stalk domain could facilitate various orientations and rotation of the spike head (Figure S5A).
Cryo-EM reconstruction of S protein of SARS-CoV2 at pH 6.5
Similarly, cryo-EM reconstruction of S protein at near-endosomal pH 6.5 was investigated to further probe the conformational flexibilities of RBD and NTD. Similar strategies were implemented for the structural characterization of S protein. We also calculated the 3D reconstruction and variability of the RBD of S protein for this particular dataset. We performed 3D classifications using RELION 3.0 to identify the open and closed conformations of RBD of S protein (Figure S4). An interesting phenomenon was noted in the 3D reconstruction, whereby a significant amount of structural variability was observed in the RBD position in the closed states of S protein (Figures 3A and 3B). From 3D classification, two closed states were isolated from the dataset whereby a compact closed state and a loosely packed closed state was observed (Figures 3A, 3B, and S4). The NTD of S protein was shifted outward in the loosely packed S protein. In both of the closed conformations, the NTD and RBD were slightly displaced from each other, and overall displacement of the NTD and RBD was <7.5 Å and <7 Å, respectively (Figure 3C). However, no displacement was observed in the S2 domain in both of the closed states. As denoted by the 3D volumes, outward shift in the NTD results in concomitant crevice formation between the adjacent RBDs. This could indicate a plausible intermediate where transition between a closed to open form commences (Figure 4D). Therefore, a compact closed state and a loosely packed closed state of S protein structures indicated that the RBD and NTD are significantly flexible, like the closed structures of S protein at pH 7.4. At this same pH two conformations were identified, whereby 1-RBD up-open conformations were characterized at resolutions of 4.9 Å and 5.44 Å. Due to the low resolution, we were unable to perform model building to determine the atomic model of up conformations. However, we calculated the morph map wherein we observed a significant amount of movement in RBD domain and adjacent NTD between the two 1-RBD up-open conformations (Figures 3A and 3B). Furthermore, all four high-resolution structures (two closed and two open conformations) showed the variable length and orientation of the stalk domain, which could be designated as the “hip,” “knee,” and “ankle” of the stalk region. These were visible in all of the 3D reconstructions of S protein at pH 6.5 and control the orientation of the spike head (Figure S5A).
Discussion
The viral S protein of SARS-CoV2 is one of the primary targets for vaccine development, and several studies of viral S protein complexed with neutralizing antibodies have been published in the last couple of months (Du et al., 2009; Elshabrawy et al., 2012; Jiang et al., 2014; Li et al., 2015; Wang et al., 2020; Ying et al., 2015; Yu et al., 2015). Several published data also suggest that coronaviruses are able to mask the receptor-binding site and employ the glycan chains to protect themselves from the host immune responses (Walls et al., 2016, 2019, 2020; Xiong et al., 2017). Many prior studies have pinpointed the structural flexibility and variability of S protein, where RBDs exist in several discrete conformational states. However, except for a recent cryo-ET study, most single-particle cryo-EM analyses of recombinant S protein reveal that 1-RBD up-open and all-RBD down-closed conformations of S protein co-exist at pH 8.0 (Walls et al., 2020; Wrapp et al., 2020). These distinct open conformational states of RBD are essential for hACE2 receptor engagement (Figure S6). However, the functional and mechanistic significance of 1-RBD up-open and 3-RBD down-closed states is ambiguous, demonstrating our limited understanding of the interaction of RBD with hACE2 (Beniac et al., 2006; Gui et al., 2017; Kirchdoerfer et al., 2016; Pallesen et al., 2017; Shang et al., 2020; Song et al., 2018; Walls et al., 2016; Yuan et al., 2017). This raises some important questions. What is the role of pH? How does pH regulate the conformational changes of S protein? How flexible are RBD and NTD of S protein? Do intermediate conformations of RBD and NTD exist? In the current study, we explored the possible existence of several distinct conformational states of SARS-CoV2 S protein at physiological pH 7.4 and near-physiological pH 6.5 and pH 8.0.
At all three different pH values, two discrete conformational states, 1-RBD up-open and all-RBD down-closed conformations, were observed in this study. However, the percentage of open and closed conformations varied as a function of pH, whereby we noticed that around 68% of S protein molecules exist as an open conformation at pH 7.4 and that the percentage of closed conformations increases at pH 6.5 and pH 8.0 (Figure 4E). Most surprisingly, we observed various relative localizations of RBD and NTD in 1-RBD up and all-RBD down conformations. Additionally, overall movement and displacement were observed in both 1-RBD up-open and all-RBD down-closed conformations captured in our dataset. A similar feature was also noticed in the NTD, where the NTD is shifted 10 Å toward the S2 domain (Figures 2C, 2D, 3C, and 3D). Due to this movement of NTD, two distinct open conformations were observed. In one conformation, NTD is close to RBD, named as a compact 1-RBD up conformation, and in another conformation NTD is separated from RBD, termed as a loosely packed 1-RBD up-open conformation. Simultaneously, a movement was also noticed in both the closed conformations, where NTD moves either toward or away from the RBD. This also results in either a loosely packed or compact closed-state S structure. Both these conformations indicate NTD and RBD are extremely flexible, even when the majority of the population exists as either open or closed state. Intermediate, distinct conformational states of RBD obtained from this study are in accordance with previous structural characterizations indicating a remarkable flexible nature of the RBD and NTD (Ke et al., 2020; Melero et al., 2020; Turoňová et al., 2020; Walls et al., 2020; Yuan et al., 2017).
Several previous studies acknowledged that the RBD up conformation is important for binding with hACE2 (Benton et al., 2020; Toelzer et al., 2020; Zhou et al., 2020b). Additionally, it is reported that pH-dependent switches also play a vital role for switching between open and closed RBD conformations of S protein. Therefore, it is reasonable that a majority of the S protein should be in open conformation at physiological pH to accommodate the hACE2 receptor. This is indeed observed in our study, where in the range of 64%–68% S protein molecules are in the open conformation and available to interact with hACE2 at pH 7.4. Similarly, a previous study reported that around 50% of S protein is in open conformation and 41% is undefined at pH 5.0 (Zhou et al., 2020b). It was further shown that at and below pH 4.5, S protein adopts more homogeneous stable closed conformation. This is supported by our observation of trends in increasing percentage of closed conformations at pH 6.5 as compared with pH 7.4. Several other published studies suggest that open-closed conformations of S protein are highly variable and that 50%–70% of S protein is in open conformation at pH 8 (Toelzer et al., 2020; Walls et al., 2020; Wrapp et al., 2020). It is also documented that D830, D839, D843, and D848 and a disulfide bond between C840 and C851 undergo a pH-induced protonated/deprotonated switch to regulate the open and closed conformations of S protein. Furthermore, amino acid residues F833, Y837, A846, L849, and P855 provide extra stability to one particular S protein conformation through the hydrophobic core due to this protonated/deprotonated switch on-off mechanism (Zhou et al., 2020b). Similar types of change in conformational distribution are observed in S protein in our study as the pH is varied from 8.0 to 7.4 to 6.5. A recent computational study (Warwicker, 2020) discusses the role of ambient pH on the pKa of D398 in mediating the open-closed RBD equilibrium, which also intriguingly correlates with our results. Our solvent-accessibility results also show significant variability in the packing of amino acid residues with respect to solvent environment, even with small changes in pH (Figure 5). Therefore, we hypothesize that the distribution of S protein conformations may be precisely orchestrated by a combinatorial impact of pH-regulated switches that lead to an increased population of 1-RBD up S protein at physiological pH, conducive to host cell receptor binding, and a concomitant gradual decrease of open conformations on either side of neutral pH.
The flexible nature of RBD and NTD undoubtedly will facilitate the accommodation of drug molecules, large Fab fragments, or antibodies within the grooves of RBD and NTD. Relative solvent-accessible surface area of representative amino acids varied even in different closed conformations, which may result in difference in antibody interaction. We observed the same in each open conformation (Figure 5). Therefore, we speculate that there is a continuous movement and rearrangement of RBD residues that could influence the strength of antibodies binding to open and closed S trimers. From our study, we propose that RBD and NTD co-exist in various distinct conformational states even in a predominantly open or closed architecture.
Flexibility of the stalk domain was highlighted in recent cryo-ET and MD simulation studies (Ke et al., 2020; Turoňová et al., 2020). Our single-particle cryo-EM study can distinguish the aforementioned “hip,” “knee,” and “ankle” arrangement of the stalk region, which display the rotation of the spike head. Therefore, the presence of viral membrane is not essential for various conformational states of the stalk domain, which facilitates the rotation of the bulky S1 subunit.
The observation of various intermediate conformational states of the stalk, NTD, and RBD as well as the high proportion of open conformations at pH 7.4 are the salient highlights of this study. The high proportion of open conformations at physiological pH helps explain why it is possible to elicit several neutralizing antibodies that recognize epitopes inaccessible in the closed conformation (Barnes et al., 2020; Liu et al., 2020). Our future plan, therefore, is to explore the interaction of drug candidates, neutralizing antibodies, and hACE2 with S protein at physiological pH. This will further assist us to understand the dynamics of S protein and discover new therapeutics against this disease.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Ni Sepharose 6 Fast Flow resin | G-Biosciences | Cat# 786-940 |
| Imidazole | Sigma Aldrich | Cat# 792527-500G |
| Tris Base | HiMedia | Cat# TC072M |
| Amicon Ultra-15 Centrifugal filter unit (100K) | Merck | Cat# UFC910024 |
| Amicon Ultra-0.5 Centrifugal filter unit (100K) | Merck | Cat# UFC510024 |
| Sodium Chloride | Qualigens | Cat# Q15918 |
| Dialysis Tubing Cellulose Membrane (14kDa cut-off) | Merck | Cat# D9527-100FT |
| Amicon Ultra-4 Centrifugal filter unit (10K) | Merck | Cat# UFC801024 |
| Phenylmethanesulfonyl Fluoride | Sigma Aldrich | Cat# P7626 |
| Uranyl Acetate 98%, ACS Reagent | Polysciences, Inc | Cat# 21447-25 |
| EM grid (Quantifoil R 1.2/1.3 300 Mesh, Gold) | Electron Microscopy Sciences | Cat# Q3100AR1.3 |
| EM grid (Carbon flim 300 mesh, Copper) | Electron Microscopy Sciences | Cat# CF300-CU |
| Potassium Chloride | Sigma Aldrich | Cat# P3911 |
| Potassium di-hydrogen orthophosphate | Qualigens | Cat# Q19465 |
| Di-sodium hydrogen orthophosphate | SD Fine Chemicals | Cat# S40158 K05 |
| Critical commercial assays | ||
| ExpiFectamine 293 Transfection kit | Gibco, Thermo Fisher | Cat# A14524 |
| Expi293™ Expression Medium | Gibco, Thermo Fisher | Cat# A1435101 |
| Deposited data | ||
| SARS-CoV2 Spike Protein structure at pH 6.5 with C1 Symmetry (Class 2) | This paper | EMD: 31092 |
| SARS-CoV2 Spike Protein structure at pH 6.5 with C1 Symmetry (Class 3) | This paper | EMD: 31093 |
| SARS-CoV2 Spike Protein structure at pH 6.5 with C1 Symmetry (Class 4) | This paper | EMD: 31094 |
| SARS-CoV2 Spike Protein structure at pH 6.5 with C1 Symmetry (Class 5) | This paper | EMD: 31095 |
| SARS-CoV2 Spike Protein structure at pH 7.4 with C1 Symmetry (Class 3) | This paper | EMD: 31096 |
| SARS-CoV2 Spike Protein structure at pH 7.4 with C1 Symmetry (Class 5) | This paper | EMD: 31097 |
| SARS-CoV2 Spike Protein structure at pH 7.4 with C1 Symmetry (Class 8) | This paper | EMD: 31101 |
| SARS-CoV2 Spike Protein structure at pH 7.4 with C1 Symmetry (Class 9) | This paper | EMD: 31100 |
| SARS-CoV2 Spike Protein structure at pH 7.4 with C3 Symmetry | This paper | EMD: 31098 |
| SARS-CoV2 Spike Protein structure at pH 8.0 with C1 Symmetry (Class 1) | This paper | EMD: 31099 |
| SARS-CoV2 Spike Protein structure at pH 8.0 with C1 Symmetry (Class 2) | This paper | EMD: 31102 |
| SARS-CoV-2 spike ectodomain structure (open state) | Walls et al., 2020 | PDB: 6VYB |
| Cryo-EM structure of SARS-CoV-2 Spike Proteins on intact virions: 3 Closed RBDs | Ke et al., 2020 | PDB: 6ZWV |
| SARS-CoV-2 Spike Glycoprotein with 1 ACE2 Bound | Benton et al., 2020 | PDB: 7A94 |
| Fully-glycosylated full-length S protein complex models | Woo et al., 2020 | PDB: 6VXX_1_1_1 |
| Cryo-EM structure of single ACE2-bound SARS-CoV-2 trimer spike at pH 7.4 | Zhou et al., 2020b | PDB: 7KNB |
| Experimental models: cell lines | ||
| Expi293F™ Cells (Female fetal kidney cell) | Gibco, Thermo Fisher | Cat# A14527 |
| Recombinant DNA | ||
| SARS-CoV-2 spike clone | Genscript USA | N/A |
| HRV-3C Protease | Genscript USA | N/A |
| Software and algorithms | ||
| RELION 3.0 | Scheres, 2012 | https://www3.mrc-lmb.cam.ac.uk/relion/index.php/Main_Page |
| MotionCor2 1.2.1 | Li et al., 2013 | https://emcore.ucsf.edu/ucsf-software |
| CTFFIND 4.1.13 | Rohou and Grigorieff, 2015 | https://grigoriefflab.umassmed.edu/ctffind4 |
| PHENIX 1.18.2-3874 | Adams et al., 2010 | https://www.phenix-online.org/ |
| PyMOL, The Molecular Graphics System 2.4.0 | Schrödinger, 2021 | https://pymol.org/2/ |
| Chimera 1.12 | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera/ |
| cisTEM 1.0.0 | Grant et al., 2018 | https://cistem.org/ |
| EMRinger | Barad et al., 2015 | https://www.phenix-online.org/ |
| NACCESS 2.1.1 | Hubbard and Thornton, 1993 | http://wolf.bms.umist.ac.uk/naccess/ |
| R-Programming Language 3.6.0 | R Core Team, 2017 | https://www.r-project.org/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Somnath Dutta (somnath@iisc.ac.in)
Materials availability
This study did not generate new unique reagents.
Data and code availability
Cryo-EM electron density maps of SARS-CoV2 spike proteins have been deposited in Electron Microscopy Data Bank (EMDB). Data identifier at EMDB for SARS-CoV2 Spike Protein structure at pH 7.4 with C1 Symmetry (Class 3) is EMD: 31096, SARS-CoV2 Spike Protein structure at pH 7.4 with C1 Symmetry (Class 9) is EMD: 31100, SARS-CoV2 Spike Protein structure at pH 7.4 with C1 Symmetry (Class 5) is EMD: 31097, SARS-CoV2 Spike Protein structure at pH 7.4 with C1 Symmetry (Class 8) is EMD: 31101, SARS-CoV2 Spike Protein structure at pH 7.4 with C3 Symmetry is EMD: 31098, SARS-CoV2 Spike Protein structure at pH 6.5 with C1 Symmetry (Class 2) is EMD: 31092, SARS-CoV2 Spike Protein structure at pH 6.5 with C1 Symmetry (Class 5) is EMD: 31095, SARS-CoV2 Spike Protein structure at pH 6.5 with C1 Symmetry (Class 4) is EMD: 31094, SARS-CoV2 Spike Protein structure at pH 6.5 with C1 Symmetry (Class 3) is EMD: 31093, SARS-CoV2 Spike Protein structure at pH 8.0 with C1 Symmetry (Class 1) is EMD: 31099, SARS-CoV2 Spike Protein structure at pH 8.0 with C1 Symmetry (Class 2) is EMD: 31102.
Experimental model and subject details
Cell lines
Expi293F cells (Female fetal kidney cell) were transfected using the manufacturer’s guidelines (Gibco, Thermo Fisher) to purify Spike ectodomain protein. Passaging of the cells was conducted at a density of 2x106 cells/ml in pre-warmed Expi293F expression media (Expi293F™ Expression Medium, Gibco, Thermo Fisher). After 24 hours, a fresh dilution of 4x106 cells/ml was prepared for the transient transfection with plasmid of interest.
Method details
Purification of recombinant S protein from mammalian cell culture
An S protein construct was prepared containing two proline mutations in the S2 subunit (S-2P) at residues 986 and 987 was synthesized at Genscript, USA (Walls et al., 2020) and expressed under control of a CMV promoter in mammalian cell culture (Expi293F™ Cells, Gibco, Thermo Fisher). Transfection of the mammalian cells (1μg Plasmid DNA per 1ml of Expi293F cells) (ExpiFectamine 293 Transfection kit, Gibco, Thermo Fisher) was performed according to the protocol of Gibco, Thermo Fisher. Culture supernatant was collected from transfected cells after 5 days and S-2P was purified by immobilized metal affinity chromatography (IMAC) using Ni-Sepharose 6 Fast flow resin (G-Biosciences). Ni-NTA beads were equilibrated with PBS (pH 7.4) (137 mM Sodium Chloride (Qualigens), 2.7 mM Potassium Chloride (Sigma Aldrich), 10 mM Di-sodium hydrogen orthophosphate (SD Fine Chemicals), 1.8 mM Potassium di-hydrogen orthophosphate (Qualigens)) followed by incubation with culture supernatant in presence of 1 mM PMSF (Phenylmethanesulfonyl Fluoride, Sigma Aldrich). The protein was eluted with a gradient concentration of Imidazole (200 mM – 500 mM) (Imidazole, Sigma Aldrich). The Ni-NTA purified protein was dialysed against Imidazole-free PBS (pH 7.4) using dialysis membrane (14 kDa cutoff, Merck). The His-tag from S protein was removed by HRV-3C protease digestion (HRV-3C clone synthesized at Genscript, USA, purified in-house). Extended protocol is described in recent literature (Malladi et al., 2020). Initially, S protein was purified in PBS buffer at pH 7.4. The protein was dialysed (Amicon Ultra-15 Centrifugal filter unit 100K, Dialysis Tubing Cellulose Membrane 14 kDa cutoff, Merck) with 25 mM Tris (pH 8.0, 7.4 and 6.5) (Tris Base, HiMedia) and concentrated (Amicon Ultra-4 Centrifugal filter unit 10K, Amicon Ultra-0.5 Centrifugal filter unit 100K) for cryo-EM grid freezing at 250C.
Sample preparation for negative staining transmission electron microscope
S protein samples at three different pH conditions (pH 8.0, pH 7.4 and pH 6.5) were analysed for overall homogeneity and distribution using conventional Negative Staining (NS) TEM. Carbon coated Cu grids (EM grid, 300 mesh, Electron Microscopy Sciences) were glow discharged for 30 sec and 3.5 μl of samples (0.1mg/ml) were added and incubated on the grid for 1.5 min. Excess buffer was blotted and negative staining was performed using 1% uranyl acetate (Uranyl Acetate 98%, ACS Reagent, Polysciences, Inc.). The samples were checked, and data acquisition was performed at room temperature using a 120 kV Tecnai T12 electron microscope. Data were collected at 120 kV using side-mounted Olympus VELITA (2Kx2K) CCD camera calibrated pixel size as 2.96 Å/pixel.
Negative staining TEM data processing
The evaluation of micrographs was done with EMAN 2.1 (Tang et al., 2007). Particles were picked manually as well as automatically. Particle co-ordinates were extracted using e2boxer.py in EMAN2.1 software. Reference free 2D classification of different projections of particle were performed using RELION 2.0 (Scheres, 2012).
Cryo-EM sample preparation and data collection
R1.2/1.3 300 mesh gold grids (Quantifoil) (Electron Microscopy Sciences) were glow discharged for 130 seconds at 20 mA before sample preparation. Freshly prepared S protein samples (3 μl) at three different pH conditions (pH 8.0, pH 7.4 and pH 6.5) were added to the glow discharged grids, incubated for 10 seconds, followed by blotting for 7.5 seconds at 100% humidity and then quickly plunged into liquid ethane using FEI Vitrobot IV plunger.
Cryo-EM data acquisition
Cryo-EM data acquisition was performed using Thermo Scientific™ Talos Arctica transmission electron microscope at 200 kV equipped with K2 Summit Direct Electron Detector. Images were collected automatically using LatitudeS automatic data collection software (Gatan Inc) at nominal magnification 42000x at the pixel size 1.17Å at specimen level. Total electron dose of about 80 e-/Å2 at the defocus range of −0.75 μm and −2.25 μm and calibrated dose of about 4 e-/Å2 per frame was subjected to the specimen. Data were recorded for 8 sec for a total of 20 frames. Around 2405, 4504 and 3166 movies were collected for S protein at pH 8.0, pH 7.4 and pH 6.5, respectively, for further data processing (Table S1).
Cryo-EM data processing
Data processing was primarily performed using RELION 3.0 (Scheres, 2012). Initially, beam-induced motion correction of the individual movies was performed using MotionCor2 software (Li et al., 2013). All the motion-corrected micrographs were manually screened using cisTEM software package (Grant et al., 2018), and the best micrographs were considered for further processing. Contrast transfer function (CTF) was estimated using CTFFIND 4.1.13 (Rohou and Grigorieff, 2015). Initially, particles were manually picked using RELION 3.0 for all three different pH (pH 8.0, pH 7.4 and pH 6.5) followed by 2D classification. Best 2D class averages were then selected as template for auto-picking for all three datasets. Automatically picked particles were extracted with box size 256 pixel, calibrated pixel size of 1.17 Å. Several rounds of 2D classification were run subsequently to clean all three datasets (Figures S2–S4). Around 10% particles from best 4-5 class averages were selected for ab-initio model generation. After several rounds of 2D classification, about 303,194 (for pH 8.0), 723,229 (for pH 7.4), and 330,534 particles (for pH 6.5) were selected for 3D classification without imposing any symmetry (C1) (Figures S2–S4).
For pH 8.0, 303194 particles were selected for subsequently three rounds of 3D Classification without imposing any symmetry (C1) to distinguish different S protein conformation (with Tikhonov regularization parameter 3) using RELION 3.0. Finally, two different conformations were observed - (i) 1-RBD up (34904 particles) and (ii) 3-RBD down (54153 particles) (Figure S2).
For pH 7.4, 723,229 particles were selected for 3D Classification without imposing any symmetry (C1) to distinguish different S protein conformation into 15 classes (with Tikhonov regularization parameter 3) using RELION 3.0. After first round of 3D classification, two 1-RBD up conformation (Class 5 – 217396 particles and Class 8 – 140411 particles) and two 3-RBD down conformation (Class 3 – 85701 particles and Class 9 – 132606 particles) were observed (Figure S3). Other 11 classes from first round of 3D Classification were merged (147115 particles) for another round of 3D Classification without imposing any symmetry (C1) to separate different conformations using RELION 3.0. Second round of 3D Classification resulted three 1-RBD up model (Class 3 – 31199 particles, Class 4 – 22068 particles and Class 6 – 62109 particles) and one 3-RBD down model (class 1- 11150 particles) (Figure S3).
To achieve a high-resolution model, at pH 7.4, 723229 particles were again selected for 3D classification with C3 Symmetry using RELION 3.0. 3D Classification resulted one 3-RBD down model (347752 particles) (Figure 4A).
For pH 6.5, 330534 particles were selected for 3D classification without imposing any symmetry (C1) (with Tikhonov regularization parameter 3) to observe different S-protein conformation. 3D Classification result showed two 1-RBD up model (Class 3 – 49730 particles and class 4 – 94237particles) and two 3-RBD down model (Class 2 – 78742 particles and Class 5 – 87829 particles) (Figure S4).
3D auto-refinement was carried out for best classes obtained from 3D classification for all three datasets (pH 8.0, pH 7.4 and pH 6.5) using soft mask in RELION 3.0. Followed by 3D auto-refinement, per particle defocus refinement with correcting beam tilt were done for each model from all three dataset (pH 8.0, pH 7.4 and pH 6.5). Particles were subjected for Bayesian polishing followed by another round of 3D auto-refinement with polished particle set (for all different models of pH 8.0, pH 7.4 and pH 6.5) using RELION 3.0.
Cryo-EM map sharpening and local resolution estimation
3D auto-refined maps were sharpened using RELION 3.0 and PHENIX (Adams et al., 2010). Fourier shell correlation (FSC) were estimated for all the maps (at pH 8.0, pH 7.4 and pH 6.5) at 0.143 (Figure S5B). Local resolution estimation was performed using unfiltered auto refine maps with ResMap (Kucukelbir et al., 2014) (Figures S5C–S5E).
Real space refinement and structural assessment
Cryo-EM maps were docked with PDB: 6vyb (Walls et al., 2020) – for 1 RBD up and PDB: 6zwv (Ke et al., 2020) - 3 RDB down). PDB: 6vyb and PDB: 6zwv models were used as initial model for phenix.dock_in_map in PHENIX. The resultant model was refined with phenix.real_space_refine with respect to the cryo-EM map in PHENIX. The structural assessment of the final phenix.real_space_refine model was done in PHENIX (Table S2). The phenix.real_space_refine models at pH 7.4 (both C1 and C3 symmetry) and pH 6.5 were fitted to respective cryo-EM density maps using UCSF Chimera (Pettersen et al., 2004). Further, phenix.real_space_refine models at pH 7.4 and pH 6.5 were aligned separately using UCSF Chimera to evaluate the difference between 1-RBD up and 3-RBD down conformations of S at different pH.
EMRinger scores and RMSD calculation
Cryo-EM maps were sharpened and atomic models were refined in PHENIX (Adams et al., 2010). EMRinger scores (Barad et al., 2015) for all cryo-EM maps were calculated using PHENIX. RMSD (aligning Cα) between different conformational states atomic models were calculated using PyMOL (Table S3). All structural analyses were performed using USCF Chimera (Pettersen et al., 2004).
Solvent accessible surface area calculation of each residue at RBD region
Solvent Accessible Surface Area (SASA) of each conformer was calculated through NACCESS (http://wolf.bms.umist.ac.uk/naccess/). Relative solvent accessible surface area calculation of each side chain residue was calculated with atomic models at pH 7.4, 6.5 and 8.0 to identify the possibility of neutralizing antibody binding. The solvent accessibility of each residue was calculated using the NACCESS program (http://wolf.bms.umist.ac.uk/naccess/). Representative amino acids responsible for neutralizing antibody binding are marked in the atomic model (monomer) using PyMOL (The PyMOL Molecular Graphics System, Version 2.0 Schrodinger, LLC.). The difference of relative solvent accessibility of neutralizing antibody binding site of different cryo-EM models represented in a heatmap generated using R (https://www.r-project.org/).
Identification of glycosylation residues
Cryo-EM map generated with C3 symmetry at pH 7.4 was docked with 6VXX_1_1_1 (Woo et al., 2020) using UCSF Chimera. The glycosylated residues were identified in the cryo-EM map and marked.
Quantification and statistical analysis
The best cryo-EM movies from pH 6.5, 7.4, and 8.0 were selected for beam-induced motion correction and beam-induced motion correction were performed using MotionCor2 software (Li et al., 2013). Micrographs were screened using cisTEM software package (Grant et al., 2018) and high-resolution (3.7-6Å) data were selected for image processing. CTF parameters were calculated for all the micrographs using CTFFIND 4.1.13 (Rohou and Grigorieff, 2015). Cryo-EM data processing was carried out using RELION 3.0 (Scheres, 2012) as described in “Cryo-EM data processing” section. Particles sorted into different classes using relion_refine 3D classification were used to determine the percentage of 1-RBD up and 3-RBD down conformations of S protein. Cryo-EM maps were sharpened using RELION 3.0 and PHENIX (Adams et al., 2010). Local resolution for high resolution maps were estimated using ResMap (Kucukelbir et al., 2014). Real space refinement was carried out in PHENIX (Adams et al., 2010). All the cryo-EM maps and PHENIX refined models were visualized in UCSF Chimera (Pettersen et al., 2004) and PyMOL (Schrödinger, 2021).
RMSD values (aligning Cα) were calculated using PyMOL (Schrödinger, 2021). Solvent Accessible Surface Area (SASA) was calculated using NACCESS (http://wolf.bms.umist.ac.uk/naccess). The Heat maps for SASA results were generated using R (https://www.r-project.org/). EM Ringer scores were calculated in PHENIX (Adams et al., 2010). The details was described in Cryo-EM map assessment.
Acknowledgments
We acknowledge the Department of Biotechnology, Department of Science and Technology (DST) and Science, and Ministry of Human Resource Development, India for funding and the cryo-EM facility at IISc-Bangalore. We acknowledge DBT-BUILDER Program (BT/INF/22/SP22844/2017) and DST-FIST (SR/FST/LSII-039/2015) for the National Cryo-EM facility at IISc, Bangalore. We acknowledge the financial support from the Science and Engineering Research Board (grant nos. SB/S2/RJN-145/2015, SERB-EMR/2016/000608, and SERB-IPA/2020/000094), DBT (grant no. BT/PR25580/BRB/10/1619/2017) for financial support for consumables, and DBT-IISc partnership program phase II for the negative staining TEM facility at Biological Sciences Division, IISc. We thank Prof. Raghavan Varadarajan for his input and suggestions in this study. We thank Ms. Sneha Bheemireddy for assisting us with the solvent-accessibility experiment.
Author contributions
S.P. and N.G. purified S protein. I.P. and S.D. prepared cryo-EM samples and data acquisition. I.P., N.S., S.M., and A.D. carried out cryo-EM image processing and model building. I.P., N.S., S.M., and S.D. analyzed the data, interpreted results, and wrote the manuscript. S.M. helped to write the manuscript and prepare the figures. S.D. designed research.
Declaration of interests
The authors declare no competing interests.
Published: April 30, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.str.2021.04.006.
Supplemental information
References
- Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barad B.A., Echols N., Wang R.Y.-R., Cheng Y., DiMaio F., Adams P.D., Fraser J.S. EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy. Nat. Methods. 2015;12:943–946. doi: 10.1038/nmeth.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes C.O., Jette C.A., Abernathy M.E., Dam K.-M.A., Esswein S.R., Gristick H.B., Malyutin A.G., Sharaf N.G., Huey-Tubman K.E., Lee Y.E., et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature. 2020;588:682–687. doi: 10.1038/s41586-020-2852-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batishchev O.V., Shilova L.A., Kachala M.V., Tashkin V.Y., Sokolov V.S., Fedorova N.V., Baratova L.A., Knyazev D.G., Zimmerberg J., Chizmadzhev Y.A. pH-dependent formation and disintegration of the influenza A virus protein scaffold to provide tension for membrane fusion. J. Virol. 2016;90:575–585. doi: 10.1128/JVI.01539-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beniac D.R., Andonov A., Grudeski E., Booth T.F. Architecture of the SARS coronavirus prefusion spike. Nat. Struct. Mol. Biol. 2006;13:751–752. doi: 10.1038/nsmb1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton D.J., Wrobel A.G., Xu P., Roustan C., Martin S.R., Rosenthal P.B., Skehel J.J., Gamblin S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature. 2020;588:327–330. doi: 10.1038/s41586-020-2772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosten C., Günther S., Preiser W., van der Werf S., Brodt H.-R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A.M., et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- Du L., He Y., Zhou Y., Liu S., Zheng B.-J., Jiang S. The spike protein of SARS-CoV—a target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009;7:226–236. doi: 10.1038/nrmicro2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elshabrawy H.A., Coughlin M.M., Baker S.C., Prabhakar B.S. Human monoclonal antibodies against highly conserved HR1 and HR2 domains of the SARS-CoV spike protein are more broadly neutralizing. PLoS One. 2012;7:e50366. doi: 10.1371/journal.pone.0050366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant T., Rohou A., Grigorieff N. cisTEM, user-friendly software for single-particle image processing. eLife. 2018;7:e35383. doi: 10.7554/eLife.35383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui M., Song W., Zhou H., Xu J., Chen S., Xiang Y., Wang X. Cryo-electron microscopy structures of the SARS-CoV spike glycoprotein reveal a prerequisite conformational state for receptor binding. Cell Res. 2017;27:119–129. doi: 10.1038/cr.2016.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson R., Edwards R.J., Mansouri K., Janowska K., Stalls V., Gobeil S.M.C., Kopp M., Li D., Parks R., Hsu A.L., et al. Controlling the SARS-CoV-2 spike glycoprotein conformation. Nat. Struct. Mol. Biol. 2020;27:925–933. doi: 10.1038/s41594-020-0479-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.-H., Nitsche A., et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard S.J., Thornton J.M. Department of Biochemistry and Molecular Biology, University College London; 1993. ‘NACCESS’, Computer Program. [Google Scholar]
- Jaimes J.A., Millet J.K., Whittaker G.R. Proteolytic cleavage of the SARS-CoV-2 spike protein and the role of the novel S1/S2 site. IScience. 2020;23:101212. doi: 10.1016/j.isci.2020.101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L., Wang N., Zuo T., Shi X., Poon K.-M.V., Wu Y., Gao F., Li D., Wang R., Guo J., et al. Potent neutralization of MERS-CoV by human neutralizing monoclonal antibodies to the viral spike glycoprotein. Sci. Transl. Med. 2014;6:234ra59. doi: 10.1126/scitranslmed.3008140. [DOI] [PubMed] [Google Scholar]
- Ke Z., Oton J., Qu K., Cortese M., Zila V., McKeane L., Nakane T., Zivanov J., Neufeldt C.J., Cerikan B., et al. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature. 2020;588:498–502. doi: 10.1038/s41586-020-2665-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchdoerfer R.N., Cottrell C.A., Wang N., Pallesen J., Yassine H.M., Turner H.L., Corbett K.S., Graham B.S., McLellan J.S., Ward A.B. Pre-fusion structure of a human coronavirus spike protein. Nature. 2016;531:118–121. doi: 10.1038/nature17200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber B., Fischer W.M., Gnanakaran S., Yoon H., Theiler J., Abfalterer W., Hengartner N., Giorgi E.E., Bhattacharya T., Foley B., et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020;182:812–827.e19. doi: 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukelbir A., Sigworth F.J., Tagare H.D. Quantifying the local resolution of cryo-EM density maps. Nat. Methods. 2014;11:63–65. doi: 10.1038/nmeth.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Mooney P., Zheng S., Booth C.R., Braunfeld M.B., Gubbens S., Agard D.A., Cheng Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods. 2013;10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Wan Y., Liu P., Zhao J., Lu G., Qi J., Wang Q., Lu X., Wu Y., Liu W., et al. A humanized neutralizing antibody against MERS-CoV targeting the receptor-binding domain of the spike protein. Cell Res. 2015;25:1237–1249. doi: 10.1038/cr.2015.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Wang P., Nair M.S., Yu J., Rapp M., Wang Q., Luo Y., Chan J.F.-W., Sahi V., Figueroa A., et al. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature. 2020;584:450–456. doi: 10.1038/s41586-020-2571-7. [DOI] [PubMed] [Google Scholar]
- Malladi S.K., Singh R., Pandey S., Gayathri S., Kanjo K., Ahmed S., Khan M.S., Kalita P., Girish N., Upadhyaya A., et al. Design of a highly thermotolerant, immunogenic SARS-CoV-2 spike fragment. J. Biol. Chem. 2020;296:100025. doi: 10.1074/jbc.RA120.016284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melero R., Sorzano C.O.S., Foster B., Vilas J.-L., Martínez M., Marabini R., Ramírez-Aportela E., Sanchez-Garcia R., Herreros D., del Caño L., et al. Continuous flexibility analysis of SARS-CoV-2 spike prefusion structures. IUCrJ. 2020;7:1059–1069. doi: 10.1107/S2052252520012725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallesen J., Wang N., Corbett K.S., Wrapp D., Kirchdoerfer R.N., Turner H.L., Cottrell C.A., Becker M.M., Wang L., Shi W., et al. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc. Natl. Acad. Sci. U S A. 2017;114:E7348–E7357. doi: 10.1073/pnas.1707304114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y.-J., Walls A.C., Wang Z., Sauer M.M., Li W., Tortorici M.A., Bosch B.-J., DiMaio F., Veesler D. Structures of MERS-CoV spike glycoprotein in complex with sialoside attachment receptors. Nat. Struct. Mol. Biol. 2019;26:1151–1157. doi: 10.1038/s41594-019-0334-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera?A visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- R Core Team . R Foundation for Statistical Computing; 2017. R: A Language and Environment for Statistical Computing. [Google Scholar]
- Rohou A., Grigorieff N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 2015;192:216–221. doi: 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres S.H.W. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger LLC . Schrödinger LLC; 2021. The PyMOL Molecular Graphics System, Version 2.4.0. [Google Scholar]
- Shang J., Ye G., Shi K., Wan Y., Luo C., Aihara H., Geng Q., Auerbach A., Li F. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;581:221–224. doi: 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W., Gui M., Wang X., Xiang Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog. 2018;14:e1007236. doi: 10.1371/journal.ppat.1007236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturman L.S., Ricard C.S., Holmes K.V. Conformational change of the coronavirus peplomer glycoprotein at pH 8.0 and 37 degrees C correlates with virus aggregation and virus-induced cell fusion. J. Virol. 1990;64:3042–3050. doi: 10.1128/jvi.64.6.3042-3050.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G., Peng L., Baldwin P.R., Mann D.S., Jiang W., Rees I., Ludtke S.J. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Toelzer C., Gupta K., Yadav S.K.N., Borucu U., Davidson A.D., Kavanagh Williamson M., Shoemark D.K., Garzoni F., Staufer O., Milligan R., et al. Free fatty acid binding pocket in the locked structure of SARS-CoV-2 spike protein. Science. 2020;370:725–730. doi: 10.1126/science.abd3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortorici M.A., Veesler D. Structural insights into coronavirus entry. Adv. Virus Res. 2019;105:93–116. doi: 10.1016/bs.aivir.2019.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turoňová B., Sikora M., Schürmann C., Hagen W.J.H., Welsch S., Blanc F.E.C., von Bülow S., Gecht M., Bagola K., Hörner C., et al. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science. 2020;370:203–208. doi: 10.1126/science.abd5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A.C., Tortorici M.A., Frenz B., Snijder J., Li W., Rey F.A., DiMaio F., Bosch B.-J., Veesler D. Glycan shield and epitope masking of a coronavirus spike protein observed by cryo-electron microscopy. Nat. Struct. Mol. Biol. 2016;23:899–905. doi: 10.1038/nsmb.3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A.C., Xiong X., Park Y.-J., Tortorici M.A., Snijder J., Quispe J., Cameroni E., Gopal R., Dai M., Lanzavecchia A., et al. Unexpected receptor functional mimicry elucidates activation of coronavirus fusion. Cell. 2019;176:1026–1039.e15. doi: 10.1016/j.cell.2018.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A.C., Park Y.-J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181:281–292.e6. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Shi W., Joyce M.G., Modjarrad K., Zhang Y., Leung K., Lees C.R., Zhou T., Yassine H.M., Kanekiyo M., et al. Evaluation of candidate vaccine approaches for MERS-CoV. Nat. Commun. 2015;6:7712. doi: 10.1038/ncomms8712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Zhang Y., Wu L., Niu S., Song C., Zhang Z., Lu G., Qiao C., Hu Y., Yuen K.-Y., et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell. 2020;181:894–904.e9. doi: 10.1016/j.cell.2020.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warwicker J. A model for pH coupling of the SARS-CoV-2 spike protein open/closed equilibrium. BioRxiv. 2020 doi: 10.1101/2020.10.31.363176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo H., Park S.-J., Choi Y.K., Park T., Tanveer M., Cao Y., Kern N.R., Lee J., Yeom M.S., Croll T.I., et al. Developing a fully glycosylated full-length SARS-CoV-2 spike protein model in a viral membrane. J. Phys. Chem. B. 2020;124:7128–7137. doi: 10.1021/acs.jpcb.0c04553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.-L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X., Tortorici M.A., Snijder J., Yoshioka C., Walls A.C., Li W., McGuire A.T., Rey F.A., Bosch B.-J., Veesler D. Glycan shield and fusion activation of a deltacoronavirus spike glycoprotein fine-tuned for enteric infections. J. Virol. 2017;92 doi: 10.1128/JVI.01628-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X., Qu K., Ciazynska K.A., Hosmillo M., Carter A.P., Ebrahimi S., Ke Z., Scheres S.H.W., Bergamaschi L., Grice G.L., et al. A thermostable, closed SARS-CoV-2 spike protein trimer. Nat. Struct. Mol. Biol. 2020;27:934–941. doi: 10.1038/s41594-020-0478-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.-Y., Huang Y., Ganesh L., Leung K., Kong W.-P., Schwartz O., Subbarao K., Nabel G.J. pH-dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC-SIGN. J. Virol. 2004;78:5642–5650. doi: 10.1128/JVI.78.11.5642-5650.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying T., Prabakaran P., Du L., Shi W., Feng Y., Wang Y., Wang L., Li W., Jiang S., Dimitrov D.S., et al. Junctional and allele-specific residues are critical for MERS-CoV neutralization by an exceptionally potent germline-like antibody. Nat. Commun. 2015;6:8223. doi: 10.1038/ncomms9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X., Zhang S., Jiang L., Cui Y., Li D., Wang D., Wang N., Fu L., Shi X., Li Z., et al. Structural basis for the neutralization of MERS-CoV by a human monoclonal antibody MERS-27. Sci. Rep. 2015;5:13133. doi: 10.1038/srep13133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y., Cao D., Zhang Y., Ma J., Qi J., Wang Q., Lu G., Wu Y., Yan J., Shi Y., et al. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat. Commun. 2017;8:15092. doi: 10.1038/ncomms15092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki A.M., van Boheemen S., Bestebroer T.M., Osterhaus A.D.M.E., Fouchier R.A.M. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012;367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.-R., Zhu Y., Li B., Huang C.-L., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T., Tsybovsky Y., Gorman J., Rapp M., Cerutti G., Chuang G.-Y., Katsamba P.S., Sampson J.M., Schön A., Bimela J., et al. Cryo-EM structures of SARS-CoV-2 spike without and with ACE2 reveal a pH-dependent switch to mediate endosomal positioning of receptor-binding domains. Cell Host Microbe. 2020;28:867–879.e5. doi: 10.1016/j.chom.2020.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Cryo-EM electron density maps of SARS-CoV2 spike proteins have been deposited in Electron Microscopy Data Bank (EMDB). Data identifier at EMDB for SARS-CoV2 Spike Protein structure at pH 7.4 with C1 Symmetry (Class 3) is EMD: 31096, SARS-CoV2 Spike Protein structure at pH 7.4 with C1 Symmetry (Class 9) is EMD: 31100, SARS-CoV2 Spike Protein structure at pH 7.4 with C1 Symmetry (Class 5) is EMD: 31097, SARS-CoV2 Spike Protein structure at pH 7.4 with C1 Symmetry (Class 8) is EMD: 31101, SARS-CoV2 Spike Protein structure at pH 7.4 with C3 Symmetry is EMD: 31098, SARS-CoV2 Spike Protein structure at pH 6.5 with C1 Symmetry (Class 2) is EMD: 31092, SARS-CoV2 Spike Protein structure at pH 6.5 with C1 Symmetry (Class 5) is EMD: 31095, SARS-CoV2 Spike Protein structure at pH 6.5 with C1 Symmetry (Class 4) is EMD: 31094, SARS-CoV2 Spike Protein structure at pH 6.5 with C1 Symmetry (Class 3) is EMD: 31093, SARS-CoV2 Spike Protein structure at pH 8.0 with C1 Symmetry (Class 1) is EMD: 31099, SARS-CoV2 Spike Protein structure at pH 8.0 with C1 Symmetry (Class 2) is EMD: 31102.