

Abstract
Objective:
To provide a comprehensive review of the ocular manifestations, outcomes, and genetic findings in patients with Coats-like retinitis pigmentosa (RP).
Design:
Multi-center, retrospective, non-consecutive case series.
Subjects:
Patients with a diagnosis of RP presenting with Coats-like exudative vitreoretinopathy between January 1, 2008 and October 1, 2019.
Methods:
Evaluation of ocular findings at RP diagnosis and at time of presentation of Coats-like exudative vitreoretinopathy, pedigree analysis, genetic testing, retinal imaging, and anatomic outcomes after treatment.
Main Outcome Measures:
Visual acuity, ophthalmoscopy, optical coherence tomography, fluorescein angiography, and identification of genetic mutations.
Results:
9 patients diagnosed with RP and presenting with Coats-like exudative vitreoretinopathy were included. Median age at time of RP diagnosis was 8 years (range, 1 – 22 years), and median age at presentation of Coats-like exudative vitreoretinopathy was 18 years (range 1 – 41 years). 7 patients were female, and 2 were male. The genetic cause of disease was identified in 6 patients. 3 patients presented with Coats-like fundus findings at the time of RP diagnosis. Exudative retinal detachment (ERD) localized to the infratemporal periphery was present in all cases, with bilateral disease observed in 7 patients. In all treated patients, focal laser photocoagulation (LP) was used to treat leaking telangiectasias and to limit further ERD expansion. Cystoid macular edema refractory to carbonic anhydrase inhibitor therapy, and ultimately amenable to treatment with intravitreal anti-vascular endothelial growth factor injection was observed in 4 patients.
Conclusion:
Coats-like vitreoretinopathy is present in up to 5% of all RP patients. The term “Coats-like RP” is colloquially used to describe this disease state which can present at the time of RP diagnosis, or more commonly develops late during the clinical course of patients with longstanding RP. Coats-like RP is distinct from Coats disease in that exudative pathology occurs exclusively in the setting of a coexisting RP diagnosis, is restricted to the infratemporal retina, can affect both eyes, and does not demonstrate a male sex bias. Given the risk of added vision loss posed by exudative vitreoretinopathy in patients with RP, a heightened awareness of this condition is critical in facilitating timely intervention.
Précis
Coats-like findings are present in 5% of patients with retinitis pigmentosa. A comprehensive characterization of this rare condition through the use of retinal imaging, and a literature review on management and patient outcomes are presented.
Introduction
Retinitis pigmentosa (RP) is a spectrum of inherited retinal disorders characterized by the progressive, widespread, and heterogenous degeneration of photoreceptors and retinal pigment epithelium (RPE).1 Coats disease is an idiopathic retinal vasculopathy characterized by unilateral aneurysmal dilation, telangiectasias, and extravascular lipid exudation within the retina and subretinal space. In 1956, Zamorani first reported pathology characteristic of both disease states affecting a single patient.2 Khan et al later coined the term “Coats-like retinitis pigmentosa” to describe this disease state,3 implicating that the exudative vitreoretinopathy observed in patients with RP is a distinct condition from that of Coats disease.
Since then, there has been a substantial increase in the number of Coats-like RP cases reported. Coats-like fundus findings are present in up to 5% of patients diagnosed with RP.3–6 Reports of serous retinal detachments complicating RP clinical course, associations with certain genotypes, and studies of affected twins add increasing evidence to support the existence of a relationship between RP and Coats-like exudative vitreoretinopathy.6–12 However, data and literature on this rare condition remain limited.
Herein, we report a series evaluating the clinical course and outcomes of 9 RP patients presenting with Coats-like exudative vitreoretinopathy. To the best of our knowledge, this study represents the largest case series and longest documented follow-up of patients with Coats-like RP reported in the literature. Moreover, we present a comprehensive characterization of the disease state through the use of multimodal retinal imaging, as well as a relevant review of the literature on the management of this rare condition in the era of modern vitreoretinal surgery and anti-VEGF therapy.
Methods
Research conducted was in compliance with the Health Insurance Portability and Accountability Act (HIPAA) and the Declaration of Helsinki, while abiding to all regional, national, and international laws of the institutions involved in this study. Every effort was made by the investigators to protect the rights of patients and families during the course of this study. This study is in concordance with the tenets of the ethics committee of each contributing center and was approved by the Institutional Review Board of the University of Michigan. Informed patient and/or parental consent was obtained for genetic testing.
Patients with a diagnosis of RP and presenting with fundus findings characteristic of Coats-like exudative vitreoretinopathy during clinical course between January 1, 2008 and October 1, 2019 were selectively included in this study. Collected data included age, anatomic sex, past medical and family history, results of genetic testing and pedigree analysis, findings from fundoscopy and examination under anesthesia, performance testing results from visual field testing and electroretinography, multimodal ocular imaging, and treatment outcomes.
Results
A total of 9 patients are included in this study, of which seven (78%) are females and two (22%) are males (Table 1). The median length of follow-up for cases in this series is 151 months (range, 45 – 349 months). All patients were born full-term and with an unremarkable birth history. Two of the patients included in this series are twin siblings and are the offspring of non-consanguineous parents.
Table 1:
Patient Demographics (n=9)
| Demographic | Number |
|---|---|
| Sex | |
| Male | 2 |
| Female | 7 |
| Age at time of RP diagnosis, years | |
| Mean (median, range) | 9.5 (7.5, 1 −22) |
| Age at time of Coats-like exudative vitreoretinopathy, years | |
| Mean (median, range) | 18.1 (18, 1 – 41) |
| Length of Follow-up, months | |
| Mean (median, range) | 166 (151, 45 – 349) |
All patients in this series demonstrated arteriolar attenuation, retinal hyperpigmentation and/or hypopigmentation, waxy optic disc pallor, and spotty RPE irregularities in both eyes on ophthalmic examination, characteristic of RP. In each case, the diagnosis of RP was made from a combined approach based on presenting patient history, examination findings, multimodal ocular imaging, visual field testing, electroretinography (ERG), pedigree analysis, and genetic testing. Seven of the nine patients (78%) presented with symptoms of nyctalopia and progressive visual field constriction. The results of visual performance testing were available for five patients, and confirmed significant peripheral constriction on Goldmann visual field testing and a substantial reduction in a-wave and b-wave amplitudes on ERG. The median age at the time of RP diagnosis was eight years (range, 1 – 22 years). A medical history of vitamin and/or mineral supplementation, including various combinations of high-dose vitamin A, vitamin C, vitamin E, lutein, beta-carotene, docosahexaenoic acid, and zinc, beginning shortly after RP diagnosis was elicited in six patients.
A positive family history of RP diagnosed in a sibling, parent, or grandparent was elicited in five patients (56%). Results of genetic testing were available for 6 patients, and pathogenic mutations were identified in 5 different genes (Table 2). Mutations in the Pre-mrna-processing-splicing factor 8 (PRPF8), Crumbs homolog 1 (CRB1), Centrosomal protein of 290 kDa (CEP290), and Retinitis pigmentosa GTPase regulator (RPGR) genes were identified in one patient each. A mutation in Cone-rod homeobox (CRX) was identified in two patients (twin female infants).
Table 2:
Clinical Characteristics of Retinitis Pigmentosa Patients with Coats-like Exudative Vitreoretinopathy (n=9)
| Patient | Genetic Cause | Age (Years) at RP Diagnosis | VA at RP Diagnosis | Age (Years) at Coats-like Changes | Disease Symmetry | VA at Coats-like Changes | Exudative Retinal Detachment | Area of Exudative Retinal Detachment | Focal Laser Y/N (#) | Cryotherapy Y/N (#) | PPV Y/N (#) | Treatment Response |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | PRPF8 c. 5804G>A, p.Arg1935His | 7.5 | 20/60 OD 20/250 OS | 9 | Bilateral (OD > OS) | 20/80 OD 20/125 OS | Y (OD) | Inferotemporal | Y (2) | N | N | Improved exudation, and small inferotemporal ERD OD |
| 2 | unknown | 14 | 20/30 OD 20/30 OS | 41 | Bilateral (OS > OD) | 20/40 OD 20/125 OS | Y (OS) | Inferotemporal | Y (6) | Y (1) | N | Improved exudation, and large inferotemporal ERD OS |
| 3 |
CRB1 c. 2290C>T: p. Arg764Cys c. 1946T>C: p. Cys460Arg |
7 | - - |
18 | Bilateral | 20/100 OD 20/125 OS | Y (OU) | Inferotemporal | Y (2) | N | N | Improved exudation, and large inferotemporal ERD with increasing SRF OU |
| 4 |
CEP290 c. 1666del, p. Ile556fs; c. 2991+1665 A>G |
9 | 20/30 OD 20/40 OS | 34 | Bilateral (OD > OS) | LP OD LP OS | Y (OD) | Inferotemporal | Y (1) | N | N | Improved exudation, and large inferotemporal ERD involving the macula OD |
| 5 |
RPGR c. 2714_2715del, p. Glu905fs |
6 | 20/100 OD 20/60 OS | 13 | Unilateral (OD) | 20/60 OD 20/50 OS | Y (OD) | Inferotemporal | Y (1) | N | N | Near completely resolved exudation |
| 6 | CRX Del of CRX gene (hom) | 1 | F + F OD F + F OS | 1 | Bilateral (OS > OD) | LP OD LP OS | Y (OS) | Inferotemporal | Y (1) | N | N | Improved exudation, and small inferotemporal ERD OS |
| 7 | CRX Del of CRX gene (hom) | 1 | F + F OD F + F OS | 1 | Bilateral | LP OD LP OS | Y (OU) | Inferotemporal | Y (1) | N | N | Improved exudation, and small inferotemporal ERD OS |
| 8 | Unknown | 18 | - - |
18 | Bilateral (OS > OD) | LP OD 20/40 OS | Y (OD) | Inferotemporal | Y (6) | Y (2) | N | Improved exudation, and retinal apposition achieved |
| 9 | Unknown | 22 | 20/40 OD 20/60 OS | 28 | Unilateral (OD) | 20/100 OD 20/50 OS | Y (OD) | Infratemporal → Expansion to Supratemporal | N | N | N | Patient elected to decline treatment |
A reported family history of Coats disease was absent in each case. However, Coats-like exudative vitreoretinopathy affecting one or both eyes was documented during the clinical course of all patients included in this series. The median age of RP patients at the time of Coats-like presentation was eighteen years (range, 1 – 41 years). Three patients (33%) presented with Coats-like fundus findings at the time of RP diagnosis. Six RP patients (67%) later developed Coats-like exudative vitreoretinopathy at a median interval of nine years (range, 1.5 – 28 years) after RP diagnosis. In this series, a Coats-like fundus is defined by the presence of aneurysmal dilation, vascular telangiectasia, intraretinal or subretinal lipid exudation, and peripheral exudative retinal detachment (ERD) on examination (Figure 1). Fluorescein angiography (FA) depicted diffuse RPE window defects, localized blockage with hypofluorescence in the area of ERD, and late leakage of telangiectatic vasculature, indicative of both retinal degeneration and exudative vitreoretinopathy (Figure 2).
Figure 1.

Wide-field fundus photographs of the right eye from patient 1 at presentation of Coats-like RP, and after treatment with focal laser photocoagulation. Fundus photograph (A) at presentation demonstrates exudative retinal detachment with overlying telangiectasias and retinal hemorrhages, with marked improvement in size of exudative retinal detachment and regression of vascular tortuosity after laser photocoagulation to the infratemporal periphery (B) at 6 month follow-up.
Figure 2.

Mid-phase and late-phase fluorescein angiography images of the left eye from patient 3 at presentation of Coats-like RP, and after treatment with laser photocoagulation and intravitreal anti-VEGF injection. Fluorescein angiography (A,B) at presentation demonstrates macular leakage and peripheral non-perfusion with hyperfluorescence of telangiectatic vessels. Fluorescein angiography at 6 month follow-up demonstrates improvement in petalloid macular leakage and near complete resolution of peripheral hyperfluorescence.
At the time of Coats-like presentation, exudative pathology was predominantly restricted to the inferior and temporal retinal quadrants in all patients. Aneurysmal dilation and intraretinal exudate were noted in seven patients (78%). Subretinal exudation, as well as telangiectasias predominantly in the retinal periphery and mid-periphery, were each present in eight patients (89%). Five of nine (56%) patients demonstrated peripheral retinal neovascularization.
All, except one patient, elected to undergo treatment at the recommendation of their respective ophthalmologist. Focal laser photocoagulation (LP) was used to treat telangiectasias, leakage, exudation, and as well as to limit the expansion of ERD. Cryotherapy after LP was performed in two patients to further limit the progression of subretinal fluid (SRF). Improved but persistent telangiectasias and exudation were noted in all treated patients, with near complete regression observed in only one case. In all treated eyes, retinal detachment remains localized to the infratemporal quadrant. The sole patient in this series who declined treatment developed worsening telangiectasias, intraretinal exudation, and eventual expansion of ERD to the superior fundus, ultimately progressing to residual light perception in the affected eye.
Of the eight treated subjects, four patients developed clinically significant cystoid macular edema (CME) impairing central vision (Table 3). Each of these patients was subsequently treated with oral acetazolamide and topical dorzolamide. Re-emergence of CME was observed in all cases. Two patients were additionally treated with topical prednisolone and intravitreal triamcinolone acetonide injection, and one patient received subtenon’s triamcinolone acetonide injection. After only subtle improvement in macular thickening observed in these three patients, steroid therapy was ultimately discontinued due to significant elevations in intraocular pressure (IOP). In light of continued progression of CME, we elected to treat with intravitreal anti-VEGF injection. All 4 patients with CME were treated with intravitreal bevacizumab injection (IVB) or intravitreal aflibercept injection (IAI) at intervals (range, six weeks - one year) determined based on the severity of observed pathology and clinical judgement of the treating ophthalmologist. A significant reduction in CME was ultimately achieved in all treated patients at most recent follow-up. Two patients treated with IVB demonstrated an eventual tachyphylaxis response with observed re-emergence of CME (Figure 3). Management in these two patients was then changed to IAI administered at the same interval, with noted improvement in CME and continued reduction in macular thickness at most recent follow-up.
Table 3:
Intravitreal anti-VEGF for the Management of Cystoid Macular Edema (n=4)
| Patient | Eye | Pre-treatment BCVA | Pre-treatment Macular Thickness | Intravitreal anti-VEGF Agent (#) | Average Treatment Frequency | BCVA at Most Recent Follow-Up | Macular Thickness at Most Recent Follow-Up |
|---|---|---|---|---|---|---|---|
| 1* | OD OS |
20/300 20/150 |
1029μm 829μm |
Bevacizumab (3), Aflibercept (3) Bevacizumab (2), Aflibercept (4) | Q 6 Weeks Q 6 Weeks |
20/80 20/25 |
795μm 353μm |
| 2 | OS | 20/800 | 981μm | Bevacizumab (3) | Q 6 Weeks | LP | 464μm |
| 3* | OD OS |
20/200 20/300 |
750μm 875μm |
Bevacizumab (9), Aflibercept (4) Bevacizumab (8), Aflibercept (4) | Q 5 Weeks | 20/125 20/150 |
612μm 437μm |
| 4 | OS | 20/40 | 311μm | Bevacizumab (5), Aflibercept (1) | Q Yearly | 20/40 | 220μm |
Initially achieved near complete resolution of macular edema with Bevacizumab treatment, followed by tachyphylaxis and eventual reemergence of CME. Treatment switched to Aflibercept at the same frequency, and CME continues to improve.
Figure 3.
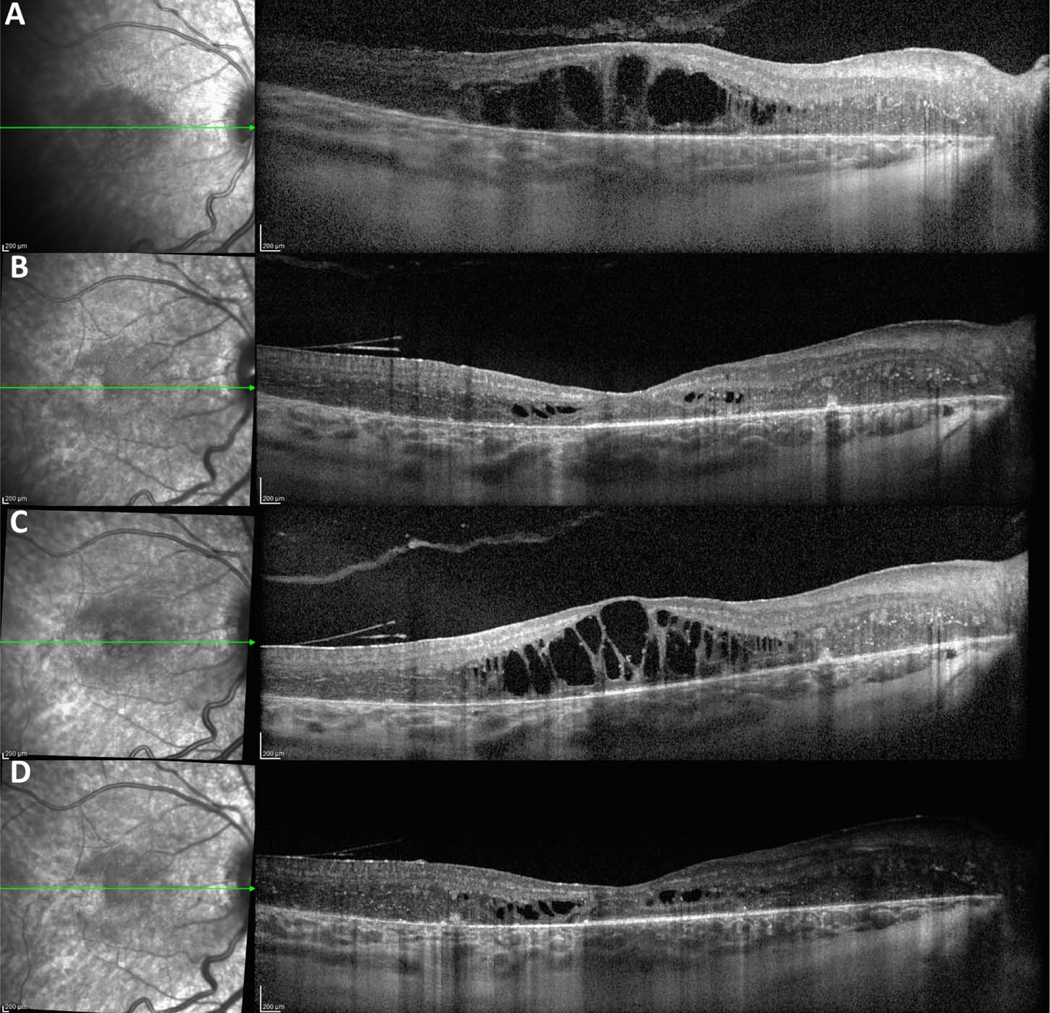
Optical coherence tomography of the right eye from patient 3 before and after treatment with intravitreal anti-VEGF injection at six week intervals. Optical coherence tomography demonstrates (A) severe cystoid macular edema at presentation of Coats-like RP, (B) near complete resolution after treatment with intravitreal bevacizumab observed at 6 month follow-up, (C) emergence of tachyphylaxis to bevacizumab and recurrence of worsening cystoid macular edema at 1 year follow-up, and (D) continued improvement at 2 year follow-up after switching to intravitreal aflibercept.
Discussion
RP is a clinically and genetically heterogeneous group of inherited retinal disorders characterized by the primary degeneration of rod photoreceptors, and the progressive secondary degeneration of cone photoreceptors and RPE.13,14 It is the most commonly inherited retinal dystrophy with an estimated prevalence of 1 in 4,000, and currently affects more than 1 million people worldwide.14,15 RP is a bilateral disease that presents with nyctalopia followed by peripheral visual field constriction, which slowly progresses to legal blindness, often by mid-adulthood, and sometimes complete blindness.14,16 On examination, arteriolar attenuation and pallor of the optic disc are commonly noted in the context of granular, bone-spiculated, and/or clumped pigmentary abnormalities observed throughout the fundus. A diagnosis of RP is often further confirmed by extinguished photoreceptor electrical activity observed on ERG, as well as genetic testing. To date, mutations in more than 80 gene loci have been identified and mapped in patients with non-syndromic RP.17,18 RP is likewise established as an association with certain syndromic disorders, believed to be attributed to aberrancies in common molecular signaling pathways during organogenesis.19,20
Interestingly, RP is also rarely associated with Coats disease. First described by George Coats in 1908, Coats disease is an idiopathic retinal disorder characterized by vascular telangiectasia, aneurysmal dilation, and lipid exudation that classically occurs unilaterally in young males.21 Coats disease is believed to stem from a breakdown in the blood-retinal-barrier of endothelial cells lining the abnormal tortuous and dilated vasculature, resulting in the subsequent spillage of blood, cholesterol, and inflammatory milieu into the retinal and subretinal space.22,23 The first reported association between RP and exudative vitreoretinopathy dates back to the previous century when Coats disease was diagnosed in a sibling of a patient with RP.3 Since Zamorani’s first description of a Coats-like RP patient in 1926,2 this rare condition has been infrequently documented in the literature.3,4,7–9,11,24–26 Studies from North America, Asia, and Europe approximate Coats-like exudative vitreoretinopathy is found in between 1% - 5% of patients with RP.3,4,27,28
Similar to that of RP itself, the precise pathophysiology of Coats-like RP remains obscure.29,30 Several mechanisms have been proposed to explain the Coats-like exudative response observed in patients with RP. Pruett proposed the accumulation of toxins produced as a byproduct of degenerating photoreceptors in RP induces a secondary vasodilatory response leading to the formation of hyperpermeable leaky vasculature.4 Khan et al theorized that early subtle microvascular leakage inherently present throughout the retinal vascular bed in patients with RP can progress to ERD, with subsequent retinal hypoxia due to separation from the choroid inducing the development of telangiectasias and neovascularization.3 More recently, Ndulue et al advanced that a Coats-like phenotype may develop as a late response to chronic retinal ischemia due to early arteriolar attenuation observed in RP eyes.31
While the nomenclature “Coats-like RP” is based on the remarkable appearance of lipid rich serous retinal detachments with accompanying overlying tortuous vasculopathy,3 it is important to consider that this condition is functionally and morphologically distinct from Coats disease. Coats disease typically presents with asymmetric vision loss in prepubertal males,32 and patients with this idiopathic disorder demonstrate diffuse exudation, telangiectasias, and subtotal ERD often affecting multiple quadrants of the retina.32,33 Conversely, Coats-like RP is a more commonly bilateral entity that occurs exclusively in the setting of a coexisting diagnosis of RP.3,4 The timing of onset of exudative vitreoretinopathy ranges from early infancy to late adulthood,2–4,7,8,24 and does not demonstrate a male sex bias. Rather, the condition demonstrates an indeterminate sex profile6 or even a slight female preponderance as presented in this series and previously reported by Khan et al.3 In consensus with our findings and previous reports,2–4,7,8,24 the presenting exudative pathology in Coats-like RP eyes is specifically localized to the inferior and temporal retinal quadrants.3,6,14 Pruett hypothesized that the preferential inferior RD observed is due to the gravitational pooling of fluid from chronically leaking vasculature in patients with Coats-like RP.4
Past studies theorized there exists a genetic predisposition towards the development of non-rhegmatogenous Coats-like retinal detachments in patients with RP.4 Jacobsen et al demonstrated disruption of CRB1 functioning impedes laminar organization of the developing human retina due to disruption of naturally occurring retinal apoptosis.34 Hollander et al later identified a CRB1 mutation spectrum in 7 patients with Coats-like RP, 24 and CRB1 mutation has since been considered to be a positive risk factor for the development of a Coats-like exudative response in RP.6,8,24 However, previous investigations have reported that CRB1 mutations are not found in the majority of patients with Coats-like RP.3,8,11,26,35–37 This is in concordance with our observation, as CRB1 mutations were identified in only two patients included in this series.
In addition, pathogenic mutations in CEP290, CRX, and RPGR have not been previously reported in patients with Coats-like RP. The finding that pathogenic mutations are present in every patient that underwent genetic testing in our series supports the idea that there exists an inherited component to Coats-like RP. The variety of genetic loci identified further demonstrates that many different mutations can result in a commonly presenting Coats-like RP phenotype. One explanation for this complex genetic phenomenon is that various mutations may individually result in the downstream disruption of a central molecular signaling event during retinal embryogenesis. Such an intricate genetic interrelationship is well established in other inherited pediatric vitreoretinopathies, such as Familial exudative vitreoretinopathy (FEVR).38,39 In FEVR, many different mutations disrupting common downstream β-catenin signaling in the nucleus have been identified as the causative etiology of impaired retinal angiogenesis and secondary exudative vitreoretinopathy.40–42 However, unlike with FEVR, cases of affected parent and offspring with Coats-like RP are yet to be reported. The absence of identifiable vertical transmission suggests the development of Coats-like RP follows a two-hit hypothesis, in which a secondary genetic event or environmental insult must take place in order for the exudative phenotype to be observed.
Patients with Coats-like RP can present with varying degrees of visual dysfunction. Peripheral vision loss may be fairly rapid due to the onset of newfound leaking vasculature and ERD, as exemplified by five patients presenting before the age of 20 in this series. In other cases, no further clinical vision loss is reported in patients with RP.43 Coats-like findings may simply be discovered incidentally, or identified late in the RP disease course as observed in six patients in our cohort. This absence of additional clinical vision loss may be attributed to ERD occurring at a peripheral site that has already undergone significant neurodegeneration and vision loss due to longstanding RP.3
Patients with Coats-like RP also often describe a steep decline in reading ability.43 The cause of this acute on chronic vision loss may be due to the development of clinically significant CME, as observed in nearly one-half of the patients in this series. The exact cause of macular edema in Coats-like RP is unknown, though it is likely due to a combination effect of the pathophysiology of CME in both Coats disease and RP. We hypothesize CME plausibly results from disruption of the blood-retinal-barrier and subsequent hyperpermeability of Coats-like tortuous vasculature,32 supplemented by the progressive failure of degenerating RPE cells to efficiently pump fluid out of the subretinal space in patients with RP.44
The early identification of Coats-like exudative vitreoretinopathy facilitates timely intervention and can therefore impede the development of early and added vision loss in patients with RP. However, complete resolution of vascular pathology and lipid exudation after treatment remains yet to be reported. Both Ide et al and Sarao et al describe marginal improvement in retinal telangiectasias at subsequent follow-up after LP treatment.8,45 Pruett et al also reported little improvement in vascular tortuosity after LP, but noted that laser treatment facilitated retinal flattening when coupled with scleral buckling (SB) and subretinal drainage.4 Sato initially reported improved exudation shortly after cryotherapy, but documented the continued development of vascular anomalies and progression to ERD 4 weeks later.35 The present case series is novel from previous literature in that we document the outcomes of LP and cryotherapy at long-term follow-up (> 6 months) in all treated patients. We report significant improvement in the degree of vascular tortuosity and exudation observed in the retinal periphery, and observe that angiographic regression is most pronounced in eyes that underwent multiple LP or cryotherapy treatments. Moreover, we report that ERD remains confined to the infratemporal periphery in treated eyes, and expands throughout the fundus if left untreated.
In regard to surgical management, both SB and pars plana vitrectomy (PPV) have been relatively unsuccessful in treating ERD in patients with Coats-like RP. Pruett reported final retinal apposition in only two out of five eyes treated with LP followed by SB and subretinal drainage.4 Kajiwara likewise reported achieving only transient retinal apposition after SB, and observed the continued progression of exudation and vascular leakage.3 Sato et al described multiple failed PPV attempts in a 5-year-old child with Coats-like RP, and documented the onset of recurrent retinal detachment and secondary glaucoma.35 We therefore refrained from using surgical intervention as a primary treatment, and instead elected to manage our Coats-like RP patients with close follow-up and interval treatment with LP and cryotherapy. We find that early intervention with LP and/or cryotherapy to further limit the progression of SRF is successful in limiting ERD to the infratemporal retina. This finding is supported by our observation that expansion of ERD beyond the inferior and temporal retinal quadrants occurred only in the single patient who declined treatment in this series.
With respect to the management of CME, there exists limited data to guide treatment selection despite the fact that macular edema is found in up to 50% of patients with RP. 46,47 Few reports have documented the course of CME specifically in Coats-like RP. Sarao described subtle improvement in CME after LP treatment at the site of inferior retinal vasculopathy.8 Demirci et al similarly administered multiple LP treatments to leaking vasculature in the temporal retinal periphery, and observed continual improvement in macular thickness on follow-up.37 De Salvo et al and Horn et al opted to combine different forms of therapies used to treat CME in RP patients. De Salvo et al initially reported minimal improvement after treatment with oral acetazolamide, topical dorzolamide, and steroid injection, but documented substantial reduction in CME after cryotherapy.11 Horn et al likewise reported no effect with IVB, but achieved significant reduction in central retinal thickness after treatment with verteporfin photodynamic therapy and intravitreal acetonide injection.48
We similarly managed CME in our treated patients using a step-wise treatment approach targeting blood-retinal-barrier dysfunction and aimed at improving fluid transport across the RPE. In consideration of evidence from prospective studies supporting the use of carbonic anhydrase inhibitors (CAI) as first-line therapy for CME in RP,49,50 we initiated treatment with oral acetazolamide and topical dorzolamide. The inhibition of intraocular carbonic anhydrase presumably results in the acidification of the subretinal space, which triggers the transport of chloride ions and fluid in to the choroidal space.44 Despite initial improvement after beginning CAI therapy, we observed CME re-emergence in all eyes. In light of this rebound macular thickening, we treated with intraocular steroids to suppress the synthesis of pro-inflammatory cytokines and inflammatory cell migration. Two patients were each treated with topical prednisolone and single intravitreal acetonide injection, and another patient received only subtenon’s triamcinolone acetonide injection. Minimal improvement in CME was observed, and corticosteroid treatment was ultimately discontinued due to steadily rising IOP. We then elected to switch to anti-VEGF therapy, given its concurrent anti-permeability and anti-angiogenic properties. All 4 patients treated with anti-VEGF injection demonstrated progressive reduction in macular thickness on OCT on continued follow-up. Intravitreal anti-VEGF injection reduced CME likely by impeding the pleiotropic impact of VEGF in mediating inflammation, vascular endothelial cell fenestration, and hemodynamic instability within the subretinal space.51
Two patients treated with anti-VEGF demonstrated an eventual tachyphylaxis response to IVB, with noted resurgence and worsening of CME. Management was switched to IAI, which resulted in improving CME at most recent follow-up. Tachyphylaxis to IVB, followed by improvement in central retinal thickness after switching to IAI, has been previously reported in patients with neovascular age-related macular degeneration52,53 and CME associated with retinal vein occlusion.54,55 However, this case series represents the first report of this phenomenon occurring in patients with Coats-like RP. Few explanations have been proposed to understand tachyphylaxis observed in patients undergoing treatment with IVB. Foorohigan et al theorized frequent injection treatment leads to the activation of macrophages that can upregulate the release of pro-angiogenic cytokines stored in vesicles, and therefore negate the impact of anti-VEGF therapy.56 Past investigations have also demonstrated that bevacizumab is detectable in serum following intravitreal injection,57 and that the amount of neutralizing antibodies present correlates positively with the duration IVB treatment.58,59 Schaal et al alternatively posed that IVB treatment leads to a reactive hyperactivation of fibroblast growth factor, amongst other inflammatory and angiogenic signaling pathways activated under retinal stress, that can overcompensate for diminished VEGF activity.60
We further advance that CME reduction after switching from bevacizumab to aflibercept may reasonably be attributed to differences in molecular structure and binding affinity between the two anti-VEGF agents. Unlike bevacizumab which is a variant of mouse anti-human VEGF,61 aflibercept is a recombinant protein composed by fusing together the binding regions of two human VEGF receptors.62,63 In light of its entirely human structure, aflibercept plausibly yields a lesser immunogenic response and therefore possess a greater likelihood of successfully reaching its molecular target. Aflibercept also has a near 100 fold greater binding affinity for VEGF-A 165,62,63 the most abundant variant of VEGF present in the eye.64 Given this exponentially greater binding affinity, Aflibercept presumably has a longer lasting therapeutic effect. Aflibercept is also unique in that it can simultaneously bind placental growth factor-2 (PIGF-2), a molecule that acts synergistically with VEGF to promote vascular leakage and inflammation.65 Aflibercept’s ability to simultaneously trap PIGF-2 provides the added advantage of suppressing other signaling pathways involved in mediating macular edema, particularly when refractory response to a VEGF specific inhibitor, such as is bevacizumab, is observed.
Despite the many findings and conclusions presented, there are notable limitations to our study. In addition to this study’s retrospective nature, the selection of patients was non-randomized and non-consecutive. Patients were identified and included only at the discretion of select ophthalmologists. Genetic testing was limited only to patients, and did not include family members. It is furthermore difficult to determine the degree of contribution of genetic mutations to the presenting Coats-like phenotypes in patients with RP. These mutations may be a chance finding, and the onset of exudative vitreoretinopathy may be attributed to undiscovered confounders in patient clinical course. Lastly, the data presented are from highly specialized tertiary eyecare referral centers, and thus the results of this research may not be generalizable to all patients with RP.
In summary, clinical literature and data on Coats-like RP are limited. Given the increased burden of blindness posed by the development of exudative vitreoretinopathy in patients with RP, a heightened awareness of this rare condition is essential. We recommend providers continue close follow up in patients diagnosed with RP, with particular attention to the temporal and inferior retinal periphery to observe for the development of new onset vascular telangiectasia, aneurysmal dilation, and ERD. We demonstrate Coats-like phenotype can present in RP patients with a wide variety of genotypes. We highlight that the time course of exudative vitreoretinopathy is variable amongst affected patients, ranging from onset in infancy to several decades after established RP diagnosis. Treatment should be initiated early and guided by the severity and location of visualized exudative pathology. Both LP and cryotherapy are effective in mediating regression of telangiectasia and restricting the expansion of ERD beyond the infratemporal periphery. In Coats-like RP patients presenting with acute on chronic vision loss, providers should maintain a high-suspicion for the development of CME. We advise beginning treatment with oral acetazolamide and topical dorzolamide. Eyes with recurrent CME can be treated with IVB injection, and providers should maintain a low-threshold for switching to IAI if tachyphylaxis is observed.
Acknowledgments
Financial Support: None
Footnotes
Conflict(s) of Interest: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Weleber RG, Gregory-Evans K. Retinitis Pigmentosa and Allied Disorders. Retina. 2006:395–498.
- 2.Zamorani G. Una rara associazone di retinite di Coats con retinite pigmentosa. G Ital Oftalmol. 1956;9:429–443. [Google Scholar]
- 3.Khan JA, Ide CH, Strickland MP. Coats’-type retinitis pigmentosa. Surv Ophthalmol. 1988;32:317–332. [DOI] [PubMed] [Google Scholar]
- 4.Pruett RC. Retinitis pigmentosa: clinical observations and correlations. Trans Am Ophthalmol Soc. 1983;81:693–735. [PMC free article] [PubMed] [Google Scholar]
- 5.Egerer I, Tasman W, Tomer TT. Coats disease. Arch Ophthalmol. 1974;92:109–112. [DOI] [PubMed] [Google Scholar]
- 6.Kan E, Yilmaz T, Aydemir O, et al. Coats-like retinitis pigmentosa: reports of three cases. Clin Ophthalmol. 2007;1:193. [PMC free article] [PubMed] [Google Scholar]
- 7.Kim RY, Kearney JJ. Coats-type retinitis pigmentosa in a 4-year-old child. Am J Ophthalmol. 1997;124:846–848. [DOI] [PubMed] [Google Scholar]
- 8.Sarao V, Veritti D, Prosperi R, et al. A case of CRB1-negative Coats-like retinitis pigmentosa. J AAPOS. 2013;17:414–416. [DOI] [PubMed] [Google Scholar]
- 9.Chebil A, El Matri L. [Retinitis pigmentosa associated with coats-like fundus]. J Fr Ophtalmol. 2016;39:235–236. [DOI] [PubMed] [Google Scholar]
- 10.Ghassemi F, Akbari-Kamrani M. Retinitis Pigmentosa Associated with Vasoproliferative Tumors and Coats-like Fundus. J Ophthalmic Vis Res. 2013;8:268–270. [PMC free article] [PubMed] [Google Scholar]
- 11.Salvo GD, De Salvo G, Gemenetzi M, et al. Cystoid macular oedema successfully treated by cryotherapy in retinitis pigmentosa with Coats’-like retinal exudation. Eye. 2011;25:821–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee S-Y, Yoon YH. Pars plana vitrectomy for exuduative retinal detachment in coats-type retinitis pigmentosa. Retina. 2004;24:450–452. [DOI] [PubMed] [Google Scholar]
- 13.Milam AH, Li ZY, Fariss RN. Histopathology of the human retina in retinitis pigmentosa. Prog Retin Eye Res. 1998;17:175–205. [DOI] [PubMed] [Google Scholar]
- 14.Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis. 2006;1:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. [DOI] [PubMed] [Google Scholar]
- 16.Fishman GA, Farber MD, Derlacki DJ. X-linked retinitis pigmentosa. Profile of clinical findings. Arch Ophthalmol. 1988;106:369–375. [DOI] [PubMed] [Google Scholar]
- 17.Xu Y, Guan L, Shen T, et al. Mutations of 60 known causative genes in 157 families with retinitis pigmentosa based on exome sequencing. Hum Genet. 2014;133:1255–1271. [DOI] [PubMed] [Google Scholar]
- 18.Anasagasti A, Irigoyen C, Barandika O, et al. Current mutation discovery approaches in Retinitis Pigmentosa. Vision Res. 2012;75:117–129. [DOI] [PubMed] [Google Scholar]
- 19.Pierrottet CO, Zuntini M, Digiuni M, et al. Syndromic and non-syndromic forms of retinitis pigmentosa: a comprehensive Italian clinical and molecular study reveals new mutations. Genet Mol Res. 2014;13:8815–8833. [DOI] [PubMed] [Google Scholar]
- 20.Bonnet C, El-Amraoui A. Usher syndrome (sensorineural deafness and retinitis pigmentosa): pathogenesis, molecular diagnosis and therapeutic approaches. Curr Opin Neurol. 2012;25:42–49. [DOI] [PubMed] [Google Scholar]
- 21.COATS G. Forms of retinal diseases with massive exudation. Roy Lond Ophthalmol Hosp Rep. 1908;17:440–525. [Google Scholar]
- 22.Chang MM, McLean IW, Merritt JC. Coats’ disease: a study of 62 histologically confirmed cases. J Pediatr Ophthalmol Strabismus. 1984;21:163–168. [DOI] [PubMed] [Google Scholar]
- 23.Shields JA, Shields CL. Review: coats disease: the 2001 LuEsther T. Mertz lecture. Retina. 2002;22:80–91. [DOI] [PubMed] [Google Scholar]
- 24.den Hollander AI, den Hollander AI, Davis J, et al. CRB1 mutation spectrum in inherited retinal dystrophies. Human Mutation. 2004;24:355–369. [DOI] [PubMed] [Google Scholar]
- 25.Jiang Y, Lim J, Janowicz M. Cholesterol Crystals Secondary to Coats-Like Response With Retinitis Pigmentosa. JAMA Ophthalmol. 2017;135:e173132. [DOI] [PubMed] [Google Scholar]
- 26.Urgancioglu B, Ozdek S, Hasanreisoglu B. Coats’-like retinitis pigmentosa variant and nanophthalmos. Canadian Journal of Ophthalmology. 2007;42:877–878. [DOI] [PubMed] [Google Scholar]
- 27.Pagon RA. Retinitis pigmentosa. Surv Ophthalmol. 1988;33:137–177. [DOI] [PubMed] [Google Scholar]
- 28.Fogle JA, Welch RB, Green WR. Retinitis pigmentosa and exudative vasculopathy. Arch Ophthalmol. 1978;96:696–702. [DOI] [PubMed] [Google Scholar]
- 29.Berson EL. Retinitis Pigmentosa and Allied Diseases. Albert & Jakobiec’s Principles & Practice of Ophthalmology. 2008:2225–2252.
- 30.Portera-Cailliau C, Sung CH, Nathans J, Adler R. Apoptotic photoreceptor cell death in mouse models of retinitis pigmentosa. Proc Natl Acad Sci U S A. 1994;91:974–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ndulue JK, Stathopoulos C, Shields CL. Retinal Vasoproliferative Tumor Secondary to Retinitis Pigmentosa. Retina Today. 2017:41–43.
- 32.Shields JA, Shields CL, Honavar SG, Demirci H. Clinical variations and complications of Coats disease in 150 cases: the 2000 Sanford Gifford Memorial Lecture. Am J Ophthalmol. 2001;131:561–571. [DOI] [PubMed] [Google Scholar]
- 33.Shields JA, Shields CL, Honavar SG, et al. Classification and management of Coats disease: the 2000 Proctor Lecture. Am J Ophthalmol. 2001;131:572–583. [DOI] [PubMed] [Google Scholar]
- 34.Jacobson SG, Cideciyan AV, Aleman TS, et al. Crumbs homolog 1 (CRB1) mutations result in a thick human retina with abnormal lamination. Hum Mol Genet. 2003;12:1073–1078. [DOI] [PubMed] [Google Scholar]
- 35.Sato T, Mimura M, Sugiyama T, et al. SENIOR–LOKEN SYNDROME COMPLICATED WITH SEVERE COATS DISEASE–LIKE EXUDATIVE RETINOPATHY. Retin Cases Brief Rep. 2007;1:172. [DOI] [PubMed] [Google Scholar]
- 36.Kıratlı H, Öztürkmen C. Coats-like lesions in Usher syndrome type II. Graefes Arch Clin Exp Ophthalmol. 2004;242:265–267. [DOI] [PubMed] [Google Scholar]
- 37.Demirci FYK, Rigatti BW, Mah TS, Gorin MB. A Novel RPGR Exon ORF15 Mutation in a Family With X-linked Retinitis Pigmentosa and Coats’-like Exudative Vasculopathy. American Journal of Ophthalmology. 2006;141:208–210. [DOI] [PubMed] [Google Scholar]
- 38.Toomes C, Bottomley HM, Jackson RM, et al. Mutations in LRP5 or FZD4 underlie the common familial exudative vitreoretinopathy locus on chromosome 11q. Am J Hum Genet. 2004;74:721–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Criswick VG, Schepens CL. Familial exudative vitreoretinopathy. Am J Ophthalmol. 1969;68:578–594. [DOI] [PubMed] [Google Scholar]
- 40.Ranchod TM, Ho LY, Drenser KA, et al. Clinical presentation of familial exudative vitreoretinopathy. Ophthalmology. 2011;118:2070–2075. [DOI] [PubMed] [Google Scholar]
- 41.Drenser KA. Wnt signaling pathway in retinal vascularization. Eye Brain. 2016;8:141–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Warden SM, Andreoli CM, Mukai S. The Wnt signaling pathway in familial exudative vitreoretinopathy and Norrie disease. Semin Ophthalmol. 2007;22:211–217. [DOI] [PubMed] [Google Scholar]
- 43.Khan JA, Ide CH, Strickland MP. Coats’-type retinitis pigmentosa. Survey of Ophthalmology. 1988;32:317–332. [DOI] [PubMed] [Google Scholar]
- 44.Strong S, Liew G, Michaelides M. Retinitis pigmentosa-associated cystoid macular oedema: pathogenesis and avenues of intervention. Br J Ophthalmol. 2017;101:31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ide CH, Khan JA, Podolsky TL, et al. Morbus Coats Typus Retinopathia pigmentosa. Klinische Monatsblätter für Augenheilkunde. 1987;190:205–206. [DOI] [PubMed] [Google Scholar]
- 46.Hajali M, Fishman GA, Anderson RJ. The prevalence of cystoid macular oedema in retinitis pigmentosa patients determined by optical coherence tomography. Br J Ophthalmol. 2008;92:1065–1068. [DOI] [PubMed] [Google Scholar]
- 47.Adackapara CA, Sunness JS, Dibernardo CW, et al. Prevalence of cystoid macular edema and stability in oct retinal thickness in eyes with retinitis pigmentosa during a 48-week lutein trial. Retina. 2008;28:103–110. [DOI] [PubMed] [Google Scholar]
- 48.Horn R, Bernstein PS. Treatment of Coats Reaction and Cystoid Macular Edema in a Retinitis Pigmentosa Patient. Invest Ophthalmol Vis Sci. 2009;50:985–985. [Google Scholar]
- 49.Fishman GA, Gilbert LD, Fiscella RG, et al. Acetazolamide for treatment of chronic macular edema in retinitis pigmentosa. Arch Ophthalmol. 1989;107:1445–1452. [DOI] [PubMed] [Google Scholar]
- 50.Fishman GA, Gilbert LD, Anderson RJ, et al. Effect of Methazolamide on Chronic Macular Edema in Patients with Retinitis Pigmentosa. Ophthalmology. 1994;101:687–693. [DOI] [PubMed] [Google Scholar]
- 51.Lux A, Llacer H, Heussen FMA, Joussen AM. Non-responders to bevacizumab (Avastin) therapy of choroidal neovascular lesions. British Journal of Ophthalmology. 2007;91:1318–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Griffin DR, Richmond PP, Olson JC. Intravitreal Aflibercept Outcomes in Patients with Persistent Macular Exudate Previously Treated with Bevacizumab and/or Ranibizumab for Neovascular Age-Related Macular Degeneration. J Ophthalmol. 2014;2014:497178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kumar N, Marsiglia M, Mrejen S, et al. visual and anatomical outcomes of intravitreal aflibercept in eyes with persistent subfoveal fluid despite previous treatments with ranibizumab in patients with neovascular age-related macular degeneration. retina. 2013;33:1605–1612. [DOI] [PubMed] [Google Scholar]
- 54.Tagami M, Sai R, Fukuda M, Azumi A. Prolongation of injection interval after switching therapy from ranibizumab to aflibercept in Japanese patients with macular edema secondary to branch retinal vein occlusion. Clinical Ophthalmology. 2017;11:403–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wirth MA, Becker MD, Graf N, Michels S. Aflibercept in branch retinal vein occlusion as second line therapy: clinical outcome 12 months after changing treatment from bevacizumab/ranibizumab—a pilot study. International Journal of Retina and Vitreous. 2016;2. [DOI] [PMC free article] [PubMed]
- 56.Forooghian F, Cukras C, Meyerle CB, et al. Tachyphylaxis after intravitreal bevacizumab for exudative age-related macular degeneration. Retina. 2009;29:723–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bakri SJ, Snyder MR, Reid JM, et al. Pharmacokinetics of intravitreal bevacizumab (Avastin). Ophthalmology. 2007;114:855–859. [DOI] [PubMed] [Google Scholar]
- 58.Rosenfeld PJ, Brown DM, Heier JS, et al. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. 2006;355:1419–1431. [DOI] [PubMed] [Google Scholar]
- 59.Forooghian F, Chew EY, Meyerle CB, et al. Investigation of the role of neutralizing antibodies against bevacizumab as mediators of tachyphylaxis. Acta Ophthalmol. 2011;89:e206–7. [DOI] [PubMed] [Google Scholar]
- 60.Schaal S, Kaplan HJ, Tezel TH. Is there tachyphylaxis to intravitreal anti-vascular endothelial growth factor pharmacotherapy in age-related macular degeneration? Ophthalmology. 2008;115:2199–2205. [DOI] [PubMed] [Google Scholar]
- 61.Gerber H-P, Wu X, Yu L, et al. Mice expressing a humanized form of VEGF-A may provide insights into the safety and efficacy of anti-VEGF antibodies. Proc Natl Acad Sci U S A. 2007;104:3478–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Papadopoulos N, Martin J, Ruan Q, et al. Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF Trap, ranibizumab and bevacizumab. Angiogenesis. 2012;15:171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stewart MW, Rosenfeld PJ. Predicted biological activity of intravitreal VEGF Trap. Br J Ophthalmol. 2008;92:667–668. [DOI] [PubMed] [Google Scholar]
- 64.Soker S, Takashima S, Miao HQ, et al. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell. 1998;92:735–745. [DOI] [PubMed] [Google Scholar]
- 65.Nguyen QD, De Falco S, Behar-Cohen F, et al. Placental growth factor and its potential role in diabetic retinopathy and other ocular neovascular diseases. Acta Ophthalmol. 2018;96:e1–e9. [DOI] [PMC free article] [PubMed] [Google Scholar]