

Abstract
Type I collagen membranes with tailored fibril nanoarchitectures were fabricated through a vitrification processing, which mimicked to a degree, the collagen maturation process of corneal stromal extracellular matrix in vivo. Vitrification was performed at a controlled temperature of either 5 °C or 39 °C at a constant relative humidity of 40% for various time periods from 0.5 wk up to 8 wk. During vitrification, the vitrified collagen membranes (collagen vitrigels, CVs) exhibited a rapid growth in fibrillar density through the evaporation of water and an increase in fibrillar stiffness due to the formation of new and/or more-stable interactions. On the other hand, the collagen fibrils in CVs maintained their D-periodicity and showed no significant difference in fibrillar diameter, indicating preservation of the native states of the collagen fibrils during vitrification. Keratocyte phenotype was maintained on CVs to varying degrees that were strongly influenced by the collagen fibril nanoarchitectures. Specifically, the vitrification time of CVs mainly governed the keratocyte morphology, showing significant increases in the cell protrusion number, protrusion length, and cell size along with CV vitrification time. The CV vitrification temperature affected the regulation of the gene expressions of keratocyte fibroblasts, including keratocan and aldehyde dehydrogenase (ALDH), demonstrating a unique way to control the expression of specific genes in vitro.
Keywords: Collagen maturation, fibril nanoarchitecture, keratocyte phenotype, corneal repair
1. Introduction
The extracellular matrix (ECM) is a complex mixture of macromolecules that provide an environment for cellular development and homeostasis unique to each tissue and organ in the body [1–3]. The cornea, in particular, has a unique ECM structure that primarily composed of type I collagen. The collagen molecules in cornea are organized at many length scales from the aggregation of triple helices followed by fibril formation at the nanoscale. Collagen fibrils are then assembled into layered lamellar plates arranged in an orthogonal orientation [4]. This fibrillar lamellar organization in the cornea contributes to the unique clarity of the tissue [5].
One remarkable feature of the ECM is a continuous change of the hierarchical structures with time as cellular development or differentiation evolves. In the embryonic corneal stroma, the matrix begins as a water-rich state and then progressively transforms into a densely packed collagen fibrillar ECM characteristic of adult corneas [6]. In contrast, progressive degeneration of the ECM in corneal stroma leads to keratoconus, which is associated with localized corneal thinning and protrusion, representing a leading reason for corneal transplantation [7, 8]. On the other hand, when the corneal stroma is injured, keratocytes, the corneal stromal primary cell type, respond readily but transition towards a fibroblast and myofibroblast phenotype and generate scar tissue [9, 10]. Therefore, the ECM architecture is both a consequence and a cause for the development, degeneration, and regeneration of the tissue.
Maintaining the keratocyte phenotype during wound healing represents a significant challenge in corneal repair and regeneration [11, 12]. Type I collagen is a natural choice employed frequently as an engineered ECM substitute for corneal repair [13]. However, conventional collagen gels are composed of loose networks of type I collagen fibrils with little organization, which is hardly comparable to the collagen structure in cornea. Compression of conventional collagen gels has been employed to increase collagen density and stiffness by applying a load onto the gel [14]. This plastically compressed collagen matrices improved cell expansion and stratification of corneal limbal epithelial cells, but still showed limited success in maintaining the cellular phenotype [15, 16]. In contrast, amniotic membrane (AM), a naturally derived ECM product used clinically to aid in corneal re-epithelialization, was able to preserve the characteristic morphology and keratocan expression of keratocytes [17]. However, amniotic membrane lacks standardization in its sources and processing method [15].
Recently, we used vitrified collagen biomaterials (collagen vitrigel, CV) developed by Takezawa et al. [18] to culture various cell types found in the cornea with the ultimate goal of developing corneal substitutes [19, 20]. CVs are synthesized from type I collagen, and could be standardized with straightforward manufacturing and handling procedures. During vitrification, collagen gels are exposed to a specific temperature and humidity that cause controlled evaporation of the water and organization of collagen fibrils. This vitrification process mimics, to some degree, the collagen maturation that occurs during the developmental formation of the corneal stromal ECM [6, 21]. More recently, we completed a design-of-experiments to evaluate the effects of time, temperature and humidity variables on collagen fibril structure, optical, mechanical, and thermal of CVs [22]. Ultimately, a variety of fibrillar structures were obtained that had a significant impact on strength and transparency. However, the effects of collagen nanoarchitectures created through vitrification process on keratocyte responses remain unknown.
The purpose of this study was to evaluate the biological response of keratocytes to CVs with varying fibrillar content and organization. We hypothesize that CVs with controlled fibrillar collagen may provide an optimal environment for maintaining the keratocyte phenotype. CV properties were manipulated by using different temperatures and processing times during vitrification. Keratocyte response was characterized by cell morphology and gene expression under either serum-free or serum-based cultures. Typically, in previous studies, keratocyte phenotype was analyzed qualitatively [11]. Here, we quantitatively examined keratocyte morphology in terms of cell size, protrusion number, and protrusion length, using a custom-designed program written in MATLAB (MathWorks®, Natick, MA) [23–25]. We also evaluated the effect of the CVs’ collagen nanoarchitectures on various keratocyte gene expressions, including the expressions of keratocan, aldehyde dehydrogenase (ALDH), and biglycan.
2. Materials and methods
2.1. Preparation of collagen membranes
Collagen membranes were prepared in a three-step procedure, as previously described [19]. First, conventional collagen gel was formed by mixing equal volumes of 0.5% type-I collagen solution (native collagen from bovine dermis, pH = 3.0, AteloCell®, Cosmo Bio Co., Ltd., Tokyo, Japan) and culture medium containing Dulbecco’s Modified Eagle’s Medium (DMEM), 10% fetal bovine serum (FBS), 20 mM hydroxyethyl piperazineethanesulfonic acid (HEPES), 100 units/mL penicillin, and 100 μg/mL streptomycin. The mixed solution of 2.0 mL was quickly poured into one well of a 6-well plate, and then incubated at 37 °C for 2 h to complete the gelation of the collagen. Second, the collagen gel was aseptically vitrified by drying under a controlled temperature and relative humidity for a prescribed time period from half week up to 8 weeks. The relative humidity of 40% was used unless otherwise specified. The collagen gel was first vitrified over 3 days and then rinsed thoroughly with PBS buffer three times for ten min each to completely remove the serum-based culture medium. The collagen gel was further vitrified until the predetermined time was achieved, leading to the formation of a rigid glasslike collagen membrane. Third, before use, the collagen membrane was thoroughly rehydrated with PBS buffer before use. The thicknesses of CVs right before and after rehydration were calculated from measuring the weight and assuming a constant density of 1 g/mL.
2.2. Transmission electron microscopy (TEM)
The nanostructure of collagen membranes was examined by TEM. Samples were fixed in 3% paraformaldehyde, 1.5% glutaraldehyde, 5mM MgCl2, 5mM CaCl2, 2.5% sucrose, and 0.1% tannic acid in 0.1 M sodium cacodylate buffer at pH 7.2 overnight at 4 °C. After buffer rinse, samples were post-fixed on ice in the dark in 1% osmium tetroxide for 1 h. Following a DH2O rinse, plates were stained with 2% aqueous uranyl acetate (0.22 μm filtered, 1 h, dark) and dehydrated in a graded series of ethanol before being embedded in Eponate 12™ resin (Ted Pella, Inc., Redding, CA). Plates were polymerized for two to three days at 37 °C before transferring them to 60 °C overnight. Thin sections, 60–90 nm, were cut with a diamond knife on the Reichert-Jung Ultracut E ultramicrotome and then placed on naked copper grids. Grids were stained with 2% uranyl acetate in 50% methanol. They were imaged with a Hitachi 7600 TEM at 80 kV. Images were captured with an AMT CCD (1k x 1k) camera (Advanced Microscopy Techniques, Woburn, MA). TEM images of both transverse and coronal planes in the CV central portions were obtained. ImageJ (NIH, Bethesda, MD) were used to analyze the collagen fibril organization in CVs, including the fibril diameter, fibril density, and D-periodicity of collagen banding. Specifically, both fibril diameter and D-periodicity of collagen banding were evaluated from ~50 collagen fibrils in the transverse plane view of the TEM images. The fibril density was obtained based on the measurements of four different locations in the coronal plane view of the TEM images.
2.3. Keratocytes isolation and culture
Full-thickness corneas were harvested from bovine eyes within 36 h after slaughter. Keratocytes were isolated using a sequential collagenase (Type 2, Worthington Biochemical Corp., Lakewood, NJ) digestion. Specifically, bovine corneas were collected and cut into small pieces with a scalpel and then transferred to a 50-mL centrifuge tube containing 3 mg/mL collagenase in DMEM/F-12. The corneal pieces were incubated on a rotary shaker at 240 rpm at 37 °C for 60 min and then collected using a 70 μm cell strainer (BD Falcon, BD Biosciences, San Jose, CA). The partially digested corneas were transferred to a fresh collagenase solution for a second digestion for 60 min. This process was repeated for a third digestion for 120 min, and the digested cells from the third digestion were collected and centrifuged at 1400 rpm for 10 min. The cell pellet was resuspended and cultured on general tissue culture plates (TCP) or CV-covered plates at 37 °C and 5% CO2. Either serum-free or serum based culture medium was used. The serum-free medium consisted of DMEM/F-12, 1% of 10 U/mL penicillin–streptomycin, and 0.5% of 1.25 mg/mL amphotericin B (Life Technologies, Carlsbad, CA). The serum-based medium contained DMEM/F-12, 10% FBS, 1% of 10 U/mL penicillin–streptomycin, and 0.5% of 1.25 mg/mL amphotericin B. The cells were plated at a concentration of 5000 cells/cm2 using the serum-free medium, while at a concentration of 1000 cells/cm2 using the serum-based medium. Before imaging and gene expression analyses, keratocytes were cultured over 3 wk in serum-free medium or 6 d in serum-based medium. Keratocyte morphologies were examined by staining with the LIVE/DEAD® Viability/Cytotoxicity Kit (Life Technologies).
2.4. AlamarBlue® assay
Proliferation of keratocytes was measured by AlamarBlue® Assay (Invitrogen, Frederick, MD). This assay employs a fluorometric/colorimetric growth indicator that changes from an oxidized (non-fluorescent, blue) form to a reduced (fluorescent, red) form when reduced by mitochondrial respiration of the cells. In this assay, the percent reduction of alamarBlue, which is determined by the ratio of the concentration of the reduced form to the total concentration of alamarBlue, is proportional to the amount of cells. Primary keratocytes were plated in 24-well plates at a concentration of 5000 cells/cm2 using the serum-free medium, or 1000 cells/cm2 using the serum-based medium. At predetermined time points, the medium was refreshed and cells were incubated with 10% alamarBlue at 37 °C and 5% CO2 for 4 hr. A sample of 200 μL of alamarBlue solution was then transferred into a 96-well plate for fluorescence reading. The absorbance was monitored at 570 nm and 600 nm using a fluorescence plate reader (Synergy 2, Biotek, Seattle, WA). The proliferation of keratocytes was determined by the increase of alamarBlue reduction at Day 4 relative to Day 2. This assay was performed in four independent experiments.
2.5. Quantification of cellular morphology
Image processing for quantification of the cellular morphological features from fluorescent images was carried out using a custom program developed in MATLAB [24, 25]. In brief, we first segmented individual cells in images and measured the cell spreading area. For the quantification of protrusion morphology, we first determined the morphological skeleton of individual cell contours and identified the main body region of cells [26]. The protrusions were identified as the skeleton structures that were extended beyond the main body region. The protrusions were further classified into two subtypes, 1st order protrusions, and 2nd order protrusions. The 1st order protrusions were considered to be the protrusions stemming directly from the cell body, and 2nd order protrusions were the ones branching from other protrusions. The length of each protrusion was measured and the total number of protrusions for individual cells was determined as the summation of 1st order and 2nd order protrusions. The morphology of keratocytes, including cell size, total number of protrusions, mean length of protrusions, primary protrusion number, secondary protrusion number, and the ratio of secondary to primary protrusions, was evaluated based on the measurements of ~60 cells.
2.6. Gene expression analysis
Total RNA was isolated from cultured keratocytes using TRIzol reagent (Life Technologies) and reverse transcribed into cDNA using SuperScript II First Strand Synthesis Kit (Life Technologies). cDNA was used for real-time PCR reactions using a StepOnePlus Real-Time PCR System (Applied Biosystems®, Life Technologies). The PCR primer sequences are listed in Table 1. GAPDH was used as a reference gene. The relative gene expression was determined using the 2−ΔΔC(T) method [27].
Table 1.
RT-PCR Primer Sequences
| Gene | Sequence (5’−3’) | Annealing Temp (°C) |
|---|---|---|
| ALDH | 60.4 | |
| Forward | GGAAGCCATCCAGTTCATCA | |
| Reverse | GTCTCCGCGATCATCTTCTT | |
| Biglycan | 62.4 | |
| Forward | ACCTCCCTGAGACCCTCAAT | |
| Reverse | TTGTTGTCCAAGTGCAGCTC | |
| Keratocan | 60.4 | |
| Forward | TGACCTGCAGCACAATAAGC | |
| Reverse | TGAGTCTCAGGAAGGCCACT | |
| GAPDH | 62.4 | |
| Forward | GGGTCATCATCTCTGCACCT | |
| Reverse | GGTCATAAGTCCCTCCACGA |
2.7. Statistics
Data were represented as averages ± standard deviations, unless otherwise specified. Two-tailed unpaired t-tests were performed to determine significance using Graphpad Prism (Graphpad Software Version 5, Graphpad Software, Inc., La Jolla, CA). P value of <0.05 was considered statistically significant.
3. Results
3.1. Collagen vitrigel preparation and nanoarchitecture
CVs were prepared following a three-stage sequence: gelation, vitrification, and rehydration (Figure 1A) [19, 22]. When the collagen solution containing 2.5 mg/mL type I collagen was first gelled at 37 °C for 2 hr, it produced a collagen gel with a thickness of 2.0 mm. This collagen gel was translucent and soft. During vitrification, the collagen gel was dried slowly at a controlled temperature of either 5 °C or 39 °C under a relative humidity of 40%. This resulted in CV formation with a dramatically decreased thickness. After vitrification at 5 °C for 2 wk, the vitrified gel had a thickness of 9 ± 1 μm, which is a 220-fold decrease in the thickness of the membrane. To compare, the CV vitrified at 39 °C for 2 wk exhibited a smaller thickness of 6 ± 1 μm, which is a 330-fold decrease in the thickness. After rehydration, the CV still maintained its shape and swelled in PBS buffer to form a hydrated membrane with a thickness of approximately 50 μm.
Fig. 1.
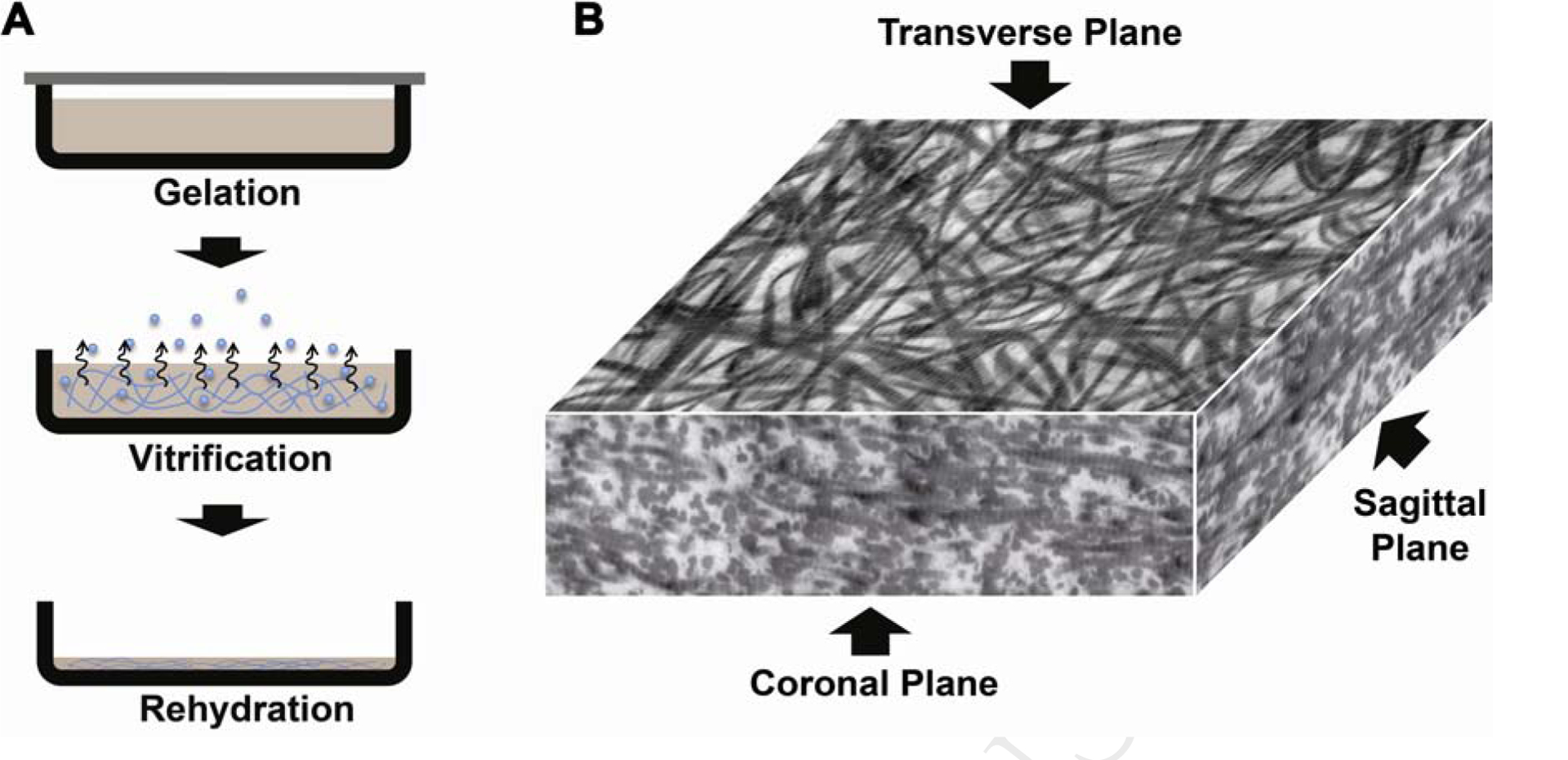
Collagen vitrigel preparation and alignment: (A) three-step procedures, and (B) TEM image of CV central portions showing 2D alignment of matured collagen fibrils by vitrification.
CVs reconstructed from highly hydrated collagen gels showed a distinctive nanoarchitecture. Fiber morphology depended on the CV conditions as visualized through TEM images in the middle portions. A three-dimensional view of a CV (5 °C, 2 wk) is shown in Figure 1B. Unlike conventional collagen gel showing homogeneous distribution of the collagen fibrils, this CV demonstrated a random fibrillar organization in the transverse plane, but showed a layer-by-layer lamellar structure in the coronal and sagittal planes. Similar results were also observed in all the other CVs, even for the CV 5 °C, 0.5 wk that was vitrified for a short time period (Figure 2A–H).
Fig. 2.
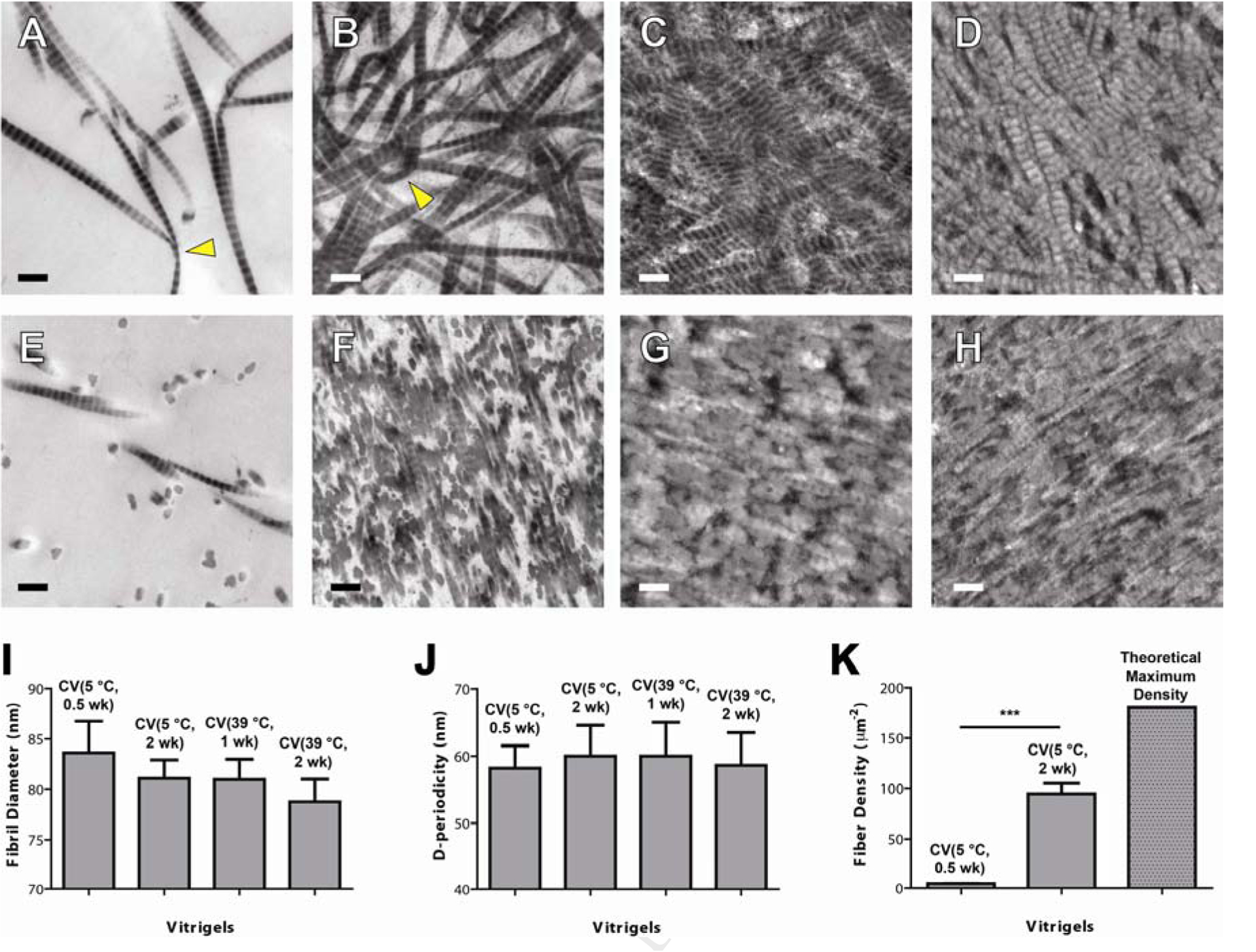
TEM images of CVs and collagen fibril analysis: (A-D) Transverse plane, (E-H) coronal plane, and (I-K) collagen fibril analyses. Four CVs were used: (A, E) CV 5 °C, 0.5 wk, (B, F) CV 5 °C, 2 wk, (C, G) CV 39 °C, 1 wk, and (D, H) CV 39 °C, 2 wk. Collagen fibril analyses include (I) fibril diameter, (J) d-periodicity (banding), (K) fibril density. The TEM samples were obtained from the center portions of the collagen membranes. Collagen fibrils with large bending curvature were indicated by triangle. The error bars in (I, J) represent the standard deviation of the mean. Scale bar in (A-H): 200 nm.
The organization of the collagen fibrils was strongly affected by the vitrification processing conditions, including the temperature and the vitrification time. First, no significant difference in collagen fibrillar diameter was observed despite of varying vitrification temperature and vitrification time (Figure 2I). Second, all of the samples clearly showed the characteristic collagen banding with spacing around 60 nm (Figure 2J). No significant difference in D-periodicity was observed at different temperatures and vitrification times. Third, at a constant temperature, the collagen fibril density dramatically increased along with the vitrification time (Figure 2K). The fibril density of the CV at 5 °C for 0.5 wk is as low as 5 fibers/μm2. After vitrification at 5 °C for 2 wk, the fibril density was dramatically increased to 95 fibers/μm2. For the CVs prepared at 39 °C, the fibril density was very high and close to the maximum density of the fiber in the collagen matrix, which was estimated to be 180 fibers/μm2 based on an assumption of hexagonally arranged collagen fibrils without fibril-fibril spacing for a CV with fibril diameter of 80 nm. Therefore, the collagen fibril density increased with vitrification temperature, which is consistent with the decreased membrane thickness as we observed above. Furthermore, collagen fibrils with large bending curvature decreased in the CVs prepared at a higher temperature and/or longer vitrification time when comparing CV 5 °C, 2wk vs. CV 39 °C, 2wk; CV 5 °C, 0.5 wk vs. CV 5 °C, 2 wk; and CV 39 °C, 1 wk vs. CV 39 °C 2 wk (Figure 2A–D).
3.2. Morphology and proliferation of subcultured keratocytes
The CV nanoarchitecture modulated keratocyte morphological response. Typical morphologies of keratocytes cultured on these CVs are shown in Figure 3A–H. Characteristic stellar phenotype of cultured keratocytes was observed using serum-free medium (Figure 3A–D). Compared to the TCP control, the keratocytes cultured on CVs had more and longer protrusions, as well as more intercellular connections. The number and length of keratocyte protrusions increased on the CVs processed for longer vitrification times. Moreover, more pinning points were observed in the extended protrusions of the keratocytes cultured on the CV processed for a longer time, especially on the CV 39 °C, 8 wk (Figure 3A–D).
Fig. 3.
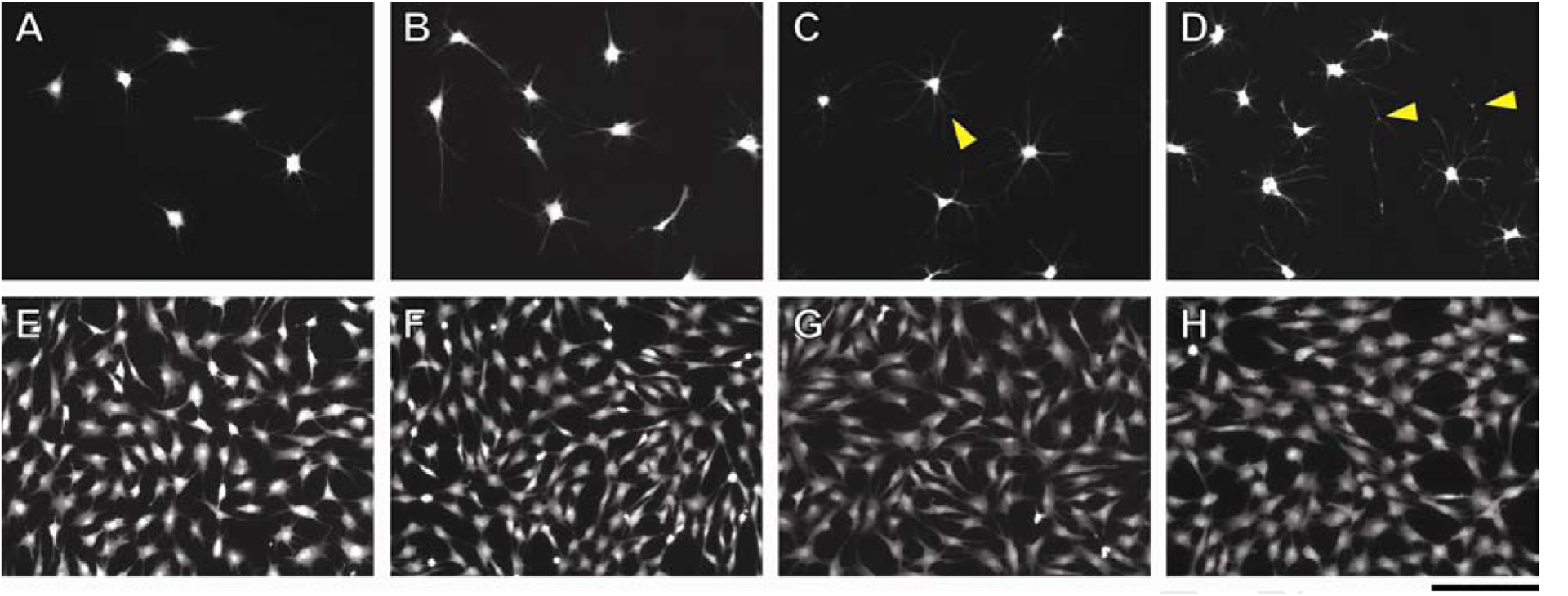
Keratocyte morphologies cultured on CVs using two different culture media: (A-D) serum-free culture medium, and (E-H) culture medium with 10% FBS. Four cell culture substrate conditions were used: (A, E) TCP control (B, F) CV 39 °C, 1 wk, (C, G) CV 39 °C, 2 wk, and (D, H) CV 39 °C, 8 wk. Pinning points in cell extensions were highlighted by triangle. Scale bar: 200 μm.
Keratocyte morphology under serum-free culture was analyzed quantitatively with a custom-designed program written in MATLAB [24, 25]. Figure 4A–C illustrates an example of a keratocyte cultured on CV 39 °C, 8 wk. Individual cellular contours were first obtained identification of the cell body and protrusions (Figure 4B). The cell size was determined as the area within the cellular contour, including cell body and protrusions. Based on the cellular contour, the morphological skeleton was determined and the protrusions were classified into two subtypes depending on their starting position. Primary protrusions, or 1st order protrusions, were defined as the longest protrusions stemming directly from the cell body, while secondary protrusions, or 2nd order protrusions, were those branching from the primary protrusions.
Fig. 4.
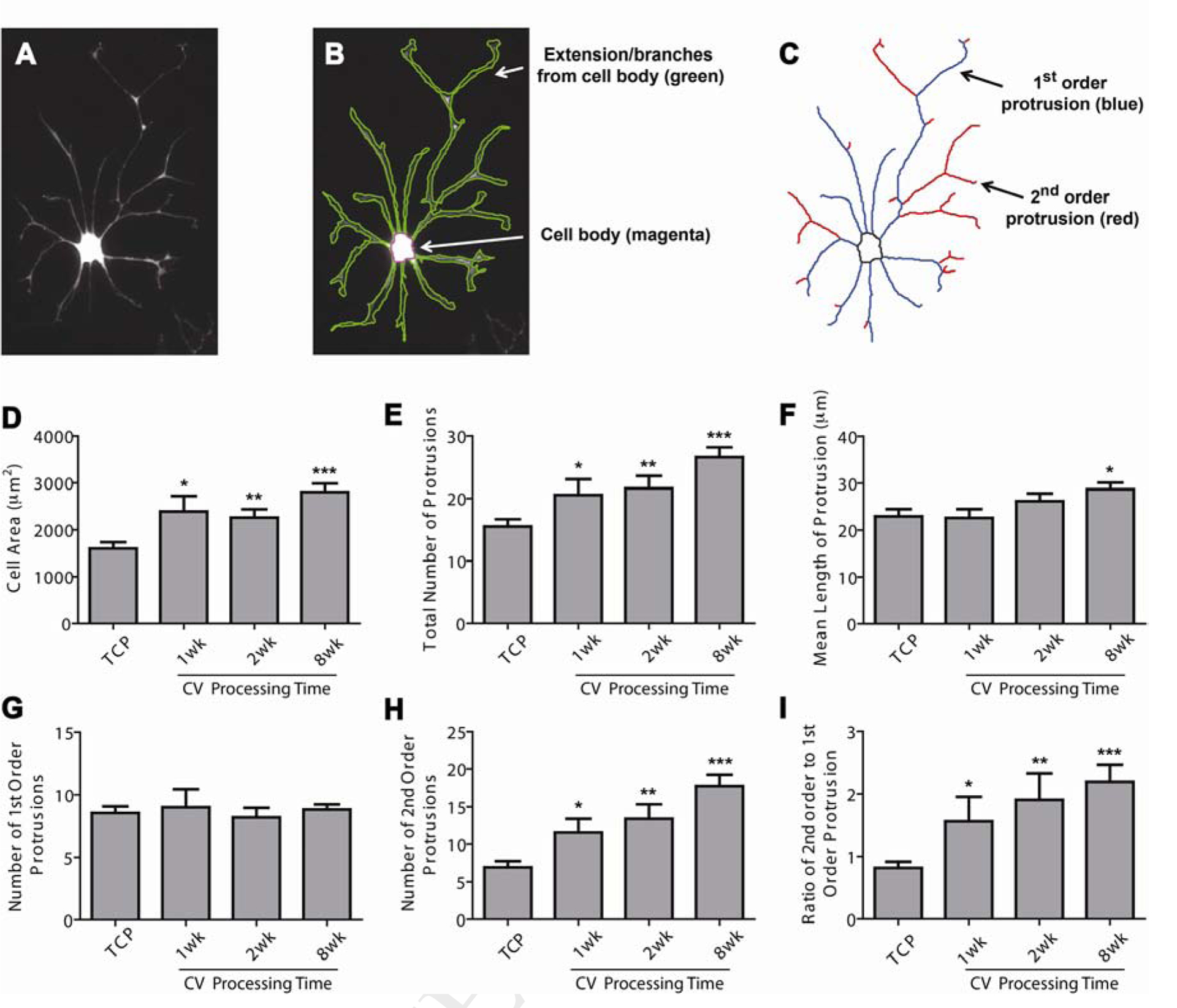
Morphological analysis of keratocytes cultured on CV vitrified at 39 °C using serum-free medium. (A-C) A keratocyte example cultured on CV (39 °C, 8 wk) (A) with cellular contour identified in (B) and morphological skeleton acquired in (C). Based on cellular contour and skeleton, the dependence of keratocyte morphologies on CV vitrification processing time was examined (D-I), including cell size (D), total number of protrusions (E), mean length of protrusions (F), primary protrusion number (G), secondary protrusion number (H), and the ratio of secondary to primary protrusions (I). The error bars in (D-I) represent the standard deviation of the mean. Keratocytes cultured on the TCP were tested as a control. All statistical differences against the TCP control are indicated. *p < 0.05, **p < 0.01, ***p < 0.001.
Keratocyte morphologies were carefully quantified, including cell size, total number of protrusions, mean length of protrusions, number of primary protrusions, number of secondary protrusions, and the ratio of secondary to primary protrusions. The cell sizes of keratocytes cultured on CVs were significantly higher than those cultured on TCP (Figure 4D), which is related to the increased number and length of protrusions (Figure 4E–F). Figure 4E shows that the number of protrusions per each keratocyte increased with the vitrification time of the CVs. Keratocytes cultured on CV 39 °C, 8 wk exhibited on average 27 protrusions per cell, a 70% increase over the average number of protrusions of those cells cultured on TCP. A similar, but smaller trend was also observed in the mean length of keratocyte protrusions. Interestingly, no significant difference was observed in the primary, or 1st order, protrusion number of the keratocytes cultured on TCP and CVs (Figure 4G). In contrast, the average number of secondary, or 2nd order, protrusions of the keratocytes increased markedly from 7 protrusions in TCP, to 12 protrusions in CV (39 °C, 1 wk), to 14 protrusions in CV (39 °C, 2 wk), and to 18 protrusions in CV (39 °C, 8 wk) (Figure 4H). A similar trend to the secondary protrusion number was also observed in the ratio of secondary to primary protrusions, further demonstrating that extent of branching/number of secondary protrusions per primary protrusion increased as a function of vitrification time (Figure 4I).
The cellular proliferation rate, determined by alamar blue, changed depending on the CV structure/morphology (Figure 5). Under serum-free culture and compared to the TCP control, a significant increase in the keratocyte population was indicated on the three CVs with highly condensed collagen fibrils, including CV (5 °C, 2 wk), CV (39 °C, 1 wk), and CV (39 °C, 2 wk). Moreover, when comparing CV (5 °C, 0.5 wk) vs CV (5 °C, 2 wk), and CV (39 °C, 1 wk) vs. CV (39 °C, 2 wk), it is evident that the cells tended to proliferate more on the CVs that had a longer vitrification time. Under serum-based culture, the keratocytes exhibit a fibroblastic phenotype (Figure 3E–H). No significant differences in the cellular morphology or proliferation were observed in any of CVs. Nevertheless, these keratocyte fibroblasts maintained their phenotypic markers to varying degrees based on the collagen nanoarchitectures of CVs.
Fig. 5.
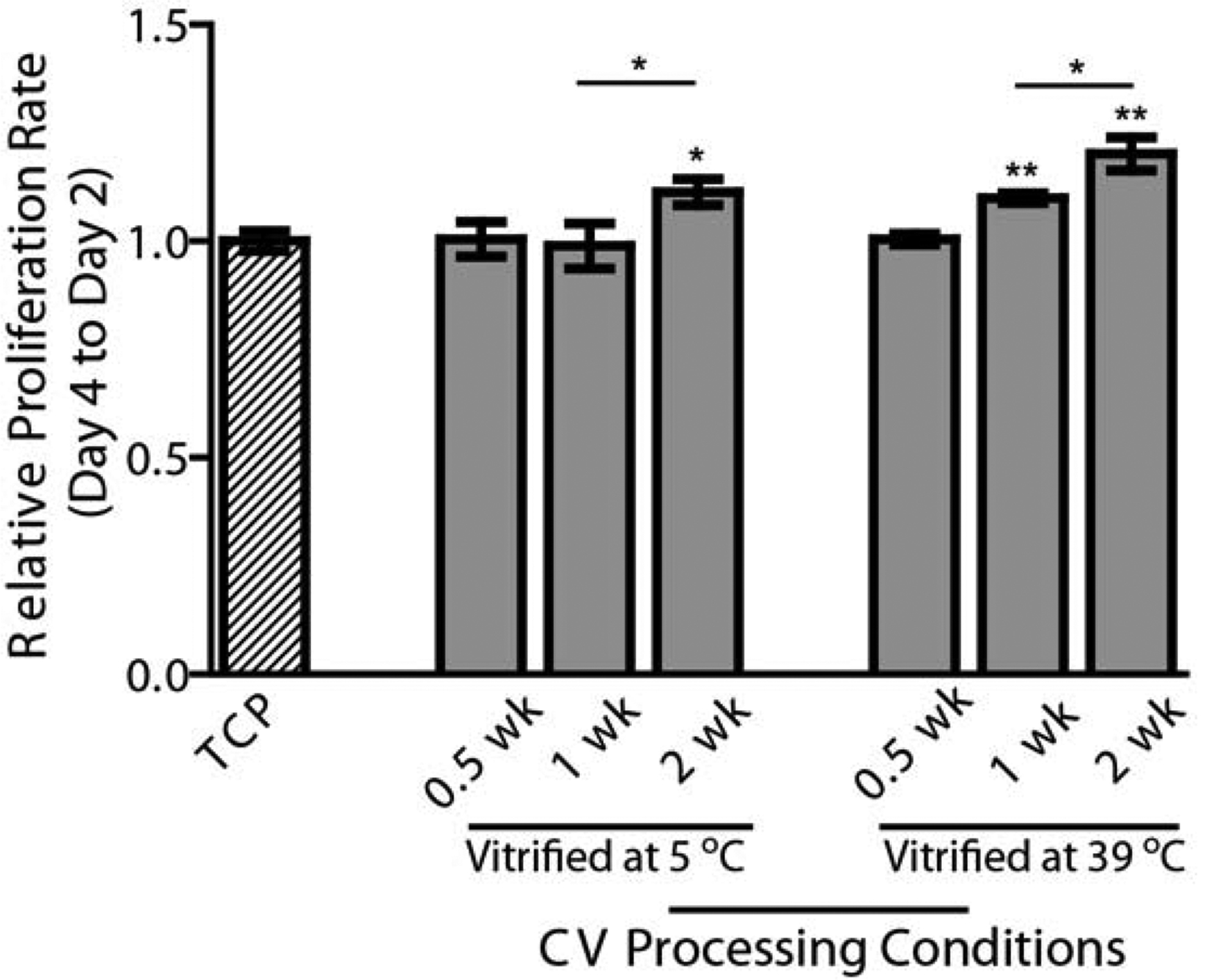
Effects of CV processing conditions on the proliferation of primary keratocytes cultured in serum-free medium. The CVs were processed from 0.5 wk up to 2 wk at two different temperatures (5 °C and 39 °C). The proliferation of keratocytes at Day 4 relative to Day 2 was assessed using an AlamarBlue assay and normalized by the TCP control. *p < 0.05, **p < 0.01.
3.3. Keratocyte gene expression dependence on collagen nanoarchitecture
Keratocyte gene expression, including keratocan, ALDH, and biglycan, was tested using CVs with different nano-architectures. Under serum-based culture, keratocytes lost characteristics of their native phenotype in TCP. Compared to TCP, the gene expression of biglycan was reduced significantly using all four types of CVs (Figure 6). For CVs prepared at 5 °C, the expression of keratocan was slightly lower using the CV processed for half week than in TCP, but markedly increased when using the CV processed for a longer period of two weeks. The expressions of ALDH in cells cultured on both CVs vitrified at 5 °C were dramatically increased compared to in those cultured on TCP. For CVs prepared at 39 °C, the expression of ALDH was slightly lower when employing CV vitrified for one week than that on TCP, but markedly increased when using CV vitrified for a longer period of two weeks. The expressions of keratocan in cells using both CVs prepared at 39 °C were greatly raised compared to those using TCP.
Fig. 6.
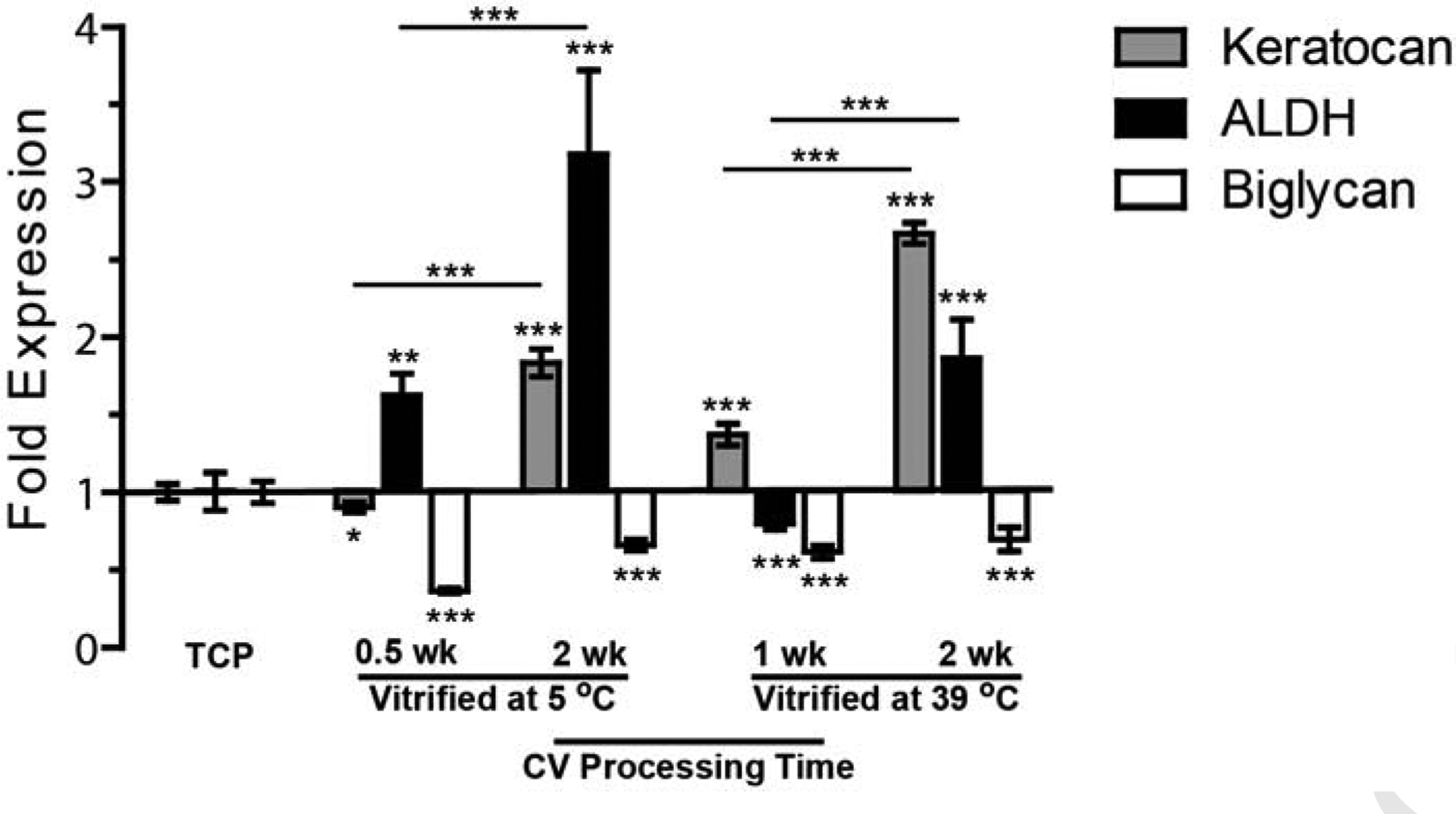
Gene expression of keratocytes fibroblasts seeded on CVs processed under different conditions and cultured in serum-based medium. All statistical differences are indicated with the ones against TCP control showing above each bar, and the others comparing CV groups portrayed on lines. *p < 0.05, **p < 0.01, ***p < 0.001.
4. Discussion
Vitrification provides a convenient means to manipulate the nanoarchitecture of collagen membranes. Lamellar structures were formed in these vitrified collagen membranes. During vitrification, collagen fibril density was dramatically increased, which led to a decrease of fibril-to-fibril distance and contributed to the enhanced transparency and mechanical properties reported previously [22]. In addition, fewer collagen fibrils with large bending curvature were observed in the CVs prepared under longer vitrification times, indicating that the collagen fibrils became stiffer during vitrification. This may be explained by new and/or more stable interactions formed in the collagen fibrils through the vitrification process, which is consistent with our previous results showing increased thermal stability of CVs with vitrification time [22, 28]. No significant difference was observed in the collagen fibril dimension and periodic banding distance. The maintenance of collagen banding periodicity suggests that the CVs were not denatured during the vitrification process, even under the high temperature of 39 °C that is above the normal human body temperature range. This is because the denaturing temperatures of these CVs we observed previously were between 54–59 °C, much higher than the CV processing temperature [22]. The processing of collagen membranes by vitrification likely resembled, to some extent, the collagen maturation in embryonic corneal stromal development because condensed collagen lamellae composed of collagen fibrils were formed with new and stable interactions [6].
The flexibility in collagen ultrastructure of CVs provided a vehicle to investigate the effect of nanofibrillar matrix on cellular responses. In this study, the organized collagen membranes supported the growth of keratocytes in serum-free medium when cells generally do not proliferate. On the other hand, cells proliferating under serum-based culture on the CVs were able to maintain a keratocyte phenotype instead of dedifferentiating into fibroblasts. CVs prepared using longer vitrification times were preferable, as they offered an extracellular environment that more closely mimicked the natural corneal stroma. Human corneal stroma has been reported to consist of collagen fibrils with a diameter of 36 nm at a high density of about 700 fibers/μm2 [29]. In our study, the highest collagen fibril density in the four CVs studied almost reached a maximum of 180 fibers/μm2, assuming no fibril-fibril spacing. Our fibril diameter, at about 80 nm or above (Figure 2), was more than twice that in naturally occurring corneal stroma. Thus, creating a further increase in collagen fibril density will require significantly reducing the collagen fibril diameter. Collagen fibril condensation could also be achieved by plastic compression of conventional collagen gels, but to an even lesser degree as compared to vitrification [14].
Quantitative analysis of the effect of collagen nanoarchitecture on keratocyte phenotype is a potentially powerful method to correlate cell morphology and gene expression/phenotype. No systematic studies have been reported in which the effect of different extracellular environments on the keratocyte phenotype was quantified. This is important when designing and testing new biomaterials for corneal reconstruction. We found that the secondary protrusions of keratocytes played a critical role in defining the phenotype. In contrast, their primary protrusion numbers remained constant at different CV processing conditions. Both total protrusion number and length increased significantly with increased vitrification time. This suggests that the collagen membranes consisting of highly condensed collagen fibrils provided a proper environment for the formation of characteristic dendritic morphology of keratocytes.
Keratocytes also maintained their phenotype characteristics in CVs under serum-based culture. Typically, primary keratocytes express a low level of biglycan, but high levels of keratocan and ALDH. In particular, keratocan is one of the four proteoglycans found in the adult corneal stromal ECM that plays an important role in maintaining fibrillar ECM nanostructures and stromal transparency [6]. ALDH is the main water-soluble protein in mammalian corneas and provides protection against ultraviolet radiation and reactive oxygen-induced damages in ocular tissues [30]. In contrast, biglycan is a sensitive marker that is associated with the fibroblastic transition of the keratocytes during pathological remodeling of the corneal stroma [9]. Both down-regulation of biglycan and up-regulation of keratocan and ALDH were observed using CVs, especially in those with longer vitrification times. The CVs prepared at 5 °C promoted higher gene expression of ALDH higher than of keratocan, whereas the CVs prepared at 39 °C favored the up-regulation of keratocan more than of ALDH. This may result from different types of interactions that were formed between the collagen molecules during the vitrification process at different temperatures, which is a unique phenomenon observed in collagen processing. Many techniques have been reported to manipulate collagen organizations. For instance, collagen matrix stiffness has been modified by changing collagen concentration [31], tuning crosslinking density [32, 33], or employing a compressive load [16]. Collagen fibril alignment has been controlled by applying a shear force [12, 34]. Nevertheless, none of these techniques mimic the process of collagen maturation as observed in vitrification of collagen membranes showing continuous formation of new interactions between collagen molecules inside collagen fibrils.
The utility of vitrification processing to tailor the nanoarchitecture of ECM or biomimetic biomaterials may be useful for various clinical applications. For instance, keratoconus is a disease where the corneal stromal matrix becomes disrupted and loses this fine structural organization. The application of local vitrification in the diseased cornea might offer a route to reorganizing the stromal matrix and eliminating corneal protrusion. In addition, the CV with a biomimetic collagen nanoarchitecture holds great potential as a corneal substitute that supports the growth of keratocytes and preserves the cellular phenotype. Similar biomaterials can also be designed to use as potential substitutes for other connective tissues such as cartilage, skin, and muscle.
5. Conclusion
This study fabricated CVs with unique engineered biomaterial scaffolds that provided an ex vivo template mimicking the native environment of corneal stroma. The well-organized collagen matrices developed in CVs were obtained through a highly controlled vitrification processing method. This collagen vitrification process resembled, to some extent, the normal collagen maturation in corneal stroma. This processing technique also provided collagen scaffolds with variable nanoarchitectures that may offer a method to study collagen degeneration and thereby be helpful in elucidating the etiology of the diseases related to a changing ECM. As illustrated in this study, the highly condensed collagen matrices in CVs allowed the proliferation of keratocytes, even without serum; and with serum, they helped to maintain the characteristic phenotypes of the cells. We suggest that with further development, CVs may be used to create a corneal substitute and guide corneal repair by placing an acellular scaffold into a defect, allowing migration of the patient’s cells into this nanostructure.
Acknowledgments
We acknowledge the Congressionally Directed Medical Research Program under the U.S. Army Medical Research and Materiel Command (Contract No. W81XWH-09-2-0173, Program Manager Dr. Charmaine Richman), Arthritis Foundation Postdoctoral Fellowship (QG), the Jules Stein Professorship from Research to Prevent Blindness (RPB) Foundation, and the NIH (CA143868 and CA85839). We thank Vince Beachley for his critical review of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Rosso F, Giordano A, Barbarisi M, Barbarisi A. From cell-ECM interactions to tissue engineering. J Cell Physiol 2004;199:174–80. [DOI] [PubMed] [Google Scholar]
- [2].Berrier AL, Yamada KM. Cell-matrix adhesion. J Cell Physiol 2007;213:565–73. [DOI] [PubMed] [Google Scholar]
- [3].Guilak F, Cohen DM, Estes BT, Gimble JM, Liedtke W, Chen CS. Control of stem cell fate by physical interactions with the extracellular matrix. Cell Stem Cell 2009;5:17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ruberti JW, Zieske JD. Prelude to corneal tissue engineering - gaining control of collagen organization. Prog Retin Eye Res 2008;27:549–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Meek KM. Corneal collagen - its role in maintaining corneal shape and transparency. Biophys Rev 2009;1:83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hassell JR, Birk DE. The molecular basis of corneal transparency. Exp Eye Res 2010;91:326–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Romero-Jimenez M, Santodomingo-Rubido J, Wolffsohn JS. Keratoconus: a review. Cont Lens Anterior Eye 2010;33:157–66; quiz 205. [DOI] [PubMed] [Google Scholar]
- [8].Sugar J, Macsai MS. What causes keratoconus? Cornea 2012;31:716–9. [DOI] [PubMed] [Google Scholar]
- [9].Funderburgh JL, Mann MM, Funderburgh ML. Keratocyte phenotype mediates proteoglycan structure: a role for fibroblasts in corneal fibrosis. J Biol Chem 2003;278:45629–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].West-Mays JA, Dwivedi DJ. The keratocyte: corneal stromal cell with variable repair phenotypes. Int J Biochem Cell Biol 2006;38:1625–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Beales MP, Funderburgh JL, Jester JV, Hassell JR. Proteoglycan synthesis by bovine keratocytes and corneal fibroblasts: maintenance of the keratocyte phenotype in culture. Invest Ophthalmol Vis Sci 1999;40:1658–63. [PubMed] [Google Scholar]
- [12].Muthusubramaniam L, Peng L, Zaitseva T, Paukshto M, Martin GR, Desai TA. Collagen fibril diameter and alignment promote the quiescent keratocyte phenotype. J Biomed Mater Res A 2012;100:613–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Liu Y, Gan L, Carlsson DJ, Fagerholm P, Lagali N, Watsky MA, et al. A simple, cross-linked collagen tissue substitute for corneal implantation. Invest Ophthalmol Vis Sci 2006;47:1869–75. [DOI] [PubMed] [Google Scholar]
- [14].Brown RA, Wiseman M, Chuo CB, Cheema U, Nazhat SN. Ultrarapid engineering of biomimetic materials and tissues: fabrication of nano- and microstructures by plastic compression. Adv Funct Mater 2005;15:1762–70. [Google Scholar]
- [15].Mi S, Chen B, Wright B, Connon CJ. Ex vivo construction of an artificial ocular surface by combination of corneal limbal epithelial cells and a compressed collagen scaffold containing keratocytes. Tissue Eng Part A 2010;16:2091–100. [DOI] [PubMed] [Google Scholar]
- [16].Jones RR, Hamley IW, Connon CJ. Ex vivo expansion of limbal stem cells is affected by substrate properties. Stem Cell Res 2012;8:403–9. [DOI] [PubMed] [Google Scholar]
- [17].Espana EM, He H, Kawakita T, Di Pascuale MA, Raju VK, Liu CY, et al. Human keratocytes cultured on amniotic membrane stroma preserve morphology and express keratocan. Invest Ophthalmol Vis Sci 2003;44:5136–41. [DOI] [PubMed] [Google Scholar]
- [18].Takezawa T, Ozaki K, Nitani A, Takabayashi C, Shimo-Oka T. Collagen vitrigel: a novel scaffold that can facilitate a three-dimensional culture for reconstructing organoids. Cell Transplant 2004;13:463–73. [DOI] [PubMed] [Google Scholar]
- [19].McIntosh Ambrose W, Salahuddin A, So S, Ng S, Ponce Marquez S, Takezawa T, et al. Collagen vitrigel membranes for the in vitro reconstruction of separate corneal epithelial, stromal, and endothelial cell layers. J Biomed Mater Res B Appl Biomater 2009;90:818–31. [DOI] [PubMed] [Google Scholar]
- [20].Puleo CM, McIntosh Ambrose W, Takezawa T, Elisseeff J, Wang TH. Integration and application of vitrified collagen in multilayered microfluidic devices for corneal microtissue culture. Lab Chip 2009;9:3221–7. [DOI] [PubMed] [Google Scholar]
- [21].Bailey AJ, Paul RG, Knott L. Mechanisms of maturation and ageing of collagen. Mech Ageing Dev 1998;106:1–56. [DOI] [PubMed] [Google Scholar]
- [22].Calderon-Colon X, Xia Z, Breidenich JL, Mulreany DG, Guo Q, Uy OM, et al. Structure and properties of collagen vitrigel membranes for ocular repair and regeneration applications. Biomaterials 2012;33:8286–95. [DOI] [PubMed] [Google Scholar]
- [23].Wu PH, Hung SH, Ren T, Shih Ie M, Tseng Y. Cell cycle-dependent alteration in NAC1 nuclear body dynamics and morphology. Phys Biol 2011;8:015005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chen WC, Wu PH, Phillip JM, Khatau SB, Choi JM, Dallas MR, et al. Functional interplay between the cell cycle and cell phenotypes. Integr Biol 2013;5:523–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chambliss AB, Wu PH, Chen WC, Sun SX, Wirtz D. Simultaneously defining cell phenotypes, cell cycle, and chromatin modifications at single-cell resolution. FASEB J 2013;27:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gonzalez RC, Woods RE. Digital image processing. 2nd ed. Boston, MA: Addison-Wesley Longman Publishing Co., Inc; 1992. [Google Scholar]
- [27].Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- [28].Xia ZY, Calderon-Colon X, Trexler M, Elisseeff J, Guo QY. Thermal denaturation of type I collagen vitrified gels. Thermochim Acta 2012;527:172–9. [Google Scholar]
- [29].Muller LJ, Pels E, Vrensen GF. The effects of organ-culture on the density of keratocytes and collagen fibers in human corneas. Cornea 2001;20:86–95. [DOI] [PubMed] [Google Scholar]
- [30].Chen Y, Thompson DC, Koppaka V, Jester JV, Vasiliou V. Ocular aldehyde dehydrogenases: protection against ultraviolet damage and maintenance of transparency for vision. Prog Retin Eye Res 2013;33:28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Plant AL, Bhadriraju K, Spurlin TA, Elliott JT. Cell response to matrix mechanics: focus on collagen. Biochim Biophys Acta 2009;1793:893–902. [DOI] [PubMed] [Google Scholar]
- [32].Rafat M, Li F, Fagerholm P, Lagali NS, Watsky MA, Munger R, et al. PEG-stabilized carbodiimide crosslinked collagen-chitosan hydrogels for corneal tissue engineering. Biomaterials 2008;29:3960–72. [DOI] [PubMed] [Google Scholar]
- [33].Mason BN, Starchenko A, Williams RM, Bonassar LJ, Reinhart-King CA. Tuning three-dimensional collagen matrix stiffness independently of collagen concentration modulates endothelial cell behavior. Acta Biomater 2013;9:4635–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Saeidi N, Sander EA, Zareian R, Ruberti JW. Production of highly aligned collagen lamellae by combining shear force and thin film confinement. Acta Biomater 2011;7:2437–47. [DOI] [PMC free article] [PubMed] [Google Scholar]