

Abstract
Abnormalities in CHRNA7, the alpha7-nicotinic receptor gene, have been reported in autism spectrum disorder. These genetic abnormalities potentially decrease the receptor’s expression and diminish its functional role. This double-blind, placebo-controlled crossover study in two adult patients investigated whether an investigational receptor-specific partial agonist drug would increase the inhibitory functions of the gene and thereby increase patients’ attention. An electrophysiological biomarker, P50 inhibition, verified the intended neurobiological effect of the agonist, and neuropsychological testing verified a primary cognitive effect. Both patients perceived increased attention in their self-ratings. Alpha7-nicotinic receptor agonists, currently the target of drug development in schizophrenia and Alzheimer Disease, may also have positive clinical effects in autism spectrum disorder.
Introduction
An emerging body of genetic and molecular research indicates that CHRNA7, the chromosome 15 gene that codes for the alpha7-nicotinic cholinergic receptor, is involved in autism spectrum disorder (ASD). Noteworthy genomic features include genome-wide significant linkage at the CHRNA7 locus in 70 multi-generational Utah autism pedigrees (Allen-Brady et al. 2010), significant homozygous haplotype sharing at the same locus in autism spectrum disorder, consistent with the recessive model used in the Allen-Brady et al. analysis (Casey et al. 2012), as well as the finding of the significant prevalence of chromosome 15q copy number variants that encompass CHRNA7 in autism patients (Pinto et al. 2010). Promoter single nucleotide polymorphisms, which we had previously characterized in schizophrenia, are now reported in autism spectrum disorder patients as well (Leonard et al. 2002; Bacchelli et al. 2015). Molecular features include significant decrease in CHRNA7 mRNA in post mortem specimens of frontal cortex (Yasui et al. 2011) and significant decrease of alpha-bungarotoxin binding, a ligand for alpha7-nicotinic receptors, in the thalamus (Ray et al. 2005).
In schizophrenia, where there are an identical range of deficits involving CHRNA7, alpha 7-nicotinic receptor activation by an investigational agonist results in increasing performance on tasks requiring focused attention (Olincy et al. 2006). The goal of this initial Phase 1 investigation was to determine if similar effects occur in ASD and to assess preliminarily whether the patient’s self-rating of his attention and social abilities would improve.
Neuropsychological deficits in attention on tasks ranging from the continuous performance test to digit symbol coding appear to be one of the basic cognitive deficits of ASD as well as schizophrenia, with deficits of similar order of magnitude in ASD to deficits in social performance (Mayes and Dickerson 2008; Souliéres et al. 2011; Spek et al. 2011). Deficits in attention-dependent tasks are generally greater than in any other neurocognitive domain (Mayes and Dickerson 2008; Souliéres et al. 2011). Spek et al. noted that covariance with patients’ deficit in neuropsychological tests of basic attention accounted nearly completely for their increased performance on the Embedded Figure Test, a higher level cognitive test frequently used in ASD (Baron-Cohen 2004). Yoran-Hegesh et al. (2009) interpreted deficits on digit symbol coding as consistent with Turner’s (1999) hypothesis of impaired inhibition in ASD.
As this trial was the first to administer an alpha7-nicotinic agonist in patients with ASD, a biomarker was chosen to assess whether the experimental drug indeed was having its intended biological effect. Decreased response to repeated stimuli is a fundamental property of the nervous system, which demonstrates the engagement of inhibitory neuronal mechanisms. A cerebral evoked response (P50, a positive potential 50 ms post stimulus) was used to assess nervous system response. Specifically, alpha 7-nicotinic receptors on cerebral inhibitory neurons, when activated by the agonist, increase the activity of the interneurons, which in turn inhibit the response of cerebral pyramidal neurons to auditory stimuli (Freedman 2014). If the drug failed to engage this inhibitory mechanism, then assessment of its effects on cognitive and more complex symptoms would be meaningless. Inhibition of P50 to repeated stimuli is abnormal in some but not all patients with ASD (Kemner et al. 2002; Orekhova et al. 2008; Magnée and Oranje 2009; Stroganova et al. 2013). Its use in this study was solely to detect the neurobiological effect of the drug, not as an underlying phenotype for the illness itself.
Methods
Subjects
Both patients were receiving psychological services at a specialized ASD clinic. Patients completed Module 4 of the Autism Diagnostic Observation Schedule, Second Edition (ADOS-2) and cognitive testing within 3 months of entry into the study. Patient 1 was a 50-year-old male with 12th grade education. Medications included metformin, gabapentin, levothyroxine, lovestatin, perphenazine, and glipizide. The Wechsler Abbreviated Scale of Intelligence, Second Edition was administered and the following standard scores were obtained: Verbal Comprehension, 89; Perceptual Reasoning, 75; and Full Scale IQ, 81. Patient 2 was a 24-year-old male with 9th grade education. Medications were escitalopram and mirtazapine. The Wechsler Adult Intelligence Scale, Fourth Edition was administered and the following standard Scores were obtained: Verbal Comprehension, 93; Perceptual Reasoning, 111; Working Memory, 86; Processing Speed, 81; and General Ability Index, 98. Both had received this initial evaluation as part of their ongoing clinical treatment, not for research purposes. Neither met clinical criteria for Attention Deficit Disorder.
Drug Substance
3-(2,4-dimethoxybenzylidene)anabaseine (DMXB-A) is a selective partial agonist for alpha7-nicotinic receptors. It has a favorable safety profile and can be given orally. DMXB-A was synthesized at the University of Florida and encapsulated in Colorado as previously described (Olincy et al. 2006). The clinical trial was conducted under Food and Drug Administration Investigational Drug Exemption (FDA IND 57,710).
Procedure
After granting informed consent, the patients received physical examinations, screening clinical chemistry, hematology, electrocardiography, medical history, urine toxicology, and physical examination. The study is registered on ClinicalTrials.gov (NCT02111551).
On each experimental day they received DMXB-A 75, 150 mg, or placebo. All treatments were administered as identical-appearing capsules, and only the pharmacist was aware of drug identity. The order of administration was randomized. Subject 1 received DMXB-A 75 mg, DMXB-A 150 mg, and placebo on successive days. Patient 2 received placebo DMXB-A 75 mg, and DMXB-A 150 mg on successive days. Patients received a second half dose of 37.5 or 75 mg at 2 h, the half-life of the first dose. All experimental procedures were performed within 3 h of administration, when previous studies using this regimen have shown that the plasma levels are in a plateau phase of elimination.
In addition to the safety of a single administration of a drug in its early human trials, single day administration of DMXB-A has biological rationale. The alpha7-nicotinic receptor is a ligand-gated ion channel, and therefore it is fully activated by a single dose of drug to open an ion channel into the neuron and thereby to activate it. Constant exposure to the drug inactivates the receptor by causing it to enter a semi-stable desensitized state. Therefore, all primary biological effects are expected in a single dose trial, and none are expected to increase with increasing drug exposure.
Assessments
Because the adaptation of the subjects to the experimental environment was unknown and the elimination of DMXB-A is relatively quick, a brief testing session was planned. The primary neurocognitive measure of outcome was the Attention Scale on the Repeatable Battery for the Assessment of Neuropsychological Status (RBANS). This instrument was chosen because of its previously demonstrated responsiveness to DMXB-A in the Phase 1 schizophrenia trial (Olincy et al. 2006). Versions A, B, and C were used in random order. The testing was limited to Attention to shorten the battery, because Attention is a primary cognitive deficit in ASD, and because it is also the Index that was most responsive to DMXB-A in prior clinical trials (Olincy et al. 2006). The electroencephalographic recording of P50 auditory evoked responses was then performed, followed by the Continuous Performance Test-Identical Pairs version (CPT-IP). The patients completed two self-rating scales, the Social Responsive Scale-2 (SRS-2) and the Connors Adult Attention Rating Scale-Self Report: Screening Version (CAARS-S:SV).
P50 evoked response recording followed the protocol used in the Phase 1 trial of DMXB-A in schizophrenia (Olincy et al. 2006). Electroencephalographic (EEG) activity was recorded from a single gold-disc electrode affixed to the vertex of the head, referenced to the right ear. Activity was filtered (−3 dB) below 1 and above 1000 Hz, with a 60 Hz notch filter. Simultaneously, electro-oculographic (EOG) activity was recorded from the left lateral canthus, referenced to the forehead. The activity was digitized at 1000 Hz. The auditory stimulus was generated by a 0.04 ms square wave amplified from 20 Hz to 12 kHz and delivered through earphones. The resultant sound lasts about 10 ms. The subject’s threshold for this stimulus was determined in each ear, and the stimulus for each ear was set to 50 dB above this threshold. Neither subject had hearing abnormalities. Stimuli were delivered with intra-pair interval 0.5 s and inter-pair interval 10 s. The patients were recorded nearly supine in a relaxation chair with their necks full supported. Five sets of 16 pairs of stimuli were delivered. A technician, blind to drug assignment, monitored the EEG for slow waves indicative of drowsiness, and for signs of startle or eye movement in response to the sound. The recording was then interrupted, and the patient was aroused or the sound level was decreased as necessary. Any trial in which either signal exceeded baseline by more than 30 mcv was automatically discarded.
Each of the five sets of 16 trials was averaged separately. Any set of trials in which the magnitude of the vertex EEG P50 response failed to exceed the magnitude of the EOG response was discarded. Primate experiments have demonstrated that different cerebral pathways are engaged by stimuli which elicit eye movements, compared to those which do not, and therefore we limit the intensity of the sound to prevent this orienting response (Griffith et al. 1995). The common technique of covarying the EEG signal for the EOG signal does not correct for this difference in physiological response.
The remaining sets of trials were then averaged together, 3–5 per subject, to obtain a grand average, which was subjected to recursive digital filtering (−10 dB below 10 Hz and above 100 Hz). A computer algorithm detected P50 as the most positive wave between 30 and 70 ms following each stimulus and measured its amplitude relative to the immediate preceding more negative wave.
Following the experimental procedures on each day, the subjects received physical examinations and medical testing to ensure that there were no adverse effects requiring medical attention. The patients also completed a side effects questionnaire for any symptoms.
Results
Tolerability and Safety
Patients felt well on all 3 days of their study. Both patients were able to tolerate the medication and completed all three arms of the study. There were no serious adverse effects and no changes in laboratory values, the electrocardiogram, vital signs or physical examination. The only side effect was moderate nausea at the higher dose for Patient 1, which responded to food.
Biomarker Response
The primary biological marker of DMXB-A’s effect was inhibition of the P50 response to repeated sounds, measured as the ratio of the amplitude of the P50 response to the second sound relative to the first (Fig. 1). The ratio is more closely related to CHRNA7 and to the effects of DMXB-A than the amplitude of response to either the first (S1) or second (S2) stimulus (Freedman et al. 1997; Olincy et al. 2006). Ninety-five percent of healthy adults have ratios ≤0.50 (Freedman et al. 1997). Both patients had elevated ratios in response to placebo [Patient 1, ratio 0.99 (S1 2.34 mcV, S2 mcV 2.32); Patient 2, ratio 0.61 (S1 2.92 mcV, S2 1.77 mcV)]. DMXB-A 75 mg decreased Patient 1’s ratio to 0.62 (S1 2.79 mcV, S2 1.74 mcV), still above the healthy range, and did not change Patient 2’s ratio (ratio 0.63, S1 1.68 mcV, S2 1.05 mcV). DMXB-A 150 mg decreased both ratios into the healthy adult range [Patient 1 ratio 0.29 (S1 5.50 mcV, S2 mcV 1.58); Patient 2 ratio 0.03 (S1 1.58 mcV, S2 mcV 0.05)]. The decrease in ratio included decrease in the amplitude of response to the second stimulus, consistent with an increase in neuronal inhibition.
Fig. 1.
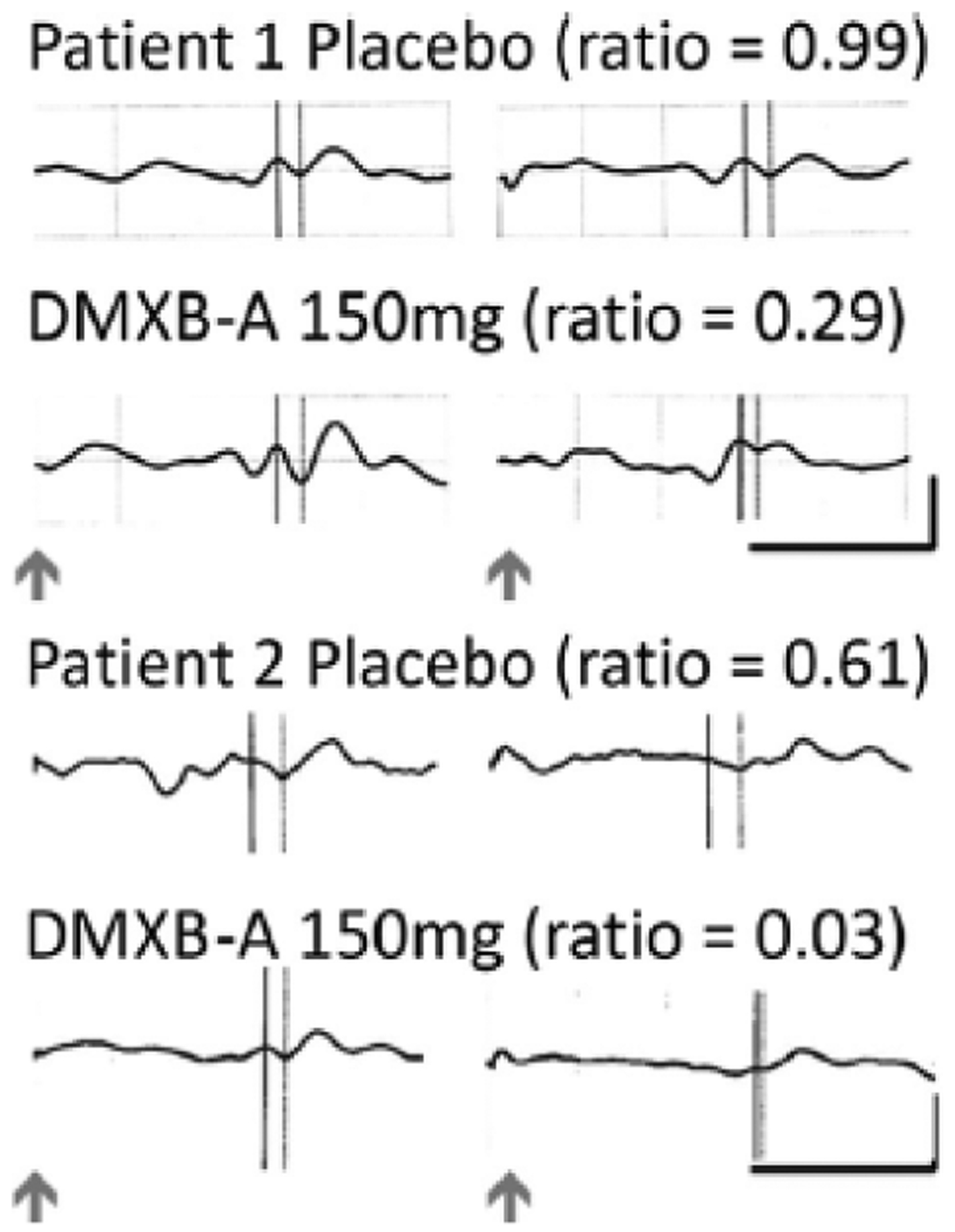
P50 audtiory-evoked responses at the vertex (Cz) to repeated sounds for Patients 1 and 2 demonstrate increased inhibition of the response to the second stimulus during DMXB-A administration. The inhibitory ratio is the amplitude of the P50 response to the second sound divided by the first. Arrows indicate delivery of the sounds, which were 0.5 s apart. P50 was measured at the dotted line relative to the preceding negativity shown by the solid line. The vertical calibration is −5 mcV and the horizontal is 100 ms
Neuropsychological Performance and Self-Rating
The RBANS Attention Standard Score was selected as a measure of attention. The score increased during DMXB-A administration for both patients, reflecting improved performance. As with the P50 ratio biomarker, the effect was more prominent at the 150 mg DMXB-A dose. Improvement with DMXB-A 150 mg was observed in both the Digit Span and Symbol Coding tests that comprise the Index (Table 1). Patients rated their ability to pay attention on the CCARS-S:SV. CAARS-S:SV T-scores decreased with increasing dose of DMXB-A, consistent with decreasing self-appraisal of attentional dysfunction. Improvement was more marked in the self-rating of symptoms of inattention.
Table 1.
Neuropsychological performance and self-ratings
| DMXB-A mg | RBANS attention | CAARS-S:SV | SRS-2 | |
|---|---|---|---|---|
| Patient 1 | 0 | 82 | 90 | 76 |
| 75 | 91 | 82 | 72 | |
| 150 | 91 | 73 | 70 | |
| Patient 2 | 0 | 60 | 71 | 72 |
| 75 | 68 | 63 | 71 | |
| 150 | 82 | 61 | 72 |
The CPT-IP showed significant order effects, with considerable improvement in signal detection over the three testing days, regardless of treatment. This order effect was not observed on any other measure.
Self-Rating of Social Dysfunction
Patient 1 slightly decreased the self-rating of social responsiveness symptoms with increasing DMXB-A dose, with changes in all areas, most notably in Social Cognition, Social Motivation, and Restricted Interests and Repetitive Behavior. Patient 2 had no decrease in self-ratings.
Discussion
DMXB-A exhibited the biological and neuropsychological effect in two adult patients with ASD that it had previous exhibited in pre-clinical animal models and in Phase 1 trials in schizophrenia. The inhibition of the cerebral evoked response to repeated sounds in animal models is produced by activation during the first response of inhibitory interneurons, whose prolonged activity then inhibits the response to the second sound. Alpha7-nicotinic receptors are abundantly expressed on interneurons of the hippocampus in both rodents and humans, where the P50 evoked response and its rodent analogue are partly generated. Receptor activation by the alpha7-nicotinic agonist increases the interneurons’ inhibitory activity. Presumably the increased sensory inhibition allows the patients to increase the focused attention required in the Digit Span and Symbol Coding tests of the RBANS Attention Index (Freedman 2014). However, alpha7-nicotinic receptors in human brain are also prominently expressed on the inhibitory neurons of the nucleus reticularis thalamis, and therefore the effects on attention may also reflect more widespread effects on the thalamo-cortical circuitry (Ray et al. 2005).
With the establishment of effects on both the biomarker and a related basic neurocognitive function in both subjects, the question of relevance to patients with autism spectrum disorder could then be addressed. More subjective effects were not expected to change dramatically after a single day’s administration even with full biological effects, but they are the ultimate therapeutic goals. Both patients reported an increased ability to pay attention, but there was little change in social responsiveness.
The dose-responsiveness, side effects, and safety profile were quite similar to what was observed in schizophrenia, which indicates no specific safety or effectiveness concerns related to this drug in autism spectrum disorder. In schizophrenia, these effects in Phase 1 were predictive on longer-term effects on negative symptoms—anhedonia, alogia, avolition, and apathy—in Phase 2 (Freedman et al. 2008). Whether or not longer-term effects of alpha7-agonist therapy would occur in ASD requires similar longer-term testing. The generalizability of any therapeutic effects on attention to the social dysfunction of ASD patients is unknown. There was little change in ratings of social function during the brief exposure to drug in this trial. It is noteworthy that passive insensitivity to the stimulus of faces, rather than gaze aversion, has been documented as a cause of diminished attention to others’ eyes in a recent study of toddlers with ASD (Moriuchi et al. 2016). Thus, it is possible that the effects of cognitive deficits in attention have a developmental role that would not be addressed by a pharmacological intervention in adulthood.
Nicotinic receptors have received only limited consideration as a therapeutic target in ASD. Patients with ASD have long been held not to smoke as excessively as patients with schizophrenia, although a case example of nicotine-seeking pica, also seen in schizophrenia, has been reported (Bejerot and Nylander 2003; Piazza et al. 1996). The nicotinic receptor agonist varenicline has been observed to have beneficial effects on behavior, and nicotine transdermal patches have been used for agitated patients (Mostafavi et al. 2016; Van Schalkwyk et al. 2015). The non-receptor specific, channel-blocking agent mecamylamine, which blocks primarily non-alpha7 nicotinic receptors, had no therapeutic effect (Arnold et al. 2012). A theoretical paper proposed that alpha7-nicotinic agonists like DMXB-A might be effective in ASD (Deutsch et al. 2010). The effects of DMXB-A on ASD are not unexpected, given the considerable evidence for pro-cognitive effects of nicotine in other conditions, such as schizophrenia and minimal cognitive impairment related to aging (Harris et al. 2004; Newhouse et al. 2012).
The trial was stopped after 8 months with the enrollment of only two subjects for lack of funding and subject interest. Generalizability of these observations to other patients with ASD is therefore limited. We initiated the trial with adults rather than children, because the drug is experimental and possible effects in ASD patients were unknown. No federal agency or dedicated national ASD fund would consider the trial, which limited its resources. Fewer adult ASD patients are in treatment with psychologists and physicians who might refer them, and there was little interest among support groups. The patients themselves were reported by their treating clinicians to be highly reluctant to volunteer to receive an experimental drug. All these factors together seemed to be responsible for the difficulties recruiting ASD patients, compared to our experience with schizophrenia patients.
Acknowledgments
Funding for the study came from an unrestricted gift from the Institute of Children’s Mental Disorders.
Footnotes
Ethical Approval
All procedures performed in studies involving human participants were in accord with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed Consent
Informed consent was obtained from all individual participants included in the study.
Contributor Information
Ann Olincy, Department of Psychiatry, University of Colorado School of Medicine, Aurora, CO, USA.
Audrey Blakeley-Smith, Department of Psychiatry, University of Colorado School of Medicine, Aurora, CO, USA; Department of Behavioral Pediatrics and JFK Partners, Children’s Hospital Colorado, Aurora, CO, USA.
Lynn Johnson, Department of Psychiatry, University of Colorado School of Medicine, Aurora, CO, USA.
William R. Kem, Department of Pharmacology and Toxicology, University of Florida School of Medicine, Gainesville, FL, USA
Robert Freedman, Department of Psychiatry, University of Colorado School of Medicine, Aurora, CO, USA.
References
- Allen-Brady K, Robison R, Cannon D, Varvil T, Villalobos M, et al. (2010). Genome-wide linkage in Utah autism pedigrees. Molecular Psychiatry, 15, 1006–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold LE, Aman MG, Holloway J, Hurt E, Bates B, Xiaobai L, et al. (2012). Placebo-controlled pilot trial of mecamylamine for treatment of autism spectrum disorders. Journal of Child and Adolescent Psychopharmacology, 22, 198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacchelli E, Battaglia A, Cameil C, Lomartire S, Tancredi R, Thomson S, et al. (2015). Analysis of CHRNA7 rare variants in autism spectrum disorder susceptibility. Amercian Journal of Medical Genetics Part A, 167A, 715–723. [DOI] [PubMed] [Google Scholar]
- Baron Cohen S (2004). The cognitive neuroscience of autism. Journal of Neurology, Neurosurgery and Psychiatry, 75, 945–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejerot S, & Nylander L (2003). Low prevalence of smoking in patients with autism spectrum disorders. Psychiatry Research, 119, 177–182. [DOI] [PubMed] [Google Scholar]
- Casey JP, Magalhaes T, Conroy JM, Regan R, Shah N, Anney R, et al. (2012). A novel approach of homozygous haplotype sharing identifies candidate genes in autism spectrum disorder. Human Genetics, 131, 565–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch SL, Urbando MR, Neumann SA, Burket JA, & Katz E (2010). Cholinergic abnormalities in autism: Is there a rationale for selective nicotinic agonist interventions? Clinical Neuropharmacology, 33, 114–120. [DOI] [PubMed] [Google Scholar]
- Freedman R (2014). Alpha7-nicotinic receptor agonists for cognitive enhancement in schizophrenia. Annual Review of Medicine, 65, 245–256. [DOI] [PubMed] [Google Scholar]
- Freedman R, Coon H, Myles-Worsley M, Orr-Urtreger A, Olincy A, Davis A, et al. (1997). Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proceedings of the National Academy of Sciences, 94, 587–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman R, Olincy A, Buchanan RW, Harris JG, Gold JM, Johnson L, et al. (2008). Initial phase 2 trial of a nicotinic agonist in schizophrenia. American Journal of Psychiatry, 165, 1040–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith J, Hoffer LD, Adler LE, Zerbe GO, & Freedman R (1995). Effects of sound intensity on a midlatency evoked response to repeated auditory stimuli in schizophrenic and normal subjects. Psychophysiology, 32, 460–466. [DOI] [PubMed] [Google Scholar]
- Harris JG, Kongs S, Allensworth DA, Sullivan B, Zerbe G, & Freedman R (2004). Effects of nicotine on cognitive deficits in schizophrenia. Neuropsychopharmacology, 29, 1378–1385. [DOI] [PubMed] [Google Scholar]
- Kemner C, Oranje B, Verbaten MN, & van Engeland H (2002). Normal P50 gating in children with autism. Journal of Clinical Psychiatry, 63, 214–217. [DOI] [PubMed] [Google Scholar]
- Leonard S, Gault J, Hopkins J, Logel J, Vianzon R, Short M, et al. (2002). Association of promoter variants in the alpha7 nicotinic acetylcholine receptor subunit gene with an inhibitory deficit found in schizophrenia. Archives of General Psychiatry, 59, 1085–1096. [DOI] [PubMed] [Google Scholar]
- Magnée MJCM, & Oranje B (2009). Cross-sensory gating in schizophrenia and autism spectrum disorder: EEG evidence for impaired brain connectivity? Neuropsychologia, 47, 1728–1732. [DOI] [PubMed] [Google Scholar]
- Mayes SD, & Dickerson S (2008). WISC-IV and WIAT-II in children with high-functioning autism. Journal of Autism and Developmental Disorders, 3, 428–439. [DOI] [PubMed] [Google Scholar]
- Moriuchi JM, Klin A, & Jones W (2016). Mechanisms of diminished attention to eyes in autism. American Journal of Psychiatry, 174. doi: 10.1176/appi.ajp.2016.15091222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafavi M, Hardy P, & Arnold LE (2016). Varenicline in autism: theory and case report of clinical and biochemical changes. Journal of Child and Adolescent Psychopharmacology,. doi: 10.1002/ajmg.a.37795. [DOI] [PubMed] [Google Scholar]
- Newhouse P, Kellar K, Aisen P, White H, Wesnes K, Coderre E, et al. (2012). Nicotine treatment of mild cognitive impairment. Neurology, 78, 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olincy A, Harris JG, Johnson L, Pender V, Kongs S, Allensworth D, et al. (2006). Proof-of-concept trial of an α7-nicotinic agonist in schizophrenia. Archives of General Psychiatry, 63, 630–638. [DOI] [PubMed] [Google Scholar]
- Orekhova EV, Stroganova TA, Prokofyev AO, Nygren G, Gillberg C, & Elam M (2008). Sensory gating in young children with autism: Relation to age, IQ, and EEG gamma oscillations. Neuroscience Letters, 434, 218–223. [DOI] [PubMed] [Google Scholar]
- Piazza C, Hanley GP, & Fisher WW (1996). Functional analysis and treatment of cigarette pica. Journal of Applied Behavior Analysis, 29, 437–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, & Regan R (2010). Functional impact of global rare copynumber variation in autism spectrum disorders. Nature, 466, 368–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray MA, Graham AJ, Lee M, Perry RH, Court JA, & Perry EK (2005). Neuronal nicotinic acetylcholine receptor subunits in autism: An immunohistochemical investigation in the thalamus. Neurobiology of Disease, 19, 366–377. [DOI] [PubMed] [Google Scholar]
- Souliéres I, Dawson M, Gernsbacher MA, & Mottron L (2011). The level and nature of autistic intelligence II: What about Asperger Syndrome? PLoS One, 6, e25372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spek AA, Scholte EM, & Van Berckelaer-Onnes IA (2011). Local information processing in adults with high functioning autism and Asperger Syndrome: The usefulness of neuropsychological tests and self-reports. Journal of Autism and Developmental Disorders, 41, 859–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroganova TA, Kozunov VV, Posikera IN, Galuta IA, Gratchev VV, & Orekhova EV (2013). Autism spectrum disorder contributes to their atypical auditory behavior: An ERP study. PLoS One, 8(e69100), 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner MA (1999). Generating novel ideas: Fluency performance in high-functioning and learning disabled individuals with autism. Journal of Child Psychology and Psychiatry, 40, 189–201. [PubMed] [Google Scholar]
- Van Schalkwyk GI, Lewis AS, Qayyum Z, Koslosky K, Picciotto MR, & Volkmar FR (2015). Reduction of aggressive episodes after repeated transdermal nicotine administration in a hospitalized adolescent with autism spectrum disorder. Journal of Autism and Developmental Disorders, 45, 3061–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui DH, Scoles HA, Shin-ichi H, Meguro-Horike M, Dunaway KW, Schroeder DI, et al. (2011). 15q11.2–13.3 chromatin analysis reveals epigenetic regulation of CHRNA7 with deficiencies in Rett and autism brain. Human Molecular Genetics, 20, 4311–4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoran-Hegesh R, Kertzman S, Vishne T, Weizman A, & Kotler M (2009). Neuropsychological mechanisms of digit symbol substitution test impairment in Asperger disorder. Psychiatry Research, 166, 35–45. [DOI] [PubMed] [Google Scholar]