

Abstract
CAR (Chimeric Antigen Receptor) T cells have demonstrated clinical success for the treatment of multiple lymphomas and leukaemias, but not for various solid tumours, despite promising data from murine models. Lower effective CAR T-cell delivery rates to human solid tumours compared to haematological malignancies in humans and solid tumours in mice might partially explain these divergent outcomes. We used anatomical and physiological data for human and rodent circulatory systems to calculate the typical perfusion of healthy and tumour tissues, and estimated the upper limits of immune cell delivery rates across different organs, tumour types and species. Estimated maximum delivery rates were up to 10 000-fold greater in mice than humans yet reported CAR T-cell doses are typically only 10–100-fold lower in mice, suggesting that the effective delivery rates of CAR T cells into tumours in clinical trials are far lower than in corresponding mouse models. Estimated delivery rates were found to be consistent with published positron emission tomography data. Results suggest that higher effective human doses may be needed to drive efficacy comparable to mouse solid tumour models, and that lower doses should be tested in mice. We posit that quantitation of species and organ-specific delivery and homing of engineered T cells will be key to unlocking their potential for solid tumours.
Keywords: trafficking, CAR, T cell, immunotherapy, failure, modelling
1. Introduction
Cellular therapies such as CAR (Chimeric Antigen Receptor) T cells have shown clinical efficacy against several leukaemias and lymphomas [1,2]. This success has not yet been matched for solid tumours, despite the efficacy seen in pre-clinical models, and a suitable dosing strategy to maximize efficacy remains uncertain [3–8]. Typical response curves (amount of CAR T-cell transgene observed in blood versus time) in patients with haematological disorders are marked by an initial cellular expansion (typically 100–1000-fold [9]), due to the large numbers of CAR T and target cells colocalizing in readily accessible tissues. Cellular expansion increases the effective cellular dose entering and proliferating within compartments with lower perfusion or less efficient access, which can drive the clearance of target cells required to achieve complete responses in these compartments. In solid tumours, relatively few target cells are in readily accessible compartments, whether due to poor perfusion or barriers to extravasation, preventing a strong initial expansion of CAR T cells. Tumour regression is achieved when the rate of tumour clearance is greater than that of tumour growth, including in the least perfused/accessible tumour lesions. In this context, tumour clearance is a numbers game and the relative lack of success for solid tumours may in some cases be due to lower effective CAR T-cell doses, since the number of accessible target cells is too low to drive the early cellular expansion that, in the case of haematological malignancies, increases the effective dose.
The amount of cellular expansion depends on tumour burden, patient and cell-product-specific factors, which results in non-intuitive observations. Increasing cellular dosage does not always increase efficacy [10,11], and patients with a high tumour burden may only require a low dosage, due to greater cellular expansion [12]. This has frustrated dose selection and the definition of a maximum tolerated dose. Early clinical studies of CAR T cells found that high dosages (1010–1011 cells) were required for efficacy, but no dose-response relationship was found beyond this minimum level [13–15]. Later studies with next-generation CARs began to favour lower doses (109 or fewer) to balance efficacy with toxicity, which also increases with dosage [16]. Dosages in mice have trended similarly: early studies noted a need for high dosages (108 cells) [13], and more recent studies favour lower doses (106–108, with limited success seen in a ‘stress-test’ study with 105 cells [17]). Experimental mice are hardy, short-lived and several strains are immunodeficient (which reduces early anti-CD19 activity, for example), so there is comparatively less focus on reduction of toxicity such as cytokine release syndrome. Scaling of dosages of any therapy between animals and humans is often assumed to depend on body mass or (erroneously [18]) surface area [19,20], but this is complicated for CAR T cells by the impact of tumour burden and the lack of a well-defined maximum tolerated dose. Additional factors such as tumour mass and receptor expression may also be considered, but to the best of our knowledge, there is no single standard for dosage scaling of CAR T-cell therapies.
Haematological and solid cancers in humans and mice may be compared by considering early-time kinetics and dynamics of the adoptively transferred cells. Inflamed tissues exhibit increased regional blood flow, and it has been observed that localization of lymphocytes is proportional to the regional blood flow of the tissue [21]. The delivery rate of cells to different compartments of the body will likely be of importance in CAR T cell or eTCR (engineered T-cell receptor) responses. For intravenous administration, cells are delivered by the circulatory system. Only a small proportion of cells leave the vasculature and extravasate into tissues, but the effective delivery rate cannot exceed the vascular delivery rate. Systematic quantitation of the variation of vascular delivery rates across organs, tumour types and species will improve understanding of comparative preclinical and clinical outcomes and inform improved dosing and dosage scaling strategies. Physiologically based pharmacokinetic modelling (PBPK) has been used extensively to predict drug concentration profiles and their variability across different tissues and individuals, to estimate the efficacy of clinical dosing regimens (for recent reviews, see [22–24]). PBPK models have also been used in drug development since 2000 and are readily accepted as providing supporting information by both the US Food and Drug Administration and the European Medicines Agency. They have been further implemented in the investigation of T-cell trafficking, for example to determine the strength of the abscopal effect and influence of metastases on the primary tumour [25,26] and to study localization of adoptively transferred T cells or cellular therapies [27–32]. However, we have not seen such models be used for quantitative exploration of the simpler consequences of differences between anatomical parameters in different species, nor an attempt to quantify and compare the maximum likely values of delivery rates of immune cells across organs and species, the aim of the present work.
We have made simple comparisons of the human, mouse and rat circulatory systems, using relevant organ, tumour and anatomical data [33–38]. We have calculated the upper bounds of cellular delivery from the circulation into each organ, considering only tissue perfusion and not factors that subsequently reduce rates of T-cell entry or engagement, such as tissue-specific extravasation probabilities or inflammation (figure 1b), or immunosuppression. The validity of predictions was tested through comparison to published positron emission tomography (PET) imaging data [39–41] taken shortly after cellular transfer and radiological data 24 h after cellular transfer, and the validity of maximum delivery rates for tumour tissue was found by comparing the typical perfusion of tumour and normal tissues [21]. Predicted maximum delivery rates exhibited extreme differences by species. The delivery rate of cells per minute per mm3 to lungs is 20 000-fold higher in mice than humans, yet typical doses of CAR T cells given to experimental mice are only 100-fold less than those in the clinic. This may partially explain the lack of success seen against solid tumours reported to date.
Figure 1.

(a) A visual summary of a model of the circulatory system. Solid and dotted lines represent blood and lymph flow, respectively. Cells flow from the heart to each organ, from which a proportion enters the interstitial space. Cells from the interstitium flow via the lymphatics back to the heart. A tumour (tmr) can be represented by choosing a tumour bearing organ (the skin in this example) from which proportions of its volume and blood supply are occupied by the tumour. (b) Only a fraction of cells delivered by the vasculature extravasate into a given organ, but the entry rate can be no higher than the vascular delivery rate. Calculation of these maximum delivery rates yields insight into inter-species and inter-organ delivery of cellular therapies. (c,d) The perfusion of different organs can differ substantially between humans (c) and mice (d). Multiplying cellular concentrations by perfusion gives maximum delivery per volume (or mass) of tissue. Anatomical values given are examples; these parameters differ by experimental reference used.
2. Methods
2.1. Model summary
Most studies of physiologically based pharmacokinetics (PBPK) or cellular kinetics (PBCK) make use of an ordinary differential equation (ODE) model representing the anatomy. A schematic of the anatomy appropriate for such equations is shown in figure 1a. T cells are assumed to flow from the heart to the vasculature of different organs, where they then return or extravasate into that organ's interstitial space. Extravasated cells return to circulation via the lymphatics, except for the spleen and the pulmonary circuit, from which cells return directly. To calculate maximum delivery rates, we require the rate at which cells are delivered by the vasculature, as shown graphically in figure 1b. This is equal to organ perfusion (blood flow B over total organ volume ) multiplied by blood concentration C. More precisely,
| 2.1 |
where Ntot is the total number of cells of interest, Vo and are, respectively, the vascular and total volumes of organ o, and Vh is the volume in the heart and interconnecting blood vessels. This expression can be shown to be equivalent to a special case of standard PBPK/PBCK models, see electronic supplementary material, §§A.1 and A.1.4. To compare tumour and healthy tissue in different organs, we define one organ as the tumour bearing organ, containing a 1 mm3 tumour (tmr) tissue volume. We consider this volume either as healthy or tumour tissue, to find how predicted delivery rates to each differ across organs and species.
2.2. Parameter selection from literature
Predicted T-cell delivery rates are dependent on assumed anatomical parameters (blood flow, blood volume and organ volume). We collected several anatomical reference banks from the literature [33–38], in particular the compilations by the ICRP and Shah and colleagues [36,38]. Each source has slightly differing fractional blood flows and volumes. To remove selection bias, delivery rates were calculated with many random values of anatomical parameters (n = 100 per organ per species), selected uniformly from the range of literature values, after which the means and standard deviations of estimated delivery rates were taken. This also serves as a proxy for population variability. To avoid using data from different studies for a single model animal, datasets that are as complete as possible were chosen. In particular, the total blood flow and blood volume, the volume of each organ, and the fractional blood flow and blood volume of each organ were recorded from each reference. These data are shown in electronic supplementary material, tables S2–S4. Presented results are the mean and standard deviation of predictions obtained by choosing random values from the literature. Random parameter values are selected from the range of literature values. We cannot be more confident in any one report than another, so we choose the random values for all parameters (for each organs and species) uniformly. This process is repeated 100 times to yield the presented results. When considering tumour perfusion distinct from healthy organ perfusion, we use measurements of tumour perfusion from the literature (see electronic supplementary material, table S3) and suppose that, since these are all measurements of different tumours, the data should follow a normal distribution. Thus, we choose normally distributed random values of tumour perfusion.
2.3. Generation of presented results
Presented data are maximum delivery rates in each species for each organ o, calculated using equation (2.1), with some deviation due to details of the vasculature. For example, the portal vein blood flow must be added to Bo for the liver (see electronic supplementary material, §A.1 for further information). The results of table 1 are obtained by applying data reported by Shah et al. [36] to equation (2.1). This is presented graphically in figure 1c, d. The results of table 2 are obtained by multiplying the ratio of mouse to human delivery rates by the dose administered to mice, 107.
Table 1.
Left: predicted absolute maximum CAR T-cell delivery rates per volume (in cells/min/mm3) to non-tumour tissue in organs in humans, mice and rats, using previously compiled physiological parameter values [36]. It is assumed that organ perfusion is homogenous and 108 CAR T cells are introduced to each species. The interspecies differences in absolute delivery rates per volume depend only on organ perfusion and cell blood concentration. Right: organ perfusion (blood flow / organ volume; BF/V) and the total blood volume in each species, obtained by summing relevant volume data from [36].
| max delivery rate/(cells/min/mm3) |
BF/V/min−1 |
|||||
|---|---|---|---|---|---|---|
| organ | human | mouse | rat | human | mouse | rat |
| lungs | 177.0 | 3 728 321 | 441 221 | 5.51 | 55.4 | 63.7 |
| kidneys | 106.7 | 266 052 | 31 767 | 3.32 | 3.95 | 4.59 |
| thymus | 53.6 | 269 612 | 26 656 | 1.67 | 4.01 | 3.85 |
| small intestine | 31.3 | 162 734 | 16 729 | 0.97 | 2.42 | 2.42 |
| pancreas | 28.6 | 131 174 | 13 298 | 0.89 | 1.95 | 1.92 |
| spleen | 27.9 | 131 336 | 13 554 | 0.87 | 1.95 | 1.96 |
| large intestine | 22.8 | 112 344 | 11 547 | 0.71 | 1.67 | 1.67 |
| liver (+ portal vein) | 22.5 | 113 382 | 12 310 | 0.70 | 1.68 | 1.78 |
| heart vasculature | 22.1 | 489 648 | 31 051 | 0.69 | 7.28 | 4.49 |
| brain | 14.4 | 49 611 | 6007 | 0.45 | 0.74 | 0.87 |
| lymph node | 13.0 | 29 774 | 3064 | 0.41 | 0.44 | 0.44 |
| stomach | 12.3 | 6687 | 19 437 | 0.38 | 0.10 | 2.81 |
| red marrow | 9.3 | 117 661 | 7012 | 0.29 | 1.75 | 1.01 |
| skin | 3.3 | 11 292 | 841 | 0.10 | 0.17 | 0.12 |
| skeletal muscle | 1.1 | 15 537 | 1590 | 0.03 | 0.23 | 0.23 |
| fat | 0.8 | 13 800 | 1419 | 0.03 | 0.21 | 0.21 |
| total blood volume/ml | 3110a | 1.49 | 14.4 | |||
aNote that the total blood volume from this reference is an underestimate, but it is expected to be underestimated by a similar amount in each species. The left table can be generated from the right by the formula , where B and are the organ blood flow and volume and Vtot is the total blood volume in each species; see §2.1.
Table 2.
Human-equivalent dosages for delivery to non-tumour tissue: The dosage of CAR T cells in humans predicted to be required to give the same absolute delivery rate per mm3 as in a mouse given 107 cells. The numbers required are much larger than many clinical dosages [42,43].
| organ | equivalent dose | (continued) | |
|---|---|---|---|
| lungs | 1.7 × 1011 | heart vasculature | 2.2 × 1011 |
| kidneys | 2.5 × 1010 | brain | 3.4 × 1010 |
| thymus | 5.0 × 1010 | lymph node | 2.3 × 1010 |
| small intestine | 5.2 × 1010 | stomach | 5.4 × 109 |
| pancreas | 4.6 × 1010 | red marrow | 1.3 × 1011 |
| spleen | 4.7 × 1010 | skin | 3.4 × 1010 |
| large intestine | 4.9 × 1010 | skeletal muscle | 1.4 × 1011 |
| liver | 5.0 × 1010 | fat | 1.7 × 1011 |
Random results in figure 2 are obtained by drawing uniformly random values of organ parameters (n = 100), calculating the maximum delivery rate per volume with equation (2.1) for each set of values, and subsequently finding their mean and standard deviation. n = 100 values were chosen for each organ to generate an indication of delivery rate variability, while ensuring that the mean of selected random parameters was within 5% of the actual mean of experimental parameter values.
Figure 2.

Species-scaled predicted delivery rates to non-tumour tissue in organs in humans, rats and mice. Rates are normalized so that the sum across organs for each species is 100. See table 1 for a comparison of absolute rates. Scatter points and bar extents indicate the mean and standard deviation of delivery rates over 100 repeats, with each repeat using different random fractional blood flows and volumes, uniformly drawn from experimental data in the literature. Text labels give the ratio of mean predicted delivery rates in rats and mice to human rates. The relative distribution of rates across the major organs differs by species, and, consequently, inter-species scaling of delivery rates is organ-specific. Note that the horizontal axis is a log scale.
Random results in figure 3 are obtained similarly, by drawing uniformly random values of organ parameters and normally distributed values of tumour perfusion Ptmr. The maximum delivery to tumour tissue is calculated from , and the maximum delivery rate to non-tumour tissue is calculated using equation (2.1) for comparison. As before, n = 100 values were chosen for each organ.
Figure 3.
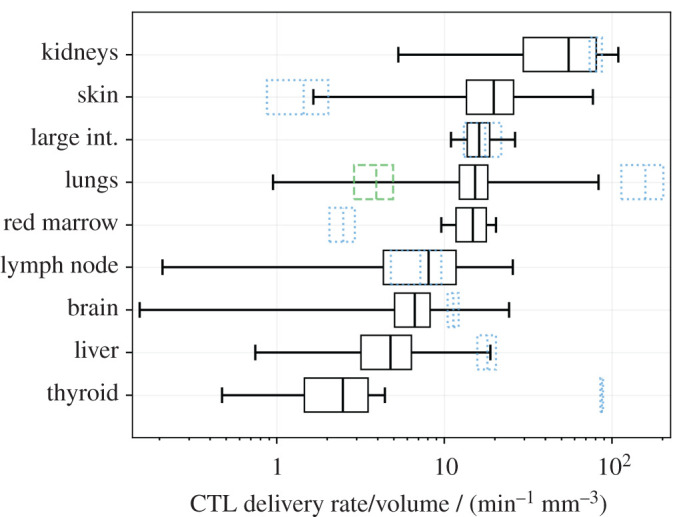
Absolute predicted delivery rates to human tumours, compared with non-tumour tissue, assuming 108 CAR T cells are administered intravenously. Predictions are presented as a mean and standard deviation over 100 repeats, with random anatomical parameters and tumour perfusion drawn from experimental data in the literature. Black boxes represent the mean and standard deviation of predicted tumour delivery rates, and whiskers indicate predictions using the extremes of possible tumour perfusion according to the literature. Blue dotted boxes indicate the mean and standard deviation of predicted delivery to non-tumour tissue, i.e. the data used to generate figure 2. The green dashed box indicates delivery rates per mm3 to healthy lung tissue when the pulmonary circuit is assumed not to contribute. Note that now the kidneys and skin have the highest predicted tumour delivery rates, and that the horizontal axis is a log scale.
3. Results
3.1. CAR T-cell delivery to organs in humans, mice and rats
We calculated and compared predictions for the vascular delivery rate per volume (cells/min/mm3) of a typical number of CAR T cells used in the clinic (108 [42,43]) to non-tumour tissues in different human, rat and mouse organs. These rates are equal to the product of the organ perfusion and CAR T-cell blood concentration, as shown graphically in figure 1b–d. Results calculated from a single anatomical dataset [36] are shown in table 1. Flow from both the hepatic artery and portal vein are included in delivery rates to the liver, and the pulmonary circuit and lung blood supply are both included for lung rates. The difference in delivery rates to the same organ in different species can be extreme, with predicted absolute lung delivery rates per volume in the mouse 21 000 times higher than in humans if the same number of CAR T cells is administered to each species (obtained by dividing 3 700 000/180 from table 1). Should a known blood concentration of endogenous cells be considered instead of a constant number, then rates per volume depend only on organ perfusion, and the absolute delivery rates for mice are up to 10 times higher than in humans. These data suggest that a more appropriate approach for scaling murine dosages to humans (or vice-versa) is to ensure that the same cellular delivery rate to tissues of interest is achieved. The results of table 1 were used to calculate the CAR T-cell doses (introduced cell numbers) required to obtain the same delivery rates in humans as in mice given a typical pre-clinical dose of 107 CAR T cells. Equivalent doses are organ-specific, and most are of the order 1010–1011 cells (table 2).
The mean and standard deviation of predicted delivery rates obtained by random selection of anatomical parameters from all datasets [33–38] are plotted in figure 2. To illustrate organ-specific scaling and to allow interspecies comparison of the distribution of delivery rates across organs, rates are scaled by species such that the sum of the mean predictions within each species is 100. The distributions share similarities but otherwise the relative rates exhibit organ-specific scaling. For each species, the lung has the highest delivery rate, followed by the kidneys.
3.2. CAR T-cell delivery to human tumours
Predicted maximum delivery rates per mm3 of tissue described above assume that perfusion is homogeneous within a given organ. However, a tumour may have perfusion different to normal tissues. The literature was surveyed to quantify the variability of human tumour perfusion (electronic supplementary material, figure S3) for incorporation into estimates of maximum delivery rates. As before, delivery rates were calculated with many random values of parameters (), drawn uniformly for all organ parameters and from a Gaussian distribution for tumour perfusion. The mean and standard deviation of predicted delivery rates for CAR T cells to human tumours are shown in figure 3, along with the corresponding delivery rates under the assumption of homogeneous perfusion (or equivalently, to non-tumour tissue; blue dotted boxes). The rank order of delivery rates to tumour and normal tissues are very different. In most cases, the average of predicted delivery rates for tumour tissue is similar to or less than that for normal tissue, but in some cases (e.g. the skin) it is considerably greater. However, their variation is considerable; extreme values (whiskers in the plot) vary over many orders of magnitude above and below that of the corresponding normal tissue, for most organs.
3.3. Maximum delivery estimates are consistent with PET imaging and radiography data
The validity of ‘maximum delivery rates’ to organs can be tested by comparing data from PET imaging and radiography studies in humans and rodents, in which cell localization at early time points has been recorded. The use of an early time point is critical, as it shows the location of cells that are still in the blood or recently extravasated into an organ, before they drain back into the blood and recirculate. At later time points, localization is a function of both cell delivery to organs, return to circulation, and other factors that modulate these, such as antigen recognition. The delivery of radiolabelled natural killer cells from the bloodstream into individual organs has been studied in rats [39] and in human patients [40,41]. These data are presented in figure 4 and compared to predictions from table 1. Patients in the human study were given 108–109 cells; the average fraction found in the liver at the first time point (30 min) was 8.9%. This corresponds to approximately 4.5 × 107 cells. The rats were given 106 to 107 cells; the average fraction found in the liver at the first time point (30 min) was 23.0%, or 1.2 × 106 cells. Adjusting the rat numbers to the human dose gives 1.2 × 108 cells. If we then assume a liver volume of 1700 ml in humans and 10 ml in rats, we obtain cell number per unit volume in the liver: 2.6 × 104 in humans and 1.1 × 107 in rats, a ratio of 429. The ratio of predicted maximum delivery rates is 546 (table 1), 1.27-fold larger than expected from the data. Repeating this analysis for the lungs and spleen gives experimental ratios 2.0-fold less than predicted from maximum delivery rates (figure 4).
Figure 4.

Comparison of reported localization of radiolabelled natural killer (NK) cells in rats and humans to predicted maximum delivery rates [39–41]. (a) Reproductions of the reported data, after normalizing data at each time point such that the total radioactivity (localization) is 1.0 at all time points. Annotations indicate the initial count of cells in the lung in each species. (b) Analysis of the data. The dosage and fractional localization in each organ can be used to calculate the number of NK cells present in each organ at each time point. By accounting for the different dose given to each species and choosing an appropriate estimate for organ volumes in each species, the number of cells per volume in each species can be calculated. The rat/human ratio of the number of cells in each organ can be compared to the ratio of predicted maximum delivery rates per volume, obtained from table 1.
Further confirmation that localization of lymphocytes is dependent upon blood flow can be obtained by measuring how the localization of labelled cells depends on the regional blood flow for a given tissue. Ottaway & Parrott [21] measured how cell localization and regional blood flow to the inflamed ear and various lymph nodes of experimental mice change in response to oxazolone-induced inflammation. They found a significant correlation between the localization of lymphocytes after 24 h and regional blood flow in most of their analyses, and that the increased blood flow occurs regardless of the applied antigen. One of their figures is replotted in figure 5.
Figure 5.
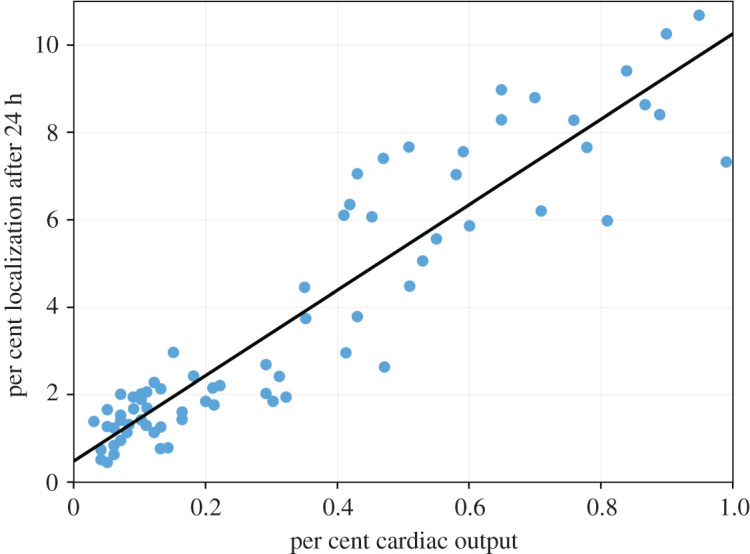
Figure replotted from Ottaway & Parrott [21]. The percentage localization of Cr-51-labelled lymphocytes after 24 h against the regional blood flow to auricular, inguinal and mesenteric lymph nodes. A regression line has been fitted to the data.
4. Discussion
4.1. Vascular delivery and cell proliferation
This study aimed to quantify physiological constraints on the rate of CAR T-cell delivery by the blood to target tissues in different species, to better predict appropriate clinical CAR T-cell doses from pre-clinical data. It has focused on adoptive T-cell cancer therapies, though the methodology may also apply to other therapeutic areas, including immune-related adverse event prediction. Values were calculated assuming that 108 T cells are introduced; delivery rates due to any other desired number or blood concentration of cells can be calculated by multiplying results by the ratio of the desired number to 108 or multiplying blood concentration by the total blood volume in the target species. Although models to predict expansion of a T-cell population have been studied in the past [9,44], it is difficult to quantify cellular proliferation in or fractional recirculation from a given tissue. However, proliferation itself depends on exposure of transferred T cells to their target antigen, so early responses are expected to be constrained by delivery. Several studies have established a relationship between dose or effective early target engagement and response for cellular therapies, despite proliferation increasing the effective dose over time [17,43,45,46]. Furthermore, delivery of cells that proliferate outside of a given tumour site would also be constrained by vascular delivery. The maximum rate of delivery due to the anatomy can be estimated with greater confidence and wider applicability than can an estimated time-course of T-cell concentration that considers proliferation and contraction, so proliferation was not considered in this work and will be the focus of future studies.
4.2. Organ-specific delivery rates and their variation
Results predict that the highest CAR T-cell delivery rates are in organs with the highest perfusion: the lungs and kidneys in humans (figure 2). When measurements of tumour-specific perfusion are considered (figure 3), it is the kidneys, skin, large intestine and lungs that are predicted to have the highest delivery rates per mm3, consistent with non-cellular immunotherapies (IL-2 and checkpoint blockade) having the highest efficacy in kidney, skin, colon and lung tumours [47–52], and the hypothesis that efficacy is driven in part by tissue perfusion. For cellular therapies including CAR T cells, vascular delivery should similarly correlate with efficacy, with the additional factor that T cells must extravasate into target tissues. Both naive and ex vivo T cells preferentially extravasate into lymph nodes, spleen and liver [53–55], consistent with CAR T-cell efficacy in haematological disorders but not solid tumours [1,42,43]. Tumour tissue may be vascular or avascular, and inflamed or uninflamed, meaning that predicted ‘maximum’ delivery rates for normal tissues may not be applicable to tumours. However, predicted mean delivery rates into tumours exceed those to normal tissue for only a minority of organs (figure 3), including the skin. Predicted delivery rates to tumours in the skin vary over many orders of magnitude but are usually greater than those for normal tissue. Healthy skin is not usually highly perfused and contains shunts to control blood flow in response to temperature. Most anatomical data for the skin describes the organ at rest and at room temperature with no inflammation, meaning most shunts will be open. Tumour tissue can increase its perfusion through inflammation or angiogenesis and likely subverts these shunts, which could explain the greater mean and variation in predicted delivery rates for skin tumours. Liver and kidney tissues are highly perfused at rest, which are unlikely to be improved by random tumour angiogenesis; accordingly, predicted delivery rates to tumours in these organs do not exceed normal tissue. Predictions for red bone marrow indicate that tumour perfusion can greatly outstrip normal tissue perfusion. Though surprising, the bone red bone marrow result is consistent with studies in which bone perfusion was measured in healthy control bone and tumour sites in patients with bone cancers and metastases [56]. Predicted rates to bone marrow are particularly interesting because many haematological malignancies exist partially within this tissue; the increased tumour perfusion shown in figure 3 may explain why the relatively low delivery rate predicted in table 1 does not contradict the relative success seen for CAR T cells against haematological malignancies.
Finally, predicted delivery rates to lung tumours may or may not exceed that of normal tissue, depending on whether the pulmonary circuit is assumed to contribute to tumour blood supply (blue dotted box) or not (green dashed box). Aside from these exceptions, results suggest that predicted maximum delivery rates to normal tissue are greater than those to tumour tissue of the same origin in most cases, and so appropriate to use as a guideline to compare species.
Both figures 2 and 3 show that predicted delivery rates are highly variable, which may be caused by differences in experimental techniques or individual variation. Physiological differences and behaviour both impact blood flow distributions; blood flow to the mesentery increases after a meal, muscles during exercise, or the skin in response to temperature. This effect is used in the clinic to prevent hair loss in chemotherapy patients by cooling the scalp. CAR T-cell therapies could be targeted to organs such as the mesentery or skin through meal consumption or temperature control, and tumour-specific blood flow could be increased with vessel normalization associated with anti-angiogenic therapies (e.g. Avastin). In patients with advanced metastatic disease, CAR T-cell dosage must be sufficient to drive tumour regression at the least perfused and/or the fastest growing site. To avoid dosage-linked increases in adverse events such as cytokine release or encephalopathy syndromes, methods to increase the effective dose on-site and not elsewhere should be considered, including alternate modes of administration, triggering proliferation at sites of interest, coadministration of inhibitors (e.g. anti-IL6), or interventions to alter blood flows should be used. Both human and rodent anatomical parameters vary, impacting any results that depend on anatomical parameters. If variability is not captured and/or care is not taken to control factors that alter blood flows (e.g. anaesthesia, exercise or the time of day [57]), then comparison of datasets may be invalid. Ideally, any study making use of blood flows and organ volumes should consider multiple measurements and include ‘error’ bars to indicate variation.
4.3. Species-specific delivery rates and dosage scaling
Relative delivery rates are distributed differently across organs in each species, meaning that dose scaling is organ-specific (figure 2 and table 1). Predicted absolute delivery rates of the same dose of CAR T cells (108) exhibited surprisingly extreme differences between species, with delivery per unit tissue volume to mouse lungs 21 000 times higher than in humans, largely because of the difference in total blood volumes between mice (2 ml) and humans (5 l). To test the relevance of these ‘maximum delivery rates’ and validate the model, we analysed published PET imaging and radiography studies of natural killer (NK) cells in humans and rats [39–41] and calculated the cell numbers present in various organs at early time points (§3.3). The human/rat ratios of NK unit volume in the lungs, liver and spleen 30 min after infusion were compared to the human/rat ratios of predicted maximum delivery rates. The measured localization ratios are 1.3–2.0-fold greater than predictions for delivery rate ratios. Such small discrepancies are not unexpected, as delivery rate ratios would only equal localization ratios if the blood concentration of NK cells and hence delivery rates were constant. However, the earliest experimental time point is 30 min, providing sufficient time for blood recirculation (as cardiac output/minute is greater than total blood volume in humans and rats). The rates of extravasation and return in each organ may differ between humans and rats, and the experimental technique and total amount of radioactivity at the first time point differs between the two studies. Regardless of these potentially confounding factors, the observations are consistent with predictions. This validation was made using NK cells rather than T cells, which may have different homing receptors or trafficking. However, we have compared maximum delivery rates, which depend only on anatomical factors, not cell-specific factors, so maximum rates are identical for NK and T cells with equal blood concentrations. Similarly, this validation was made using rats and not mice, though mouse–human comparison is the greater focus of this work. The comparison was made through anatomical parameters, which are well characterized for both mice and rats. Given that the ratios of measured cell localization in humans and rats were consistent with the differences in their cardiovascular systems, there is no reason to believe this would not be the case between humans and mice, particularly as rats are physically similar to mice.
To further confirm that cell localization depends in part on local tissue perfusion, we have replotted a figure by Ottaway & Parrott [21], figure 5, who showed that localization of lymphocytes in the ear and various lymph nodes after 24 h correlates with regional blood flow. As many lymphocytes arrive in the lymph nodes from the lymphatics rather than directly from the blood, one might have expected that the correlation be poorest for this tissue type, but there is a significant correlation between localization and regional blood flow for most of Ottaway and Parrott’s analyses. The increase in regional blood flow is due to local inflammation, so it may be that local inflammation correlates both with regional blood flow and with lymphocyte localization, i.e. that there is no causal relationship between blood flow and lymphocyte localization. However, this increased localization was shown to occur regardless of applied antigen, and so it is unlikely that the increased localization is due to an increase in the fraction of cells that extravasate, supporting the hypothesis that delivery of lymphocytes depends in part on their delivery by the vasculature.
Despite the considerably greater delivery rates of cells in mice than humans, typical doses (cell numbers) introduced to mice are not considerably lower than those given to humans. Most patients are given CAR T-cell dosages between 107 and 109 cells [42,43], while mouse studies have used (for example) two doses of 1– a week apart [6], two doses of 107 cells a week apart [3], and a single dose of 107 cells [4]. To illustrate how large these doses are, we calculated equivalent human dosages that would yield the same absolute delivery rates in humans as in a mouse given 107 CAR T cells (table 2). The resulting doses range between 1010 and 1011 T cells, much higher than typical clinical doses and many dose escalation studies [43]. This may explain why pre-clinical success does not always translate to the clinic. A pre-clinical study of a carcinoembryonic antigen (CEA) CAR T-cell therapy resulted in regression of subcutaneous tumours in mice with a dose of 5 × 106 cells (equivalent to 1.7 × 1010 in humans) [58]. In another study, a CAR T-cell therapy restricted the growth of pancreatic tumours in all treated mice to below the limit of detection with a dose of (equivalent to 4.6 × 1010 in humans) [59]. A study in which lower doses of around 105 anti-CD19 cells (human equivalent, using total blood volume only, of ) were given to mice as a ‘stress-test’ was associated with poor tumour control [17]. In the clinic, a study of CEA CAR T cells against colorectal cancer [46] escalated doses between 107 and 1010 cells. The authors found that the lower doses did not stop tumour progression (in 3 of 14 of presented patients) and higher doses achieved only stable disease. Our results suggest that dosages of order 1010 cells would be required to drive tumour regression at the primary site, and 1011 would be required for the lung metastases. Clinical studies in which Tumour Infiltrating Lymphocytes (TILs) were introduced in greater numbers (109–1011) [60–63] and in which CAR T cells were introduced regionally (bypassing trafficking via the bloodstream) [42] are associated with greater efficacy. An important caveat of the simple comparisons made here is that some of the studies lymphodepleted the mice or patients before infusing T cells, which aids proliferation, and some did not. Other differences in study design may also impact the choice of dosage. The relative human and mouse dosages reported in pre-clinical and clinical work with matching authors or centres are also similar to those discussed above: 106 in mice and 107–1010 in humans for anti-CEA CAR T cells [46,64], 105–106 cells in mice and 109 cells in humans for studies of anti-mesothelin CAR T cells [65,66], and 106 cells in mice and 109–1011 cells in humans for TIL studies by Rosenberg and colleagues [62,63,67].
We used natural killer cell localization data to validate the model, by confirming that early localization of cells correlates with predicted maximum delivery rates and assuming that natural killer and T cells behave similarly to each other at short time scales. A more appropriate validation would compare predictions to the localization of adoptively transferred cells to solid tumours in mice and humans, however, such data are sparsely published, and we have found no reported data for humans that includes organ and tumour localization at an early time point (of the order of minutes). Such data would be useful for further work, as would a time course that could be used to quantify the subsequent constraints imposed by homing and proliferation of cells.
The numbers presented here compare organs like-for-like between mice and humans, but many mouse studies use subcutaneous tumours, and scaling of perfusion is more uncertain for xenografts than for ordinary tissue. The ratio of the maximum delivery rate per volume to skin tissue between mice and humans is 2 if the same blood concentration of immune cells is assumed, or 3400 if the same number of immune cells is assumed (calculated from table 1). The ratio of delivery rates per volume to mouse skin versus human kidney tissue, for example, is 0.05 if the same concentration of cells is used, or 100 if the same number of immune cells is assumed. A previous study [68] has shown that small xenografts have similar local perfusion to the original tissue, but larger xenografts have reduced perfusion relative to the original tissue. This nonlinearity further confounds extrapolation of preclinical results and highlights some of the historically observed difficulties in the clinical translation of preclinical mouse xenograft model results [69]. However, the predicted maximum delivery rate to mouse skin is still orders of magnitude above predicted rates for any human tissue, suggesting that subcutaneous mouse models would still show greater efficacy than human studies. Additionally, mouse studies of orthotopic tumour grafts report success with similar doses to those of subcutaneous tumours: orthotopic, species-matched pancreatic tumours were eradicated by 107 CAR T cells [59], orthotopic glioblastoma xenografts were markedly slowed (but not eradicated) by 2 × 106 CAR T cells [70], pontine glioma xenografts introduced to the pons, spinal cord or thalamus of mice were eliminated by 107 CAR T cells [71] and orthotopic hepatocellular carcinoma xenografts were eliminated or substantially reduced in mice by 5–20 × 105 CAR T cells [72]. These considerations suggest that interpretation of pre-clinical therapeutic success requires dosages to be appropriately scaled to humans, in addition to consideration of physiological and immunological differences (such as the adhesion molecules required for T-cell extravasation). A model that considers organ-specific blood flow and volumes across species can be used to estimate likely efficacious human doses more precisely than allometric scaling.
4.4. Prediction refinement by T-cell homing and further considerations
The presented results are the predicted maximum delivery rates of CAR T cells per unit volume (cells/min/mm3) to organs and tumours, based on only organ blood flows and volumes. Refining these predictions requires quantification of CAR T-cell proliferation and organ-specific homing. The probability of T-cell extravasation differs by location and cell type. Naive T cells extravasate mainly into the lymph nodes or spleen and activated cells have a higher probability of extravasating into non-lymphoid tissues [53,73], distributed according to upregulated homing receptors (e.g. L-selectin or CCR7 [74]). These probabilities may differ across species (e.g. homing receptor CXCR1 is present in humans but not mice [75]), further limiting inter-species extrapolation of pre-clinical results. Homing receptor density, vessel normalization and hence homing probabilities may further differ in tumour tissue, particularly following therapies such as Avastin [76,77]. It is possible to quantify organ-specific homing by fitting ODE models (like the model shown in electronic supplementary material, §A.1) to T-cell localization data in experimental animals, as previous authors have done, e.g. [31]. Such quantification of homing probabilities would allow application of T-cell trafficking models to case reports in which the final localization of transgenic cells was measured (e.g. [78]), for diagnostic purposes. However, we have found limited human data with multiple, early time-points for cross-species comparison, which is the primary aim of this work. Parameters obtained from fits to multiple experiments would differ due to differences in the animals and the cells, so several datasets would be required to quantify the variation of and/or a confidence interval for parameter estimates. Additionally, the focus of this study is on anti-tumour therapies, where tumour homing would be further affected by factors such as inflammation. For this reason, we chose to quantify maximum delivery rates by examining the case where T cells have a 100% probability of extravasation in the target organ, and no extravasation elsewhere. Species comparisons are made by implicitly assuming that homing probabilities to each organ or tumour tissue would be similar between species. Expected variation in predictions was quantified by using the variation among anatomical reference values as a proxy. Both maximum values and this variation could be improved by more precise measurements of blood flows and volumes using the same techniques in each species, or else finding anatomical parameters for a precise experimental animal of interest.
Another challenge for CAR T cells in solid tumours is the identification of suitable target antigen. The ideal antigen is highly expressed on tumour cells and not expressed on healthy cells elsewhere. A typical target for B-cell malignancies is CD19 [43], as it is expressed by the entire pool of B cells and is limited almost exclusively to B cells. Several different antigens have been targeted for solid tumours, but with limited success (for example, GD2 has had encouraging results [43]). Target antigen may only be expressed by a subset of tumour cells and may not be sufficiently rare elsewhere in the body. For example, CAIX is expressed in some renal cell carcinomas, but it is also expressed in the liver bile duct resulting in on-target, off-tumour toxicities in a phase III trial [79]. Tumours may evolve to reduce expression of target antigen in response to successful T-cell killing, reducing the rate of tumour elimination or promoting outgrowth of therapy-resistant cells. Although these considerations are a barrier to treatment success, the rate at which cells can be delivered is a parallel and important factor. CAR T cells that are specific for an antigen that is expressed on most tumour cells will not drive tumour regression if their kill rate is lower than the tumour growth rate, given the combined rates of T-cell delivery and proliferation. On the other hand, CAR T cells specific for a rarer antigen may drive tumour regression if they arrive in sufficient numbers to eliminate all cells carrying that antigen, subsequently proliferating to greater numbers to drive regression at more restricted sites and/or drive a secondary response against one or more other antigens (i.e. epitope spread). Like T-cell delivery rates and T-cell extravasation probabilities, typical tumour growth rates are species, organ and individual specific. Together, these considerations show that tumour immunotherapy is a numbers game and hence more generally quantitative studies can be a useful tool for understanding the translational gap between pre-clinical and clinical outcomes.
5. Conclusion
Details of the human, rat and mouse circulatory systems were considered to predict CAR T-cell delivery to human tumours, and to human, rat and mouse organs. Predictions show up to an order of 10 000-fold increased CAR T-cell delivery per unit volume of target tissue in mice than humans, while typical clinical cell therapy dosages are 100-fold less than typical pre-clinical doses. These numbers are consistent with experimental studies of NK cell localization and various clinical observations. These predictions could partially explain why pre-clinical models of solid tumour clearance by CAR T cells show greater efficacy than in humans. Dosage scaling was found to be organ-specific and is particularly hard to quantify for xenografts, confounding the interpretation of pre-clinical results and lowering their potential clinical value, which is an important consideration in the context of the reduction and replacement of animal experiments. Control of tumour and organ-specific blood flow through exercise, circadian timing or food consumption could increase cellular delivery to tumour sites without raising the prospect of adverse outcomes, while vascular normalization may also induce such benefits, though with accompanying risk. More generally, cellular kinetic and dynamic models will lead to better understanding of how pre-clinical outcomes translate to the clinic, and hence better determination of appropriate clinical dosages and treatment strategies for cell-based therapies.
Data accessibility
Code and parameter data for humans, mice and rats used to generate the results of this work are available as electronic supplementary materials.
Authors' contributions
L.V.B. designed the model code, did the analysis and wrote the manuscript. All other authors supervised, advised and edited the manuscript.
Competing interests
J.W. has previously been an employee and shareholder of Hoffmann-La Roche. L.V.B. has previously completed an internship at that same company. J.W. is an employee of AC Immune SA.
Funding
This research was supported by funding from a Clarendon Scholarship, Hoffmann-La Roche and the Engineering and Physical Sciences Research Council (EPSRC), grant no. EP/L016044/1. Further support was given by Linacre College, Oxford.
References
- 1.Pettitt D, Arshad Z, Smith J, Stanic T, Holländer G, Brindley D. 2018. CAR-T cells: a systematic review and mixed methods analysis of the clinical trial landscape. Mol. Ther. 26, 342-353. ( 10.1016/j.ymthe.2017.10.019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Graham C, Hewitson R, Pagliuca A, Benjamin R. 2018. Cancer immunotherapy with CAR-T cells–behold the future. Clin. Med., J. R. Coll. Physicians London 18, 324-328. ( 10.7861/clinmedicine.18-4-324) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li H et al. 2018. Antitumor activity of EGFR-specific CAR T cells against non-small-cell lung cancer cells in vitro and in mice. Cell Death Dis. 9, 177. ( 10.1038/s41419-017-0238-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, Bouchier-Hayes L, Savoldo B, Dotti G. 2014. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. 74, 5195-5205. ( 10.1158/0008-5472.CAN-14-0697) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Newick K, Moon E, Albelda SM. 2016. Chimeric antigen receptor T-cell therapy for solid tumors. Mol. Ther. Oncolytics 3, 16006. ( 10.1038/mto.2016.6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Avanzi MP et al. 2018. Engineered tumor-targeted T cells mediate enhanced anti-tumor efficacy both directly and through activation of the endogenous immune system. Cell Rep. 23, 2130-2141. ( 10.1016/j.celrep.2018.04.051) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sachs JR, Mayawala K, Gadamsetty S, Kang SP, de Alwis DP. 2016. Optimal dosing for targeted therapies in oncology: drug development cases leading by example. Clin. Cancer Res. 22, 1318-1324. ( 10.1158/1078-0432.CCR-15-1295) [DOI] [PubMed] [Google Scholar]
- 8.Ji Y, Jin JY, Hyman DM, Kim G, Suri A. 2018. Challenges and opportunities in dose finding in oncology and immuno-oncology. Clin. Transl. Sci. 11, 345. ( 10.1111/cts.12540) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stein AM et al. 2019. Tisagenlecleucel model-based cellular kinetic analysis of chimeric antigen receptor–T cells. CPT Pharmacometrics Syst. Pharmacol. 8, 285-295. ( 10.1002/psp4.12388) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li DH, Whitmore JB, Guo W, Ji Y. 2017. Toxicity and efficacy probability interval design for phase I adoptive cell therapy dose-finding clinical trials. Clin. Cancer Res. 23, 13-20. ( 10.1158/1078-0432.CCR-16-1125) [DOI] [PubMed] [Google Scholar]
- 11.Abramson JS et al. 2020. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet 396, 839-852. ( 10.1016/S0140-6736(20)31366-0) [DOI] [PubMed] [Google Scholar]
- 12.Wages NA, Chiuzan C, Panageas KS. 2018. Design considerations for early-phase clinical trials of immune-oncology agents. J. ImmunoTher. Cancer 6, 1-10. ( 10.1186/s40425-018-0389-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosenberg SA, Spiess P, Lafreniere R. 1986. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science 233, 1318-1321. ( 10.1126/science.3489291) [DOI] [PubMed] [Google Scholar]
- 14.Dudley ME et al. 2002. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science (New York, N.Y.) 298, 850. ( 10.1126/science.1076514) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dudley ME. 2005. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J. Clin. Oncol. 23, 2346. ( 10.1200/JCO.2005.00.240) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Majzner RG, Mackall CL. 2019. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 25, 1341-1355. ( 10.1038/s41591-019-0564-6) [DOI] [PubMed] [Google Scholar]
- 17.Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, Plotkin J, Sadelain M. 2015. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell 28, 415-428. ( 10.1016/j.ccell.2015.09.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blanchard OL, Smoliga JM. 2015. Translating dosages from animal models to human clinical trials—revisiting body surface area scaling. FASEB J. 29, 1629-1634. ( 10.1096/fj.14-269043) [DOI] [PubMed] [Google Scholar]
- 19.Nair AB, Jacob S. 2016. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 7, 27. ( 10.4103/0976-0105.177703) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wiegel FW, Perelson AS. 2004. Some scaling principles for the immune system. Immunol. Cell Biol. 82, 127-131. ( 10.1046/j.0818-9641.2004.01229.x) [DOI] [PubMed] [Google Scholar]
- 21.Ottaway CA, Parrott DMV. 1979. Regional blood flow and its relationship to lymphocyte and lymphoblast traffic during a primary immune reaction. J. Exp. Med. 150, 218-230. ( 10.1084/jem.150.2.218) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoshida K, Budha N, Jin J. 2017. Impact of physiologically based pharmacokinetic models on regulatory reviews and product labels: frequent utilization in the field of oncology. Clin. Pharmacol. Ther. 101, 597-602. ( 10.1002/cpt.622) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saeheng T, Na-Bangchang K, Karbwang J. 2018. Utility of physiologically based pharmacokinetic (PBPK) modeling in oncology drug development and its accuracy: a systematic review. Eur. J. Clin. Pharmacol. 74, 1365-1376. ( 10.1007/s00228-018-2513-6) [DOI] [PubMed] [Google Scholar]
- 24.Shebley M et al. 2018. Physiologically based pharmacokinetic model qualification and reporting procedures for regulatory submissions: a consortium perspective. Clin. Pharmacol. Ther. 104, 88-110. ( 10.1002/cpt.1013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poleszczuk JT, Luddy KA, Prokopiou S, Robertson-Tessi M, Moros EG, Fishman M, Djeu JY, Finkelstein SE, Enderling H. 2016. Abscopal benefits of localized radiotherapy depend on activated T-cell trafficking and distribution between metastatic lesions. Cancer Res. 76, 1009-1018. ( 10.1158/0008-5472.CAN-15-1423) [DOI] [PubMed] [Google Scholar]
- 26.Walker R, Poleszczuk J, Pilon-Thomas S, Kim S, Anderson AARA, Czerniecki BJ, Harrison LB, Moros EG, Enderling H. 2018. Immune interconnectivity of anatomically distant tumors as a potential mediator of systemic responses to local therapy. Sci. Rep. 8, 1-11. ( 10.1038/s41598-018-27718-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu H, Melder RJ, Baxter LT, Jain RK. 1996. Physiologically based kinetic model of effector cell biodistribution in mammals: implications for adoptive immunotherapy. Cancer Res. 56, 3771-3781. [PubMed] [Google Scholar]
- 28.Melder RJ, Munn LL, Stoll BR, Marecos EM, Baxter LT, Weissleder R, Jain RK. 2002. Systemic distribution and tumor localization of adoptively transferred lymphocytes in mice: comparison with physiologically based pharmacokinetic model. Neoplasia 4, 3-8. ( 10.1038/sj/neo/7900209) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Friedrich SW, Lin SC, Stoll BR, Baxter LT, Munn LL, Jain RK. 2002. Antibody-directed effector cell therapy of tumors: analysis and optimization using a physiologically based pharmacokinetic model. Neoplasia 4, 449-463. ( 10.1038/sj.neo.7900260) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khot A, Matsueda S, Thomas VA, Koya RC, Shah DK. 2019. Measurement and quantitative characterization of whole-body pharmacokinetics of exogenously administered T cells in mice. J. Pharmacol. Exp. Ther. 368, 503-513. ( 10.1124/jpet.118.252858) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ganusov VV, Auerbach J. 2014. Mathematical modeling reveals kinetics of lymphocyte recirculation in the whole organism. PLoS Comput. Biol. 10, e1003586. ( 10.1371/journal.pcbi.1003586) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu H et al. 2011. Modeling of influenza-specific CD8+ T cells during the primary response indicates that the spleen is a major source of effectors. J. Immunol. 187, 4474-4482. ( 10.4049/jimmunol.1101443) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, Beliles RP. 1997. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol. Ind. Health 13, 407-84. ( 10.1177/074823379701300401) [DOI] [PubMed] [Google Scholar]
- 34.Graf JF, Scholz BJ, Zavodszky MI. 2012. BioDMET: a physiologically based pharmacokinetic simulation tool for assessing proposed solutions to complex biological problems. J. Pharmacokinet. Pharmacodyn. 39, 37-54. ( 10.1007/s10928-011-9229-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baxter LT, Zhu H, Mackensen DG, Jain RK. 1994. Physiologically based pharmacokinetic model for specific and nonspecific monoclonal antibodies and fragments in normal tissues and human tumor xenografts in nude mice. Cancer Res. 54, 1517-1528. (ISSN 0008–5472) [PubMed] [Google Scholar]
- 36.Shah DK, Betts AM. 2012. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J. Pharmacokinet. Pharmacodyn. 39, 67-86. ( 10.1007/s10928-011-9232-2) [DOI] [PubMed] [Google Scholar]
- 37.Peters SA. 2012. Physiologically-based pharmacokinetic (PBPK) modeling and simulations: principles, methods, and applications in the pharmaceutical industry. New York, NY: Wiley. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Valentin J. 2002. Basic anatomical and physiological data for use in radiological protection reference values. ICRP Publication 89. Ann. ICRP 32, 1-277. [PubMed] [Google Scholar]
- 39.Rolstad B, Herberman RB, Reynolds CW. 1986. Natural killer cell activity in the rat. V. The circulation patterns and tissue localization of peripheral blood large granular lymphocytes (LGL). J. Immunol. 136, 2800-2808. (https://www.ncbi.nlm.nih.gov/pubmed/3485674) [PubMed] [Google Scholar]
- 40.Brand J-M, Meller B, Von Hof K, Luhm J, Bähre M, Kirchner H, Frohn C. 2004. Kinetics and organ distribution of allogeneic natural killer lymphocytes transfused into patients suffering from renal cell carcinoma. Stem Cells Dev. 13, 307-314. ( 10.1089/154732804323099235) [DOI] [PubMed] [Google Scholar]
- 41.Meller B, Frohn C, Brand J-M, Lauer I, Schelper LF, von Hof K, Kirchner H, Richter E, Baehre M. 2004. Monitoring of a new approach of immunotherapy with allogenic (111)In-labelled NK cells in patients with renal cell carcinoma. Eur. J. Nucl. Med. Mol. Imaging 31, 403-407. ( 10.1007/s00259-003-1398-4) [DOI] [PubMed] [Google Scholar]
- 42.Sridhar P, Petrocca F. 2017. Regional delivery of chimeric antigen receptor (CAR) T-cells for cancer therapy. Cancers 9, 1-10. ( 10.3390/cancers9070092) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hartmann J, Schüßler-Lenz M, Bondanza A, Buchholz CJ. 2017. Clinical development of CAR T cells–challenges and opportunities in translating innovative treatment concepts. EMBO Mol. Med. 9, e201607485. ( 10.15252/emmm.201607485) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vibert J, Thomas-Vaslin V. 2017. Modelling T cell proliferation: dynamics heterogeneity depending on cell differentiation, age, and genetic background. PLoS Comput. Biol. 13, 1-25. ( 10.1371/journal.pcbi.1005417) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gattinoni L et al. 2005. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J. Clin. Invest. 115, 1616-1626. ( 10.1172/JCI24480) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang C et al. 2017. Phase I escalating-dose trial of CAR-T therapy targeting CEA+metastatic colorectal cancers. Mol. Ther. 25, 1248-1258. ( 10.1016/j.ymthe.2017.03.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiao Y, Freeman GJ. 2015. The microsatellite instable subset of colorectal cancer is a particularly good candidate for checkpoint blockade immunotherapy. Cancer Discov. 5, 16-18. ( 10.1158/2159-8290.CD-14-1397) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gina Columbus. 2016 Expert Discusses Microsatellite Instability, Immunotherapy in CRC. See http://www.onclive.com/web-exclusives/expert-discusses-microsatellite-instability-immunotherapy-in-crc. (accessed 6 December 2016).
- 49.Rosenberg SA. 2007. Interleukin 2 for patients with renal cancer. Nat. Clin. Pract. Oncol. 4, 497. ( 10.1038/ncponc0926) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reck M et al. 2016. Pembrolizumab versus chemotherapy for PD-L1–positive non-small-cell lung cancer. N. Engl. J. Med. 375, 1823-1833. ( 10.1056/NEJMoa1606774) [DOI] [PubMed] [Google Scholar]
- 51.Fellner C. 2012. Ipilimumab (yervoy) prolongs survival in advanced melanoma. P&T 37, 503-512. [PMC free article] [PubMed] [Google Scholar]
- 52.Amin A, White RL. 2014. Interleukin-2 in renal cell carcinoma: a has-been or a still-viable option? J. Kidney Cancer and VHL 1, 74-83. ( 10.15586/jkcvhl.2014.18) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith ME, Ford WL. 1983. The recirculating lymphocyte pool of the rat: a systematic description of the migratory behaviour of recirculating lymphocytes. Immunology 49, 83-94. [PMC free article] [PubMed] [Google Scholar]
- 54.Kershaw MH et al. 2006. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 12, 6106-6115. ( 10.1158/1078-0432.CCR-06-1183) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Parente-Pereira AC et al. 2011. Trafficking of CAR-engineered human T cells following regional or systemic adoptive transfer in SCID beige mice. J. Clin. Immunol. 31, 710-718. ( 10.1007/s10875-011-9532-8) [DOI] [PubMed] [Google Scholar]
- 56.Lahtinen T, Karjalainen P, Alhava EM. 1979. Measurement of bone blood flow with a 133Xe washout method. Eur. J. Nuclear Med. 4, 437. ( 10.1007/BF00300841) [DOI] [PubMed] [Google Scholar]
- 57.Scheiermann C, Gibbs J, Ince L, Loudon A. 2018. Clocking in to immunity. Nat. Rev. Immunol. 18, 423-437. ( 10.1038/s41577-018-0008-4) [DOI] [PubMed] [Google Scholar]
- 58.Wang L et al. 2016. Efficient tumor regression by adoptively transferred CEA-specific CAR-T cells associated with symptoms of mild cytokine release syndrome. OncoImmunology 5, 1-13. ( 10.1080/2162402X.2016.1211218) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chmielewski M, Hahn O, Rappl G, Nowak M, Schmidt-Wolf IH, Hombach AA, Abken H. 2012. T cells that target carcinoembryonic antigen eradicate orthotopic pancreatic carcinomas without inducing autoimmune colitis in mice. Gastroenterology 143, 1095-1107.e2. ( 10.1053/j.gastro.2012.06.037) [DOI] [PubMed] [Google Scholar]
- 60.Jiang S-S et al. 2015. A phase I clinical trial utilizing autologous tumor-infiltrating lymphocytes in patients with primary hepatocellular carcinoma. Oncotarget 6, 41 339-41 349. ( 10.18632/oncotarget.5463) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Foppen MHG, Donia M, Svane IM, Haanen JBAG. 2015. Tumor-infiltrating lymphocytes for the treatment of metastatic cancer. Mol. Oncol. 9, 1918-1935. ( 10.1016/j.molonc.2015.10.018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morgan RA et al. 2007. Cancer regression in patients after transfer of genetically engineered lymphocytes. In Vitro 126, 126-130. ( 10.1126/science.1129003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tran E et al. 2016. T-cell transfer therapy targeting mutant KRAS in cancer. N. Engl. J. Med. 375, 2255-2262. ( 10.1056/NEJMoa1609279) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schirrmann T, Pecher G. 2002. Human natural killer cell line modified with a chimeric immunoglobulin T-cell receptor gene leads to tumor growth inhibition in vivo. Cancer Gene Ther. 9, 390-398. ( 10.1038/sj.cgt.7700453) [DOI] [PubMed] [Google Scholar]
- 65.Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, Jones DR, Sadelain M. 2014. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci. Transl. Med. 6, 261ra151. ( 10.1126/scitranslmed.3010162) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.20202. Malignant Pleural Disease Treated With Autologous T Cells Genetically Engineered to Target the Cancer-Cell Surface Antigen Mesothelin - ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT02414269. Clinical trial NCT02414269 (accessed May 2020).
- 67.Spiess PJ, Yang JC, Rosenberg SA. 1987. In vivo antitumor activity of tumor-infiltrating lymphocytes expanded in recombinant interleukin-2. J. Natl Cancer Inst. 79, 1067-1075. [PubMed] [Google Scholar]
- 68.Kallinowski F, Schlenger KH, Runkel S, Kloes M, Stohrer M, Okunieff P, Vaupel P. 1989. Blood flow, metabolism, cellular microenvironment, and growth rate of human tumor xenografts. Cancer Res. 49, 3759-3764. [PubMed] [Google Scholar]
- 69.Willyard C. 2018. The mice with human tumours: growing pains for a popular cancer model. Nature 560, 156-157. ( 10.1038/d41586-018-05890-8) [DOI] [PubMed] [Google Scholar]
- 70.Shiina S et al. 2016. CAR T cells targeting podoplanin reduce orthotopic glioblastomas in mouse brains. Cancer Immunol. Res. 4, 259-268. ( 10.1158/2326-6066.CIR-15-0060) [DOI] [PubMed] [Google Scholar]
- 71.Mount CW et al. 2018. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M+ diffuse midline gliomas. Nat. Med. 24, 572-579. ( 10.1038/s41591-018-0006-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li D et al. 2020. Persistent polyfunctional chimeric antigen receptor T cells that target glypican 3 eliminate orthotopic hepatocellular carcinomas in mice. Gastroenterology 158, 2250-2265. ( 10.1053/j.gastro.2020.02.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mohammed RN, Watson HA, Vigar M, Ohme J, Thomson A, Humphreys IR, Ager A. 2016. L-selectin is essential for delivery of activated CD8+ T cells to virus-infected organs for protective immunity. Cell Rep. 14, 760-771. ( 10.1016/j.celrep.2015.12.090) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Masopust D, Schenkel JM. 2013. The integration of T cell migration, differentiation and function. Nat. Rev. Immunol. 13, 309-320. ( 10.1038/nri3442) [DOI] [PubMed] [Google Scholar]
- 75.Mestas J, Hughes CCW. 2004. Of mice and not men: differences between mouse and human immunology. J. Immunol. 172, 2731-2738. ( 10.4049/jimmunol.172.5.2731) [DOI] [PubMed] [Google Scholar]
- 76.Dings RPM, Loren M, Heun H, McNiel E, Griffioen AW, Mayo KH, Griffin RJ. 2007. Scheduling of radiation with angiogenesis inhibitors anginex and avastin improves therapeutic outcome via vessel normalization. Clin. Cancer Res. 13, 3395-3402. ( 10.1158/1078-0432.CCR-06-2441) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Amit L, Ben-Aharon I, Vidal L, Leibovici L, Stemmer S. 2013. The impact of bevacizumab (avastin) on survival in metastatic solid tumors – a meta-analysis and systematic review. PLoS ONE 8, e51780. ( 10.1371/journal.pone.0051780) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. 2010. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 18, 843-851. ( 10.1038/mt.2010.24) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lamers CHJ, Klaver Y, Gratama JW, Sleijfer S, Debets R. 2016. Treatment of metastatic renal cell carcinoma (mRCC) with CAIX CAR-engineered T-cells—a completed study overview. Biochem. Soc. Trans. 44, 951-959. ( 10.1042/BST20160037) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Code and parameter data for humans, mice and rats used to generate the results of this work are available as electronic supplementary materials.