

Abstract
Protein kinase signalling, which transduces external messages to mediate cellular growth and metabolism, is frequently deregulated in human disease, and specifically in cancer. As such, there are 77 kinase inhibitors currently approved for the treatment of human disease by the FDA. Due to their historical association as the receptors for the tumour-promoting phorbol esters, PKC isozymes were initially targeted as oncogenes in cancer. However, a meta-analysis of clinical trials with PKC inhibitors in combination with chemotherapy revealed that these treatments were not advantageous, and instead resulted in poorer outcomes and greater adverse effects. More recent studies suggest that instead of inhibiting PKC, therapies should aim to restore PKC function in cancer: cancer-associated PKC mutations are generally loss-of-function and high PKC protein is protective in many cancers, including most notably KRAS-driven cancers. These recent findings have reframed PKC as having a tumour suppressive function. This review focusses on a potential new mechanism of restoring PKC function in cancer – through targeting of its negative regulator, the Ser/Thr protein phosphatase PHLPP. This phosphatase regulates PKC steady state levels by regulating the phosphorylation of a key site, the hydrophobic motif, whose phosphorylation is necessary for the stability of the enzyme. We also consider whether the phosphorylation of the potent oncogene KRAS provides a mechanism by which high PKC expression may be protective in KRAS-driven human cancers.
Introduction
Protein Kinase C and Phorbol Esters
Protein kinase C (PKC) isozymes are members of the AGC family of Ser/Thr protein kinases. They are activated in response to extracellular signals and phosphorylate a wide variety of substrates to regulate cell growth, metabolism and also cell death and senescence. PKC isozymes were initially studied in cancer following their identification as the ‘receptors’ for tumour promoting phorbol esters. These compounds embed in cell membranes and mimic binding of the natural agonist diacylglycerol (DAG) to acutely activate PKC (1,2). Consequently, PKC became a target of cancer therapies, with many trials focussing on PKC inhibition. Unlike DAG, which is transiently elevated in response to extracellular signals, phorbol esters are metabolically stable, and therefore cause sustained signalling of PKC. However, chronic activation of PKC by these compounds ultimately promotes the degradation of PKC, resulting in paradoxical inhibition of the pathway (3). Indeed, before the development of targeted genetic knockdown approaches, overnight treatment with the phorbol ester, phorbol myristate acetate (PMA),was a routine approach to deplete cells of PKC (4). Recent meta-analysis of the use of PKC inhibitors in clinical trials determined that not only were these treatments ineffective, in some cases they may have been detrimental. This meta-analysis focussed on randomised clinical trials for treatment of recurrent or malignant non-small cell lung cancer (NSCLC), in which combination of PKC inhibitors enzastaurin, an ATP competitive inhibitor (https://pubmed.ncbi.nlm.nih.gov/12749884/) or aprinocarsen, a PKCa antisense oligonucleotide (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC45312/), with standard chemotherapy was compared to chemotherapy alone (pemetrexed, gemcitabine, cisplatin). Patients receiving PKC inhibitors in addition to standard chemotherapy exhibited a significantly decreased response rate and disease control rate, with a significant increase in adverse effects, compared to patients receiving standard chemotherapy (5). These data suggested that our understanding of the role of PKC in oncogenic signalling was incomplete (6), and that inhibition of PKC does not generally show therapeutic efficacy. PKC is subject to complex regulatory mechanisms and understanding these mechanisms provide the foundation for understanding how to appropriately target PKC in cancer.
PKC is basally phosphorylated, autoinhibited and stable
There are 10 major PKC isozymes, encoded by 9 genes, categorised into 3 subtypes based on their domain structure and mechanism of activation (Fig 1). All PKC isozymes contain a conserved C-terminal Kinase domain, with an AGC protein kinase regulatory C tail, and variable N-terminal regulatory region which binds second messengers to mediate activation of PKC. Conventional PKC isozymes (cPKC isozymes; PKCα, the alternatively spliced βI and βII, γ) contain two tandem C1 domains (C1A, C1B), which bind DAG on the plasma membrane and on internal membranes such as the Golgi, and a C2 domain which upon binding to Ca2+ associates with the plasma membrane where it binds PIP2. Novel PKC isozymes (nPKC isozymes; PKCδ, ε, η, θ) contain a novel C2 domain which does not bind Ca2+, and instead activation is mediated solely by binding of DAG to the C1 domains. Atypical PKC isozymes (aPKC isozymes; PKCξ, ι) do not contain these membrane targeting modules, and instead have an N-terminal PB1 protein interaction domain, which allows their scaffolding to substrates. Importantly, all PKC isozymes contain an N-terminal pseudosubstrate segment, which sits in the active site of the kinase, maintaining the enzyme in an inactive, stable conformation until its activation by second messengers. With the exception of aPKC family members, PKC isozymes are activated by DAG and, in the case of cPKCs, also Ca2+, produced by type C phospholipases in response to extracellular signals (reviewed in (7)).
Figure 1: Primary domain structure of conventional, novel and atypical PKC isozymes.
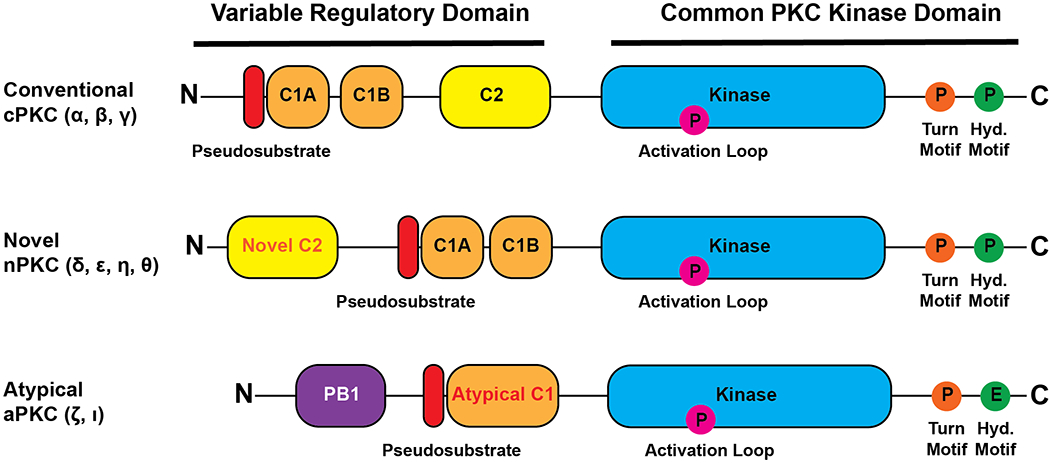
The conventional, novel and atypical PKC isozymes share a common C-terminal PKC Kinase domain (blue), with regulatory C-tail. The family members differ in their variable N-terminal regulatory modules. All isozymes contain an autoinhibitory pseudosubstrate (red). Conventional and novel PKC isozymes contain tandem DAG-binding C1 domains (orange) and a C2 domain (yellow). The C2 domain of conventional PKC isozymes, but not novel PKC isozymes, is a Ca2+-regulated plasma membrane sensor, with determinants for PIP2 binding. Atypical PKC isozymes contain a PB1 protein interaction module (purple) and an atypical C1 domain which does not bind DAG.
PKC processing and life cycle
Upon initial translation, PKC is in an open state with its domains exposed (Fig 2A). Phosphorylation at three conserved sites ‘prime’ the enzyme to adopt an autoinhibited and stable conformation. Without these phosphorylations, the enzyme is unstable and subject to proteasomal degradation, described below. The first two phosphorylations are catalysed by upstream kinases: the activation loop (T500 for PKCβI/II) and turn motif (T641 for PKCβII) residues are phosphorylated by PDK1 and mTORC2 respectively. These phosphorylations allow for the low-level activation of PKC, triggering the final phosphorylation, autophosphorylation of its hydrophobic motif (S660 for PKCβII (reviewed in (7)). These priming phosphorylations align residues in the active site for catalysis and promote the positioning of its regulatory pseudosubstrate segment within its active site, ‘clamped’ in place by the C2 domain (Fig 2B) (8,9). This autoinhibited, primed form of PKC is exceptionally stable, with a half-life of days in the cell. If PKC fails to form this ‘primed’ state, it remains in an open state, with its phosphorylation sites labile to dephosphorylation (Fig 2C). Note that once ‘primed’, the enzyme retains activity following dephosphorylation of the hydrophobic motif, but it is unstable (10). It is this ‘primed’ PKC that transduces signals upon stimulation with second messengers.
Figure 2: Life cycle of PKC and quality control by PHLPP1.
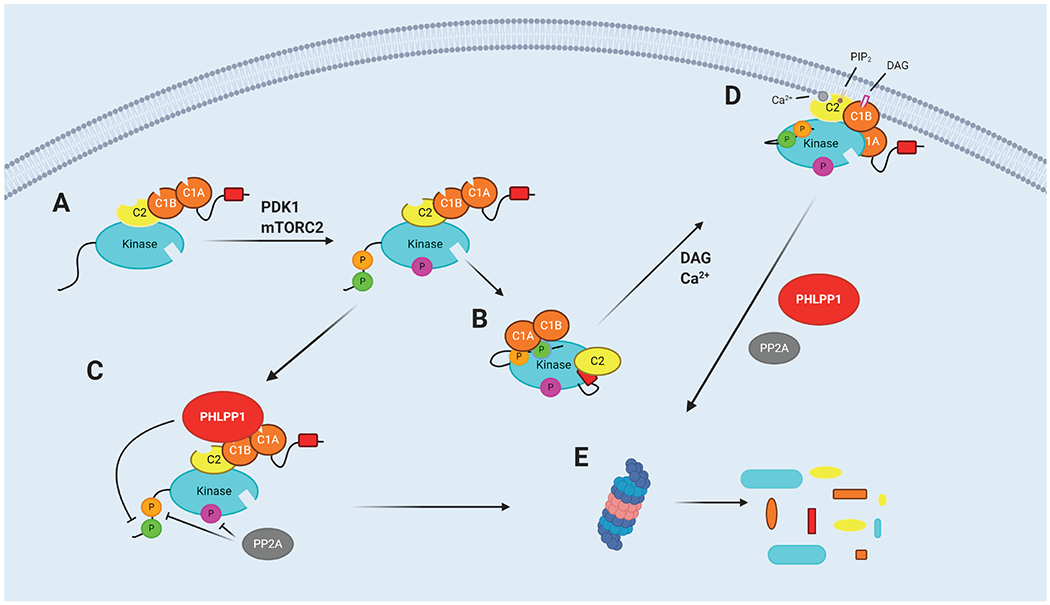
A. Newly translated PKC is unphosphorylated and present in an open state. B. Upon phosphorylation by PDK1 and mTORC2, PKC adopts an autoinhibited state with its pseudosubstrate in the active site. C. PKC that does not properly autoinhibit remains in an open conformation and is labile to dephosphorylation by PHLPP1. D. In response to second messengers DAG (pink rectangle) and Ca2+ (grey circle), PKC translocates to the plasma membrane, a conformational change removes the pseudosubstrate from the active site, and PKC is active to phosphorylate substrates E. PKC which is dephosphorylated by PHLPP1/2 is sensitive to polyubiquitylation and proteasomal degradation. Created with BioRender.com
Conventional and novel PKC isozymes are activated by receptor-mediated hydrolysis of phospholipids (11). One major pathway is through activation of phospholipase C (PLC) which both generates DAG and elevates intracellular Ca2+. This phospholipase is activated in response to extracellular signals such as hormones and growth factors via G-protein-coupled receptors (GPCR) and receptor tyrosine kinases (RTKs). PLC catalyses the hydrolysis of PtdIns(4,5)P2 (PIP2) on the plasma membrane to release cytosolic IP3 and membrane-bound DAG. IP3 binds to IP3 receptors on, for example, the endoplasmic reticulum, triggering the release of Ca2+ from intracellular stores and increasing Ca2+ concentrations within cells. The PKC C2 domain binds Ca2+, facilitating C2 binding to PIP2 on the membrane (12,13). At the plasma membrane, the membrane-embedded allosteric activator, diacylglycerol, binds one of the tandem C1 domains. Novel PKC isozymes, containing a novel C2 domain which does not bind Ca2+, are instead reliant on solely their C1 domains for activation; a single amino acid difference between the C1B domains of conventional and novel PKCs (Tyr to Trp, respectively) results in nPKC isozymes binding to DAG with two orders of magnitude higher affinity (14). As such, novel PKC isozymes are often activated on Golgi membranes (15,16), which contain significant amounts of DAG (https://www.sciencedirect.com/science/article/pii/0968000488900825?via%3Dihub) https://pubmed.ncbi.nlm.nih.gov/9695805/. Importantly, membrane binding causes a conformational change in PKC that results in release of the pseudosubstrate from the active site of the kinase and favours an open, active conformation allowing phosphorylation of cellular substrates (Fig 2D).
However, this active membrane-bound conformation of PKC is also labile to dephosphorylation, with biochemical studies revealing that the rate of dephosphorylation is accelerated by two orders of magnitude upon membrane binding (17). Under situations of prolonged activation by chronic signals or by, for example, the tumorigenic phorbol esters, PKC is dephosphorylated on its hydrophobic motif by the PPM phosphatase PHLPP described in the following section, and its turn motif and activation loop by PP2A (18,19). It is specifically the dephosphorylation of the hydrophobic motif which signals for degradation of PKC (3,10). Open, dephosphorylated PKC is polyubiquitylated and degraded via the proteasome (Fig 2E) (20,21), although one report suggests that phosphorylated PKC activated at the plasma membrane can also be degraded by a distinct pathway (BLACK 2003). Thus, protein-level downregulation of PKC results from upon chronic activation.
Degradation of PKC
E3 ligases have been identified that selectively regulate the degradation of PKC under basal conditions or the degradation following phorbol ester stimulation, suggesting at least two distinct degradation mechanisms. Basal levels of PKC are regulated by the E3 ligase TRIM41/RINCK (21). RINCK has been demonstrated to directly ubiquitylate PKC both in vitro and in cells, an event that reduces steady state levels of PKCβII by a proteosome-dependent mechanism. Conversely, genetic knockdown of RINCK by siRNA was shown to increase the steady state levels of PKCα, PKCβII and PKCζ in HeLa cells. However, siRNA knockdown of RINCK did not influence the phorbol ester induced degradation of PKC, indicating that this downregulation occurs through a different pathway. Nonetheless, phorbol ester induced downregulation of PKC also occurs through ubiquitylation and proteasomal degradation (22–25), albeit through a different E3 ligase. The E3 ligase complex LUBAC, comprised of HOIP and HOIL-1L, has been reported to preferentially bind and ubiquitylate the activated, open species of cPKC isozymes, triggering their degradation (26). Whereas steady-state levels of PKCa were reported to be equal in WT and HOIL-1L−/− MEFS, PKCα was relatively resistant to PMA-induced down-regulation in MEFs lacking this E3 ligase complex. PKCλ has similarly been demonstrated to be degraded through the ubiquitin-proteasome system through the E3 ligase pVHL (27). Whether additional E3 ligases regulate both the basal and phorbol ester dependent downregulation of PKC remains to be established.
While different E3 ligases may mediate the ubiquitylation of PKC isozymes under basal conditions and activation-induced downregulation, both pathways are dependent on the dephosphorylation of the hydrophobic motif site by PHLPP. siRNA knockdown of PHLPP1 or PHLPP2 inhibited the phorbol ester-induced dephosphorylation of endogenous PKCα at the hydrophobic motif, and also increased PKC protein expression in cancer cells (20). Similarly, PHLPP1−/− MEFS have greater basal PKCα expression and a reduced rate of PDBu-induced PKCα downregulation (10). By dephosphorylation of poorly primed or chronically activated PKC, PHLPP can carefully tune signalling output by regulation of protein levels.
PHLPP1 and Protein Kinase C Quality Control
The PH domain leucine-rich repeat protein phosphatase (PHLPP) isozymes, PHLPP1 and PHLPP2, are Ser-Thr phosphatases within the protein phosphatase magnesium/manganese-dependent (PPM) “shrub” of the phosphatome (28). PHLPP1/2 signalling has been identified as an important regulator of many disease processes, such as in oncogenic signalling and regulation of immunity. To confer this array of functions, PHLPP dephosphorylates a broad range of substrates. Many phosphatases such as the PP1/PP2A class phosphatases elicit selectivity towards substrates through interaction with additional regulatory modules; however PPM phosphatases such as PHLPP1/2 encode the catalytic and any regulatory domains within one polypeptide chain (29). PHLPP1/2 are encoded by two separate genes, yet they share a similar structure with several conserved domains including a pleckstrin homology (PH) domain, a hydrophobic leucine-rich repeat (LRR) region, a PP2C family phosphatase domain and a C-terminal PDZ-binding motif (Fig 3A) (30). In addition to these conserved regions, PHLPP1 contains an additional N-terminal extension (NTE), whose function has not yet been fully characterised, however has been demonstrated to contain a bipartite Nuclear Localisation Signal (NLS) that drives PHLPP1’s function in the nucleus (31). PHLPP1 also contains a nuclear export sequence (NES) between the LRR and PP2C domains.
Figure 3: Hydrophobic motif phosphorylation regulates Akt activity and PKC protein levels.
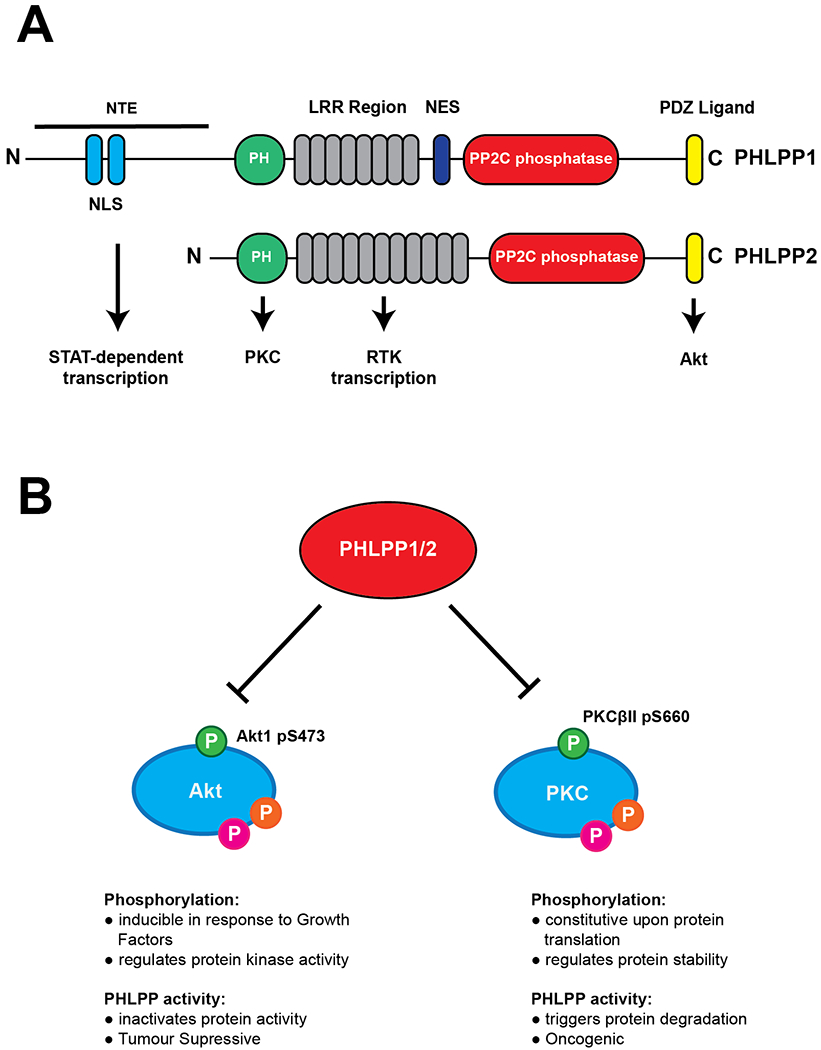
A. PHLPP1 and PHLPP2 phosphatases contain a conserved PP2C phosphatase domain (red) and a number of regulatory modules. The PH (green), Leucine-rich repeats (grey) and PDZ-ligand (yellow) domain all mediate binding of a myriad of substrates. PHLPP1 differs by an N-terminal Extension (NTE), which contains a bipartite Nuclear Localisation Signal and allows targeting of PHLPP1 to its nuclear substrates, and a Nuclear Export Signal (NES). B. Phosphorylation state of the hydrophobic motif has different functional effects on Akt and PKC. Ser473 phosphorylation of Akt is inducible, regulates activity of Akt towards certain substrates, but does not affect protein stability. In contrast, Ser660 phosphorylation of PKCβII regulates protein stability and total protein levels. Activation loop (Magenta), Turn Motif (Orange) and Hydrophobic Motif (Green) phosphorylation sites are shown.
PHLPP1/2 have relatively low catalytic activity, with the activity of their isolated phosphatase domains measured to be on the order of one reaction per second in an in vitro phosphatase assay toward phosphopeptide substrates (32) or purified Akt (33). Therefore intracellular targeting to substrates via these regulatory regions is critical for protein function (34). For example, the LRR region is critical for the regulation of receptor tyrosine kinase transcription, through modification of histone phosphorylation and acetylation (35), and the PDZ-binding motif is critical for scaffolding of PHLPP to the plasma membrane near Akt, to drive its dephosphorylation (33). PHLPP1 and PHLPP2 are divergent in their PDZ-binding motif (DTPL and DTAL for PHLPP1/2 respectively), and regulate different Akt isoforms and, therefore, different Akt substrates (36). The PH domain, which as an isolated domain has been shown to have weak affinity both in cells and in vitro for phosphoinositides (37,38), is required for PHLPP to bind and dephosphorylate PKC in cells (20) (Fig 3A).
The first identified substrate of PHLPP was the hydrophobic motif site of Akt (Ser473 for Akt1) (33). This phosphorylation site in the regulatory C-tail of the kinase permits its full activation and phosphorylation of downstream substrates. This phosphorylation is inducible, by mTORC2 downstream of PI3K signalling, and is also very phosphatase labile and easily removed by PHLPP. PHLPP was later demonstrated to dephosphorylate the hydrophobic motif of other AGC protein kinases such as S6K1 (39), to modulate its activity, and PKC. Unlike the inducible, stimulus-sensitive phosphorylation of Akt, the hydrophobic motif site of PKC (S660 for PKCβII) is one of three stable, priming phosphorylations which occur during initial translation and processing of PKC, maintaining the protein in a stable, autoinhibited state (Fig 3B). Atypical PKC isozymes contain a glutamate residue instead of a serine at their hydrophobic motif site and therefore are not directly regulated by PHLPP, although PHLPP has been reported to indirectly regulate these isozymes (40).
Balance of PHLPP and PKC expression in human cancer
As c/nPKC isozymes are stable in an autoinhibited state, the PKC signalling output in cells is regulated at the level of protein expression. PKC protein expression levels are therefore important biomarkers for activity of the signalling pathway in cancer. Recent data have demonstrated that PKC protein levels, mediated by PHLPP1 activity, may play a key, protective role in certain cancers.
Consistent with PKC protein levels being controlled by hydrophobic motif modulation by PHLPP, Reverse Phase Protein Array (RPPA) analysis of >5000 tumour samples revealed a statistically significant 1:1 correlation between PKCα/β protein levels and hydrophobic motif phosphorylation (10). Total PKC expression levels were also negatively correlated with PHLPP1 expression. This confirmed that PKC in tumour samples is phosphorylated, and that dephosphorylated PKC is degraded. Whereas this correlation was clear for PKC, the same was not true for Akt or S6K1, whose hydrophobic motif phosphorylation is inducible, regulates activity, and does not regulate protein stability (Fig 3C) (10). It is interesting that the phosphorylation on the same site, the hydrophobic motif, has divergent roles in these related kinases: it controls the chronic stability (but not activity) of PKC and the acute activity (but not stability) of Akt.
Tumour suppressive functions of Protein Kinase C
As discussed above, PKC isozymes were targeted with inhibitors for many years in clinical trials, however this approach showed no therapeutic benefit (5). More recent studies from our lab and others have reframed PKC as having a tumour suppressive function, suggesting that instead of inhibiting PKC, therapies should aim to increase PKC signalling in cancer. Analysis of cancer-associated mutations revealed that the majority of these mutations were in fact loss of function, either inactivating kinase activity or critically impacting stability and ultimately protein levels (41). Further, correction of a PKCβ A509T mutation in the DLD-1 colorectal cancer cell line both restored PKC kinase activity and increased PKCβII protein levels, and the resulting cells had reduced cell growth in vitro and tumour development in vivo (41). A heterozygous knockout of the mutant PKCβ in these cells had a partial effect, indicating that PKCβ was haploinsufficient and that total protein levels are important in the tumour suppressive function of PKC.
Two particular cancers in which high PKC expression has been demonstrated to be protective are colorectal carcinoma (CRC) and pancreatic adenocarcinoma (PDAC). In a study of protein expression in PDAC tumour samples, high levels of PKCα and PKCβ, measured by their hydrophobic motif phosphorylation by RPPA, were associated with increased survival. Patients with high PKCβ had a remarkable 5 year survival rate of 50%, whereas those with low PKCβ had no survival past 5 years (Fig 4B) (10). Activation of PKC with prostratin, a weak PKC activator (42), was sufficient to inhibit the mutant KRAS-driven growth of pancreatic cancer cells both in vitro and in an orthotopic transplant model (43)
Figure 4: High expression of PKC is associated with improved survival in PDAC and CRC.
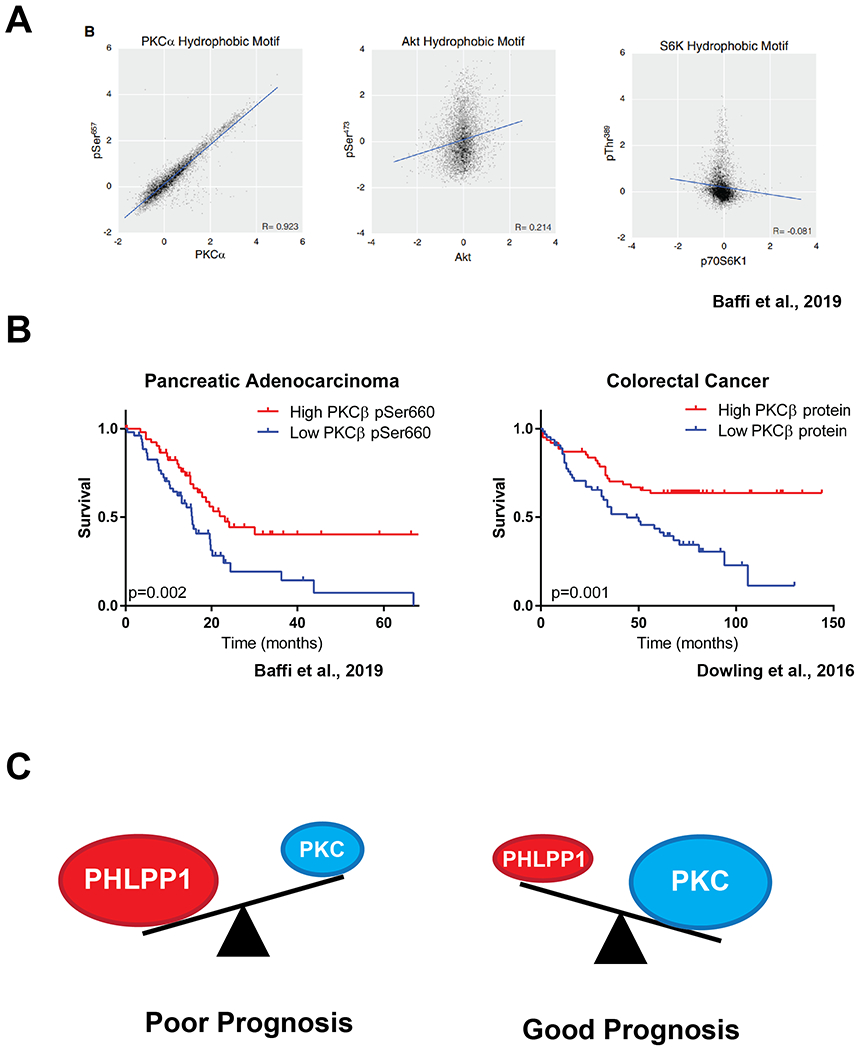
A. RPPA analysis of hydrophobic motif phosphorylation and total protein levels of PKCα, Akt and S6K1 (from (10)). B. RPPA analysis of 105 PDAC tumour samples revealed high levels of PKCβII hydrophobic motif phosphorylation correlates with significantly improved survival outcomes. From (10). Tissue microarray analysis of CRC patients with increased PKCβII protein expression in tissue distant from the tumour had increased survival outcomes. From (44). C. Model: The balance of PKC and PHLPP1 levels in PDAC/CRC correlate with survival outcomes. High PHLPP1, resulting in low PKC, is a marker of poor prognosis, whereas low PHLPP1, resulting in high PKC, is marker of good prognosis.
Dowling et al. demonstrated that PKCβII functions as a tumour suppressor in colorectal cancer, and that low expression of PKCβII was associated with decreased disease-free survival. Patients with low levels of PKCβII, measured by microarray analysis of tissue distal to the tumour, had a low (10%) 10 year survival, whereas patients with high expression had a 10 year survival rate of 60% (Fig 4C) (44). In a separate study, immunohistochemical analysis of >200 human CRC patient samples determined that low expression of PKCα, and high expression of KRAS, was a biomarker of poor prognosis in colorectal cancer patients (45). PKCα also has a demonstrated tumour suppressive role in an APCmin model of intestinal tumorigenesis (46). PKCα/δ phosphorylate Sur8, a scaffold of MAPK signalling overexpressed in CRC, and trigger its ubiquitylation and degradation (47). Sur8 and PKCα/δ protein levels were also negatively correlated in human CRC tumour samples (47). Dupasquier et al. investigated whether enhancing PKCα function could be beneficial in CRC. They determined, in both in vitro and transgenic mouse models, that enhancing PKCα signalling inhibited growth and triggered CRC cell death, whereas overexpression of PKCα was not deleterious in normal intestinal epithelium, suggesting PKCα could be therapeutically targeted in CRC (48).
Within three transgenic mouse models of KRAS-driven lung cancer, loss of PKCα resulted in an increase in tumour number, burden and grade. The PKCα-mediated suppression of tumour growth was regulated through control of oncogene-induced senescence (OIS) by activation of a p38 MAPK-TGFβ signalling axis which induced cellular growth arrest in vitro (49). Ohm et al. demonstrated that within KRAS-mutated lung adenocarcinoma high PRKCD (PKCδ) mRNA expression correlated with improved survival outcomes (50). However, the authors also discovered that PKCδ could perform pro-apoptotic or pro-tumorigenic functions, and this role of PKCδ in lung adenocarcinoma was segregated according to KRAS-dependence of the chosen cell line. In a panel of 17 KRAS-mutant NSCLC cell lines, KRAS-dependent cell lines required PKCδ for survival, whereas in KRAS-independent cell lines PKCδ had a pro-apoptotic and tumour-suppressive role (50). While mouse models and human patient samples suggest a tumour suppressive role for PKCα and PKCδ, a very recent study from Garg et al., suggested that PKCε expression is required for KRAS-driven lung tumorigenesis (51). Stratification of patient primary lung adenocarcinomas according to KRAS mutation status and PRKCE expression revealed increased survival in patients with low PRKCE expression. PKCα and PKCδ may both be tumour suppressive in endometrial cancer. Hsu et al. determined that PKCα functioned as a tumour suppressor in endometrium, by regulating Akt through PP2A (52). Further Reno et al. demonstrated that PKCδ expression was negatively correlated with increasing tumour grade in endometrial cancer (53).
In the skin-tumour model of tumour promotion by phorbol esters, tumours develop after a tumour-initiation event, such as application of the carcinogen DMBA, and repeated application of phorbol ester to the skin for tumour-promotion. Such experiments attributed an oncogenic role to PKC, however under our new understanding of PKC downregulation in response to chronic phorbol exposure, these treatments would result in tumorigenesis through loss of PKC. In support of this new model, transgenic mice which overexpress PKCδ were resistant to phorbol-induced tumorigenesis (54,55).
Overall, it is clear from recent studies that high PKC expression dampens oncogenic signalling in a range of cancer types. In particular, PDAC and CRC patient data show that higher PKC expression is associated with improved survival outcomes. In a pan-cancer analysis, PKC expression was tightly correlated with hydrophobic motif phosphorylation, and negatively correlated with expression of PHLPP1. Therefore, strategies to target PKC signalling in PDAC and CRC should focus on enhancing PKC levels, which can be achieved through the targeting of PHLPP.
Targeting the balance of PKC and PHLPP in cancer
Reframing PKC as being tumour suppresive provides an explanation for why previous clinical trials inhibiting PKC have failed and suggest that therapeutic strategies should focus on restoration of PKC signalling. Development of novel approaches to block the dampening of PKC signalling by PHLPP may represent a viable new therapeutic option in these cancers. PP2C phosphatases such as PHLPP are resistant to common Ser/Thr phosphatase inhibitors such as okadaic acid and microcystin (56). Two compounds, NSC117079 and NSC-45586, have been identified which inhibit the phosphatase activity of PHLPP (57). These tool compounds have been used to demonstrate that inhibition of PHLPP can be neuroprotective, and also protective in osteoarthritis (58,59). PHLPP2 dephosphorylates Myc at T58, regulating its stability as a driver of prostate cancer progression, which was prevented by inhibition of PHLPP2 with NSC-45586 (60).
The PHLPP inhibitors were screened from the NCI Diversity set and have been useful as proof of principle compounds in determining cellular roles of PHLPP. However, development of optimised compounds is required. Firstly, they have low efficacy and require micromolar concentrations to elicit activity. Additionally, NSC-45586 has been reported to bind to albumin in serum, requiring even greater concentrations to be used in cells (58). In the studies described above, between 5-300 μM compound was required for inhibition in cells, dependent on the cell type (57,58,60). In the murine model of osteoarthritis, an intra-articular injection equivalent to 4 μM was sufficient to impact on PHLPP biology (59). Further, an important consideration is that these compounds inhibit the phosphatase activity and therefore all functions of PHLPP. In the above study which determined the neuroprotective role of PHLPP1, it was noted by authors that PHLPP inhibition was detrimental in astrocytes, specifically through a pro-survival signalling mechanism of PHLPP2, and called for PHLPP1 specific inhibitors. PHLPP1 is known to play a critical role in the function of the immune system, for example through regulation of STAT1 transcriptional activity (31), and PHLPP also functions as a tumour suppressor by dampening signalling through Akt and ERK (61,62). Developing effective PHLPP inhibitors for specific substrates/functions is highlighted by the finding that Phlpp1−/− mice are protected from sepsis (31). In the case of PKC, it is therefore desirable to develop new inhibitors that can specifically target the binding of the PHLPP PH domain to PKC. As yet, regulation of PKC protein levels is the only known role of this domain, therefore allosterically targeting this specific region will allow a blockade of PKC downregulation by PHLPP whereas maintaining other functions. The PHLPP1 PH domain has very weak affinity for phosphoinositides (38), however has been demonstrated to directly bind the C1A domain of PKC to induce its dephosphorylation (20). Therefore, this PH domain represents a novel, unique druggable space for targeting PHLPP1.
An alternative to inhibition of PHLPP1 to rescue PKC expression could be at the level of targeting the proteasomal degradation of PKC. An early study of phorbol-ester induced degradation of PKC determined that not only did treatment with a proteasome inhibitor block PMA-induced downregulation of the protein, it also prevented the tumour-promoting effects of PMA (22).
Does PKC suppress the function of oncogenic KRAS?
The above recent data provide a possible mechanism by which PKC may play a tumour suppressive role in cancer. Two cancer types in which PKC is known to be protective, CRC and PDAC, are primarily driven by mutant KRAS. KRAS mutations drive 95% of PDAC cases and 40% of CRC cases (63–66). Analysis of cancer-associated PKC mutations also demonstrated that a majority of these mutations co-occur with KRAS mutations (41). Is it therefore possible that one role of PKC is to suppress the oncogenic signalling of mutant KRAS, with loss of PKC protein required to unleash its full oncogenic potential?
Recent work from Wang et al. has demonstrated that growth of mutant KRAS-driven pancreatic tumours could be reduced by treatment with the PKC activator prostratin (43). This compound induces PKC activity at a low level and therefore does not induce its protein-level downregulation. As discussed above, PKCα was found to supress KRAS-mediated lung tumour formation in transgenic mouse models of lung cancer. Tumorigenesis in this model was strictly dependent on KRAS activation, as no tumours were observed in Prkca−/− mice in the absence of tumour initiators (49).
One mechanism through which PKC may inhibit oncogenic KRAS signalling is through phosphorylation of Ser181 in its C-terminal hypervariable region (HVR). This region, unique to KRAS4B over the other Ras isoforms, has a highly basic patch which aids membrane binding of KRAS (Fig 5A). Two papers from the Philips lab proposed that phosphorylation of Ser181 within the HVR by PKC may disrupt membrane binding and function as a farnesyl-electrostatic switch to shuttle KRAS to intracellular membranes. Phosphorylated KRAS limited cell survival of mutant KRAS-transformed NIH3T3 cells or CRC cells by binding BcL-xL and inducing apoptosis (67,68).
Figure 5: KRAS4B and MARCKS are regulated by a similar polybasic, PKC-dependent, membrane binding domain.
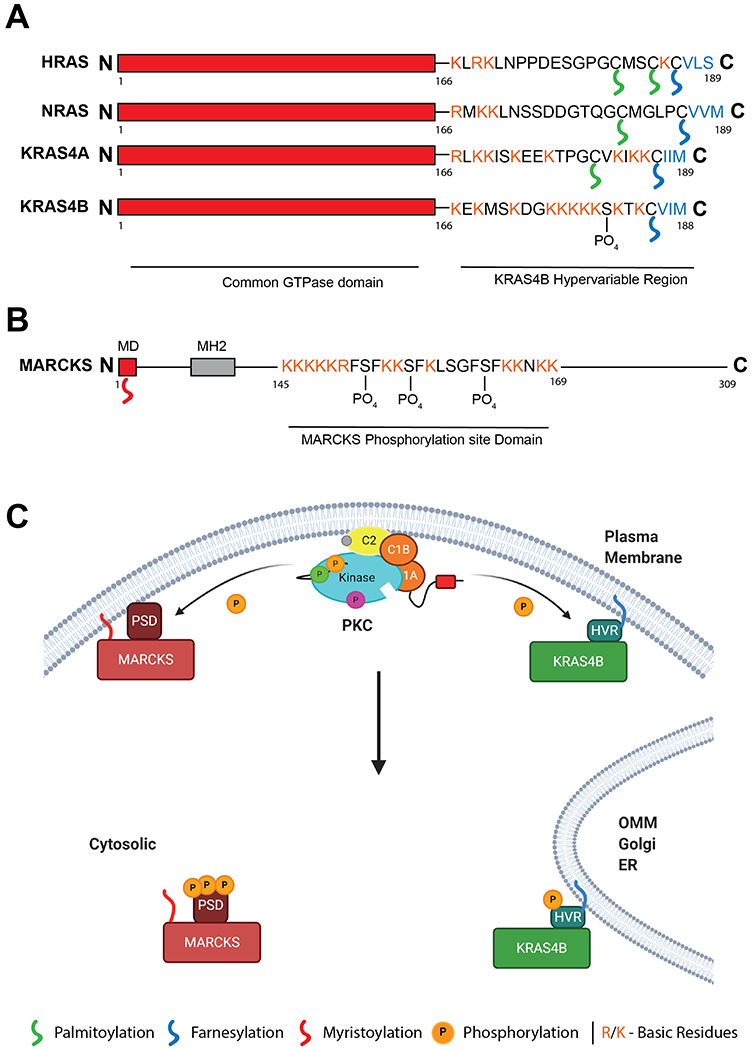
A. Domain structure of HRAS, NRAS and KRAS4A/B, differing in their C-terminal hypervariable region (HVR). KRAS4B HVR contains a single lipid modification however is binds the membrane through a polybasic sequence. This sequence can be phosphorylated by PKC at Ser181. B. Domain structure of PKC substrate MARCKS. N-terminal myristoylation domain (MD) is modified upon translation. A polybasic stretch in the middle of the protein enhances membrane binding and is modified by PKC at 3 sites. C. Model of KRAS4B and MARCKS response to intracellular Ca2+. Activation of PKC by intracellular Ca2+ signals leads to phosphorylation of the MARCKS PSD and the KRAS4B HVR. Upon phosphorylation both proteins translocate from the plasma membrane to intracellular localisations such as the Outer Mitochondrial Membrane (OMM), Golgi, or Endoplasmic Reticulum (ER). Created with BioRender.com
The phosphorylation of a farnesyl-electrostatic switch in KRAS4B, proposed by Philips and colleagues, remarkably mirrors the well characterised myristoyl-electrostatic switch of the best-defined cellular substrate of PKC, MARCKS (69). PKC is a basophilic kinase, with a substrate specificity motif of RxxS, and typically phosphorylates polybasic regions of proteins. Myristoylated Alanine Rich C Kinase Substrate (MARCKS) is phosphorylated by PKC on a number of sites within its highly basic phosphorylation site domain (PSD) (Fig 5B). MARCKS is typically situated on the plasma membrane through N-terminal myristoylation and the highly hydrophobic PSD enhances membrane binding through interaction with PIP2 and actin-binding. Upon the release of intracellular Ca2+ stores, MARCKS dissociates from the plasma membrane into the cytosol, through either binding of Calmodulin (CaM) or phosphorylation by PKC in the PSD (69,70). Importantly, although both these mechanisms release MARCKS from the membrane in response to Ca2+, CaM binding and PSD phosphorylation are mutually exclusive (Fig 5C).
The intracellular localisation of KRAS appears to be similarly modulated in response to Ca2+. Whereas other Ras isoforms contain multiple lipid moieties for membrane binding, the HVR of KRAS4B is only farnesylated on its C-terminal CAAX box, and membrane binding enhanced by a string of basic residues in its unique HVR (71) (Fig 5A). In response to Ca2+ signals, either phosphorylation or CaM binding of the HVR causes KRAS translocation from the membrane to the cytosol. Modelling membrane binding of the HVR found that the farnesyl group spontaneously inserts into the lipid bilayers, however the insertion is restricted by phosphorylation due to an altered conformation of the HVR (72). In cells, it was found that Ser181 phosphorylation substantially weakens but does not fully inhibit membrane binding and clustering of KRAS4B (73). KRAS4B also translocates from the plasma membrane to bind CaM in response to Ca2+. The structure of KRAS HVR binding to CaM was recently solved, and whereas the conformations of CaM binding were distinct from CaM in complex with canonical peptides, they were remarkably similar to the mechanism by which MARCKS binds CaM (74). As with MARCKS, phosphorylation and CaM binding both disengage KRAS from the membrane in response to Ca2+, however phosphorylation of the HVR is mutually exclusive with CaM, and modulates activity, function and localisation of KRAS4B (75,76). Prostratin treatment in PDAC cell lines, activating PKC, compromised the interaction of KRAS with CaM and prevented tumorigenicity (43).
In identification of the farnesyl-electrostatic switch of KRAS, Bivona et al. proposed that K-Ras and MARCKS represent a shared class of proteins that are anchored to the plasma membrane via lipid modification in conjunction with a stretch of polybasic residues (68). In a recent review, Grant and colleagues proposed a common motif they termed the Singly Lipidated Polybasic Terminus (SLIPT) which mediates plasma membrane binding and when phosphorylated reduces affinity for both CaM and plasma membrane (77). Similar motifs have also been reported in the small GTPases Rap1A, RhoA, RalA and RalB (78–81). This supports the concept of a common mechanism by which phosphorylation of polybasic stretches by PKC could critically impact protein localisation and interactors. PKCα has also been reported to phosphorylate the hypervariable (HV) sequence of RalB, but not RalA, at Ser192 and Ser198 (82). PKCα-dependent phosphorylation was observed to relocate RalB from the plasma membrane to late endosomes, positively regulate RalBP1 binding and abolish binding to Sec5 and the exocyst complex. RalB has been demonstrated to antagonise anchorage-independent growth of CRC cells, which required interaction with Sec5 (83) A phosphomimetic mutation of the PKCα-dependent sites of RalB impaired α5 trafficking, impairing CRC cell attachment to fibronectin. By regulating effector-binding, localisation and trafficking of RalB, PKCα may regulate its effect in CRC through phosphorylation of these sites.
However, although the above evidence suggests KRAS phosphorylation dampens mutant KRAS signalling, other data suggest that this phosphorylation may drive oncogenesis. It has been proposed that the change in localisation upon phosphorylation favours activation of Raf-1 and PI3K, and this phosphorylation is in fact required for subcutaneous tumour growth of mutant KRAS-transformed NIH3T3 cells (84,85). Ribonucleoprotein HNRNPA2B1 interacted with and regulated oncogenic signalling of phosphorylated mutant KRAS in PDAC cells (86).
As well as acting directly on KRAS, PKC may act to dampen oncogenic KRAS signalling further down the pathway. In transgenic mouse models of lung carcinoma, PKCα blocked KRAS-mediated oncogenesis through p38 MAPK-TGFβ pathway (49). PKCα/δ have also been identified to act downstream of Ras in colorectal cancer cells by regulating protein levels of the Ras scaffold Sur8 (47). PKC was also shown to have a tumour suppressive role in an APCmin driven mouse model of colorectal cancer. This suggests other functions of PKC outside of the KRAS pathway.
Conclusions
PKC belongs to a growing number of kinases with tumour suppressive function (87), which initially complicated the study and therapeutic targeting of PKC isozymes in human cancer. As described above, total protein levels of PKC are important for its tumour suppressive function. As protein levels are tightly controlled via posttranslational mechanisms by PHLPP, mRNA levels are not necessarily representative of total PKC protein expression. A recent proteomic study of the CCLE cancer cell line panel found a very poor correlation between mRNA expression and protein levels for most PKC isozymes (88). Although a large amount of data exists concerning genomic and transcriptomic phenotypes of human cancers through large-scale collaborative efforts such as The Cancer Genome Atlas (TCGA, (89)) and curated in databases such as cBioPortal and COSMIC (90,91), these data may not correlate well to PKC protein expression and its role in these cancers. With technological advances, large scale proteomic analyses of these cancers have begun to be characterised, and as we further develop resources such as The Cancer Proteome Atlas (TCPA, (92)) we will have a better understanding of how PKC protein levels correlate with disease prognosis. What is becoming clear is that the levels of PKC protein in the cell are exquisitely regulated and deregulation of any of the steps that control these levels result in pathophysiologies. For cancers such as colon and pancreatic cancer, restoring PKC levels may be beneficial.
Compounds that increase PKC activity slightly, without inducing its downregulation, may have a therapeutic advantage. For example, low levels of bryostatin-1 have been observed to activate the kinase without inducing downregulation, whereas higher levels will induce degradation of the protein (4). Further, as discussed in this review, prostratin is a non-tumorigenic activator of PKC which has been demonstrated to inhibit the growth of mutant KRAS-driven tumours (43). However, compounds that activate PKC, even weakly, destabilize the enzyme and risk reducing steady state levels.
A more attractive therapeutic avenue may be to target the negative regulators of PKC. PHLPP is one such negative regulator that plays an important role in dictating the amount of PKC in at least one cancer, PDAC. In this cancer, not only are PHLPP1 and PKC levels inversely correlated, but patients with low PHLPP1 and high PKC have significantly improved survival. Because PHLPP is recruited to or localized near its substrates through its various domains, designing molecules to interfered with the PHLPP:PKC interface may be a promising therapeutic avenue to enhance the steady-state levels of PKC without interfering with other functions of PHLPP. Such an approach may circumvent the paradoxical loss of function of PKC resulting from prolonged activation.
Abbreviations:
- DAG
diacylglycerol
- PIP2
phosphatidylinositol-4,5-bisphosphate
- PKC
protein kinase C
- cPKC
conventional protein kinase C
- nPKC
novel protein kinase C
- PMA
phorbol myristate acetate
References
- 1.Castagna M, Takai Y, Kaibuchi K, Sano K, Kikkawa U, Nishizuka Y. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J Biol Chem. 1982;257(13):7847–51. [PubMed] [Google Scholar]
- 2.Nishizuka Y The role of protein kinase C in cell surface signal transduction and tumour promotion. Nature. 1984. April;308(5961):693–8. [DOI] [PubMed] [Google Scholar]
- 3.Jaken S, Tashjian AH, Blumberg PM. Characterization of Phorbol Ester Receptors and Their Down-Modulation in GH<sub>4</sub>C<sub>1</sub> Rat Pituitary Cells. Cancer Res 1981. June 1;41(6):2175 LP – 2181. [PubMed] [Google Scholar]
- 4.Szallasi Z, Smith CB, Pettit GR, Blumberg PM. Differential regulation of protein kinase C isozymes by bryostatin 1 and phorbol 12-myristate 13-acetate in NIH 3T3 fibroblasts. J Biol Chem. 1994. January;269(3):2118–24. [PubMed] [Google Scholar]
- 5.Zhang LL, Cao FF, Wang Y, Meng FL, Zhang Y, Zhong DS, et al. The protein kinase C (PKC) inhibitors combined with chemotherapy in the treatment of advanced non-small cell lung cancer: meta-analysis of randomized controlled trials. Clin Transl Oncol Off Publ Fed Spanish Oncol Soc Natl Cancer Inst Mex. 2015. May;17(5):371–7. [DOI] [PubMed] [Google Scholar]
- 6.Garg R, Benedetti LG, Abera MB, Wang H, Abba M, Kazanietz MG. Protein kinase C and cancer: what we know and what we do not. Oncogene. 2014. November;33(45):5225–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newton AC. Protein kinase C: perfectly balanced. Vol. 53, Critical Reviews in Biochemistry and Molecular Biology. Taylor and Francis Ltd; 2018. p. 208–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Antal CE, Callender JA, Kornev AP, Taylor SS, Newton AC. Intramolecular C2 Domain-Mediated Autoinhibition of Protein Kinase C βII. Cell Rep. 2015. August;12(8):1252–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones AC, Taylor SS, Newton AC, Kornev AP. Hypothesis: Unifying Model of Domain Architecture for Conventional and Novel Protein Kinase C Isozymes. IUBMB Life Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baffi TR, Van A-AN, Zhao W, Mills GB, Correspondence ACN. Protein Kinase C Quality Control by Phosphatase PHLPP1 Unveils Loss-of-Function Mechanism in Cancer. Mol Cell. 2019;74:378–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nishizuka Y Studies and perspectives of protein kinase C. Science. 1986. July;233(4761):305–12. [DOI] [PubMed] [Google Scholar]
- 12.Corbalán-García S, García-García J, Rodríguez-Alfaro JA, Gómez-Fernández JC. A new phosphatidylinositol 4,5-bisphosphate-binding site located in the C2 domain of protein kinase Calpha. J Biol Chem. 2003. February;278(7):4972–80. [DOI] [PubMed] [Google Scholar]
- 13.Evans JH, Murray D, Leslie CC, Falke JJ. Specific Translocation of Protein Kinase Cα to the Plasma Membrane Requires Both Ca2+ and PIP2 Recognition by Its C2 Domain. Mol Biol Cell. 2005. October 19;17(1):56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dries DR, Gallegos LL, Newton AC. A single residue in the C1 domain sensitizes novel protein kinase C isoforms to cellular diacylglycerol production. J Biol Chem. 2007. January;282(2):826–30. [DOI] [PubMed] [Google Scholar]
- 15.Gallegos LL, Kunkel MT, Newton AC. Targeting protein kinase C activity reporter to discrete intracellular regions reveals spatiotemporal differences in agonist-dependent signaling. J Biol Chem. 2006. October;281(41):30947–56. [DOI] [PubMed] [Google Scholar]
- 16.Carrasco S, Merida I. Diacylglycerol-dependent binding recruits PKCtheta and RasGRP1 C1 domains to specific subcellular localizations in living T lymphocytes. Mol Biol Cell. 2004. June;15(6):2932–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dutil EM, Keranen LM, DePaoli-Roach AA, Newton AC. In vivo regulation of protein kinase C by trans-phosphorylation followed by autophosphorylation. J Biol Chem. 1994. November;269(47):29359–62. [PubMed] [Google Scholar]
- 18.Hansra G, Garcia-Paramio P, Prevostel C, Whelan RD, Bornancin F, Parker PJ. Multisite dephosphorylation and desensitization of conventional protein kinase C isotypes. Biochem J. 1999. September;342 ( Pt 2(Pt 2):337–44. [PMC free article] [PubMed] [Google Scholar]
- 19.Hansra G, Bornancin F, Whelan R, Hemmings BA, Parker PJ. 12-O-Tetradecanoylphorbol-13-acetate-induced dephosphorylation of protein kinase Calpha correlates with the presence of a membrane-associated protein phosphatase 2A heterotrimer. J Biol Chem. 1996. December;271(51):32785–8. [DOI] [PubMed] [Google Scholar]
- 20.Gao T, Brognard J, Newton AC. The phosphatase PHLPP controls the cellular levels of protein kinase C. J Biol Chem. 2008. March 7;283(10):6300–11. [DOI] [PubMed] [Google Scholar]
- 21.Chen D, Gould C, Garza R, Gao T, Hampton RY, Newton AC. Amplitude control of protein kinase C by RINCK, a novel E3 ubiquitin ligase. J Biol Chem. 2007. November;282(46):33776–87. [DOI] [PubMed] [Google Scholar]
- 22.Lu Z, Liu D, Hornia A, Devonish W, Pagano M, Foster DA. Activation of protein kinase C triggers its ubiquitination and degradation. Mol Cell Biol. 1998. February;18(2):839–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee H- W, Smith L, Pettit GR, Smith JB. Bryostatin 1 and Phorbol Ester Down-Modulate Protein Kinase C-α and -ε via the Ubiquitin/Proteasome Pathway in Human Fibroblasts. Mol Pharmacol. 1997. March 1;51(3):439 LP – 447. [PubMed] [Google Scholar]
- 24.Lee HW, Smith L, Pettit GR, Vinitsky A, Smith JB. Ubiquitination of protein kinase C-alpha and degradation by the proteasome. J Biol Chem. 1996. August;271(35):20973–6. [PubMed] [Google Scholar]
- 25.Kang B- S, French OG, Sando JJ, Hahn CS. Activation-dependent degradation of protein kinase Cη. Oncogene. 2000;19(37):4263–72. [DOI] [PubMed] [Google Scholar]
- 26.Nakamura M, Tokunaga F, Sakata S, Iwai K. Mutual regulation of conventional protein kinase C and a ubiquitin ligase complex. Biochem Biophys Res Commun. 2006;351(2):340–7. [DOI] [PubMed] [Google Scholar]
- 27.Okuda H, Saitoh K, Hirai S, Iwai K, Takaki Y, Baba M, et al. The von Hippel-Lindau tumor suppressor protein mediates ubiquitination of activated atypical protein kinase C. J Biol Chem. 2001. November;276(47):43611–7. [DOI] [PubMed] [Google Scholar]
- 28.Chen MJ, Dixon JE, Manning G. Genomics and evolution of protein phosphatases. Sci Signal. 2017. April 11;10(474):eaag1796. [DOI] [PubMed] [Google Scholar]
- 29.Shi Y Serine/Threonine Phosphatases: Mechanism through Structure. Cell. 2009;139(3):468–84. [DOI] [PubMed] [Google Scholar]
- 30.Grzechnik AT, Newton AC. PHLPPing through history: a decade in the life of PHLPP phosphatases. Biochem Soc Trans. 2016. December;44(6):1675–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cohen-Katsenelson K, Stender JD, Kawashima AT, Lordén G, Uchiyama S, Nizet V, et al. PHLPP1 counter-regulates STAT1-mediated inflammatory signaling. Elife. 2019. August;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sierecki E, Newton AC. Biochemical characterization of the phosphatase domain of the tumor suppressor PH domain leucine-rich repeat protein phosphatase. Biochemistry. 2014. June;53(24):3971–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005. April;18(1):13–24. [DOI] [PubMed] [Google Scholar]
- 34.Baffi TR, Cohen-Katsenelson K, Newton AC. PHLPPing the Script: Emerging Roles of PHLPP Phosphatases in Cell Signaling. Annu Rev Pharmacol Toxicol. 2020. September 30; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reyes G, Niederst M, Cohen-Katsenelson K, Stender JD, Kunkel MT, Chen M, et al. Pleckstrin homology domain leucine-rich repeat protein phosphatases set the amplitude of receptor tyrosine kinase output. Proc Natl Acad Sci. 2014. Sep 23;111(38):E3957 LP–E3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brognard J, Sierecki E, Gao T, Newton AC. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol Cell. 2007. March;25(6):917–31. [DOI] [PubMed] [Google Scholar]
- 37.Lemmon MA. Pleckstrin homology (PH) domains and phosphoinositides. Biochem Soc Symp. 2007;(74):81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park WS, Heo W Do, Whalen JH, O’Rourke NA, Bryan HM, Meyer T, et al. Comprehensive identification of PIP3-regulated PH domains from C. elegans to H. sapiens by model prediction and live imaging. Mol Cell. 2008. May;30(3):381–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu J, Stevens PD, Li X, Schmidt MD, Gao T. PHLPP-mediated dephosphorylation of S6K1 inhibits protein translation and cell growth. Mol Cell Biol. 2011. December;31(24):4917–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xiong X, Li X, Wen Y- A, Gao T. Pleckstrin Homology (PH) Domain Leucine-rich Repeat Protein Phosphatase Controls Cell Polarity by Negatively Regulating the Activity of Atypical Protein Kinase C. J Biol Chem. 2016. November;291(48):25167–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Antal CE, Hudson AM, Kang E, Zanca C, Wirth C, Stephenson NL, et al. Cancer-associated protein kinase C mutations reveal kinase’s role as tumor suppressor. Cell. 2015. January 29;160(3):489–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Szallasi Z, Blumberg PM. Prostratin, a Nonpromoting Phorbol Ester, Inhibits Induction by Phorbol 12-Myristate 13-Acetate of Ornithine Decarboxylase, Edema, and Hyperplasia in CD-1 Mouse Skin. Cancer Res. 1991. October 1;51(19):5355 LP – 5360. [PubMed] [Google Scholar]
- 43.Wang MT, Holderfield M, Galeas J, Delrosario R, To MD, Balmain A, et al. K-Ras Promotes Tumorigenicity through Suppression of Non-canonical Wnt Signaling. Cell. 2015. November 19;163(5):1237–51. [DOI] [PubMed] [Google Scholar]
- 44.Dowling CM, Phelan J, Callender JA, Cathcart MC, Mehigan B, McCormick P, et al. Protein kinase C beta II suppresses colorectal cancer by regulating IGF-1 mediated cell survival. Oncotarget. 2016. April 12;7(15):20919–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen S, Wang Y, Zhang Y, Wan Y. Low expression of PKCα and high expression of KRAS predict poor prognosis in patients with colorectal cancer. Oncol Lett. 2016. September;12(3):1655–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oster H, Leitges M. Protein kinase C alpha but not PKCzeta suppresses intestinal tumor formation in ApcMin/+ mice. Cancer Res. 2006. July;66(14):6955–63. [DOI] [PubMed] [Google Scholar]
- 47.Lee KH, Jeong W-J, Cha P-H, Lee S-K, Min DS, Choi K-Y. Stabilization of Sur8 via PKCα/δ degradation promotes transformation and migration of colorectal cancer cells. Oncotarget. 2017. December;8(70):115596–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dupasquier S, Blache P, Picque Lasorsa L, Zhao H, Abraham J-D, Haigh JJ, et al. Modulating PKCα Activity to Target Wnt/β-Catenin Signaling in Colon Cancer. Cancers (Basel). 2019. May;11(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hill KS, Erdogan E, Khoor A, Walsh MP, Leitges M, Murray NR, et al. Protein kinase Cα suppresses Kras-mediated lung tumor formation through activation of a p38 MAPK-TGFβ signaling axis. Oncogene. 2014;33(16):2134–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ohm AM, Tan A- C, Heasley LE, Reyland ME. Co-dependency of PKCδ and K-Ras: inverse association with cytotoxic drug sensitivity in KRAS mutant lung cancer. Oncogene. 2017. July;36(30):4370–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garg R, Cooke M, Benavides F, Abba MC, Cicchini M, Feldser DM, et al. PKC epsilon is required for KRAS-driven lung tumorigenesis. Cancer Res. 2020. September; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hsu AH, Lum MA, Shim K- S, Frederick PJ, Morrison CD, Chen B, et al. Crosstalk between PKCα and PI3K/AKT Signaling Is Tumor Suppressive in the Endometrium. Cell Rep. 2018. July;24(3):655–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reno EM, Haughian JM, Dimitrova IK, Jackson TA, Shroyer KR, Bradford AP. Analysis of protein kinase C delta (PKC delta) expression in endometrial tumors. Hum Pathol. 2008. January;39(1):21–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reddig PJ, Dreckschmidt NE, Ahrens H, Simsiman R, Tseng CP, Zou J, et al. Transgenic mice overexpressing protein kinase Cdelta in the epidermis are resistant to skin tumor promotion by 12-O-tetradecanoylphorbol-13-acetate. Cancer Res. 1999. November;59(22):5710–8. [PubMed] [Google Scholar]
- 55.Aziz MH, Wheeler DL, Bhamb B, Verma AK. Protein kinase C delta overexpressing transgenic mice are resistant to chemically but not to UV radiation-induced development of squamous cell carcinomas: a possible link to specific cytokines and cyclooxygenase-2. Cancer Res. 2006. January;66(2):713–22. [DOI] [PubMed] [Google Scholar]
- 56.Fjeld CC, Denu JM. Kinetic analysis of human serine/threonine protein phosphatase 2Calpha. J Biol Chem. 1999. July;274(29):20336–43. [DOI] [PubMed] [Google Scholar]
- 57.Sierecki E, Sinko W, McCammon JA, Newton AC. Discovery of Small Molecule Inhibitors of the PH Domain Leucine-Rich Repeat Protein Phosphatase (PHLPP) by Chemical and Virtual Screening. J Med Chem. 2010. October 14;53(19):6899–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jackson TC, Verrier JD, Drabek T, Janesko-Feldman K, Gillespie DG, Uray T, et al. Pharmacological inhibition of pleckstrin homology domain leucine-rich repeat protein phosphatase is neuroprotective: differential effects on astrocytes. J Pharmacol Exp Ther. 2013. November;347(2):516–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hwang SM, Feigenson M, Begun DL, Shull LC, Culley KL, Otero M, et al. Phlpp inhibitors block pain and cartilage degradation associated with osteoarthritis. J Orthop Res. 2018. May 1;36(5):1487–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nowak DG, Cohen-Katsenelson K, Watrud KE, Chen M, Mathew G, D’Andrea VD, et al. The PHLPP2 phosphatase is a druggable driver of prostate cancer progression. J Cell Biol. 2019. June 3;218(6):1943–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.O’Neill AK, Niederst MJ, Newton AC. Suppression of survival signalling pathways by the phosphatase PHLPP. FEBS J. 2013. January;280(2):572–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smith AJ, Wen Y-A, Stevens PD, Liu J, Wang C, Gao T. PHLPP negatively regulates cell motility through inhibition of Akt activity and integrin expression in pancreatic cancer cells. Oncotarget. 2016. February;7(7):7801–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moore AR, Rosenberg SC, McCormick F, Malek S. RAS-targeted therapies: is the undruggable drugged? Nat Rev Drug Discov. 2020;19(8):533–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Raphael BJ, Hruban RH, Aguirre AJ, Moffitt RA, Yeh JJ, Stewart C, et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2017;32(2):185–203.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Muzny DM, Bainbridge MN, Chang K, Dinh HH, Drummond JA, Fowler G, et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Collisson EA, Campbell JD, Brooks AN, Berger AH, Lee W, Chmielecki J, et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sung PJ, Tsai FD, Vais H, Court H, Yang J, Fehrenbacher N, et al. Phosphorylated K-Ras limits cell survival by blocking Bcl-xL sensitization of inositol trisphosphate receptors. Proc Natl Acad Sci U S A. 2013. December;110(51):20593–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bivona TG, Quatela SE, Bodemann BO, Ahearn IM, Soskis MJ, Mor A, et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol Cell. 2006. February 17;21(4):481–93. [DOI] [PubMed] [Google Scholar]
- 69.McLaughlin S, Aderem A. The myristoyl-electrostatic switch: a modulator of reversible protein-membrane interactions. Trends Biochem Sci. 1995. July;20(7):272–6. [DOI] [PubMed] [Google Scholar]
- 70.Yamauchi E, Nakatsu T, Matsubara M, Kato H, Taniguchi H. Crystal structure of a MARCKS peptide containing the calmodulin-binding domain in complex with Ca2+-calmodulin. Nat Struct Biol. 2003. March;10(3):226–31. [DOI] [PubMed] [Google Scholar]
- 71.Laude AJ, Prior IA. Palmitoylation and localisation of RAS isoforms are modulated by the hypervariable linker domain. J Cell Sci. 2008. February 15;121(4):421 LP – 427. [DOI] [PubMed] [Google Scholar]
- 72.Jang H, Abraham SJ, Chavan TS, Hitchinson B, Khavrutskii L, Tarasova NI, et al. Mechanisms of membrane binding of small GTPase K-Ras4B farnesylated hypervariable region. J Biol Chem. 2015. April;290(15):9465–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang S-Y, Sperlich B, Li F-Y, Al-Ayoubi S, Chen H-X, Zhao Y-F, et al. Phosphorylation Weakens but Does Not Inhibit Membrane Binding and Clustering of K-Ras4B. ACS Chem Biol. 2017. June 16;12(6):1703–10. [DOI] [PubMed] [Google Scholar]
- 74.Grant BMM, Enomoto M, Back S-I, Lee K-Y, Gebregiworgis T, Ishiyama N, et al. Calmodulin disrupts plasma membrane localization of farnesylated KRAS4b by sequestering its lipid moiety. Sci Signal. 2020. March 31;13(625):eaaz0344. [DOI] [PubMed] [Google Scholar]
- 75.Lopez-Alcalá C, Alvarez-Moya B, Villalonga P, Calvo M, Bachs O, Agell N. Identification of Essential Interacting Elements in K-Ras/Calmodulin Binding and Its Role in K-Ras Localization. J Biol Chem . 2008. April 18;283(16):10621–31. [DOI] [PubMed] [Google Scholar]
- 76.Alvarez-Moya B, López-Alcalá C, Drosten M, Bachs O, Agell N. K-Ras4B phosphorylation at Ser181 is inhibited by calmodulin and modulates K-Ras activity and function. Oncogene. 2010;29(44):5911–22. [DOI] [PubMed] [Google Scholar]
- 77.Grant BMM, Enomoto M, Ikura M, Marshall CB. A Non-Canonical Calmodulin Target Motif Comprising a Polybasic Region and Lipidated Terminal Residue Regulates Localization. Int J Mol Sci. 2020. April;21(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sawada N, Itoh H, Yamashita J, Doi K, Inoue M, Masatsugu K, et al. cGMP-dependent protein kinase phosphorylates and inactivates RhoA. Biochem Biophys Res Commun. 2001. January;280(3):798–805. [DOI] [PubMed] [Google Scholar]
- 79.Wang H, Owens C, Chandra N, Conaway MR, Brautigan DL, Theodorescu D. Phosphorylation of RalB is important for bladder cancer cell growth and metastasis. Cancer Res. 2010. November;70(21):8760–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lim K- H, Brady DC, Kashatus DF, Ancrile BB, Der CJ, Cox AD, et al. Aurora-A phosphorylates, activates, and relocalizes the small GTPase RalA. Mol Cell Biol. 2010. January;30(2):508–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wilson JM, Prokop JW, Lorimer E, Ntantie E, Williams CL. Differences in the Phosphorylation-Dependent Regulation of Prenylation of Rap1A and Rap1B. J Mol Biol. 2016. December;428(24 Pt B):4929–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Martin TD, Mitin N, Cox AD, Yeh JJ, Der CJ. Phosphorylation by protein kinase Cα regulates RalB small GTPase protein activation, subcellular localization, and effector utilization. J Biol Chem. 2012. April;287(18):14827–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Martin TD, Samuel JC, Routh ED, Der CJ, Yeh JJ. Activation and involvement of Ral GTPases in colorectal cancer. Cancer Res. 2011. January;71(1):206–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Barceló C, Paco N, Beckett AJ, Alvarez-Moya B, Garrido E, Gelabert M, et al. Oncogenic K-ras segregates at spatially distinct plasma membrane signaling platforms according to its phosphorylation status. J Cell Sci. 2013. October 15;126(20):4553 LP–4559. [DOI] [PubMed] [Google Scholar]
- 85.Barceló C, Paco N, Morell M, Alvarez-Moya B, Bota-Rabassedas N, Jaumot M, et al. Phosphorylation at Ser-181 of Oncogenic KRAS Is Required for Tumor Growth. Cancer Res. 2014. February 15;74(4):1190 LP–1199. [DOI] [PubMed] [Google Scholar]
- 86.Barceló C, Etchin J, Mansour MR, Sanda T, Ginesta MM, Sanchez-Arévalo Lobo VJ, et al. Ribonucleoprotein HNRNPA2B1 interacts with and regulates oncogenic KRAS in pancreatic ductal adenocarcinoma cells. Gastroenterology. 2014. October;147(4):882–892.e8. [DOI] [PubMed] [Google Scholar]
- 87.An E, Brognard J. Orange is the new black: Kinases are the new master regulators of tumor suppression. IUBMB Life. 2019;71(6):738–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nusinow DP, Szpyt J, Ghandi M, Rose CM, McDonald III ER, Kalocsay M, et al. Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell. 2020. January 23;180(2):387–402.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Retrieved from www.cancer.gov/tcga.
- 90.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012. May;2(5):401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sondka Z, Bamford S, Cole CG, Ward SA, Dunham I, Forbes SA. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat Rev Cancer. 2018;18(11):696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li J, Akbani R, Zhao W, Lu Y, Weinstein JN, Mills GB, et al. Explore, Visualize, and Analyze Functional Cancer Proteomic Data Using the Cancer Proteome Atlas. Cancer Res. 2017. November;77(21):e51–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dowling CM, Hayes SL, Phelan JJ, Cathcart MC, Finn SP, Mehigan B, et al. Expression of protein kinase C gamma promotes cell migration in colon cancer. Oncotarget. 2017. September;8(42):72096–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Reina-Campos M, Diaz-Meco MT, Moscat J. The Dual Roles of the Atypical Protein Kinase Cs in Cancer. Cancer Cell. 2019. September;36(3):218–35. [DOI] [PMC free article] [PubMed] [Google Scholar]