

SUMMARY
Purpose
Congenital disorders of glycosylation (CDG) are a growing group of inborn metabolic disorders with multiorgan presentation. SLC39A8-CDG is a severe subtype caused by biallelic mutations in the manganese transporter SLC39A8, reducing levels of this essential cofactor for many enzymes including glycosyltransferases. The current diagnostic standard for disorders of N-glycosylation is the analysis of serum transferrin.
Patients and methods
Exome and Sanger sequencing were performed in two patients with severe neurodevelopmental phenotypes suggestive of CDG. Transferrin glycosylation was analyzed by high performance liquid chromatography (HPLC) and isoelectric focusing (IEF), in addition to comprehensive N-glycome analysis using MALDI-TOF MS. Atomic absorption spectroscopy was used to quantify whole blood manganese levels.
Results
Both patients presented with a severe, multisystem disorder and a complex neurological phenotype. MRI revealed a Leigh-like syndrome with bilateral T2 hyperintensities of the basal ganglia. In patient 1, exome sequencing identified the previously undescribed homozygous variant c.608T>C [p.F203S] in SLC39A8. Patient 2 was found to be homozygous for c.112G>C [p.G38R]. Both individuals showed a reduction of whole blood manganese, though transferrin glycosylation was normal. N-glycome using MALDI-TOF MS identified an increase of the asialo-agalactosylated precursor N-glycan A2G1S1 and a decrease in bisected structures. In addition, analysis of heterozygous CDG-allele carriers identified similar but less severe glycosylation changes.
Conclusions
Despite its reliance as a clinical gold standard, analysis of transferrin glycosylation cannot be categorically used to rule out SLC39A8-CDG. These results emphasize that SLC39A8-CDG presents as a spectrum of dysregulated glycosylation, and mass spectrometry is an important tool for identifying deficiencies not detected by conventional methods.
Synopsis:
MALDI-TOF MS identifies subtle dysglycosylation not detected by conventional methods of glycosylation analysis in SLC39A8-CDG.
Keywords: glycosylation, MALDI-TOF MS, SLC39A8, congenital disorders of glycosylation, manganese
INTRODUCTION
Rare, highly penetrant mutations in SLC39A8 (HGNC:20862), the gene encoding the eponymous manganese channel, cause a severe metabolic disorder marked by manganese deficiency and multisystem dysfunction.1,2
In contrast, a common missense variant in SLC39A8 is associated with a range of common, polygenic disorders and phenotypes through genome-wide association studies (GWAS) including idiopathic scoliosis3, schizophrenia4 and Crohn’s disease and decreased manganese.5,6
Few detailed reports exist of SLC39A8-CDG (OMIM: 616721).1,2,7 The pleiotropic effects of manganese deficiency are likely related to its function as a cofactor for a multitude of enzymes including arginase, glutamine synthetase (GS), pyruvate carboxylase, manganese superoxide dismutase (MnSOD, SOD2)8,9 and many Golgi glycosyltransferases.10
Severe hypoglycosylation was among the initial observations in individuals with SLC39A8 deficiency, leading to the characterization of the disease as a type II congenital disorder of glycosylation.2 Analysis of serum transferrin in affected individuals using HPLC and/or IEF showed pronounced dysglycosylation while ESI-TOF MS revealed a pronounced hypogalactosylation of Asn (N)-linked glycoproteins, which was attributed to the role of manganese as an essential cofactor for β−1,4 galactosyltransferase.11,12 As in many other glycosylation disorders, patients display a multisystem involvement marked by severe neurological manifestations including seizures, profound psychomotor retardation, and dysmorphic features(Table 1).1,2
Table 1.
Phenotype and clinical characteristics of currently known SLC39A8-CDG patients
| Case | Patient 1 | Patient 2 | Park et al. 2015 Individual A | Park et al. 2015 Individual B | Boycott et al. 2015 Individual 1 (family A) | Boycott et al. 2015 Individual 2 (family B) | Boycott et al. 2015 Individual 3 (family 3) | Boycott et al. 2015 Individual 4 (family D) | Boycott et al. 2015 Individual 5 (family D) | Boycott et al. 2015 Individual 6 (family E) | Boycott et al. 2015 Individual 7 (family F) | Boycott et al. 2015 Individual 8 (family F) | Riley et al. 2016 Patient 1 | Riley et al. 2016 Patient 2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variant | F203S homozygous | G38R homozygous | [G38R]; [I340N] | [V33M;S335T];[G204C] | G38R homozygous | G38R homozygous | G38R homozygous | G38R homozygous | G38R homozygous | G38R homozygous | G38R homozygous | G38R homozygous | C113S homozygous | C113S homozygous |
| Gender | F | M | F | F | F | M | M | F | F | M | F | M | F | F |
| Age | 3 years | 2 years | 4 months (presentation) | 19 years | 18 years | 23 years | 10 years | 6 years | 6 years | 9 years | 9 years | 3 years | died aged 26 months following infection | 16 years |
| Manganese | 3.7 (NR 7–11) ng/ml | initially 1.5 (4–15) ng/ml, undetectable in later samples | undetectable | undetectable | ND | erythrocyte: 20 nmol/L (NR 273–728) | 20 nmol/l (NR 78–289) | 14.2 nmol/L (NR 5.3–40.8) | 5.5 nmol/L (NR 5.3–40.8) | 18.4 nmol/l (NR 5.3–40.8) | 1.1 μg/L (NR 5–12.4) | 1.1 μg/L (NR 5–12.4) | ND | undetectable |
| Tf-IEF | normal | normal | type II pattern | type II pattern | ND | type II pattern (mild) | ND | type II pattern (mild) | type II pattern (mild) | ND | ND | ND | ND | type II pattern |
| cMRI | Leigh-like basal ganglia T2 hyperintensities, delayed myelination | Leigh-like basal ganglia T2 hypertintensities | Leigh-like basal ganglia T2 hyperintensities, asymmetric cerebral atrophy | cerebellar atrophy | severe cerebellar atrophy (vermis and hemispheres) | severe cerebellar atrophy (vermis and hemispheres) | severe cerebellar atrophy (vermis and hemispheres) | severe cerebellar atrophy (vermis and hemispheres) | ND | progressive cerebellar atrophy (vermis and hemispheres) | severe cerebellar atrophy (vermis and hemispheres) | frontal cerebral atrophy, mild cerebellar atrophy (vermis and hemispheres) | Leigh-like basal ganglia T2 hyperintensities | Leigh-like basal ganglia T2 hyperintensities, mild cerebral atrophy |
| failure to thrive, atrial septal defect, hepatomegaly | failure to thrive, | failure to thrive, hepatopathy | osteopenia | osteopenia | failure to thrive | failure to thrive, osteopenia |
Manganese is a cofactor for a plethora of enzymes in addition to its pivotal role for several galactosyltransferases that modify glycoproteins.12–14 From a metabolic perspective, its involvement in mitochondrial redox metabolism as a cofactor of SOD2 (MnSOD, EC 1.15.1.1) is intriguing and suggests broader consequences of manganese deficiency. This hypothesis was supported by Riley et al. who identified patients with SLC39A8 mutations and Leigh-syndrome like presentation.7 In these individuals, MRI signal alterations of the basal ganglia as well as impairment of the respiratory chain implied SOD2 dysfunction secondary to manganese deficiency. Both galactose and manganese-II-sulfate supplementation were shown to be effective in correcting glycosylation abnormalities in cases of SLC39A8 deficiency. The effect of these interventions on mitochondrial involvement – if any – is unknown.
Mass spectrometry (MS) has been used in the diagnosis of CDG since the early 1990s15 and initially focused on selected marker glycoproteins.16 N-glycome profiling using matrix assisted laser desorption ionization time of flight (MALDI-TOF) MS broadens the scope of this technique by analyzing the entirety of N-glycans in a sample and is increasingly used in CDG diagnosis.17,18
To our knowledge, apparently normal glycosylation has not been described in cases of SLC39A8 deficiency where glycosylation analysis has been performed. We recently published the first MALDI-TOF MS N-glycome profiles of two SLC39A8-CDG cases who also had abnormal transferrin IEF, identifying glycosylation abnormalities that were reversed after a year of manganese supplementation.19 Here, we describe two patients who show no detectable abnormalities as assessed by conventional glycosylation analysis. By contrast, we identified changes in the serum N-glycome profiling by MALDI-TOF consistent with our prior studies on SLC39A8-CDG. Further, we report the first analysis of serum from parents of SLC39A8-CDGs and identified similar but less severe dysglycosylation. These findings underscore the value of mass spectrometry in the assessment of suspected glycosylation disorders. These more subtle changes hint at the possibility of underdiagnosing CDG when relying solely on conventional methods of glycosylation assessment as well as the unknown impact hypofunctioning SLC39A8-CDG alleles have in heterozygous carriers.
METHODS
Subjects
The patients’ guardians gave their informed consent prior to inclusion in this study. Investigations on patients of SLC39A8-CCD were approved by the institutional review boards of Shodair Children’s Hospital (IRB00008287, PKU-016) and the University of Münster (2019–199f-s). Glycomic analysis for research purposes of previously obtained serum/plasma samples was approved by the Massachusetts General Hospital institutional review board (2017P000115),
Genetic analysis
All patients and probands underwent exome sequencing followed by Sanger sequencing as outlined previously.2
Trace element analysis
Whole blood manganese levels were evaluated using atomic absorption spectroscopy as described previously.20
Glycosylation analysis
Transferrin glycosylation analysis was performed using both high-performance liquid chromatography (HPLC) and isoelectric focusing (IEF) following previously published protocols.21
Purification of serum protein N-glycans was performed following previously described protocols (https://ncfg.hms.harvard.edu). In brief, 5 μL of serum were treated with PNGase F (New England Biolabs, P0710S), the released N-glycans were purified and eluted from a C18 Sep-Pak (50 mg) column (Waters Corp, WAT054955), permethylated, and analyzed by MALDI-TOF MS (Bruker). The relative abundance of 57 serum N-glycans was determined; N-glycans were grouped into categories based on shared components for analysis (Supp. Table 1). Glycan profiles were compared to a control cohort.
Statistical analysis
Though limited by the number of cases, statistical analysis of serum N-glycomics data from SLC39A8-CDG cases and heterozygote carriers were compared to controls using individual unpaired t-tests assuming unequal variance. Groups included a) controls (33 healthy adults homozygous for the major allele (C) at rs13107325 of SLC39A8, b) parents of P1 and P2 who are heterozygous mutations carriers and c) patients P1, P2 as well as two previously described cases of SLC39A8-CDG also analyzed by MALDI-TOF by our group.19 Experimental analyses were performed at least in duplicates and results are presented as mean with standard error of mean. Statistical analyses were performed on GraphPad Prism Version 8.0 (GraphPad Software, San Diego, CA, USA).
RESULTS
SLC39A8-CDG can present with a phenotype suggestive of mitochondrial dysfunction.
The clinical presentations of both patients and of previously reported cases is summarized in Table 1 (Tab.1).
Patient 1 is a three-year-old girl of Turkish origin presenting with a complex phenotype with severe psychomotor retardation, dystonia, seizures, and dysmorphic features. She is the second daughter of consanguineous parents (first degree cousins). An older sister was diagnosed with congenital hypothyroidism while an uncle was reported to have been diagnosed with type I diabetes. There are no known inherited disorders in the family. The patient was born at term after an uneventful pregnancy with a low birthweight (2365 g, Z-score −2.15), postnatally showing a weak sucking reflex. Psychomotor retardation and failure to thrive were noted in the first year of life while an atrial septal defect was recognized shortly after birth. Seizures with a semiology suggesting infantile spasms began within the first months of life and were initially treated with phenobarbital and later clonazepam. This led to a reduction in both severity and frequency with a normalized EEG.
When first assessed at a specialized facility at the age of 32 months, the girl presented with pronounced muscular hypotonia and dystonic movement patterns accompanied by dysphagia. Severe growth restriction was noted with a Z-score of −3.64 for weight, −3.35 for length, and −2.88 for head circumference. Dysmorphic features such as low set ears, retrognathia, and an arched palate as well as strabismus were noted. Cranial MRI revealed bilateral T2-hyperintense lesions of the caudate and lentiform nuclei while the basal ganglia were generally hypotrophic (Fig. 1 A). Furthermore, delayed myelination was described. These were at the time described as Leigh-like lesions and mitochondrial disease was suspected. Extensive metabolic analyses (acylcarnitines, plasma and urinary amino acids, urinary organic acids and oligosaccharides) yielded no abnormal results. Of note, lactate and ammonia levels were not elevated in repeated measurements.
Figure 1.
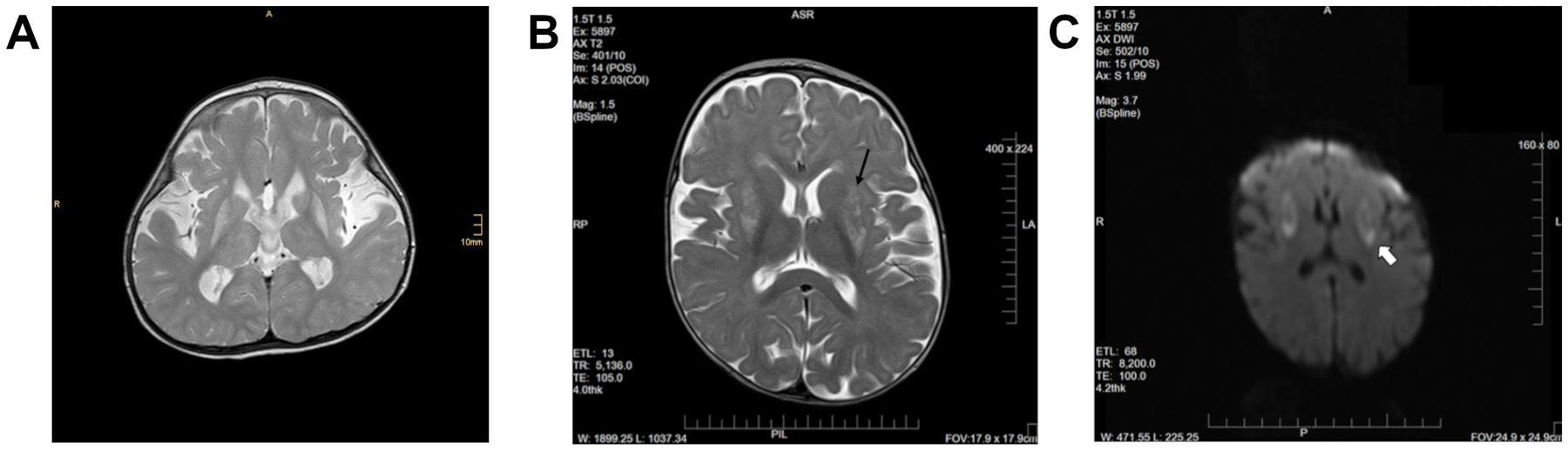
Leigh-like lesions of the basal ganglia in SLC39A8-CDG. Cranial MRI performed in both patients revealed symmetric bilateral T2 hyperintense lesion. In patient 1, pronounced symmetric bilateral hyperintensities, especially of the putamen were noted along with volume reduction of the entire basal ganglia (A). Similarly, patient 2 showed T2 hyperintense lesions of the putamen and caudate nucleus along with abnormal perfusion restrictions (B, C).
Patient 2 is a two year old boy of Hutterite ancestry who presented with a complex disorder of growth restriction, mild dysmorphic features, delayed neurodevelopment, and neurological manifestation.
He is the youngest of 5 children born to a union without known consanguinity. Three sisters and a brother are healthy. He was born at 37 weeks gestation by spontaneous delivery to a 32-year-old Gravida 6, Para 4 with a previous history of spontaneous miscarriage. Prenatal history was notable for maternal gestational diabetes and maternal hypothyroidism. After a reportedly unremarkable neonatal period, he continued to have poor head control and lagging motor development. Around 5 months of age he developed seizures that initially were suggestive of infantile spasms. An EEG showed multifocal and generalized epileptiform discharges of high amplitude similar to modified hypsarrhythmia and a suppression burst pattern, prompting a trial of prednisone (and pyridoxine) without clear improvement. Subsequent EEGs displayed seizure activity and/or encephalopathy without hypsarrhythmia and his seizures became predominantly tonic responding to levetiracetam both clinically and electrically.
At 7 months of age he was hospitalized for failure to thrive and protein malnutrition accompanied by feeding difficulties with oropharyngeal dysphagia and oral motor discoordination. Placement of a gastrostomy-tube and enteral tube feedings improved his nutritional status but he continued to develop short stature and proportionate microcephaly with anthropometric parameters at 9 months showing a Z score of −3.84 for length −1.52 for weight, and −2.16 for head circumference.
He displayed distinct physical features including a prominent forehead with frontal bossing, a prominent nasal root with a mildly depressed nasal bridge, low set posteriorly rotated ears with decreased complexity and overfolded helix, a characteristic horizontal crease with an associated dimple above both knees as well as ridging and dimpling of the plantar surface of his feet, and a capillary formation over the glabella. His neurological phenotype was complex and included axial hypotonia with persistently poor head control, peripheral spasticity, and increased neuroexcitability. He had frequent escalating crying episodes responding to benzodiazepines but not opioids. He displayed periodically increased tone with back arching and stiffening of extremities strongly suggestive of dystonia. Notably, he had improved symptom control after initiation of carbidopa levodopa.
An MRI of the brain without contrast at 7 months showed abnormal diffusion restriction and T2 hyperintensity in the bilateral putamen/lentiform nuclei (Fig. 1 B, C). Metabolic studies at the time of the brain MRI were normal including acylcarnitine profile, plasma amino acids, lactate, ammonia, and urine organic acids, purines and pyrimidines, urine sulfite screening as well as urine panel for creatine disorders.
Whole blood manganese levels were severely reduced in both patients. Patient 1 showed levels ranging from 1–3 ng/ml [reference: 7–11 ng/ml], while patient 2 had an initial serum manganese of 1.5 ng/ml [reference: 4–15 ng/ml] and subsequently undetectable whole blood manganese. Urinary manganese levels were undetectable in patient 1 and within normal ranges in patient 2 (2.1 μg/L [reference: 0.0–5.0 μg/L]).
F203S is a novel disease-causing variant in SLC39A8.
Exome sequencing revealed the previously undescribed homozygous missense variant c.608T>C [p.F203S] in SLC39A8 (Ref Seq NM_001135146.2) in patient 1, which was confirmed by Sanger sequencing. Both parents were heterozygous for this variant. It lies within the highly conserved fourth transmembrane domain of the ion channel (Supplementary Figure 1) and one amino acid upstream from the previously described pathogenic variant of c.610G>T (p.G204C).2 It is not listed in variant and/or genome databases such as gnomAD or ClinVar. The variant was predicted to be deleterious by multiple in silico prediction tools including PolyPhen2 (score 0.998), SIFT (score 0.01), and Provean (score −4.899).
Patient 2 was homozygous for the previously described variant1,2 c.112G>C [p.G38R], while both parents were heterozygous. Prior to patient 2, we had identified this homozygous variant in two Hutterite families that were not knowingly related which, in the context of the previous report, establishes this variant as a pathogenic founder variant in this population.
Serum protein N-glycomics identifies dysglycosylation despite normal transferrin glycosylation analysis.
Based on clinical presentation, genetic results, and hypomanganesemia, CDG was suspected. However, standard glycosylation analyses from samples taken at the ages of three years (patient 1) and 16 months (patient 2), i.e. HPLC and IEF of serum transferrin, did not reveal abnormalities in either patient (Tab. 2, Fig. 2). Both studies were within normal limits and neither patient fulfilled the diagnostic criteria for CDG. A slight elevation in trisialo-transferrin was noted in a heterozygous carrier of the F203S variant (P1.1).
Table 2.
Glycosylation analysis using high-performance liquid chromatography (HPLC) of serum transferrin and N-glycan mass spectrometry
| Transferrin isoforms | P1 patient 1 |
P1.1 heterozygous mother |
P1.2 heterozygous father |
P2 patient2 |
P2.1 heterozygous mother |
P2.2 heterozygous father |
Reference (area%) |
|---|---|---|---|---|---|---|---|
| Asialo-Tf | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Monosialo-Tf | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Disialo-Tf | 1.30 | 0.49 | 0.80 | 0.75 | 0.71 | 1.15 | < 1.8 |
| Trisialo-Tf | 3.60 | 6.63 | 2.82 | 1.73 | 2.90 | 1.84 | <6.5 |
| Tetrasialo-Tf | 88.00 | 89.18 | 91.40 | 94.53 | 94.34 | 95.22 | >85.0 |
| Pentasialo-Tf | 7.10 | 3.71 | 4.98 | 2.99 | 2.06 | 1.79 | <15 |
| Glycomic structure | P1 patient 1 |
P1.1 heterozygous mother |
P1.2 heterozygous father |
P2 patient2 |
P2.1 heterozygous mother |
P2.2 heterozygous father |
Controls (% abundance) |
| High-mannose | 2.6 | 5.1 | 2.4 | 2.0 | 3.6 | 2.9 | 3.6 |
| Mono-antennary | 1.9 | 2.3 | 2.3 | 2.4 | 1.9 | 1.7 | 1.1 |
| Bi-antennary | 85.9 | 88.8 | 87.5 | 85.4 | 87.5 | 87.2 | 88.0 |
| Tri-antennary | 8.8 | 3.7 | 7.4 | 9.5 | 6.7 | 7.7 | 6.8 |
| Tetra-antennary | 0.8 | 0.2 | 0.3 | 0.8 | 0.3 | 0.4 | 0.5 |
| Sialyated | 79.5 | 65.4 | 75.1 | 91.4 | 77.9 | 74.8 | 74.7 |
| Fucosylated | 28.7 | 37.5 | 34.6 | 16.4 | 29.8 | 38.4 | 31.6 |
| Bisecting | 4.1 | 9.2 | 6.4 | 1.8 | 6.1 | 4.5 | 7.9 |
| 2227.25 | 2.0 | 0.6 | 0.9 | 2.8 | 1.5 | 1.0 | 0.4 |
| 3603.01 | 4.9 | 2.0 | 4.5 | 5.0 | 3.8 | 3.0 | 3.7 |
| 3777.11 | 1.7 | 0.3 | 1.2 | 2.5 | 1.1 | 2.7 | 1.6 |
Figure 2.
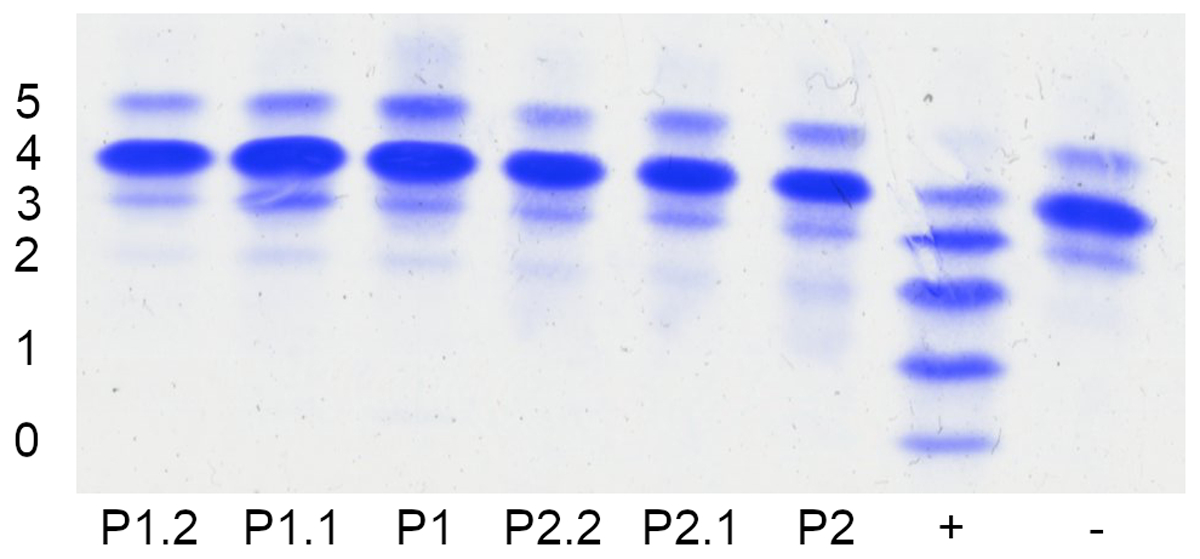
Normal glycosylation pattern of serum transferrin in two cases of SLC39A8-CDG. Conventional glycosylation analysis of serum transferrin of probands in comparison to a wildtype control (−) and PMM2-CDG (+) using isoelectric focusing (IEF). Numbers indicate the transferrin isoforms carrying the respective amount of sialic acid residues. No indication of the type II dysglycosylation pattern previously observed in SLC39A8-CDG can be seen in either patient (P1, P2) or their heterozygous parent carriers (P1.1, P1.2, P2.1, P2.2). A small elevation of trisialo-transferrin was noted in a heterozygous carrier of the F203S variant.
Given our prior report of glycomics changes in SLC39A8-CDG,22 MALDI-TOF MS glycomics were performed from the same serum samples used for conventional glycosylation studies. Analysis of serum N-glycans revealed a distinct pattern consistent with prior studies in plasma of individuals with SLC39A8-CDG. The hypogalactosylated precursor monosialo-monogalacto-biantennary glycan (A2G1S1, m/z 2227) was increased in both P1 and P2, and pooled analysis of all previously analyzed homozygous SLC39A8-CDG patients (mean: 2.15 % abundance, SEM: 0.25, n=4) analyzed by our lab was significantly increased, irrespective of the underlying variant, when compared to heterozygous carriers (p < 0.01, mean: 1.0 %, SEM: 0.19, n=4) and wildtype controls (p < 0.005, mean: 0.44 %, SEM: 0.04, n=33) (Fig. 3, A). Bisected N-glycans are produced by a single glycosyltransferase (MGAT3) predicted to bind manganese based on presence of a conserved DxD domain.23 Pooled analysis of bisected glycans in all SLC39A8 homozygous variant carriers (mean: 2.58 %, SEM: 0.68, n=4) were significantly different compared to controls (p < 0.001, mean: 7.83 %, SEM: 0.69, n=33), and heterozygous carriers (p < 0.05, mean: 6.55 %, SEM: 0.98, n=4) (Fig. 3, B), consistent with decreased activity of MGAT3. We performed additional analyses of glycan traits including branching, fucosylation, sialylation, hypogalactosylation (terminal GlcNAc), and hyposialylation (terminal galactose). Although a few classes were different in SLC39A8-CDGs, consistent with a broad disruption of plasma glycosylation, the effect sizes and ability to distinguish between groups, particularly heterozygous carriers, were smaller than for the abundance of A2G1S1 and bisected N-glycans (Tab. 2, Fig 3C, 3D, S2–5). Full MALDI-TOF MS data for serum N-glycans of heterozygous and homozygous SLC39A8 variant carriers as well as the classifications scheme used is available in supplemental material (Tab. S1, Tab. S2).
Figure 3.
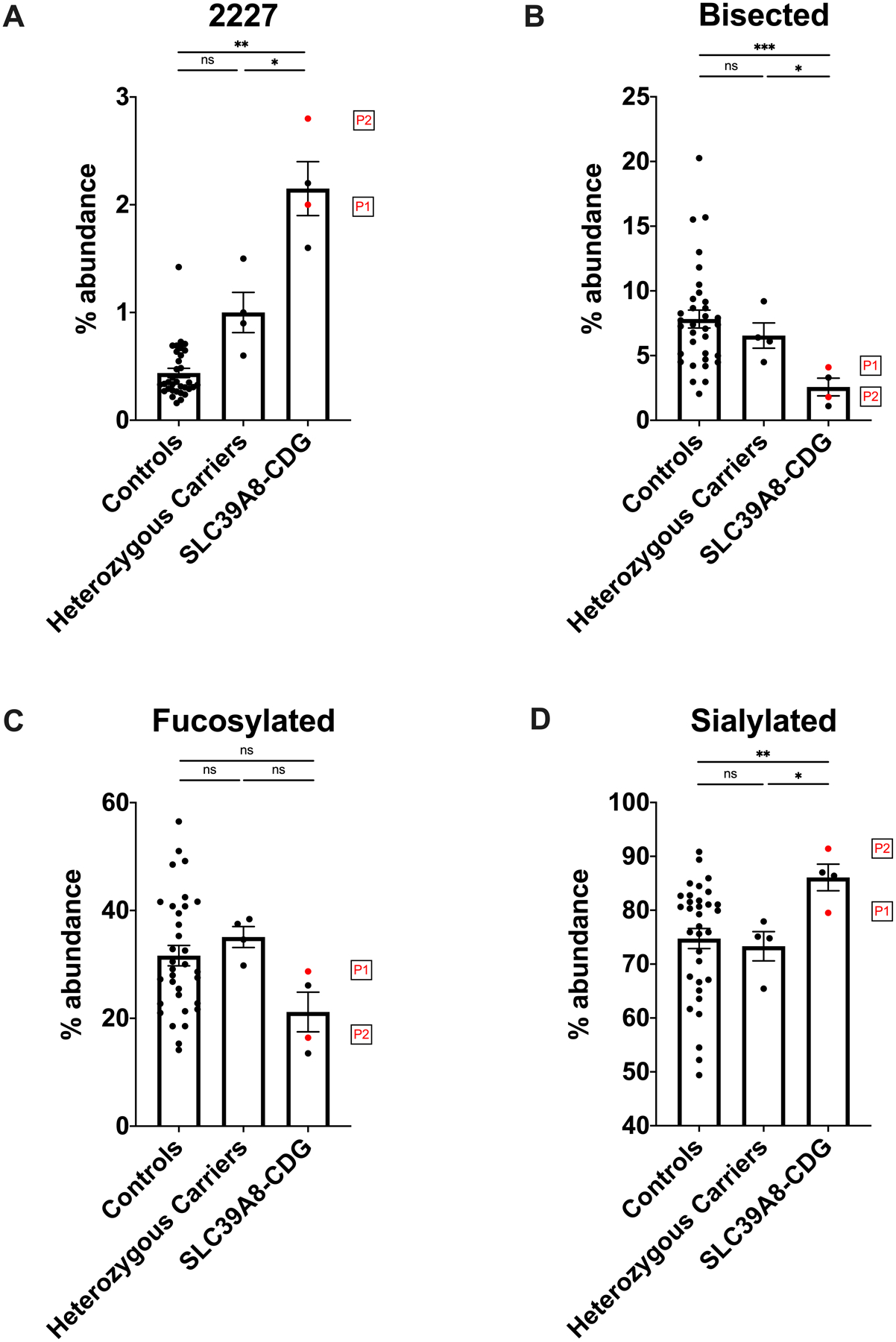
N-glycome analysis detects dysglycosylation in samples from patients with homozygous F203S and G38R mutations with normal transferrin. N-glycans from serum samples were analyzed by MALDI-TOF MS. Results are presented as relative abundance of glycans within the full spectrum. The precursor glycan A2G1S1 (m/z 2227) showed a significant increase in the newly identified SLC39A8-CDG patients (P1 and P2) and historical SLC39A8-CDG cases (subject A and B presented in Park et al. 2015) when compared to heterozygous carriers (p < 0.05) and reference controls (p < 0.01) (A). A2G1S1 abundance between heterozygous carriers and controls was approaching statistical significance (p 0.054). The abundance of bisected glycans was significantly reduced in SLC39A8-CDG patients compared to reference controls (p < 0.001) or heterozygous carriers (p < 0.05) (B). In contrast, no significant difference in the abundance of fucosylated glycan species could be observed (C), whereas sialylated glycans allowed a differentiation between SLC39A8-CDG cases and both controls (p < 0.01) and heterozygous carriers (p < 0.05). (n.s.: not significant, *: p ≤ 0.05, **: p ≤ 0.01, ***: p ≤ 0.001, error bars signify standard error of mean).
DISCUSSION
Here, we describe two patients with SLC39A8-CDG identified by exome sequencing and confirmed using MALDI-TOF MS glycomics. Both were homozygous for rare missense variants, patient 1 for the novel variant p.F203S affecting a highly conserved amino acid residue in the third transmembrane domain, and patient 2 for the p.G38R variant. Both had severely reduced or undetectable blood concentrations of manganese consistent with systemic manganese deficiency caused by impaired ion channel function of SLC39A8. However, neither of the patients revealed any abnormalities in serum transferrin glycosylation by HPLC (Tab. 2) or IEF (Fig. 2), both regarded as gold standard tools of CDG diagnostics. In contrast, serum N-glycome profiling identified dysglycosylation not detected by conventional methods. This distinctive pattern was consistent between all SLC39A8-CDG cases independent of transferrin glycosylation abnormalities and characterized by an increase in the precursor glycan A2G1S1 (m/z 2227) and a decrease of bisected N-glycans (Fig. 3). These changes can be attributed to impaired enzyme function of manganese-dependent glycosyltransferases, which include galactosyltransferases and MGAT3,23 though the intricacies of how the many glycosyltransferases with manganese-binding DxD motifs are affected by the loss their cofactor, in a tissue and cell-specific manner, remains to be determined.
The reason for apparently normal glycosylation of serum transferrin in SLC39A8-CDG is unclear. Transferrin, mainly derived from liver, represents only a fraction of the total glycosylated proteins in plasma, and decreased manganese in SLC39A8 may affect different glycosyltransferases uniquely based on their affinity for this cofactor and their target specificity.24 Furthermore, differentially expressed SLC39A8 protein isoforms represent another possible cause for the apparently normal transferrin glycosylation observed in these patients. There are three known protein coding transcript variants, one of which (Uniprot identifier: Q9C0K1–2) is lacking the first 67 amino acid residues of the canonical sequence25 and thus the residue affected in patient 2. However, dysglycosylation has been observed in homozygous carriers of the variant2, albeit to a lesser degree than in other variants. In addition, the variant identified in patient 1 is located in a protein domain present in all splice variants, rendering selective expression of different transcripts an unlikely driver of the findings presented herein.
Similarly, our results argue against a linear relationship between blood manganese and transferrin glycosylation, as patients 1 and 2 with low or even undetectable blood manganese have normal transferrin glycosylation. Similarly, variation in transferrin glycosylation exists even in patients homozygous for the same variant (p.G38R), highlighting its limitation in evaluating SLC39A8-CDG. In contrast, all SLC39A8-CDG patients displayed a consistent pattern on MALDI-TOF glycomics. Although low sample size and individual variation limit definitive conclusions, our findings across an allelic series of SLC39A8 variants including the common p.A391T variant22 and otherwise normal heterozygous carriers of SLC39A8-CDG mutations suggest that a spectrum of SLC39A8 dysfunction might lead to varying degrees of disease. Given the association of p.A391T with a broad range of phenotypes, determining how “subclinical” dysglycosylation contributes to disease pathogenesis may elucidate novel mechanisms in schizophrenia, scoliosis, and Crohn’s disease.3–5,26 Dysglycosylation in heterozygous SLC39A8-CDG-mutation carriers has not been previously reported. Given that the less severe p.A391T mutation is associated with multiple diseases through GWAS and does not cause a CDG in homozygous carriers, we hypothesize that heterozygous carriers of CDG-causing SLC39A8 mutations may have previously unappreciated health consequences due to their hypofunctioning allele and subsequent dysglycosylation. Clinical data and manganese levels for these individuals was not available for our study, though this information may be worth considering when treating populations with pathogenic founder variants such as the Hutterite community. Although generally considered unaffected, a recent study including heterozygote carriers of CDG-causing mutations in ALG6 and PMM2 identified LDL changes that mirrored those found in affected homozygous CDG patients.27
The findings presented here have broad implications for the diagnostic work-up of suspected CDG cases. In our patients, transferrin glycosylation analysis failed to detect dysglycosylation in SLC39A8-CDG, prompting the possibility of this being an underdiagnosed entity. It is not clear whether subtle glycosylation changes in other CDG subtypes might equally be missed using conventional methods and should be further investigated. Thus, we advocate for the use of N-glycome profiling on a broader scale as a screening method in suspected CDG cases given its potential to detect even minute changes in glycosylation. Further studies would undoubtedly detect specific dysglycosylation patterns in CDG subtypes and advance the diagnostic potential of glycome profiling. In addition, O-glycans can be easily isolated and processed from these same samples, furthering the potential of this method for CDGs.
Tracking small changes in glycosylation might be especially valuable in the context of therapeutic interventions. SLC39A8-CDG is one of the few treatable glycosylation disorders, showing normalization of transferrin glycosylation by HPLC/IEF as well as A2G1S1 and bisected N-glycans by glycomics following galactose and/or manganese-II-sulfate substitution.20,22 Other treatable CDGs include MPI-CDG, SLC35C1-CDG, SLC35A2-CDG, and PGM1-CDG,28–31 and MALDI-TOF MS glycome profiling would likely provide a more detailed evaluation of treatment effects . Indeed, previously attempted interventions that were deemed unsuccessful32,33 could be reevaluated with special focus on changes missed by the study of transferrin glycosylation alone or other glycomic techniques such as ESI-QTOF. Though every technique for serum or plasma glycomics has its own unique strengths and limitations, we find that MALDI-TOF generates reliable and reproducible results including the identification of nearly 60 individual N-glycans from plasma.
Both patients showed a distinct mitochondriopathic phenotype with Leigh-like lesions identified on MRI and dystonia, mirroring previously reported findings of a Leigh-like syndrome associated with mutations in SLC39A8.7 However, unlike these cases, the patients presented here did not show any abnormalities in common markers of mitochondrial dysfunction, illustrating the inconsistency of this finding in SLC39A8-CDG.1 This suggests a dual phenotype may result from manganese deficiency, with glycosylation abnormalities or mitochondrial dysfunction (Fig. 4) influenced by the causal mutation, susceptibility of different enzymes to a lack of manganese, or additional environmental and genetic factors. Several manganese-dependent enzymes are directly or indirectly involved in mitochondrial metabolism.34–36 Since all have a close relation to redox metabolism, involvement in SLC39A8 mutation-related mitochondrial impairment is possible. Among these, SOD2 is a promising candidate as discussed previously by Riley et al.7 and supported by in vitro data from a cellular model expressing known human SLC39A8 mutations that demonstrated impaired SOD2 and mitochondrial function. Of note, these data demonstrate altered mitochondrial function in all variants tested, despite lack of clinical evidence in human patients. Why some patients do not show signs of mitochondrial involvement despite considerable manganese deficiency remains unknown. As the MRI signal can be affected by local paramagnetic properties of divalent cations including manganese, further imaging studies in SLC39A8-CDG focused on metal transport vs volume loss secondary to apoptotic lesions would be informative.
Figure 4.
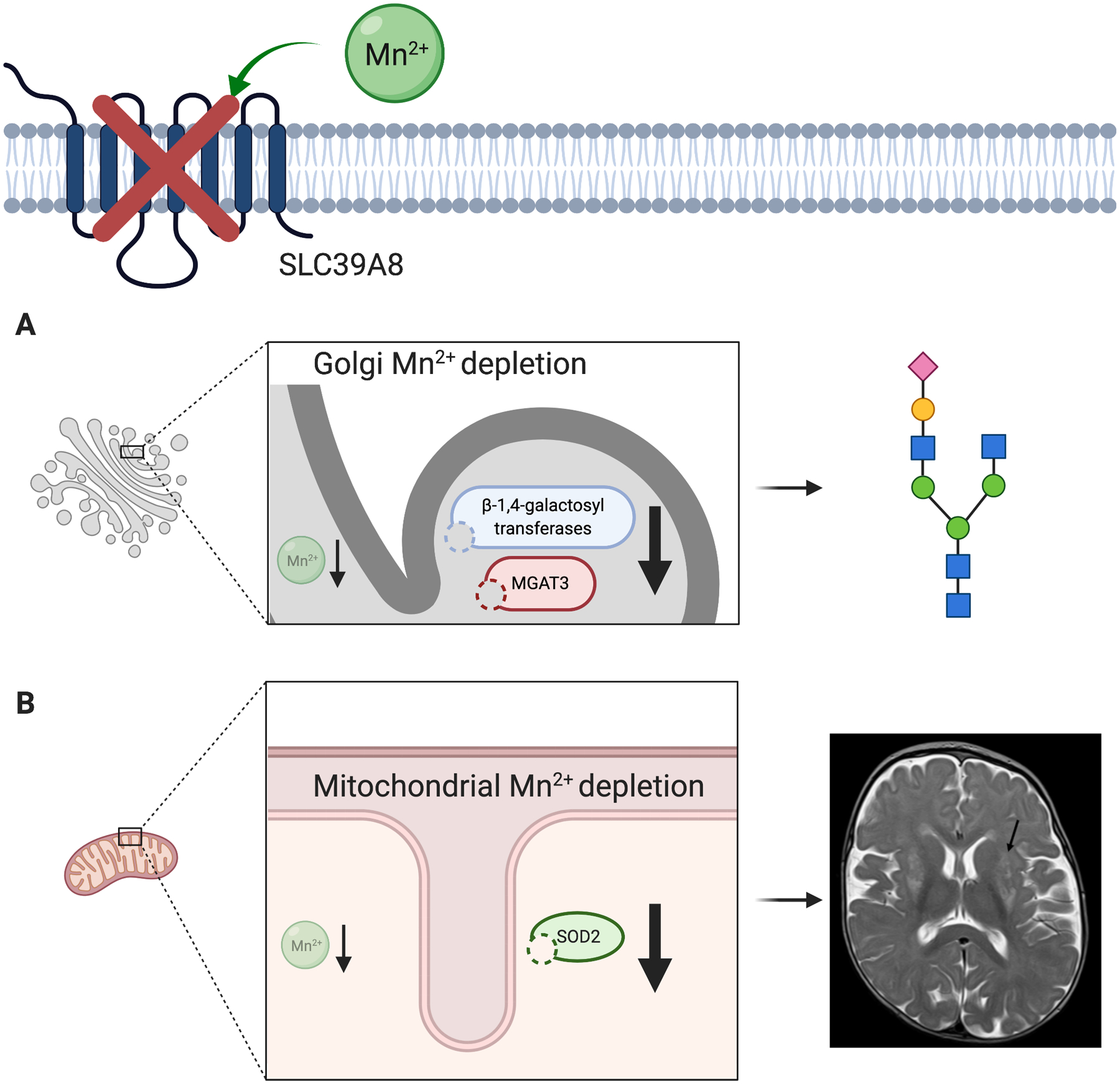
Manganese deficiency is the key driver of pathogenesis in SLC39A8 deficiency. The defective ion channel leads to intracellular manganese depletion in the Golgi apparatus and mitochondria. (A) In the Golgi apparatus, manganese dependent glycosyltransferases exhibit decreased activity due to lack of their obligatory cofactor. Hypogalactosylation and decreased bisection are a core finding in SLC39A8-CDG. (B) Mitochondrial dysfunction is likely mediated by impaired function of manganese superoxide dismutase (SOD2), likely contributing to a clinical presentation suggestive of mitochondrial disease including T2-hyperintense “Leigh-like” lesions of the basal ganglia. The glycan depiction in this figure follows the current international symbol nomenclature for glycans. Figure created with BioRender.com.
In conclusion, our findings show that conventional glycosylation analyses using transferrin glycosylation is insufficient to detect dysglycosylation in SLC39A8-CDG. N-glycome profiling readily identifies subtle dysglycosylation and is thus a valuable tool both in screening and therapy monitoring. Further research is needed to identify and validate specific patterns of dysglycosylation and to delineate the nature of mitochondrial involvement in SLC39A8 deficiency.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
The authors would like to thank the patients as well as their families for their participation and ongoing support during the conception of this study. The expert technical assistance of Maria Plate is gratefully acknowledged. We thank the National Center for Functional Glycomics at Harvard for use of the mass spectrometry resources. This work was partly supported by the fund “Innovative Medical Research” of the University of Münster Medical School (to JHP; Project PA 5 2 19 01). The Montana Genetics Program at Shodair Children’s Hospital is supported by the Montana Department of Public Health and Human Services (PHH18-0157JT MT Clinical Genetics Program). RGM is supported by The Stanley Center for Psychiatric Research at Broad Institute of Harvard/MIT.
Footnotes
Conflict of Interest:
The authors declare that they have no conflict of interest.
Informed Consent:
All procedures were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients prior to inclusion in the study.
Ethics approval:
Clinical care and research investigations on patients of SLC39A8-CDG were approved by the institutional review boards of Shodair Children’s Hospital (IRB00008287, PKU-016) and the University of Münster (2019-199f-s). Glycomic analysis for research purposes of previously obtained serum/plasma samples was approved by the Massachusetts General Hospital institutional review board (Protocol #: 2017P000115).
IACUC:
No animals were used in this study.
References
- 1.Boycott KM, Beaulieu CL, Kernohan KD, et al. Autosomal-Recessive Intellectual Disability with Cerebellar Atrophy Syndrome Caused by Mutation of the Manganese and Zinc Transporter Gene SLC39A8. Am J Hum Genet 2015;97:886–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park JH, Hogrebe M, Gruneberg M, et al. SLC39A8 Deficiency: A Disorder of Manganese Transport and Glycosylation. Am J Hum Genet 2015;97:894–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haller G, McCall K, Jenkitkasemwong S, et al. A missense variant in SLC39A8 is associated with severe idiopathic scoliosis. Nat Commun 2018;9:4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McCoy TH Jr., Pellegrini AM, Perlis RH. Using phenome-wide association to investigate the function of a schizophrenia risk locus at SLC39A8. Transl Psychiatry 2019;9:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li D, Achkar JP, Haritunians T, et al. A Pleiotropic Missense Variant in SLC39A8 Is Associated With Crohn’s Disease and Human Gut Microbiome Composition. Gastroenterology 2016;151:724–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ng E, Lind PM, Lindgren C, et al. Genome-wide association study of toxic metals and trace elements reveals novel associations. Human Molecular Genetics 2015;24:4739–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Riley LG, Cowley MJ, Gayevskiy V, et al. A SLC39A8 variant causes manganese deficiency, and glycosylation and mitochondrial disorders. J Inherit Metab Dis 2017;40:261–9. [DOI] [PubMed] [Google Scholar]
- 8.Nebert DW, Liu Z. SLC39A8 gene encoding a metal ion transporter: discovery and bench to bedside. Human Genomics 2019;13:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fujishiro H, Himeno S. New Insights into the Roles of ZIP8, a Cadmium and Manganese Transporter, and Its Relation to Human Diseases. Biological and Pharmaceutical Bulletin 2019;42:1076–82. [DOI] [PubMed] [Google Scholar]
- 10.Breton C, Šnajdrová L, Jeanneau C, Koča J, Imberty A. Structures and mechanisms of glycosyltransferases. Glycobiology 2005;16:29R–37R. [DOI] [PubMed] [Google Scholar]
- 11.Kuhn NJ, Ward S, Leong WS. Submicromolar manganese dependence of Golgi vesicular galactosyltransferase (lactose synthetase). Eur J Biochem 1991;195:243–50. [DOI] [PubMed] [Google Scholar]
- 12.Hennet T The galactosyltransferase family. Cell Mol Life Sci 2002;59:1081–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wedler FC, Denman RB. Glutamine synthetase: the major Mn(II) enzyme in mammalian brain. Curr Top Cell Regul 1984;24:153–69. [DOI] [PubMed] [Google Scholar]
- 14.Keen CL, Ensunsa JL, Watson MH, et al. Nutritional aspects of manganese from experimental studies. Neurotoxicology 1999;20:213–23. [PubMed] [Google Scholar]
- 15.Wada Y, Nishikawa A, Okamoto N, et al. Structure of serum transferrin in carbohydrate-deficient glycoprotein syndrome. Biochem Biophys Res Commun 1992;189:832–6. [DOI] [PubMed] [Google Scholar]
- 16.Wada Y Mass spectrometry of transferrin and apolipoprotein C-III for diagnosis and screening of congenital disorder of glycosylation. Glycoconj J 2016;33:297–307. [DOI] [PubMed] [Google Scholar]
- 17.Abu Bakar N, Lefeber DJ, van Scherpenzeel M. Clinical glycomics for the diagnosis of congenital disorders of glycosylation. J Inherit Metab Dis 2018;41:499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bruneel A, Fenaille F. Integrating mass spectrometry-based plasma (or serum) protein N-glycan profiling into the clinical practice? Annals of translational medicine 2019;7:S225–S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mealer RG, Jenkins BG, Chen C-Y, et al. The schizophrenia risk locus in SLC39A8 alters brain metal transport and plasma glycosylation. Scientific Reports 2020;10:13162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park JH, Hogrebe M, Fobker M, et al. SLC39A8 deficiency: biochemical correction and major clinical improvement by manganese therapy. Genet Med 2018;20:259–68. [DOI] [PubMed] [Google Scholar]
- 21.Zuhlsdorf A, Park JH, Wada Y, et al. Transferrin variants: pitfalls in the diagnostics of Congenital disorders of glycosylation. Clin Biochem 2015;48:11–3. [DOI] [PubMed] [Google Scholar]
- 22.Mealer RG, Jenkins BG, Chen C-Y, et al. The schizophrenia risk locus in SLC39A8 alters brain metal transport and plasma glycosylation. bioRxiv 2020:757088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagae M, Yamaguchi Y, Taniguchi N, Kizuka Y. 3D Structure and Function of Glycosyltransferases Involved in N-glycan Maturation. Int J Mol Sci 2020;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clerc F, Reiding KR, Jansen BC, Kammeijer GS, Bondt A, Wuhrer M. Human plasma protein N-glycosylation. Glycoconj J 2016;33:309–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ota T, Suzuki Y, Nishikawa T, et al. Complete sequencing and characterization of 21,243 full-length human cDNAs. Nat Genet 2004;36:40–5. [DOI] [PubMed] [Google Scholar]
- 26.Mealer RG, Williams SE, Daly MJ, Scolnick EM, Cummings RD, Smoller JW. Glycobiology and schizophrenia: a biological hypothesis emerging from genomic research. Molecular Psychiatry 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boogert MAWvd, Larsen LE, Ali L, et al. N-Glycosylation Defects in Humans Lower Low-Density Lipoprotein Cholesterol Through Increased Low-Density Lipoprotein Receptor Expression. Circulation 2019;140:280–92. [DOI] [PubMed] [Google Scholar]
- 28.Niehues R, Hasilik M, Alton G, et al. Carbohydrate-deficient glycoprotein syndrome type Ib. Phosphomannose isomerase deficiency and mannose therapy. J Clin Invest 1998;101:1414–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marquardt T, Luhn K, Srikrishna G, Freeze HH, Harms E, Vestweber D. Correction of leukocyte adhesion deficiency type II with oral fucose. Blood 1999;94:3976–85. [PubMed] [Google Scholar]
- 30.Witters P, Tahata S, Barone R, et al. Clinical and biochemical improvement with galactose supplementation in SLC35A2-CDG. Genetics in Medicine 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tegtmeyer LC, Rust S, van Scherpenzeel M, et al. Multiple phenotypes in phosphoglucomutase 1 deficiency. N Engl J Med 2014;370:533–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kjaergaard S, Kristiansson B, Stibler H, et al. Failure of short-term mannose therapy of patients with carbohydrate-deficient glycoprotein syndrome type 1A. Acta Paediatrica 1998;87:884–8. [DOI] [PubMed] [Google Scholar]
- 33.Grünert SC, Marquardt T, Lausch E, et al. Unsuccessful intravenous D-mannose treatment in PMM2-CDG. Orphanet J Rare Dis 2019;14:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang C-C, Lee C-C, Lin H-H, Chen M-C, Lin C-C, Chang J-Y. Autophagy-Regulated ROS from Xanthine Oxidase Acts as an Early Effector for Triggering Late Mitochondria-Dependent Apoptosis in Cathepsin S-Targeted Tumor Cells. PLOS ONE 2015;10:e0128045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baly DL, Keen CL, Hurley LS. Effects of manganese deficiency on pyruvate carboxylase and phosphoenolpyruvate carboxykinase activity and carbohydrate homeostasis in adult rats. Biological Trace Element Research 1986;11:201. [DOI] [PubMed] [Google Scholar]
- 36.Flynn JM, Melov S. SOD2 in mitochondrial dysfunction and neurodegeneration. Free radical biology & medicine 2013;62:4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.