

Abstract
Within the broad field of synthetic biology, genetic code expansion (GCE) techniques enable creation of proteins with an expanded set of amino acids. This may be invaluable for applications in therapeutics, bioremediation, and biocatalysis. Central to GCE are aminoacyl-tRNA synthetases (aaRSs) as they link a non-canonical amino acid (ncAA) to their cognate tRNA, allowing ncAA incorporation into proteins on the ribosome. The ncAA-acylating aaRSs and their tRNAs should not cross-react with 20 natural aaRSs and tRNAs in the host, i.e., they need to function as an orthogonal translating system. All current orthogonal aaRS•tRNA pairs have been engineered from naturally occurring molecules to change the aaRS’s amino acid specificity or assign the tRNA to a liberated codon of choice. Here we discuss the importance of orthogonality in GCE, laboratory techniques employed to create designer aaRSs and tRNAs, and provide an overview of orthogonal aaRS•tRNA pairs for GCE purposes.
1. Overview of orthogonal translation systems in genetic code expansion
GCE encompasses numerous techniques that allow co-translational installation of ncAAs into proteins within living organisms. Two general strategies have been developed for achieving GCE. In one strategy, ncAAs that are isostructural analogs of canonical amino acids (cAAs) are incorporated into proteins by endogenous aaRSs, which are unable to distinguish between the analog and their natural substrate. Using this methodology, known as residue-specific GCE or sense codon reassignment, all instances of the AA within a protein may be replaced by the non-canonical analog. In the second strategy, known as site-specific GCE or stop codon suppression, exogenous aaRS and tRNA pairs are expressed within a host organism and facilitate the incorporation of ncAAs in response to reassigned codons. Typically, site-specific GCE utilizes nonsense suppressor tRNAs that introduce the ncAA in response to reassigned stop codons. Therefore, unlike residue-specific GCE, with site-specific GCE the position of the ncAA within a protein can be precisely defined by introducing a nonsense mutation into the protein coding gene.
To be useful for site-specific GCE an aaRS•tRNA pair must fulfill the following criteria: (1) the aaRS must be able to be expressed in its active form within the host organism, (2) the tRNA must be correctly processed within the host organism, (3) the tRNA must be compatible with the translational machinery (ribosome, elongation factors, etc.) of the host, (4) the aaRS•tRNA pair must not cross-react with endogenous aaRSs and tRNAs, i.e., it must be orthogonal, and (5) the aaRS must selectively recognize an ncAA substrate over cAAs. In this chapter we describe several aaRS•tRNA pairs that meet these criteria and have been used to site-specifically install ncAAs into proteins in various host organisms.
2. Orthogonality of aaRSs•tRNA pairs and ncAAs
Translation of the genetic code requires aaRSs to attach amino acids to their cognate tRNAs. In general, all aaRSs interact with the acceptor stem of the tRNA in a similar way, with class I synthetases approaching from the minor groove side and class II synthetases from the major groove. Amino acids in class I synthetases are found to make direct or water-mediated interactions with the second and third base pairs of the corresponding tRNA [1]. Although aaRSs’ interaction with the tRNA acceptor stem can be generalized, other features of tRNA recognition are more complex and can only be extrapolated to closely related systems. The tRNA specificity of aaRSs is dictated by a set of idiosyncratic features which are embedded in each tRNA. These features are known as tRNA identity elements [2].
Identity elements can be residues which promote (determinants) or prevent false (anti-determinants) aminoacylation. They can include isolated nucleotides, single-stranded regions, base pairs, or structural motifs. These identity elements can be found across the tRNA L-shape structure but are generally found in the acceptor stem and anticodon loop of the tRNA. Specifically the discriminator base N73 in the acceptor stem and N35 and N36 in the anticodon loop are common tRNA identity elements [2]. For example, all aaRSs from E. coli, except for glutamyl-tRNA synthetase (GluRS) and threonyl-tRNA synthetase (ThrRS), rely on the identity of N73, whereas only alanyl-, histidyl-, seryl- and leucyl-tRNA synthetases (AlaRS, HisRS, SerRS, and LeuRS, respectively) do not use anticodon bases for specific recognition [3].
A widely used strategy to create an orthogonal translating system (OTS), is to import an aaRS•tRNA pair from a phylogenetically distant organism. Phylogenetic distance can create divergence in the tRNA identity elements which prevents such pairs from cross-reacting even if they are specific for the same AA [4]. This strategy is useful to identify possible OTSs, however further engineering is usually required to improve orthogonality [5].
Another consideration is AA orthogonality, whether the ncAA is recognized by endogenous aaRSs or the orthogonal aaRS (o-aaRS) recognizes cAAs as substrates. AA orthogonality ensures that the ncAA is incorporated only at the desired position and not replaced by a cAA. This becomes especially important when introducing multiple orthogonal systems into a host (described in detail below) [3] (Fig. 1).
Fig. 1.
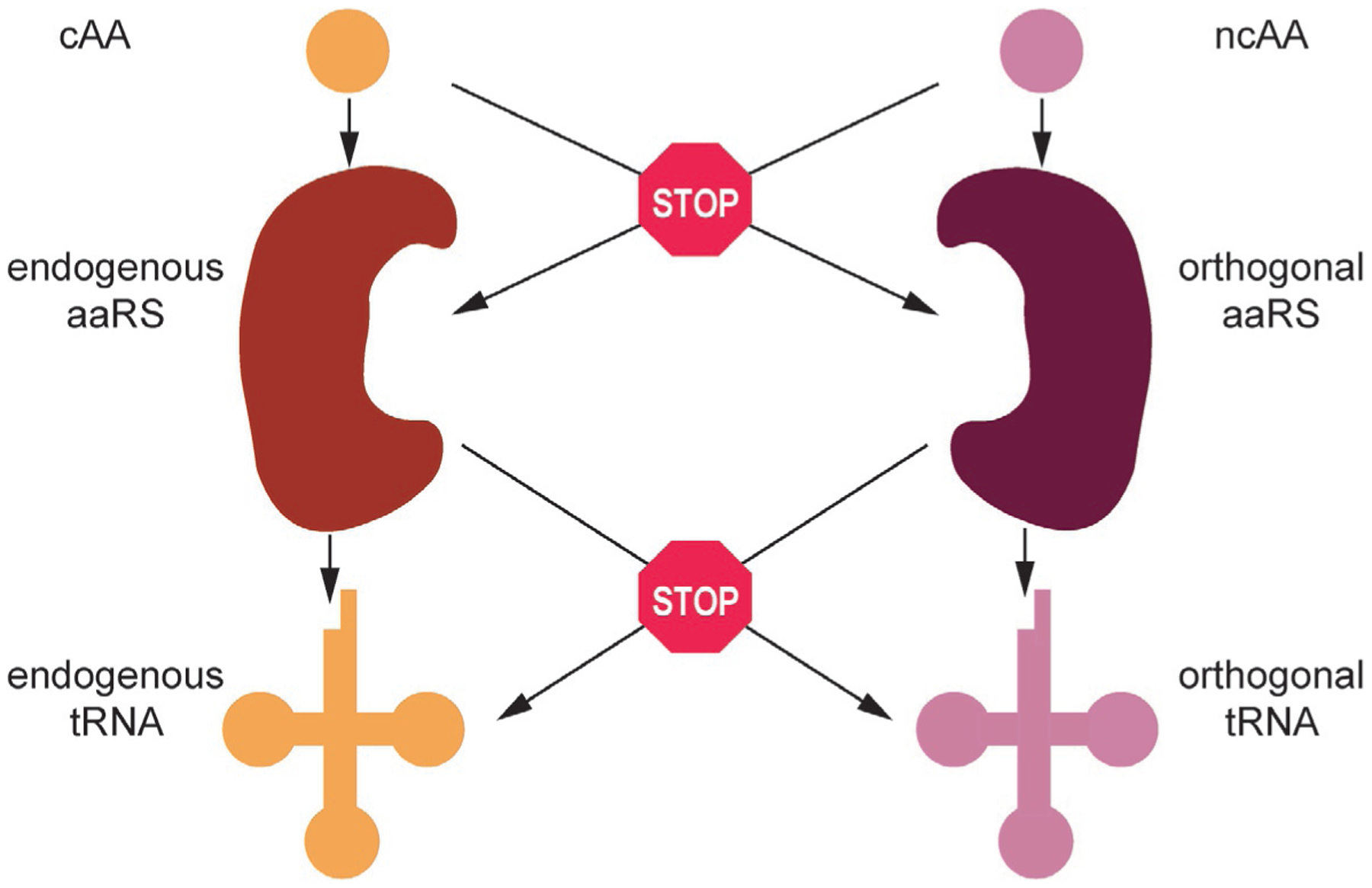
Orthogonality requirements from the level of amino acid (AA) to tRNA. A fully orthogonal translation system cannot cross-talk at any level.
3. General approaches in aaRS and tRNA engineering
3.1. Common aims of orthogonal aaRS and tRNA engineering
The majority of OTSs require further optimization once they have been introduced into the host. These may include optimization of aaRS solubility, aaRS and tRNA expression, specific mutagenesis of aaRS residues involved with ncAA/AA recognition, and specific nucleotide mutations in the tRNA. With natural suppressors such as tRNAPyl, no modifications of the anticodon are necessary. Other orthogonal tRNAs (o-tRNAs) require, at the very least, mutation of the original anticodon sequence in order to decode stop codons of choice. Since the vast majority of aaRSs recognize the cognate tRNA anticodon with high specificity (see above), mutations disrupt these interactions and decrease the aaRS’s affinity for the mutated o-tRNA. In such instances, it may be necessary to randomize interacting residues in the anticodon-binding domain of the aaRS in question.
The second major consequence of an anticodon mutation may be unwanted recognition by host aaRSs. In E. coli, GlnRS and LysRS, tend to misacylate various o-tRNAs [6–8] due to similarities in the anticodon sequence between the amber suppressor and their cognate tRNAs. In such instances nucleotides that could act as anti-determinants for these aaRSs are targeted for mutagenesis or complete randomization. Fully optimized tRNA should achieve high orthogonality but retain the capacity to be well recognized by the cognate aaRS, as well as the host’s elongation factor, ribosome, and tRNA modifying enzymes.
To direct ncAA insertion, the aaRS’s active site usually needs to be altered. Residues that participate in hydrogen bonds and salt bridges with the AA substrate, as well as those that establish hydrophobicity of the active site or determine steric properties of the AA binding cavity may need to be mutated. In the case of ncAAs containing unconventional chemistry at the position of the original α-amino group, conserved acidic residues devoted to its recognition may need to be mutated. Simultaneously, the aaRS should improve its ncAA-acylation efficiency and lose the ability to charge its original cognate substrate. This is achieved through multiple rounds of positive and negative selection of the aaRS variants both in the presence of an ncAA (positive) and in its absence (negative). However, because the negative selection step is rarely executed in the presence of other ncAA substrates, the vast majority of evolved aaRSs show a broad substrate range with respect to various ncAAs, and are therefore polyspecific [9].
Some aaRSs possess a distinct domain that contains an additional catalytic center devoted to the hydrolysis of misacylated tRNAs. Due to sterical and chemical similarities, almost half of the aaRSs misacylate their tRNAs with noncognate natural AAs. Upon aminoacylation in the synthetic active site, the acylated 3′-end of the tRNA enters the hydrolytic, editing site, able to hydrolyze the ester bond between the noncognate AA substrate and tRNA, but not the one with the cognate AA. Through evolution, this proofreading mechanism developed in response to noncognate natural AAs and it may or may not be sensitive to ncAA misacylation [10]. Therefore, aaRS engineering to improve ncAA specificity may include the editing site, in addition to the synthetic site of the aaRS [11]. Engineering with fused editing domains (to clear the natural AA substrate) has also been reported [12,13].
3.2. Methods employed for the directed evolution of aaRSs
Optimization of aaRS activity usually includes very large libraries (≤109 members) generated by complete randomization of appropriate active site or anticodon-binding domain residues. Given the size of an average aaRS library, variants with desired acylation properties are most often identified using in vivo (e.g., cat- or GAL-4 mediated assays [5,14]) or ex vivo selection platforms (phage-assisted (non)continuous evolution, PA(N)CE, [15,16]). In silico and in vitro selection approaches, although holding great promise, are much less present in the field [17,18].
To identify an optimized aaRS in vivo, an aaRS library is first generated in vitro, through focused mutagenesis or error-prone PCR. In these instances, the quality of the starting library (i.e., balanced distribution of each unique DNA sequence or a clone) becomes intertwined with transformation efficiency. For this reason, selection platforms using E. coli as a host are still the most predominant ones.
Focused mutagenesis in vivo can be undertaken with multiplex automated genome engineering (MAGE) and has been utilized to generate a TyrRS variant that can mediate ncAA insertion and stop codon suppression at 30 unique positions of a reporter protein [19]. In this strategy, the o-aaRS is introduced into the E. coli genome and single-stranded DNA oligonucleotides containing mutations at desired sites are supplied by electroporation. During outgrowth, a λ-phage protein involved in homologous recombination mediates annealing of the electroporated DNA to their genomic target. Iterative transformation of mutagenic oligonucleotides is needed to generate a fully randomized o-aaRS library [19,20].
Introduction of random mutations into o-aaRSs in vivo can also be executed using mutator strains, such as XL1-red E. coli [21], or with mutagenic plasmids. The latter contain various combinations of previously identified mutator genes placed under an inducible promoter [22]. Because mutagenic plasmids offer tunable mutation rates in vivo they have been employed in combination with PACE and allowed identification of PylRS variants with improved acylation properties toward Nε-Boc-l-lysine [15,16].
After generating a library of mutated aaRS variants, those with improved activity toward an ncAA of choice (or cognate suppressor tRNA) must be selected (Fig. 2). This is accomplished through the observed suppression of a nonsense mutation placed within a reporter gene upon addition of the ncAA to the growth media. Reporter proteins typically include those that offer resistance to certain antibiotics (such as β-lactamase [23], and chloramphenicol acetyltransferase, CAT [5]) or give rise to other selectable features (such as fluorescence [24,25], or growth on selective media [14]). In general, reporter genes contain one or several in-frame stop codons at permissive positions. However, under this type of selection, the observed stop codon read-through may not signal specific ncAA incorporation. Therefore, negative selection needs to be undertaken to eliminate the clones that still possess significant affinity for the cognate AA, as well as those that have acquired ability to acylate a noncognate, canonical AA. To this end, the population of aaRS variants collected in the first round of positive selection is co-transformed with a second reporter gene containing a missense mutation; this step is conducted in ncAA absence. Reporter proteins employed for the purposes of negative selection include various toxins, such as barnase [5], CcdB toxin [26], TolC [19], or a toxic variant of E. coli PheRS [27]. Because the surviving clones are collected in the absence of the ncAA, aaRS variants that mediate read-through using natural AAs are eliminated.
Fig. 2.
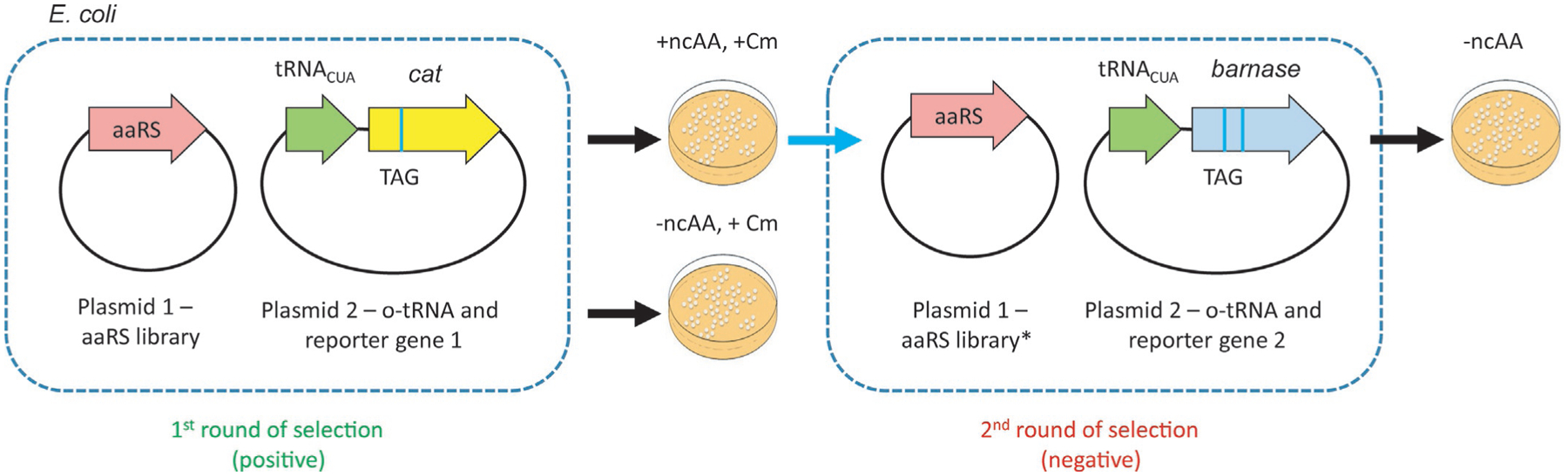
General scheme of aaRS selection. In the first round of selection the aaRS library is co-transformed with a plasmid harboring the orthogonal tRNA suppressor (tRNACUA) and reporter gene (cat). Suppression, and therefore growth in the presence of chloramphenicol (Cm) may occur when the tRNA is acylated both with the ncAA, as well as cAA. Surviving clones are isolated and co-transformed with a second reporter plasmid containing a toxic gene with two stop codons (barnase). In the absence of an ncAA, only those clones that do not facilitate read-through using cAAs are selected.
Another strategy for selection is phage-assisted evolution. Here, the M13 phage protein pIII required for phage infectivity serves as a reporter [15,16]. A permissive E. coli strain harboring a plasmid with gene III containing one or several in-frame TAG codons is infected with an M13 derivative containing an aaRS gene in place of gene III. Phages containing aaRS variants capable of mediating stop codon read-through in gene III can synthesize full-length pIII and are able to infect E. coli cells and propagate. To eradicate those variants that mediate incorporation of cAAs into pIII, a dominant-negative variant of pIII (pIII-neg) is used as a second reporter [28]. The pIII-neg reduces the phage’s infectivity thereby preventing false positives (AA-acylating phages) from accumulating within the population.
3.3. Polyspecificity of evolved aaRSs
Almost all evolved aaRS variants show some activity with chemically similar ncAA substrates, and in some cases even retain activity with the original substrate. This is in part due to the fact that the vast majority of positive selection reporters are not specific for the ncAA in question (one notable exception being selenocysteine selection markers [29,30]). While ncAA polyspecificity can be tolerated with systems where only one ncAA is introduced, insertion of multiple ncAAs mandates use of OTSs that remain orthogonal to each other, and do not possess an overlapping ncAA substrate range. A recent report shows that protein degradation machinery can be exploited in such a way to signal specific insertion of the ncAA [31]. In the case of azide- or alkyne-bearing ncAAs, successive rounds of positive selection can be undertaken, the first to demonstrate the UAG read-through, and a second to demonstrate selective ncAA labeling. Fluorescence-activated cell sorting (FACS) is especially useful for this purpose, as it allows simultaneous monitoring of GFP synthesis (UAG read-through) and chemoselective modification (azide-alkyne click reaction with a fluorescent dye). This approach was used to increase specificity of an originally polyspecific aaRS variant [32]. We expect that in the future, novel strategies will allow identification of ncAA insertion in order to accelerate the development of mutually orthogonal aaRS•tRNA pairs, which are able to facilitate simultaneous insertion of different ncAAs (Fig. 2).
4. The PylRS•tRNAPyl pair
In 1998 it was discovered that the gene encoding Mtmb, a monomethylamine methyltransferase in the archaeon Methanosarcina barkeri, was disrupted by a single, in-frame amber (UAG) nonsense codon that did not terminate translation [33]. Shortly after, a number of in-frame amber codons were also found in the genes encoding both dimethyl- and trimethylamine methyltransferases in M. barkeri and in the related Methanosarcina thermophila [34]. To investigate the source of nonsense suppression in these organisms the crystal structure of MtmB was solved and reported in 2002. At the position encoded by UAG was found a unique residue—a lysine modified at the Nε-amine with (4R,5R)-4-substituted-pyrroline-5-carboxylate. This new AA was dubbed l-pyrrolysine (Pyl, Fig. 3A) [36]. Near the methyltransferase gene cluster, genes encoding an amber suppressor tRNA, tRNAPyl, and a novel class II aaRS, PylRS, were also found (pylT and pylS, respectively) [37]. Using chemically synthesized Pyl [9] acylation of Pyl to tRNAPyl by PylRS was demonstrated [38,39]. These results established Pyl to be the 22nd co-translationally installed AA.
Fig. 3.
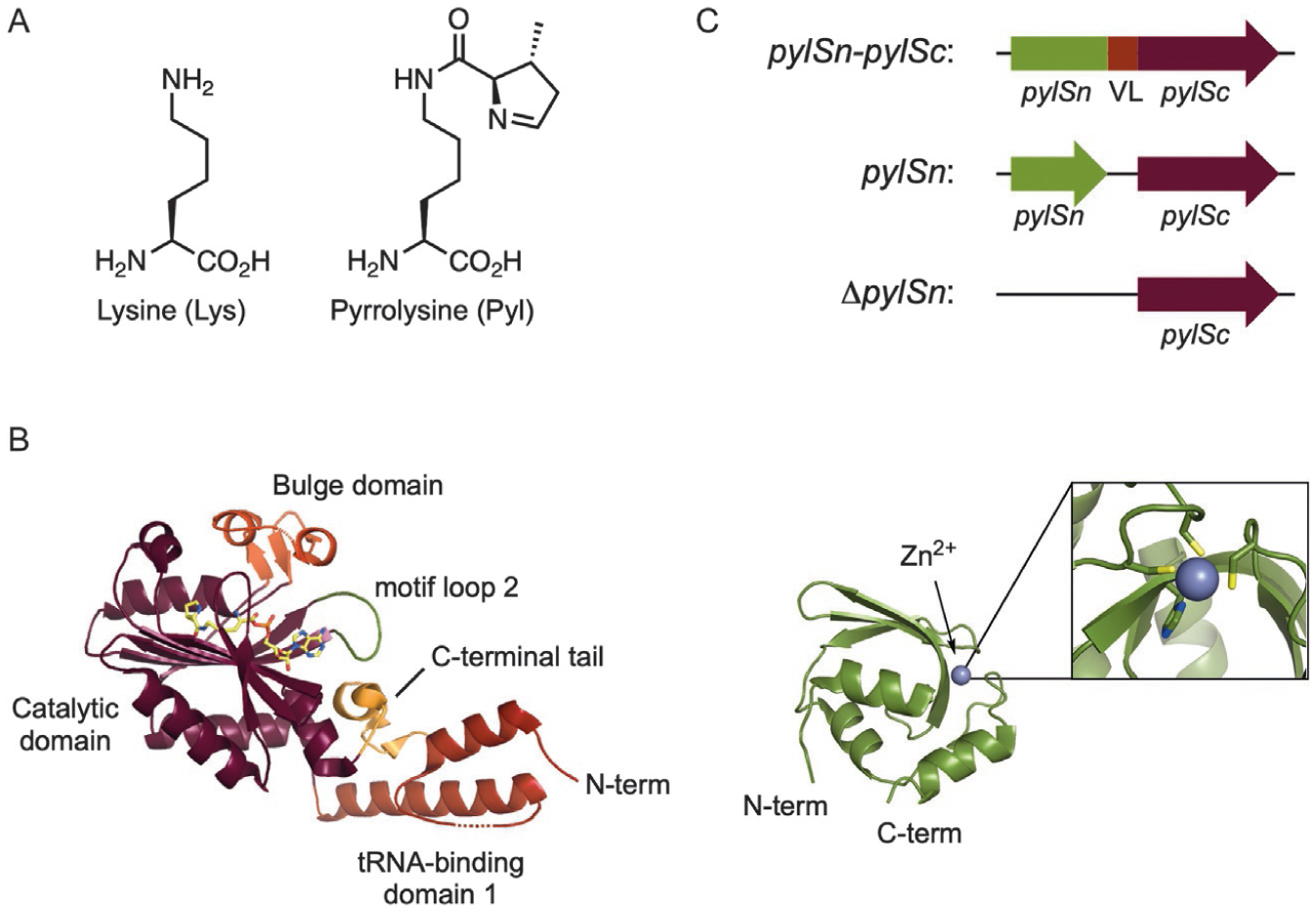
(A) The structures of l-lysine and l-pyrrolysine. (B) The structures of the C-terminal (left) and N-terminal (right) domains of PylRS from M. mazei. (PDB: 2Q7H, 5UD5) [15,35]. (C) Organization of the N-terminal (pylSn) and C-terminal (pylSc) domains of PylRS from the three defined classes (VL=variable linker).
At the time that Pyl was discovered in M. barkeri, a bioinformatic analysis of available genomes revealed homologs of PylRS and tRNAPyl in the Gram-positive bacterium Desulfitobacterium hafniense [37]. More recently, homologs of these genes have been revealed in the genomes of >95 archaea and bacteria, including those found in humans [40]. It is assumed that Pyl incorporation evolved for the specific purpose of methylamine metabolism, the reactive imine of Pyl serving to bind and activate the methylamine substrate [41]. In at least one case, Pyl is biosynthesized directly in response to the presence of methylamines in the growth media [42]. In other cases, Pyl can be found incorporated at non-essential positions within proteins unrelated to methylamine metabolism, suggesting deeper integration of Pyl into the proteome and a more general expansion of the genetic code in nature [43].
4.1. Structure of PylRS and tRNAPyl
4.1.1. PylRS
Crystal structures of bacterial and archaeal PylRS have revealed that the overall enzyme architecture resembles that of other class II aaRSs with the characteristic motifs 1, 2, and 3 [44,45]. The C-terminal domain of PylRS contains the conserved catalytic domain (a seven-stranded antiparallel β-sheet surrounded by several α-helices), a bulge domain, and tRNA-binding domain 1 (Fig. 3B). In the crystal structure, the enzyme forms a homodimer with residues from motif 1 mediating the interface between PylRS monomers. Along with the C-terminal catalytic domain, PylRS enzymes from some organisms also possess a unique N-terminal RNA-binding domain, dissimilar to any known RNA-binding protein [46]. PylRS enzymes can be subdivided into three classes (pylSn-pylSc fusion, pylSn, or ΔpylSn) based on the arrangement of their C- and N-terminal domains (Fig. 3C). Enzymes in the pylSn-pylSc fusion class, such as those from the Methanosarcina, contain an N-terminal RNA-binding domain that is connected to the C-terminal catalytic domain by a variable linker [40,47]. The fused N-terminal domain is absolutely required for enzymatic activity in vivo [48]. Poor solubility of the N-terminal domain has prevented crystallization of full-length PylRS enzymes from the pylSn-pylSc fusion class [49]; however, the crystal structure of the isolated N-terminal domain of the PylRS from Methanosarcina mazei (MmPylRS) was recently solved in complex with tRNAPyl [15]. This structure revealed that the N-terminal domain folds into a compact globule, stabilized by a coordinated zinc ion, and contacts the T- and variable loops of tRNAPyl (Fig. 3B). Furthermore, the structure revealed that PylRS makes extensive contact with tRNAPyl, wrapping around the tRNA with the N- and C-terminal domains binding on opposite sides [15].
PylRS enzymes in the pylSn class originate from bacteria, the most well-studied being from D. hafniense (DhPylRS). In these enzymes, homologs of the C-terminal catalytic domain and N-terminal RNA-binding domain are expressed as two distinct proteins encoded by separate genes. Unlike enzymes in the pylSn-pylSc fusion class, the catalytic domain of DhPylRS displays significant activity in vivo without expression of the RNA-binding domain [50,51]. It was therefore postulated that the biological function of the RNA-binding domain may be to help recruit tRNAPyl to the catalytic domain. This is supported by the high binding affinity of the RNA-binding domain for tRNAPyl [46,48]. However, co-expression of the D. hafniense RNA-binding domain does not improve in vivo activity [50,51]. Therefore, the exact physiological role of the N-terminal domain of PylRS enzymes in the pylSn class is unclear.
PylRS enzymes in the ΔpylSn class contain the conserved C-terminal catalytic domain yet lack an N-terminal RNA-binding domain. This class is comprised of enzymes that originate from a recently described, and largely uncultured, 7th order of methylotrophic methanogens dubbed the Methanomassiliicoccales [52–55]. Due to the lack of an RNA-binding domain and the bizarre structure of tRNAPyl in these organisms (discussed below), it was not immediately clear whether PylRS was active in these organisms. However, as with other known Pyl-incorporating organisms, the gene encoding MtmB maintained an in-frame UAG codon, strongly supporting genetic encoding of Pyl [47,54]. Recently, several PylRS•tRNAPyl pairs of the ΔpylSn class were cloned and their in vivo activity was confirmed in E. coli with the widely used Pyl analog Nε-Boc-l-lysine (4) [40]. The most active among these new PylRS enzymes was that from “Candidatus Methanomethylophilus alvus” (CMaPylRS) which displayed significantly higher in vivo activity than the widely used MmPylRS [40]. Since this initial report, the activity of CMaPylRS has been corroborated by several studies [56–59] and the crystal structure of the enzyme has been deposited by two independent groups (PDB: 6EZD, 6JP2) [60,61]. Interestingly, despite the high activity of this enzyme in the absence of an RNA-binding domain, no remarkable features were apparent in the crystal structure, with the CMaPylRS showing good sequence and structural alignment with MmPylRS and DhPylRS. Therefore, the high activity of the stand-alone catalytic domain might result from the distinct features of tRNAPyl from these organisms.
4.1.2. tRNAPyl
Despite relatively low sequence similarity, tRNAPyl from bacteria and the Methanosarcinaceae share numerous unique features that distinguish them from most cytosolic tRNAs. These include only one base between the acceptor and D-stems, an anticodon stem containing five instead of six base pairs, a short three-base variable loop, five-base D-loop, and the absence of the highly conserved GG and TΨC sequences in the D- and T-loops, respectively (Fig. 4A) [37,62]. Whereas tRNAPyl from the pylSn and pylSn-pylSc fusion classes share these unique features, tRNAPyl within the ΔpylSn class are more disparate and contain further structural anomalies. In tRNAPyl from the ΔpylSn class, the acceptor and D-stems are separated by one or two bases, and in some cases none at all, the D-loop is shortened further to four or three bases, and, intriguingly, the anticodon stem has additional unpaired residues that form a loop of five to seven bases (Fig. 4A) [47,54]. Despite these structural deviations tRNAPyl adopts the L-shape of canonical tRNA and behaves as a typical elongator tRNA in both bacteria and eukaryotes [15,51,63].
Fig. 4.
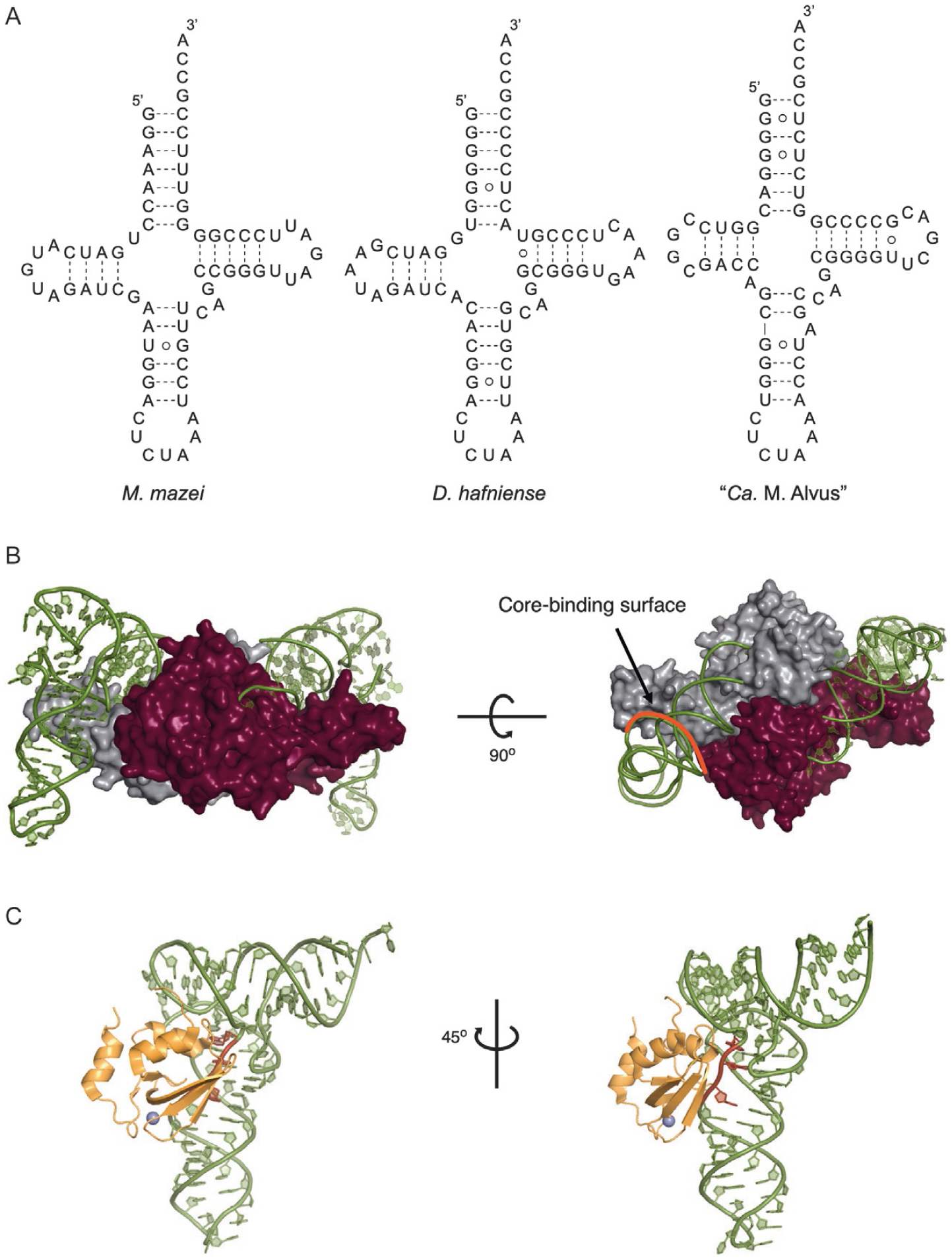
(A) The cloverleaf structures of tRNAPyl from M. mazei, D. hafniense, and “Ca. M. alvus”. (B) The structure of the PylRS C-terminal domain from D. hafniense in complex with tRNAPyl. The two enzyme monomers are colored gray and maroon. The core-binding surface is highlighted red (PDB: 2ZNI) [51]. (C) The structure of the PylRS N-terminal domain from M. mazei in complex with tRNAPyl. The variable loop of tRNAPyl is highlighted red (PDB: 5UD5) [15].
The PylRS•tRNAPyl pair is highly orthogonal in bacteria and eukaryotes which has allowed for it to be used for GCE in numerous model organisms. Crystal structures of tRNAPyl in complex with the catalytic and RNA-binding domains have revealed how the unique features of tRNAPyl and PylRS contribute to this exceptional orthogonality. Orthogonality can largely be attributed to two features. First, the shortened variable and D-loops, together with the shortened spacing between the acceptor and D-stems cause tRNAPyl to adopt a tightly compacted core structure [51]. The complementary-shaped core-binding surface on PylRS makes extensive contact with the compacted core of tRNAPyl and sterically occludes bulkier tRNAs (Fig. 4B) [4,51]. Second, the N-terminal domain of PylRS recognizes the shortened variable loop of tRNAPyl and rejects tRNAs with larger variable loops (Fig. 4C) [15]. Therefore, by artificially elongating the variable loop in the ΔpylSn class tRNAPyl, it becomes orthogonal to pylSn-pylSc fusion enzymes. This has been exploited to make mutually orthogonal PylRS enzymes that can be used to encode two distinct ncAAs into one protein in the same host [40,56]. A key feature of the PylRS•tRNAPyl interaction is that the enzyme does not interact with the anticodon of tRNAPyl. This allows tRNAPyl to be used for suppression of codons aside from UAG including opal and ochre nonsense codons, as well as four base and reassigned sense codons [64–66].
4.2. The use of PylRS•tRNAPyl in genetic code expansion
The PylRS•tRNAPyl pair has emerged as the most widely used aaRS•tRNA pair for GCE. Several features of PylRS make it an excellent choice for this purpose. First, high substrate side chain promiscuity, low selectivity of the substrate α-amine, and lack of an editing domain, allow PylRS and its mutants to recognize a variety of structurally diverse ncAAs. Second, tRNAPyl is a natural amber suppressor. Therefore, the system can be directly used for incorporation of ncAAs in response to amber codons without optimization. Further, the lack of recognition of the tRNA anticodon by PylRS allows for facile reassignment of other nonsense, sense, and four base codons. Third, the PylRS•tRNAPyl pair is highly orthogonal in bacteria and eukaryotes, displaying no cross-reactivity with endogenous aaRSs or tRNAs [67].
4.3. Engineering the PylRS substrate binding pocket
Crystal structures of MmPylRS in complex with Pyl, together with biochemical data, have identified residues in PylRS that govern substrate recognition (Fig. 5). Three residues in the substrate binding pocket of PylRS (Arg330, Asn346, and Tyr384) form a hydrogen-bonding network with the Pyl substrate. The side chain of Arg330 hydrogen bonds with the α-carboxyl oxygen of Pyl. The side chain amide of Asn346 makes direct hydrogen-bonding interaction with the side chain amide oxygen of Pyl, along with a water-mediated interaction with the substrate α-amine. Tyr384 sits at the tip of a dynamic β7-β8 hairpin. In the apo-form Tyr384 is distal to the active site and is only weakly ordered. However, upon Pyl binding the hairpin swings closed and Tyr384 makes hydrogen-bonding interactions with the nitrogen of the pyrrole ring and the α-amine of Pyl [35,68,69]. This interaction is not critical as Tyr384Phe or Tyr384Trp mutation increases the activity of PylRS with both Pyl, as well as ncAAs [70–72]. It has been speculated that Tyr384 hydrogen-bonding closes the active site, which serves to protect the unstable pyrrolysyl-adenylate when the cellular concentration of tRNAPyl is low [68]. A key feature of all PylRS enzymes is a deep, hydrophobic binding pocket that accommodates the pyrrole ring of the substrate. In MmPylRS this pocket is comprised of residues Tyr306, Tyr384, Trp417, Cys348, and Val401 [35].
Fig. 5.
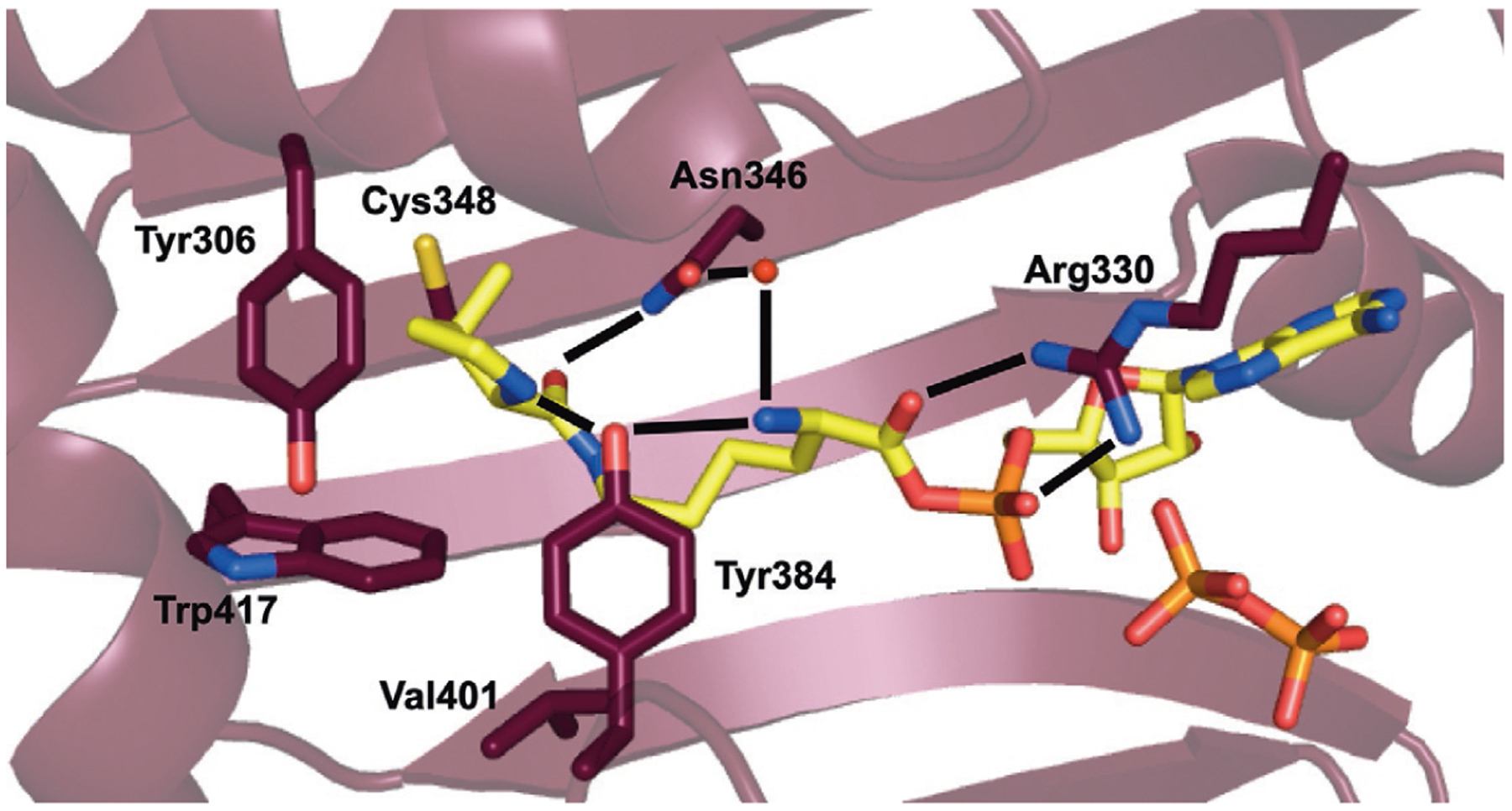
The structure of MmPylRS in complex with adenylated Pyl (PDB: 2Q7H) [35]. Hydrogen bonds that govern substrate recognition are shown.
Synthetases are inherently promiscuous enzymes [10,73]. The ability of aaRSs to recognize various AA substrates that are isostructural analogs of their native substrate has long been used to incorporate ncAAs into proteins for various purposes [73–75]. In the same manner, wild-type PylRS has been used to install numerous AAs that are structural analogs of Pyl (Fig. 6, 1–12). Due to low specific recognition of the pyrrole ring by PylRS, and the relatively large size of the hydrophobic pocket into which this moiety fits, the pyrrole ring can be replaced with a number of non-native hydrophobic constituents while maintaining high enzymatic activity [67,76]. Indeed for most analogs simply maintaining the Nε-amide moiety, which hydrogen bonds to Asn346, is sufficient to maintain binding to PylRS [77]. However, analogs with small substituents, such as Nε-acetyl-l-lysine, are not recognized by the wild-type enzyme requiring significant enzyme engineering to generate a smaller, more hydrophobic binding pocket [9,26,78]. For analogs with larger substituents, widening of the hydrophobic pocket accommodating the pyrrole ring can greatly improve their incorporation; e.g., the Tyr306Ala mutation significantly expands the pocket and allows for incorporation of a range of Pyl analogs containing large substituents in place of the pyrrole ring (Fig. 6, 13–24) [71,79].
Fig. 6.
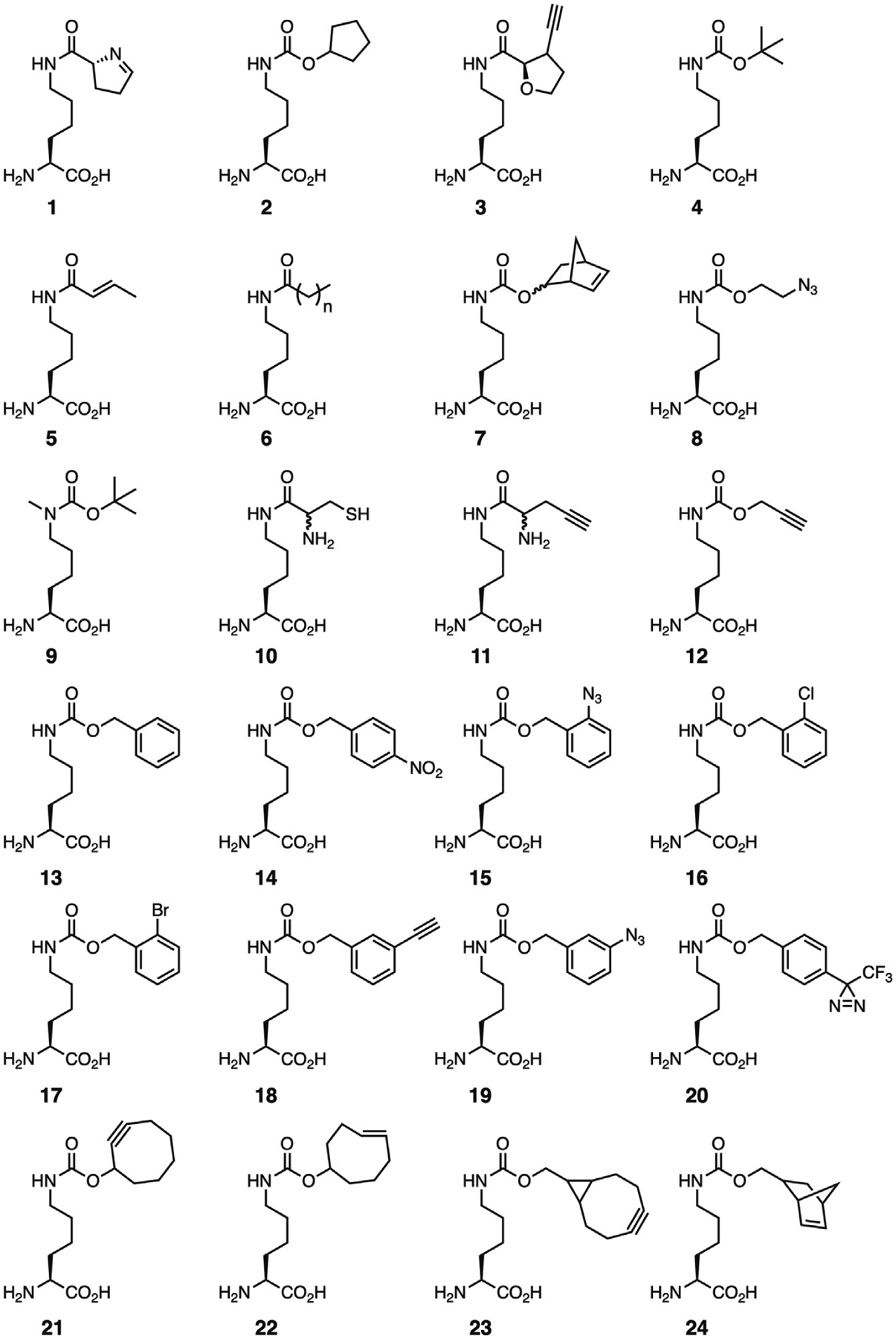
Representative lysine derivatives recognized by wild-type PylRS and the Tyr306Ala/Tyr384Phe mutant.
Two other features of PylRS substrate recognition have allowed genetic incorporation of diverse Pyl analogs. First, in contrast to most aaRSs, PylRS only weakly recognizes the α-amine of its substrate via the non-essential Tyr384 and a water-mediated hydrogen bond to Asn346. This lack of strong backbone recognition allows PylRS to accept α-hydroxy, Nα-methyl, and d-Pyl derivatives [80,81]. Second, PylRS does not utilize the side chain amide nitrogen for substrate recognition. This has allowed for the use of PylRS to direct the genetic encoding of several Nε-modified AAs [82,83].
Along with Pyl derivatives, PylRS also facilitates genetic encoding of l-phenylalanine derivatives. The change in AA substrate recognition from Pyl to Phe, at first glance, appears to be a radical shift. However, altering PylRS to recognize Phe substrates requires surprisingly few modifications. In fact, only two mutations are required to convert MmPylRS to an enzyme with high activity toward Phe [84]. Mutating Asn346 to the smaller hydrophobic residues Ala or Val abrogates steric clashes with Phe, while mutating Cys348 to a larger residue, such as Met or Trp, reduces the size of the pocket to accommodate the new substrate [84,85].
The observation that the Cys348Met mutation decreases the size of the substrate binding pocket to accommodate Phe, led to the speculation that mutating Cys348 to a smaller residue would allow recognition of Phe derivatives with large para substituents [86]. On this basis an MmPylRS variant containing mutations Asn346Ala and Cys348Ala (PylRS-AA) was developed and tested for recognition of a variety of Phe derivatives. While PylRS-AA was able to efficiently utilize several Phe derivatives containing large para substituents, the enzyme lost most of its activity toward Phe [86]. Comparison of the crystal structure of PylRS-AA to that of E. coli PheRS revealed an enlarged substrate pocket with few hydrophobic contacts available to mediate Phe recognition [87]. Since then, PylRS-AA has been tested with a number of substrates and found to have a remarkably broad substrate spectrum recognizing Phe derivatives with a diverse range of ortho-, meta-, and para substituents, as well as histidine derivatives (Fig. 7, 25–64) [86,88–91]. Polyspecificity appears to be a general feature of evolved aaRSs with many examples of engineered enzymes with diverse substrate ranges reported in the literature [9,92–94]. In one sense, polyspecificity is a desirable feature as it reduces engineering efforts by removing the necessity to evolve unique aaRS variants to incorporate structurally diverse ncAAs. However, overlapping substrate recognition of o-aaRSs will significantly hamper future efforts to encode multiple distinct ncAAs at defined sites [95]. In response to the need for more specific aaRS enzymes, recent efforts in synthetase evolution have focused on improving negative selections or utilizing ncAA-specific positive selections to narrow the substrate scope of the evolved aaRS variant [16,31,32].
Fig. 7.
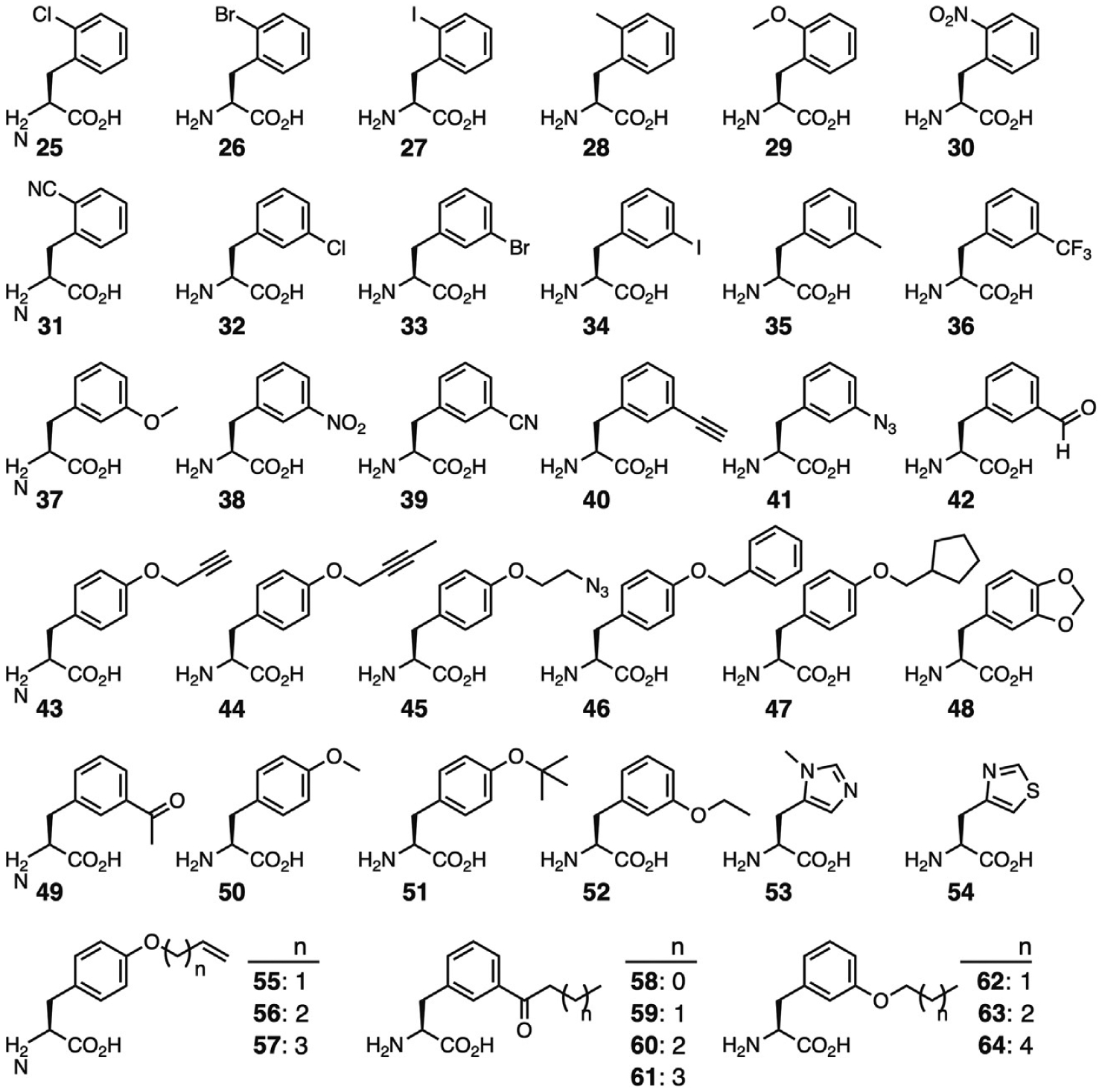
Phenylalanine derivatives recognized by the Asn346Ala/Cys348Ala MmPylRS mutant.
Aside from the widely used M. mazei and M. barkeri PylRSs, enzymes from D. hafniense and Ca. M. alvus have also been employed for GCE. However, due to the relatively poor activity of DhPylRS in E. coli, and the only recent discovery of CMaPylRS, examples of ncAAs recognized by these enzymes are limited. Nonetheless, wild-type DhPylRS has been shown to be able to aminoacylate tRNAPyl with several Pyl derivatives in vitro with a substrate range similar to MmPylRS [96]. Recently a DhPylRS mutant containing mutations at positions Asn176Ala and Thr178Leu (corresponding to residues Asn346 and Cys348 in MmPylRS) was shown to mediate incorporation of Phe in response to UAG codons [50]. Furthermore, introducing a Thr178Gly mutation afforded an enzyme with preference for para-azido-l-phenylalanine over Phe. To our knowledge, this is the only report of GCE with DhPylRS.
Like MmPylRS, wild-type CMaPylRS recognizes a number of lysine derivatives with small substituents in place of the pyrrole ring [40,56,57]. In E. coli, most of these substrates show higher incorporation by CMaPylRS compared to MmPylRS and MbPylRS [57]; whereas, MmPylRS is more active in HEK293T [56,58]. This discrepancy is possibly the result of inefficient recognition of the unique Ca. M. alvus tRNAPyl by eukaryotic elongation factors (discussed below) [97]. The substrate binding pocket of CMaPylRS is highly similar to MmPylRS differing by only two residues. Residues corresponding to Leu309 and Cys348 in MmPylRS are Met and Val in CMaPylRS, respectively. Because of their high similarity, several mutations that expand the substrate range of MmPylRS have been successfully transplanted into CMaPylRS, e.g., the aforementioned Tyr306Ala mutation (Tyr126Ala in CMaPylRS), which increases the size of the binding pocket, allows CMaPylRS (Tyr126) to recognize Lys derivatives with more bulky substituents [40,56,57].
4.4. Enhancing PylRS-mediated ncAA incorporation
While most studies toward engineering PylRS target the substrate binding pocket, mutations elsewhere in the enzyme can also improve ncAA incorporation. As mentioned above, the N-terminal domain of PylRS is poorly soluble which not only hinders crystallization of the full-length enzyme, but also limits soluble expression in vivo. To improve the solubility of this domain, a PylRS N-terminal domain library was screened affording a mutant enzyme with up to threefold improvement in in vivo ncAA incorporation [98]. Using phage-assisted evolution, enzyme variants have been isolated with mutations in the N-terminal domain that directly improve catalytic efficiency by decreasing the Km of tRNAPyl [15]. Importantly, because mutations in the N-terminal domain do not affect AA substrate recognition, they can be transplanted into other previously identified PylRS variants to afford similar improvements in activity.
In addition to the catalytic and RNA-binding domains, mutations in tRNAPyl can also improve ncAA incorporation. Because the PylRS•tRNAPyl pair originate from archaea, tRNAPyl has not co-evolved with the translational components in bacteria and eukaryotes. Suboptimal recognition of tRNAPyl by the translational machinery within these organisms leads to decreased protein yields. Protein yields can be improved, however, using tRNAPyl variants engineered for better recognition by the host translation system. For example, introducing mutations into the acceptor and T-stems of tRNAPyl improve its recognition by E. coli EF-Tu resulting in a tRNAPyl variant with up to a fivefold increase in in vivo UAG suppression [97]. Similarly by introducing base substitutions into tRNAPyl that are conserved in human cytosolic tRNAs, tRNAPyl variants have been developed that show enhanced UAG suppression in eukaryotes [99].
5. The TyrRS•tRNATyr pair from Methanocaldococcus jannaschii
In parallel to the research going on using PylRS for GCE, other orthogonal aaRS•tRNA pairs are sought out for their use in E. coli. In 2000, biochemical analysis of the TyrRS•tRNATyr pairs from various organisms revealed that the TyrRS•tRNATyr pair from the archaeon Methanocaldococcus jannaschii (MjTyrRS) had a unique potential for GCE in E. coli [100]. First, the tRNATyr from archaea and eukaryotes have distinct identity elements from bacteria. Archaeal and eukaryotic tRNATyr contain a C1:G72 pair and short variable arm which distinguish them from bacterial tRNATyr which have a G1:C72 pair and a long variable arm. Both archaeal and bacterial tRNATyr share the A73 identity element (Fig. 8) [101]. This suggested that an archaeal TyrRS•tRNATyr could potentially be used as an OTS in bacteria. Not only does MjtRNATyr have the archaeal tRNA identity elements as described, but MjTyrRS is lacking an editing domain [102] and the anticodon loop-binding domain which is found in most enzymes (Fig. 9A) [104]. Without an editing domain MjtRNATyr can be aminoacylated with an ncAA without concern that it will be removed by the editing mechanism. Additionally, absence of the anticodon loop-binding domain suggests that the anticodon could be mutated without affecting the aminoacylation activity [103,105–107]. These three aspects highlight MjTyrRS•MjtRNATyr as a potential candidate for GCE in bacteria.
Fig. 8.
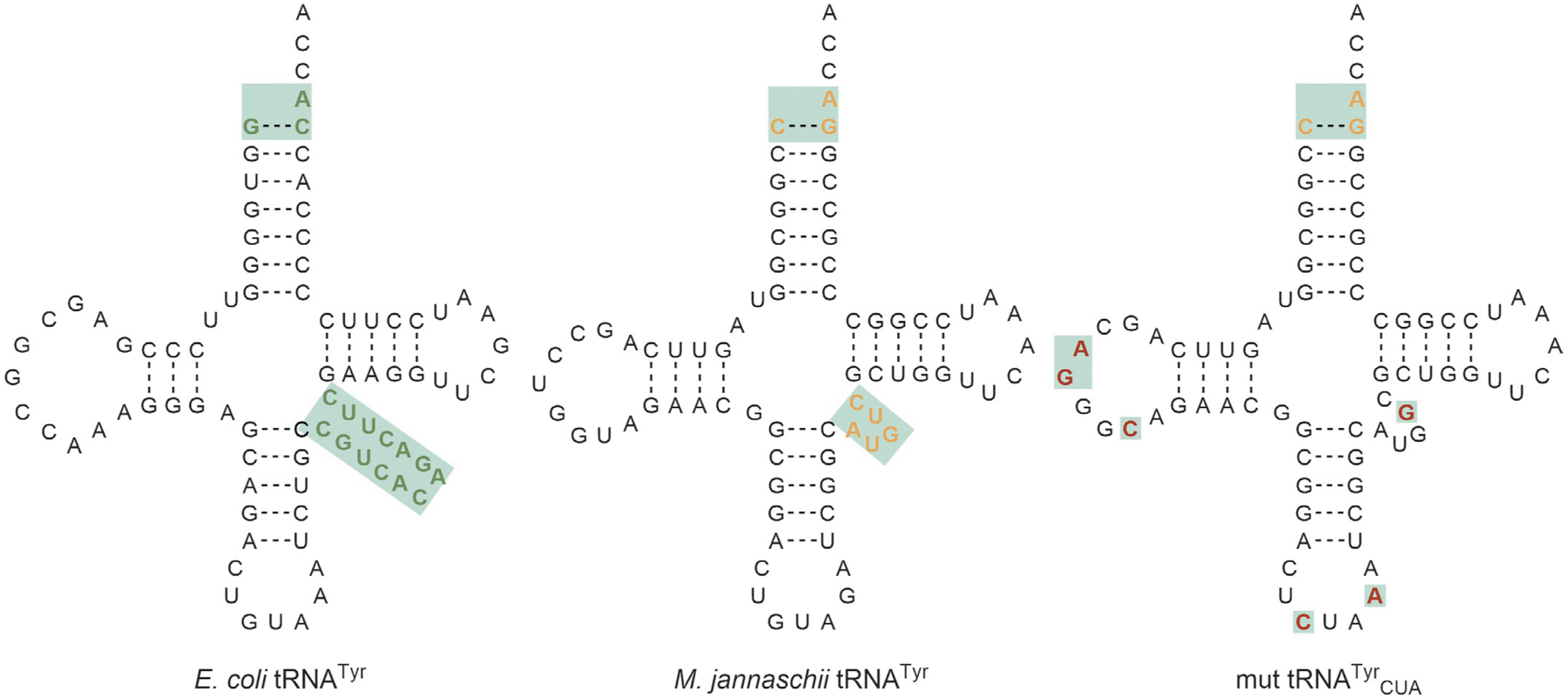
Secondary structures of tRNATyr from E. coli and M. jannaschii highlight the identity elements which suggested that the two were orthogonal. M. jannaschii tRNATyr was further mutated for efficient amber suppression and to enhance orthogonality in E. coli. The differences in the mut MjtRNATyrCUA from MjtRNATyr are highlighted in red.
Fig. 9.
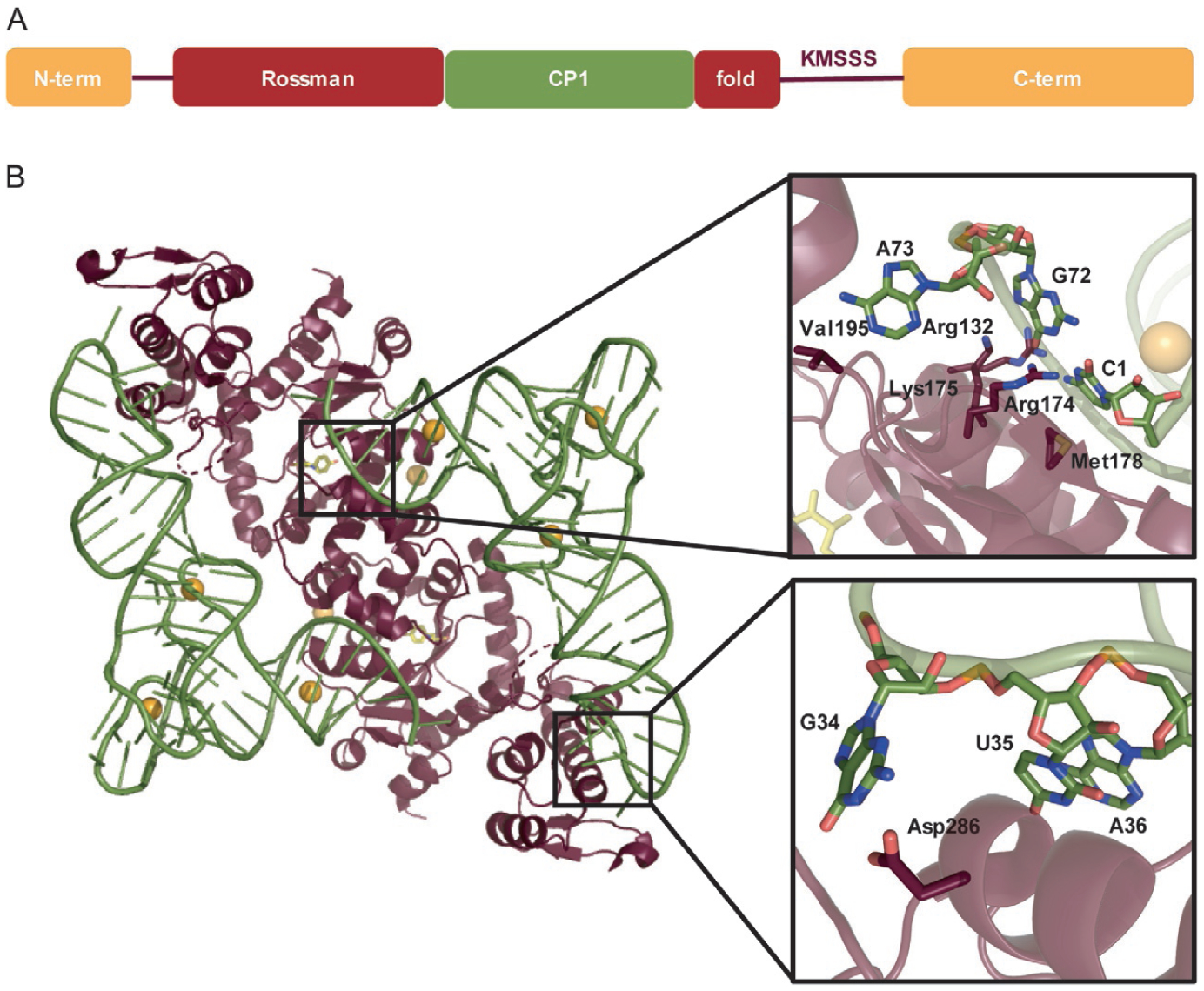
(A) Domain structure of MjTyrRS. (B) Interaction of MjTyrRS dimer with two MjtRNATyr molecules (PDBID: 1J1U) [103]. Closer look at the acceptor stem (upper box) and anticodon loop (lower box) displays the specific residues of MjTyrRS (maroon) and MjtRNATyr (green) involved in the interaction.
5.1. Structure of MjTyrRS and MjtRNATyr
5.1.1. MjTyrRS
Crystal structures of MjTyrRS show that it forms a homodimer and is more like apo human mini-TyrRS [108] than bacterial TyrRSs [102,109,110]. MjTyrRS is a class I aaRS which is divided into five regions. The Rossman-fold domain follows a short N-terminal region and binds to l-tyrosine. Inserted into the Rossman-fold domain is the connective-polypeptide 1 (CP1) domain which forms the dimer interface. Finally, the consensus KMSKS loop (in this case KMSSS) links the Rossman-fold domain to the C-terminal domain [103] (Fig. 9A). The C-terminal domain of MjTyrRS is different from that of the Thermus thermophilus TyrRS although the two enzymes recognize the same anticodon. In M. jannaschii there is an inserted β-310-β structure while in T. thermophilus the extra C-terminal domain follows the anticodon-binding domain to recognize the long variable arm. In the crystal structure, the two MjtRNATyr molecules span the two MjTyrRS subunits: the acceptor stem of MjtRNATyr binds to one subunit while the anticodon loop binds to the other. As with bacterial and eukaryotic TyrRS enzymes, its cognate tRNA is recognized by the major groove of the acceptor stem; a characteristic of class II aaRSs (Fig. 9B) [110,111].
5.1.2. MjtRNATyr
The first base pair in the acceptor stem (C1:G72) is the most important element for orthogonality of the MjTyrRS•MjtRNATyr pair. The crystal structure shows that the C1 base is twisted and tilted by ~20° compared to G72 which is positioned in the regular base pair plane. It is this twist which is specifically recognized by MjTyrRS; the O2 and N3 atoms of C1 form hydrogen bonds with the guanidino group of Arg174, the 4-amino group forms water-mediated hydrogen bonds with Arg132, and the C1 base hydrophobically interacts with Met178 (Fig. 9B, upper box). While recognition of C1:G72 by MjTyrRS is majorly through C1, the N7 of G72 is found to form hydrogen bonds with the amino group of Lys175. These three residues (Arg174, Arg132, and Lys175) are highly conserved among archaeal and eukaryotic TyrRSs which emphasizes their importance in recognition of their cognate tRNA. Although both archaeal and bacterial tRNATyr share the A73 identity element, its positioning differs. In MjtRNATyr it is projected away from the helical axis to interact with Val195 of MjTyrRS but in T. thermophilus is stacked on the C72 to extend the helix [103]. This evidence supports biochemical analyses that show archaeal TyrRS specifically recognizes C1:G72 and rejects bacterial tRNATyr [112].
5.2. The use of MjTyrRS•MjtRNATyr in genetic code expansion
In order to use MjtRNATyr as an amber suppressor tRNA for GCE purposes, the anticodon was mutated from GUA to CUA. It was hypothesized that this anticodon change would have minimal impact on MjtRNATyr recognition because MjTyrRS is missing the anticodon loop-binding domain. However, the crystal structure showed significant interaction of G34 in the anticodon of MjtRNATyr with Asp286 of MjTyrRS and less so with U35 and A36 (Fig. 9B, lower box). Therefore, mutation of the anticodon from the native GUA to the amber suppressor CUA caused a significant loss of activity [103,112], which was partially rescued (eightfold increase in activity) by an Asp286Arg mutation in MjTyrRS [100,103]. It was found that numerous residues interact with the anticodon loop of MjtRNATyr and that the Asp286Arg mutation was not enough to suppress AGA codons. However, further optimization of the anticodon-binding region of MjTyrRS made this possible [113].
Furthermore, to use the MjTyrRS•MjtRNATyrCUA system in E. coli, its predicted orthogonality had to be empirically demonstrated. MjtRNATyrCUA was found to not be fully orthogonal in E. coli with low recognition by endogenous E. coli aaRSs [100]. To optimize the orthogonality of MjtRNATyrCUA in E. coli, a tRNA library was created by randomizing 11 nucleotides which do not directly interact with the enzyme. Using a double-sieve selection method, a mutant suppressor (mut MjtRNATyrCUA; substitutions C17A, U17aG, U20C, G37A, U47G) was identified which affords a fourfold decrease in the background aminoacylation activity of endogenous E. coli aaRSs compared to the wild-type MjtRNATyrCUA (Fig. 8) [106].
5.3. Engineering the MjTyrRS substrate binding pocket
With MjTyrRS missing an editing domain the question to be investigated is how flexible the substrate binding pocket is to recognize ncAAs. Due to the absence of an MjTyrRS crystal structure, first attempts to engineer its substrate binding pocket utilized the TyrRS crystal structure from Bacillus stearothermophilus (PDB ID: 1TYD) [102]. The resulting library of MjTyrRS mutants was selected for its capacity to mediate stop codon read-through in the presence of the desired ncAA, followed by negative selection in the absence of ncAA [106]. The double-sieve selection approach is very effective and allowed for incorporation of many Tyr and Phe derivatives. After the MjTyrRS crystal structure was solved, structural details for l-Tyr recognition were revealed [103]. This allowed for a more detailed analysis of the MjTyrRS active site, and consequently novel libraries of MjTyrRS mutants to be screened. Higher quality of the MjTyrRS libraries allowed identification of variants that can acylate more complex AAs such as l-3-(2-naphthyl)alanine (NpAla, 65) [114], l-(7-hydroxycoumarin-4-yl) ethylglycine (Cou, 67) [115] and photocaged AAs (70–72) [17,116] (Fig. 10).
Fig. 10.
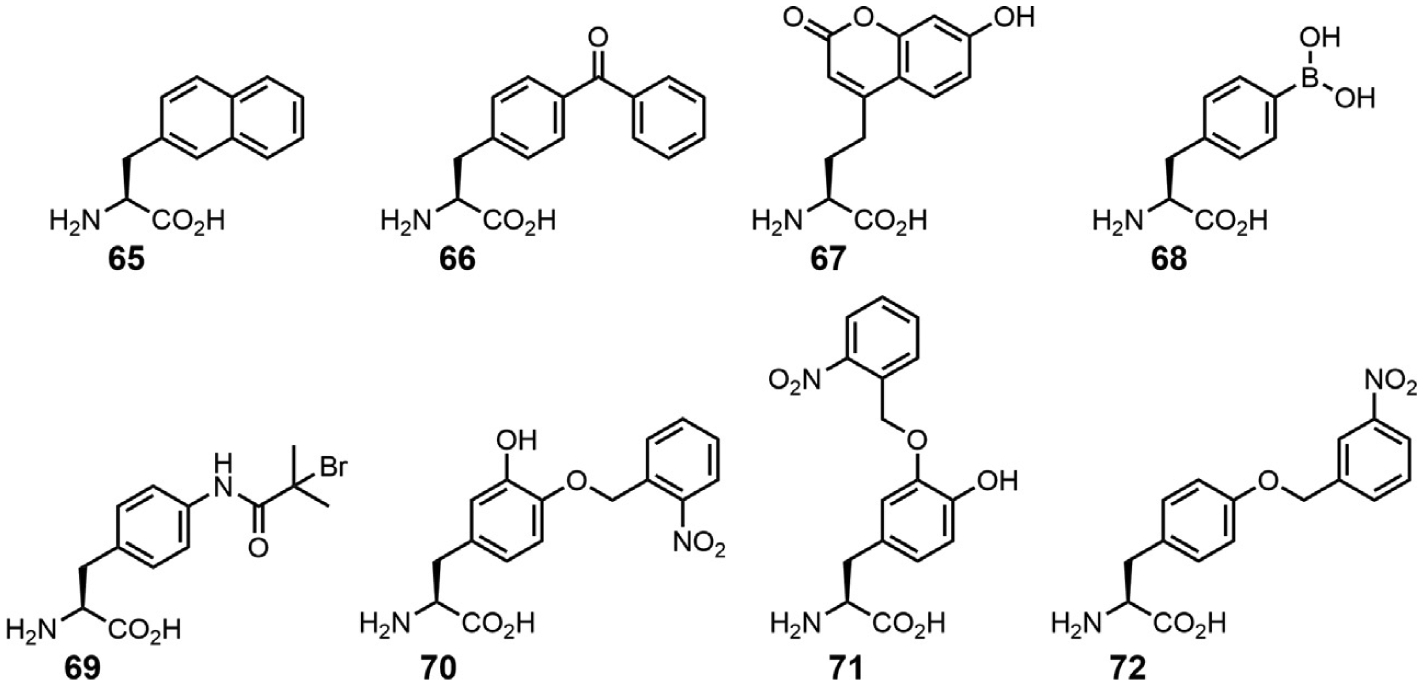
A subset of tyrosine derivatives that can be incorporated by engineered MjTyrRS variants.
The MjTyrRS crystal structure also revealed that the residues in the l-Tyr binding pocket are well conserved in archaeal, eukaryotic and bacterial TyrRSs. Residues Tyr32, Asp158, Gln155, Gln173, and Tyr151 in M. jannaschii correspond to Tyr34, Asp176, Gln173, Gln195, and Tyr169 in the B. stearothermophilus active site [103]. In the MjTyrRS binding pocket, l-Tyr hydrogen bonds with Tyr151, Gln75 and Gln173, but it is Tyr32 and Asp158 that ultimately provide the specificity for l-Tyr (Fig. 11A). These residues create a strong polar environment at the bottom of a deep cleft which favors the polar phenol of l-Tyr over the structurally similar benzene of Phe [118]. Therefore it is not surprising that the engineered MjTyrRS variants consistently contained mutations in Tyr32 and Asp158 to remove specificity for l-Tyr [119,120]. Other commonly occurring mutations (Glu107, Ile159, and Leu162) were found to open the substrate binding pocket to accommodate and favor incorporation of bulkier ncAAs [106,119]. Details of these mutations and how residues interact with the ncAA through hydrogen bonds or van der Waals interactions can be gathered from their crystal structures. For example, in the case of p-bromophenylalanine (p-BrPhe) the Asp158Pro mutation specifically disrupts the hydrogen bonds in the α8-helix resulting in a short 310-helix [117]. This causes translational and rotational disruptions of several active site residues; side chains of mutated residues at positions 158, 159, and 162 move away from the active site to become solvent exposed, resulting in the formation of van der Waals contacts between AA positions 160 and 161 and p-BrPhe (Fig. 11B). These rearrangements are optimized for binding of p-BrPhe and simultaneously disallow l-Tyr accommodation [117]. On the other hand, for O-allyl-l-tyrosine (O-AlTyr), Tyr32 which is a key residue for binding to l-Tyr is not mutated. Therefore the substrate binding pocket in this case has increased its affinity to the ncAA over l-Tyr, rather than completely losing its ability to bind to its cognate AA (Fig. 11C) [121].
Fig. 11.
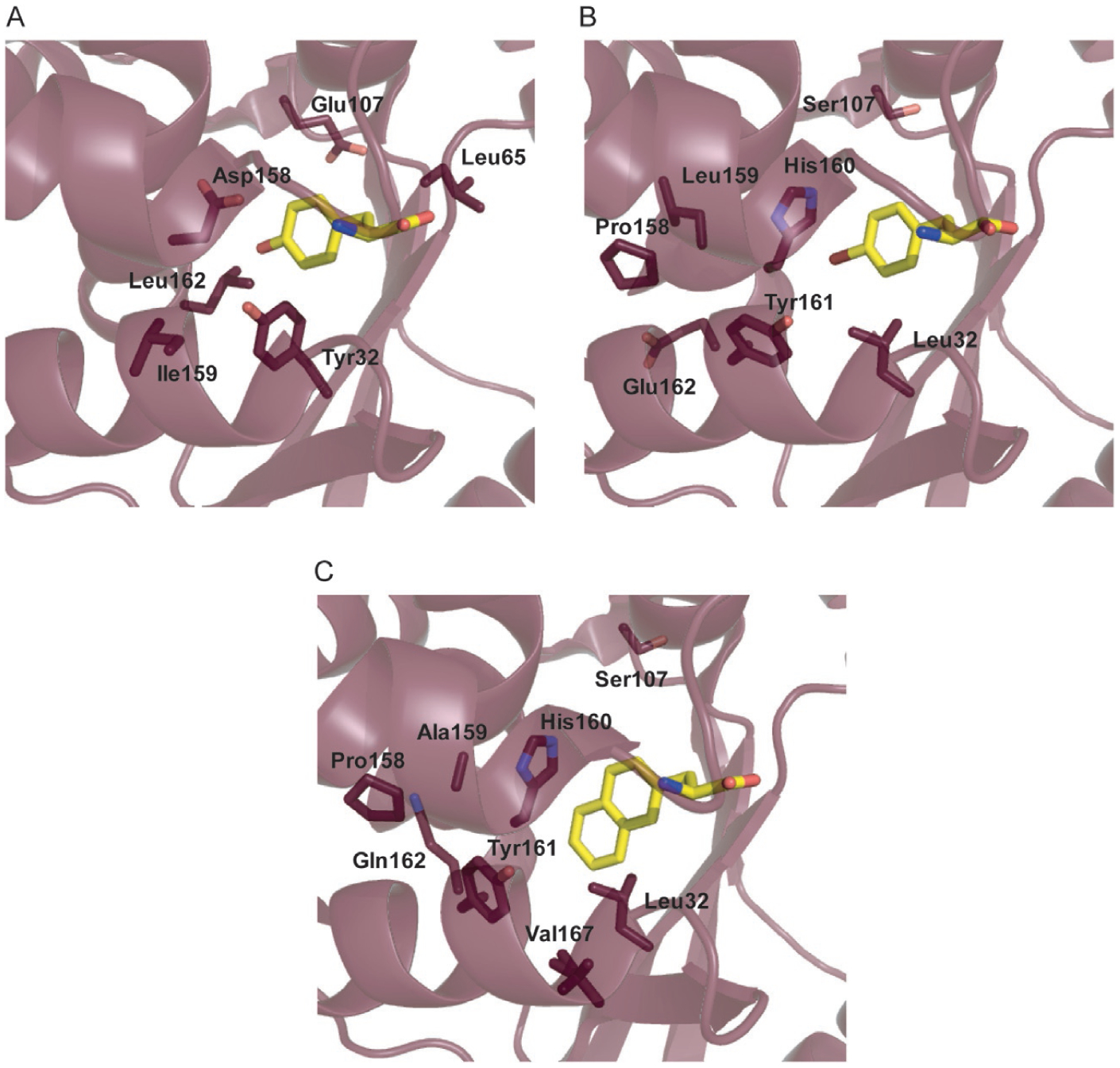
Substrate binding pocket of (A) wild-type MjTyrRS bound to l-tyrosine (PDB: 1J1U) [103], (B) MjTyrRS variant engineered for binding to p-BrPhe (PDB: 2AG6) [117], and (C) MjTyrRS variant engineered for binding to NpAla (PDB: 1ZH0) [117].
5.4. Applications of MjTyrRS•MjtRNATyr
Initial studies with MjTyrRS focused on evolving the enzyme to incorporate ncAAs which were structurally similar to tyrosine (e.g., O-methyl-l-tyrosine (O-MeTyr, 29) and p-BrPhe) [106,117]. Identification of NpAla-acylating variants greatly increased the ncAA substrate range of MjTyrRS, including interesting alkyl-, aryl-, acyl-, and azido-substituted amino acids [114]. With only five mutations (Tyr32Leu, Asp158Pro, Ile159Ala, Leu162Gln, and Ala167Val), the substrate binding pocket of MjTyrRS was engineered to bind NpAla. As with p-BrPhe, the Asp158Pro mutation removes the hydrogen bonds to l-Tyr and creates the rotational and translational disruptions of Ile159Ala and Leu162Gln to prevent l-Tyr binding. Unlike p-BrPhe, the Tyr32Leu mutation not only expands the active site but creates favorable van der Waals interactions to occur with the aromatic ring of NpAla. Furthermore, the Ala167Val mutation has an indirect effect, causing a rotation of Tyr161 by ~45° to that of the p-BrPhe MjTyrRS structure for accommodating the sidechain of NpAla [117]. These results demonstrate the versatility of MjTyrRS to incorporate a wide range of ncAAs and expanded its applications. For example, chemical groups with a wide breadth of reactivity (pAcF, O-AlTyr) [121,122], photo-crosslinking groups (p-benzoylphenylalanine (pBpa, 66), pAzF) [123,124], biochemical probes (p-nitro-l-phenylalanine (pNO2pa)) [125], polymerization initiators [126], and fluorescent groups (Cou, 67) [115] can all be co-translationally installed into proteins using engineered variants of MjTyrRS.
5.4.1. Amber suppression in Mycobacterium
Since MjTyrRS is not orthogonal in eukaryotes, most of its applications have been in commonly used laboratory strains of E. coli. However, from the information described above, the MjTyrRS•MjtRNATyr pair should theoretically be orthogonal in all prokaryotes. This was found to be true for Mycobacterium tuberculosis and Mycobacterium smegmatis. It was determined that the MjTyrRS variants engineered in E. coli for incorporation of ncAAs (pBpa, pAzF, pNO2pa, Bpa, and p-iodo-l-phenylalanine (pIpa)) could be directly transferred for use in M. tuberculosis and M. smegmatis. Unlike E. coli, M. tuberculosis contains over three times more genes which use the amber stop codon (1194 compared to 365). With that being said, suppression efficiency using MjtRNATyrCUA was only 9% that of wild-type protein in the best scenario (incorporation of pIpa). Due to the increase in amber stop codons in the genome, toxicity of amber suppression was a concern. However, with such low suppression, MjtRNATyrCUA would not compete well with the release factor and therefore it was presumed that endogenous protein expression was not significantly affected [127].
5.5. Enhancing MjTyRS-mediated ncAA incorporation
Alterations in the substrate binding pocket needed to improve ncAA activation, may not be enough for high levels of ncAA incorporation into proteins. Although the Asp286Arg mutation was found to enhance efficiency of MjTyrRS for MjtRNATyrCUA [100], to our knowledge no further studies were done to optimize recognition of MjtRNATyrCUA. The pAzFRS crystal structure (MjTyrRS variant which incorporates pAzF) revealed at least 10 residues in the tRNA-recognition domain that could improve this interaction. After rounds of selection, a pAzFRS variant was identified which had a threefold higher efficiency than the Asp286Arg mutant of pAzFRS [128].
As mentioned above, most mutant enzymes have been generated through libraries of MjTyrRS variants with up to six amino acid positions mutated. AaRS libraries are limited to this size by the transformation efficiency of E. coli with a practical upper limit of 109 sequence variants. Residues are typically chosen which are in direct contact with the substrate, limiting the scope of variants that are screened. To obtain a highly efficient aaRS for a given ncAA, mutations beyond the residues directly contacting the substrate may be necessary. To scan through all residues in the first two shells of the active site (<9 Å), it would require randomization of approximately 30 amino acids or 2030=1039 total sequences, far exceeding the transformation efficiency of E. coli. By using computational approaches, this large sequence space can be brought down by identifying AA replacements that are most likely to result in increased activity, rather than employing full randomization of every identified residue. Such a strategy was used to improve MjONBYRS efficiency sixfold [116] and to generate MjTyrRS variants to incorporate p-ortho-nitrobenzyl (ONB)-l-3,4-dihydroxyphenylalanine (DOPA) and m-ONB-DOPA (70–71) [17].
6. Other orthogonal aaRS•tRNA pairs applied for genetic code expansion
6.1. E. coli TyrRS•tRNA pair
This OTS (which uses a suppressor variant of tyrT, supF [129]) has been employed for GCE in eukaryotic organisms. This is possible because the bacterial TyrRS recognizes its tRNATyr using a different set of identity elements compared to the archaeal and eukaryotic counterparts [130] (Section 5.1). The orthogonality of the E. coli TyrRS•tRNATyr was established in 1990, in the pioneering work of Edwards and Schimmel. Expression of E. coli tRNATyrCUA in S. cerevisiae showed no suppression in three different reporter genes, demonstrating that it is not a substrate for the host TyrRS [131]. In contrast, EcTyrRS recognized EctRNATyr efficiently in vivo. Not surprisingly, the orthogonality is dependent on the expression level. Increasing the expression from 40 to 300% with respect to average abundance of yeast tRNATyr led to suppression of amber codon containing alleles in vivo. Analysis of the AA composition of the amber-containing test enzyme showed that leucine was also inserted at the internal UAG codon. This indicates that, at least to some extent, E. coli tRNATyrCUA may be aminoacylated by S. cerevisiae LeuRS [132].
The first account of ncAA insertion using EcTyrRS•EctRNATyrCUA in S. cerevisiae reported the successful incorporation of five ncAAs: pAcF, pBpa, pAzF, O-MeTyr, and pIpa [133]. An aaRS library was selected using a GAL-4 mediated activation of HIS3, URA3, or lacZ reporter genes [14]. To remove clones where E. coli TyrRS still retained specificity for its natural substrate l-Tyr, a negative selection step was undertaken using 0.1% 5-fluorootic acid, which, upon expression of URA3, is converted in vivo to 5-fluorouracil, resulting in cell death [134].
Improvements of this system showed that the increased expression of both EctRNATyrCUA and EcTyrRS, as well as the expression and codon optimization of the target gene leads to improved yields [135]. Additional improvements were also made with respect to tRNA expression and processing [136]. RNA polymerase III which transcribes tRNAs in eukaryotes relies on A- and B-box identity elements present within the sequences of eukaryotic tRNA genes. Because these elements may be absent in bacterial tRNAs, a strategy to assemble such sequences upstream of the tyrT gene was developed. Because the transcription results with A- and B-box fused to heterologous E. coli tRNATyr, the transcript needs to be matured post-transcriptionally. Yeast genes SNR52 and RPR1 possess such promoter organization, present in the primary transcript but absent in the mature RNA. Although transcription levels of E. coli tRNATyr that use a 5′-flanking sequence of SUP4 are ~100-fold higher than those using SNR52 or RPR1 promoter, higher levels of amber codon suppression occur with the latter system. This indicates that the SUP4 driven transcripts may not be correctly processed [136]. In addition to S. cerevisiae, GCE with EcTyrRS•EctRNATyrCUA was successfully undertaken in Pichia pastoris, Candida albicans and Schizosaccharomyces pombe [137–140].
As in yeast, the lack of internal promoter sequences in EctRNATyr has proven challenging for its use in higher eukaryotes. As a result, in mammalian cell lines, and whole organisms such as Danio rerio and Mus musculus, EcTyrRS was used in combination with B. stearothermophilus tRNATyrCUA. EctRNATyr normally possesses the B-box internal promoter, but lacks the conserved sequence of box A. Therefore, mutations U9A and C10G were introduced to create an artificial A-box promoter, along with a G25C mutation to compensate for the G10-G25 mismatch. However, these mutations rendered EctRNATyr unusable, as no UAG suppression was observed with the reporter in CHO cells [141]. In contrast, BsttRNATyrCUA, a substrate for EcTyrRS [142], led to a significant amount of read-through. In this report, the authors used a previously developed EcTyrRS variant (EcTyrRS-Val37/Cys195) that efficiently charges 3-iodo-l-tyrosine, as demonstrated by in vitro aminoacylation assays and UAG suppression in a wheat germ cell-free system [141,143]. For the purpose of introducing light-induced crosslinks between proteins in CHO cell lines, pBpa-specific EcTyrRS variant [133] was used in conjunction with BsttRNATyr [144]. To study G protein-coupled receptors, EcTyrRS•BsttRNATyrCUA was used to mediate insertion of pAcF and pBpa in HEK293T cells [145].
In Caenorhabditis elegans EcTyrRS was used with its cognate EctRNATyrCUA [146]. Supplementation of ncAAs in the form of dipeptides and genomic integration of the UAG-containing reporter were shown to be critical for faithful incorporation of O-MeTyr [146]. In D. rerio and M. musculus the pAzF charging E. coli TyrRS [133] was used with heterologous BsttRNATyrCUA [147]. A cassette containing EcTyrRS under the human EF1α promoter, as well as four copies of BsttRNATyrCUA under the U6 promoter, were integrated into the mouse genome. Astoundingly, transgenic mice were morphologically and histologically indistinguishable from wild-type counterparts and the EcTyrRS-BsttRNATyr cassette was transmitted to succeeding generations. In the liver, where EcTyrRS was expressed the most, RNA-seq analysis identified minor changes in the transcriptome of the transgenic mice [147].
Since it is difficult to generate large stable libraries in mammalian cell lines, majority of OTSs are first evolved in E. coli or yeast (e.g., [148]). The engineered OTS pair is usually introduced by transient transfection which normally has low efficiency and further varies between cell lines. To combat this issue, a baculovirus-based transduction system was developed, capable of delivering OTSs, as well as other necessary genetic material to a variety of cell lines [149]. Interestingly, both EctRNATyrCUA and BsttRNATyrCUA were used in conjunction with EcTyrRS variant originally developed for incorporation of O-MeTyr [133]. This aaRS was selected for its increased polyspecificity, as the authors determined that it is able to acylate 10 different ncAAs [149].
As an alternative to evolution of EcTyrRS in yeast, strains of E. coli have been created to allow facile selection of ncAA-acylating variants of this enzyme [150,151]. In these strains EcTyrRS and three EctRNATyr isoacceptors are removed from the genome, and regular translation maintained by MjTyrRS•MjtRNATyr [150,151]. Importantly, both MjTyrRS•MjtRNATyr and ScTyrRS•SctRNATyr pairs, when over-produced, can complement the conditionally lethal EcTyrRS mutant [150]. Both reports demonstrate the importance of the engineering strategy with respect to the deletions of endogenous tRNATyr isoacceptors, as different strategies result in strains with very different fitness. Specifically, tRNA deletions are better tolerated when accomplished by insertion of suppressor tRNAs, rather than protein coding genes or as clean deletions, presumably because the inserted tRNA genes allow better preservation of the local chromosomal architecture [150,151]. Using these strains, UAG-selective incorporation of 3-azido-l-tyrosine was achieved [150], as well as the directed evolution of polyspecific EcTyrRS variants with improved affinity for O-MeTyr and Bpa [151].
6.2. E. coli LeuRS•tRNA pair
Both prokaryotes and eukaryotes employ tRNALeu isoacceptors with a long variable arm. In vitro cross-species aminoacylation experiments have shown that EcLeuRS does not recognize yeast tRNAs, but that ScLeuRS aminoacylates not only EctRNALeu, but also EctRNATyr [152]. In vivo, however, a suppressor variant of EctRNALeu retains sufficient orthogonality for GCE experiments [153]. Thus, the EcLeuRS•EctRNALeu pair was used to evolve EcLeuRS with improved affinity for O-MeTyr, α-aminocaprylic acid (73), and O-nitrobenzyl-l-cysteine (74) using a GAL4-mediated synthesis of HIS3 and URA3 reporters in S. cerevisiae [153]. In a separate report, the same pair was used to encode a fluorescent amino acid dansylalanine (75) (Fig. 12) [154]. This dansylalanine charging EcLeuRS variant was later employed for GCE in C. elegans [146]. The same pair was also expressed in HeLa cells where dansylalanine was incorporated with 13% efficiency [155]. Detailed study of bacterial tRNA expression in yeast revealed that although 5′-flanking sequence of SUP4 affords EctRNALeuCUA expression, the expression levels of amber-containing GFP reporter increase fourfold when this tRNA is expressed from the SNR52 promoter [136]. This is interesting because EctRNALeu contains the A- and B-box identity elements necessary for transcription by RNA Pol III, in contrast to EctRNATyr (see above) [136].
Fig. 12.
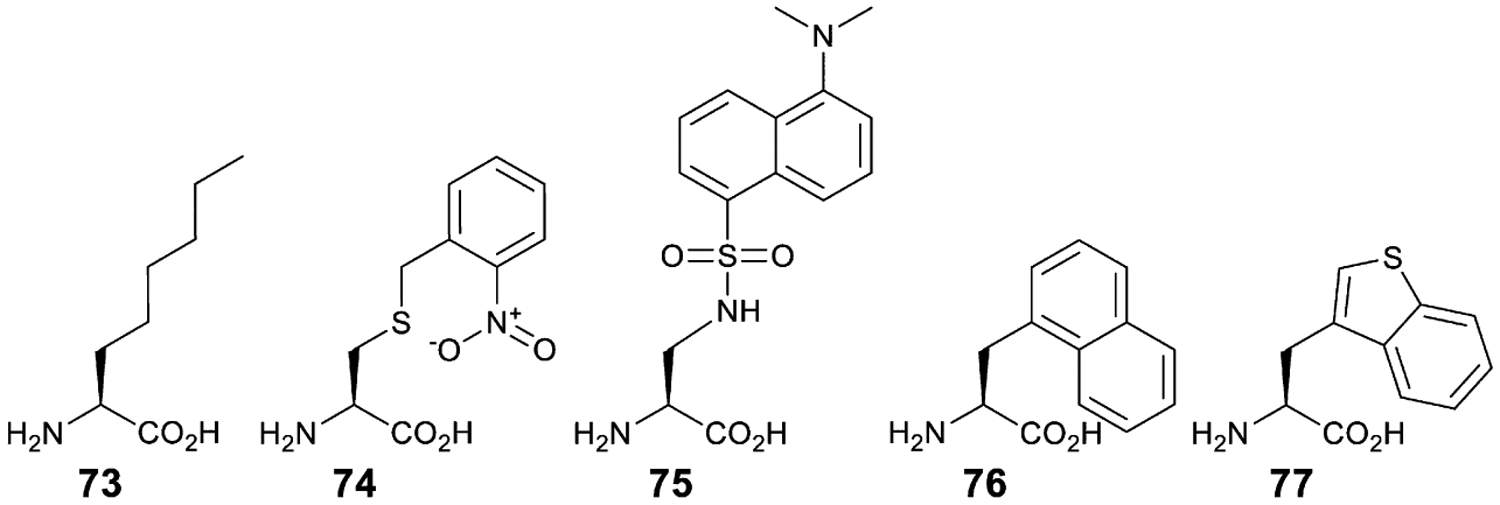
Non-canonical amino acids employed in GCE using EcLeuRS (73–75) and ScTrpRS (76, 77).
6.3. E. coli and S. cerevisiae TrpRS•tRNA pairs
Both E. coli and S. cerevisiae TrpRS•tRNA pairs use the CCA anticodon as identity elements but depend on a distinct set of nucleotides at the top of the acceptor stem. Comparison of tRNATrp sequences reveals conservation of G73 in bacterial tRNAs, while archaeal and eukaryotic tRNAs possess an A73 and G1:C72 base pair. Although the exact distribution of the bacterial vs archaeal/eukaryotic-type tRNATrp may be slightly more complex [156], it has been established that the bacterial TrpRS relies on the G73 discriminator base, and the first three base pairs of the acceptor stem to a lesser extent. As a result, a G73A mutation, as well as mutations of the G1:C72 base pair abolish aminoacylation capacity of E. coli TrpRS [157]. In order to use E. coli tRNATrp for amber suppression a C35U mutation is necessary. However, this mutation has been shown to reduce this tRNA’s orthogonality making it a suitable substrate for EcGlnRS [6].
In contrast, when C35U mutation is introduced into SctRNATrp it does not influence its aminoacylation by ScTrpRS [158]. However, application of SctRNATrp in E. coli is complicated due to its similarity with EctRNALys and ensuing aminoacylation by EcLysRS [159]. Furthermore, EcLysRS appears to accept the C34 base in place of U34, which is normally modified to 5-[(methylamino)-methyl] 2-thiouridine. Analysis of a number of acceptor stem and anticodon stem variants showed that the latter, in some cases, increase orthogonality without compromising aminoacylation with ScTrpRS [157]. Two superior anticodon stem variants were later used to further improve their activity through complete randomization of the first five base pairs of the acceptor stem [160]. Using one of the identified variants, a ScTrpRS variant specific to 3-(1-naphthyl)-l-alanine was selected (76). While this variant is selective against natural amino acids, it is polyspecific with regards to other ncAAs; it has the capacity to charge l-methyl-l-tryptophan, 3-benzothienyl-l-alanine (77), and 6-methyl-l-tryptophan (Fig. 12) [160].
Analogous to liberation of EcTyrRS•EctRNATyrCUA, it was shown that plasmid-borne ScTrpRS•SctRNATrpCCA can allow deletion of endogenous counterparts in E. coli [161]. In this strain, engineered SctRNATrpCCA decodes sense Trp codons, and the E. coli tRNATrp suppressor decodes the stop codon in the target gene. To avoid misacylation of EctRNATrp amber suppressor by EcGlnRS, the authors opted for the opal, UGA suppressor. In wild-type strains, UGA read-through is frequently non-specific, due to near-cognate recognition by EctRNATrpCCA. Interestingly, this phenomenon was not observed in the engineered strain, indicating decreased ability of SctRNATrpCCA to pair with UGA codons. As a result, several polyspecific EcTrpRS variants were evolved which can acylate 5-hydroxytryptophan, as well as numerous 5-substituted tryptophan derivatives. These variants were also introduced to HEK293T cells, and used to incorporate the ncAAs in response to an amber stop codon, demonstrating the feasibility of TrpRS evolution in E. coli, and subsequent employment in mammalian cell lines [161].
6.4. M. jannaschii SepRS•tRNA pair
Some archaeal species lack CysRS, and synthesize Cys-tRNACys via an indirect route employing O-phosphoseryl-tRNA synthetase (SepRS) and Sep-tRNA:Cys-tRNA synthase (SepCysS) [162]. SepRS aminoacylates tRNACys with O-phospho-l-serine, which is subsequently converted to Cys by SepCysS. Structural analyses of Methanococcus maripaludis enzyme [163] and Archaeoglobus fulgidus SepRS•tRNACys complex revealed extensive contacts with the tRNACys anticodon, but also reported successful engineering of the anticodon-binding domain to allow recognition of stop codon suppressor variants [164]. Interestingly, SepRS can be engineered to recognize unnatural bases placed at the anticodon [165].
An amber suppressor derived from M. jannaschii tRNACys, tRNASep, was first employed to genetically encode phosphoserine in E. coli [166,167]. The efficiency of SepRS•tRNASep mediated phosphoserine incorporation was further enhanced by introducing a dedicated, evolved version of EF-Tu, which contains a substrate binding pocket optimized for phosphoserine acceptance [166]. Further optimizations to the system include introduction of additional mutations in the anticodon-binding domain of SepRS [168,169], and variations in the anticodon stem-loop structure [168] and acceptor stem [170], to increase translational efficiency and tRNA orthogonality, respectively. While wild-type SepRS can be employed to introduce a nonhydrolyzable phosphoserine analog [168], mutations in the active site were necessary to allow genetic incorporation of phosphothreonine [170]. Importantly, SepRS•tRNASep pair is orthogonal to PylRS and its variants. Therefore, these systems have been employed to simultaneously introduce Sep and Nε-acetyl-l-lysine which establishes a methodology to create proteins with two posttranslational modifications at desired positions [171].
6.5. P. horikoshii LysRS•tRNA pair
In nature there are two unrelated classes of LysRSs. The less abundant class I LysRS is present in some archaeal and, to a lesser extent, bacterial species [172]. Interestingly, class I LysRS can rescue growth in an E. coli strain with a conditionally lethal LysRS variant. The crystal structure of the class I Pyrococcus horikoshii LysRS revealed that it contacts the same elements in tRNALys as the class II enzyme (albeit in a different manner) [173].
Despite potential cross-reactivity with the endogenous class II LysRS, the P. horikoshii LysRS•tRNALys pair was employed for GCE in E. coli [21]. Instead of a stop codon, the authors reassigned a quadruplet codon, AGGA. Attempts to constitutively express PhLysRS in E. coli revealed its toxicity. However, using the E. coli mutator strain XL1-red the authors were able to isolate a nontoxic variant, truncated at the C-terminal end (the part responsible for anticodon recognition). Using a complementation assay in a LysRS-deficient E. coli strain, it was shown that the truncated PhLysRS is orthogonal to the E. coli translational machinery. Based on the analysis of all archaeal tRNALys that were known at the time, the consensus sequence was used to generate the suppressor tRNALys. While an amber derivative of this tRNALys was orthogonal, tRNALysUCCU was not. Upon randomization of the first four base pairs in its acceptor stem, an orthogonal tRNALysUCCU variant was selected, containing a U1:A72, and U4:A69 mutation, as well as wobble pair G2:U71 and a peculiar A37C mutation. This tRNA was subsequently used to evolve PhLysRS that acylates l-homoglutamine. The authors also showed its orthogonality to MjTyrRS, thereby demonstrating its usefulness in simultaneous insertion of two ncAAs in E. coli [21].
6.6. P. horikoshii ProRS•tRNA pair
In nature, two distinct types of ProRS enzymes rely on different elements of tRNAPro for recognition and show very little cross-reactivity [174,175]. Encouraged by this fact, a ProRS•tRNAPro pair was developed, orthogonal in E. coli, and consisting of A. fulgidus tRNAPro and P. horikoshii ProRS. Amber and quadruplet (CCCU) codon suppressors created from AftRNAPro afforded cell survival in a cat-based assay. Using a combination of positive selection and screening, the authors were able to identify mutually orthogonal PhProRS variants that selectively accept amber and quadruplet tRNAPro suppressor variants [176].
6.7. S. cerevisiae PheRS•tRNA pair
Early in vitro work demonstrated that E. coli PheRS aminoacylates yeast tRNAPheAAA poorly (<0.1% aminoacylation rate of its cognate E. coli tRNAPheGAA) [177]. In pioneering work by Furter, the amber suppressor derived from yeast tRNAPhe and its cognate PheRS were shown to direct insertion of the para fluorinated Phe analog in a reporter protein [178]. In an alternative approach, ScPheRS•SctRNAPheAAA pair was used in E. coli to selectively outcompete endogenous tRNAPheGAA in decoding UUU codons and insert NpAla [179]. Based on previously developed non-orthogonal variants of EcPheRS generated through computational design [180,181], the authors were able to show preferential insertion of NpAla at UUU codons [179].
Acknowledgments
Work in the authors’ laboratories was supported by the Center for Genetically Encoded Materials, an NSF Center for Chemical Innovation (NSF CHE-1740549), and the National Institute of General Medical Sciences (R35GM122560).
References
- [1].Cavarelli J, Moras D, Recognition of tRNAs by aminoacyl-tRNA synthetases, FASEB J. 7 (1) (1993) 79–86. [DOI] [PubMed] [Google Scholar]
- [2].Giege R, Sissler M, Florentz C, Universal rules and idiosyncratic features in tRNA identity, Nucleic Acids Res. 26 (22) (1998) 5017–5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Reynolds NM, Vargas-Rodriguez O, Söll D, Crnković A, The central role of tRNA in genetic code expansion, Biochim. Biophys. Acta Gen. Subj 1861 (11 Pt. B) (2017) 3001–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Melnikov SV, Söll D, Aminoacyl-tRNA synthetases and tRNAs for an expanded genetic code: what makes them orthogonal? Int. J. Mol. Sci 20 (8) (2019) 1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wang L, Schultz PG, A general approach for the generation of orthogonal tRNAs, Chem. Biol 8 (9) (2001) 883–890. [DOI] [PubMed] [Google Scholar]
- [6].Yaniv M, Folk WR, Berg P, Söll L, A single mutational modification of a tryptophan-specific transfer RNA permits aminoacylation by glutamine and translation of the codon UAG, J. Mol. Biol 86 (2) (1974) 245–260. [DOI] [PubMed] [Google Scholar]
- [7].Fukunaga J, Yokogawa T, Ohno S, Nishikawa K, Misacylation of yeast amber suppressor tRNATyr by E. coli lysyl-tRNA synthetase and its effective repression by genetic engineering of the tRNA sequence, J. Biochem 139 (4) (2006) 689–696. [DOI] [PubMed] [Google Scholar]
- [8].Normanly J, Kleina LG, Masson JM, Abelson J, Miller JH, Construction of Escherichia coli amber suppressor tRNA genes. III. Determination of tRNA specificity, J. Mol. Biol 213 (4) (1990) 719–726. [DOI] [PubMed] [Google Scholar]
- [9].Guo LT, Wang YS, Nakamura A, Eiler D, Kavran JM, Wong M, Kiessling LL, Steitz TA, O’Donoghue P, Söll D, Polyspecific pyrrolysyl-tRNA synthetases from directed evolution, Proc. Natl. Acad. Sci. U. S. A 111 (47) (2014) 16724–16729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Crnković A, Vargas-Rodriguez O, Söll D, Plasticity and constraints of tRNA aminoacylation define directed evolution of aminoacyl-tRNA synthetases, Int. J. Mol. Sci 20 (9) (2019) 2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Voller JS, Dulic M, Gerling-Driessen UI, Biava H, Baumann T, Budisa N, Gruic-Sovulj I, Koksch B, Discovery and investigation of natural editing function against artificial amino acids in protein translation, ACS Cent. Sci 3 (1) (2017) 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Richardson CJ, First EA, Altering the enantioselectivity of tyrosyl-tRNA synthetase by insertion of a stereospecific editing domain, Biochemistry 55 (10) (2016) 1541–1553. [DOI] [PubMed] [Google Scholar]
- [13].Kartvelishvili E, Peretz M, Tworowski D, Moor N, Safro M, Chimeric human mitochondrial PheRS exhibits editing activity to discriminate nonprotein amino acids, Protein Sci. 25 (3) (2016) 618–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chin JW, Cropp TA, Chu S, Meggers E, Schultz PG, Progress toward an expanded eukaryotic genetic code, Chem. Biol 10 (6) (2003) 511–519. [DOI] [PubMed] [Google Scholar]
- [15].Suzuki T, Miller C, Guo LT, Ho JML, Bryson DI, Wang YS, Liu DR, Söll D, Crystal structures reveal an elusive functional domain of pyrrolysyl-tRNA synthetase, Nat. Chem. Biol 13 (12) (2017) 1261–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bryson DI, Fan C, Guo LT, Miller C, Söll D, Liu DR, Continuous directed evolution of aminoacyl-tRNA synthetases, Nat. Chem. Biol 13 (12) (2017) 1253–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hauf M, Richter F, Schneider T, Faidt T, Martins BM, Baumann T, Durkin P, Dobbek H, Jacobs K, Moglich A, Budisa N, Photoactivatable mussel-based underwater adhesive proteins by an expanded genetic code, ChemBioChem 18 (18) (2017) 1819–1823. [DOI] [PubMed] [Google Scholar]
- [18].Uyeda A, Watanabe T, Kato Y, Watanabe H, Yomo T, Hohsaka T, Matsuura T, Liposome-based in vitro evolution of aminoacyl-tRNA synthetase for enhanced pyrrolysine derivative incorporation, ChemBioChem 16 (12) (2015) 1797–1802. [DOI] [PubMed] [Google Scholar]
- [19].Amiram M, Haimovich AD, Fan C, Wang YS, Aerni HR, Ntai I, Moonan DW, Ma NJ, Rovner AJ, Hong SH, Kelleher NL, Goodman AL, Jewett MC, Söll D, Rinehart J, Isaacs FJ, Evolution of translation machinery in recoded bacteria enables multi-site incorporation of nonstandard amino acids, Nat. Biotechnol 33 (12) (2015) 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ellis HM, Yu D, DiTizio T, Court DL, High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides, Proc. Natl. Acad. Sci. U. S. A 98 (12) (2001) 6742–6746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Anderson JC, Wu N, Santoro SW, Lakshman V, King DS, Schultz PG, An expanded genetic code with a functional quadruplet codon, Proc. Natl. Acad. Sci. U. S. A 101 (20) (2004) 7566–7571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Badran AH, Liu DR, Development of potent in vivo mutagenesis plasmids with broad mutational spectra, Nat. Commun 6 (2015) 8425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liu DR, Schultz PG, Progress toward the evolution of an organism with an expanded genetic code, Proc. Natl. Acad. Sci. U. S. A 96 (9) (1999) 4780–4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Santoro SW, Wang L, Herberich B, King DS, Schultz PG, An efficient system for the evolution of aminoacyl-tRNA synthetase specificity, Nat. Biotechnol 20 (10) (2002) 1044–1048. [DOI] [PubMed] [Google Scholar]
- [25].Kuhn SM, Rubini M, Fuhrmann M, Theobald I, Skerra A, Engineering of an orthogonal aminoacyl-tRNA synthetase for efficient incorporation of the non-natural amino acid O-methyl-L-tyrosine using fluorescence-based bacterial cell sorting, J. Mol. Biol 404 (1) (2010) 70–87. [DOI] [PubMed] [Google Scholar]
- [26].Umehara T, Kim J, Lee S, Guo LT, Söll D, Park HS, N-acetyl lysyl-tRNA synthetases evolved by a CcdB-based selection possess N-acetyl lysine specificity in vitro and in vivo, FEBS Lett. 586 (6) (2012) 729–733. [DOI] [PubMed] [Google Scholar]
- [27].Maranhao AC, Ellington AD, Evolving orthogonal suppressor tRNAs to incorporate modified amino acids, ACS Synth. Biol 6 (1) (2017) 108–119. [DOI] [PubMed] [Google Scholar]
- [28].Carlson JC, Badran AH, Guggiana-Nilo DA, Liu DR, Negative selection and stringency modulation in phage-assisted continuous evolution, Nat. Chem. Biol 10 (3) (2014) 216–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Haruna K, Alkazemi MH, Liu Y, Söll D, Englert M, Engineering the elongation factor Tu for efficient selenoprotein synthesis, Nucleic Acids Res. 42 (15) (2014) 9976–9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Thyer R, Robotham SA, Brodbelt JS, Ellington AD, Evolving tRNASec for efficient canonical incorporation of selenocysteine, J. Am. Chem. Soc 137 (1) (2015) 46–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kunjapur AM, Stork DA, Kuru E, Vargas-Rodriguez O, Landon M, Söll D, Church GM, Engineering posttranslational proofreading to discriminate nonstandard amino acids, Proc. Natl. Acad. Sci. U. S. A 115 (3) (2018) 619–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kwok HS, Vargas-Rodriguez O, Melnikov SV, Söll D, Engineered aminoacyl-tRNA synthetases with improved selectivity toward noncanonical amino acids, ACS Chem. Biol 14 (4) (2019) 603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Burke SA, Lo SL, Krzycki JA, Clustered genes encoding the methyltransferases of methanogenesis from monomethylamine, J. Bacteriol 180 (13) (1998) 3432–3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Paul L, Ferguson DJ Jr., J.A. Krzycki, The trimethylamine methyltransferase gene and multiple dimethylamine methyltransferase genes of Methanosarcina barkeri contain in-frame and read-through amber codons, J. Bacteriol 182 (9) (2000) 2520–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kavran JM, Gundllapalli S, O’Donoghue P, Englert M, Soll D, Steitz TA, Structure of pyrrolysyl-tRNA synthetase, an archaeal enzyme for genetic code innovation, Proc. Natl. Acad. Sci. U. S. A 104 (27) (2007) 11268–11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hao B, Gong W, Ferguson TK, James CM, Krzycki JA, Chan MK, A new UAG-encoded residue in the structure of a methanogen methyltransferase, Science 296 (5572) (2002) 1462–1466. [DOI] [PubMed] [Google Scholar]
- [37].Srinivasan G, James CM, Krzycki JA, Pyrrolysine encoded by UAG in Archaea: charging of a UAG-decoding specialized tRNA, Science 296 (5572) (2002) 1459–1462. [DOI] [PubMed] [Google Scholar]
- [38].Blight SK, Larue RC, Mahapatra A, Longstaff DG, Chang E, Zhao G, Kang PT, Green-Church KB, Chan MK, Krzycki JA, Direct charging of tRNACUA with pyrrolysine in vitro and in vivo, Nature 431 (7006) (2004) 333–335. [DOI] [PubMed] [Google Scholar]
- [39].Polycarpo C, Ambrogelly A, Berube A, Winbush SM, McCloskey JA, Crain PF, Wood JL, Söll D, An aminoacyl-tRNA synthetase that specifically activates pyrrolysine, Proc. Natl. Acad. Sci. U. S. A 101 (34) (2004) 12450–12454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Willis JCW, Chin JW, Mutually orthogonal pyrrolysyl-tRNA synthetase/tRNA pairs, Nat. Chem 10 (8) (2018) 831–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gaston MA, Jiang R, Krzycki JA, Functional context, biosynthesis, and genetic encoding of pyrrolysine, Curr. Opin. Microbiol 14 (3) (2011) 342–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Prat L, Heinemann IU, Aerni HR, Rinehart J, O’Donoghue P, Söll D, Carbon source-dependent expansion of the genetic code in bacteria, Proc. Natl. Acad. Sci. U. S. A 109 (51) (2012) 21070–21075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Heinemann IU, O’Donoghue P, Madinger C, Benner J, Randau L, Noren CJ, Söll D, The appearance of pyrrolysine in tRNAHis guanylyltransferase by neutral evolution, Proc. Natl. Acad. Sci. U. S. A 106 (50) (2009) 21103–21108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cusack S, Berthet-Colominas C, Hartlein M, Nassar N, Leberman R, A second class of synthetase structure revealed by X-ray analysis of Escherichia coli seryl-tRNA synthetase at 2.5 Å, Nature 347 (6290) (1990) 249–255. [DOI] [PubMed] [Google Scholar]
- [45].Eriani G, Delarue M, Poch O, Gangloff J, Moras D, Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs, Nature 347 (6289) (1990) 203–206. [DOI] [PubMed] [Google Scholar]
- [46].Jiang R, Krzycki JA, PylSn and the homologous N-terminal domain of pyrrolysyl-tRNA synthetase bind the tRNA that is essential for the genetic encoding of pyrrolysine, J. Biol. Chem 287 (39) (2012) 32738–32746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Tharp JM, Ehnbom A, Liu WR, tRNAPyl: structure, function, and applications, RNA Biol. 15 (4–5) (2018) 441–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Herring S, Ambrogelly A, Gundllapalli S, O’Donoghue P, Polycarpo CR, Söll D, The amino-terminal domain of pyrrolysyl-tRNA synthetase is dispensable in vitro but required for in vivo activity, FEBS Lett. 581 (17) (2007) 3197–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yanagisawa T, Ishii R, Fukunaga R, Nureki O, Yokoyama S, Crystallization and preliminary X-ray crystallographic analysis of the catalytic domain of pyrrolysyl-tRNA synthetase from the methanogenic archaeon Methanosarcina mazei, Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun 62 (Pt. 10) (2006) 1031–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Fladischer P, Weingartner A, Blamauer J, Darnhofer B, Birner-Gruenberger R, Kardashliev T, Ruff AJ, Schwaneberg U, Wiltschi B, A semi-rationally engineered bacterial pyrrolysyl-tRNA synthetase genetically encodes phenyl azide chemistry, Biotechnol. J 14 (3) (2019) e1800125. [DOI] [PubMed] [Google Scholar]
- [51].Nozawa K, O’Donoghue P, Gundllapalli S, Araiso Y, Ishitani R, Umehara T, Söll D, Nureki O, Pyrrolysyl-tRNA synthetase-tRNAPyl structure reveals the molecular basis of orthogonality, Nature 457 (7233) (2009) 1163–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Borrel G, Gaci N, Peyret P, O’Toole PW, Gribaldo S, Brugere JF, Unique characteristics of the pyrrolysine system in the 7th order of methanogens: implications for the evolution of a genetic code expansion cassette, Archaea 2014 (2014) 374146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Borrel G, O’Toole PW, Harris HM, Peyret P, Brugere JF, Gribaldo S, Phylogenomic data support a seventh order of methylotrophic methanogens and provide insights into the evolution of methanogenesis, Genome Biol. Evol 5 (10) (2013) 1769–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Borrel G, Parisot N, Harris HM, Peyretaillade E, Gaci N, Tottey W, Bardot O, Raymann K, Gribaldo S, Peyret P, O’Toole PW, Brugere JF, Comparative genomics highlights the unique biology of Methanomassiliicoccales, a Thermoplasmatales-related seventh order of methanogenic archaea that encodes pyrrolysine, BMC Genomics 15 (2014) 679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Mihajlovski A, Alric M, Brugere JF, A putative new order of methanogenic Archaea inhabiting the human gut, as revealed by molecular analyses of the mcrA gene, Res. Microbiol 159 (7–8) (2008) 516–521. [DOI] [PubMed] [Google Scholar]
- [56].Meineke B, Heimgartner J, Lafranchi L, Elsasser SJ, Methanomethylophilus alvus Mx1201 provides basis for mutual orthogonal pyrrolysyl tRNA/aminoacyl-tRNA synthetase pairs in mammalian cells, ACS Chem. Biol 13 (11) (2018) 3087–3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Yamaguchi A, Iraha F, Ohtake K, Sakamoto K, Pyrrolysyl-tRNA synthetase with a unique architecture enhances the availability of lysine derivatives in synthetic genetic codes, Molecules 23 (10) (2018) 2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Beranek V, Willis JCW, Chin JW, An evolved Methanomethylophilus alvus pyrrolysyl-tRNA synthetase/tRNA pair is highly active and orthogonal in mammalian cells, Biochemistry 58 (5) (2019) 387–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Tharp JM, Ad O, Amikura K, Ward FR, Garcia EM, Cate JHD, Schepartz A, Söll D, Initiation of protein synthesis with non-canonical amino acids in vivo, Angew. Chem. Int. Ed. Engl 59 (8) (2020) 3122–3126. [DOI] [PubMed] [Google Scholar]
- [60].Pavkov-Keller T, Schweiger K, Gruber K, Pyrrolysyl-tRNA synthetase from Canditatus Methanomethylophilus alvus (MmaPylRS). 2017. 10.2210/pdb6EZD/pdb. [DOI]
- [61].Yanagisawa T, Kuratani M, Yokoyama S, Crystal structure of pyrrolysyl-tRNA synthetase from Methanomethylophilus alvus. 2019. 10.2210/pdb6JP2/pdb. [DOI] [PubMed]
- [62].Herring S, Ambrogelly A, Polycarpo CR, Söll D, Recognition of pyrrolysine-tRNA by the Desulfitobacterium hafniense pyrrolysyl-tRNA synthetase, Nucleic Acids Res. 35 (4) (2007) 1270–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Theobald-Dietrich A, Frugier M, Giege R, Rudinger-Thirion J, Atypical archaeal tRNA pyrrolysine transcript behaves towards EF-Tu as a typical elongator tRNA, Nucleic Acids Res. 32 (3) (2004) 1091–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Wan W, Huang Y, Wang Z, Russell WK, Pai PJ, Russell DH, Liu WR, A facile system for genetic incorporation of two different noncanonical amino acids into one protein in Escherichia coli, Angew. Chem. Int. Ed. Engl 49 (18) (2010) 3211–3214. [DOI] [PubMed] [Google Scholar]
- [65].Niu W, Schultz PG, Guo J, An expanded genetic code in mammalian cells with a functional quadruplet codon, ACS Chem. Biol 8 (7) (2013) 1640–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ho JM, Reynolds NM, Rivera K, Connolly M, Guo LT, Ling J, Pappin DJ, Church GM, Söll D, Efficient reassignment of a frequent serine codon in wild-type Escherichia coli, ACS Synth. Biol 5 (2) (2016) 163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Wan W, Tharp JM, Liu WR, Pyrrolysyl-tRNA synthetase: an ordinary enzyme but an outstanding genetic code expansion tool, Biochim. Biophys. Acta 1844 (6) (2014) 1059–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Yanagisawa T, Ishii R, Fukunaga R, Kobayashi T, Sakamoto K, Yokoyama S, Crystallographic studies on multiple conformational states of active-site loops in pyrrolysyl-tRNA synthetase, J. Mol. Biol 378 (3) (2008) 634–652. [DOI] [PubMed] [Google Scholar]
- [69].Yanagisawa T, Sumida T, Ishii R, Yokoyama S, A novel crystal form of pyrrolysyl-tRNA synthetase reveals the pre- and post-aminoacyl-tRNA synthesis conformational states of the adenylate and aminoacyl moieties and an asparagine residue in the catalytic site, Acta Crystallogr. D Biol. Crystallogr 69 (Pt. 1) (2013) 5–15. [DOI] [PubMed] [Google Scholar]
- [70].Lee YJ, Wu B, Raymond JE, Zeng Y, Fang X, Wooley KL, Liu WR, A genetically encoded acrylamide functionality, ACS Chem. Biol 8 (8) (2013) 1664–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Yanagisawa T, Ishii R, Fukunaga R, Kobayashi T, Sakamoto K, Yokoyama S, Multistep engineering of pyrrolysyl-tRNA synthetase to genetically encode Nε-(O-azidobenzyloxycarbonyl) lysine for site-specific protein modification, Chem. Biol 15 (11) (2008) 1187–1197. [DOI] [PubMed] [Google Scholar]
- [72].Kaya E, Vrabel M, Deiml C, Prill S, Fluxa VS, Carell T, A genetically encoded norbornene amino acid for the mild and selective modification of proteins in a copper-free click reaction, Angew. Chem. Int. Ed. Engl 51 (18) (2012) 4466–4469. [DOI] [PubMed] [Google Scholar]
- [73].Fan C, Ho JML, Chirathivat N, Söll D, Wang YS, Exploring the substrate range of wild-type aminoacyl-tRNA synthetases, ChemBioChem 15 (12) (2014) 1805–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Hoesl MG, Budisa N, Recent advances in genetic code engineering in Escherichia coli, Curr. Opin. Biotechnol 23 (5) (2012) 751–757. [DOI] [PubMed] [Google Scholar]
- [75].Voloshchuk N, Montclare JK, Incorporation of unnatural amino acids for synthetic biology, Mol. Biosyst 6 (1) (2010) 65–80. [DOI] [PubMed] [Google Scholar]
- [76].Crnković A, Suzuki T, Söll D, Reynolds NM, Pyrrolysyl-tRNA synthetase, an aminoacyl-tRNA synthetase for genetic code expansion, Croat. Chem. Acta 89 (2) (2016) 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Flugel V, Vrabel M, Schneider S, Structural basis for the site-specific incorporation of lysine derivatives into proteins, PLoS One 9 (4) (2014) e96198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Neumann H, Peak-Chew SY, Chin JW, Genetically encoding Nε-acetyllysine in recombinant proteins, Nat. Chem. Biol 4 (4) (2008) 232–234. [DOI] [PubMed] [Google Scholar]
- [79].Yanagisawa T, Kuratani M, Seki E, Hino N, Sakamoto K, Yokoyama S, Structural basis for genetic-code expansion with bulky lysine derivatives by an engineered pyrrolysyl-tRNA synthetase, Cell Chem. Biol 26 (7) (2019) 936–949.e13. [DOI] [PubMed] [Google Scholar]
- [80].Kobayashi T, Yanagisawa T, Sakamoto K, Yokoyama S, Recognition of non-alpha-amino substrates by pyrrolysyl-tRNA synthetase, J. Mol. Biol 385 (5) (2009) 1352–1360. [DOI] [PubMed] [Google Scholar]
- [81].Li YM, Yang MY, Huang YC, Li YT, Chen PR, Liu L, Ligation of expressed protein α-hydrazides via genetic incorporation of an α-hydroxy acid, ACS Chem. Biol 7 (6) (2012) 1015–1022. [DOI] [PubMed] [Google Scholar]
- [82].Nguyen DP, Garcia Alai MM, Kapadnis PB, Neumann H, Chin JW, Genetically encoding Nε-methyl-L-lysine in recombinant histones, J. Am. Chem. Soc 131 (40) (2009) 14194–14195. [DOI] [PubMed] [Google Scholar]
- [83].Wang YS, Wu B, Wang Z, Huang Y, Wan W, Russell WK, Pai PJ, Moe YN, Russell DH, Liu WR, A genetically encoded photocaged Nε-methyl-L-lysine, Mol. Biosyst 6 (9) (2010) 1557–1560. [DOI] [PubMed] [Google Scholar]
- [84].Wang YS, Russell WK, Wang Z, Wan W, Dodd LE, Pai PJ, Russell DH, Liu WR, The de novo engineering of pyrrolysyl-tRNA synthetase for genetic incorporation of L-phenylalanine and its derivatives, Mol. Biosyst 7 (3) (2011) 714–717. [DOI] [PubMed] [Google Scholar]
- [85].Takimoto JK, Dellas N, Noel JP, Wang L, Stereochemical basis for engineered pyrrolysyl-tRNA synthetase and the efficient in vivo incorporation of structurally divergent non-native amino acids, ACS Chem. Biol 6 (7) (2011) 733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Wang YS, Fang X, Wallace AL, Wu B, Liu WR, A rationally designed pyrrolysyl-tRNA synthetase mutant with a broad substrate spectrum, J. Am. Chem. Soc 134 (6) (2012) 2950–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Lee YJ, Schmidt MJ, Tharp JM, Weber A, Koenig AL, Zheng H, Gao J, Waters ML, Summerer D, Liu WR, Genetically encoded fluorophenylalanines enable insights into the recognition of lysine trimethylation by an epigenetic reader, Chem. Commun. (Camb.) 52 (85) (2016) 12606–12609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Tharp JM, Wang YS, Lee YJ, Yang Y, Liu WR, Genetic incorporation of seven ortho-substituted phenylalanine derivatives, ACS Chem. Biol 9 (4) (2014) 884–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Tuley A, Lee YJ, Wu B, Wang ZU, Liu WR, A genetically encoded aldehyde for rapid protein labelling, Chem. Commun. (Camb.) 50 (56) (2014) 7424–7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Wang YS, Fang X, Chen HY, Wu B, Wang ZU, Hilty C, Liu WR, Genetic incorporation of twelve meta-substituted phenylalanine derivatives using a single pyrrolysyl-tRNA synthetase mutant, ACS Chem. Biol 8 (2) (2013) 405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Sharma V, Wang YS, Liu WR, Probing the catalytic charge-relay system in alanine racemase with genetically encoded histidine mimetics, ACS Chem. Biol 11 (12) (2016) 3305–3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Young DD, Young TS, Jahnz M, Ahmad I, Spraggon G, Schultz PG, An evolved aminoacyl-tRNA synthetase with atypical polysubstrate specificity, Biochemistry 50 (11) (2011) 1894–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Stokes AL, Miyake-Stoner SJ, Peeler JC, Nguyen DP, Hammer RP, Mehl RA, Enhancing the utility of unnatural amino acid synthetases by manipulating broad substrate specificity, Mol. Biosyst 5 (9) (2009) 1032–1038. [DOI] [PubMed] [Google Scholar]
- [94].Hohl A, Karan R, Akal A, Renn D, Liu X, Ghorpade S, Groll M, Rueping M, Eppinger J, Engineering a polyspecific pyrrolysyl-trna synthetase by a high throughput FACS screen, Sci. Rep 9 (1) (2019) 11971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Zheng Y, Addy PS, Mukherjee R, Chatterjee A, Defining the current scope and limitations of dual noncanonical amino acid mutagenesis in mammalian cells, Chem. Sci 8 (10) (2017) 7211–7217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Katayama H, Nozawa K, Nureki O, Nakahara Y, Hojo H, Pyrrolysine analogs as substrates for bacterial pyrrolysyl-tRNA synthetase in vitro and in vivo, Biosci. Biotechnol. Biochem 76 (1) (2012) 205–208. [DOI] [PubMed] [Google Scholar]
- [97].Fan C, Xiong H, Reynolds NM, Söll D, Rationally evolving tRNAPyl for efficient incorporation of noncanonical amino acids, Nucleic Acids Res. 43 (22) (2015) e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Sharma V, Zeng Y, Wang WW, Qiao Y, Kurra Y, Liu WR, Evolving the N-terminal domain of pyrrolysyl-tRNA synthetase for improved incorporation of noncanonical amino acids, ChemBioChem 19 (1) (2018) 26–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Serfling R, Lorenz C, Etzel M, Schicht G, Bottke T, Morl M, Coin I, Designer tRNAs for efficient incorporation of non-canonical amino acids by the pyrrolysine system in mammalian cells, Nucleic Acids Res. 46 (1) (2018) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Wang L, Magliery TJ, Liu DR, Schultz PG, A new functional suppressor tRNA/aminoacyl–tRNA synthetase pair for the in vivo incorporation of unnatural amino acids into proteins, J. Am. Chem. Soc 122 (20) (2000) 5010–5011. [Google Scholar]
- [101].Marck C, Grosjean H, tRNomics: analysis of tRNA genes from 50 genomes of Eukarya, Archaea, and Bacteria reveals anticodon-sparing strategies and domain-specific features, RNA 8 (10) (2002) 1189–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Brick P, Bhat TN, Blow DM, Structure of tyrosyl-tRNA synthetase refined at 2.3 Å resolution. Interaction of the enzyme with the tyrosyl adenylate intermediate, J. Mol. Biol 208 (1) (1989) 83–98. [DOI] [PubMed] [Google Scholar]
- [103].Kobayashi T, Nureki O, Ishitani R, Yaremchuk A, Tukalo M, Cusack S, Sakamoto K, Yokoyama S, Structural basis for orthogonal tRNA specificities of tyrosyl-tRNA synthetases for genetic code expansion, Nat. Struct. Biol 10 (6) (2003) 425–432. [DOI] [PubMed] [Google Scholar]
- [104].Jakubowski H, Goldman E, Editing of errors in selection of amino acids for protein synthesis, Microbiol. Rev 56 (3) (1992) 412–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Steer BA, Schimmel P, Major anticodon-binding region missing from an archaebacterial tRNA synthetase, J. Biol. Chem 274 (50) (1999) 35601–35606. [DOI] [PubMed] [Google Scholar]
- [106].Wang L, Brock A, Herberich B, Schultz PG, Expanding the genetic code of Escherichia coli, Science 292 (5516) (2001) 498–500. [DOI] [PubMed] [Google Scholar]
- [107].Wang L, Schultz PG, Expanding the genetic code, Chem. Commun. (Camb.) (1) (2002) 1–11. [DOI] [PubMed] [Google Scholar]
- [108].Yang XL, Skene RJ, McRee DE, Schimmel P, Crystal structure of a human aminoacyl-tRNA synthetase cytokine, Proc. Natl. Acad. Sci. U. S. A 99 (24) (2002) 15369–15374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Qiu X, Janson CA, Smith WW, Green SM, McDevitt P, Johanson K, Carter P, Hibbs M, Lewis C, Chalker A, Fosberry A, Lalonde J, Berge J, Brown P, Houge-Frydrych CS, Jarvest RL, Crystal structure of Staphylococcus aureus tyrosyl-tRNA synthetase in complex with a class of potent and specific inhibitors, Protein Sci. 10 (10) (2001) 2008–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Yaremchuk A, Kriklivyi I, Tukalo M, Cusack S, Class I tyrosyl-tRNA synthetase has a class II mode of cognate tRNA recognition, EMBO J. 21 (14) (2002) 3829–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Bedouelle H, Recognition of tRNATyr by tyrosyl-tRNA synthetase, Biochimie 72 (8) (1990) 589–598. [DOI] [PubMed] [Google Scholar]
- [112].Fechter P, Rudinger-Thirion J, Tukalo M, Giege R, Major tyrosine identity determinants in Methanococcus jannaschii and Saccharomyces cerevisiae tRNATyr are conserved but expressed differently, Eur. J. Biochem 268 (3) (2001) 761–767. [DOI] [PubMed] [Google Scholar]
- [113].Wang Y, Tsao ML, Reassigning sense codon AGA to encode noncanonical amino acids in Escherichia coli, ChemBioChem 17 (23) (2016) 2234–2239. [DOI] [PubMed] [Google Scholar]
- [114].Wang L, Brock A, Schultz PG, Adding L-3-(2-naphthyl)alanine to the genetic code of E. coli, J. Am. Chem. Soc 124 (9) (2002) 1836–1837. [DOI] [PubMed] [Google Scholar]
- [115].Wang J, Xie J, Schultz PG, A genetically encoded fluorescent amino acid, J. Am. Chem. Soc 128 (27) (2006) 8738–8739. [DOI] [PubMed] [Google Scholar]
- [116].Baumann T, Hauf M, Richter F, Albers S, Moglich A, Ignatova Z, Budisa N, Computational aminoacyl-tRNA synthetase library design for photocaged tyrosine, Int. J. Mol. Sci 20 (9) (2019) 2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Turner JM, Graziano J, Spraggon G, Schultz PG, Structural plasticity of an aminoacyl-tRNA synthetase active site, Proc. Natl. Acad. Sci. U. S. A 103 (17) (2006) 6483–6488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Brick P, Blow DM, Crystal structure of a deletion mutant of a tyrosyl-tRNA synthetase complexed with tyrosine, J. Mol. Biol 194 (2) (1987) 287–297. [DOI] [PubMed] [Google Scholar]
- [119].Zhang Y, Wang L, Schultz PG, Wilson IA, Crystal structures of apo wild-type M. jannaschii tyrosyl-tRNA synthetase (TyrRS) and an engineered TyrRS specific for O-methyl-L-tyrosine, Protein Sci. 14 (5) (2005) 1340–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Turner JM, Graziano J, Spraggon G, Schultz PG, Structural characterization of a p-acetylphenylalanyl aminoacyl-tRNA synthetase, J. Am. Chem. Soc 127 (43) (2005) 14976–14977. [DOI] [PubMed] [Google Scholar]
- [121].Zhang Z, Wang L, Brock A, Schultz PG, The selective incorporation of alkenes into proteins in Escherichia coli, Angew. Chem. Int. Ed. Engl 41 (15) (2002) 2840–2842. [DOI] [PubMed] [Google Scholar]
- [122].Wang L, Zhang Z, Brock A, Schultz PG, Addition of the keto functional group to the genetic code of Escherichia coli, Proc. Natl. Acad. Sci. U. S. A 100 (1) (2003) 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Chin JW, Martin AB, King DS, Wang L, Schultz PG, Addition of a photo-crosslinking amino acid to the genetic code of Escherichia coli, Proc. Natl. Acad. Sci. U. S. A 99 (17) (2002) 11020–11024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Chin JW, Santoro SW, Martin AB, King DS, Wang L, Schultz PG, Addition of p-azido-L-phenylalanine to the genetic code of Escherichia coli, J. Am. Chem. Soc 124 (31) (2002) 9026–9027. [DOI] [PubMed] [Google Scholar]
- [125].Tsao ML, Summerer D, Ryu Y, Schultz PG, The genetic incorporation of a distance probe into proteins in Escherichia coli, J. Am. Chem. Soc 128 (14) (2006) 4572–4573. [DOI] [PubMed] [Google Scholar]
- [126].Peeler JC, Woodman BF, Averick S, Miyake-Stoner SJ, Stokes AL, Hess KR, Matyjaszewski K, Mehl RA, Genetically encoded initiator for polymer growth from proteins, J. Am. Chem. Soc 132 (39) (2010) 13575–13577. [DOI] [PubMed] [Google Scholar]
- [127].Wang F, Robbins S, Guo J, Shen W, Schultz PG, Genetic incorporation of unnatural amino acids into proteins in Mycobacterium tuberculosis, PLoS One 5 (2) (2010) e9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Gan R, Perez JG, Carlson ED, Ntai I, Isaacs FJ, Kelleher NL, Jewett MC, Translation system engineering in Escherichia coli enhances non-canonical amino acid incorporation into proteins, Biotechnol. Bioeng 114 (5) (2017) 1074–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Goodman HM, Abelson J, Landy A, Brenner S, Smith JD, Amber suppression: a nucleotide change in the anticodon of a tyrosine transfer RNA, Nature 217 (5133) (1968) 1019–1024. [DOI] [PubMed] [Google Scholar]
- [130].Bonnefond L, Giege R, Rudinger-Thirion J, Evolution of the tRNATyr/TyrRS aminoacylation systems, Biochimie 87 (9–10) (2005) 873–883. [DOI] [PubMed] [Google Scholar]
- [131].Edwards H, Schimmel P, A bacterial amber suppressor in Saccharomyces cerevisiae is selectively recognized by a bacterial aminoacyl-tRNA synthetase, Mol. Cell. Biol 10 (4) (1990) 1633–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Edwards H, Trezeguet V, Schimmel P, An Escherichia coli tyrosine transfer RNA is a leucine-specific transfer RNA in the yeast Saccharomyces cerevisiae, Proc. Natl. Acad. Sci. U. S. A 88 (4) (1991) 1153–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Chin JW, Cropp TA, Anderson JC, Mukherji M, Zhang Z, Schultz PG, An expanded eukaryotic genetic code, Science 301 (5635) (2003) 964–967. [DOI] [PubMed] [Google Scholar]
- [134].Boeke JD, LaCroute F, Fink GR, A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance, Mol. Gen. Genet 197 (2) (1984) 345–346. [DOI] [PubMed] [Google Scholar]
- [135].Chen S, Schultz PG, Brock A, An improved system for the generation and analysis of mutant proteins containing unnatural amino acids in Saccharomyces cerevisiae, J. Mol. Biol 371 (1) (2007) 112–122. [DOI] [PubMed] [Google Scholar]
- [136].Wang Q, Wang L, New methods enabling efficient incorporation of unnatural amino acids in yeast, J. Am. Chem. Soc 130 (19) (2008) 6066–6067. [DOI] [PubMed] [Google Scholar]
- [137].Simoes J, Bezerra AR, Moura GR, Araujo H, Gut I, Bayes M, Santos MA, The fungus Candida albicans tolerates ambiguity at multiple codons, Front. Microbiol 7 (2016) 401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Palzer S, Bantel Y, Kazenwadel F, Berg M, Rupp S, Sohn K, An expanded genetic code in Candida albicans to study protein-protein interactions in vivo, Eukaryot. Cell 12 (6) (2013) 816–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Young TS, Ahmad I, Brock A, Schultz PG, Expanding the genetic repertoire of the methylotrophic yeast Pichia pastoris, Biochemistry 48 (12) (2009) 2643–2653. [DOI] [PubMed] [Google Scholar]
- [140].Shao N, Singh NS, Slade SE, Jones AM, Balasubramanian MK, Site specific genetic incorporation of azidophenylalanine in Schizosaccharomyces pombe, Sci. Rep 5 (2015) 17196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Sakamoto K, Hayashi A, Sakamoto A, Kiga D, Nakayama H, Soma A, Kobayashi T, Kitabatake M, Takio K, Saito K, Shirouzu M, Hirao I, Yokoyama S, Site-specific incorporation of an unnatural amino acid into proteins in mammalian cells, Nucleic Acids Res. 30 (21) (2002) 4692–4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Bedouelle H, Guez-Ivanier V, Nageotte R, Discrimination between transfer-RNAs by tyrosyl-tRNA synthetase, Biochimie 75 (12) (1993) 1099–1108. [DOI] [PubMed] [Google Scholar]
- [143].Kiga D, Sakamoto K, Kodama K, Kigawa T, Matsuda T, Yabuki T, Shirouzu M, Harada Y, Nakayama H, Takio K, Hasegawa Y, Endo Y, Hirao I, Yokoyama S, An engineered Escherichia coli tyrosyl-tRNA synthetase for site-specific incorporation of an unnatural amino acid into proteins in eukaryotic translation and its application in a wheat germ cell-free system, Proc. Natl. Acad. Sci. U. S. A 99 (15) (2002) 9715–9720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Hino N, Okazaki Y, Kobayashi T, Hayashi A, Sakamoto K, Yokoyama S, Protein photo-cross-linking in mammalian cells by site-specific incorporation of a photoreactive amino acid, Nat. Methods 2 (3) (2005) 201–206. [DOI] [PubMed] [Google Scholar]
- [145].Ye S, Kohrer C, Huber T, Kazmi M, Sachdev P, Yan EC, Bhagat A, Raj Bhandary UL, Sakmar TP, Site-specific incorporation of keto amino acids into functional G protein-coupled receptors using unnatural amino acid mutagenesis, J. Biol. Chem 283 (3) (2008) 1525–1533. [DOI] [PubMed] [Google Scholar]
- [146].Parrish AR, She X, Xiang Z, Coin I, Shen Z, Briggs SP, Dillin A, Wang L, Expanding the genetic code of Caenorhabditis elegans using bacterial aminoacyl-tRNA synthetase/tRNA pairs, ACS Chem. Biol 7 (7) (2012) 1292–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Chen Y, Ma J, Lu W, Tian M, Thauvin M, Yuan C, Volovitch M, Wang Q, Holst J, Liu M, Vriz S, Ye S, Wang L, Li D, Heritable expansion of the genetic code in mouse and zebrafish, Cell Res. 27 (2) (2017) 294–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Chen PR, Groff D, Guo J, Ou W, Cellitti S, Geierstanger BH, Schultz PG, A facile system for encoding unnatural amino acids in mammalian cells, Angew. Chem. Int. Ed. Engl 48 (22) (2009) 4052–4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Chatterjee A, Xiao H, Bollong M, Ai HW, Schultz PG, Efficient viral delivery system for unnatural amino acid mutagenesis in mammalian cells, Proc. Natl. Acad. Sci. U. S. A 110 (29) (2013) 11803–11808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Iraha F, Oki K, Kobayashi T, Ohno S, Yokogawa T, Nishikawa K, Yokoyama S, Sakamoto K, Functional replacement of the endogenous tyrosyl-tRNA synthetase-tRNATyr pair by the archaeal tyrosine pair in Escherichia coli for genetic code expansion, Nucleic Acids Res. 38 (11) (2010) 3682–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Italia JS, Latour C, Wrobel CJJ, Chatterjee A, Resurrecting the bacterial tyrosyl-tRNA synthetase/tRNA pair for expanding the genetic code of both E. coli and eukaryotes, Cell Chem. Biol 25 (10) (2018) 1304–1312.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Soma A, Himeno H, Cross-species aminoacylation of tRNA with a long variable arm between Escherichia coli and Saccharomyces cerevisiae, Nucleic Acids Res. 26 (19) (1998) 4374–4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Wu N, Deiters A, Cropp TA, King D, Schultz PG, A genetically encoded photocaged amino acid, J. Am. Chem. Soc 126 (44) (2004) 14306–14307. [DOI] [PubMed] [Google Scholar]
- [154].Summerer D, Chen S, Wu N, Deiters A, Chin JW, Schultz PG, A genetically encoded fluorescent amino acid, Proc. Natl. Acad. Sci. U. S. A 103 (26) (2006) 9785–9789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Wang W, Takimoto JK, Louie GV, Baiga TJ, Noel JP, Lee KF, Slesinger PA, Wang L, Genetically encoding unnatural amino acids for cellular and neuronal studies, Nat. Neurosci 10 (8) (2007) 1063–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Mukai T, Reynolds NM, Crnković A, Söll D, Bioinformatic analysis reveals archaeal tRNATyr and tRNATrp identities in Bacteria, Life (Basel) 7 (1) (2017) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Himeno H, Hasegawa T, Asahara H, Tamura K, Shimizu M, Identity determinants of E. coli tryptophan tRNA, Nucleic Acids Res. 19 (23) (1991) 6379–6382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Yesland KD, Nelson AW, Six Feathers DM, Johnson JD, Identity of Saccharomyces cerevisiae tRNATrp is not changed by an anticodon mutation that creates an amber suppressor, J. Biol. Chem 268 (1) (1993) 217–220. [PubMed] [Google Scholar]
- [159].Hughes RA, Ellington AD, Rational design of an orthogonal tryptophanyl nonsense suppressor tRNA, Nucleic Acids Res. 38 (19) (2010) 6813–6830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Chatterjee A, Xiao H, Yang PY, Soundararajan G, Schultz PG, A tryptophanyl-tRNA synthetase/tRNA pair for unnatural amino acid mutagenesis in E. coli, Angew. Chem. Int. Ed. Engl 52 (19) (2013) 5106–5109. [DOI] [PubMed] [Google Scholar]
- [161].Italia JS, Addy PS, Wrobel CJ, Crawford LA, Lajoie MJ, Zheng Y, Chatterjee A, An orthogonalized platform for genetic code expansion in both bacteria and eukaryotes, Nat. Chem. Biol 13 (4) (2017) 446–450. [DOI] [PubMed] [Google Scholar]
- [162].Sauerwald A, Zhu W, Major TA, Roy H, Palioura S, Jahn D, Whitman WB, Yates JR 3rd, Ibba M, Söll D, RNA-dependent cysteine biosynthesis in archaea, Science 307 (5717) (2005) 1969–1972. [DOI] [PubMed] [Google Scholar]
- [163].Kamtekar S, Hohn MJ, Park HS, Schnitzbauer M, Sauerwald A, Söll D, Steitz TA, Toward understanding phosphoseryl-tRNACys formation: the crystal structure of Methanococcus maripaludis phosphoseryl-tRNA synthetase, Proc. Natl. Acad. Sci. U. S. A 104 (8) (2007) 2620–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Fukunaga R, Yokoyama S, Structural insights into the first step of RNA-dependent cysteine biosynthesis in archaea, Nat. Struct. Mol. Biol 14 (4) (2007) 272–279. [DOI] [PubMed] [Google Scholar]
- [165].Fukunaga R, Harada Y, Hirao I, Yokoyama S, Phosphoserine aminoacylation of tRNA bearing an unnatural base anticodon, Biochem. Biophys. Res. Commun 372 (3) (2008) 480–485. [DOI] [PubMed] [Google Scholar]
- [166].Park HS, Hohn MJ, Umehara T, Guo LT, Osborne EM, Benner J, Noren CJ, Rinehart J, Söll D, Expanding the genetic code of Escherichia coli with phosphoserine, Science 333 (6046) (2011) 1151–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Hohn MJ, Park HS, O’Donoghue P, Schnitzbauer M, Söll D, Emergence of the universal genetic code imprinted in an RNA record, Proc. Natl. Acad. Sci. U. S. A 103 (48) (2006) 18095–18100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Rogerson DT, Sachdeva A, Wang K, Haq T, Kazlauskaite A, Hancock SM, Huguenin-Dezot N, Muqit MM, Fry AM, Bayliss R, Chin JW, Efficient genetic encoding of phosphoserine and its nonhydrolyzable analog, Nat. Chem. Biol 11 (7) (2015) 496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Lee S, Oh S, Yang A, Kim J, Söll D, Lee D, Park HS, A facile strategy for selective incorporation of phosphoserine into histones, Angew. Chem. Int. Ed. Engl 52 (22) (2013) 5771–5775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Zhang MS, Brunner SF, Huguenin-Dezot N, Liang AD, Schmied WH, Rogerson DT, Chin JW, Biosynthesis and genetic encoding of phosphothreonine through parallel selection and deep sequencing, Nat. Methods 14 (7) (2017) 729–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Venkat S, Sturges J, Stahman A, Gregory C, Gan Q, Fan C, Genetically incorporating two distinct post-translational modifications into one protein simultaneously, ACS Synth. Biol 7 (2) (2018) 689–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Ibba M, Losey HC, Kawarabayasi Y, Kikuchi H, Bunjun S, Söll D, Substrate recognition by class I lysyl-tRNA synthetases: a molecular basis for gene displacement, Proc. Natl. Acad. Sci. U. S. A 96 (2) (1999) 418–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Terada T, Nureki O, Ishitani R, Ambrogelly A, Ibba M, Söll D, Yokoyama S, Functional convergence of two lysyl-tRNA synthetases with unrelated topologies, Nat. Struct. Biol 9 (4) (2002) 257–262. [DOI] [PubMed] [Google Scholar]
- [174].Stehlin C, Burke B, Yang F, Liu H, Shiba K, Musier-Forsyth K, Species-specific differences in the operational RNA code for aminoacylation of tRNAPro, Biochemistry 37 (23) (1998) 8605–8613. [DOI] [PubMed] [Google Scholar]
- [175].Burke B, Lipman RS, Shiba K, Musier-Forsyth K, Hou YM, Divergent adaptation of tRNA recognition by Methanococcus jannaschii prolyl-tRNA synthetase, J. Biol. Chem 276 (23) (2001) 20286–20291. [DOI] [PubMed] [Google Scholar]
- [176].Chatterjee A, Xiao H, Schultz PG, Evolution of multiple, mutually orthogonal prolyl-tRNA synthetase/tRNA pairs for unnatural amino acid mutagenesis in Escherichia coli, Proc. Natl. Acad. Sci. U. S. A 109 (37) (2012) 14841–14846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Peterson ET, Uhlenbeck OC, Determination of recognition nucleotides for Escherichia coli phenylalanyl-tRNA synthetase, Biochemistry 31 (42) (1992) 10380–10389. [DOI] [PubMed] [Google Scholar]
- [178].Furter R, Expansion of the genetic code: site-directed p-fluoro-phenylalanine incorporation in Escherichia coli, Protein Sci. 7 (2) (1998) 419–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Kwon I, Kirshenbaum K, Tirrell DA, Breaking the degeneracy of the genetic code, J. Am. Chem. Soc 125 (25) (2003) 7512–7513. [DOI] [PubMed] [Google Scholar]
- [180].Datta D, Wang P, Carrico IS, Mayo SL, Tirrell DA, A designed phenylalanyl-tRNA synthetase variant allows efficient in vivo incorporation of aryl ketone functionality into proteins, J. Am. Chem. Soc 124 (20) (2002) 5652–5653. [DOI] [PubMed] [Google Scholar]
- [181].Kast P, Hennecke H, Amino acid substrate specificity of Escherichia coli phenylalanyl-tRNA synthetase altered by distinct mutations, J. Mol. Biol 222 (1) (1991) 99–124. [DOI] [PubMed] [Google Scholar]