

Abstract
In structural biology, cryo-electron microscopy (cryo-EM) has emerged as the main technique for determining the atomic structures of macromolecular complexes. This has largely been due to the introduction of direct electron detectors, which have allowed for routinely reaching a near-atomic resolution when imaging such complexes. In chemistry and materials science, the applications of cryo-EM have been much more limited. A recent paper (J. Am. Chem. Soc. 2019, 141, 19448−19457) has used low resolution cryo-EM to analyze polymorphic helical tubes formed by a tetrameric protein, and has made detailed models for the interfaces between the tetramers in these assemblies. Due to intrinsic ambiguities in determining the correct helical symmetry, we show that many of the models are likely to be wrong. This note highlights both the enormous potential for using cryo-EM, and also the pitfalls possible for helical assemblies when a near-atomic level of resolution is not reached.
The main technique responsible for most atomic structures of macromolecules has been x-ray crystallography. However, over the past eight years there has been a revolution in cryo-electron microscopy (cryo-EM) with an exponential growth (Fig. 1) in the number of atomic structures being determined by cryo-EM1–3. This revolution has largely been driven by the introduction of direct electron detectors4, which have provided a huge improvement in both signal-to-noise and detected quantum efficiency over film, CCD or CMOS detectors. As a result, cryo-EM is now the method of choice for structural studies of macromolecular assemblies5 and has led to the deposition in the Electron Microscopy Data Base of more than 6,500 three-dimensional reconstructions at better than 5 Å resolution. For most of these (~ 5,000 at better than 4.0 Å resolution) building atomic models ab initio, with no prior knowledge of the structure of the subunits6, is not only possible but quite routine. While cryo-EM is now becoming the dominant technique in structural biology, it has made far fewer inroads in chemistry and materials science, with only a limited number of publications at near-atomic resolution of peptide assemblies7–10. One reason for this disparity is that due to the incredibly poor signal-to-noise ratio (SNR) in cryo-EM (where most of the contrast in images is due to noise, and not signal), averaging of images from thousands to millions of particles is necessary to achieve a reasonable resolution and SNR. But these particle images need to be aligned, and that sets a lower bound on the size of the individual particle that can be studied by cryo-EM. It is useful to understand that the ribosome, which has a molecular weight of several million Daltons, became a model system in the development of cryo-EM11. But when relatively small molecules assemble into helical polymers, the internal symmetry in such structures can be exploited to greatly boost the SNR and allow for the alignment and averaging of an asymmetric unit that may only be hundreds of Daltons or less.
Figure 1.
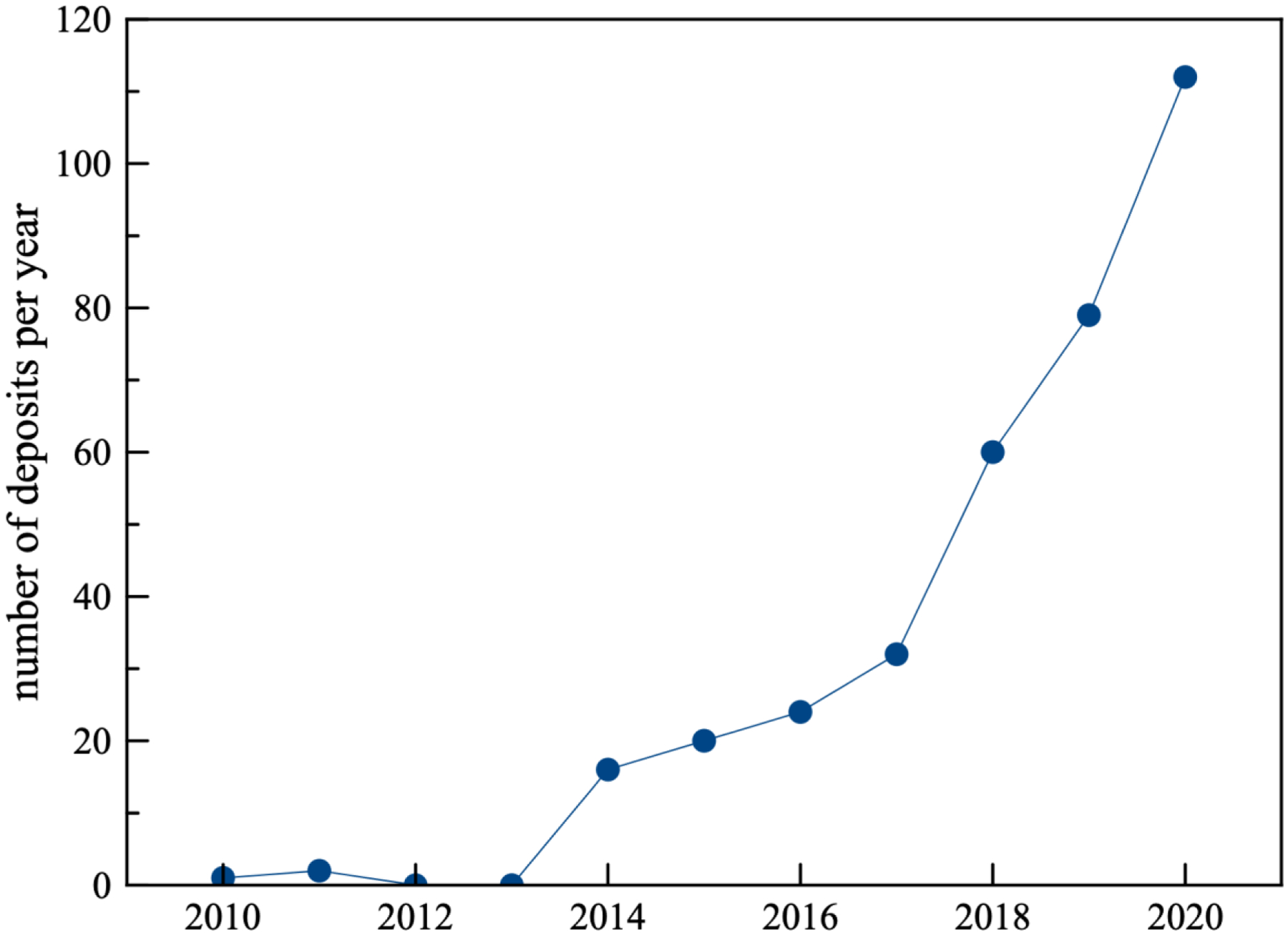
The number of deposits per year in the Electron Microscopy Data Bank (EMDB) for helical assemblies reconstructed by cryo-EM at a resolution better than 5 Å. The number of helical assemblies is still small compared to 6,137 single particle structures at better than 5 Å deposited over this time period.
The purpose of this short communication is to show that when a near-atomic level of resolution for helical polymers is not achieved by cryo-EM, there are numerous possibilities for wrong or inconclusive results. A recent paper12 has used low resolution cryo-EM to generate three-dimensional reconstructions of polymorphic tubes formed by a protein tetramer, soybean agglutinin, whose crystal structure is known13. As with an earlier three-dimensional reconstruction from such tubes14, imaging was done without a direct electron detector, greatly limiting the resolution possible. For helical polymers, there can be intrinsic ambiguities in the helical symmetry15–17, such that for a given resolution, there may be multiple different helical symmetries that will all give rise to indistinguishable projection images, which are what is recorded in cryo-EM. The worse the resolution, the greater are the possible ambiguities. For example, it was shown that for the rod-like tobacco mosaic virus, at 10 Å resolution one would be unable to determine the correct symmetry15. This means that one could create multiple three dimensional volumes that look different, but the projections of these volumes onto two dimensions would look the same.
Given that an atomic model exists for the protomer, soybean agglutin, that is forming these polymorphic tubes, a reality test exists in terms of the correspondence between the three-dimensional reconstruction and the atomic model. This can be easily measured by either an overall correlation coefficient (which will be in the range 0.0 to 1.0) or as a Fourier Shell Correlation (FSC), which measures the correlation in frequency space shells between the reconstruction and the model. The resolution may thus be assessed by asking where the correlation between the reconstruction and the model falls below some arbitrary threshold, such as FSC=0.38. An alternative approach measures the resolution by looking at internal consistency or reproducibility. That is, if one creates two reconstructions independently from different images, at what resolution does the correlation between these two reconstructions fall below another arbitrary threshold, frequently taken18 as FSC=0.143. In the presence of symmetry, this measure of consistency can be very different from the actual resolution, as one can generate two completely artifactual maps (reconstructions) that agree with each other to fairly high resolution but have no relation to reality19.
We have therefore looked not at the internal consistency determined by comparing two independent maps, as was done for the published soybean agglutinin maps12, 14, but at the correlations between the maps and the atomic models for the published three-dimensional volumes (Table 1). These correlations are expressed two different ways: one as simply an overall coefficient of correlation, while the other (using the FSC) involves asking at what resolution the correlation between the map and the model loses significance. A visual comparison between one such volume and the atomic model (Fig. 2a) suggests that the actual resolution of the volume cannot be the 10.9 Å claimed, as the atomic model filtered to 10.9 Å (Fig. 2b) looks quite different from the volume. Thus, the map:model resolution found for this volume of 20 Å (Table 1) is consistent with the visual comparison, while the internal consistency metric yielding a value of 10.9 Å is inconsistent with the visual comparison. The discrepancy between the internal consistency map:map FSC and the reality-based map:model FSC can most simply be explained by the use of a wrong helical symmetry17. A simple experiment to validate this is doing reconstructions for all possible symmetries and examine the volumes. The best match between the map:map FSC and the map:model FSC is for EMDB-8065, published in the original paper14. Does this mean that the symmetry used in this case is validated as correct, given the agreement between the map and model at ~ 9 Å resolution? We think not, as it has been shown at 5 Å resolution that two different helical symmetries for a bacterial mating pilus can generate volumes that are almost indistinguishable20. However, the use of the correct symmetry resulted in a final resolution of 3.9 Å for the mating pilus, while the incorrect symmetry never improved beyond 5 Å.
Table 1.
Cryo-EM maps vs. 2.8 Å crystal structure 1SBE
| Map EMBD ID | Claimed resolution (Å) | Model vs. map correlation coefficient | Model vs. map FSC (Å, 0.38 cutoff) |
|---|---|---|---|
| EMD-0735 | 8.2 | 0.44 | 16 |
| EMD-0736 | 10.9 | 0.74 | 13 |
| EMD-0737 | 13.1 | 0.76 | 18 |
| EMD-0738 | 8.8 | 0.62 | 15 |
| EMD-0739 | 10.9 | 0.69 | 20 |
| EMD-8065 | 7.8 | 0.66 | 9 |
Statistics for the deposited volumes and comparisons with the atomic structure of the protomer (PDB 1SBE). The program PHENIX was used to generate these metrics22.
Figure 2.

(a) A transparent surface of the EMDB-0739 volume, with a ribbon representation of the crystallographic 1SBE.PDB tetramer fit into it. While the stated resolution of the volume is 10.9 Å, we find from the map:model FSC that the resolution is 20 Å. For comparison (b), we show the tetramer fit into a volume generated from the atomic model that has been filtered to 10.9 Å resolution.
The power of cryo-EM continues to grow, with true atomic resolution having now been achieved21. We expect that there will be a rapidly increasing number of applications of cryo-EM to assemblies formed by peptides and small molecules, yielding a vastly greater amount of information than techniques such as SAXS (small angle x-ray scattering) that are very model-dependent in their interpretation. However, as with any technique, errors in execution and interpretation can be made, and readers and reviewers need to understand both the strengths and limitations in using cryo-EM.
Acknowledgements
This work was supported by NIH GM122510.
Footnotes
Conflict of Interest
The authors declare no conflicts of interest.
References
- 1.Egelman EH, The Current Revolution in Cryo-EM. Biophys J 2016, 110 (5), 1008–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bai XC; McMullan G; Scheres SH, How cryo-EM is revolutionizing structural biology. Trends Biochem Sci 2015, 40 (1), 49–57. [DOI] [PubMed] [Google Scholar]
- 3.Kuhlbrandt W, Biochemistry. The resolution revolution. Science 2014, 343 (6178), 1443–4. [DOI] [PubMed] [Google Scholar]
- 4.Li X; Mooney P; Zheng S; Booth CR; Braunfeld MB; Gubbens S; Agard DA; Cheng Y, Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nature Methods 2013, 10 (6), 584–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramakrishnan V, The ribosome emerges from a black box. Cell 2014, 159 (5), 979–84. [DOI] [PubMed] [Google Scholar]
- 6.DiMaio F; Song Y; Li X; Brunner MJ; Xu C; Conticello V; Egelman E; Marlovits TC; Cheng Y; Baker D, Atomic-accuracy models from 4.5-A cryo-electron microscopy data with density-guided iterative local refinement. Nature Methods 2015, 12 (4), 361–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang F; Gnewou O; Modlin C; Beltran LC; Xu C; Su Z; Juneja P; Grigoryan G; Egelman EH; Conticello VP, Structural analysis of cross α-helical nanotubes provides insight into the designability of filamentous peptide nanomaterials. Nature Communications 2021, 12 (1), 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hughes SA; Wang F; Wang S; Kreutzberger MAB; Osinski T; Orlova A; Wall JS; Zuo X; Egelman EH; Conticello VP, Ambidextrous helical nanotubes from self-assembly of designed helical hairpin motifs. Proc Natl Acad Sci U S A 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Egelman EH; Xu C; DiMaio F; Magnotti E; Modlin C; Yu X; Wright E; Baker D; Conticello VP, Structural plasticity of helical nanotubes based on coiled-coil assemblies. Structure 2015, 23 (2), 280–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feng Z; Wang H; Wang F; Oh Y; Berciu C; Cui Q; Egelman EH; Xu B, Artificial Intracellular Filaments. Cell Rep Phys Sci 2020, 1 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frank J, Generalized single-particle cryo-EM--a historical perspective. Microscopy (Oxf) 2016, 65 (1), 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Z; Chen S; Gao C; Yang Z; Shih K-C; Kochovski Z; Yang G; Gou L; Nieh M-P; Jiang M; Zhang L; Chen G, Chemically Controlled Helical Polymorphism in Protein Tubes by Selective Modulation of Supramolecular Interactions. Journal of the American Chemical Society 2019, 141 (49), 19448–19457. [DOI] [PubMed] [Google Scholar]
- 13.Olsen LR; Dessen A; Gupta D; Sabesan S; Sacchettini JC; Brewer CF, X-ray crystallographic studies of unique cross-linked lattices between four isomeric biantennary oligosaccharides and soybean agglutinin. Biochemistry 1997, 36 (49), 15073–80. [DOI] [PubMed] [Google Scholar]
- 14.Yang G; Zhang X; Kochovski Z; Zhang Y; Dai B; Sakai F; Jiang L; Lu Y; Ballauff M; Li X; Liu C; Chen G; Jiang M, Precise and Reversible Protein-Microtubule-Like Structure with Helicity Driven by Dual Supramolecular Interactions. Journal of the American Chemical Society 2016, 138 (6), 1932–1937. [DOI] [PubMed] [Google Scholar]
- 15.Egelman EH, Reconstruction of helical filaments and tubes. Methods in Enzymology 2010, 482, 167–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Egelman EH, Three-dimensional reconstruction of helical polymers. Arch Biochem Biophys 2015, 581, 54–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Egelman EH, Ambiguities in Helical Reconstruction. eLife 2014, 3:e04969 doi: 10.7554/eLife.04969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosenthal PB; Henderson R, Optimal Determination of Particle Orientation, Absolute Hand, and Contrast Loss in Single-particle Electron Cryomicroscopy. Journal of Molecular Biology 2003, 333 (4), 721–745. [DOI] [PubMed] [Google Scholar]
- 19.Subramaniam S; Earl LA; Falconieri V; Milne JL; Egelman EH, Resolution advances in cryo-EM enable application to drug discovery. Curr Opin Struct Biol 2016, 41, 194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng W; Pena A; Low WW; Wong JLC; Frankel G; Egelman EH, Cryoelectron-Microscopic Structure of the pKpQIL Conjugative Pili from Carbapenem-Resistant Klebsiella pneumoniae. Structure 2020, 28 (12), 1321–1328 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakane T; Kotecha A; Sente A; McMullan G; Masiulis S; Brown P; Grigoras IT; Malinauskaite L; Malinauskas T; Miehling J; Uchanski T; Yu L; Karia D; Pechnikova EV; de Jong E; Keizer J; Bischoff M; McCormack J; Tiemeijer P; Hardwick SW; Chirgadze DY; Murshudov G; Aricescu AR; Scheres SHW, Single-particle cryo-EM at atomic resolution. Nature 2020, 587 (7832), 152–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Afonine PV; Klaholz BP; Moriarty NW; Poon BK; Sobolev OV; Terwilliger TC; Adams PD; Urzhumtsev A, New tools for the analysis and validation of cryo-EM maps and atomic models. Acta Crystallogr D Struct Biol 2018, 74 (Pt 9), 814–840. [DOI] [PMC free article] [PubMed] [Google Scholar]