

Abstract
Proper localization of Ras proteins at the plasma membrane (PM) is crucial for their functions. To get to the PM, KRas4B and some other Ras family proteins bind to the PDEδ shuttling protein through their farnesylated hypervariable regions (HVRs). The docking of their farnesyl (and to a lesser extent geranylgeranyl) in the hydrophobic pocket of PDEδ’s stabilizes the interaction. At the PM, guanosine 5′-triphosphate (GTP)-bound Arf-like protein 2 (Arl2) assists in the release of Ras from the PDEδ. However, exactly how is still unclear. Using all-atom molecular dynamics simulations, we unraveled the detailed mechanism of Arl2-mediated release of KRas4B, the most abundant oncogenic Ras isoform, from PDEδ. We simulated ternary Arl2–PDEδ–KRas4B HVR complexes and observed that Arl2 binding weakens the PDEδ–farnesylated HVR interaction. Our detailed analysis showed that allosteric changes (involving β6 of PDEδ and additional PDEδ residues) compress the hydrophobic PDEδ pocket and push the HVR out. Mutating PDEδ residues that mediate allosteric changes in PDEδ terminates the release process. Mutant Ras proteins are enriched in human cancers, with currently no drugs in the clinics. This mechanistic account may inspire efforts to develop drugs suppressing oncogenic KRas4B release.
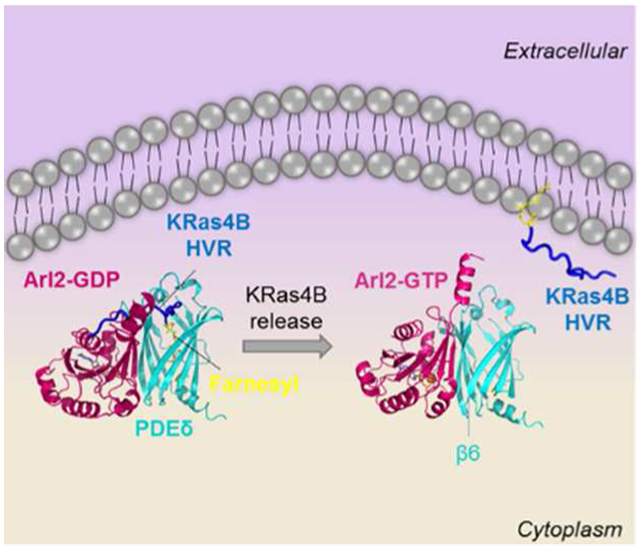
Graphical abstract
INTRODUCTION
Small GTPases including Ras isoforms KRas (KRas4A and KRas4B), HRas, and NRas regulate multiple cellular processes such as intracellular trafficking, gene expression, cell proliferation, transformation, and motility.1,2 Ras isoforms are mutated in approximately 20–30% of human cancers and KRas4B is the most frequently mutated.3-5 Membrane anchorage is a common feature of the Ras superfamily and plasma membrane (PM) attachment is essential for their function.6 They are synthesized as soluble precursors and their C-terminal CAAX sequences (where C stands for cysteine, A is an aliphatic residue, and X denotes any amino acids) undergo a series of post-translational modifications.7 Prenylation (addition of the prenyl group to the cysteine residue) of Ras proteins, involving farnesylation or geranylgeranylation, is a significant modification for trafficking and membrane insertion.8,9
Guanosine nucleotide dissociation inhibitors (GDIs) such as RhoGDI are responsible for the shuttling of Rho proteins from the Ras superfamily of GTPases.10 The high structural similarity of phosphodiesterase-δ (PDEδ) to RhoGDI suggests that PDEδ has a role in binding to and shuttling of small GTPases.11,12 Studies support the role of PDEδ in solubilizing and trafficking prenylated KRas, HRas, and Rheb.11,13,14 PDEδ interacts with Ras proteins (KRas4B and likely KRas4A),15,16 through their hypervariable regions (HVRs), embedding the prenyl group in the hydrophobic cavity.17,18 Biochemical studies indicate that PDEδ binding to small GTPases is more favorable for farnesylated than geranylgeranylated proteins.12,13
Studies show that PDEδ can also associate with two nonprenylated Arf-like proteins, Arl2 and Arl3. GTP-dependent interaction of Arl2/Arl3 with PDEδ indicates that PDEδ is an Arl2/Arl3 effector.11,19,20 PDEδ stabilizes Arl2/Arl3 in their GTP-bound form by blocking guanosine 5′-triphosphate (GTP) hydrolysis.19 Arl2 and Arl3 are responsible for the regulation of the release and/or uptake of farnesylated cargos and small inhibitory molecules from PDEδ.11,21 However, the exact mechanism of the KRas4B release from PDEδ to facilitate PM anchorage is unknown and the role of Arl2/Arl3 in the release is unclear. Ras is a major drug target,22,23 and to date, no drugs are in the clinic. PM localization is essential for its proper function. Localization is regulated by membrane trafficking/solubilizing factors such as PDEδ and Arl2/Arl3, respectively.24 Interfering with proper Ras localization can be one possible therapeutic approach.25,26 To this end, the exact mechanism of Ras protein transport to the PM and release from the shuttling factor should be fully understood.17,18,27
To reveal the release mechanism, we carried out molecular dynamics (MD) simulations of ternary guanosine diphosphate (GDP)- and GTP-bound Arl2–PDEδ–farnesylated/geranyl-geranylated HVR complexes and in silico mutagenesis in PDEδ. We observed that the interaction between the farnesylated HVR of KRas4B and PDEδ is weakened by binding of Arl2–GTP to PDEδ, with some important interactions between β6 of PDEδ (involving Glu88) and the HVR disrupted. Moreover, upon Arl2–GTP binding, the farnesylated HVR is pushed upward, out of the hydrophobic pocket. It has been previously reported that some residues in the hydrophobic PDEδ pocket such as Met20, Arg61, and Ile129 conformationally change and mediate the Rheb (a small GTPase from the Ras superfamily) release by clashing with the farnesyl group of Rheb.28 We observed similar conformational changes in these residues as well as in Phe133 upon Arl2 binding, which bounce the KRas4B HVR back and facilitate release from the pocket. Arl2–GTP binding induces allosteric changes in the PDEδ, mainly involving β6 residues that enable farnesylated KRas4B release. We further observed a gradually stabilizing energy profile for the Arl2 and PDEδ interaction pointing to the formation of a fast-dissociating ternary complex. When we mutated the allosteric region of PDEδ, we observed that the HVR is not pushed upward, Met20, Arg61, and Ile129 are not shifted toward the inside of the pocket, and a gradual stabilizing energy profile cannot be detected. We also observed that geranylgeranylated KRas4B does not follow a farnesylated KRas4B-like regulation scenario, suggesting a different release mechanism. Here, we focus on unraveling the underlying mechanism of the Arl2–GTP-mediated release of farnesylated KRas4B from the PDEδ hydrophobic pocket.
METHODS
Initial Configurations of Arl2–PDEδ and Arl2–PDEδ–HVR Complexes.
PDEδ–KRas4B HVR complex structures were adopted from our previous work.17 In that work, two PDEδ structures were used with state 1 and state 2 Phe133 side chains (PDB code: 5E8F and 5F2U, respectively) (Figure S1). The C-terminal KRas4B HVR (residues 167–185) was post-transitionally modified with the methylation and the farnesyl/geranylgeranyl groups. Because PDEδ interacting with geranylgeranylated HVR adopts state 1, only state 1 was modeled for the PDEδ–geranylgeranylated HVR complex. PDEδ–farnesylated HVR complexes were modeled for both state 1 and state 2. In this study, Arl2–PDEδ crystal structures (PDB codes: 1KSH with Arl2–GDP and 1KSG with Arl2–GTP) were used to obtain Arl2–PDEδ–HVR complexes. Arl2 sequences were modified into the human. For Arl2–GDP, the missing N-terminus portion (Arl21–16) was modeled with MODELLER29 as an α-helix using Arl2–GTP as a template. The Ile residue was added to the C-terminus to avoid charged terminus. The simulation inputs were built using CHARMM all-atom additive force field (version C36) programming package.30,31 Bonded parameters for the GTP and the prenylated and geranylgeranylated cysteine were adopted from previous works.32,33
Atomistic MD Simulations.
MD simulations in an aqueous environment were carried out for a total of 10 initial configurations: two Arl2–PDEδ complexes (systems 1 and 5), six Arl2–PDEδ–HVR complexes (systems 2–4 and 6–8), and two Arl2-mutated PDEδ (F94A/I98A double mutation)–farnesylated HVR complexes (systems 6m and 7m). Several rigid body minimization steps were performed for the systems. The TIP3P water model was used to create the isometric unit cell box containing the protein complex. For each system, the unit cell box of 90 × 90 × 90 A3 contains around 75 000 atoms. In the production runs, the Langevin temperature control maintained the constant temperature at 310 K and the pressure was maintained at 1 atm. A total of 5.0 μs simulations, each with 500 ns, were performed using the NAMD parallel-computing code34 on a Biowulf cluster at the National Institutes of Health (Bethesda, MD). To monitor the systems to reach equilibration, root-mean-square deviation (rmsd) and free energy ΔGsol + ΔGgas) were calculated as a function of time (Figures S2 and S3). Averages were taken after 100 ns, discarding initial transient trajectories. The simulated trajectories were analyzed using CHARMM.30 In addition to analyses performed using CHARMM, the WISP35 plugin of VMD 1.9.236 was used for allosteric pathway analysis.
Molecular Mechanics Combined with the Generalized Born and Surface Area Continuum Solvation Calculations.
The average binding free energy is derived from the Gibbs free energy,
| (1) |
where Gsol and Ggas denote the solvation energy and gas-phase contributions, respectively, and TΔS is the entropic contribution. The change in the binding free energy due to the complex formation is calculated as follows
| (2) |
Further details of the calculation protocol have been reported in our previous studies.37 The CHARMM program was used to obtain the electrostatic and nonpolar contributions to the solvation free energy and to calculate the entropic contribution.30
RESULTS
PDEδ Affinity To KRas4B is Reduced by Arl2–GTP Binding.
We used the crystal structures of the Arl2–PDEδ complex (PDB codes: 1KSH with Arl2–GDP and 1KSG with Arl2–GTP)11 and the modeled structures of the PDEδ–KRas4B HVR complex.17 PDEδ has two different states (state 1 and state 2) relating to the different positions of the Phe133 side chain in the PDEδ hydrophobic pocket. In state 1, the Phe133 side chain faces downward; in state 2, the Phe133 side chain faces upward (Figure S1). State 1 configuration of PDEδ provides a deeper hydrophobic cavity for HVR binding; in state 2, it is shallower.17,18 Because the geranylgeranyl group is relatively longer, it flips the Phe133 side chain downward, similar to state 1. We considered both PDEδ states for farnesylated KRas4B HVR systems. The Arl2 simulations included GTP-bound (active) and GDP-bound (inactive) states. We also simulated complexes of Ras-free PDEδ with Arl2. Altogether, atomistic MD simulations were initially performed on eight systems for 500 ns (systems 1–8). Table 1 and Figure 1 summarize the initial structures and systems.
Table 1.
Initial Configurations of the Simulated Systems
| Arl2 |
KRas4B HVR |
PDEδ |
|||
|---|---|---|---|---|---|
| systems | PDB | state (GDP/GTP) | prenylation | PDB | Phe133 side chain/state |
| system 1 | 1KSH | GDP | |||
| system 2 | 1KSH | GDP | farnesyl | 5E8F | down/1 |
| system 3 | 1KSH | GDP | farnesyl | 5F2U | up/2 |
| system 4 | 1KSH | GDP | geranylgeranyl | 5E8F | down/1 |
| system 5 | 1KSG | GTP | |||
| system 6 | 1KSG | GTP | farnesyl | 5E8F | down/1 |
| system 7 | 1KSG | GTP | farnesyl | 5F2U | up/2 |
| system 8 | 1KSG | GTP | geranylgeranyl | 5E8F | down/1 |
Figure 1.

Initial configurations of the simulated systems. In the upper panel, systems with Arl2–GDP (systems 1–4) are shown, and in the lower panel, systems with Arl2–GTP (systems 5–8) are represented. The pink and cyan molecules are Arl2 and PDEδ, respectively. The KRas4B HVR is represented by the dark blue tube, and HVR’s prenyl groups and the Phe133 residue of PDEδ are shown by yellow sticks. Both GDP and GTP are shown by blue sticks, and the Mg2+ molecule is represented by the orange sphere.
PDEδ binds to Arl2 in a GTP-specific manner and inhibits the dissociation of GTP from Arl2. Arl2 regulation of PDEδ-mediated transport of prenylated cargos is expected only upon Arl2–GTP binding. To compare the interaction strengths of the HVR and PDEδ in GTP- and GDP-bound Arl2, we calculated the binding free energies of PDEδ–HVR in systems 2–4 and systems 6–8 using the molecular mechanics combined with the generalized Born and surface area continuum solvation (MM–GBSA) method.38 Our calculated binding free energies of the PDEδ–HVR interaction are −69.9 ± 8.2, −49.8 ± 6.0, and −62.3 ± 8.6 kcal/mol for systems 2–4 with Arl2–GDP, respectively, and −46.5 ± 9.2, −45.4 ± 8.3, and −41.6 ± 5.0 kcal/mol for systems 6–8 with Arl2–GTP, respectively (Figure 2). PDEδ–HVR interaction in systems with Arl2–GTP (systems 6–8) is weaker than that with Arl2–GDP (systems 2–4). This suggests that Arl2–GTP can mediate KRas4B release from PDEδ. The difference between the interaction strengths in system 3 and system 7 is not large compared to the other systems (system 2 vs system 6 and system 4 vs system 8). It was previously reported that the interaction of farnesylated HVR with PDEδ in state 1 causes some clashes.17 PDEδ in state 2 is more favorable for farnesylated cargo binding and more stable interactions can be established between farnesylated proteins and PDEδ in state 2, making state 2 more challenging for Arl2-stimulated HVR release from PDEδ.
Figure 2.

Binding free energies of PDEδ–HVR in systems 2–4 and systems 6–8 in kcal/mol calculated by MM–GBSA method. In the calculation, gas-phase contribution, ⟨ΔGgas⟩, the solvation energy contribution, ⟨ΔGsol⟩, and the entropic contribution, −TΔS, combine the average binding free energy, ⟨ΔGb⟩. In the box graphs, the red dot and red line denote the mean and median values, respectively.
Additional evidence that PDEδ–HVR interaction strength is reduced upon Arl2–GTP binding to PDEδ is the decrease in the number of stable interactions between PDEδ and the HVR. There are fewer stable salt bridges and hydrogen bonds (H-bonds) between PDEδ and HVR in systems 6 and 7 compared to systems 2 and 3, respectively (Figure 3). Previously, it has been reported that Glu88 of PDEδ plays a major role in KRas binding and inhibitors that can bind Glu88 impair oncogenic KRas-driven tumor growth.21 In our systems 2 and 3, stable interactions between Glu88 and HVR residues are established; however, they are disrupted in systems 6 and 7 (Figure 3). On the other hand, in the geranylgeranylated systems (systems 4 and 8), even though the binding free-energy calculations indicate that the PDEδ–geranylgeranylated HVR interaction is weakened by binding of Arl2–GTP, the number of stable interactions between PDEδ and HVR increases. This suggests that although farnesylated KRas4B release is mediated by Arl2, the mechanism of geranylgeranylated cargo release may differ. Because PDEδ binding to farnesylated HVR is more favorable than geranylgeranylated HVR, revealing the release mechanism of farnesylated KRas4B from PDEδ may provide insight into KRas4B trafficking to the PM.
Figure 3.

Interactions between Arl2, PDEδ, and HVR. Stable salt bridges and H-bond formed between Arl2, PDEδ, and HVR in systems 2–4 (left hand side) and systems 6–8 (right hand side) can be seen. The pink and cyan molecules/sticks show Arl2 and PDEδ molecules/residues, respectively, whereas the dark blue molecules/sticks represent KRas4B HVR molecules/residues. The black dashed lines show salt bridges, and the orange dashed lines show H-bond.
Arl2–Stimulated KRas4B HVR Slips out of the Hydrophobic PDEδ Cavity.
KRas4B HVR should be released from the PDEδ pocket to bind the PM. To follow the release process, we analyzed the initial and final positions of the farnesyl group inside the hydrophobic PDEδ pocket. We measured the distance between Cα atoms of Phe133 and the farnesyl/geranylgeranyl groups at 10 and 500 ns. The distance is larger at 500 ns in each system except system 8 (Table 2 and Figure 4A). The changes in the distance between systems 2–4 and systems 6–8 were compared. We observed that upon Arl2–GTP binding to PDEδ, the farnesylated HVR slips out further compared to the Arl2–GDP (Table 2). In systems 6 and 7 with Arl2–GTP, the distance changes are 5.10 Å and 1.62 Å for PDEδ in states 1 and 2, respectively. However, the changes in the distance are smaller in systems 2 and 3 with Arl2–GDP, which are 3.86 and 0.64 Å for PDEδ in states 1 and 2, respectively. For PDEδ in state 1, both systems 2 and 6 with Arl2–GDP and Arl2–GTP, respectively, yield larger distance changes than the corresponding systems 3 and 7 with PDEδ in state 2. Because for PDEδ in state 2, the Phe133 side chain is initially upward and already closer to the farnesyl groups in systems 3 and 7, smaller difference between Phe133 and farnesyl groups are expected. However, we observed that downward Phe133 side chains in systems 2 and 6 flip upward during the simulations (Figure 4A). Thus, even though the Phe133 side chain moves closer to the farnesyl group in systems 2 and 6, the distance between Phe133 and the farnesyl group at the end of the simulation is still larger. The movement of downward Phe133 to an upward position provides a repulsive force for the HVR to be released from the PDEδ pocket. PDEδ residues Met20, Arg61, and Ile129 have been reported to move upon Arl2 binding, clashing with the farnesyl group inside of the pocket.28 These changes facilitate farnesylated protein release. We also observed conformational changes of these residues throughout the simulation. Residues which were initially positioned lower, shift upward and toward the inside of the pocket in the farnesylated systems (Figure 4B). Like Phe133, these residues help HVR ejection from the hydrophobic pocket by compressing the pocket and pushing the HVR upward. However, geranylgeranylated systems again show an opposite behavior. We observed that for system 8 with Arl2–GTP, the geranylgeranyl group of KRas4B HVR is intact in the hydrophobic PDEδ pocket (Table 2 and Figure 4A). Also, the conformations of Met20, Arg61, and Ile129 do not change much compared to the change in farnesylated systems (Figure 4B), in line with geranylgeranylated cargo release by a variant mechanism.
Table 2.
Distances between Phe133 and CYF/CYGa at 10 ns, D10, and at 500 ns, D500, and the Change in Distance, ΔD = D500, − D10
| systems | D10 (Å) | D500 (Å) | ΔD (Å) |
|---|---|---|---|
| system 2 | 17.00 | 20.86 | 3.86 |
| system 3 | 20.31 | 20.95 | 0.64 |
| system 4 | 18.99 | 20.69 | 1.70 |
| system 6 | 15.51 | 20.61 | 5.10 |
| system 7 | 19.23 | 20.85 | 1.62 |
| system 8 | 20.77 | 20.37 | −0.40 |
CYF and CYG stand for farnesyl and geranylgeranyl groups, respectively.
Figure 4.

Initial and final positions of the KRas4B HVR and some key residues. (A) Initial and final positions of the farnesyl/geranylgeranyl group and Phe133 side chain in systems 2–4 and 6–8. (B) Initial and final positions of Met20, Arg61, and Ile129 of PDEδ in systems 6–8. The structures at 10 ns are represented by orange, whereas the structures at 500 ns are shown by gray. The HVR is removed in panel (B) to be able to clearly see the movement of the residues. The β4/β7 and β6 interfaces are shown with blue labels.
The PDEδ open conformation is more favorable for the farnesylated cargo binding.28,39 The open conformation has a larger cavity volume compared to the closed PDEδ conformation. A closed conformation is induced by Arl2–GTP binding to PDEδ.28 The conformational changes in PDEδ Met20, Arg61, Ile129, and Phe133 decrease the volume of the cavity and facilitate the shift to the closed conformation.28 Arl2 interacts with β4 (residue 59–67) and mainly β7 (residues 101–109) at one side of the β-sandwich and β6 (residues 88–97) at the other side,11 which is on the opposite PDEδ face (Figures S1 and 4B). Formation of these interactions lead to the movement of β4/β7 and β6 toward each other, which further promotes the closed conformation. During the simulations, we also observed that as expected, new interactions are established between these β-strands and Arl2–GTP (Table 3). The β6 conformation changes and the interaction between Glu88 on β6 and the HVR is disrupted upon Arl2–GTP binding (Figure 3). Therefore, Arl2–GTP binding induces KRas4B release from PDEδ by changing the PDEδ conformation to the unfavorable closed state. Because of this conformational change, the interactions between PDEδ and farnesylated HVR are also impaired, altogether resulting in the HVR slipping out from the pocket.
Table 3.
New Interactions Established between Arl2 and PDEδ upon Arl2–GTP Binding
| systems | interactionsa |
|---|---|
| system 5 | Lys29-Asp42, Glu110-Lys13, Lys57-Glu16, Lys57-Glu56, Glu62b-Lys35, Glu88b-Lys9 |
| system 7 | Lys73-Glu82, Arg23-Asp42, Glu93b-Arg18 |
| system 8 | Lys57-Glu56, Glu62b-Lys34, Arg23-Asp40, Arg23-Asp42 |
First residues in the residue pairs belong to PDEδ and the second ones belong to Arl2.
PDEδ residues from β4, β7 or β6.
Allosteric Regulation of PDEδ by Arl2 Facilitates KRas4B Release.
Previous experimental studies showed that PDEδ binding sites to farnesylated cargos and Arl2 do not overlap.28,39 This suggests that Arl2 allosterically regulates PDEδ to promote the release of the farnesylated cargos. Phe94 and Ile98 of PDEδ are at the Arl2–PDEδ interaction interface and undergo considerable conformational changes upon Arl2–GTP binding to PDEδ.28 These residues are also identified as hot spots, which contribute to the binding free energy most,40 making them candidates for the interaction and allosteric regulation of PDEδ. Experimental studies showed that the release of farnesylated Rheb from PDEδ is less efficient for PDEδF94A/I98A double mutant than the wild type. In line with this, the ternary complex formed by Arl2–PDEδ–farnesylated cargos is more stable in the double mutant.28 HVR position in systems 2 and 3 shows that when Arl2–GDP is bound, the HVR is folded, covering the β6 (residues 88–97) Arl2 interaction site of PDEδ, which involves Phe94 and Ile98. However, upon Arl2–GTP binding in systems 6 and 7, the HVR is released from β6 (Figure 5A). Glu93 of PDEδ, which forms salt bridges with the HVR in systems with Arl2–GDP, establishes interactions with Arl2 and other PDEδ residues exposing the β6 interface in systems with Arl2–GTP. Differences in the HVR movement in the systems can be observed. To reveal the allosteric mechanism of Arl2, we conducted a dynamical network analysis using weighted implementation of the suboptimal paths (WISP) algorithm.35 WISP can identify the shortest primary communicating pathway through nodes. Nodes refer to the protein residues of interest. Hundred optimal paths between selected residue pairs on Arl2 and PDEδ were calculated. The residue pairs are selected considering Arl2 residues interacting with Phe94PDEδ and Ile98PDEδ, which are identified as Tyr76Arl2 and Tyr80Arl2. PDEδ residue pairs are selected among the residues showing a higher fluctuation profile upon Arl2–GTP binding (Figure 5B), which comprise residues between 33–50 and 93–118. Residues 93–118 involve β6 and a flexible loop (residues 111–117), which are known to play a role in the farnesylated cargo interactions.28 Root-mean-square fluctuation (RMSF) plots alone are not enough to determine allosteric change sites because they show the average fluctuation of each residue through the simulations. Therefore, further analysis such as WISP and mutagenesis should be performed to validate allosteric changes. Among the selected residue pairs, we could obtain only one pathway in system 7 (Figure 5C). This possible pathway obtained by WISP starts with the Arl2 residue Tyr80 and ends in the PDEδ residue Phe96. Frequently involved residues in the 100 different paths of this pathway are Gln70, Met71, Gly95, Val97, and Ile98 of PDEδ. The fluctuations observed in PDEδ residues 93–118 result from allosteric regulation induced by Arl2–GTP binding. Allosteric changes in β6 and the flexible loop, which are initiated by Tyr80Arl2 and involve Ile98PDEδ, facilitate KRas4B HVR release.
Figure 5.

Allosteric changes in PDEδ induced by Arl2–GTP. (A) Position of KRas4B HVR in systems 2 and 3 with Arl2–GDP and in systems 6 and 7 with Arl2–GTP. The cyan molecules are superimposed PDEδs. The dark blue, blue, and light blue tubes represent the HVR conformations at 10, 150, and 500 ns, respectively. (B) RMSF plots of PDEδ in systems 2 and 6 (upper panel) and systems 3 and 7 (lower panel). The blue lines cover residues of systems 2 and 3, and the green lines cover residues of systems 6 and 7. The residues between 33–50 and 93–118, which are used in WISP analysis, are represented with black lines in systems 6 and 7. (C) Allosteric pathway between the selected residue pair on Arl2 and PDEδ, Tyr80Arl2/Ile98PDEδ, in system 7 with Arl2–GTP.
To compare the KRas4B HVR release mechanism for the wild-type and the double mutant structures, we simulated systems 6 and 7 with double mutant PDEδF94A/I98A. We observed that in mutant system 6 (systems 6m), the HVR cannot slip out as much as in the wild-type system 6 (Figure 6A, the left side of the upper panel). The distance between Phe133 and the farnesyl group at 10 ns is 16.80 Å and at 500 ns, it is 20.44 Å, a difference of 3.64 Å. This difference is smaller than that between both system 2 (3.86 Å) and the wild-type system 6 (5.10 Å) (Table 2). Therefore, in system 6m, the HVR is even more embedded in the hydrophobic pocket than in systems with Arl2–GDP. Also, the conformation changes observed in Met20, Arg61, and Ile129 in the wild-type system 6 cannot be observed in system 6m (Figure 6A, the right side of the upper panel). Especially, Met20 and Ile129 are at almost the same position at the end of the 500 ns simulation as in the initial configuration. We compared the RMSF plots of system 2 and system 6m. In wild-type system 6, some PDEδ residues (residues 33–50 and 93–118) fluctuate more than system 2, indicating allosteric changes in the PDEδ conformation. However, mutant system 6 exhibits a fluctuation profile similar to system 2 (Figure 6B, upper panel), suggesting that allosteric regulation cannot occur. This further confirms that in mutant system 6, the HVR cannot slip out. To compare the wild-type and mutant system 7 (system 7m), we monitored the HVR and observed that surprisingly, in system 7m, it can also be pushed upward (Figure 6A, the left side of the lower panel). Met20 and Arg61 also undergo conformational changes and point upward, similar to wild-type system 7 (Figure 6A, the right side of the lower panel); however, Ile129 does not change considerably. The effect of mutations on system 7 is not as significant as that in system 6, possibly because in system 7, the double mutations also affect the favorable bonds between PDEδ and the farnesylated HVR. Thus, even in the presence of the mutations, HVR release from PDEδ is not affected in system 7 as much as in system 6. The fluctuation profiles of system 3 and system 7m are mostly similar (Figure 6B, lower panel); the fluctuations observed between residues 33–50 and 93–118 in the wild-type system 7 cannot be seen in system 7m. This indicates that the allosteric changes cannot be induced in system 7m. The HVR bounce in system 7m is not caused by allosteric changes but by the disruption of favorable bonds between PDEδ and the HVR. Also, WISP analysis does not result in any allosteric paths unlike wild-type system 7. Overall, allosteric changes initiated by Arl2–GTP binding and involving the Phe94, Ile98, Gln70, Met71, Gly95, and Val97 residues of PDEδ play an important role in farnesylated KRas4B release from PDEδ and these allosteric changes are more efficient in the release of KRas4B HVR in system 6 compared to system 7. Furthermore, the number of stable interactions between PDEδ and HVR is higher in mutant systems 6 and 7 compared to wild-type Arl2–GTP-bound systems (Figure 6C). In Figure 3, we observed that the Arl2–GDP-bound systems have higher number of stable interactions, whereas Arl2–GTP binding induces dissociation of some interactions and leading fewer stable interactions. However, PDEδF94A/I98A mutations, which inhibit allosteric changes upon Arl2–GTP binding, increase the number of stable interaction between PDEδ and HVR.
Figure 6.

Effect of PDEδF94A/I98A double mutation on farnesylated KRas4B HVR release in the mutated systems (systems 6m and 7m). (A) Initial and final positions of the farnesyl group and Phe133 side chain (left panels) and some key residues (Met20, Arg61, and Ile129) of PDEδ (right panels) in system 6m (upper panels) and in system 7m (lower panels). The structures at 10 ns are represented by orange, whereas structures at 500 ns are shown by gray. The HVR is removed in the right panels to be able to clearly see the movement of the residues. (B) RMSF plots of PDEδ in systems 2 and system 6m (upper panel panel) and systems 3 and system 7m (lower panel). The blue lines cover residues of systems 2 and 3, and the green lines cover residues of systems 6m and 7m. (C) Interactions between Arl2, PDEδ, and HVR in systems 6m and 7m. The pink and cyan molecules/sticks show Arl2 and PDEδ molecules/residues, respectively, whereas the dark blue molecules/sticks represent HVR molecules/residues. The black dashed lines show salt bridges, and the orange dashed lines show H-bond.
Arl2–GTP, PDEδ, and Farnesylated KRas4B Form an Unstable Ternary Complex.
PDEδ binds to Arl2 in a GTP-dependent manner.11 Arl2–GDP binding to free PDEδ is much weaker than Arl2–GTP binding to free PDEδ (Figure 7A). Our calculated binding free energies are 7.7 ± 5.9 kcal/mol for system 1 and −19.8 ± 12.4 kcal/mol for system 5. The steady energy profile of system 5 shows that the Arl2–GTP–PDEδ complex is stable throughout the simulation. The binding energy profile of Arl2 to PDEδ drastically changes in the presence of the farnesylated KRas4B HVR in the ternary complex. In Figure 7B, the lower panels present step-like binding energy profiles between Arl2–GTP and PDEδ in systems 6 and 7. However, the energy profiles of systems 2 and 3 are smooth (Figure 7B, upper panel), as in the case of the Arl2–free PDEδ interaction. This indicates that the interaction of Arl2–GTP with PDEδ is unsteady and hardly reaches equilibrium after binding of farnesylated KRas4B to PDEδ. Experimental studies showed that the binding sites for Arl2 and farnesylated cargos do not overlap and ternary complex formation is structurally feasible. However, it is unstable and mutually exclusive.28,39 Allosteric displacement of the farnesylated cargo from PDEδ by Arl2 is mediated by the intermediate ternary complex.28 The Arl2–PDEδ–farnesylated cargo ternary complex dissociates into Arl2–PDEδ over time and the stability of Arl2–PDEδ interaction gradually increases (Figure 7B, lower panel). This indicates that the Arl2–PDEδ complex is not steady and stable in the presence of the farnesylated KRas4B; however, as the release of KRas4B proceeds, the Arl2–PDEδ interaction is stabilized. In line with the experimental studies, we observed that upon binding of Arl2–GTP and farnesylated HVR to their allosterically coupled PDEδ binding sites, a fast-dissociating ternary complex is formed. Our previous observations showed that HVR affinity to PDEδ decreases and the HVR moves toward the outside of the pocket. Figure 7B shows that the Arl2–PDEδ interaction strength increases during the HVR release. The energy profiles of systems 6 and 7 have been changed by PDEδF94A/I98A double mutations. Instead of a step-like profile between Arl2–GTP and PDEδ in systems 6 and 7 (Figure 7B, the lower panel), we observed a smooth profile in the corresponding mutant systems (systems 6m and 7m) (Figure 7C), similar to systems 2 and 3. Rather than fast-dissociating, the mutations stabilize the ternary complexes. The allosteric changes are necessary for the formation of the fast-dissociating intermediate complex. In geranylgeranylated systems 4 and 8, the energy profiles of Arl2–PDEδ interaction are smooth and stable (Figure 7B, right hand side of upper and lower panels), supporting KRas4B geranylgeranylation regulation differing from the farnesylated Ras case. The Arl2–PDEδ interaction is not affected by geranylgeranylated KRas4B binding, in contrast to farnesylated KRas4B. The geranylgeranylated HVR did not slip out from the pocket as much as the farnesylated HVR did and forms even more interactions with PDEδ upon Arl2–GTP binding, suggesting that its release is not coupled with Arl2. The ternary complex of Arl2–PDEδ-geranylgeranylated KRas4B is not fast-dissociating but steady.
Figure 7.

Binding energy profiles of Arl2–PDEδ complexes. (A) Binding free energies of Arl2–PDEδ in systems 1 and 5 in kcal/mol calculated by MM–GBSA method (left) and energy profiles of Arl2–PDEδ over time (ns) in systems 1 and 5 in kcal/mol (right). (B) Energy profiles of Arl2–PDEδ over time (ns) in systems 2–4 (upper panel) and in systems 6–8 (lower panel) in kcal/mol. (C) Energy profiles of Arl2–PDEδ over time (ns) in the mutant systems (systems 6m and 7m) in kcal/mol. Only gas-phase and solvation energy contributions are considered and entropic contribution is excluded in the energy profiles.
DISCUSSION
Subcellular localization critically affects prenylated Ras signaling capacity and activation.41,42 PM localization has been shown as increasing Ras activity through nanoclustering or dimerization42-45 and playing a role in signaling selectivity of Ras isoforms.32,41,46 Ras locates at the PM in the vicinity of receptors, effectors, and nucleotide exchange factors, which facilitate its signaling.41,45,47 Ras localization requires farnesylation-dependent interaction with PDEδ. PDEδ enriches its localization at the PM and promotes its activity.48,49 Inhibiting Ras–PDEδ interaction may target oncogenic Ras.50
Here, we describe a possible mechanism for the release of KRas4B from PDEδ by Arl2 in atomistic detail. We simulated eight Arl2–PDEδ–KRas4B HVR (including two mutant systems) and two Arl2–PDEδ complex systems. We observed that binding of Arl2–GTP to PDEδ decreases KRas4B affinity to PDEδ, supporting KRas4B release being mediated by Arl2 in a GTP-dependent manner. The number of stable salt bridges and H-bonds between PDEδ and farnesylated KRas4B HVR decreases and the farnesylated KRas4B HVR slips out from the hydrophobic PDEδ pocket upon Arl2–GTP binding. Our detailed atomistic level analysis showed that some of the critical interactions between PDEδ and KRas4B HVR are disrupted by Arl2–GTP–PDEδ interaction. PDEδ Glu88 interaction with the HVR is abolished by Arl2–GTP binding. The disruption of Glu88-involved interactions can considerably affect the binding affinity of KRas4B to PDEδ, in line with this residue being a main drug target.21 Disruption of these critical interactions is mostly caused by shifting of PDEδ β4/β7 and β6.28 The shift of these regions toward each other decreases the hydrophobic cavity volume. As we have shown here, the shift results in dissociating the critical interactions between β6 (involving Glu88) and KRas4B. We also showed that upon farnesylated KRas4B-bound systems interacting with Arl2–GTP, additional residues, Met20, Arg61, Ile129, and Phe133, of PDEδ shift toward the inside of the pocket and point upward. These changes facilitate the closed PDEδ conformation, which is not favorable for KRas4B binding. Moreover, once these residues point upward, the farnesylated KRas4B HVR is pushed outside of the PDEδ pocket. Together, these findings suggest that Arl2–GTP facilitates the release of farnesylated KRas4B HVR from PDEδ by decreasing the KRas4B HVR affinity to PDEδ.
Despite having distinct binding sites, binding of Arl2 and farnesylated cargos to PDEδ is mutually exclusive.28 This, together with substantial conformational changes discussed above, implies that Arl2 binding allosterically regulates KRas4B release. We identified that allosteric pathways are initiated by Arl2–GTP residues and are mostly composed of β6 residues of PDEδ In Arl2–GDP-bound systems, β6 residues are covered by the HVR; however, upon Arl2–GTP binding, the HVR swings and the β6 site is exposed. Consequently, β6 residues fluctuate more upon Arl2–GTP binding, which also indicates the conformational changes. Among the β6 residues, PDEδ Phe94 and Ile98 were experimentally reported as playing a role in allosteric regulation inducing farnesylated Rheb release from PDEδ.28 Ismail et al. experimentally constructed a PDEδF94A/I98A double mutant and compared its affinity to Arl2 and Rheb with wild-type PDEδ.28 They concluded that after addition of Arl2, dissociation of Rheb from PDEδF94A/I98A is not as efficient as in the wild-type case. On the basis of this experimental study, we tried to validate whether these PDEδ residues are also crucial in KRas4B HVR release. We further confirmed the effect of these residues on allosteric regulation and KRas4B HVR release by analyzing double mutant (PDEδF94A/I98A) farnesylated systems. The allosteric changes observed in wild-type farnesylated systems cannot be detected in the mutant systems and the HVR cannot slip out from the hydrophobic pocket in some systems. In some cases, allosteric changes can generate new binding sites and increase the number of binding partners;51 however, in others, one of the binding partners can be released, as in the case of the Arl2–PDEδ–KRas4B HVR ternary complex.
Experimental studies demonstrated that Arl2–GTP binding induces the formation of a closed PDEδ conformation and farnesylated Rheb release from PDEδ by generating the fast-dissociating ternary complex.28,39 Also, double mutation (PDEδF94A/I98A) decreases the release efficiency of Rheb and increases the stability of the ternary complex.28 We monitored the binding energy profiles of Arl2–free PDEδ and Arl2–GDP–PDEδ–KRas4B HVR systems. Both are steady throughout the simulations. However, the Arl2–GTP–PDEδ interaction is unsteady in farnesylated KRas4B HVR systems but is gradually stabilized. This shows that the formation of the fast-dissociating intermediate ternary complex is required for the release of the farnesylated KRas4B HVR; then, as release occurs, the Arl2–PDEδ system is equilibrated. Our observations are parallel to the experimental results suggesting formation of a fast-dissociating ternary complex required for farnesylated cargo release. Our PDEδF94A/I98A mutant systems present a behavior similar to that of Arl2–GDP-bound systems. This indicates that without allosteric changes, the fast-dissociating intermediate ternary complex cannot be formed. The ternary complex is stabilized and the farnesylated KRas4B release is inhibited.
With respect to geranylgeranylated cargo shuttling and release, our analysis showed that KRas4B geranylgeranylated systems behave in an opposite manner. HVR dislocation, the stable interactions, and the binding energy profile of Arl2–PDEδ in geranylgeranylated systems all differ, with PDEδ farnesylated cargos being more favorable. Experimentally, it has been shown that PDEδ can interact with both farnesyl and geranylgeranyl groups.12 However, the binding affinity between PDEδ and the geranylgeranyl group is much weaker12,52 implying those differences in the binding and release mechanisms. Releasing the long geranylgeranyl group from PDEδ is more likely to require more drastic conformational changes. Notably, in Rap1 members of the Ras subfamily of small GTPases, prenylation was recently observed to be required for high-affinity interactions with CAP1 in a geranylgeranyl-specific manner.53
Thus, the mechanism of the Arl2-mediated release of farnesylated KRas4B from PDEδ can now be understood. We combined MD simulations with in silico mutagenesis to obtain realistic models of farnesylated KRas4B release. Here, we reveal that Arl2–GTP binding to β4/β7 and β6 of PDEδ shifts these regions toward each other. This shifting decreases the PDEδ cavity volume and compresses the cavity, coupled with conformational changes in the β6 residues. These changes disrupt some stable bonds between PDEδ and HVR, allosterically regulating HVR release. The compression of the cavity, which is unfavorable for farnesylated cargo binding, and allosteric changes in PDEδ residues push the HVR upward, toward the outside of the cavity. The HVR then dissociates from the fast-dissociating ternary complex and the Arl2–PDEδ complex is stabilized (Figure 8).
Figure 8.

Release mechanism of KRas4B HVR from PDEδ. When Arl2–GDP is bound to PDEδ, the HVR is embedded in the hydrophobic PDEδ pocket and covers part of the Arl2 binding site preventing allosteric changes. However, upon Arl2–GTP binding, the allosteric site is revealed. PDEδ undergoes allosteric and conformation changes resulting in compressing the hydrophobic pocket and pushing the HVR outside. Although the HVR is bound, a fast-dissociation ternary complex is formed. Following HVR release, the Arl2–PDEδ complex is stabilized. In the middle panel, Arl2 and HVR are shaded with lighter colors to indicate the unsteady interactions.
To conclude, highly oncogenic Ras GTPases, particularly KRas, have drawn the attention of the community. However, their intracellular trafficking mechanisms have been enigmatic. The role of PDEδ in the farnesylated cargo shuttling was established as essential but the exact release mechanism from PDEδ has been obscure. Here, we elucidate this mechanism at the conformational level. Disruption of membrane localization of KRas4B might be one therapeutic approach to target KRas-driven tumor growth. Unraveling the Arl2-mediated release mechanism of farnesylated KRas4B from PDEδ may facilitate therapeutic targeting. Targeting the Arl2 interaction interface of PDEδ helps to impair PDEδ–Arl2 interaction and therapeutically aiming at allosteric sites of PDEδ may block formation of its closed form. These approaches might be promising for drug design to prevent KRas4B release from PDEδ and inhibit proper membrane localization.
Supplementary Material
ACKNOWLEDGMENTS
All simulations had been performed using the high-performance computational facilities of the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov). ESO acknowledges TUBITAK (The Scientific and Technological Research Council of Turkey) for financial support (Scholarship 2211-E). This work has been partially supported by TUBITAK Research grant no. 114M196. This project has been funded in whole or in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN261200800001E. This research was supported (in part) by the Intramural Research Program of NIH, Frederick National Lab, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcb.8b04347.
Structures and domains of PDEδ and Arl2, rmsd, and energy plots of all systems (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Hall A Signal transduction: G Proteins and Small GTPases: Distant Relatives Keep in Touch. Science 1998, 280, 2074–2075. [DOI] [PubMed] [Google Scholar]
- (2).Vojtek AB; Der CJ Increasing complexity of the Ras signaling pathway. J. Biol. Chem 1998, 273, 19925–19928. [DOI] [PubMed] [Google Scholar]
- (3).Bos JL ras oncogenes in human cancer: a review. Cancer Res. 1989, 49, 4682–4689. [PubMed] [Google Scholar]
- (4).Prior IA; Lewis PD; Mattos C A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Wittinghofer A Signal transduction via Ras. Biol. Chem 1998, 379, 933–937. [PubMed] [Google Scholar]
- (6).Willumsen BM; Norris K; Papageorge AG; Hubbert NL; Lowy DR Harvey murine sarcoma virus p21 ras protein: biological and biochemical significance of the cysteine nearest the carboxy terminus. EMBO J. 1984, 3, 2581–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Lowy D; Willumsen BM Function and regulation of ras. Annu. Rev. Biochem 1993, 62, 851–891. [DOI] [PubMed] [Google Scholar]
- (8).Ray A; Jatana N; Thukral L Lipidated proteins: Spotlight on protein-membrane binding interfaces. Prog. Biophys. Mol. Biol 2017, 128, 74–84. [DOI] [PubMed] [Google Scholar]
- (9).Cox A; Der CJ Farnesyltransferase inhibitors: promises and realities. Curr. Opin. Pharmacol 2002, 2, 388–393. [DOI] [PubMed] [Google Scholar]
- (10).Wu S; Zeng K; Wilson I; Balch W Structural insights into the function of the Rab GDI superfamily. Trends Biochem. Sci 1996, 21, 472–476. [DOI] [PubMed] [Google Scholar]
- (11).Hanzal-Bayer M; Renault L; Roversi P; Wittinghofer A; Hillig RC The complex of Arl2-GTP and PDEdelta: from structure to function. EMBO J. 2002, 21, 2095–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zhang H; Liu X-H; Zhang K; Chen C-K; Frederick JM; Prestwich GD; Baehr W Photoreceptor cGMP Phosphodiesterase δ Subunit (PDEδ) Functions as a Prenyl-binding Protein. J. Biol. Chem 2004, 279, 407–413. [DOI] [PubMed] [Google Scholar]
- (13).Nancy V; Callebaut I; El Marjou A; de Gunzburg J The δ Subunit of Retinal Rod cGMP Phosphodiesterase Regulates the Membrane Association of Ras and Rap GTPases. J. Biol. Chem 2002, 277, 15076–15084. [DOI] [PubMed] [Google Scholar]
- (14).Hanzal-Bayer M; Linari M; Wittinghofer A Properties of the Interaction of Arf-like Protein 2 with PDEδ. J. Mol. Biol 2005, 350, 1074–1082. [DOI] [PubMed] [Google Scholar]
- (15).Nussinov R; Tsai C-J; Chakrabarti M; Jang H A new view of Ras isoforms in cancers. Cancer Res. 2016, 76, 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Chakrabarti M; Jang H; Nussinov R Comparison of the conformations of KRAS isoforms, K-Ras4A and K-Ras4B, points to similarities and significant differences. J. Phys. Chem. B 2016, 120, 667–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Muratcioglu S; Jang H; Gursoy A; Keskin O; Nussinov R PDEδ Binding to Ras Isoforms Provides a Route to Proper Membrane Localization. J. Phys. Chem. B 2017, 121, 5917–5927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Dharmaiah S; Bindu L; Tran TH; Gillette WK; Frank PH; Ghirlando R; Nissley DV; Esposito D; McCormick F; Stephen AG; Simanshu DK Structural basis of recognition of farnesylated and methylated KRAS4b by PDEδ. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, E6766–E6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Linari M; Hanzal-Bayer M; Becker J The delta subunit of rod specific cyclic GMP phosphodiesterase, PDE δ, interacts with the Arf-like protein Arl3 in a GTP specific manner. FEBS Lett. 1999, 458, 55–59. [DOI] [PubMed] [Google Scholar]
- (20).Veltel S; Kravchenko A; Ismail S; Wittinghofer A Specificity of Arl2/Arl3 signaling is mediated by a ternary Arl3-effector-GAP complex. FEBS Lett. 2008, 582, 2501–2507. [DOI] [PubMed] [Google Scholar]
- (21).Martín-Gago P; Fansa EK; Klein CH; Murarka S; Janning P; Schürmann M; Metz M; Ismail S; Schultz-Fademrecht C; Baumann M; Bastiaens PIH; Wittinghofer A; Waldmann H A PDE6δ-KRas Inhibitor Chemotype with up to Seven H-Bonds and Picomolar Affinity that Prevents Efficient Inhibitor Release by Arl2. Angew. Chem., Int. Ed 2017, 56, 2423–2428. [DOI] [PubMed] [Google Scholar]
- (22).Lu S; Jang H; Gu S; Zhang J; Nussinov R Drugging Ras GTPase: a comprehensive mechanistic and signaling structural view. Chem. Soc. Rev 2016, 45, 4929–4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Lu S; Banerjee A; Jang H; Zhang J; Gaponenko V; Nussinov R GTP binding and oncogenic mutations may attenuate hypervariable region (HVR)-catalytic domain interactions in small GTPase K-Ras4B, exposing the effector binding site. J. Biol. Chem 2015, 290, 28887–28900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Vartak N; Bastiaens P Spatial cycles in G-protein crowd control. EMBO J. 2010, 29, 2689–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Cox AD; Der CJ; Philips MR Targeting RAS membrane association: Back to the future for anti-RAS drug discovery? Clin. Cancer Res. 2015, 21, 1819–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Gysin S; Salt M; Young A; McCormick F Therapeutic strategies for targeting ras proteins. Genes Cancer 2011, 2, 359–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zhou M; Wiener H; Su W; Zhou Y; Liot C; Ahearn I; Hancock JF; Philips MR VPS35 binds farnesylated N-Ras in the cytosol to regulate N-Ras trafficking. J. Cell Biol 2016, 214, 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ismail SA; Chen Y-X; Rusinova A; Chandra A; Bierbaum M; Gremer L; Triola G; Waldmann H; Bastiaens PIH; Wittinghofer A Arl2-GTP and Arl3-GTP regulate a GDI-like transport system for farnesylated cargo. Nat. Chem. Biol 2011, 7, 942–949. [DOI] [PubMed] [Google Scholar]
- (29).Webb B; Sali A Comparative Protein Structure Modeling Using MODELLER. Methods Mol. Biol 2014, 1137, 1–15. [DOI] [PubMed] [Google Scholar]
- (30).Brooks BR; Brooks CL 3rd; Mackerell AD Jr.; Nilsson L; Petrella RJ; Roux B; Won Y; Archontis G; Bartels C; Boresch S; et al. CHARMM: the biomolecular simulation program. J. Comput. Chem 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Klauda JB; Venable RM; Freites JA; O’Connor JW; Tobias DJ; Mondragon-Ramirez C; Vorobyov I; MacKerell AD Jr.; Pastor RW Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Jang H; Abraham SJ; Chavan TS; Hitchinson B; Khavrutskii L; Tarasova NI; Nussinov R; Gaponenko V Mechanisms of membrane binding of small GTPase K-Ras4B farnesylated hypervariable region. J. Biol. Chem 2015, 290, 9465–9477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Chavan TS; Jang H; Khavrutskii L; Abraham SJ; Banerjee A; Freed BC; Johannessen L; Tarasov SG; Gaponenko V; Nussinov R; Tarasova NI High-affinity interaction of the K-Ras4B hypervariable region with the Ras active site. Biophys. J 2015, 109, 2602–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid D; Villa E; Chipot C; Skeel RD; Kalé L; Schulten K Scalable molecular dynamics with NAMD. J. Comput. Chem 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Van Wart AT; Durrant J; Votapka L; Amaro RE Weighted Implementation of Suboptimal Paths (WISP): An optimized algorithm and tool for dynamical network analysis. J. Chem. Theory Comput 2014, 10, 511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Humphrey W; Dalke A; Schulten K VMD: visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- (37).Jang H; Muratcioglu S; Gursoy A; Keskin O; Nussinov R Membrane-associated Ras dimers are isoform-specific: K-Ras dimers differ from H-Ras dimers. Biochem. J 2016, 473, 1719–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Sun H; Li Y; Tian S; Xu L; Hou T Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys. Chem. Chem. Phys 2014, 16, 16719–16729. [DOI] [PubMed] [Google Scholar]
- (39).Wätzlich D; Vetter I; Gotthardt K; Miertzschke M; Chen Y-X; Wittinghofer A; Ismail S The interplay between RPGR, PDEδ and Arl2/3 regulate the ciliary targeting of farnesylated cargo. EMBO Rep. 2013, 14, 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Cukuroglu E; Gursoy A; Keskin O HotRegion: a database of predicted hot spot clusters. Nucleic Acids Res. 2012, 40, D829–D833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Nussinov R; Tsai C-J; Jang H Oncogenic Ras isoforms signaling specificity at the membrane. Cancer Res. 2018, 78, 593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Nussinov R; Tsai C-J; Jang H Is Nanoclustering essential for all oncogenic KRas pathways? Can it explain why wild-type KRas can inhibit its oncogenic variant? Semin. Cancer Biol 2018, DOI: 10.1016/j.semcancer.2018.01.002, in press. [DOI] [PubMed] [Google Scholar]
- (43).Muratcioglu S; Chavan TS; Freed BC; Jang H; Khavrutskii L; Freed RN; Dyba MA; Stefanisko K; Tarasov SG; Gursoy A; et al. GTP-dependent K-Ras dimerization. Structure 2015, 23, 1325–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Nan X; Tamgüney TM; Collisson EA; Lin L-J; Pitt C; Galeas J; Lewis S; Gray JW; McCormick F; Chu S Ras-GTP dimers activate the Mitogen-Activated Protein Kinase (MAPK) pathway. Proc. Natl. Acad. Sci. U.S.A 2015, 112, 7996–8001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Nussinov R; Tsai C-J; Jang H Oncogenic KRas mobility in the membrane and signaling response. Semin. Cancer Biol 2018, DOI: 10.1016/j.semcancer.2018.02.009, in press. [DOI] [PubMed] [Google Scholar]
- (46).Erwin N; Patra S; Dwivedi M; Weise K; Winter R Influence of isoform-specific Ras lipidation motifs on protein partitioning and dynamics in model membrane systems of various complexity. Biol. Chem 2017, 398, 547–563. [DOI] [PubMed] [Google Scholar]
- (47).Nussinov R; Tsai C-J; Muratcioglu S; Jang H; Gursoy A; Keskin O Principles of K-Ras effector organization and the role of oncogenic K-Ras in cancer initiation through G1 cell cycle deregulation. Expert Rev. Proteomics 2015, 12, 669–682. [DOI] [PubMed] [Google Scholar]
- (48).Schmick M; Vartak N; Papke B; Kovacevic M; Truxius DC; Rossmannek L; Bastiaens PIH KRas localizes to the plasma membrane by spatial cycles of solubilization, trapping and vesicular transport. Cell 2014, 157, 459–471. [DOI] [PubMed] [Google Scholar]
- (49).Chandra A; Grecco HE; Pisupati V; Perera D; Cassidy L; Skoulidis F; Ismail SA; Hedberg C; Hanzal-Bayer M; Venkitaraman AR; et al. The GDI-like solubilizing factor PDEδ sustains the spatial organization and signalling of Ras family proteins. Nat. Cell Biol 2011, 14, 148–158. [DOI] [PubMed] [Google Scholar]
- (50).Spiegel J; Cromm PM; Zimmermann G; Grossmann TN; Waldmann H Small-molecule modulation of Ras signaling. Nat. Chem. Biol 2014, 10, 613–622. [DOI] [PubMed] [Google Scholar]
- (51).Ozdemir ES; Jang H; Gursoy A; Keskin O; Li Z; Sacks DB; Nussinov R Unraveling the molecular mechanism of interactions of the Rho GTPases Cdc42 and Rac1 with the scaffolding protein IQGAP2. J. Biol. Chem 2018, 293, 3685–3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Chen Y-X; Koch S; Uhlenbrock K; Weise K; Das D; Gremer L; Brunsveld L; Wittinghofer A; Winter R; Triola G; Waldmann H Synthesis of the Rheb and K-Ras4B GTPases. Angew. Chem., Int. Ed 2010, 49, 6090–6095. [DOI] [PubMed] [Google Scholar]
- (53).Zhang X; Cao S; Barila G; Edreira MM; Wankhede M; Naim N; Buck M; Altschuler DL Cyclase-Associated Protein 1 (CAP1) is a prenyl-binding partner of Rap1 GTPase. J. Biol. Chem 2018, 293, 7659–7673. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.