

Abstract
Plasmodium falciparum proteasome (Pf20S) inhibitors are active against Plasmodium at multiple stages – erythrocytic stages, gametocyte stages, liver stages and gamete activation, indicating that selective Pf20S inhibitors possess the potential to be therapeutic, prophylactic and transmission-blocking antimalarials. Starting from a reported compound, we developed a noncovalent, macrocyclic peptide inhibitor of the malarial proteasome with high species selectivity and improved pharmacokinetic properties. The compound demonstrates specific, time-dependent inhibition of the β5 subunit of the Plasmodium falciparum proteasome, kills artemisinin-sensitive and artemisinin-resistant P. falciparum isolates in vitro and reduces parasitemia in humanized, P. falciparum-infected mice.
Keywords: malaria, proteasome, inhibitor, selectivity, therapy
Graphical Abstract
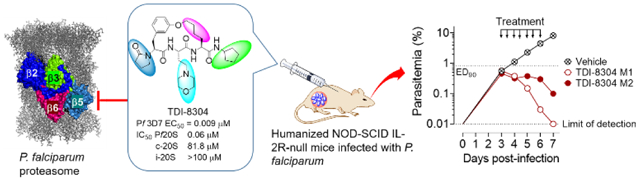
Pf20S inhibitors possess the potential to be therapeutic, prophylactic and transmission-blocking antimalarials. We present a novel and highly species-selective malaria proteasome inhibitor that potently reduced parasitemia in humanized mice infected with P. falciparum, the most deadly plasmodium parasites.
The spread of drug resistance threatens recent gains in malaria control even as the current burden of disease remains intolerable, with 228 million new cases and 405,000 deaths, primarily in children, estimated in 2018.[1] The limited range of clinically validated antimalarial targets and the danger of increasing resistance to current antimalarial regimens highlight the need for novel antimalarial agents. Proteasomes of pathogens have recently become targets of interest in tuberculosis, malaria, Chagas disease, leishmaniasis and schistosomiasis. The Plasmodium falciparum proteasome (Pf20S) has served as a promising target for parasite-selective proteasome inhibitors acting against erythrocytic, gametocyte, liver and mosquito stages, indicating that Pf20S inhibitors can potentially have therapeutic, prophylactic and transmission-blocking indications.[2] Given that human hematopoietic cells, plasma cells and plasmacytoid dendritic cells are highly susceptible to proteasome inhibition,[3] proteasome inhibitors for treatment of malaria must be highly selective for the malarial proteasome over the three proteolytic subunits of the human constitutive proteasome (c-20S) and the three proteolytic subunits of the human immunoproteasome (i-20S) to avoid toxicity and interference with immune responses.
We recently reported noncovalent Pf20S inhibitors that showed marked antimalarial activity against multiple stages of the parasites’ life cycle.[2d, 4] However, these inhibitors all demonstrated fast clearance in vivo (Figure S1). To identify a compound for use in mouse proof of concept studies, we explored an alternative scaffold for Pf20S-selective inhibitors. Bogyo and coworkers recently reported a macrocyclic peptide, compound 1, that shows potent proteasome inhibition, selectivity and effective antimalarial activity.[5] However, compound 1 suffers from poor solubility and fast clearance by liver microsomes (Table S1).
We conducted an extensive structure-activity relationship (SAR) study to improve the pharmacokinetic properties of this scaffold (Figure 1). The full SAR study will be described in a separate paper. In brief, starting from compound 1, we first explored substitution at P5 to reduce hydrogen bond donor count and improve passive permeability, which yielded compound 2. Compound 2 is highly potent against P. falciparum strain Pf 3D7 (EC50 1 nM) and has activity against c-20S (IC50 0.34 μM) and i-20S (IC50 > 34 μM) (Figure 1). However, the poor solubility and mouse microsomal stability of compound 2 needed additional optimization. Substitutions at P1, P3, and the tether were structure-guided using a docking workflow where macrocycles underwent extended conformation sampling followed by docking with a constraint to align the core of the query molecule with that of a reference pose model.[6] This pose model for the macrocycle series in Pf20S was initially developed for Compound 2 based on the acyclic analog bound in the yeast proteasome crystal structure (PDB: 3MG4) (Figure 2).[7] This docking workflow was implemented into LiveDesign for automated docking pose generation. We modified the P3 moiety to reduce LogD by replacing the phenyl with a morpholino group, and further modified the P1 moiety to reduce a lability to hydrolysis by replacing 2,2,2-trifluoroethylamino with cyclopropylmethyl, yielding compound 3. Compound 3 showed a comparable EC50 against P falciparum and demonstrated improved selectivity for c-20S (Figure 1). This optimization also improved metabolic stability and solubility (> 130 μg/mL). Further improvements in metabolic stability were realized by substitution at P1 with a cyclopentyl moiety and replacement of the biaryl ether with a mono phenyl alkyl chain. The final compound, TDI-8304, was highly potent against Pf 3D7 with marked selectivity for inhibition of Pf20S over human proteasomes (Figure 3A).
Figure 1.
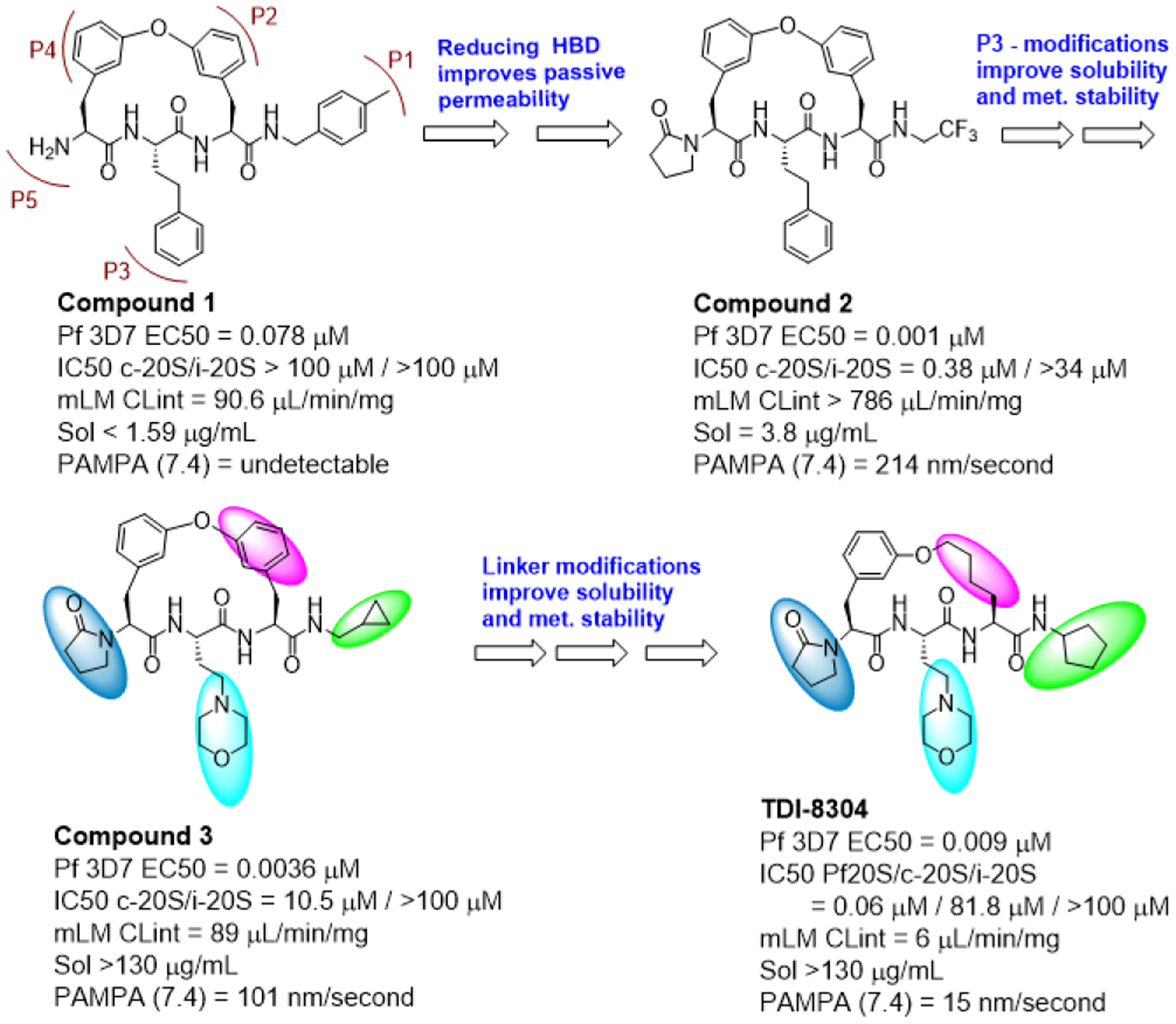
SAR evolution from compound 1 to TDI-8304.
Figure 2.
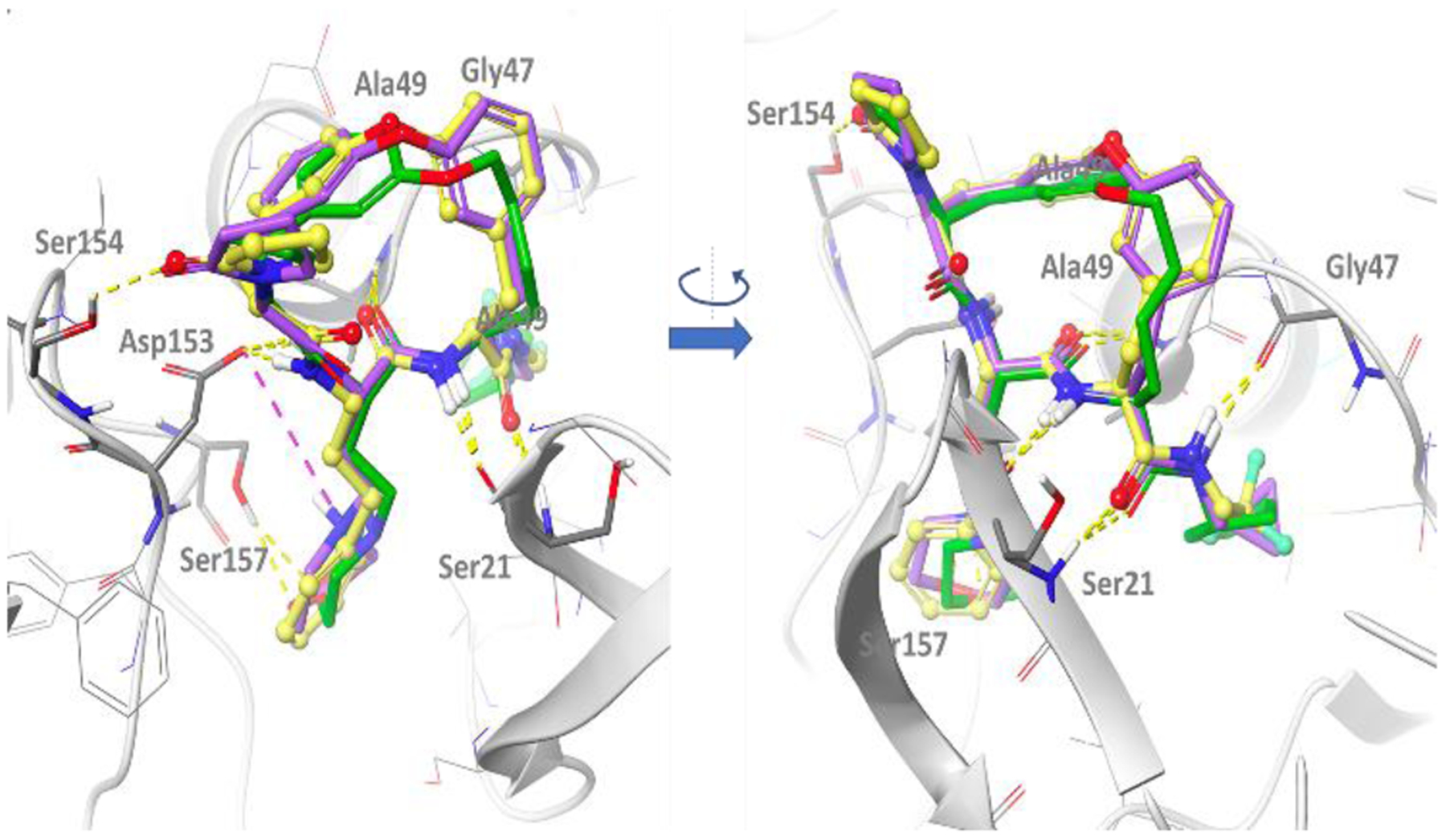
Docking model of macrocyclic peptides in Pf20S. The structure of Pf20S β5–6 was obtained by homology modeling based on yeast 20S β5–6 (3MG4), and then refined using molecular dynamics simulations. The amide bonds of all compounds form 5 well conserved hydrogen bonds with the backbone of Ser21, Gly47 and Ala49 as well as the side chain of Asp153. Additionally, the pyrrolidone in P5 of compounds 2, 3 and TDI-8304 forms a hydrogen bond with Ser154, and the morpholino oxygen in compound 3 and TDI-8304 form a hydrogen bond with hydroxyl group of Ser157 in β6. For compound 3, the protonated form of the morpholine has been represented to illustrate the possible salt bridge with Asp153, but the neutral form might predominate at relevant pH (predicted pKa ~ 6.6). Compound 2 is shown in yellow, compound 3 in purple and TDI-8304 in green.
Figure 3.
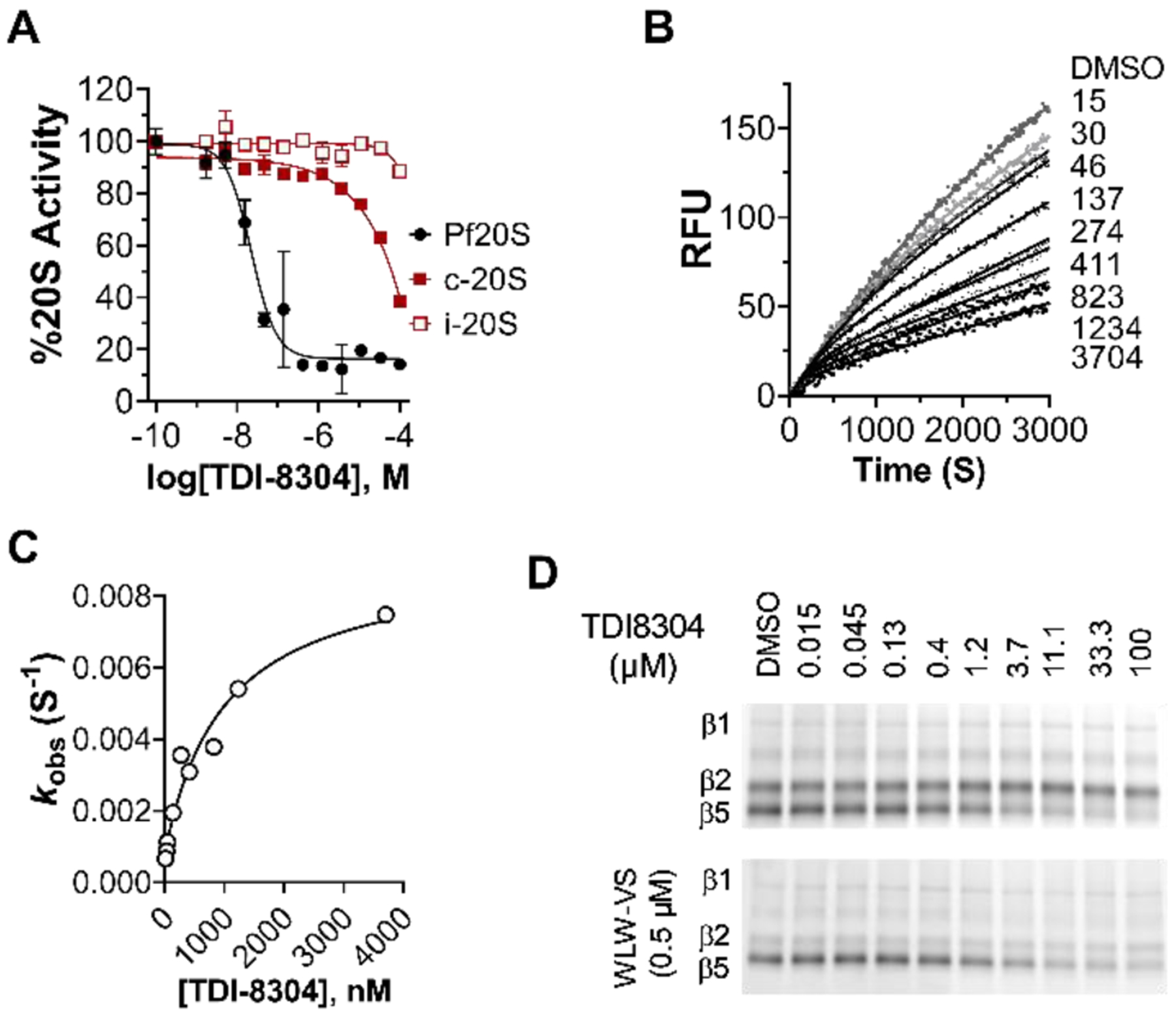
TDI-8304 selectively inhibits Pf20S. A) Dose-dependent inhibition of chymotryptic β5 subunits of Pf20S, human c-20S and i-20S. B) Time courses of inhibition of Pf20S β5 by TDI-8304 at indicated concentrations (nM). C) Plot of kobs values obtained from the time course curves with varied TDI-8304 concentrations yields KIapp = 1007 nM, Ki* = 89.6 nM and koff = 0.0008 S−1. D) TDI-8304 specifically inhibited Pf20S β5 but not β2 or β1, from being labeled by MV151, either in the absence of a Pf20S β2 specific inhibitor WLW-VS3 (top) or in the presence of WLW-VS (bottom).
To determine the inhibition modality of TDI-8304, we monitored the hydrolysis of the proteasome substrate suc-LLVY-AMC by Pf20S in the presence of 0.5 μM β2-specific inhibitor WLW-VS and TDI-8304 at indicated concentrations (Figure 3B). TDI-8304 demonstrated time-dependent inhibition with an induced-fit mechanism.[8] The hyperbolic correlation of kobs as a function of TDI-8304 concentrations was fit to equation kobs = k6 + (k5 / (1 + (Kiapp / [I]) and the values of Kiapp 1,007 nM, Ki*app 89.6 nM and t1/2 14.4-minute half-life for the Pf20S:TDI-8304 complex were calculated by equation Ki* = Ki / (1 + K5 / k6) (Figure 3C). TDI-8304 dose-dependently blocked labelling of Pf20S β5, but not of Pf20S β1 or β2 (Figure 3D), by MV151, suggesting that TDI-8304 is a highly specific inhibitor of Pf20S β5.
TDI-8304 is nontoxic to HepG2 cells (Figure 4A) and potent not only against Pf 3D7 (Figure 4A) but also against the P. falciparum lab strain Dd2 and two Dd2-derived strains that are resistant to our previous noncovalent proteasome inhibitors PKS21004 and TDI4258 by virtue of mutation Pf20Sβ6A117D in the adjacent β6 subunit (which changes the conformation of S3 and S4 binding pockets of the β5 active site) and mutation Pf20Sβ5A49S in the active site of the β5 catalytic domain, respectively.[2d, 4] The EC50 values of TDI-8304 against Dd2 and Dd2β5A49S were comparable, suggesting that its binding mode, which is stabilized by its macrocyclic tether structure, is unaffected by the mutation. However, an ~18-fold reduction in potency against Dd2β6A117D (Figure 4B) suggests that the conformational changes caused by the β6A117D mutation affect the binding of TDI8304 more than the conformational changes caused by the β5A49S mutation. Supporting on-target whole-cell activity, a 6-hour exposure to TDI-8304 led to accumulation of polyubiquitinated proteins in Pf trophozoites (Figure 4C). TDI-8304 showed comparable activity against Pf strains HB3 (chloroquine-sensitive), 3663 (ART-sensitive) and 4884 (ART-resistant)[9] (Figure 4D), indicating no apparent cross-resistance.
Figure 4.
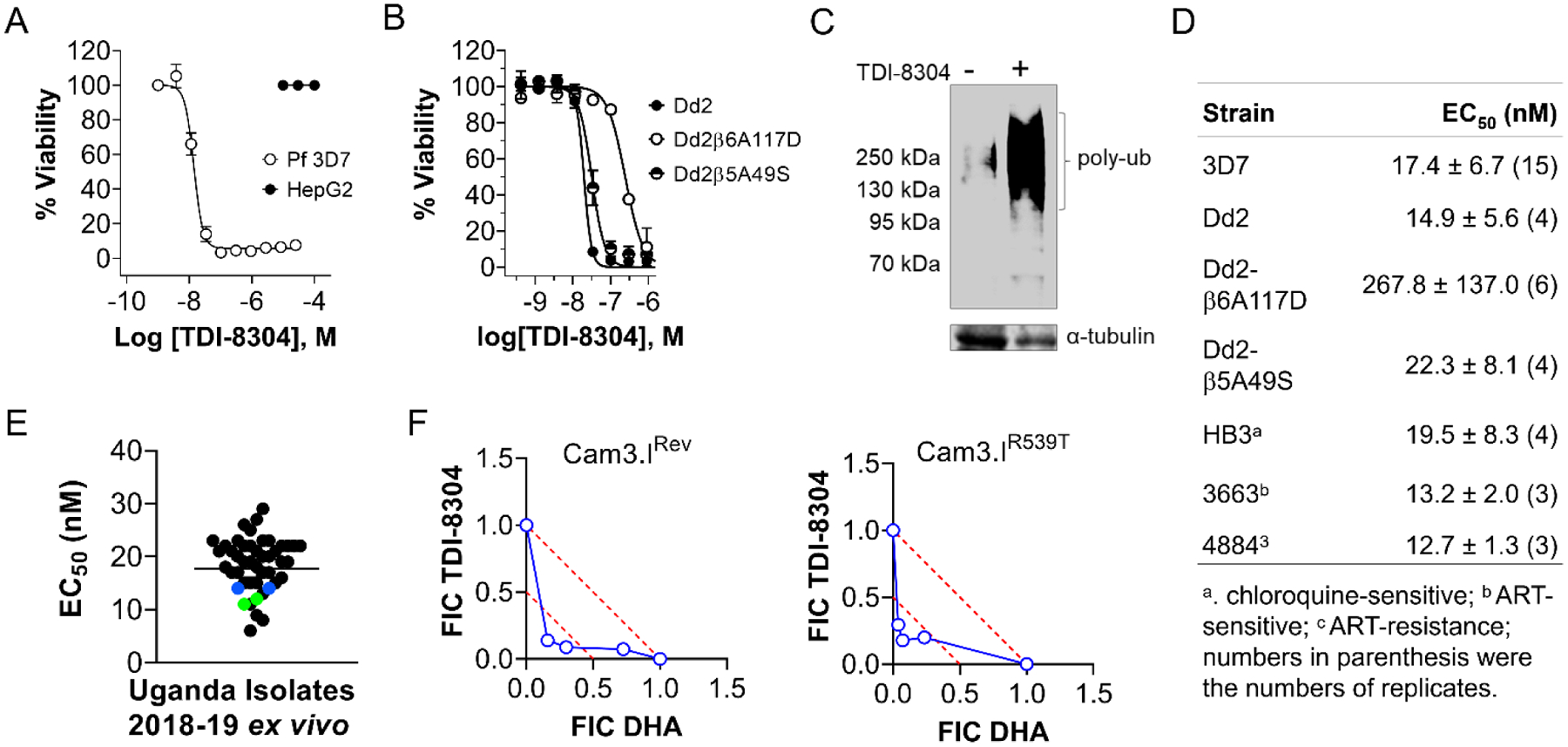
TDI-8304 inhibits growth of erythrocytic stage P. falciparum. A) TDI-8304 potently inhibited Pf3D7 growth (open black circles) but is not cytotoxic for HepG2 human hepatoma cells (solid black circles); B) TDI-8304 inhibits growth of Pf Dd2 and two proteasome inhibitor-resistant strains, Pf Dd2β6A117D and Pf Dd2β5A49S. C) TDI-8304 at 1 μM led to accumulation of polyubiquitinated proteins after treatment of P. falciparum schizonts for 6 hours. D) EC50 values of TDI-8304 against P. falciparum strains with various drug resistance profiles; E) Ex vivo activity of TDI-8304 against 38 clinical isolates from Uganda in years 2018 and 2019. The geometric mean EC50 was 18 nM. Each black spot represents a single P. falciparum isolate. Blue and green spots represent Dd2 and 3D7 laboratory controls, respectively. F) In vitro synergy of TDI-8304 and DHA against artemisinin-sensitive P. falciparum Dd2 Cam3.IRev and artemisinin-resistant Cam3.IR539T. Data were average of two independent experiments and each in duplicates. FIC: fractional inhibitory concentration.
TDI-8304 was further tested for ex vivo activity against fresh falciparum isolates from 38 malaria patients in Uganda. The EC50s ranged from 5–30 nM, with a mean of 18 nM, in agreement with the results from P. falciparum laboratory strains (Figure 4E). There was no observed association between sensitivities to TDI-8304 and chloroquine resistance among the clinical isolates.
Several studies have established that proteasome inhibitors are synergistic with ARTs.[2a, 2b, 2d] To investigate the synergy of TDI-8304 with ART, we tested TDI-8304 in combination with dihydroartemisinin (DHA) using a modified ring-stage survival assay (RSA) with both artemisinin-sensitive Pf Cam3.IRev and artemisinin-resistant Pf Cam3.IR539T in a multidrug-resistant Dd2 strain background. We exposed early (1- to 3-h) ring-stage parasites to a 3-h pulse of DHA and continuous TDI-8304 for further 69 h, as described.[2d] Parasitemia was measured at 72 h post initial exposure. Isobologram analyses clearly demonstrated synergistic killing of artemisinin–sensitive (Figure 4F Left) and artemisinin-resistant (Figure 4F right) parasites by the combination of TDI-8304 and DHA.
TDI-8304 is metabolically stable upon incubation with human, mouse and rat microsomes and cell-permeable (Figure 5A). It inhibited by less than 10% a panel of CYP enzymes, and showed no cytotoxicity to HepG2 cells. TDI-8304 further showed no inhibition of the hERG (Ether-à-go-go) channel, implying low risk of cardiac arrhythmia through action on this ion channel. TDI-8304 is rapidly cleared when administrated to mice by i.v. (Figure 5B) or p.o. routes (Figure S1), but shows prolonged clearance when administrated s.c. (Figure 5B). This allowed twice-daily dosing (b.i.d) to afford sufficient exposure for efficacy experiments in mice. We tested TDI-8304 in a humanized mouse model of Pf infection[10] with s.c. administration at 100 mg/kg b.i.d., from days 3 – 6 post infection and monitoring parasitemia from day 3 until day 7 (Figure 5C). TDI-8304 reduced the parasitemia by a factor of 2 log10 in one mouse and cleared the parasitemia in another mouse. Pharmacokinetics of TDI-8304 from the infected SCID mice during the course of the assay were also determined (Table S2), and the data showed that the whole blood concentration of TDI-8304 was constantly greater than the EC90 (29 nM) of TDI-8304 in both mice, except for one data point at the 24h time-point in mouse 2 (Figure S2), in agreement with the observed reduction of parasitemia in infected mice.
Figure 5.
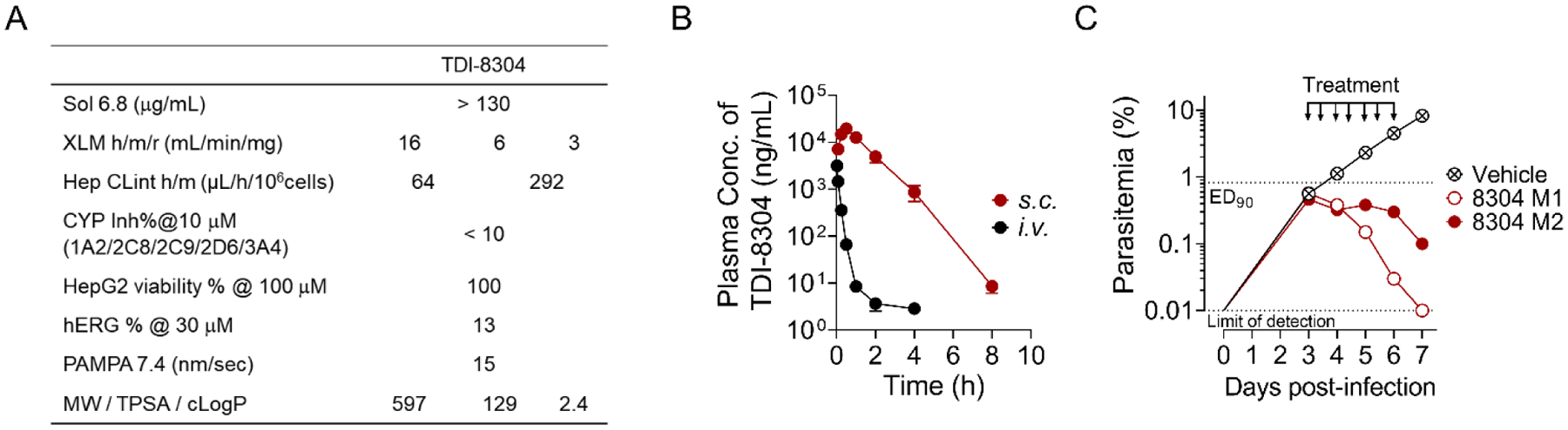
Pharmacokinetic properties of TDI-8304 and its efficacy in humanized, P. falciparum-infected mice. A) PK properties of TDI-8304. B) In vivo PK of TDI-8304 administrated i.v. (1 mg/kg) or s.c. (100 mg/kg). C) Parasitemia reduction of P. falciparum Pf3D70087/N9 in NOD-SCID IL-2R-null mice transfused with human erythrocytes. TDI-8304 was given twice-daily by s.c. (100 mg/kg) on days 3 through 6. Two mice were used in each group, each serially sampled on days 3 to 7. Percentage parasitemia was calculated by acquiring a minimum number of 500 parasitized erythrocytes.
In conclusion, we developed a preclinical and species-selective P. falciparum proteasome inhibitor with marked antimalarial activity in a humanized mouse model of P. falciparum infection. To our knowledge, this is the first noncovalent Pf20S inhibitor shown to have anti-malaria activity in Pf infected mice.
Supplementary Material
Acknowledgements
This work is supported by NIH grants R01AI143714 (G.L.), R21AI123794 (G.L. and L.A.K.), AI139179 (P.J.R. and R.A.C.), and T37MD003407 (S.C.); The Brockman Foundation (L.A.K.); Department of Medicine, Weill Cornell Medicine Seed fund (L.A.K.); Tri-Institutional Therapeutics Discovery Institute; Weill Cornell Medicine Matching Fund (G.L.); Milstein Program in Chemical Biology and Translational Medicine; and Medicines for Malaria Venture RD/15/0001 (P.J.R. and R.A.C.). We gratefully acknowledge in-kind support of the Tri-Institutional Therapeutics Discovery Institute (TDI), a 501(c)(3) organization. TDI receives financial support from TDI’s owners (Memorial Sloan Kettering Cancer Center, The Rockefeller University and Weill Cornell Medicine), Takeda Pharmaceutical Company, and generous contributions from Mr. Lewis Sanders, Mr. Howard Milstein and other philanthropic sources. We thank Drs. Stacia Kargman and Leigh Baxt at TDI for their constructive suggestions. The Department of Microbiology and Immunology is supported by the William Randolph Hearst Foundation.
References
- [1].WHO, World Health Organization, Geneva: World Health Organization, 2018. [Google Scholar]
- [2].a) Dogovski C, Xie SC, Burgio G, Bridgford J, Mok S, McCaw JM, Chotivanich K, Kenny S, Gnadig N, Straimer J, Bozdech Z, Fidock DA, Simpson JA, Dondorp AM, Foote S, Klonis N, Tilley L, PLoS Biol 2015, 13, e1002132; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li H, O’Donoghue AJ, van der Linden WA, Xie SC, Yoo E, Foe IT, Tilley L, Craik CS, da Fonseca PC, Bogyo M, Nature 2016, 530, 233; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhang M, Wang C, Otto TD, Oberstaller J, Liao X, Adapa SR, Udenze K, Bronner IF, Casandra D, Mayho M, Brown J, Li S, Swanson J, Rayner JC, Jiang RHY, Adams JH, Science 2018, 360; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Kirkman LA, Zhan W, Visone J, Dziedziech A, Singh PK, Fan H, Tong X, Bruzual I, Hara R, Kawasaki M, Imaeda T, Okamoto R, Sato K, Michino M, Alvaro EF, Guiang LF, Sanz L, Mota DJ, Govindasamy K, Wang R, Ling Y, Tumwebaze PK, Sukenick G, Shi L, Vendome J, Bhanot P, Rosenthal PJ, Aso K, Foley MA, Cooper RA, Kafsack B, Doggett JS, Nathan CF, Lin G, Proc Natl Acad Sci U S A 2018, 115, E6863–E6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Hirai M, Kadowaki N, Kitawaki T, Fujita H, Takaori-Kondo A, Fukui R, Miyake K, Maeda T, Kamihira S, Miyachi Y, Uchiyama T, Blood 2011, 117, 500; [DOI] [PubMed] [Google Scholar]; b) Neubert K, Meister S, Moser K, Weisel F, Maseda D, Amann K, Wiethe C, Winkler TH, Kalden JR, Manz RA, Voll RE, Nat Med 2008, 14, 748; [DOI] [PubMed] [Google Scholar]; c) Santos R. d. L. A., Bai L, Singh PK, Murakami N, Fan H, Zhan W, Zhu Y, Jiang X, Zhang K, Assker JP, Nathan CF, Li H, Azzi J, Lin G, Nat Commun 2017, 8, 1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zhan W, Visone J, Ouellette T, Harris JC, Wang R, Zhang H, Singh PK, Ginn J, Sukenick G, Wong TT, Okoro JI, Scales RM, Tumwebaze PK, Rosenthal PJ, Kafsack BFC, Cooper RA, Meinke PT, Kirkman LA, Lin G, J Med Chem 2019, 62, 6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Li H, Tsu C, Blackburn C, Li G, Hales P, Dick L, Bogyo M, J Am Chem Soc 2014, 136, 13562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sindhikara D, Spronk SA, Day T, Borrelli K, Cheney DL, Posy SL, J Chem Inf Model 2017, 57, 1881. [DOI] [PubMed] [Google Scholar]
- [7].Blackburn C, Gigstad KM, Hales P, Garcia K, Jones M, Bruzzese FJ, Barrett C, Liu JX, Soucy TA, Sappal DS, Bump N, Olhava EJ, Fleming P, Dick LR, Tsu C, Sintchak MD, Blank JL, Biochem J 2010, 430, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Copeland RA, in Evaluation of enzyme inhibitors in drug discovery: A guide for medicinal chemists and pharmacologists, Second Edition ed., John Wiley & Sons, Inc., Hoboken, 2013. [PubMed] [Google Scholar]
- [9].Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois AC, Khim N, Kim S, Duru V, Bouchier C, Ma L, Lim P, Leang R, Duong S, Sreng S, Suon S, Chuor CM, Bout DM, Menard S, Rogers WO, Genton B, Fandeur T, Miotto O, Ringwald P, Le Bras J, Berry A, Barale JC, Fairhurst RM, Benoit-Vical F, Mercereau-Puijalon O, Menard D, Nature 2014, 505, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Angulo-Barturen I, Jimenez-Diaz MB, Mulet T, Rullas J, Herreros E, Ferrer S, Jimenez E, Mendoza A, Regadera J, Rosenthal PJ, Bathurst I, Pompliano DL, Gomez de las Heras F, Gargallo-Viola D, PLoS One 2008, 3, e2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.