

Abstract
Ifitm3 was previously identified as an endosomal protein that blocks viral infection1–3. Studying clinical cohorts of B-cell leukemia and lymphoma patients, we identified IFITM3 as a strong predictor of poor outcome. In normal resting B-cells, Ifitm3 was minimally expressed and mainly localized in endosomes. However, B-cell receptor (BCR) engagement induced expression of Ifitm3 and phosphorylation at Y20, resulting in accumulation at the cell surface. In B-cell leukemia, oncogenic kinases phosphorylate IFITM3-Y20, causing constitutive plasma membrane localization. Ifitm3ˉ/ˉ naïve B-cells developed at normal numbers; however, germinal center formation and production of antigen-specific antibodies were compromised. Oncogenes that induce development of leukemia and lymphoma failed to transform Ifitm3ˉ/ˉ B-cells. Conversely, the phospho-mimetic IFITM3-Y20E induced oncogenic PI3K-signaling and initiated transformation of pre-malignant B-cells. Mechanistic experiments revealed that Ifitm3 functions as PIP3-scaffold and central amplifier of PI3K signaling. PI3K signal-amplification depends on Ifitm3 scaffolding PIP3-accumulation via two lysine residues (K83 and K104) in its conserved intracellular loop. In Ifitm3ˉ/ˉ B-cells, lipid rafts were depleted of PIP3, resulting in defective expression of >60 lipid raft-associated surface receptors, impaired BCR-signaling and cellular adhesion. We conclude that phosphorylation of IFITM3 upon B-cell antigen-encounter induces a dynamic switch from antiviral effector functions in endosomes to a PI3K-amplification loop at the cell surface. IFITM3-dependent amplification of PI3K-signaling in part downstream of the BCR is critical to enable rapid expansion of B-cells with high affinity to antigen. In addition, multiple oncogenes depend on IFITM3 to assemble PIP3-dependent signaling complexes and amplify PI3K-signaling for malignant transformation.
Interferon-induced transmembrane protein 3 (IFITM3) is a small antiviral membrane protein (133 amino acids) with activity against influenza A virus (IAV), SARS coronavirus (SARS-CoV) and many other viruses1–5. Endocytosis and preferential endosomal localization of IFITM3 are mediated by its N-terminal AP2 sorting motif (YEML)1. Phosphorylation of this motif (Y20) by Src-kinases prevents not only endocytosis but also proteasomal degradation6–7. Individuals with the IFITM3 SNP rs12252-C allele show increased morbidity and mortality from H1N1 strains of IAV8 and SARS-CoV-24. IFITM3 SNP rs12252-C was predicted to encode a 21-amino acid truncation of the N-terminus including Y20 through mRNA splicing, although a recent transcript analysis found no evidence for such processed mRNA9. IFITM3 likely functions as an antiviral effector molecule by limiting viral fusion through increasing ‘rigidity’ of endosomal cell membranes10–11. IFITM3 is overexpressed in multiple cancer types, and its expression levels correlated with histopathological grading and staging12–13. Although IFITM3-dependent antiviral effector functions are restricted to endosomal compartments14, we report here on IFITM3-dependent functions at the cell surface of B-cells where IFITM3 functions as amplifier of PI3K signaling.
IFITM3 expression is induced by B cell receptor engagement and PI3K-activation
While minimally expressed in normal resting B-cells, BCR-engagement and acute activation of the PI3K pathway strongly induced expression of Ifitm3 (Extended Data Figure 1a–c). Expression of IFITM proteins was recently established as a diagnostic biomarker of high-risk B-cell lineage acute lymphoblastic leukemia (B-ALL) expressing the oncogenic BCR-ABL1 tyrosine kinase or related kinases15, while BCR-ABL1 kinase inhibition decreased IFITM3 expression levels (Extended Data Figure 1d). IFITM3 mRNA levels were generally higher in B-cell malignancies compared to their normal counterparts (Extended Data Figure 1f–g) and higher than median IFITM3 mRNA levels were associated with poor clinical outcomes in multiple clinical cohorts of patients with B-cell malignancies, including pediatric and adult B-ALL and mantle cell lymphoma (Extended Data Figure 1h–l). IFITM3 was previously identified as a transcriptional target of the of the transcriptional repressor IKZF116, suggesting that high expression levels of IFITM3 may be the result of genetic lesions of IKZF1. Our results confirmed that reconstitution of IKZF1 suppressed IFITM3 expression while inducible deletion of Ikzf1 relieved transcriptional repression (Extended Data Figure 1m–n). A multivariate analysis of risk factors in B-ALL, however, showed that high mRNA levels of IFITM3 were an independent predictor of poor outcome, regardless of IKZF1 deletion (Extended Data Figure 1o). We next examined how genetic deletion of Ifitm3 affects normal B-cell development. Surface expression of Cd19, a common B-cell surface receptor that promotes Src-kinase and PI3K signaling17, was substantially diminished on Ifitm3ˉ/ˉ pre-B-cells (Figure 1a). Consistent with increased apoptosis and reduced proliferation, Myc and Bcl2 expression and PI3K signaling were reduced in Ifitm3ˉ/ˉ pre-B-cells (Figure 1a–c). The development of resting Ifitm3ˉ/ˉ B-cell populations was largely unperturbed (Extended Data Figure 2), while B-1 cell and marginal zone B-cell (MZB) populations were decreased (Figure 1d, Extended Data Figure 2). To assess B-cell-specific effects of Ifitm3 during humoral immune responses, we performed adoptive transfer experiments: Ifitm3+/+ or Ifitm3ˉ/ˉ splenic B-cells were transferred into congenic μMT-mice that lack endogenous mature B-cells. After reconstitution of mature B-cell development, recipient μMT-mice were immunized with NP-KLH. Upon immunization, μMT-mice reconstituted with Ifitm3+/+ B-cells developed ~5% NP-specific GC-B cells, which were largely absent in μMT-mice reconstituted with Ifitm3ˉ/ˉ B-cells (Figure 1e–h). Likewise, the number of follicles in the spleen, PNA+ GC-B cells, and production of IgM, IgG1 and IgG2b antibodies were reduced in the absence of Ifitm3 (Figure 1i). These abnormalities in Ifitm3-deficient B-cells phenocopied B-cell defects upon deletion of Pik3cd18 and Cd1919.
Figure 1: Ifitm3 is essential for B-cell activation and affinity maturation in germinal centers.

a, CD19 surface expression (top), cell cycle progression (middle; percentages in S phase) and cell viability (bottom; percentages of Annexin V+ 7-AAD+ cells) were measured. b, Number of viable Ifitm3+/+ and Ifitm3−/− pre-B cells were counted at times indicated. c, Levels of phospho-Akt-S473, Akt, Myc, p53, p21 and Bcl2 were assessed in Ifitm3+/+ and Ifitm3−/− pre-B cells. d, Splenic B-cells from Ifitm3+/+ and Ifitm3−/− mice (n=5) were analyzed for CD21high CD23low/− marginal zone B-cells (MZB), peritoneal cavity B-cells for Mac-1+ IgM+ B-1, and CD5+ IgM+ B-1a cells. e, Splenic B-cells from Ifitm3+/+ and Ifitm3−/− mice were adoptively transferred to μMT recipient mice (n=10) followed by immunization with 0.5 mg of NP-KLH or Vehicle (Veh). Spleens were collected on day 12 after immunization and subjected to immunofluorescence staining of tissue sections with B220, CD3 and peanut agglutinin (PNA). f-g, Splenocytes harvested from μMT mice (n=10) were analyzed by flow cytometry 12 days post immunization for CD95, GL7 and NP to identify NP-specific GC-B cells. Relative fractions (f), absolute numbers (g), and representative flow cytometry plots (h) are shown. i, Levels of serum immunoglobulin isotypes in μMT recipient mice transplanted with Ifitm3+/+ or Ifitm3−/− B-cells are shown before and after immunization (n=10; day 12). Serum levels of IgM, IgG1 and IgG2b were determined by ELISA. a-c, For gel source data, see Supplementary Fig. 1. b,f–g,i, Mean ± s.d. indicated, significance determined by two-tailed t-test.
Ifitm3 is essential for oncogenic PI3K- and Src-kinase signaling and B-cell transformation
To elucidate a potential contribution of Ifitm3 to oncogenic signaling, we studied transformation of Ifitm3+/+ and Ifitm3ˉ/ˉ pre-B cells by BCR-ABL1 and NRASG12D (Figure 2a–b). As in normal B-cells, Ifitm3ˉ/ˉ B-ALL cells showed multiple defects in survival and proliferation and markedly reduced ability to form colonies in semi-solid methylcellulose (Figure 2b–c). Consistent with a 70-fold reduction of the frequency of leukemia-initiating cells (LIC; P=3.7e-07), Ifitm3ˉ/ˉ B-ALL cells failed to initiate overt leukemia in immunodeficient transplant recipient mice (Figure 2d–e). Similar to B-ALL cells, deletion of IFITM3 in mantle cell lymphoma (MCL) cells reduced competitive fitness and proliferation (Figure 2f). Both murine Ifitm3ˉ/ˉ B-cells and human IFITM3-deficient MCL cells showed defective Ca2+-release in response to BCR-engagement and partially lost CD19 surface expression (Extended Data Figure 2c–d). In addition, BCR-engagement in IFITM3ˉ/ˉ MCL cells failed to induce processive signal amplification between CD19 and LYN17, since CD19 was not phosphorylated and did not interact with LYN. IFITM3-deficient MCL cells expressed lower levels of LYN and failed to activate Src-kinases in response to BCR-engagement (Extended Data Figure 2e). RNA-seq gene expression studies revealed upregulation of Ifitm1, presumably to compensate for loss of Ifitm3. Gene set enrichment analyses showed that Ifitm3-deficiency was associated with reduced expression of components of the BCR-signaling pathway and a phenotype reminiscent of B-cell anergy (Extended Data Figure 3a–c).
Figure 2: Essential role of Ifitm3 in oncogenic signaling and B-cell transformation.

a, CD19 surface expression (top), cell cycle progression (middle; percentages in S phase) and cell viability (bottom; percentages of Annexin V+ 7-AAD+ cells) were measured. b, Ifitm3+/+ and Ifitm3−/− BCR-ABL1 or NRASG12D B-ALL cells were assayed for levels of phospho-Akt-S473, Akt, Myc, p53, p21 and Bcl2 (n=3) and c, plated in semi-solid methylcellulose. d, Kaplan-Meier analyses of NSG recipient mice injected with indicated numbers of Ifitm3+/+ and Ifitm3−/− BCR-ABL1 (left) or NRASG12D (right) B-ALL cells (n=5) e, Frequencies of leukemia-initiating cells (LIC) estimated with extreme limiting dilution analysis (ELDA; 90% CI; likelihood ratio test). f, Jeko1 mantle cell lymphoma (MCL) cells expressing doxycycline (Dox)-inducible Cas9 were transduced with IFITM3-targeting or non-targeting (NT) sgRNAs. Enrichment or depletion of targeted cells (Cas9+gRNA+) monitored by flow cytometry upon Dox-treatment (mean±s.d.), and IFITM3 levels measured by Western blot. g, Pre-malignant LSL-BcrBCR-ABL1 x Mb1-Cre pre-B cells expressing IFITM3, IFITM3Y20E (Y20E) or EV were plated for colony forming assays (7 days; representative images shown at ×1 (top) and ×10 (bottom) magnification). h, Survival analyses (P=0.0001, log-rank test) of congenic recipient mice transplanted with LSL-BcrBCR-ABL1 x Mb1-Cre B-cells transduced with EV, IFITM3 or Y20E (n=7). i, Engraftment and expansion of luciferase-labeled leukemia cells were monitored by luciferase bioimaging at times indicated. j, Effects of IFITM3 or IFITM3Y20E on oncogenic signaling in LSL-BcrBCR-ABL1 x Mb1-Cre B-cell precursors measured by Western blot and compared to EV. Phosphorylation of Cd19-Y531, Src/Lyn-Y416 and Akt -S473 were examined. a-c,f-g,j, For gel source data, see Supplementary Fig. 1. c,g, Two-tailed t-test.
IFITM3 Y20-phosphorylation initiates an amplification loop of oncogenic signaling
Combinations of surface and intracellular staining revealed a cytoplasmic localization of the N-terminus with a short extracellular portion of the C-terminus (Extended Data Figure 3d). Previous studies showed that phosphorylation of Ifitm3-Y20 prevents both endocytosis and degradation6–7. Phosphomimetic Y20E-Ifitm3 was constitutively localized at the cell surface. Likewise, phosphorylation by BCR-ABL1 induced IFITM3-accumulation at the cell membrane, which was sensitive to BCR-ABL1/Src-kinase inhibition. (Extended Data Figure 3e). Hence, phosphorylation of Y20 induces a dynamic switch from antiviral functions in endosomes to BCR- or oncogene-signaling at the cell surface (Extended Data Figure 3e–f). To test whether IFITM3-Y20 phosphorylation and constitutive membrane localization are sufficient to enable oncogenic signaling, we overexpressed wild-type and Y20E-IFITM3 in pre-malignant B-cells carrying a BCR-ABL1-knockin allele, which did not readily give rise to leukemia20. Expression of Y20E-IFITM3 increased the ability of pre-B cells to form colonies by 129-fold (P=2.1e-08; Figure 2g), induced oncogenic signaling and initiated fatal leukemia in congenic transplant recipients (Figure 2h–j). To elucidate how membrane-bound Y20E-IFITM3 promotes malignant transformation, we performed phosphoproteomic analyses of IFITM3-Y20 signaling in patient-derived B-ALL cells. Consistent with processive signal amplification17, activation of IFITM3-Y20 induced phosphorylation of CD19 on multiple residues including Y531, which mediates PI3K-activation. In addition, IFITM3-Y20 engaged multiple components of the BCR and integrin receptor signaling pathways (Figure 3a, Extended Data Figure 4a), which converge with CD19 at the level of PI3K-activation. A comprehensive analysis of phosphosites showed significant enrichment of phosphorylation in the PI3K, BCR and integrin receptor pathways, which we confirmed for Src, CD19 and PI3K-signaling by Western blot (Figure 3a–c). IFITM3-Y20E required crosslinking by a full antibody, since engagement of monomeric IFITM3-Y20 using F(ab) fragments failed to elicit PI3K-activation (Extended Data Figure 4b). Since PI3K-signaling in B-ALL cells increased Ifitm3 mRNA levels by 10-fold and protein levels by >50-fold, these observations suggest a feed forward loop of signal amplification between IFITM3 and PI3K signaling (Extended Data Figure 1b–e). Comparable to effects on BCR-signaling and CD19 surface expression in B-cells, genetic deletion of IFITM3 in human Jurkat T-cells compromised TCR-signaling and CD3 surface expression (Extended Data Figure 4c), suggesting that IFITM3 may play a similar role in T-cells.
Figure 3: IFITM3 links components of the BCR and integrin receptor pathways to PI3K signaling.

a, Phosphorylated protein levels in PDX2 B-ALL cells transduced with HA-tagged IFITM3Y20E or empty vector (EV) control identified by mass spectrometry upon IFITM3-crosslinking with anti-HA antibodies. b, Feature set enrichment analysis (FSEA) for phosphorylated proteins in the BCR-(red), integrin (blue) and PI3K- (gray) pathways, ranked by log2-fold change (Kolmogorov-Smirnov). c, Levels of phospho-SRC-Y416, LYN, phospho-CD19-Y531, CD19, phospho-AKT-S473 and AKT assessed in ICN12 cells expressing IFITM3-HA, IFITM3Y20E-HA or EV-HA upon IFITM3-crosslinking. d-e, Interactomes of BirA-IFITM3Y20E or EV control expressed in PDX2 B-ALL (d) and Jeko1 MCL (e) cells. f-g, Validation of IFITM3 interacting proteins by anti-FLAG co-immunoprecipitation in PDX2 cells transduced with FLAG-IFITM3 or FLAG-EV, followed by Western blotting for validation of interacting proteins and STAT5 and eIF4E as specificity controls (g). h-i, PLA in Jeko1 cells upon BCR-stimulation, assessed for proximity of CD79B to IFITM3. Representative images with PLA signal (red), nuclei (DAPI) and plasma membrane (WGA; green). (h). Quantitation of PLA signals per cell (two-tailed t-test). j, Structural model of IFITM3-mediated signal amplification between CD19 and LYN. In resting B-cells (left), IFITM3 is localized in endosomes. Upon antigen-encounter (right), LYN-mediated phosphorylation of IFITM3 induces membrane translocation, acting as a scaffold for CD19 and LYN in proximity with BCR-molecules. BCR-CD19-IFITM3 complexes form clusters for PI3K-activation and accumulation of PIP3 in lipid rafts. a-i, n=3, For gel source data, see Supplementary Fig. 1. a,d,e, Row-scaled protein abundance ranked by fold change, key proteins of interest are highlighted.
IFITM3 links components of the BCR and integrin receptor pathways to PI3K signaling
By enriching for cell surface proteins using N-linked glycosylation-site biotin labeling, we identified 65, mostly lipid rafts-associated, surface receptors that were downregulated in Ifitm3ˉ/ˉ B-ALL cells, including Cd19, BCR-associated and adhesion receptors. Only 5 surface proteins were upregulated, including the Ifitm-family member Ifitm2, suggesting a possible compensation mechanism (Extended Data Figure 4d). Validation was performed by flow cytometry for 17 surface receptors for Ifitm3 loss- (Ifitm3ˉ/ˉ) and gain-of-function (Y20E; Extended Data Figure 4e–g). Continuous forced expression of Cd19 (>1 week) in Ifitm3ˉ/ˉ B-cells partially restored defective survival and proliferation in Ifitm3ˉ/ˉ B-cells, which required the Cd19-Y531 PI3K-activation motif21 (Extended Data Figure 5a–e). However, inducible Cd19-membrane translocation did not restore any defects in Ifitm3ˉ/ˉ B-cells, suggesting that Ifitm3 is still required to integrate Cd19 into signaling complexes for Src-kinase and PI3K signaling (Extended Data Figure 5f–h). To study IFITM3-interacting complexes, we performed enzyme-catalyzed proximity labeling studies based on N-terminal fusions of the BirA biotin ligase with IFITM3-Y20E carrying an extracellular HA-tag in B-ALL and IgM+ MCL-cells. After IFITM3- (anti-HA) or BCR-engagement (anti-IgM), IFITM3-interacting proteins were biotinylated based on proximity to the cytoplasmic BirA-moiety, harvested using streptavidin-coated beads and identified by mass spectrometry. Consistent with phosphoproteomic analyses (Figure 3a) and across different cell types (B-ALL, MCL) and stimulations (anti-HA, anti-IgM), IFITM3-interacting proteins included BCR-, integrin receptor and PI3K signaling elements (Figure 3d–e, Extended Data Figure 6a–d). We transduced MCL cells with FLAG-IFITM3 for validation by co-immunoprecipitation and Western blotting (Figure 3f–g). In proximity ligation assays, we confirmed inducible complex formation of IFITM3 with the BCR-signaling chain CD79B upon BCR-engagement. BCR:IFITM3 interactions were induced within minutes and dissociated after 30 minutes (Figure 3h–i). Consistent with endosomal BCR-functions22, BCR:IFITM3 interactions occasionally colocalized with LAMP1+ endosomal compartments (Extended Data Figure 6e). Hence, BCR-mediated activation of Src-mediated phosphorylation of IFITM3-Y20 induces IFITM3 accumulation at the plasma membrane, where IFITM3 functions as a scaffold for CD19 and LYN in proximity of BCR-molecules to enable processive signal amplification (Figure 3j). In resting B cells, IgM-BCRs are excluded from lipid rafts and recruited upon antigen-encounter23. In contrast, IgD-BCRs are already in close proximity to CD19 within lipid rafts in resting B-cells, suggesting that IFITM3 may specifically enable IgM-BCRs.
Ifitm3 binds to PIP3 and is essential for lipid raft formation
Reduced expression of integrins and adhesion receptors was associated with deceased homotypic adhesion of Ifitm3ˉ/ˉ pre-B cells, whereas membrane-bound Ifitm3-Y20E triggered formation of large clusters (Extended Data Figure 7). Since IKZF1 negatively regulates adhesion24–25, we examined whether this is mediated by transcriptional repression of IFITM316. Overexpression of a dominant-negative IKZF1-mutant (IK6) dramatically increased colony formation, adhesion to stroma and surface expression of adhesion receptors in the presence but not in the absence of Ifitm3. While dominant-negative inhibition of IKZF1 relieved transcriptional repression of adhesion receptors, proper expression and function of these receptors at the cell surface still required Ifitm3 and its ability to link integrins and adhesion receptors to the PI3K pathway. Loss of Ifitm3 and reduced activity of Src kinase (Lyn) and CD19-PI3K signaling could broadly affect lipid rafts and short-circuit BCR-signaling. Consistent with this scenario, the lipid raft components cholesterol and ganglioside GM1 were markedly reduced in Ifitm3ˉ/ˉ B-cells (Figure 4a). In addition, BCR-engagement increased membrane rigidity in IFITM3+/+ MCL cells. However, membrane-stiffening in response to BCR-engagement was significantly reduced in IFITM3ˉ/ˉ MCL cells (Extended Data Figure 8a). Consistent with defects in PI3K signaling, the ratios of PIP3 relative to the PI3K-substrate PIP2 were reduced substantially in murine and patient-derived IFITM3ˉ/ˉ B-ALL cells. In contrast, overexpression of IFITM3 (~3-fold) and IFITM3-Y20E (~5.5-fold) increased PIP3:PIP2 ratios (Figure 4b–c). To elucidate how IFITM3 interacts with plasma membrane lipids, we probed 15 distinct lipid classes for binding to IFITM3 in vitro. IFITM3 bound selectively to PIP3 but not any of the other 15 lipids (Figure 4d). Since Ifitm3 function is required to retain normal PIP3 levels, we tested whether exogenous delivery of PIP3 could rescue Ifitm3-deficiency. Insertion of exogenous PIP3 into the cell membrane partially restored Akt- and Myc-activation and colony formation but not Cd19- and Src-phosphorylation and surface expression of lipid raft-associated receptors (Extended Data Figure 8b–d). Hence, Ifitm3 promotes PI3K signaling not only by increasing the amount of PIP3 but also by integrating PIP3 into signaling complexes in lipid rafts.
Figure 4: Structural basis of Ifitm3-mediated regulation of PIP3 signaling from lipid rafts.

a, Ganglioside GM1 and cholesterol (stained with filipin III) levels measured in Ifitm3+/+ and Ifitm3−/− pre-B (top) and B-ALL (bottom) cells. b-c, Ratios of PIP3 and PIP2 were measured in Ifitm3+/+ and Ifitm3−/− murine (b, left), and human B-ALL cells (PDX2; b, right), and upon expression of EV, IFITM3 or IFITM3Y20E (c; Y20E). d, In vitro lipid-binding assays for GST-IFITM3 and 15 lipid classes. Recombinant GST-tag used as baseline. e, Lipid-binding assays were performed for biotin-IFITM3 N-terminus (1–57), first intramembrane α-helix (IM-α; 58–70), conserved intracellular loop (CIL; 71–105) and transmembrane α-helix (TM-α; 89–105). f, Lipid-binding assays to study interactions of PIP3 with biotinylated IFITM3-CIL fragments carrying mutations of basic residues. g-i, PIP3 binding to K83/K104 and R85/R87/K88 basic residue patches (g), dashed lines indicate PIP3 contacts within 3.5 Å. h, Interaction energies of PIP3 with basic residues K83 or R85 averaged over MD simulation trajectories (median levels in red dotted line, see Methods). Binding affinity for PIP3 as shown by heatmap of contact frequencies between PIP3 and each residue (i). j-l, Levels of phospho-AKT-S473, AKT, phospho-S6K-T389/421S424, S6K, phospho-CD19-Y531, CD19, phospho-SRC-Y416, LYN, phospho-PAK1/3-S199/204, phospho-PAK2-S192/197, PAK1, phospho-FAK-Y397/576/577, FAK, phospho-CXCR4-S339, CXCR4, and Integrin-β1 in PDX2 expressing EV, IFITM3-HA, IFITM3Y20E-HA, and R85A, K83A, R85A/R87A/K88A and K83A/K104A mutants upon IFITM3-crosslinking with anti-HA antibodies. a-f,j-l, n=3, For gel source data, see Supplementary Fig.1. b–c,e,h, two-tailed t-test.
Structural basis of Ifitm3-binding to PIP3
Proteins that harbor a cluster of four or more basic residues located at the membrane–solution interface can laterally sequester PIP3, which does not require a PH-domain or any other specific structure26. To map PIP3-binding to specific portions of IFITM3, we repeated lipid-binding assays with biotin-tagged fragments including the N-terminus, the first intramembrane α-helix and the conserved intracellular loop (CIL). PIP3-binding was detected only for the CIL (Figure 4e), which contains a cluster of five basic residues in close proximity. To address why IFITM3 preferentially binds to PIP3 over PIP2, we performed multiscale molecular dynamics (MD) simulations of IFITM3 in multi-component lipid bilayers mimicking the plasma membrane. All-atom MD simulations revealed that the representative binding pose of PIP2 from its most populated conformation cluster showed a charge interaction with one single basic residue (R85; −213 kJ/mol), which was energetically weaker than the two charge interactions with PIP3 (K83; K104; −704 kJ/mol; P=0.0008). Two-residue contacts were rare for PIP2 and more common for PIP3 (P=0.011; Extended Data Figure 9). To experimentally test the predicted function of basic CIL residues, we performed PIP3 binding assays for biotinylated IFITM3-CIL fragments carrying mutations of these residues. The K85A and R85A/R87/K88A mutants largely retained ability to bind PIP3, which was abrogated for the K83A/K104A mutant (Figure 4f). To understand the structural basis of this difference, we modeled the interaction of PIP3 with each of the basic residue patch K83/K104 and R85/R87/K88 as well as K83A and R85A mutants using all-atom MD simulations. PIP3 shows three times stronger interaction energy with K83/K104 than with R85/R87/K88 (Figure 4g–i). PIP3 makes dual-pronged salt bridge interactions with K83/K104 while it only interacts with R85 but not R87 and K88 in the other basic patch. The R87 and K88 residues form intra-protein salt bridges with D92 and D86, respectively, and are not available for interaction with PIP3.
The K83-K104 CIL-residues are critical to engage IFITM3-dependent PI3K, BCR and integrin signaling
To experimentally validate this model, we globally identified signal transduction changes that depend on K83 and R85 residues in human B-ALL and MCL B-cells. The most prominent differences were enriched for phosphorylation sites associated with PI3K, BCR and adhesion receptor signaling (Extended Data Figure 10a–b). While most phosphorylation events induced by membrane-bound IFITM3-Y20E were lost upon mutation of K83, this was not the case for substitution of the R85 residue (Extended Data Figure 10c–d). PLA studies revealed IFITM3-dependent recruitment of multiple proteins in the PI3K, BCR and integrin signaling pathways. However, almost all of these interactions were lost upon mutation of K83 (Extended Data Figure 10e–h). Biochemical studies revealed that crosslinking of IFITM3-Y20E resulted in massive activation of PI3K and integrin receptor pathways, whereas CD19 and Src were constitutively phosphorylated in the presence of IFITM3-Y20E (Figure 4j–l). The K85A mutation had only minor effects, while substitution of K83 almost entirely abolished IFITM3-dependent signal transduction. In contrast to the triple-mutant R85A/R87A/K88A, the K83/K104A double-mutant lost all activity in PI3K, BCR and adhesion receptor pathways (Figure 4j–l). In summary, we revealed a previously unrecognized function of IFITM3 as PIP3-scaffold for amplification of PI3K signaling. In Ifitm3ˉ/ˉ B-cells, lipid rafts were depleted of PIP3, resulting in profound defects of lipid raft-associated BCR and receptor signaling. IFITM3-dependent amplification of PI3K-signaling in part downstream of the BCR and adhesion receptors is critical to enable rapid expansion of B-cells with high affinity to antigen. In addition, multiple oncogenes depend on IFITM3 to assemble PIP3-dependent signaling complexes and amplify PI3K-signaling for malignant transformation.
Methods
Analysis of patient gene expression and outcome data
Gene expression microarray data from three large cohorts of patients with pre-B ALL were downloaded from GSE531427 (the Eastern Cooperative Oncology Group (ECOG) Clinical Trial E2993), GSE1187728 (the Children’s Oncology Group (COG) Clinical Trial P9906), St. Jude Research Hospital pediatric ALL29 (http://www.stjuderesearch.org/site/data/ALL3/). In ECOG E2993, pretreatment bone marrow or peripheral blood samples were obtained at diagnosis before any treatment from 83 patients with Ph+ B-ALL enrolled in the Medical Research Council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG) E2993 phase III trial. In data set from St. Jude childhood, 15 cases of Ph+ B-ALL were selected from the original 327 diagnostic bone marrow aspirates. Minimal residual disease (MRD) data were available for patients with pediatric high-risk B-ALL (COG P9906). IFITM3 expression levels were measured in patients with either a MRD positive (MRD+) status or a MRD negative (MRD−) status. In COG P9906 clinical outcome, expression profiles were obtained in pre-treatment leukemic samples from 207 uniformly treated children with high-risk ALL30, a risk category largely defined by pretreatment clinical characteristics. Patients had MRD tested by flow cytometry with two combinations (CD20/CD10/CD45 or CD9/CD19/CD34/CD45), and were defined as MRD positive or MRD negative at the end of induction therapy (day 29) using a threshold of 0.01% as previously described31. Then, RNA was purified from 207 pretreatment diagnostic samples with more than 80% blasts (131 bone marrow, 76 peripheral blood) and subjected to microarrays. Log-rank test was used to assess statistical significance.
Primary human samples and cell lines
Patient samples (Supplementary Table S7) were sourced ethically from patients who gave informed consent, and were in compliance with the internal review boards of the Beckman Research Institute of City of Hope. We have complied with all relevant ethical regulations. Patient samples were harvested from biopsy of bone marrow from patients with ALL at the time of diagnosis or relapse. All samples were transplanted into sublethally irradiated NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice (NSG mice, The Jackson Laboratory) through tail-vein injection. After samples were collected, patient-derived primary human pre-B ALL xenografts were cultured on OP9 stroma in Alpha Minimum Essential Medium (MEMα; Life Technologies) with GlutaMAX containing 20% fetal bovine serum (FBS), 100 IU ml−1 penicillin, 100 μg ml−1 streptomycin and 1 mM sodium pyruvate. The human cell lines (Supplementary Table S8) were cultured in RPMI-1640 (GIBCO) with GlutaMAX containing 20%FBS, 100 IU ml−1 penicillin, 100 μg ml−1 streptomycin at 37 °C in a humidified incubator with 5% CO2. All human primary samples and cell lines were tested negative for mycoplasma by detection kit (MycoAlert PLUS, LONZA)
Genetic mouse models
Genetic mouse models used in this study were listed in Supplementary Table S9. NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) were purchased from Jackson Laboratory. Ifitm3tm1Masu 32 were backcrossed to C57BL/6J background for more than 8 generations. B6.C(Cg)-Cd79atm1(cre)Reth/EhobJ (Mb1-Cre) were purchased from Jackson Laboratory. B6.129S2-Ighmtm1Cgn/J (μMT mice) were purchased from Jackson Laboratory. C;129S4-Ptentm1Hwu/J (Ptenfl/fl) were purchased from Jackson Laboratory. For animals bred in house, littermates of the same sex were randomized to experimental groups. For in vivo leukemia initiation assay 8- to 10-week-old female NSG mice were randomly allocated before injection. To generate a model for pre-leukemic B cell precursors expressing BCR-ABL1, LSL-Bcr+/ BCR-ABL1 mice33 were crossed with Mb1-Cre strain (Mb1-Cre x LSL-Bcr+/ BCR-ABL1) for excision of a stop-cassette in early pre-B cells. For in vivo oncogenic priming assay with Mb1-Cre x LSL-Bcr+/ BCR-ABL1 B-cell precursors, 8- to 10-week-old female NSG mice were randomly allocated before injection. Temperatures of 18–23°C with 40–60% humidity were maintained with 14-hour light/10-hour dark cycle. Following score was considered as end-point. 1. Failure to eat food / drink water for 24 hours. 2. Failure to make normal postural adjustments / display normal behavior. 3. Tumor Burden (1.5 cm × 1.5c, ×1.5 cm, tumor ulceration is NOT expected). If an animal either loses 25% of the initial body weight (or reaches 16g of body weight, regardless of the initial weight) or if we observe a weight loss of 15% on two sequential weight measurements, we euthanized the mouse immediately. All mouse experiments were subject to institutional approval by the Beckman Research Institute of City of Hope Animal Care and Use Committee.
Murine primary and leukemia cells
Bone marrow cells from 6–10 weeks old mice were harvested by flushing cavities of femur and tibia with chilled PBS followed by filtering through 40 μm strainer to yield a single-cell suspension. Spleen or thymus cells were directly extracted by forcing tissues through a 40 μm strainer into chilled PBS. Filtered cells were further incubated with lysis buffer (RBC Lysis Buffer, BioLegend) to lyse erythrocytes. After washing with PBS, cells were subjected to further experiments. For IL-7-dependent pre-B cell culture, bone marrow cells were harvested and cultured in Iscove’s modified Dulbecco’s medium (IMDM; GIBCO) with GlutaMAX containing 20% FBS, 50 μM 2-mercaptoethanol, 100 IU ml−1 penicillin, 100 μg ml−1 streptomycin in the presence of 10 ng ml−1 recombinant mouse IL-7 (Peprotech). For BCR–ABL1 leukemia model, pre-B cells were retrovirally transformed by BCR-ABL1 and then IL-7 was removed to select the transduced cells. NrasG12D ALL cells were selected with puromycin (GIBCO) and were maintained with IL-7 added IMDM.
Retroviral and lentiviral transduction
Retroviral supernatant was generated by co-transfection of HEK 293FT cells with retroviral constructs together with pHIT60 (gag-pol) and pHIT123 (for mouse) or pHIT456 (for human ecotropic envelope) using Lipofectamine 2000 (Invitrogen). Lentiviral supernatant for CRISPR-mediated gene editing was produced by co-transfection of HEK 293FT cells with lentiviral constructs together with pCDNL/BH and EM140. Constructs transfected HEK 293FT cells were cultured in high glucose Dulbecco’s modified Eagle’s medium (DMEM, GIBCO) with GlutaMAX containing 10% fetal bovine serum, 100 IU ml−1 penicillin, 100 μg ml−1 streptomycin, 25 mmol L−1 HEPES, 1 mmol l−1 sodium pyruvate and 0.1 mmol l−1 non-essential amino acids for 16 h. After Sodium butyrate (10 mM) induction for 8 h, the virus supernatant was collected, filtered through a 0.45 μm filter. The virus-containing supernatants were loaded by centrifugation (2,000 g, 90 min at 32 °C) on 50 μg ml−1 Retronectin (Takara) coated non-tissue culture 6-well plates. For retroviral transduction, 3–5 × 106 cells were transduced per well by centrifugation at 600g for 30 min in the appropriate culture medium and maintained at 37 °C at 5% CO2 for 48 h. For lentiviral transduction, 3–5 × 106 cells per well were centrifuged at 600g for 30 min in the presence of lentiviral supernatant and maintained at 37 °C at 5% CO2. The lentiviral supernatant was replaced with fresh medium on the next day.
Western blotting
PBS washed cells were lysed in CelLytic buffer (Sigma-Aldrich) supplemented with 1% protease inhibitor cocktail (Roche Diagnostics), 1% phosphatase inhibitor cocktail (EMD Millipore) and 1mM PMSF on ice. A total 10 μg of cell lysates were separated on mini precast gels (Bio-Rad) and transferred on nitrocellulose membranes (Bio-Rad). Membranes were probed with the appropriate primary antibodies and were incubated with alkaline-phosphatase conjugated secondary antibodies (Invitrogen) and chemiluminescent substrate (Invitrogen) and were further detected by film exposure, UVP BioSpectrum 810 Imaging System (Thermo Fisher Scientific) or by using the ChemiDoc MP Imaging System (BioRad). Antibodies used in this study in Supplementary Table S10 with 1:750 to 1:1000 dilution in blocking buffer.
Flow cytometry
PBS washed cells were blocked with Fc blocker for 10 min on ice and then stained with the appropriate antibodies listed in Supplementary Table S10 or isotype control for 25 min on ice. Cells were then washed and resuspended in chilled PBS containing 0.75 μg ml−1 of DAPI to exclude dead cells. Acquisition was performed by LSRFortessa flow cytometer (BD Biosciences) with BD FACSDIVA™ SOFTWARE. The fluorescence based cell sorting was performed by FACSAria II (BD Biosciences) with BD FACSDIVA™ SOFTWARE. FACS data were analyzed with FlowJo software (FlowJo, LLC). For apoptosis analyses, annexin V and 7-AAD (BD Biosciences) were used. For cell cycle analysis, the BrdU flow cytometry kit (BD Biosciences) was used according to manufacturer’s instructions. For intracellular staining of cytoplasmic proteins, cells were first stained for cell surface antigens and subsequently fixed in Fixation and Permeabilization Solution (BD Biosciences) containing 4 % paraformaldehyde and the detergent saponin. Cells were then washed and resuspended in Perm/Wash Buffer (BD Biosciences) and stained with the appropriate antibodies. For the statistical quantification, data were plotted with GraphPad Prism 7 or SigmaPlot. FACS antibodies in Supplementary Table S10 were pre-diluted in 1:5–1:10. 2 uL of diluted antibody was added to 1–2 million cells/50 uL in PBS for final dilution of 1:50 for human cells or 1:200 for mouse cells.
Pharmacological inhibitors and reagents
Imatinib was purchased from LC Laboratories. Stock solutions were prepared in sterile water at 10 mmol l−1 and treated with 10 μmol l−1. Dasatinib was purchased from SelleckChem. Stock solutions were prepared in sterile DMSO at 25 μmol l−1 and treated with 25 nmol l−1 (Supplementary table S11).
Analysis of ChIP–Seq data
IKZF1 ChIP-seq was performed as described previously34 with two different patient-derived human B-ALL xenograft cells (ICN1 and LAX2), both expressing wild type full-length IKZF1, and no detectable level of dominant negative (DN) IKZF1 isoform. ChIP–seq tracks (GSE58825) for IKZF1 antibody in LAX2 on IFITM3 gene promoter regions are shown. The y axis represents the normalized number of reads per million reads for peak summit for each track. ChIP-seq tracks (GSE86897) for enrichment of RNAPII and H3K4me3 at the Ifitm3 locus in pre-B cells from Ikzf1exon5fl/fl mice upon Cre-mediated deletion of Ikzf1 are shown. The ChIP–seq peaks were called by the MACS peak-caller by comparing read density in the ChIP experiment relative to the input chromatin control reads, and are shown as bars under each wiggle track. Gene models are shown in UCSC genome browser hg19. Integrative Genomics Viewer (IGV) was used to visualize ChIP-seq tracks.
Inducible expression of IKZF1
Human Ph+ pre-B ALL cells (BV173) carrying deletions of IKZF1 were transduced with pRetroX-Tet3G-Neo. Neomycin-resistant cells were transduced with pRetroX-TRE3G-wild-type IKZF1-Puro. Expression of wild-type IKZF1 in puromycin-resistant cell was initiated by treatment of 1 μg ml−1 of Doxycycline (TetOn).
Inducible reconstitution of CD19
For inducible CD19 reconstitution, murine CD19 was fused to the ligand binding domain of a mutant estrogen receptor (ERT2) at C-terminus of CD19. BCR–ABL1-transformed murine B-lineage ALL cells were retrovirally transduced with MSCV CD19-ERT2-Puro. Puromycin-resistant cells were treated with 4-OHT (4-hydroxytamoxifen, Sigma-Aldrich) or vehicle to induce reconstitution of CD19.
Cell viability assay
One-hundred-thousand BCR–ABL1-transformed murine B-lineage ALL cells were seeded in a volume of 100 μl medium in one well of a 96-well plate (BD Biosciences). Imatinib (LC Laboratories) were added at the indicated concentration in a total volume of 150 μl. After culturing for 3 days, 15 μl of Resazurin (R&D) was added into each well and incubated for 4 h at 37 °C. Medium without cells was used as blank. The fluorescence was read at 535 nm and the reference wavelength was 590 nm. Relative viability was calculated using baseline values of vehicle treated cells as a reference.
Colony-forming assay
The methylcellulose colony-forming assays were performed with 10,000 cells. Cells were resuspended in mouse MethoCult medium (without cytokines for BCR-ABL1-transformed cells; with IL7 for NRASG12D-expressing cells) and cultured on 3-cm diameter dishes, with an extra water supply dish to prevent evaporation. Colonies were imaged and counted after 7 days using GelCount (Oxford Optronix) and packages Q-Capture pro 7 connected with Olympus IX71 microscope.
In vitro IFITM3-HA crosslinking
8 million patient-derived B-ALL cells per sample were resuspended into complete medium and treated with either 2.5 μg ml−1 polyclonal anti-HA (Abcam) or isotype control for indicated times. F(ab) fragments for the anti-HA antibody were generated by using the F(ab) preparation kit following the manufacturer’s instructions (Thermo Fisher Scientific). F(ab) fragmentation was confirmed by Criterion TGX Stain-Free Precast Gels (Bio-Rad).
Measurement of intracellular calcium mobilization
In Extended Data Figure 2f, 1 million fresh splenocytes were incubated with the 4 μM Rhod-2-AM Ca2+-binding dye (Thermo Fisher Scientific) for 15 min at room temperature in dark. Cells were then resuspended in PBS and maintained at 37 °C, and Ca2+ response was induced by adding 10 μg ml−1 of polyclonal anti-IgM (Southern Biotech) at 50 seconds after acquisition of background fluorescence. Intracellular Ca2+ mobilization in response to crosslinked IgM was measured up to 300 seconds by flow cytometry. In Extended Data Figure 4c, 1 × 106 viable Jurkat cells were incubated with the 4 μM Fluo4-AM Ca2+-binding dye (Thermo Fisher Scientific) for 15 min at room temperature in dark. Cells were then resuspended in PBS and maintained at 37 °C, and Ca2+ response was induced by adding 10 μg ml−1 of monoclonal (OKT3) purified NA/LE anti-human CD3 (Biolegend) at 50 seconds after acquisition of background fluorescence. Intracellular Ca2+ mobilization in response to crosslinked IFITM3 was measured up to 300 seconds by flow cytometry. The same procedure was performed for Extended Data Figure 6f, 1 × 106 viable JEKO1 cells were incubated with the 4 μM Fluo4-AM (Thermo Fisher Scientific). Ca2+ response was induced by adding 10 μg ml−1 of polyclonal F(ab’)2 anti-human μ chain (Jackson Immunoresearch).
Homotypic aggregation assay
Ifitm3−/− B-ALL cells expressing C-terminally HA-tagged wild-type IFITM3, IFITM3 mutant vector (Y20E) or an empty vector were incubated with 2.5 μg ml−1 of monoclonal (1D3) purified LEAF anti-mouse CD19 (Biolegend) or 2.5 μg ml−1 polyclonal anti-HA (Abcam) or isotype controls at 37 °C at 5% CO2. After culturing for 24 hours, homotypic aggregation was visualized using light microscopy.
In vivo oncogenic priming assay with Mb1-Cre; LSL-Bcr+/ BCR-ABL1 B-cell precursors
To generate a model for pre-leukemic B cell precursors expressing BCR–ABL1, BCR–ABL1 knock-in mice were crossed with Mb1-Cre strain (Mb1-Cre x LSL-Bcr+/ BCR-ABL1) for excision of a stop-cassette in early pre-B cells. Bone marrow cells collected from Mb1-Cre x LSL-Bcr+/ BCR-ABL1 mice, cultured in the presence of 10 ng ml−1 recombinant mouse IL-7 (Peprotech). IL-7 dependent for pre-leukemic B cell were labeled with retroviral firefly luciferase and selected by blasticidin for 7 days. After selection, cells were further transduced with MSCV-IFITM3-HA-IRES-Puro, MSCV-IFITM3Y20E-HA-IRES-Puro or empty vector and selected by puromycin for 3 days. 1 × 107 viable cells were injected via the tail vein into sublethally irradiated (200 cGy) NSG recipient mice. The in vivo expansion and leukemia burden were monitored by luciferase bioimaging (IVIS 100 bioluminescence/optical imaging system; Xenogen) at the indicated time points. Briefly, D-luciferin (Promega) dissolved in PBS was injected intraperitoneally at a dose of 2.5 mg per mouse 15 min before measuring luminescent. All mice were anesthetized by 5% isoflurane and continued during detection of light emission with 2% isoflurane introduced through a nose cone. A mouse was euthanized when they showed signs of leukemia burden such as a hunched back, weight loss and inability to move. Kaplan-Meier survival analysis was performed using GraphPad Prism 7 (GraphPad Software Inc.) to compare overall survival (OS). Mantel–Cox log-rank test was used as statistical analysis using GraphPad Prism 7 (Supplementary Table S4).
Cell adhesion assay
Ifitm3+/+ or Ifitm3−/− BCR-ABL1 B-ALL cells were transduced with MSCV-IK6-IRES-GFP or MSCV-IRES-GFP as negative control. 1 × 105 GFP+ cells sorted by FACS were cultured on 1.5 × 105 OP9 stroma on 6 well plate with IMDM-GlutaMAX containing 20% FBS, 50 μM 2-mercaptoethanol, 100 IU ml−1 penicillin, 100 μg ml−1 streptomycin. For calculation of the ratio of adherent cells to nonadherent cells, nonadherent cells were harvested and counted with the trypan blue exclusion method using the Countess II FL Automated Cell Counter (Life Technologies). After washing of plate with PBS twice, adherent cells were detached with trypsinization and GFP+ B-ALL cells were counted with the trypan blue exclusion using the Countess II FL Automated Cell Counter (Life Technologies) and ratios were calculated.
Adoptive transfer of purified B cells into μMT-mice
For adoptive transfer of B-cells, splenocytes from 7- to 10-week-old Ifitm3+/+ or Ifitm3−/− mice were negatively selected by MojoSort Mouse Pan B Cell Isolation Kit II (Miltenyi Biotec) using immunomagnetic beads against Cd3, Cd4, Cd8a, Cd11c, Cd49b, Ly6G/Ly6C (Gr-1), and Ter-119. 10 million flow-sorted splenic B-cells were intravenously injected into μMT mice (B6.129S2-Ighmtm1Cgn/J) that lack the Cμ exon and mature B-cell development as a result of defective surface IgM expression. Reconstitution of donor B cells was determined by flow cytometry 20 days post injection. Recipient μMT mice were immunized with the hapten NP (4-hydroxy-3-nitrophenyl-acetyl) coupled to a carrier protein (keyhole limpet hemocyanin; KLH) on day 0 and day 7 and spleens were harvested on day 12.
Immunization and immunohistology
Ifitm3+/+ and Ifitm3−/− littermates were immunized with 0.5 mg NP-KLH (Biosearch Technologies) in alum (Sigma-Aldrich) intraperitoneally. 7 days later, mice were immunized a second time for 5 days. Spleens were isolated at day 12 after immunization. Spleen was embedded in optimum cutting temperature compound and 5 μm thick cryosections were used for staining. Sections were fixed with acetone for 10 min, and nonspecific antigens were blocked in DPBS containing 2% FBS for 15 min. Sections were stained with 1:200 diluted polyclonal (RA3–6B2) anti-CD45R (B220, BD Biosciences), monoclonal anti-mouse CD3 (17A2, BioLegend) antibodies and biotinylated Peanut Agglutinin (B-1075, Vector Laboratories) for 45 minutes. Sections were washed and further stained with Alexa Fluor 647 Streptavidin (BioLegend) antibody for 45 minutes. All antibodies were diluted 1:100 to their original concentration in blocking buffer. After washing with blocking buffer, sections were mounted in ProLong™ Diamond Antifade Mountant (ThermoFisher Scientific). Images were acquired on a ZEISS LSL 880 confocal microscope and analyzed on ZEN 2.3 (Zeiss) software.
ELISA measurements
For determination of the concentrations of Ig isotypes in sera, ELISA were carried out according to the manufacturer’s protocol (Ig Isotyping Mouse Instant ELISA Kit for IgG1, IgG2b and IgM, Supplementary table S11). NP-specific antibodies were measured by ELISA using 10 μg/ml of NP(24)-BSA (Biosearch Technologies, Novato, CA) as the coating reagent. NP-specific IgM and IgG1 was detected using goat anti–mouse IgM and IgG1 Fc-specific antibodies conjugated to horseradish peroxidase and developed with tetramethylbenzidine (Sigma). Optical densities were determined by an ELISA reader at 450 nm (SpectraMax M3, Molecular Devices).
RNA-seq analysis
Total mRNA from Ifitm3+/+ and Ifitm3−/− BCR-ABL1 or NRASG12D B-ALL cells was extracted using RNeasy Kit (Qiagen) according to the manufacturer’s instructions. Sequencing was performed on an Illumina Hiseq 2500 (Illumina, San Diego, CA, USA) instrument using the TruSeq PE Cluster Kit V4-cBot-HS (Illumina®) to generate 101 bp Paired-end reads sequencing with v4 chemistry. Quality control of RNA-Seq reads was performed using FastQC. For analysis raw sequence reads were mapped to the mouse genome (mm10) using STAR v2.5.335, and the frequency of genes was counted using featureCounts v1.5.136. The raw counts were then normalized using the trimmed mean of M values (TMM) method and compared using Bioconductor package “edgeR”37. Reads per kilo base per million (RPKM) mapped reads were also calculated from the raw counts. For differential expression analysis, transcripts were quantified using Salmon v1.1.0 against gencode GRCm38 vM24 transcript annotations; normalization and statistical analysis was done in R using DESeq2 v1.28.1. Differentially expressed genes were identified if RPKM ≥1 in at least one sample, fold change ≥2, and P ≤ 0.05. RPKM data were later used in the Gene set enrichment analysis. GSEA analysis was performed using the DOSE package in R32, genes were ranked by log2 fold-change, gene sets were obtained from MSigDB or from internal data as indicated.
CRISPR-mediated gene deletion
All lentiviral constructs expressing Cas9 nuclease and guide RNA were purchased from Transomic Technologies. For gene deletion, cells were transduced with pTOL-hCMV-Tet3G-Hygromycin. Hygromycin-resistant cells were subsequently transduced with pCLIP-Tre3g-hCMV-Cas9-P2A-zsGreen. Expression of Cas9-P2A-zsGreen was induced by 1 μg ml−1 of Doxycycline for 16 h, and zsGreen+ cells were sorted by Flow cytometry. Cells were washed out to remove doxycycline to turn off Cas9 expression (TetOff) and then subsequently transduced with pCLIP-gRNA-hCMV-RFP-gRNA. Sorted RFP+ cells were subjected to further experiments. Gene deletion was initiated by treatment of 1 μg ml−1 of Doxycycline (TetOn). Non-targeting guide RNA was used as control.
Gene deletion by non-viral genome targeting
Chemically synthesized crRNAs (160 μM) and tracrRNAs (160 μM) were mixed 1:1 by volume and annealed by incubation at 37 °C for 30 min. Recombinantly produced Cas9 (40 μM) were then mixed 1:1 by volume with gRNA to produce RNA ribonucleoprotein (RNP) complexes. RNPs were freshly complexed prior to electroporation. Electroporation was performed by using pulse code EH-115 on a Lonza 4D 96-well electroporation system. Predesigned Alt-R CRISPR-Cas9 guide RNAs were purchased from IDT. Non-targeting control guide RNAs were purchased from IDT.
Co-Immunoprecipitation
Co-immunoprecipitation was performed with the Pierce Crosslink Magnetic IP/Co-IP kit according to manufacturer’s instructions (Thermo Scientific,). Briefly, patient-derived Ph+ ALL cells (PDX2) were transduced with MSCV Flag-IRES-Puro or MSCV Flag-IFITM3-IRES-Puro and selected by puromycin for 3 days. 5 × 107 viable cells were harvested and washed by PBS before lysis using IP lysis/Wash buffer. Each 5 μg of anti-Flag antibody M2 (F1804, Sigma-Aldrich) per samples were coupled to protein A/G magnetic beads and covalently cross-linked with 20 μM disuccinimidyl suberate (DSS). The antibody cross-linked beads were incubated with cell lysate, washed to remove non-bound material and eluted in a low-pH elution buffer that dissociates bound antigen from the antibody cross-linked beads. The enriched antigen in low-pH was immediately neutralized and subjected to Western blotting. JEKO1 cells electroporated with either non-targeting RNP or IFITM3-targeting RNP complex were stimulated with 10 μg ml−1 F(ab’)2 fragment goat anti-human μ chain (Jackson Immunoresearch) for indicated time points at 37 °C, 5% CO2. Cells were immediately washed with chilled PBS and subjected to co-immunoprecipitation with anti-CD19 antibody (#90176, CST).
Proximity ligation assay
For PLA of pre-BCR or BCR: IFITM3, Cells were incubated with 10 μg ml−1 F(ab’)2 fragment goat anti-human μ chain (Jackson Immunoresearch) for 5 min at 37 °C, 5% CO2. Cells were immediately washed with chilled PBS and subsequently fixed in fixation buffer (Biolegend) containing 4% paraformaldehyde for 25 min on ice and then washed with chilled PBS. For cellular membrane staining, cells were labelled with 5 μg ml−1 WGA conjugated to Alexa Fluor 488 (Thermo Fisher Scientific) for 5 min at room temperature. Cells were then permeabilized in Perm/Wash Buffer (BD Biosciences) and then blocked in Duolink Blocking buffer for 30 min at room temperature. Cells were incubated with the 1:150 diluted primary antibodies listed in Supplementary Table S10 overnight at 4 °C. For late endosome staining, cells were incubated with anti-human LAMP1 conjugated to Alexa Fluor 488 (R&D Systems) with the primary antibodies. Cells were washed and settled on Cell-Tak (Corning) coated Shandon Single Cytoslide by cytospin at 400g for 5 min. PLA reactions were carried out according to the manufacturer’s protocol (Duolink, Sigma). Briefly, primary antibodies were coupled with Duolink in situ PLA probe Plus or Minus (Sigma-Aldrich) and then probes were visualized with Duolink Detection Reagent Red (Sigma-Aldrich). Cells were mounted with Duolink in situ Mounting Medium with DAPI (Sigma-Aldrich). Microscopic images were acquired with an Olympus IX3–55 and analyzed by CellSens imaging software (Olympus) and ImageJ. For quantification of PLA signals, one dot was defined as pixel size of 5 × 5 by BlobFinder software. Statistical significance was calculated using unpaired Student’s t-test with Excel and plotted with GraphPad Prism 7.
Phospho-proteomic analysis
Patient-derived B-ALL (PDX2) cells transduced with HA-tagged IFITM3-Y20E or empty vector (EV) control were incubated with 2.5 μg ml−1 of polyclonal anti-HA (Abcam) for 5 min at 37 °C at 5% CO2. Cellular extracts were prepared in urea lysis buffer, sonicated, centrifuged, reduced with DTT, and alkylated with iodoacetamide. 15mg total protein for each sample was digested with trypsin, and 500μg total protein for each sample was digested with LysC/trypsin for IMAC analysis. Samples were purified over C18 columns and dried in a lyophilizer. Dried samples were resuspended and enriched with Fe-IMAC beads, purified over C18 STAGE tips (Rappsilber). Replicate injections of each sample were run non-sequentially on the instrument. Peptides were eluted using 150-minute (IMAC) linear gradient of acetonitrile in 0.125% formic acid delivered at 280 nL/min. Tandem mass spectra were collected in a data-dependent manner with a Thermo Orbitrap Fusion™ Lumos™ Tribrid™ mass spectrometer using a top-twenty MS/MS method, a dynamic repeat count of one, and a repeat duration of 30 sec. Real time recalibration of mass error was performed using lock mass (Olsen) with a singly charged polysiloxane ion m/z = 371.101237. MS/MS spectra were evaluated using SEQUEST and the Core platform from Harvard University (Eng, Huttlin, Villen). Files were searched against the SwissProt Homo sapiens FASTA database. A mass accuracy of +/−5 ppm was used for precursor ions and 0.02 Da for product ions. Enzyme specificity was limited to trypsin, with at least one tryptic (K- or R-containing) terminus required per peptide and up to four mis-cleavages allowed. Cysteine carboxamidomethylation was specified as a static modification, oxidation of methionine and phosphorylation on serine, threonine, and tyrosine residues were allowed as variable modifications. Reverse decoy databases were included for all searches to estimate false discovery rates, and filtered using a 1% FDR in the Linear Discriminant module of Core. Peptides were also manually filtered using a −/+ 5ppm mass error range and presence of a phosphorylated residue. All quantitative results were generated using Skyline (MacLean) to extract the integrated peak area of the corresponding peptide assignments. Accuracy of quantitative data was ensured by manual review in Skyline or in the ion chromatogram files.
Bio-ID proteomics
Patient-derived PDX2 B-ALL cells and Jeko1 MCL cells were transduced with Flag-tagged IFITM3Y20E or K83A/K104 mutant constructs with N-terminal Turbo-ID BirA (engineered biotin ligase) and selected by puromycin for 3 days. Turbo-ID BirA expressing construct tagged with Flag was used as negative control. To induce biotinylation of proteins proximal to IFITM3, cells were treated with 50 μmol l-1 biotin with anti-HA antibody for simultaneous induction of IFITM3 crosslinking for 10 min. Cells were washed three times with chilled PBS and lysed in IP/WASH buffer (#87788, Pierce) with 1 × HALT protease inhibitor (Thermo Fisher). The lysates were incubated by Streptavidin C1 MyOne Dynabeads (Invitrogen) for 16 hours at 4 °C. Unbound proteins were washed three times by 2% SDS-PBS, three times by PBS and three time with pure water. The eluted proteins were gel purified, followed by in-gel digestion and subjected to Mass Spectrometry. For LC-MS/MS analysis, peptides were analyzed using a Dionex UltiMate 3000 Rapid Separation LC (RSLC) systems and a Orbitrap mass spectrometer (ThermoFisher Scientific). 6 μl peptide samples were loaded onto the trap column, which was 150 μm × 3 cm in-house packed with 3 μm C18 beads. The analytical column was a 75 μm × 10.5 cm PicoChip column packed with 3 μm C18 beads (New Objectives). The flow rate was kept at 300 nL/min. Solvent A was 0.1% FA in water and Solvent B was 0.1% FA in ACN. The peptide was separated on a 120-min analytical gradient from 5% ACN/0.1% FA to 40% ACN/0.1% FA. The mass spectrometer was operated in data-dependent mode. The source voltage was 2.40 kV and the capillary temperature was 275 ⁰C. MS1 scans were acquired from 400–2000m/z at 60,000 resolving power and automatic gain control (AGC) set to 1×106. The fifteen most abundant precursor ions in each MS1 scan were selected for fragmentation. Precursors were selected with an isolation width of 1 Da and fragmented by collision-induced dissociation (CID) at 35% normalized collision energy in the ion trap, previously selected ions were dynamically excluded from re-selection for 60 seconds. The MS2 AGC was set to 3 ×105. For data analysis, proteins were identified from the MS raw files using Mascot search engine (Matrix science). MS/MS spectra were searched against the SwissProt human database. All searches included carbamidomethyl cysteine as a fixed modification and oxidized Met, deamidated Asn and Gln, acetylated N-term as variable modifications. Three missed tryptic cleavages were allowed. The MS1 precursor mass tolerance was set to 10 ppm and the MS2 tolerance was set to 0.6 Da. A 1% false discovery rate cutoff was applied at the peptide level. Only proteins with a minimum of two peptides above the cutoff were considered for further study. For comparison to empty vector control, background peptide abundances for missing values were imputed from a Gaussian distribution centered around the minimum observed abundance using the MinProb method from MSnbase package in R.
Cell surface proteome analyses
Cell surface proteins were labeled with biotin using the N-linked glycosylation-site biotin labeling method39. Briefly, 40 million Ifitm3+/+ or Ifitm3−/− B-ALL cells were washed twice and resuspended in 1 ml of ice-cold PBS, and treated with 1.6 mM sodium metaperiodate (VWR) at 4 °C for 20 minutes to oxidize the vicinal diols of sugar residues linked to surface proteins. The cells were then washed twice in PBS to remove excess sodium metaperiodate. Cells were resuspended in 1ml of ice-cold PBS and treated with 1 mM biocytin hydrazide (Biotium) and 10 mM Aniline (Sigma-Aldrich) at 4 °C for 90 minutes with gentle mixing in order to biotinylate free aldehydes exposed on the sugar residues. After labeling, cells were washed three times with ice-cold PBS to removed excess biotin, frozen in liquid nitrogen, and stored at −80 °C until further processing for mass spectrometry. All experiments were performed in biological triplicated with replicates harvested from consecutive passages. Frozen cell pellets were thawed on ice in 1ml of RIPA buffer (Millipore) with the addition of 1 × HALT protease inhibitors (Pierce). After incubation on ice for 10 minutes, cells were disrupted by sonication and the lysates were clarified by centrifugation at 17,000 rcf at 4 °C for 10 minutes. Clarified lysate was mixed with 500 μl of neutravidin agarose resin (Thermo Fisher Scientific) and incubated at 4 °C for 2 hours with end-over-end mixing. Neutravidin beads with captured biotinylated surface proteins were washed extensively by gravity flow to remove unbound proteins using 50mls of 1× RIPA + 1mM EDTA, followed by 50 ml of PBS + 1 M NaCl, and finally 50 ml of 50 mM ABC + 2 M Urea buffer. Washed beads were resuspended in digestion buffer (50 mM Tris pH 8.5, 10 mM TCEP, 20 mM 2-Iodoacetamide, 1.6 M Urea) with 10 μg of added Trypsin protease (Pierce, 90057) to perform simultaneous disulfide reduction, alkylation, and on-bead peptide digestion at room temperature overnight (16–20 hours). After digestion, the pH was dropped to ~2 with neat trifluoroacetic acid (Sigma-Aldrich) and the peptide mixture was desalted using a SOLA-HRP column (Thermo Fisher Scientific) on a vacuum manifold. Desalted peptides were eluted with 50 % acetonitrile (Sigma-Aldrich) and 50% water with 0.1% TFA and dried down completely in a speedvac. Dried peptides were resuspended in LC/MS grade water (Fisher) with 2% ACN and 0.1% formic acid (FA). Peptide concentration was measured using 280 nm absorbance on a Nanodrop, and the peptide concentration was adjusted to 0.2 μg μl−1 for mass spec runs.
LC-MS and data analysis for cell surface proteome
For each replicate, 1 μg of peptide was injected onto a Dionex Ultimate 3000 NanoRSLC instrument with a 15-cm Acclaim PEPMAP C18 (Thermo Fisher Scientific, 164534) reverse phase column. The samples were separated on a 3.5-hour non-linear gradient using a mixture of Buffer A (0.1% FA) and B (80% ACN/0.1% FA), from 2.4% ACN to 32% ACN. Eluted peptides were analyzed with a Thermo Q-Exactive Plus mass spectrometer. The MS survey scan was performed over a mass range of 350–1500 m/z with a resolution of 70,000, with a maximum injection time (IT) of 100 ms. We performed a data-dependent MS2 acquisition at a resolution of 17,500, AGC of 5e4, and IT of 150 ms. The 15 most intense precursor ions were fragmented in the HCD at a normalized collision energy of 27. Dynamic exclusion was set to 20 seconds to avoid over-sampling of highly abundant species. The raw spectral data files are available at the ProteomeXchange PRIDE repository (Accession number PXD014691). Raw spectral data was analyzed using MaxQuant v1.5.1.240 to identify and quantify peptide abundance and searched against the human Swiss-Prot annotated human proteome from Uniprot (downloaded with 20,303 entries). The “match-between-runs” option was selected to increase peptide identifications while the “fast LFQ” option was selected to calculate label-free quantification values (LFQ) of identified proteins. All other settings were left to the default MaxQuant values. The MaxQuant output data was analyzed using Perseus41 and the R program (version 3.4.0). Proteins annotated as “reverse”, “only identified by site”, and “potential contaminant” were filtered out as well as proteins that were quantified in less than 2 out of 3 biological replicates in at least one experimental group. Missing values were imputed based on the normal distribution of the dataset as implemented by Perseus. Volcano plots were generated using output from a two-sample t-test comparing the log2 transformed LFQ protein abundance values from different cell lines with a false discovery rate (FDR) set to 0.01. Validation was performed by flow cytometry (Supplementary Table S1).
Cholesterol and lipid raft measurement
For the depletion of cholesterol, cells were preincubated with 5 mM of methy-β-cyclodextrin (MBCD) for 30 min at 37 °C before Filipin staining. For ganglioside GM1 staining, cells were labeled with cholera toxin B (CT-B) using Vybrant lipid raft labeling kit (Molecular Probes) according to the manufacturer’s protocol. In brief, cells were labeled with cholera toxin unit B conjugated with Alexa Fluor 594 on ice for 15 min, washed twice with PBS. CT-B-labeled lipid rafts were cross-linked with anti-CT-B antibody on ice for 15 min, washed twice with PBS and analyzed by flow cytometry.
In vivo transplantation of leukemia cells
Murine pre-B ALL cells transformed by BCR–ABL1 or NRASG12D were injected into sublethally irradiated (200 cGy) NOD-scid IL2Rgnull (NSG) mice via the tail vein. 8- to 10-week-old female NSG mice were randomly allocated before injection. A mouse was euthanized when they showed signs of leukemia burden such as a hunched back, weight loss and inability to move and then the bone marrow and/or spleen were collected to test leukemia infiltration by flow cytometry. Kaplan-Meier survival analysis was performed using GraphPad Prism 7 (GraphPad Software Inc.) to compare overall survival (OS). Mantel–Cox log-rank test was used as statistical analysis using GraphPad Prism 7 (Supplementary Table S3). The minimal number of mice in each group was calculated by using the ‘cpower’ function in R/Hmisc package. No blinding was used. All mouse experiments were subject to institutional approval by the Beckman Research Institute of City of Hope Animal Care and Use Committee.
PIP3 quantification
In Figure 4, 60 million viable cells were resuspended with chilled 0.5 M TCA in a total volume of 1 ml and incubated on ice for 5 min. Cells were centrifuged at 3,000 rpm for 7 min at 4 °C and resuspended in 5% TCA (tricarboxylic acid)/1 mM EDTA in a total volume of 1 ml. After vortex for 30 seconds, cells were washed again with 5% TCA/1 mM EDTA. To extract neutral lipids, cells were resuspended in 1 ml of MeOH:CHCl3 (2:1) and vortexed for 10 min at room temperature and centrifuged at 3,000 rpm for 5 min. After one more extraction of neutral lipids, the acidic lipids were extracted by adding 750 μl MeOH:CHCl3:12 M HCl (80:40:1) with vigorous vortexing for 25 min at room temperature. After centrifugation at 3,000 rpm for 5 min, supernatant was transferred to a new 2 ml centrifuge tube and mixed with 250 μl of CHCl3 and 450 μl of 0.1 M HCl, vortexed for 30 sec, and centrifuged at 3,000 rpm for 5 min to separate organic and aqueous phases. 500 μl of lower organic phases was collected into a clean 1.5 ml vial and dried in a vacuum dryer for 1 hr. Dried lipid was stored at −20 °C. On the day of assay, lipid samples were reconstituted with 200 μl of PBS containing 0.25 % Protein Stabilizer, 20 μl was used for PI(4,5)P2 measurement while rest of 180 μl was used for PI(3,4,5)P3 measurement using ELISA kit (Echelon Biosciences) according to the manufacturer’s instructions.
Exogenous delivery of PIP3
Phosphatidylinositol 3,4,5-trisphosphate diC16 (Echelon Biosciences) were freshly reconstituted with PBS at 2 mM. Unlabeled Shuttle PIP Carrier 2 (Histone H1) were freshly reconstituted with water at 2 mM. PIP3 in a volume of 75 μl were mixed with 25 μl of PIP Carrier and incubated at room temperature for 10 min. 100 μl of PIP3-Carrier complex were added into Ifitm3+/+ or Ifitm3ˉ/ˉ B-ALL cells in a total volume of 5 ml and incubated for 24 hours. PIP3 loaded Ifitm3+/+ or Ifitm3ˉ/ˉ B-ALL cells were subjected to colony-forming assay in Extended Data Figure 8.
Lipid binding assay
To assess direct binding of IFITM3 to lipids, lipid binding assay was performed using Membrane Lipid Strips (Echelon Biosciences) according to the manufacturer’s instructions. Briefly, membranes were blocked in 5% fatty acid-free BSA (Sigma-Aldrich) in TBST (50 mM Tris-HCl,150 mM NaCl, and 0.1% Tween 20) for 1 h at room temperature in the dark followed by overnight incubation with 0.5 μg/ml of recombinant proteins in blocking buffer at 4°C with gentle agitation. After washing membranes three times for 30 min in TBST, membranes were incubated for 1 hour with anti-GST tag polyclonal Ab (Thermo Fisher Scientific) or anti-Biotin monoclonal antibody (Cell Signaling Technology) antibodies listed in Supplementary Table S10. Membranes were incubated with alkaline-phosphatase conjugated secondary antibodies (Invitrogen) and chemiluminescent substrate (Invitrogen) and were further detected by UVP BioSpectrum 810 Imaging System (Thermo Fisher Scientific). Recombinant GST-tag protein was purchased from Sigma-Aldrich. Recombinant human GST-IFITM3 protein was purchased from Abnova. Recombinant human IFITM3 fragments listed in Supplementary Table S5 were synthesized at LifeTein, South Plainfield, NJ.
Preferential PIP3 accumulation and binding to IFITM3 using multi-scale molecular dynamics simulations
The IFITM3 protein structure from residues 58 to 128 was modeled by coarse-grained (CG) molecular dynamics simulation method in GROMACS in explicit cell membrane mimic bilayer. There is no structure was available for IFITM3 or for any close homolog. We calculated the hydrophobicity for each position using the hydrophobic index of amino acids42 and averaged hydrophobicity over a sliding window of seven amino acids. Prior structural studies on IFITM3 structure43 showed there is only one transmembrane domain 2 that is inserted into the membrane. Based on this evidence we used the topology shown in Fig. S9 as the starting structure. TM1 was generated as α-helix structure, the CRAC motifs and linker region were built as fully extended chain. The linker region was relaxed using 5000 steps of CG simulation using GROMACS program44 with Martini force field45 to remove stress in the system. We then added the TM2 region as a helix and inserted TM2 into a cell membrane mimicking bilayer46. The composition and ratio of lipids for the mixed bilayer is (POPC:DOPC:POPE:DOPE:CHOL:Sph:GM1=0.2:0.2:0.05:0.05:0.25:0.15:0.1) for the outer leaflet of the bilayer and 0.05:0.05:0.20:0.2:0.08:0.08:0.25:0.03:0.03:0.03 of POPC: DOPC: POPE: DOPE: POPS: DOPS: CHOL: PIP1:PIP2:PIP3 for the inner leaflet. We generated three possible starting setup for the mixed lipid bilayer with water in the upper and inner regions with CHARMM-GUI47, 48. The charges in the system were neutralized with 0.15M NaCl. We had nine replicates of the protein IFITM3 structure in the simulation box (see Fig. S9a). The initial configuration was minimized using steepest decent method for 5000 steps, then equilibrated in NPT ensemble for 5 ns, at 303K temperature and 1 bar pressure. The temperature was controlled by Berendsen thermostat49 with coupling constant of 1 ps, and the pressure was controlled by Berendsen barostat49 in semi-isotropic type with a coupling constant of 5 ps and a compressibility of 3 × 10−4 bar−1. The reaction field coulomb50 was applied to describe electrostatics, with cutoff at 1.1 nm. The cutoff for van der Waals interactions used was also 1.1 nm. We used the Leap-frog integrator, and a 20-fs time step for integration. All systems were solvated in coarse grained water model adopted in the MARTINI forcefield water, neutralized with 0.15 M NaCl. 10 μs of production simulation was performed for each of the three-starting conformations of the lipid bilayer. The last 1 μs of the trajectory from each of the three setups were used to analyze the folded structure of the CRAC1-basicpatch-CRAC2 region of the IFITM3 model. We clustered the conformations of IFITM3 by the root mean square deviation (RMSD) cutoff of 0.3 nm in the backbone CG particles in the CG simulations. We analyzed the binding patterns of PIP2 and PIP3 in this most populated conformational cluster. We observed that both PIP2 and PIP3 compete for the “basic patch” of residues shown in the amino acid sequence in Extended Data Figure 9. We extracted three snapshots from this conformational cluster that represent the most diverse patterns in PIP2 and PIP3 binding.
Details of the all-atom MD simulations for Phospholipid binding
To better understand the detailed binding conformation of PIP2 and PIP3 to IFITM3 and to calculate their binding energies to IFITM3, we converted the three snapshots extracted from the CG simulations to an all-atom system using Martini tools51. The three chosen snapshots were cut into 9 × 9 nm2 box centered at IFITM3 unit from the CG simulations to preserve the local optimized lipid environment. The lipid and protein were converted to all atom model, and were resolvated into 9 × 9 × 9 nm3 simulation box, neutralized by 0.15 M NaCl. Extended Data Figure 9 show the detailed interaction between PIP2/PIP3 with IFITM3 protein in all atom resolution base on these snapshots. The all-atom simulations were performed using GROMACS package44 and CHARMM36 force field52, with TIP3 water model. The non-bond interactions were calculated with a cut-off of 12 Å, particle mesh Ewald method51 was applied to solve long range van der Waals interaction. Each system was gradually heated to 310K with random initial velocities sampled from Boltzmann distribution. The heating process last for 1ns, with temperature controlled by Nosé-Hoover thermostat53, followed by 30 ns equilibration in NPT ensemble with harmonic position restraints on protein heavy atoms. The pressure was maintained at 1 bar in semi-isotropic environment, controlled by Parrinello-Rahman method. The restraint force was gradually reduced from 5 kcal/mol to 0 kcal/mol with a −1 kcal/mol step per 5 ns window. The last frame of equilibration was taken for production run. The production run was performed twice for 50 ns each, with two different random initial velocities assigned to the equilibrated structure. An integration time step of 2 fs was used. The interaction energy between PIP3 with IFITM3 protein was calculated as the sum of electrostatic Coulombic energy and van der Waals potential energy averaged over the two 50 ns trajectories, totaling to 100 ns.
Interaction of PIP3 with the two basic residue patches
The PIP3 binding and AKT signaling activation assays showed that mutation of the K83/K104 patch to K83A/K104A had higher effect than mutating the R85/R87/K88 patch to R85A/R87A/K88A. To further validate the interaction strength difference between PIP3 and the two basic patches, we converted all PIP1, PIP2 in the simulation box to PIP3. We further performed simulations on the K83A/K104A IFITM3 and on R85A/R87A/K88A IFITM3 in PIP3 only simulation box. Starting from this structure we performed equilibration of the system and 5 all-atom MD production simulations each 200ns long, using the protocol for all-atom MD simulations described above. We aggregated the last 100ns of simulation trajectories from each of the 5 runs which added to 500ns of MD simulation trajectories for analysis. We calculated the interaction energy as the sum of electrostatic Coulombic and van der Waals potential energies between PIP3 and residues forming the basic patch only (K83/K104 or R85/R87/K88). The interaction energies were averaged over the 500ns of aggregated MD simulation trajectories for each system. We repeated these interaction energy calculations for the wild type IFITM3 and for the mutant K83A/K104A and R85A/R87A/K88A constructs.
Contact frequency heat map calculations from MD simulations
The important information that can be extracted from MD simulations is the temporal frequency of PIP3 contacts with the two basic residue patches. The persistence of these interactions plays an important role in the PIP3 accumulation by IFITM3. We calculated the percentage of MD snapshots that show contacts between each of the residues in the basic patch. This is termed as contact frequency. We also calculated the percentage of MD snapshots that show simultaneous contacts made by PIP3 with two or more residues in the two basic residue patches. We generated a heat map using the frequencies calculated for the wild type IFITM3 and in the alanine mutants.
Quantitative RT–PCR
BM from a healthy donor was stained and sorted as described in Buchner et al., 2015. Total RNA from cells was extracted using the RNA isolation kit from Macherey-Nagel. Complementary DNA was generated with the qScript™ cDNA SuperMix (Quanta Biosciences). Quantitative real-time PCR was performed with FAST SYBR® Green Master Mix (Applied Biosystems) and the Vii7 real-time PCR system (Applied Biosystems) according to standard PCR conditions. COX6B was used as a reference gene.
Analysis of ChIP-seq data
ChIP-seq tracks for RNAPII, H3K4me3 and IKZF1 antibodies in B cell on the IFITM3 gene promoter region are shown. The ChIP-seq peaks were called by the MACS peak caller by comparing read density in the ChIP experiment relative to the input chromatin control reads, and are shown as bars under each wiggle track.
Acoustic scattering measurements
Single-cell size-normalized acoustic scattering (SNACS) was measured using a previously established microfluidic method, which has been shown to be specifically sensitive to cell surface stiffness55. Full measurement details can be found in55. In short, cells were flowed through a standing acoustic wave generated inside a vibrating suspended microchannel resonator (SMR). The SMR is a cantilever-based mass measurement tool56, which can also detect acoustic scattering from cells when the cells transit through the acoustic wave. The cantilever vibration frequency was monitored, and its shift was used to quantify the acoustic scattering from the cells as well as the buoyant mass of the cells. Prior to a set of measurements, the SMR was cleaned with 0.25% Trypsin-EDTA for 20 min, followed by 5% bleach for 3-min and then a 5-min rinse with DI H2O, to remove persistent biological debris. After cleaning, the SMR was passivated with 1 mg/mL PLL-g-PEG in H2O for 10 min at room temperature, followed by a 5-min rinse with normal cell culture media. During the measurement, all the samples were loaded into the SMR through 0.005-inch-inner-diameter fluorinated ethylene propylene (FEP) tubing. The fluid flow across the SMR was driven by three independent electronic pressure regulators and three solenoid valves. A consistent differential pressure was applied across the SMR to maintain constant shear and data rate for cell measurement. The data displayed (Extended Data Figure 8d) was obtained using a 350 μm long cantilever with 15×20 μm sized channel inside of the cantilever and a ~200 ms transit time through the cantilever. All the regulators, valves and data acquisition were controlled by custom software coded in LabVIEW 2017 (National Instruments) as detailed in55. A parallel volume measurement using Coulter Counter (Beckman Coulter) was carried out to quantify average cell volume, which was used together with the single-cell buoyant mass measurements to calculate SNACS for each cell, as reported before55. All measurements were carried out in normal cell culture media in room temperature within 10 minutes of taking cells out of cell culture incubator. After a measurement of the untreated JEKO1 cells, a new patch of same cells was obtained from the incubator, treated with 10 μg ml−1 F(ab’)2 fragment goat anti-human μ chain (Jackson Immunoresearch) and the cells were immediately (in ~1 minute) loaded into the SMR for measurement. Antibody treated JEKO1 cells were measured for ~7 minutes to ensure that the anti-human μ chain stimulated changes were not reversed during the experiment. The SMR was briefly washed with PBS between each experiment. WT JEKO1 cells were also measured after fixation to obtain a positive control for cell stiffness. For fixation, the cells were washed twice with PBS, mixed with 8% PFA for 30 min, washed twice with PBS and stored in +4 °C prior to the acoustic scattering measurements.
Quantification and statistical analysis
Data are shown as mean ± s.d. unless stated. Statistical analysis was performed by GraphPad Prism 7 (GraphPad Software Inc.) using unpaired two-tailed t test or as indicated in figure legends. Significance was considered at P< 0.05. For in vivo transplantation experiments, the minimal number of mice in each group was calculated through use of the ‘cpower’ function in the R/Hmisc package. Kaplan-Meier survival analysis was used to estimate OS with GraphPad Prism 7 Mantel-Cox log-rank test was used to compare the difference between two groups. No animals were excluded. For patient overall survival analysis, patients in each dataset divided into two groups based on whether their expression was above or below the median level of IFITM3 levels and Kaplan–Meier survival analysis was used to estimate overall survival. The datasets used include B-ALL (COG P9906, n=207), B-ALL (ECOG E2993, n=83), B-ALL (St. Jude, n=15), mantle cell lymphoma (LLMPP, n=92) and AML (TCGA, n=200). A log-rank test was used to compare survival differences between patient groups. R package ‘survival’ Version 2.35–8 was used for the survival analysis and Cox proportional hazards regression model in R package for the multivariate analysis (https://www.r-project.org/). The investigators were not blinded to allocation during experiments and outcome assessment. Experiments were repeated to ensure reproducibility of the observations.
Data availability
Patient-outcome data for B-ALL were obtained from the National Cancer Institute TARGET DATA Matrix of the Children’s Oncology Group (COG) Clinical Trial P9906 (GSE11877; Harvey et al., 2010; Kang et al., 2010), Eastern Cooperative Oncology Group (ECOG) Clinical Trial E2993 (GSE5314; Juric et al., 2007) and St. Jude Children’s Research Hospital (https://www.stjuderesearch.org/site/data/ALL3/; Ross et al., 2003). Patient-outcome data for mantle cell lymphoma were obtained from https://llmpp.nih.gov/MCL/57. Patient-outcome data for AML were obtained from TCGA Acute Myeloid Leukemia Project (http://www.cbioportal.org/study/summary?id=laml_tcga_pub#clinical; Cancer Genome Atlas Research Network, 2013)58. Proteomics data was deposited to the ProteomeXchange Consortium via the PRIDE partner repository with following accession numbers: cell surface proteome PXD014691, phosphoproteome PXD020696 and IFITM3 interactomes PXD020697. IFITM3 mRNA levels across human normal and malignant B-lymphoid samples were obtained from http://Amazonia.transcriptome.eu/. All other data are available from the corresponding author upon reasonable request. Genome binding/occupancy profiling from WT and IKDN stromal adherent pre-B cells were obtained from GSE86897. Immunohistochemistry images for IFITM3 levels in normal or malignant B cells were obtained from The Human Protein Atlas https://www.proteinatlas.org/. ChIP–seq data of the genome wide mapping of IKZF1 binding (ChIP-Seq) in human patient-derived B-ALL xenograft cells were obtained from GSE58825. ChIP–seq data of the genetic analysis of IKZF1 target genes (ChIP-Seq) and tumor suppressor function in BCR-ABL1+ pre-B ALL were obtained from GSE90656. RNA sequencing (RNA-Seq) data with Ifitm3+/+ and Ifitm3−/− BCR-ABL1 or NRASG12D B-ALL cells are available at GSE155305. RNA sequencing (RNA-Seq) data with Ptenfl/fl pre-B cells carrying 4-OHT-inducible Cre-ERT2 or ERT2 are available at GSE155618. Supplementary Table S12 summarized the accession numbers and publicly deposited data from this study. All other data needed to evaluate the conclusions in the paper are available within the main text or supplementary materials.
Extended Data
Extended Data Figure 1: Ifitm3 expression is induced by oncogenic PI3K signaling and repressed by IKZF1.
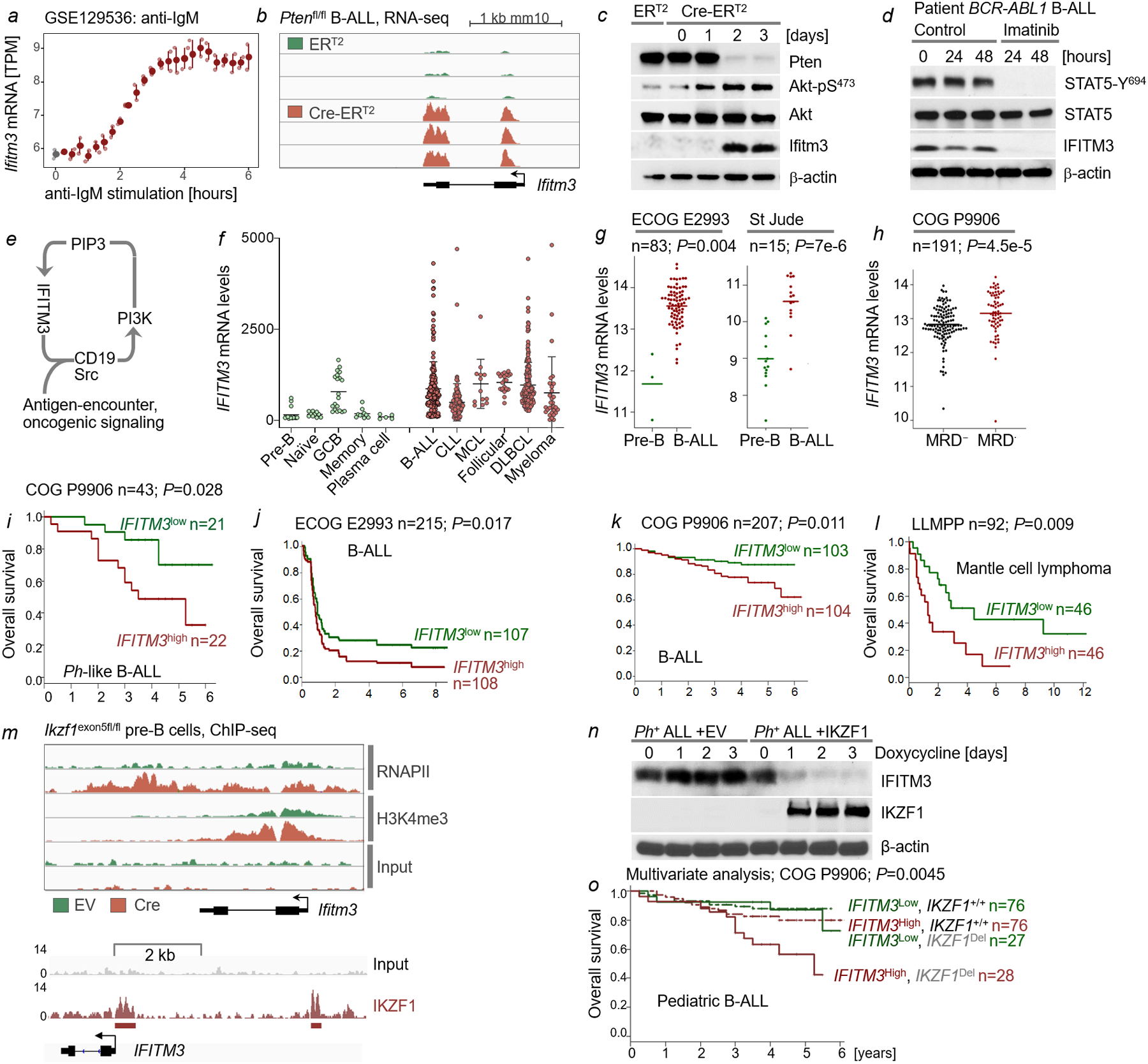
a, Changes of Ifitm3 mRNA levels were monitored in murine splenic B cells upon BCR-engagement (mean ± s.e.; n=2). Ptenfl/fl pre-B cells carrying 4-OHT-inducible Cre-ERT2 or ERT2 were treated with 4-OHT and studied by RNA-seq for Ifitm3 transcript expression (b) and Western blot for protein levels of Pten, phospho-Akt-S473, Akt and Ifitm3 (c; n=3). d, Phospho-STAT5-Y694, STAT5 and IFITM3 levels in patient-derived B-ALL cells (PDX2) measured by Western blotting upon Imatinib treatment (n=3). e, Scenario of the PI3K-pathway as positive regulator of IFITM3, which in turn amplifies BCR and oncogenic signaling. f, IFITM3 mRNA levels across human normal and malignant hematopoietic and B-lymphoid samples (source data and statistics Supplementary Table S2; two-tailed t-test). g, IFITM3 mRNA levels in pre-B cells from healthy donors and B-ALL patient samples were compared for two clinical cohorts (ECOG E2993 and St. Jude). In ECOG E2993, bone marrow samples were obtained at diagnosis before treatment from 83 adults with B-ALL with a confirmed purity of >90% leukemic blasts. For the St. Jude data sets, 15 pediatric B-ALL samples prior to treatment were compared to flow-sorted pre-B cells from bone marrow aspirates of healthy donors. (two-tailed Wilcoxon). h, Minimal residual disease (MRD) was determined in the COG P9906 cohort, IFITM3 mRNA levels were compared in MRD+ (n=67) and MRD- (n=124) patients (two-tailed Wilcoxon). i-l, Patients with leukemia and lymphoma from five clinical cohorts were segregated into two groups based on higher (IFITM3high) or lower (IFITM3low) than median IFITM3 mRNA levels. Overall survival was compared by two-tailed log-rank test. m, ChIP-seq enrichment of RNAPII and H3K4me3 at the Ifitm3 locus in pre-B cells (top) from Ikzf1exon5fl/fl mice upon Cre-mediated deletion of Ikzf1 (GSE86897). Binding of IKZF1 to the promoter region of IFITM3 was also analyzed in ChIP-seq data from patient-derived B-ALL cells (bottom, LAX2, GSE58825). n, Human B-ALL cells (BV173) carrying IKZF1-deletions were reconstituted with doxycycline-inducible IKZF1 or EV. Levels of IFITM3 were assessed by Western blotting upon doxycycline-induction (n=3). o, Multivariate analysis of established risk factors in pediatric B-ALL patients (COG P9906, n=207), including mutation or deletion of IKZF1. Patients (n=207) were separated into IKZF+/+ or IKZFDel groups, then further segregated based on higher or lower than median expression levels of IFITM3. The comparison of these four groups established IFITM3 mRNA levels as an independent risk factor regardless of IKZF1-deletion status (two-sided log-rank test; P=0.0045). c-d, n, For gel source data, see Supplementary Fig. 1.
Extended Data Figure 2: Ifitm3 is essential for the development of B1 and germinal center B-cells.

a, Hardy fractions of B-cell subsets isolated from bone marrow of Ifitm3+/+ and Ifitm3ˉ/ˉ littermates analyzed by flow cytometry (n=3). b, Surface expression of IgM, CD20, CD19, IgD, CD2 and CD21 measured by flow cytometry in enriched bone marrow (Gr-1−, Nk1.1− and B220+) and splenic B-cells (CD3− and B220+) from Ifitm3+/+ or Ifitm3ˉ/ˉ mice (n=7; mean±s.d.). Mean fluorescence intensities (MFI) values for individual measurement compared by two-tailed t-test. c, Ca2+-mobilization from cytoplasmic stores in response to BCR (IgM)-engagement was measured in Ifitm3+/+ and Ifitm3ˉ/ˉ splenic B cells. Ca2+ release was induced by addition of 10 μg ml−1 anti-mouse IgM 60 seconds after acquisition of background fluorescence. Ca2+ release was measured over 300 seconds with cell permeant Rhod-2 dye; MFI compared between replicates (n=3). d, Ca2+ mobilization in response to BCR-engagement measured upon CRISPR-Cas9-mediated deletion of IFITM3 in Jeko1 mantle cell lymphoma (MCL) cells. Ca2+-release upon addition of 10 μg ml−1 of polyclonal F(ab’)2 anti-human IgM was measured for 300 seconds with cell permeant Fluo-4 dye; MFI compared between replicates (left; n=3). Surface expression of CD19 following deletion of IFITM3 in Jeko1 MCL cells, MFIs for CD19 indicated (right; n=3). e, Jeko1 MCL cells were electroporated with non-targeting RNP (Cas9-gRNA ribonucleoproteins, gNT) or IFITM3-targeting RNP complex (gIFITM3). Following electroporation, MCL cells were treated with vehicle (DMSO) or 25 nmol l−1 of Dasatinib for 3 hours. Cells were stimulated with 10 μg ml−1 of anti-human IgM F(ab’)2 for indicated timepoints and subjected to co-immunoprecipitation with an anti-CD19 antibody. Immunoblots were performed to measure levels of CD19-tyrosine phosphorylation, and binding of LYN to CD19. Levels of IFITM3, Src-pY416 and Lyn were assessed in whole cell lysates (10% input) with β-actin as loading control. (n=3; gel source data Supplementary Fig. 1). f-h, Relative fractions (left) and absolute cell counts (right) of total B-1 (f), B-1a (g) cells in the peritoneal cavity and marginal zone B cells (h) in spleen of Ifitm3+/+ and Ifitm3−/− littermates (n=5) are shown (means ± s.d.; two-tailed t-test).
Extended Data Figure 3: Ifitm3-deficient B-ALL cells exhibit an anergic phenotype and compensatory upregulation of Ifitm1 and PI3K signaling molecules.
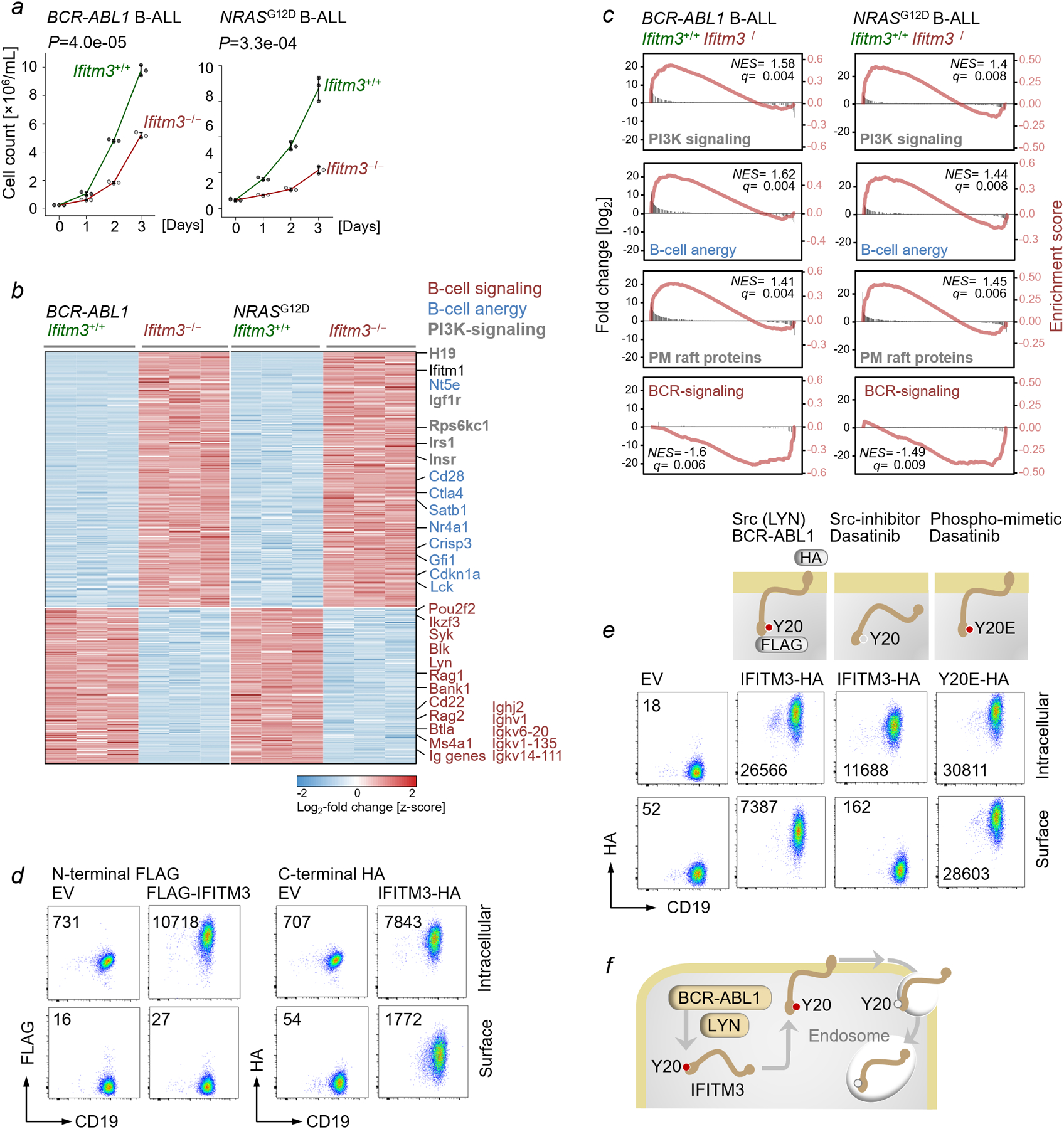
a, Numbers of viable Ifitm3+/+ and Ifitm3−/− BCR-ABL1 or NRASG12D B-ALL cells were counted by Trypan blue dye exclusion (n=3; mean±s.d.; two-tailed t-test). b, RNA-seq was performed for Ifitm3+/+ and Ifitm3−/− BCR-ABL1 and NRASG12D B-ALL cells. Relative rlog normalized gene expression values for all strongly differentially expressed genes (P < 1e-5 & L2FC > 1; Wald test with BH correction) in both BCR-ABL1 and NRASG12D conditions plotted as a heatmap with row-scaling. B-cell signaling related genes are labeled in red, anergy-related genes are labeled in blue and PI3K signaling related genes are labeled in gray. c, Gene set enrichment analysis for genes ranked by ratio of Ifitm3−/− to Ifitm3+/+ as log2 fold change; red lines indicates running enrichment score (right axis), gray bars indicates fold change (left axis). Statistical significance was determined by two-tailed Kolmogorov-Smirnov test. d, Patient-derived B-ALL cells (PDX2) were transduced with N-terminally FLAG-tagged or C-terminally HA-tagged IFITM3 constructs. Combinations of intracellular and surface staining were performed to examine IFITM3 topology at the cell membrane. e, Patient-derived B-ALL cells (PDX2) were transduced with C-terminally HA-tagged IFITM3 or the IFITM3Y20E phosphomimetic. Combinations of intracellular and surface staining, with or without Src-kinase inhibition by dasatinib, were performed to examine IFITM3 topology at the cell membrane and its regulation by Src-kinases. d-e, Representative plots from 3 independent experiments f, A scenario of the topology of IFITM3 regulated by Src (Lyn)- or oncogenic tyrosine kinases (BCR-ABL1) at the plasma membrane is shown. Phosphorylation of Y20 hinders the recognition of20YEM23 endocytosis motif by the AP-2 complex, thereby antagonizes endocytosis and endosomal trafficking of IFITM3.
Extended Data Figure 4: IFITM3 amplifies PI3K signaling downstream of BCR and integrin receptors.

a, Volcano plot of differentially phosphorylated proteins in patient-derived B-ALL (PDX2) cells transduced with IFITM3Y20E compared to empty vector (EV) control (n=3; Wald test with BH correction). b, F(ab) fragments of the anti-HA antibody or isotype control were purified and their identity confirmed by Western blot (left). 8 million patient-derived B-ALL (PDX2) cells carrying IFITM3-HA, IFITM3Y20E-HA or EV control were resuspended into complete medium and treated with either 2.5 μg ml−1 of full antibodies or F(ab) fragments of anti-HA or isotype control for the indicated times. Levels of phospho-AKT-S473, AKT and HA tagged IFITM3 were assessed by Western blots using β-actin as loading control (right). Data from three independent experiments. For gel source data, see Supplementary Fig. 1. c, Ca2+-mobilization in response to TCR-engagement using CD3ɛ-specific antibodies was measured upon CRISPR-Cas9-mediated deletion of IFITM3 in T-ALL cells (Jurkat; left). Ca2+ release from cytoplasmic stores was induced by adding 10 μg ml−1 of monoclonal (OKT3) anti-human CD3ɛ at 50 seconds after acquisition of background fluorescence. Surface expression of CD3 was measured following deletion of IFITM3 in Jurkat cells (right). MFIs for CD3 are indicated. Shown are representative plots from 3 independent biological experiments. MFI values for individual measurement were compared by two-tailed t-test. d, Surface proteins on Ifitm3+/+ and Ifitm3−/− B-ALL cells were labeled with biotin and enriched with streptavidin affinity pull-down followed by on-bead trypsin digestion, mass spectrometry and quantified with Lable-Free Quantification (LFQ). Differentially expressed cell surface proteins on Ifitm3+/+ and Ifitm3−/− B-ALL cells are shown with the mean difference of LFQ plotted against the P-value (Welch’s t-test). All experiments were performed in biological triplicates. e-g, Validation of differential expression of surface receptors between Ifitm3+/+, Ifitm3−/− and Ifitm3Y20E-overexpressing B-ALL cells. Flow cytometry analyses show surface expression of BCR-signaling components (e), integrins and adhesion receptors (f) and other surface receptors (g) in Ifitm3+/+, Ifitm3−/− and B-ALL cells expressing Ifitm3Y20E (n=3).
Extended Data Figure 5: Inducible membrane-translocation of CD19 does not rescue defective Src- and PI3K signaling in Ifitm3-deficient B-cells.

a, Surface expression of Cd19 was assessed by flow cytometry following forced expression of Cd19 for >1 week in Ifitm3+/+ and Ifitm3−/− B-ALL cells. b, Western blot analyses of phospho-Akt-S473, Akt, Myc and Bcl2 upon forced expression of Cd19 for >1 week in murine Ifitm3+/+ and Ifitm3−/− B-ALL cells. Colony forming ability (c) and cell cycle progression (d) of Ifitm3+/+ and Ifitm3−/− B-ALL cells upon forced expression of Cd19 for >1 week was examined. c, Colony numbers for individual measurement were compared by two-tailed t-test. d, Numbers indicate percentage of cells in S phase. e, Numbers of viable Ifitm3+/+ and Ifitm3−/− B-ALL cells following forced expression of Cd19 were counted by Trypan blue dye exclusion (left). Ifitm3ˉ/ˉ B-ALL cells were transduced with GFP-tagged constructs for expression of Cd19 with an intact (Y531) or mutant (Y531F) PI3K-activation motif in its cytoplasmic tail. Relative changes of GFP+ cells (transduced with Cd19-Y531 or Cd19-F531) were plotted over time (means ± s.d.). a-e, Data from three independent experiments. b, For gel source data, see Supplementary Fig. 1. f, Murine Cd19−/− B-ALL clones were generated by electroporation of murine B-ALL cells with Cd19-targeting RNP (Cas9-gRNA ribonucleoproteins, gCd19 ALL) and single-cell clones with biallelic deletion were selected. Cd19−/− B-ALL cells were transduced with Cd19-ERT2, a fusion of the ER-ligand binding domain to the C terminus of Cd19, or ERT2 as empty vector control. Reconstitution of Cd19−/− B-ALL cells with Cd19-ERT2 resulted in stable expression of the fusion proteins that were retained in complex with cytoplasmic heatshock proteins. Addition of 4-OHT released Cd19-ERT2 from its cytoplasmic heatshock chaperone and enable cell surface expression within 30 minutes of 4-OHT addition. g, To test the effect of inducible Cd19 membrane translocation in Ifitm3−/− B-ALL cells, Ifitm3+/+ and Ifitm3−/− B-ALL cells were transduced with Cd19-ERT2 or ERT2 empty vector control. 4-OHT-mediated translocation of Cd19 to the cell surface was assessed by flow cytometry for indicated times (0 to 3 hours). h, Ifitm3+/+ and Ifitm3−/− B-ALL cells were transduced with Cd19-ERT2 or ERT2 empty vector control. Cells were treated for 0, 1 and 3 hours with 4-OHT for surface-translocation of Cd19. Cell lysates from these populations were analyzed by Western blot for phospho-Cd19-Y531, Cd19, phospho-Src-Y416, Lyn, phospho-AKT-S473, AKT, Myc and Bcl2. While Cd19-ERT2 reconstitutes Cd19 protein levels in Ifitm3−/− B-ALL cells and rapid translocation to the cell surface (g), this change alone was not sufficient to induce proper phosphorylation of Cd19, Src-kinases and PI3K-signaling via Akt f-h, Data from three independent experiments. h, For gel source data, see Supplementary Fig. 1.
Extended Data Figure 6: Ifitm3 links components of the BCR and integrin receptor pathways to PI3K signaling.

a, Schematic of HA-tagged-IFITM3Y20E BirA-fusion proteins used for TurboID interactome analyses. BirA (biotin ligase) was fused to N-terminal IFITM3 carrying the phosphomimetic Y20E mutation for membrane-localization. HA-IFITM3Y20E-BirA or HA-BirA controls were expressed in PDX2 B-ALL (b) or Jeko1 MCL cells (c and d). c,d Cells were incubated with exogenous biotin for 10 min upon IFITM3- (anti-HA) or BCR- (anti-IgM) engagement. IFITM3Y20E interactome analyses identified interacting proteins by mass spectrometry, plotted based on significance and log2-fold enrichment over EV control. Essential interactors as BCR component (red), integrin (blue) and PI3K-signaling (gray) are highlighted. Data from three independent biological replicates. e, Proximity ligation assays were performed with Jeko1 MCL cells upon engagement of BCR. Jeko1 MCL cells were stimulated by BCR-engagement for 0, 3 and 30 minutes, then fixed, permeabilized and assessed for the proximity of CD79B to IFITM3. Representative microscopic images with PLA signal (red dot) and nuclei stained with DAPI as blue are shown. LAMP1 was used as a marker for endosomes to distinguish plasma membrane-bound from endosomal localization of CD79B:IFITM3 complexes. Scale bars, 5 mm. Data from three independent replicates.
Extended Data Figure 7: Ifitm3 functions as a central effector of B-cell adhesion.

a, Homotypic aggregation was studied in Ifitm3−/− B-ALL cells that were reconstituted with C-terminally HA-tagged Ifitm3, Ifitm3Y20E or empty vector (EV) and incubated with anti-CD19, anti-HA antibodies or isotype control for 24 hours. Data from three independent experiments. b, Ifitm3+/+ or Ifitm3−/− B-ALL cells were transduced with GFP-tagged IK6 or GFP alone. IK6 levels in flow-sorted GFP+ cells assessed by Western blot analysis using β-actin as loading control (right). 10,000 Ifitm3+/+ or Ifitm3−/− B-ALL cells carrying IK6 or EV were plated for colony forming assays. Colonies were imaged and counted after 7 days. Representative images are shown with colony numbers, Data from three independent experiments and assessed by two-tailed t-test (means ± s.d.). For gel source data, see Supplementary Fig. 1.c, 100,000 Ifitm3+/+ or Ifitm3−/− B-ALL cells carrying IK6 or EV were cultured on OP9 stroma cells. Ratios of adherent cells to nonadherent cells were calculated. Data are analyzed from three independent biological experiments (right) and assessed by two-tailed t-test (means ± s.d.). d, Representative images of adherent B-ALL cells on OP9 stroma are shown. Round and light-refracting cells are adherent B-ALL cells attached to stroma cells. Dark and round cells are adherent B-ALL cells incorporated into stromal layer. Data from three independent biological replicates. e, Surface expression levels of integrins on adherent B-ALL cells were measured by flow cytometry. MFI values are indicated for individual measurements. Data from three independent biological replicates.
Extended Data Figure 8: Ifitm3 functions as a PIP3 scaffold and mediated cell membrane-stiffening upon BCR-engagement.

a, Single-cell size-normalized acoustic scattering (SNACS) was measured using a previously established microfluidic method as a metric for cell surface stiffness. Jeko1 mantle cell lymphoma cells were flown through a standing acoustic wave generated inside a vibrating suspended microchannel resonator. The cantilever vibration frequency was monitored, and its shift was used to quantify the acoustic scattering from the cells as well as the buoyant mass of the cells. The data displayed was obtained using a 350 μm long cantilever with 15 × 20 μm sized channel inside of the cantilever and a ~200 ms transit time through the cantilever. All the regulators, valves and data acquisition were controlled by custom software coded in LabVIEW 2017 (National Instruments). A parallel volume measurement using Coulter Counter was carried out to quantify average cell volume, which was used together with the single-cell buoyant mass measurements to calculate SNACS for each cell. Jeko1 mantle cell lymphoma cells without (IFITM3+/+) and with (IFITM3−/−) CRISPR/Cas9-mediated deletion of IFITM3 were kept under cell culture conditions and treated with an anti-IgM antibody. Fixation with paraformaldehyde (PFA) was used as s positive control. Shown are representative plots from 3 independent experiments (median levels in red dotted line). Statistical significance was determined by two-tailed t-test. Numbers indicated cells studied for SNACS measurement. b, Ifitm3+/+ and Ifitm3−/− B-ALL cells were incubated with 30 μM of the PIP3 carrier histone H1 or PIP3-histone H1 complex for 30 minutes. Levels of phospho-CD19-Y531, CD19, phospho-Src-Y416, Lyn, phospho-Akt-S473, Akt and Myc were measured by Western blot using β-actin as loading control. Data from three independent replicates. For gel source data, see Supplementary Fig. 1. c, Colony formation assays were performed for Ifitm3+/+ and Ifitm3−/− B-ALL cells that were treated with 30 μM of PIP3-histone H1 or the shuttle protein histone H1 (H1) alone. Photomicrographs and colony numbers per 10,000 plated cells are shown. Data are presented as means ± standard derivation (s.d.) from three independent experiments. Statistical significance was determined by two-tailed t-test. d, Flow cytometry analyses of surface expression of CD19, CD44, CD25 and CD44 in Ifitm3+/+ and Ifitm3−/− B-ALL cells treated with 30 μM of PIP3-histone H1 or the shuttle protein histone H1 (H1) alone for 72 hours. Data from three independent replicates.
Extended Data Figure 9: Modeling of binding of IFITM3 to PIP2 and PIP3.

The hierarchical scheme of MD simulations (a-c) to delineate the structural and dynamical basis of PIP2 and PIP3 binding to IFITM3 (yellow). a, Coarse grained simulations of IFITM3 in composite cell membrane. The side views of the lipid bilayer are shown. The lipid raft markers are shown in color with GM1 (pink), cholesterol (green) and sphingomyelin (blue). The gray surface represents all the other lipids. b, The simulation cell extracted from the coarse grained simulation as the starting structure for all-atom MD simulations. The IFITM3 protein is shown in yellow and the PIP2 and PIP3 are shown in green and red stick representations. c, Close up view of one of the predicted binding poses of PIP2 and PIP3 in the most populated conformation of IFITM3. The dashed lines shown are the PIP2 or PIP3 contacts with the basic residues in IFITM3 (top). The amino acid sequence of IFITM3 in the stretch of residues between TM1 and TM2 from 57 to 128 modeled in this work is shown. The conserved intracellular loop (CIL) region is boxed and basic amino acids are highlighted with blue. The average interaction energy of PIP2 or PIP3 with the two basic batches measured in all-atom MD simulations is indicated (see Supplementary Table S6). d, e, The average interaction energy of PIP2 (green) or PIP3 (red) with the IFITM3 (residues 57–128) was measured in all-atom MD simulations. The 1-residue contact indicates the conformation of PIP2 or PIP3 binding with only one basic residue in the basic amino patch KSRDRK of IFITM3. The 2-residue contact refer to the binding conformation of PIP2 or PIP3 that show contacts with two basic residues. Shown are representative plots from at least 4 independent experiments (mean ± s.d.). P-values were determined by two-tailed t-test (d). Populations showing one or two-residue contacts of PIP2 (green) or PIP3 (red) to IFITM3 were quantitated from all-atom MD simulations. The population density was assessed by the normalization of number of events with the total number of frames. Shown are representative plots from 3 independent experiments (mean ± s.d.). Statistical significance was determined by two-tailed t-test (e).
Extended Data Figure 10: IFITM3-mediated PI3K signaling downstream of BCR and integrin receptors depends on K83-K104 but not on R85-R87-K88 residues.

a-c, Levels of differentially phosphorylated proteins in patient-derived B-ALL (PDX2) cells transduced with IFITM3Y20E, K83A, R85A or empty vector (EV) control were identified by mass spectrometry (n=3). Relative abundance values are plotted for all sites ranked by fold-change as indicated, phosphosites of interest are highlighted. b, Feature set enrichment analysis (FSEA) ranked by log2 fold change are shown for phosphosites in PI3K signaling (gray), BCR-signaling (red) and integrin and adhesion receptor elements (blue). Statistical significance was determined by two-tailed Kolmogorov-Smirnov test. d, Cumulative distribution frequencies for log2-fold changes in phosphosite abundance between IFITM3 transduced and EV conditions were calculated for all sites and globally increased in IFITM3Y20E over EV. The analysis was repeated for the K83A- (red) and R85A- (green) mutants of IFITM3Y20E. The light gray line indicates background variance observed between EV replicates. Shifts that were caused by the K83A and R85A mutants are indicated by arrows. Overall changes in distribution (shifts) between IFITM3Y20E and K83A and R85A mutants were measured by two-tailed Kolmogorov-Smirnov test. e, Schematic of BirA (engineered biotin ligase) was fused at N-terminal ends of HA-tagged IFITM3Y20E or its K83A mutant and expressed in PDX2 B-ALL or Jeko1 MCL cells. e-g, Interactomes of IFITM3Y20E and its K83A mutant were compared in PDX2 B-ALL cells upon IFITM3-engagement (anti-HA; e) or in Jeko1 MCL cells upon IFITM3- (anti-HA; f) or BCR- (anti-IgM; g) engagement. Phosphosites of interest, including BCR-signaling, PI3K-signaling and integrins and adhesion receptor elements were highlighted.
Supplementary Material
Acknowledgments.
We would like to thank Dr. Mark A. Lemmon (Yale University) for critical discussion and advice, and current and former members of the Müschen laboratory for their support and helpful discussions. Research in the Müschen laboratory is funded by the NIH/NCI through Outstanding Investigator Award R35CA197628, R01CA157644, R01CA213138, P01CA233412, the Dr. Ralph and Marian Falk Medical Research Trust Transformational Awards Program through MCG-18447-19, the Leukemia and Lymphoma Society through MCL-7000-18 and Blood Cancer Discoveries Grant BCDG-20327-20, as well the Howard Hughes Medical Institute HHMI-55108547 (to M.M.). This work was partly supported by National Institute of Health grants R35CA200422, R01CA251275, AI116585, AI116585S, AI140718, AI140705, DE023926, DE028521 (to J.U.J.), R01 AI104002 and R01 AI127513 (to M.S.D.), R01 GM117923 and R01 GM097261 (to N.V.), NCI R35 CA231958 (to D.M.W), Wellcome Trust 110275/Z/15/Z (to T.P.M) and the Paul G. Allen Frontiers Group and the Cancer Systems Biology Consortium funding (U54-CA217377) from the National Cancer Institute (to S.R.M). Relevant research in the A.T.-T. laboratory was supported by the Cancer Moonshot grant U01 CA232563. T.S. is a fellow of the Lymphoma Research Foundation.V.K. is supported by Career Development Fellow grant (5491-20) from the Leukemia and Lymphoma Society, and by a Young Investigator Award from Alex’s Lemonade Stand Foundation. Proteomics services were performed by the Northwestern Proteomics Core Facility, generously supported by NCI CCSG P30 CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center, instrumentation award (S10OD025194) from NIH Office of Director, and the National Resource for Translational and Developmental Proteomics supported by P41 GM108569. M.M. is a Howard Hughes Medical Institute (HHMI) Faculty Scholar. J.C. is a Leukemia & Lymphoma Society (LLS) Scholar.
Footnotes
Declaration of Interests
The authors have no competing interests.
Data and materials availability Patient-outcome data for B-ALL were obtained from the National Cancer Institute TARGET DATA Matrix of the Children’s Oncology Group (COG) Clinical Trial P9906 (GSE11877), Eastern Cooperative Oncology Group (ECOG) Clinical Trial E2993 (GSE5314) and St. Jude Children’s Research Hospital (https://www.stjuderesearch.org/site/data/ALL3). Patient-outcome data for mantle cell lymphoma were obtained from https://llmpp.nih.gov/MCL. Patient-outcome data for AML were obtained from TCGA Acute Myeloid Leukemia Project (http://www.cbioportal.org/study/summary?id=laml_tcga_pub#clinical). Proteomics data was deposited to the ProteomeXchange Consortium via the PRIDE partner repository with following accession numbers: cell surface proteome PXD014691, phosphoproteome PXD020696 and IFITM3 interactomes PXD020697. IFITM3 mRNA levels across human normal and malignant B-lymphoid samples were obtained from http://Amazonia.transcriptome.eu/. All other data are available from the corresponding author upon reasonable request. Genome binding/occupancy profiling from WT and IKDN stromal adherent pre-B cells were obtained from GSE86897. Immunohistochemistry images for IFITM3 levels in normal or malignant B cells were obtained from The Human Protein Atlas https://www.proteinatlas.org/. ChIP–seq data of the genome wide mapping of IKZF1 binding (ChIP-Seq) in human patient-derived B-ALL xenograft cells were obtained from GSE58825. ChIP–seq data of the genetic analysis of IKZF1 target genes (ChIP-Seq) and tumor suppressor function in BCR-ABL1+ pre-B ALL were obtained from GSE90656. RNA sequencing (RNA-Seq) data with Ifitm3+/+ and Ifitm3−/− BCR-ABL1 or NRASG12D B-ALL cells are available at GSE155305. RNA sequencing (RNA-Seq) data with Ptenfl/fl pre-B cells carrying 4-OHT-inducible Cre-ERT2 or ERT2 are available at GSE155618. All other data needed to evaluate the conclusions in the paper are available within the main text or supplementary materials.
References
- 1.Bailey CC, Zhong G, Huang IC & Farzan M IFITM-family proteins: the cell’s first line of antiviral defense. Annu. Rev. Virol 1, 261–283 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Diamond MS & Farzan M The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol 13, 46–57 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brass AL et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139, 1243–1254 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y, Qin L, Zhao Y, Zhang P, Xu B, Li K, Liang L, Zhang C, Dai Y, Feng Y, Sun J, Hu Z, Xiang H, Knight JC, Dong T, Jin R. Interferon-Induced Transmembrane Protein 3 Genetic Variant rs12252-C Associated with disease severity in Coronavirus Disease 2019. J Infect Dis. 222: 34–37 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao X, Sehgal M, Hou Z, Cheng J, Shu S, Wu S, Guo F, Le Marchand SJ, Lin H, Chang J, Guo JT. Identification of Residues Controlling Restriction versus Enhancing Activities of IFITM Proteins on Entry of Human Coronaviruses. J Virol. 92, e01535–17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jia R et al. The N-Terminal Region of IFITM3 Modulates Its Antiviral Activity by Regulating IFITM3 Cellular Localization. J. Virol 86, 13697–13707 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chesarino NM, McMichael TM, Hach JC, Yount JS. Phosphorylation of the antiviral protein IFITM3 dually regulates its endocytosis and ubiquitination. J. Biol. Chem 289, 11986–11992 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Everitt AR et al. IFITM3 restricts the morbidity and mortality associated with influenza. Nature 484, 519–523 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Makvandi-Nejad S, Laurenson-Schafer H, Wang L, Wellington D, Zhao Y, Jin B, Qin L, Kite K, Moghadam HK, Song C, Clark K, Hublitz P, Townsend AR, Wu H, McMichael AJ, Zhang Y, Dong T. Lack of Truncated IFITM3 Transcripts in Cells Homozygous for the rs12252-C Variant That is Associated with Severe Influenza Infection. J Infect Dis. 217, 257–262 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Kohn A Early interactions of viruses with cellular membranes. Adv. Virus Res 24, 223–276 (1979). [DOI] [PubMed] [Google Scholar]
- 11.Liu SY, Sanchez DJ, Cheng G. New developments in the induction and antiviral effectors of type I interferon. Curr. Opin. Immunol 23, 57–64 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li D, Peng Z, Tang H, Wei P, Kong X, Yan D, Huang F, Li Q, Le X, Li Q, Xie K. KLF4-mediated negative regulation of IFITM3 expression plays a critical role in colon cancer pathogenesis. Clin Cancer Res. 17, 3558–3568 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang D, Wang H, He H, Niu H, Li Y. Interferon induced transmembrane protein 3 regulates the growth and invasion of human lung adenocarcinoma. Thorac Cancer. 8, 337–343 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spence JS, He R, Hoffmann HH, Das T, Thinon E, Rice CM, Peng T, Chandran K, Hang HC. IFITM3 directly engages and shuttles incoming virus particles to lysosomes. Nat Chem Biol. 15, 259–268 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harvey RC et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling : correlation with genome-wide DNA copy number alterations , clinical characteristics , and outcome Identification of nov. Blood 116, 4874–4884 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Witkowski MT et al. Conserved IKAROS-regulated genes associated with B-progenitor acute lymphoblastic leukemia outcome. J. Exp. Med 214, 773–791 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fujimoto M et al. CD19 regulates Src family protein tyrosine kinase activation in B lymphocytes through processive amplification. Immunity 13, 47–57 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Okkenhaug K, Bilancio A, Farjot G, Priddle H, Sancho S, Peskett E, et al. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science. 297, 1031–1034 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Engel P, Zhou LJ, Ord DC, Sato S, Koller B, Tedder TF. Abnormal B lymphocyte development, activation, and differentiation in mice that lack or overexpress the CD19 signal transduction molecule. Immunity. 1995; 3, 39–50. [DOI] [PubMed] [Google Scholar]
- 20.Chan LN et al. Metabolic gatekeeper function of B-lymphoid transcription factors. Nature 542, 479–483 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y et al. The physiologic role of CD19 cytoplasmic tyrosines. Immunity 17, 501–514 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Phelan JD et al. A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature 560, 387–391 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kläsener K, Maity PC, Hobeika E, Yang J, Reth M, B cell activation involves nanoscale receptor reorganizations and inside-out signaling by Syk. Elife 3, e02069 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwickert TA, Tagoh H, Gültekin S, Dakic A, Axelsson E, Minnich M, Ebert A, Werner B, Roth M, Cimmino L, Dickins RA, Zuber J, Jaritz M, Busslinger M. Stage-specific control of early B cell development by the transcription factor Ikaros. Nat Immunol. 2014; 15: 283–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joshi I, Yoshida T, Jena N, Qi X, Zhang J, Van Etten RA, Georgopoulos K. Loss of Ikaros DNA-binding function confers integrin-dependent survival on pre-B cells and progression to acute lymphoblastic leukemia. Nat Immunol. 2014; 15: 294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McLaughlin S, Murray D (2005). Plasma membrane phosphoinositide organization by protein electrostatics. Nature 438, 605–611. [DOI] [PubMed] [Google Scholar]
- 27.Geng H et al. Integrative Epigenomic Analysis Identifies Biomarkers and Therapeutic Targets in Adult B-Acute Lymphoblastic Leukemia. Cancer Discov. 2, 1004–1023 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harvey RC et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: Correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood (2010). doi: 10.1182/blood-2009-08-239681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ross ME et al. Classification of pediatric acute lymphoblastic leukemia by gene expression profiling. Blood 102, 2951–9 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Kang H et al. Gene expression classifiers for relapse-free survival and minimal residual disease improve risk classification and outcome prediction in pediatric B-precursor acute lymphoblastic leukemia. Blood 115, 1394–405 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borowitz MJ et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: A Children’s Oncology Group study. Blood 111, 5477–5485 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poddar S, Hyde JL, Gorman MJ, Farzan M & Diamond MS The Interferon-Stimulated Gene IFITM3 Restricts Infection and Pathogenesis of Arthritogenic and Encephalitic Alphaviruses. J. Virol 90, 8780–94 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Foley SB et al. Expression of BCR/ABL p210 from a Knockin Allele Enhances Bone Marrow Engraftment without Inducing Neoplasia. Cell Rep. 5, 51–60 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schjerven H et al. Genetic analysis of Ikaros target genes and tumor suppressor function in BCR-ABL1+ pre-B ALL. J. Exp. Med 214, 793–814 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dobin A et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liao Y, Smyth GK & Shi W FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Robinson MD, McCarthy DJ & Smyth GK edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. - PubMed - NCBI Available at: https://www.ncbi.nlm.nih.gov/pubmed/25765347. (Accessed: 6th May 2020) [DOI] [PubMed]
- 39.Wollscheid B et al. Mass-spectrometric identification and relative quantification of N-linked cell surface glycoproteins. Nat. Biotechnol 27, 378–386 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tyanova S, Temu T & Cox J The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc 11, 2301–2319 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Tyanova S et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Monera OD, Sereda TJ, Zhou NE, Kay CM & Hodges RS Relationship of sidechain hydrophobicity and α-helical propensity on the stability of the single-stranded amphipathic α-helix. J. Pept. Sci 1, 319–329 (1995). [DOI] [PubMed] [Google Scholar]
- 43.Ling S et al. Combined approaches of EPR and NMR illustrate only one transmembrane helix in the human IFITM3. Sci. Rep 6, 24029 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berendsen HJC, van der Spoel D & van Drunen R GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun 91, 43–56 (1995). [Google Scholar]
- 45.de Jong DH et al. Improved Parameters for the Martini Coarse-Grained Protein Force Field. J. Chem. Theory Comput 9, 687–697 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Yen H-Y et al. PtdIns(4,5)P2 stabilizes active states of GPCRs and enhances selectivity of G-protein coupling. Nature 559, 423–427 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hsu P-C et al. CHARMM-GUI Martini Maker for modeling and simulation of complex bacterial membranes with lipopolysaccharides. J. Comput. Chem 38, 2354–2363 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qi Y et al. CHARMM-GUI Martini Maker for Coarse-Grained Simulations with the Martini Force Field. J. Chem. Theory Comput 11, 4486–4494 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A & Haak JR Molecular dynamics with coupling to an external bath. J. Chem. Phys 81, 3684–3690 (1984). [Google Scholar]
- 50.Tironi IG, Sperb R, Smith PE & van Gunsteren WF A generalized reaction field method for molecular dynamics simulations. J. Chem. Phys 102, 5451–5459 (1995). [Google Scholar]
- 51.Wassenaar TA, Pluhackova K, Böckmann RA, Marrink SJ & Tieleman DP Going Backward: A Flexible Geometric Approach to Reverse Transformation from Coarse Grained to Atomistic Models. J. Chem. Theory Comput 10, 676–690 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Huang J & MacKerell AD CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem 34, 2135–2145 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martyna GJ, Klein ML & Tuckerman M Nosé–Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys 97, 2635–2643 (1992). [Google Scholar]
- 54.Buchner M et al. Identification of FOXM1 as a therapeutic target in B-cell lineage acute lymphoblastic leukaemia. Nat. Commun 6, 6471 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kang JH et al. Noninvasive monitoring of single-cell mechanics by acoustic scattering. Nat. Methods 16, 263–269 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burg TP et al. Weighing of biomolecules, single cells and single nanoparticles in fluid. Nature 446, 1066–1069 (2007). [DOI] [PubMed] [Google Scholar]
- 57.Rosenwald A et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell 3, 185–197 (2003). [DOI] [PubMed] [Google Scholar]
- 58.Network TCGAR Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N. Engl. J. Med 368, 2059–2074 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Patient-outcome data for B-ALL were obtained from the National Cancer Institute TARGET DATA Matrix of the Children’s Oncology Group (COG) Clinical Trial P9906 (GSE11877; Harvey et al., 2010; Kang et al., 2010), Eastern Cooperative Oncology Group (ECOG) Clinical Trial E2993 (GSE5314; Juric et al., 2007) and St. Jude Children’s Research Hospital (https://www.stjuderesearch.org/site/data/ALL3/; Ross et al., 2003). Patient-outcome data for mantle cell lymphoma were obtained from https://llmpp.nih.gov/MCL/57. Patient-outcome data for AML were obtained from TCGA Acute Myeloid Leukemia Project (http://www.cbioportal.org/study/summary?id=laml_tcga_pub#clinical; Cancer Genome Atlas Research Network, 2013)58. Proteomics data was deposited to the ProteomeXchange Consortium via the PRIDE partner repository with following accession numbers: cell surface proteome PXD014691, phosphoproteome PXD020696 and IFITM3 interactomes PXD020697. IFITM3 mRNA levels across human normal and malignant B-lymphoid samples were obtained from http://Amazonia.transcriptome.eu/. All other data are available from the corresponding author upon reasonable request. Genome binding/occupancy profiling from WT and IKDN stromal adherent pre-B cells were obtained from GSE86897. Immunohistochemistry images for IFITM3 levels in normal or malignant B cells were obtained from The Human Protein Atlas https://www.proteinatlas.org/. ChIP–seq data of the genome wide mapping of IKZF1 binding (ChIP-Seq) in human patient-derived B-ALL xenograft cells were obtained from GSE58825. ChIP–seq data of the genetic analysis of IKZF1 target genes (ChIP-Seq) and tumor suppressor function in BCR-ABL1+ pre-B ALL were obtained from GSE90656. RNA sequencing (RNA-Seq) data with Ifitm3+/+ and Ifitm3−/− BCR-ABL1 or NRASG12D B-ALL cells are available at GSE155305. RNA sequencing (RNA-Seq) data with Ptenfl/fl pre-B cells carrying 4-OHT-inducible Cre-ERT2 or ERT2 are available at GSE155618. Supplementary Table S12 summarized the accession numbers and publicly deposited data from this study. All other data needed to evaluate the conclusions in the paper are available within the main text or supplementary materials.