

Abstract
Technology advance during the past decade has greatly expanded our understanding of the higher order structure of the genome. The various chromosome conformation capture (3C)-based techniques such as Hi-C have provided the most widely used tools for interrogating three-dimensional (3D) genome organization. We recently developed a Hi-C variant, DNase Hi-C, for characterizing 3D genome organization. DNase Hi-C employs DNase I for chromatin fragmentation, aiming to overcome restriction enzyme digestion-related limitations associated with traditional Hi-C methods. By combining DNase Hi-C with DNA capture technology, we further implemented a high-throughput approach, called targeted DNase Hi-C, which enables to map fine-scale chromatin architecture at exceptionally high resolution, and thereby is an idea tool for mapping the physical landscapes of cis-regulatory networks and for characterizing phenotype-associated chromatin 3D signatures. Here, I describe a detailed protocol of targeted DNase Hi-C library preparation, which covers experimental steps starting from cell crosslinking to library amplification.
Keywords: 3C, Hi-C, DNase Hi-C, chromatin, chromosome, 3D genome
1. Introduction
The development and wide spread application of 3C [3] -based high throughput techniques [2, 4] such as Hi-C [14] has demonstrated that eukaryotic genomes are hierarchically organized in the nucleus [7, 8, 13, 14, 23]. This hierarchy consists of at least four distinct levels: whole chromosome territories (CTs) [1], large-scale active or repressed compartments (A/B compartments) [14], chromatin domains, e.g. topologically associated (TAD) [5, 20], lamina associated (LAD) [11, 21] or nucleolus associate (NAD) [19, 26], and chromatin loops [24], which provide multiple regulatory layers for gene regulation and other genome functions, and thereby play pivotal roles in development and disease [8, 13, 23]. For example, distal cis-elements (e.g., enhancers and super-enhancers (SE)) regulate gene expression through long-range physical interactions via chromatin looping, which occurs much more frequently within a TAD than between TADs. TADs are considered to be both building blocks and functional units that guide, constrain, and facilitate promoter-enhancer interactions [8, 13, 23]. Each TAD often contains multiple genes and enhancers, allowing for coordinated regulation. Hence, dissecting the 3D genome and its dynamics will provide information essential for us to completely understand gene regulation and the regulation of other nuclear processes.
Mapping the spatial organization of cis-regulatory elements in the nucleus will lead to new insights into the mechanisms of how distal enhancers regulate their target genes and how regulatory elements coordinate to achieve temporal and cell type-specific transcriptional regulation during development. Many 3C derivatives, such as ChIA-PET [10], 4C [25, 27], 5C [6], Hi-C [14], Capture-C [12], Capture Hi-C [17] and HiChIP/PLAC-seq [9, 18], can be used to map chromatin interaction landscapes of cis-regulatory elements. However, the ultimate resolution of the chromatin interaction maps constructed by these methods is limited by the genomic distribution of restriction enzyme (RE) sites, as these methods all employ RE digestion to fragment chromatins. To overcome this limitation, we recently developed DNase Hi-C [15] and its in situ version in situ DNase Hi-C [22], in which chromatin fragmentation is achieved by sequence-independent digestion of DNase I. By combining DNase Hi-C (or in situ DNase Hi-C) with DNA capture technologies, we subsequently developed targeted DNase Hi-C, a method for mapping fine-scale chromatin architectures of specific genomic regions of interest at high resolution in a massively parallel fashion [15].
In brief, as outlined in Fig 1, the targeted DNase Hi-C protocol involves cross-linking cells with formaldehyde; chromatin is then randomly fragmented by DNase I. The resulting chromatin fragments are end-repaired and dA-tailed, then marked with a biotinylated internal adaptor, and proximity ligation is carried out in the intact nucleus to favor ligation events between the cross-linked DNA fragments. The resulting DNA sample contains ligation products consisting of chimeric DNA fragments that were originally in close spatial proximity in the nucleus, marked with biotin at the junction. A whole-genome chromatin interaction library is constructed by shearing the DNA, selecting the biotin-containing fragments with streptavidin magnetic beads and PCR amplification. The chromatin contacts associated with a specific group of loci of interest are then enriched by a bait library targeting the loci via in-solution hybridization-based DNA capture. Finally, the enriched chromatin interactions are quantified by massively parallel deep sequencing. Data processing and further computational analysis can be carried out as previously described [15, 16, 22].
Fig 1.
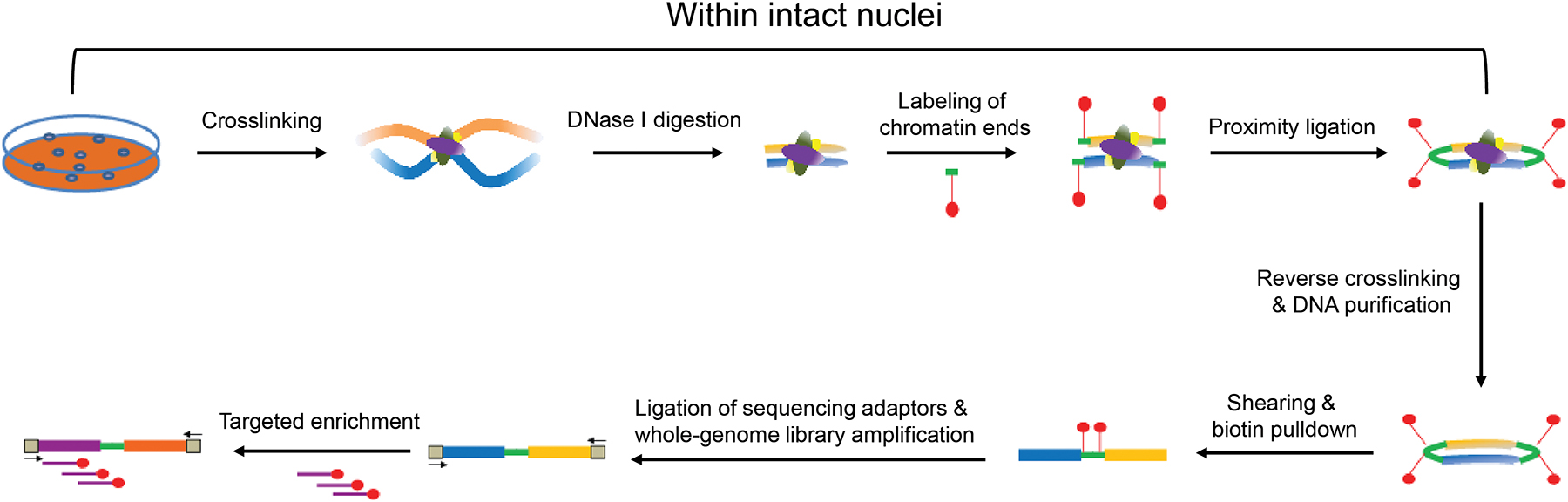
Targeted DNase Hi-C workflow.
2. Materials
2.1. Reagents
37% (vol/vol) Formaldehyde
10x NEBuffer 2 (500 mM NaCl, 100 mM Tris-HCl, 100 mM MgCl2, 10 mM DTT, pH 7.9@25°C)
10% (wt/vol) UltraPure SDS
DNase I, RNase-free (supplied with MnCl2 and reaction buffer) (1 U/μL)
RNase A, DNase and protease-free (10 mg/ml)
Klenow Fragment (10 U/μL)
Klenow Fragment (exo–) (5 U/μL)
T4 DNA Polymerase (5 U/μL))
T4 DNA Ligase (5 U/μL) provided with 50% PEG-4000
T4 Polynucleotide Kinase (10 U/μL)
10X T4 DNA Ligase Buffer w/ATP
Agencourt AMPure XP
2X KAPA HiFi HotStart PCR ReadyMix
Fast DNA End Repair Kit
Proteinase K
FastDigest® BamHI
Dynabeads® MyOne™ Streptavidin C1
dNTP Set (100 mM 4× 0.25ml)
10 mM dNTP mix
100 bp DNA ladder
GlycoBlue (15 mg/mL)
3 M Sodium acetate (pH 5.2)
100% Ethanol
100% Isopropanol
1M UltraPure Tris-HCl pH 8.0
1M UltraPure Tris-HCl pH 7.5
Buffer EB (10 mM Tris-HCl, pH 8.5)
0.5 M EDTA
Qubit dsDNA HS kit
100% IGEPAL CA-630
100% Triton X-100
1X DPBS
Protease Inhibitor Tablets
QIAquick PCR Purification Kit
Rapid DNA Ligation Kit
5M Sodium Chloride (NaCl)
SeqCap EZ Reagent Kit v2
Zymolyase 20T
2.2. Buffers
Spheroplast buffer: 1 M sorbitol, 100 mM potassium phosphate (PH 7.5)
2.5 M Glycine: Bring 9.35 g glycine to 50 ml in ddH2O and filter sterilize using Steriflip column. Store at RT for up to 6 months.
80% (vol/vol) Ethanol: Mix 8 ml 100% ethanol with 2 mL ddH2O. Make fresh for every day of experiments.
10% (vol/vol) Triton X-100: Mix 1 ml 100% Triton X-100 with 9 ml ddH2O. Store at RT for up to 6 months.
2% (vol/vol) Tween-20: Mix 1 ml 100% tween-20 with 49 ml ddH2O. Store at RT for up to 6 months.
10% Igepal CA-630: Mix 5 ml 100% Igepal CA-630 with 45 ml ddH2O.
1X Cell Lysis buffer: Mix 500 μl 1M Tris-HCl pH 8.0, 100 μl 5M NaCl, 150 μl 1M MgCl2, and 1 ml 10% Igepal CA-630 and bring up to 50 ml in ddH2O. Store at 4°C for up to 6 months.
1x TE lysis buffer: 50mM Tris.HCl (pH7.0), 1mM EDTA, 1% SDS. Store at RT
2X B&W buffer: Mix 500 μl 1M Tris-HCl pH 8.0, 100 μl 0.5M EDTA, and 20 ml 5M NaCl and bring up to 50 ml in ddH2O. Store at RT for up to 6 months.
1x B&W buffer: Mix 25 ml 2x B&W buffer with 25 ml ddH2O.
1x B&W buffer with 0.1% Tween-20: Mix 25 ml 2x B&W buffer with 2.5 ml 2% Tween-20 and 22.5 ml ddH2O.
0.5X DNase SDS Buffer: Mix 25 μl 10X DNase digestion buffer with 25 μl of 10 mM MnCl2, 10–20 μl of 10% SDS and 450 μl of ddH2O. Use immediately.
0.5X DNase digestion Buffer: Mix 25 μl 10X DNase digestion buffer with 25 μl 10 mM MnCl2, 100 μl 10% Triton X-100 and 350 μl of ddH2O. Use immediately.
20% PEG Buffer: Mix 10 g PEG-8000 in 25 ml 5M NaCl and bring to 50 ml with ddH2O. Shake vigorously to mix until PEG-8000 has completely gone into solution. Store at 4°C for up to 6 months
2.3. Equipment
Water bath
Thermocycler
DynaMag Magnetic Rack (e.g. Life Tech 12321D)
Nanodrop 1000
Qubit Fluorometer (e.g. Life Tech Q33216)
0.2 mL PCR tubes (e.g. Fisher 14-230-212)
1.5 mL microcentrifuge tubes (e.g. Fisher 05408129)
6% TBE-PAGE gels (e.g. Life Tech EC6265BOX)
Cell scraper (e.g. Fisher 08-100-241)
50 ml tube (e.g. Fisher 14-432-22)
Cell culture plates (e.g. Sigma CLS430167–100EA)
Microcentrifuge
Vacuum Concentrator
Sonicator (e.g., Covaris S220 Focused-ultrasonicator)
2.4. Oligoes and Primers
T-tailed Biotinylated Bridge Adaptor 5’:/5Phos/GCTGAGGGA/iBiodT/C (IDT)
Bridge Adaptor 3’T: CCTCAGCT (IDT)
Blunt Bridge Adaptor 5’: GCTGAGGGAC (IDT)
Blunt Bridge Adaptor 3’: CCTCAGC (IDT)
Illumina_PE_Adapt_F: ACACTCTTTCCCTACACGACGCTCTTCCGATC*T (IDT)
Illumina_PE_Adapt_R: PO4-GATCGGAAGAGCGGTTCAGCAGGAATGCCGAG (IDT)
Pre-Capture PCR primer_F: ACACTCTTTCCCTACACGACG (IDT)
Pre-Capture PCR primer_R: CGGTCTCGGCATTCCTGCTGAACC (IDT)
Illumina_PE-PCR_Primer_F: AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGAC (IDT)
Illumina_PE-PCR_Primer_R: CAAGCAGAAGACGGCATACGAGAT [8 bp barcode] CGGTCTCGGCATTCCTGCTGAACCG (IDT)
Bait library (e.g., SeqCap® EZ Prime Choice Probes, Roche NimbleDesign)
Adaptor-Hi-Block: ACACTCTTTCCCTACACGACGCTCTTCCGATCT (IDT)
Internal adaptor-Block: AGCTGAGGGATCCCTCAGCT (IDT)
Nbgn-8bp-ID-BL-B: CGGTCTCGGCATTCCTGCTGAACCGCTCTTCCG AT/3ddC/(IDT)
3. Methods
3.1. Crosslinking of cells (note 1)
3.1.1. Yeast cells
Yeast cells such as the saccharomyces cerevisiae strain BY4741 (genotype: Mata his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 bar1::KanMX) are cultured at 30° C by shaking overnight in 50 ml of YEP media plus 2% glucose.
Cultured cells are diluted the next morning to an OD600 = 0.2 in one liter of YEP plus 2% glucose. Cells are incubated with shaking at 30°C until reaching an OD600 = 1.0 (about 3–4 hours).
Cells are treated with 27.7 ml of 37% formaldehyde (final concentration = 1%) for 10 minutes at room temperature with constant stirring.
Fixation is quenched with 52.6 ml of 2.5 M glycine (final concentration = 0.125 M) for 15 minutes at room temperature with constant stirring.
Fixed cells are collected via centrifugation (1500xg - 5 minutes) and re-suspended in 50 ml of spheroplast buffer plus 30 mM dithiothreitol (DTT).
Fixed cells are recollected via centrifugation (1500xg - 5 minutes) and re-suspended in 50 ml of spheroplast buffer plus 1 mM DTT.
Fixed cells are converted to spheroplasts with Zymolyase 20T (Final concentration=0.66 g/L) treatment at 30° C with gentle rotation. Conversion to spheroplasts is confirmed by microscopy.
Fixed Spheroplasts are collected via centrifugation at 4° C (1500xg- 5 minutes).
Fixed spheroplasts are washed twice in 50 ml spheroplast buffer and collected via centrifugation at 4° C (1500xg- 5 minutes).
Fixed spheroplasts are re-suspended in 50 ml of 1x NEBuffer 2 and aliquot into 50 1.7-ml microcentrifuge tubes (1 ml per tube, containing about 1–2 × 109 spheroplasts). Collect spheroplasts via centrifugation (2000xg- 5 minutes) in a refrigerated desktop centrifuge. Spheroplasts can be rapidly frozen in liquid nitrogen and stored at −80oC until use (up to 1.5 years).
3.1.2. Suspension cell cultures
Pellet the cells (about 2–3 × 107) at 800xg for 5 min at RT.
Discard the supernatant and resuspend the pellet in 45 ml of fresh culture medium without serum. Break cell clumps by pipetting up and down.
Crosslink the cells by adding 1.25 ml of 37% formaldehyde (1% final concentration). Mix quickly by inverting the tube several times.
Incubate at RT for 10 min. Gently invert the tube every 1–2 min.
Add 2.5 ml of 2.5 M glycine (0.125M final concentration) to quench the cross-linking reaction, mix well.
Incubate for 10 min at RT to stop cross-linking.
Spin down the crosslinked cells at 800xg for 3 min at RT.
Discard the supernatant by aspiration and resuspend the cells with 1 x PBS (1ml PBS per 106 cells).
Split the crosslinked cell suspension into aliquots of 1–2 × 106 cells (in 1.7 ml microtubes).
Centrifuge the cross-linked cells at 800xg for 3 min at RT.
Discard the supernatant by aspiration.
Cells can be snap-frozen in liquid nitrogen and stored at −80°C until use (up to 1.5 years).
3.1.3. Adherent monolayer cell cultures
Aspirate the medium and add 10 ml of fresh medium without serum per 10 cm-plate.
Crosslink the cells by adding 280 μl of 37% formaldehyde (1% final concentration). Mix gently, immediately after addition of formaldehyde.
Incubate at room temperature (RT) for 10 min.
Add 560 μl of 2.5 M glycine (0.125M final concentration) to quench the crosslinking reaction, mix well.
Incubate for 10 min at RT to stop cross-linking completely.
Discard the supernatant by aspiration and wash the crosslinked cells with 1 x PBS once.
Scrape the cells from the plates with a cell scraper and transfer to a 1.7 ml microtube.
Split the crosslinked cell suspension into aliquots of 1–2 × 106 cells (in 1.7 ml microtubes).
Centrifuge the cross-linked cells at 800xg for 3 min at RT.
Cells can be snap-frozen in liquid nitrogen and stored at −80°C until use (up to 1.5 years).
3.1.4. Solid primary tissue cells
Place frozen tissue sample (~0.5 g) in 7 mL pre- chilled 7ml glass homogenizer with 6ml of cell lysis buffer.
Incubate on ice for 5 min.
Homogenize on ice until no clumps of cells persist, e.g., dounce 5–10 times with loose pestle followed by 10–15 strokes with tight pestle.
Transfer to a 15ml tube and spin at 800xg for 5 min at 4°C.
Resuspend the cell pellet in 10ml PBS.
Add 280 μl of 37% formaldehyde (1% final concentration), RT for 10 min, with occasionally inverting.
Quench formaldehyde with 125mM glycine by adding 500 μl 2.5 M glycine, RT for 5 min.
Pellet the cells at 800xg for 5 min.
Resuspend cells with 10 ml PBS and aliquot into 10 1.7 ml microtubes, 1ml per tube.
Centrifuge the cross-linked cells at 800xg for 3 min at RT.
Cells can be snap-frozen in liquid nitrogen and stored at −80°C until use (up to 1.5 years).
3.2. Cell permeabilization and chromatin digestion with DNase I
Resuspend one crosslinked cell aliquot (1–2 × 106 cells) in 1 ml of ice-cold cell lysis buffer containing protease inhibitor cocktail (Note 2).
Incubate on ice for 30 min.
Centrifuge for 1 min at 800xg at RT.
Discard the supernatant and resuspend the pellet in 200 μl 0.5 x DNase I SDS buffer (Note 3).
Incubate at 37°C for 60 min.
Add 200 μl of 0.5 x DNase I digestion buffer, mix well.
Incubate at 37°C for 10 min.
Add appropriate amount (e.g., 3–6 units) of DNase I, mix well.
Incubate at RT for 5 min.
Add 20 μl 0.5 M Ethylenediaminetetraacetic acid (EDTA), mix well. Save 20μl of lysate for assessing the DNase I digestion efficiency if desired (Note 4).
Add 220 μl 20% PEG-8000 buffer and 200 μl AMPure XP beads, mix well (Note 5).
Incubate at RT for 5 min, and place the tube in a DynaMag-Spin magnet for 2 min.
Discard the supernatant and wash the beads twice with 1 ml of 80% ethanol. Briefly spin down the beads and remove the residual ethanol as completely as possible, then air dry the beads for no more than 2 min.
Resuspend immediately the beads with 100 μl of water, ready for the next end-repair step.
3.3. End repair, dA-tailing and labelling of chromatin ends in intact nuclei (Note 6)
Prepare 100 μl End-Repair master mix: 70 μl ddH2O, 20 μl 10x T4 ligase buffer, 5 μl 10 mM dNTPs, 2 μl T4 DNA Polymerase (5U/μl), and 3 μl Klenow (5U/μl).
Add the100 μl End-Repair mastermix to the above DNase I digested sample. Mix well by pipetting up and down.
Incubate at RT for 1 h.
Place the tube in a DynaMag-Spin magnet for 2 min.
Discard the supernatant and wash the beads once with 200 μl ddH2O.
Resuspend immediately the beads in 100 μl of ddH2O, ready for the next dA-tailing step.
Prepare 50 μl dA-Tailing master: 20 μl ddH2O, 15 μl 10x NEBuffer 2, 5 μl 20 mM dATP, and 10 μl Klenow (EXO-) (5U/μl).
Add the 50 μl dA-Tailing master mix to the end-repaired sample from step 6. Mix well by pipetting up and down.
Incubate at 37°C for 1 h.
Place the tube in a DynaMag-Spin magnet for 2 min.
Discard the supernatant and wash the beads once with 200 μl ddH2O.
Resuspend immediately the beads in 30 μl of ddH2O, ready for the next step
Prepare 70 μl ligation master mix: 25 μl Bridge adaptor w/Biotin (40μM), 20 μl Blunt adaptor w/o Biotin (50 μM), 10 μl 10x T4 ligase buffer (with 10mM ATP), 10 μl PEG-4000 (50%), 5 μl T4 and DNA ligase (5 U/μl).
Add the 70 μl ligation master mix to the dA-tailed sample from step 13. Mix well by pipetting up and down.
Incubate at 16°C overnight.
Add 5 μl of 10% SDS to the reaction.
Place the tube in a DynaMag-Spin magnet for 2 min.
Discard the supernatant.
Resuspend the pellet in 200 μl of ddH2O.
Add 165 μl of 20% PEG buffer to the tube, mix thoroughly by pipetting up and down.
Incubate at RT for 5 min, and place the tube in a DynaMag-Spin magnet for 2 min.
Discard the supernatant and wash the beads once with 1 ml of 80% ethanol.
Briefly spin down the beads and remove the residual ethanol as completely as possible.
Resuspend the beads in 200 μl of ddH2O, mix thoroughly by pipetting up and down.
Add 165 μl of 20% PEG buffer to the tube, mix thoroughly by pipetting up and down.
Incubate at RT for 5 min, and place the three tubes in a DynaMag-Spin magnet for 2 min.
Discard the supernatant and wash the beads twice with 1 ml of 80% ethanol. Briefly spin down the beads and remove the residual ethanol as completely as possible, then air dry the beads for no more than 2 min.
Resuspend immediately the beads in 165 μl of ddH2O, ready for the next phosphorylation step.
3.4. In situ Phosphorylation and proximity ligation
Add 20 μl 10X T4 ligase buffer and 15 μl PNK (10 U/μl) to the 165 μl above adaptor-ligated sample. Mix well.
Incubate at 37°C for 1 hr.
Add the following reaction to the above tube: 265 μl ddH2O, 30 μl 10X T4 ligase buffer, and 5 μl T4 DNA ligase (5 U/μl).
Incubate at RT for 4 hr.
3.5. Reverse cross-linking and DNA purification
Centrifuge for 3 min at 800xg at RT.
Resuspend the pellet in 150 μl ddH2O.
Add 20 μl 10X NEBuffer 2, 10 μl 10% SDS, and 20 μl of 20 mg/ml Proteinase K.
Incubate overnight at 62°C.
Add 150 μl AMPure XP beads, mix well.
Incubate mixture at RT for 5 min, and place the tube in a DynaMag magnet for 2 min.
Discard the supernatant and wash the beads twice with 1 ml of 80% ethanol. Briefly spin down the beads and remove residual ethanol as completely as possible, then air-dry the beads for no more than 2 min.
Resuspend the beads in 100 μl nuclease-free water.
Incubate beads at RT for 1 min. Collect beads via DynaMag magnet and transfer eluent to fresh 1.5 mL tube.
Determine the concentration of the recovered DNA with a Nanodrop spectrophotometer. A typical yield is 3–5 μg if starting with 1–2×106 cells.
3.6. DNA sonication, end Repair, dA-tailing and ligation of sequencing adaptors (Note 7)
To shear the DNA with a Covaris S2 instrument, transfer the DNA to Covaris microtube (3–5 μg DNA in 100 μl 1x TE lysis buffer).
Shear the DNA to a size of 100 – 300 bp using the following parameters: Duty cycle: 2%, intensity: 5, cycles per burst: 200, set mode: frequency sweeping, continuous degassing, process time: 20 sec, number of cycles: 5.
Transfer the 100 μl sonicated DNA solution to a 1.7 ml microtube.
Bring the volume to 200 μl by adding ddH2O.
Add 200 μl of AMPure XP beads to the tube, mix well.
Incubate at RT for 5 min.
Place the tube in a DynaMag-Spin magnet for 2 min.
Discard the supernatant and wash the beads twice with 1 ml of 80% ethanol.
Briefly spin down the beads and remove the residual ethanol as completely as possible, then air dry the beads for 2 min.
Set up the End-repair reaction with the Fast DNA End Repair Kit: 86 μl ddH2O, 10 μl 10x Reaction buffer, and 4 μl Enzyme mix, mix well.
Incubate at 16 °C for 10 min.
Add 5 μl of 10% SDS to the tube to stop the reaction. Mix well.
Add 150 μl of 20% PEG buffer to the tube, mix well.
Incubate at RT for 5 min.
Place the tube in a DynaMag-Spin magnet for 2 min.
Discard the supernatant and wash the beads twice with 1 ml of 80% ethanol.
Briefly spin down the beads and remove the residual ethanol as completely as possible, then air dry the beads for 2 min.
Set up the dA-tailing reaction by adding: 81 μl ddH2O, 10 μl 10x NEBuffer 2, 4 μl 20 mM dATP and 5 μl Klenow (exo-) (5 U/μl).
Incubate at 37 °C for 30 min.
Add 5 μl of 10% SDS to each tube to stop the reaction. Mix well.
Add 150 μl of 20% PEG buffer to each tube, mix thoroughly by pipetting up and down.
Incubate at RT for 5 min and place the tubes in a DynaMag-Spin magnet.
Discard the supernatant and wash the beads twice with 1 ml of 80% ethanol.
Briefly spin down the beads and remove the residual ethanol as completely as possible, then air dry the beads for 2 min.
Set up the ligation reaction by adding: 34 μl ddH2O, 5 μl 10x T4 ligase Buffer (with 10 mM ATP), 5 μl PEG-4000 (50%), 3 μl Illumina-PE-adaptor (25 μM), and 3 μl T4 DNA ligase (5 U/μl).
Incubate at RT for 30 min or 16°C overnight.
Add 5 μl of 10% SDS to each tube to stop the reaction.
Add 145 μl of ddH2O to bring up the volume to 200 μl, Mix well.
Add 200 μl of 20% PEG buffer, mix well.
Incubate at RT for 5 min, and place the tube in a DynaMag-Spin magnet for 2 min.
Discard the supernatant and wash the beads once with 1.5 ml of 80% ethanol.
Briefly spin down the beads and remove the residual ethanol as completely as possible, then air dry the beads for 2 min.
Elude DNA from the beads with 100 μl of ddH2O, ready for the next biotin pull-down step.
3.7. Biotin pull-down and amplification of whole-genome DNase Hi-C library
Wash 30 μl of MyOne C1 Dynabeads with 100 μl of 1× B&W buffer,
Resuspend the beads in 100 μl of 2× B&W buffer.
Transfer the 100 μl eluted DNA from above to the tube containing the MyOne C1 beads, mix well.
Incubate the sample for 15 min at RT with rotation.
Reclaim beads against the DynaMag-Spin magnet for 1 min, discard the supernatant.
Wash beads four times with 200 μl of 1X B&W buffer containing 0.1% Tween-20.
Wash beads twice with 200 μl of EB buffer.
Resuspend the beads with 40 μl of EB buffer.
To generate DNA templates for targeted DNase Hi-C assays, prepare PCR master mix: 200 μl of 2x KAPA HiFi HotStart DNA Polymerase ReadyMix; 20 μl of 10 μM Pre-Capture PCR primer pairs; 140 μl of ddH2O.
Mix PCR master mix with the beads (400 μl in total), divide into 10 aliquots of 40 μl, and amplify by PCR using the following conditions: 3 min at 98°C; 8–9 cycles of: 20 sec at 98°C, 30 sec at 65°C, 30 sec at 72°C; 7 min at 72 °C (Note 8).
Collect PCR reactions into a new 1.5 ml tube.
Purify library by adding 0.8X volumes of AMPure XP beads.
Incubate mixture at RT for 5 min and place tube in a DynaMag magnet for 2 min.
Discard the supernatant and wash the beads twice with 1 ml of 80% ethanol. Briefly spin down the beads and remove residual ethanol as completely as possible, then air-dry the beads for 5 min.
Elute DNA in 30 μl EB buffer.
Determine the DNA concentration using Qubit dsDNA HS kit according to manufacturer’s instructions. The yield usually is 500–1,500 ng.
3.8. Enrichment of a subset of chromatin interactions of interest (Note 9)
Design of a bait library to enrich the chromatin interactions associated with specific genomic regions of interest using the design tool of a selected targeted DNA sequencing platform (e.g., the NimbleDesign tool of the SeqCap EZ system). Below is the protocol for the SeqCap EZ system (Note 10).
Transfer 500 ng to 1 μg of DNase Hi-C library into a new tube, add 1 μl of 1 mM Adaptor-Hi-Block, 1 μl of 1 mM Internal adaptor-Block, 1 μl of 1 mM of Nbgn-8bp-ID-BL-B and 5 μg of human Cot-1 DNA (for human samples) or murine Cot-1 DNA (for mouse samples).
Evaporate sample on a vacuum Concentrator at 60 °C until dry.
Add 7.5 μl of 2x Hybridization buffer and 3 μl of Hybridization Component A to the dried-down sample.
Vortex for 10 sec and centrifuge at maximum speed for 10 sec.
Denature DNA at 95 °C for 10 min in a thermocycler, and then centrifuge at maximum speed for 10 sec.
Add 4.5 μl aliquot of SeqCap EZ bait library to the tube, vortex for 3 sec and centrifuge at maximum speed for 10 sec.
Incubate in a thermocycler at 47 °C for 48–72 hours. The thermocycler’s heated lid should be turned on and set to maintain at 57 °C.
Set the temperature of a water bath at 47 °C.
Dilute 10x Wash Buffers (I, II, III and Stringent) and 2.5x Bead Wash Buffer from the SeqCap EZ Reagent Kit v2 to create 1x working solutions according to the manufactories’ instruction.
Pre-warm 400 μl of 1x Stringent Wash Buffer and 400 μl of 1x Wash Buffer I at 47 °C in advance (at least 2 hours before washing the captured DNA).
Add 100 μl of streptavidin Dynabeads M-270 into a new 1.7 ml microtube, place the tube in a DynaMag magnet for 2 min remove supernatant.
Wash beads with 200 μl of 1x Bead Wash Buffer. Vortex for 10 sec. Repeat two more times and discard the supernatant.
Transfer the hybridization samples into the tube with the magnetic beads. Mix well.
Incubate at 47 °C for 45 min. Mix the samples by vortex for 3 sec at 15 min intervals to ensure the beads remain in suspension.
Add 200 μl of pre-warmed 1x Wash Buffer I to the tube. Vortex for 10 sec.
Reclaim beads on the magnetic separation stand and remove the clear supernatant.
Re-suspend beads into 200 μl of the pre-warmed 1x Stringent Wash Buffer, mix well.
Incubate for 5 min at 47 °C.
Reclaim beads on the magnetic separation stand and remove the clear supernatant. Repeat one more time of wash the beads with 1x Stringent Wash Buffer.
Re-suspend beads into 200 μl of 1x Wash Buffer I, and mix by vortexing for 2 min.
Place the tubes in the magnetic separation stand and remove the liquid.
Re-suspend beads into 200 μl of 1x Wash Buffer II, and mix by vortexing for 1 min.
Place the tubes in the magnetic separation stand and remove the liquid.
Re-suspend beads into 200 μl of 1x Wash Buffer III, and mix by vortexing for 30 sec.
Place the tubes in the magnetic separation stand and remove the liquid.
Resuspend the beads in 50 μl of EB.
3.9. Library amplification and Quality control
Prepare PCR master mix: 200 μl of 2x KAPA HiFi HotStart DNA Polymerase ReadyMix; 20 μl of 10 μM Illumina PE forward primer; 20 μl of 10 μM indexed Illumina PE Reverse primer; 120 μl of ddH2O.
Mix PCR master mix with the beads (400 μl in total), divide into 10 aliquots of 40 μl, and amplify by PCR using the following conditions: 3 min at 98°C; 12–18 cycles of: 20 sec at 98°C, 30 sec at 65°C, 30 sec at 72°C; 7 min at 72 °C (Note 11).
Collect PCR reactions into a new 1.5 ml tube.
Purify library by adding 0.8X volumes of AMPure XP beads.
Incubate mixture at RT for 5 min and place tube in a DynaMag magnet for 2 min.
Discard the supernatant and wash the beads twice with 1 ml of 80% ethanol. Briefly spin down the beads and remove residual ethanol as completely as possible, then air-dry the beads for 5 min.
Elute DNA in 30 μl EB buffer.
Determine the DNA concentration using Qubit dsDNA HS kit according to manufacturer’s instructions.
Quantitate amount of dsDNA in library using Qubit dsDNA HS kit as per manufacturer’s protocols.
Digest a small aliquot of the final DNase Hi-C library (50 – 100 ng) with BamHI to estimate the portion of molecules with valid biotinylated junctions as follows: 1 μl of 10X Fast digestion buffer, 1 μl of targeted DNase Hi-C product, 1 μl of Fast digestion BamHI, and 7 μl ddH2O. Also set a control reaction without adding the BamHI enzyme.
Incubate at 37°C for 30 min.
Run both the BamHI digestion and control reactions side-by-side on a 6% TBE-PAGE gel. Digested libraries should demonstrate a marked shift in library size distribution, as shown in [16, 22]. If libraries pass this QC metric, proceed to Illumina sequencing (Note 12).
4. Notes
Different types of cells may require different crosslinking conditions with formaldehyde. Here, the experimental protocols for preparing crosslinked yeast cells, mammalian monolayer and suspension cultures, and solid tissue samples are listed. However, for other cell types, crosslinking conditions might need to be optimized. For example, efficient crosslink of mouse and human embryonic stem cells, which often aggregate to form large clumps during growth, usually requires one of two alternative approaches: preparing single-cell suspension before carrying out crosslinking under the same condition as for suspension cells, or crosslinking the cells using the same protocol as for adherent monolayer cultures but with increased formaldehyde concentration or longer fixation time [16].
To achieve optimal complexity and resolution of targeted DNase Hi-C libraries, we recommend starting with 3–5 million cells for a targeted DNase Hi-C assay (or even more cells, depending on the size of the targets and the desired resolution). When starting with more cells, usually three vials of 1–2 million cells can be proceeded in parallel.
The concentration of SDS in the 0.5 x DNase I SDS buffer can be cell type-specific. When carrying out targeted DNase Hi-C assays on a cell type for the first time, we recommend to titrate the optimal SDS concentration. In general, the SDS concentration should be within 0.2–0.5%.
To assess DNase I digestion efficiency, add 70 μl of 1x TE lysis buffer and 10 μl of Proteinase K (20 mg/ml) to 20 μl of lysed cells from this step. Incubate for 30 min at 65°C. Purify DNA by the Qiaquick PCR purification kit. Check size distribution of the DNA fragments by running the sample on 1% agarose gel. In general, efficient DNase I digestion will result in the majority of DNA fragments with a size less than 1kb [16, 22]. This QC step is recommended when applying DNase Hi-C protocol to a new cell type.
In the protocols of DNase Hi-C, in situ DNase Hi-C and targeted DNase Hi-C, SPRI (Solid Phase Reversible Immobilization) magnetic beads (e.g., Ampure XP) are employed to immobilize nuclei/chromatin complexes, which enables efficient removal of DNase I and the DNA modifying enzymes used in the of the protocols, including the Klenow enzyme, T4 DNA polymerase, Klenow fragment (3’-->5’ exo-), and T4 DNA ligase [16]. These beads are also essential for the efficient removal of the low-molecular-weight DNA fragments that escape the nucleus following cell lysis and chromatin digestion free, and more importantly, for the efficient removal of unligated internal bridge adaptors following bridge-adaptor ligation. If not removed, the unligated internal bridge adaptors will severely interfere the downstream in situ proximity ligation step and lead to high percentage of dangling ends in the final DNase Hi-C libraries. Plus, when starting with a low number of cells (e.g., less than 1 million cells), the SPRI beads (usually with a brown color) can help visualize the cell/nuclei pellets throughout the protocol.
The efficiency of these three enzymatic reactions in intact nuclei, Klenow and T4 DNA polymerase enzyme-catalyzed DNA end-repair, Klenow (Exo-)-mediated dA-tailing and ligation of the biotinylated-bridge adaptors to the dA-tailed chromatin ends by T4 DNA ligase, largely determine the success of an in situ DNase Hi-C or targeted DNase Hi-C assay. Hence, a control ligation reaction in parallel to the experimental ligation of the biotinylated-bridge adaptors can be performed to examine the efficiency of these reactions. Briefly, a small portion (~5–10%) of dA-tailed sample can be taken and ligated to the dT-tailed Illumina sequencing Y-adaptors. After the ligation, genomic DNA is then purified and the ligation efficiency examined by performing quantitative PCR (qPCR) with the appropriate Illumina PCR primers [16]. If amplification signal appears before 10 PCR cycles using 10 ng of genomic DNA as a template, it suggests that the efficiency of the upstream end-repair and dA-tailing steps are acceptable. This QC step is recommended when applying the DNase Hi-C protocol to a new cell type.
When the amount of the starting DNase Hi-C genomic DNA is low (e.g., less than 1 μg), these four steps can be replaced by using the Tagmentation-based library construction protocol with the Illumina Nextera DNA Library Preparation Kit.
The number of PCR cycles for amplifying the whole-genome library to generate sufficient templates for the downstream hybridization-mediated capture assay is dependent on both the amount of starting materials and the efficiency of the upstream enzymatic reactions. To determine the appropriate number of PCR cycles, trial PCR reactions with 6, 8, 10, or 12 cycles can be carried out as follows: 3 ul ddH2O, 5 ul 2X HotStart ReadyMix, 1 ul 10 μM Pre-Capture PCR primer pairs, 1 μl of DNA-bound streptavidin beads. Using the following PCR program: 3 min at 98°C; indicated numbers of cycles of: 20 sec at 98°C, 30 sec at 65°C, 30 sec at 72°C; 7 min at 72 °C. Then run the 10 μl of each PCR reaction product on a 2% agarose gel to determine the appropriate number of cycles and amount of input beads for the large-scale library amplification.
In principle, the enrichment of a subset chromatin interactions of interest can be achieved by the various array-based or in-solution-based DNA capture techniques, such as the Agilent Sureselect and the Roche NimbleGen SeqCap EZ technologies [16]. The protocol described here is for using the SeqCap EZ technology.
Targeted DNase Hi-C can be used to enrich chromatin interactions associated with any genomic region or regions of interest. The target regions can be continuous genomic segments (even an entire chromosome), or discrete cis-regulatory elements (e.g., promoters, enhancers, transcription factors’ binding sites and DNase I hypersensitive sites, etc.). In principle, the size of the targeted regions can also vary, ranging from a few kilobases to hundreds of megabases.
The number of PCR cycles for amplifying the targeted DNase Hi-C library to generate sufficient materials for Illumina sequencing is largely dependent on the overall size of the target genomic regions. The bigger the overall size of the targeted regions, the more chromatin interactions they associated and the fewer PCR cycles required. It is recommended to carry out trial PCR reactions to determine the appropriate number of PCR cycles.
Before the purified targeted DNase Hi-C libraries being subjected to high throughput sequencing, the capture efficiency of the libraries can be estimated using quantitative PCR (qPCR) assays. PCR primers to specific target regions can be designed and qPCR carried out by using the targeted DNase Hi-C library or the parent whole-genome DNase Hi-C library as templates. The enrichment of a specific target can be assessed by comparing its amplification rate from the target DNase Hi-C library with that from the parent whole-genome DNase Hi-C library [16]. Since the enrichment efficiency of a specific target in a targeted DNase Hi-C assay is determined by multiple factors and is likely to be uneven between individual targets, it is recommended to examine enrichment at multiple individual targets simultaneously using this qPCR assay to ensure the accuracy of the assessment.
Acknowledgments
This work was supported by the UW Bridge Fund (ZD), ASH Bridge Grant (ZD), and the NIH Common Fund U54DK107979.
References
- 1.Cremer T, Cremer M (2010) Chromosome territories. Cold Spring Harbor perspectives in biology 2:a003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Wit E, De Laat W (2012) A decade of 3C technologies: insights into nuclear organization. Genes & development 26:11–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dekker J, Rippe K, Dekker M et al. (2002) Capturing chromosome conformation. Science 295:1306–1311 [DOI] [PubMed] [Google Scholar]
- 4.Denker A, De Laat W (2016) The second decade of 3C technologies: detailed insights into nuclear organization. Genes & development 30:1357–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dixon JR, Selvaraj S, Yue F et al. (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dostie J, Richmond TA, Arnaout RA et al. (2006) Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome research 16:1299–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duan Z, Andronescu M, Schutz K et al. (2010) A three-dimensional model of the yeast genome. Nature 465:363–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duan Z, Blau CA (2012) The genome in space and time: does form always follow function? How does the spatial and temporal organization of a eukaryotic genome reflect and influence its functions? Bioessays 34:800–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fang R, Yu M, Li G et al. (2016) Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell research 26:1345–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fullwood MJ, Liu MH, Pan YF et al. (2009) An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 462:58–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guelen L, Pagie L, Brasset E et al. (2008) Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453:948–951 [DOI] [PubMed] [Google Scholar]
- 12.Hughes JR, Roberts N, Mcgowan S et al. (2014) Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nature genetics 46:205–212 [DOI] [PubMed] [Google Scholar]
- 13.Krumm A, Duan Z (2018) Understanding the 3D genome: Emerging impacts on human disease. Seminars in cell & developmental biology Epub ahead of print [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lieberman-Aiden E, Van Berkum NL, Williams L et al. (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326:289–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma W, Ay F, Lee C et al. (2015) Fine-scale chromatin interaction maps reveal the cis-regulatory landscape of human lincRNA genes. Nature methods 12:71–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma W, Ay F, Lee C et al. (2018) Using DNase Hi-C techniques to map global and local three-dimensional genome architecture at high resolution. Methods 142:59–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mifsud B, Tavares-Cadete F, Young AN et al. (2015) Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nature genetics 47:598–606 [DOI] [PubMed] [Google Scholar]
- 18.Mumbach MR, Rubin AJ, Flynn RA et al. (2016) HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nature methods 13:919–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nemeth A, Conesa A, Santoyo-Lopez J et al. (2010) Initial genomics of the human nucleolus. PLoS genetics 6:e1000889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nora EP, Lajoie BR, Schulz EG et al. (2012) Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peric-Hupkes D, Meuleman W, Pagie L et al. (2010) Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Molecular cell 38:603–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramani V, Cusanovich DA, Hause RJ et al. (2016) Mapping 3D genome architecture through in situ DNase Hi-C. Nat Protoc 11:2104–2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramani V, Shendure J, Duan Z (2016) Understanding Spatial Genome Organization: Methods and Insights. Genomics, proteomics & bioinformatics 14:7–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rao SS, Huntley MH, Durand NC et al. (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159:1665–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simonis M, Klous P, Splinter E et al. (2006) Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nature genetics 38:1348–1354 [DOI] [PubMed] [Google Scholar]
- 26.Van Koningsbruggen S, Gierlinski M, Schofield P et al. (2010) High-resolution whole-genome sequencing reveals that specific chromatin domains from most human chromosomes associate with nucleoli. Molecular biology of the cell 21:3735–3748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao Z, Tavoosidana G, Sjolinder M et al. (2006) Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nature genetics 38:1341–1347 [DOI] [PubMed] [Google Scholar]