

Abstract
COPD likely has developmental origins; however, the underlying molecular mechanisms are not fully identified. Investigation of lung tissue-specific epigenetic modifications such as DNA methylation using network approaches might facilitate insights linking in utero smoke (IUS) exposure and risk for COPD in adulthood.
We performed genome-wide methylation profiling for adult lung DNA from 160 surgical samples and 78 fetal lung DNA samples isolated from discarded tissue at 8–18 weeks of gestation. Co-methylation networks were constructed to identify preserved modules that shared methylation patterns in fetal and adult lung tissues and associations with fetal IUS exposure, gestational age and COPD.
Weighted correlation networks highlighted preserved and co-methylated modules for both fetal and adult lung data associated with fetal IUS exposure, COPD and lower adult lung function. These modules were significantly enriched for genes involved in embryonic organ development and specific inflammation-related pathways, including Hippo, phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT), Wnt, mitogen-activated protein kinase and transforming growth factor-β signalling. Gestational age-associated modules were remarkably preserved for COPD and lung function, and were also annotated to genes enriched for the Wnt and PI3K/AKT pathways.
Epigenetic network perturbations in fetal lung tissue exposed to IUS and of early lung development recapitulated in adult lung tissue from ex-smokers with COPD. Overlapping fetal and adult lung tissue network modules highlighted putative disease pathways supportive of exposure-related and age-associated developmental origins of COPD.
Introduction
COPD remains a major cause of mortality worldwide [1–3], and is caused by genetic and epigenetic risk factors [4, 5] that communicate through biological networks. Early-life factors associated with reduced maximally attained lung function [6] and accelerated lung function decline [5] might predispose to adult lung diseases such as COPD [7–9]. Investigating these factors has become a greater focus [10], as these insights may impact diagnostic, therapeutic and preventative interventions. Maternal smoking during pregnancy is prevalent, at 14–26% in the US population [11–13] and 10–35% worldwide [12, 14]. This poses severe risks to the developing fetus and fetal lung [15], including intrauterine growth abnormalities [16, 17], pre-term birth [18], low birthweight [19] and altered immune responses [20, 21]. DNA methylation has been suggested as a potential mediator between maternal smoking and low birthweight [22]. A longitudinal study identified persistently perturbed methylation marks associated with maternal smoking during pregnancy [23]. While the detrimental effects of parental smoke exposure on early-life development [24] and its associations with bronchopulmonary dysplasia [25] are widely recognised, links to asthma [26] and adult chronic diseases including COPD have been emerging more recently [27–32]. There is also evidence that maternal smoking is a risk factor for severe, early-onset COPD [33]. A recent epigenome-wide meta-analysis identified more than 6000 significant differentially methylated CpG sites in cord blood associated with maternal smoking and lung developmental processes [34]. A recent meta-analysis of 1688 children in five cohorts identified differentially methylated regions in cord blood DNA, associated with lower lung function in newborns and asthma and COPD in later life [35].
Identifying early COPD and associated mechanisms is crucial for an improved understanding of lung disease pathophysiology and defining therapies [36]. DNA methylation represents a molecular mark that is critical for pre- and post-natal lung development but perturbable by in utero exposures [37]. Such perturbations may set the fetal lung on a trajectory of altered development and risk for adult lung diseases, including COPD. Experimental data suggest that DNA methylation changes are dynamic and tissue specific during early development [38]. However, there are limited studies in lung tissue investigating epigenetic links to the developmental origins of COPD. Network approaches can be used to cluster complex biological processes most relevant for age-related respiratory diseases [39, 40]. In this study, we hypothesise that epigenetic signatures of in utero smoke (IUS) exposure and age in fetal lung tissue are associated with COPD and lung function in adult lung tissue, and would highlight functionally relevant co-methylation modules supportive of the developmental origins of adult lung disease.
Methods
Study samples
The fetal lung DNA samples were isolated from discarded tissue at 8–18 weeks of gestation at pseudoglandular and early canalicular stages of human lung development as previously described [41]. Approval was obtained from the Partners Human Research Committee Institutional Review Board (Boston, MA, USA).
Genome-wide methylation assays were performed using 750 ng bisulfite-treated DNA per sample using the Infinium HumanMethylation450 BeadChip array (Illumina, San Diego, CA, USA) [42] and further data processing was performed using Minfi [43]. Details on sample dropout and filtering of CpGs are described in supplementary table S1. Details on ancestry composition estimates [44] and adjustment for confounding using BACON [45] are included in the supplementary material. Since the fetal lung tissue samples were de-identified, data were only available for gestational age, fetal sex and cotinine levels. Sex was determined using unique Y chromosome microarray probes as previously described [46] and verified using X and Y chromosome methylation. IUS exposure was inferred by measuring placental cotinine concentrations. Exposure was treated as both a continuous and dichotomous predictor variable, with levels of cotinine ⩽7.5 ng·g−1 considered as unexposed (control group) and levels of cotinine >7.5 ng·g−1 considered as exposed; this level has been reported to have a high sensitivity and specificity for smoke exposure during pregnancy [41]. Further details are available in Chhabra et al. [47]. We used our previously published adult lung tissue methylation results (from the Lung Tissue COPD (LT-COPD) study; n=160: 114 cases and 46 healthy smoker controls) to replicate the fetal lung dataset findings [48].
Network approach: weighted gene correlation network analysis
An overview of the analytical pipeline is represented in figure 1. For fetal lung data, methylation was the dependent variable (outcome) and IUS exposure was the predictor. Quality control, pre-processing (supplementary table S1) and differential methylation analyses for fetal lung data were performed prior to performing network analyses to make the data suitable for comparison to the adult lung tissue site-specific CpG analysis [48], as detailed in the supplementary material. We further performed an independent network co-methylation analysis in both tissues.
FIGURE 1.
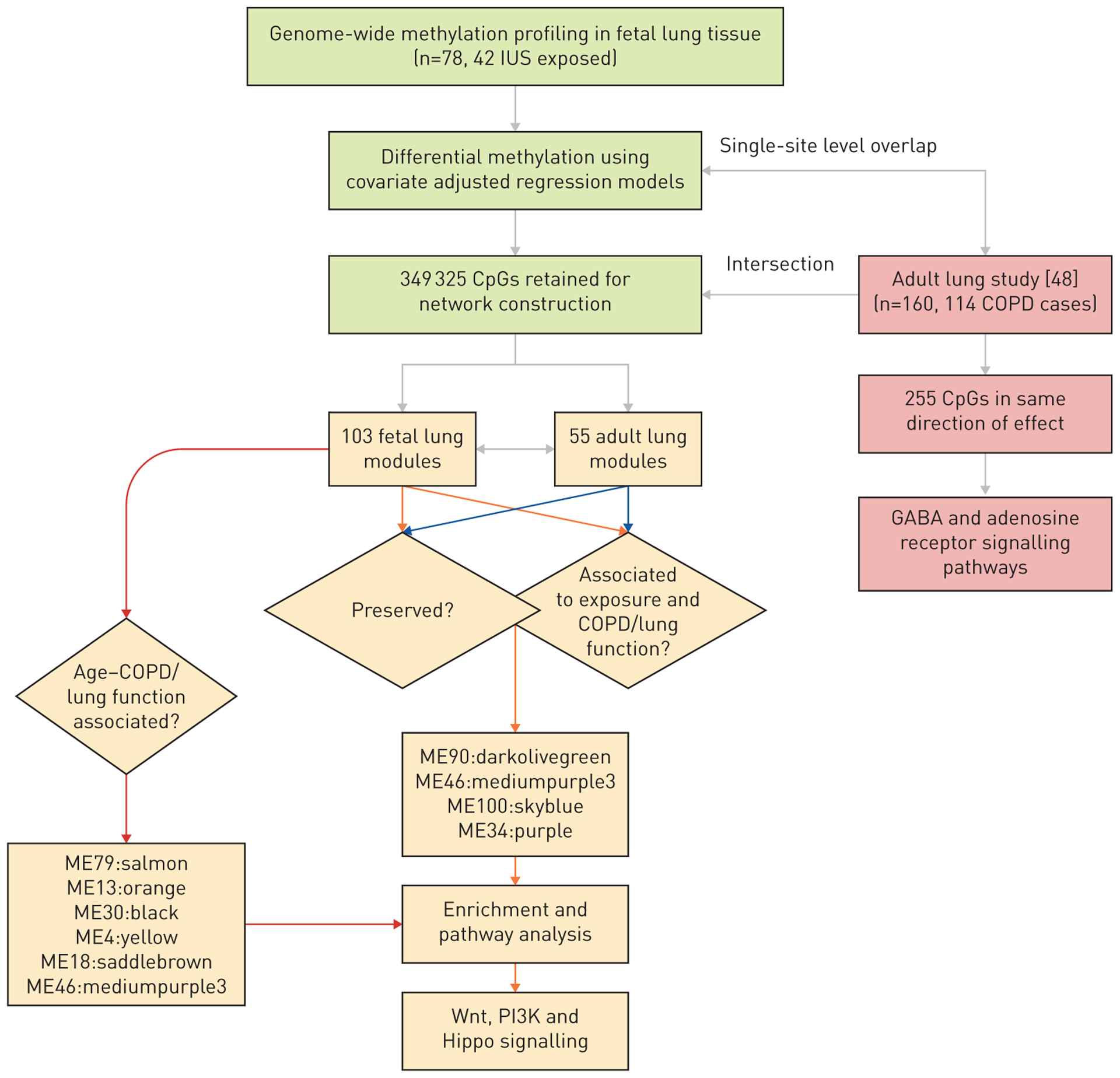
Overview of the study flowchart and analytical pipeline for pre-processing and module network creation using weighted correlation network analysis (WGCNA). GABA: γ-aminobutyric acid; ME: module eigengene; PI3K: phosphatidylinositol 3-kinase. ME90:darkolivegreen, ME46:mediumpurple3, ME100:skyblue, ME34:purple, ME79:salmon, ME13:orange, ME30:black, ME4:yellow and ME18:saddlebrown correspond to the resulting modules from the WGCNA output. The colours in boxes represent different stages of the analytical pipeline. Green, yellow and pink colours correspond to the steps for data pre-processing, downstream post-processing, and overlap of fetal and adult lung tissue single-site CpG analysis, respectively. Orange and blue arrows point to the modules and associated findings resulting from fetal lung and adult lung as reference networks, respectively.
The filtered and post-processed 349 355 CpGs from the fetal lung data were evaluated for overlap with the quality controlled CpGs from the LT-COPD study. The overlapping set of 349 325 CpGs from both the fetal lung (reference) and the adult LT-COPD (test) datasets was retained for identifying co-methylated modules (clusters of correlated CpGs) and their associations with phenotypic traits using weighted correlation network analysis (WGCNA) [49]. Modules are named based on associated numbers and colours as output from WGCNA. Phenotype variables including age, forced expiratory volume in 1 s (FEV1), forced vital capacity (FVC), FEV1/FVC ratio, quantitative markers of emphysema (low-attenuation areas <−950 HU (LAA-950) and the Hounsfield units value corresponding to the 15th percentile on the density histogram (Perc15)), time since quitting smoking (months) and pack-years (quantitative measure for smoking intensity) were analysed as continuous except for sex, IUS exposure status (1 or 0 with 1=exposed) and COPD status (1 or 0 with 1=COPD cases) for investigating module–trait associations. Module membership (MM) quantifies the contribution of each CpG in the module and allows screening for disease-related hub CpGs/genes. We also evaluated module preservation between fetal and adult lung datasets defined by Zsummary as a measure of connectivity of CpGs within co-methylated modules. As a sensitivity analysis, we used the LT-COPD dataset as the starting reference and repeated the aforementioned analyses to assess whether the LT-COPD-associated modules for COPD, lung function and age recapitulate in fetal lung tissue. Our workflow for the network analysis using WGCNA is shown in figure 1. Detailed methods and parameters used for reproducibility of the network analysis are described in the supplementary material.
Enrichment and pathway analysis
The R packages missMethyl [50, 51] and g:Profiler [52] were used to perform enrichment analysis for the differentially methylated CpGs and the annotated genes, respectively. Enrichment Map [53] in Cytoscape version 3.6.1 (cytoscape.org) was used to create a network representation of the biological processes enriched in the modules of interest (false discovery rate (FDR) <0.05), using the g:Profiler output including transcription factors, Gene Ontology terms, Reactome and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways.
Integration with prior gene expression and genome-wide association study associations
In line with our hypothesis to identify fetal origins of COPD, we investigated all CpGs with MM ⩾0.70 and with gene annotations for correlations with the previously published COPD differential expression in the LT-COPD dataset [48] and with 82 published COPD genome-wide association studies (GWASs) [54].
Results
Support for the fetal origins of COPD using network methods
Our study included 78 fetal lung (42 IUS exposed and 36 unexposed) samples and 349 355 CpGs that passed the quality control measures (supplementary table S1); of those, 349 325 CpGs overlapped with the LT-COPD dataset. We first evaluated the 535 significant differentially methylated CpG–COPD associations (FDR <5% and difference in mean methylation >1%) published from our LT-COPD study [48] and investigated their methylation patterns in the fetal lung epigenome-wide study (EWAS) findings (supplementary figures S1–S4 and supplementary tables S2–S4). Overlapping CpGs highlighted robust overlap for genes (permutation-based p-value=9.9×10−4) (supplementary figure S5) including CHRM1. CHRM1 had the highest methylation difference in the LT-COPD dataset (cg06289725: FDR 1.81×10−6; 10% hypermethylated in COPD cases) and nominal significance and same direction of effect in the fetal lung data with a relative hypermethylation of 1.9% in smoke-exposed compared with unexposed fetal lung samples (supplementary figure S6; detailed in the supplementary material).
IUS exposure–COPD associations and preserved modules
We performed an independent network analysis using WGCNA for the overlapping set of 349 325 CpGs from the fetal lung methylation data and identified 319 366 CpGs assigned to 103 co-methylation modules (supplementary figure S7a). A brief framework for selecting preserved module–trait associations between fetal and adult lung datasets, focusing on lung tissue modules with high network preservation, is highlighted in table 1.
TABLE 1.
Preserved module associations for gestational age, in utero smoke exposure and COPD/lung function with their annotated pathways
| Modules | CpGs | Fetal lung associations | LT-COPD associations | Preservation (score)# | Pathways (number of annotated genes)¶ | Genes with highest MM | |
|---|---|---|---|---|---|---|---|
| Binary | Continuous | ||||||
| Preserved and exposure-COPD/lung function-associated modules | |||||||
| ME34:purple | 5024 | Yes | Yes | FEV1 | High (18) | Hippo (n=44), Wnt (n=89), MAPK (n=13), protein serine/threonine kinase (n=69) and TGF-β (n=41) | FOXB1 |
| ME90:darkolivegreen | 543 | No | Yes | Sex, FEV1, FVC | High (15) | snRNP, scaRNP, snoRNP and telomerase biogenesis (n=4) | TSSK6 |
| ME100:skyblue | 712 | Yes | Yes | FEV1, FVC, FEV1/FVC | High (10) | ERBB4 (n=5) and cell-cell signalling (n=10) | SLC26A8 |
| Preserved and age-COPD/lung function-associated modules | |||||||
| ME13:orange | 878 | No | No | COPD, FEV1, FVC, FEV1/FVC, months quit | High (36) | Wnt signalling (n=23), PI3K/AKT (n=19) and MAPK (n=17) network | ZNF704 |
| ME79:salmon | 3348 | No | No | COPD, FEV1, FVC, FEV1/FVC, months quit | High (35) | Protein serine/threonine kinase (n=44), Wnt (n=53), PI3K (n=16), ERBB (n=21), TGF-β (n=23) and BMP (n=24) signalling | PRDM8 |
| ME30:black | 10135 | No | No | COPD, FEV1, FVC, FEV1/FVC | High (18) | Protein serine/threonine kinase signalling (n=83), Wnt signalling (n=133), BMP (n=45), PI3K (n=38) and GABA (n=9) signalling | PCOLCE |
| ME4:yellow | 29032 | No | No | COPD, FEV1, FVC, FEV1/FVC, months quit | High (16) | ERBB (n=94), PI3K/AKT (n=66), Wnt (n=191), EGFR (n=74), protein serine/threonine kinase (n=150), TGF-β (n=91) and IFN-γ-mediated (n=50) signalling | RNF213 |
| ME18:saddlebrown | 682 | No | No | COPD, FEV1, FVC, FEV1/FVC | High (13) | Trans-synaptic and synaptic signalling (n=22) | HNF1A |
| Exposure and age-COPD/ lung function-associated and preserved modules | |||||||
| ME46:mediumpurple3+ | 284 | Yes | Yes | COPD, FEV1, FVC, FEV1/FVC | Moderate (7) | RNP biogenesis (n=11), PI3K/AKT (n=8), MHC mediation (n=8),Wnt signalling (n=5), ER-phagosome (n=4) and MAPK (n=6) signalling | HARS2, GUK1 |
The table highlights associations with exposure in fetal lung and COPD/lung function in the Lung Tissue COPD (LT-COPD) study dataset, associations with gestational age in fetal lung and COPD/lung function in the LT-COPD dataset, and associations with both gestational age and exposure in fetal lung and COPD/lung function in the LT-COPD dataset. MM: module membership; FEV1: forced expiratory volume in 1 s; MAPK: mitogen-activated protein kinase; TGF: transforming growth factor; FVC: forced vital capacity; RNP: ribonucleoprotein; snRNP: small nuclear RNP; scaRNP: small Cajal body-specific RNP; snoRNP: small nucleolar RNP; PI3K/AKT: phosphatidylinositol 3-kinase/protein kinase B; BMP: bone morphogenetic protein; GABA: γ-aminobutyric acid; EGFR: epidermal growth factor receptor; IFN: interferon; MHC: major histocompatibility complex; ER: endoplasmic reticulum.
a preservation score (Zsummary) as defined by the default weighted correlation network analysis criteria [49] was used to assess the density and connectivity patterns between CpGs in a module. Zsummary 2–<10 implies weak to moderate preservation and Zsummary ⩾10 implies strong and high preservation. Additionally, modules with Zsummary 6–<10 were considered moderately preserved. Only high or moderately preserved modules are included and the scores are rounded without decimal places.
genes in the highlighted pathways for each module are detailed in supplementary table S19.
module associated with both exposure and gestational age in fetal lung and COPD/lung function in the LT-COPD dataset.
We identified seven modules, i.e. ME90:darkolivegreen, ME49:darkseagreen, ME97:indianred3, ME46: mediumpurple3, ME100:skyblue, ME34:purple and ME6:plum1, associated with (nominal p⩽0.05) either measure of IUS exposure (binary or continuous) (figure 2a and supplementary table S5). ME46: mediumpurple3, ME100:skyblue and ME34:purple were associated with both measures of exposure (figure 2a). Projecting the fetal lung module structure as reference onto the adult lung tissue methylation data (supplementary figure S7b), we observed that these IUS exposure modules identified in fetal lung associate with either COPD or with FEV1 and FEV1/FVC in adult lung tissue subjects (figure 2b). The ME100: skyblue module had a relatively suggestive association for COPD, but they all remained significant for FEV1 at FDR <0.05 (figure 2b and supplementary table S6). Based on Zsummary, three of the seven exposure modules, i.e. ME34:purple, ME90:darkolivegreen and ME100:skyblue, were highly preserved and ME46:mediumpurple3 was moderately preserved between both fetal lung and COPD datasets that we refer to as exposure–COPD modules (table 1, supplementary figure S8 and supplementary table S7). Consistent with the associations, eigengene values of the ME90:darkolivegreen module were relatively increased and positively correlated with higher cotinine values in IUS-exposed compared with unexposed fetal lung samples (supplementary figure S9a and b). The largest positive eigengene values of the ME90: darkolivegreen module were driven by lower FEV1 values in COPD adults compared with controls, with pack-years pointing towards more recent smoking cessation (supplementary figure S9c–e). For the ME34: purple and ME100:skyblue modules, eigengene values were significantly correlated with IUS-exposed subjects in the fetal lung dataset and lower FEV1 in the LT-COPD dataset (supplementary figures S10 and S11). Based on the MM criteria, FOXB1 (MM=0.881), TSSK6 (MM=0.804) and SLC26A8 (MM=0.871) were the top genes in the ME34:purple, ME90:darkolivegreen and ME100:skyblue modules, respectively (supplementary table S8).
FIGURE 2.

Module–trait association heatmaps for the selected nine in utero smoke (IUS) exposure and gestational age modules associated with COPD and lung function phenotypes highlighting fetal origins for COPD at the network level. Cat_exposure: IUS exposure (exposed versus unexposed); Cont_exposure: IUS exposure (continuous cotinine variable); COPD_ctrl: COPD (cases versus controls); FEV1: forced expiratory volume in 1 s; FVC: forced vital capacity; LAA-950: low-attenuation areas <−950 HU; Perc15: Hounsfield units value corresponding to the 15th percentile on the density histogram. a) Association of modules in the fetal lung network with the traits of interest in the fetal lung data. b) Association of Lung Tissue COPD dataset traits of interest projected on to the fetal lung module network as reference. The y-axis shows the module labels (module eigengene number:colour), followed by the number of CpGs in the respective module. The panels highlight modules associated with IUS exposure, gestational age, COPD and lung function phenotypes. The blue and yellow colours in the boxes represent the scale for significance (nominal p<0.05), with blue being not significant and yellow being highly significant. The mean β-effect estimate per module corresponding to the trait is shown below in parentheses. Missing data: FEV1 and FVC were missing for 13 subjects, LAA-950 and Perc15 were missing for 55 subjects, and months since quitting smoking was missing for one subject. Subjects with missing data were removed prior to running models for these predictors.
Fetal age–COPD modules suggestive of developmental perturbations
18 modules were associated with fetal age but not cotinine exposure (supplementary figure S7a (FDR <0.05) and supplementary table S5). 15 of those were associated with COPD in adult lung tissue (supplementary figure S7b and supplementary table S6). Most of the fetal age-associated modules were also associated with lung function phenotypes (FEV1 and FEV1/FVC) (supplementary table S6). Based on Zsummary, the highly preserved age-associated modules (figure 2a and b) included ME13:orange, ME79: salmon, ME30:black, ME4:yellow and ME18:saddlebrown, with ME46:mediumpurple3 as moderately preserved (table 1, supplementary figure S8 and supplementary table S7). The highest preserved ME13: orange module also had a suggestive association with quantitative computed tomography measures of emphysema (LAA-950; p=0.075) (figure 2b). Based on the MM criteria, PRDM8 (MM=0.91), ZNF704 (MM=0.92), PCOLCE (MM=0.92), RNF213 (MM=0.96) and HNF1A (MM=0.79) were the top genes annotated to CpGs in the ME79:salmon, ME13:orange, ME30:black, ME4:yellow and ME18:saddlebrown modules, respectively (supplementary table S9).
Module connecting exposure and fetal development to COPD
The ME46:mediumpurple3 module showed moderate preservation (supplementary table S7), and its association with cotinine exposure and age in fetal lung (nominal p<0.05) (supplementary table S5) and COPD and all lung function phenotypes in the LT-COPD dataset (figure 2 and table 1) made it an interesting module to study further. The associations with fetal age and COPD, FEV1 and FEV1/FVC in the LT-COPD dataset remained significant at FDR <0.05 (supplementary table S6).
Consistent with those associations, the ME46:mediumpurple3 module was strongly associated with IUS exposure and the methylation levels of this module were overall 1.4% higher among IUS-exposed compared with unexposed fetal lung samples (figure 3a). Moreover, smoke-exposed samples having high cotinine levels were positively correlated with the ME46:mediumpurple3 module eigengene (R=0.30, p=8.4×10−3) (figure 3b). Notably, the methylation levels were also overall 1.1% higher in COPD cases (figure 3c) and this positive eigengene association was correlated with lower FEV1 values (R=−0.32, p=1.0×10−4) (figure 3d), but not associated with cigarette smoke exposure/pack-years (R=0.09, p=0.22) (figure 3e). Based on the MM criteria, HARS2 (MM=0.79) and GUK1 (MM=0.79) were the top genes in the ME46:mediumpurple3 module (supplementary table S8). Seven significant hypermethylated CpGs were annotated to the transcription start site of multisite gene STK19 within the module (supplementary figure S12).
FIGURE 3.

Correlation plots between the ME46:mediumpurple3 methylation eigengene and traits of interest. COPD: chronic obstructive pulmonary disease; FEV1: forced expiratory volume in 1 s; IUS: in utero smoke. Eigengene values of the ME46:mediumpurple3 module were a) robustly increased and b) associated with higher cotinine values in IUS-exposed fetal lung samples compared with unexposed samples. For exposure, 0 corresponds to unexposed samples and 1 corresponds to IUS-exposed samples. Eigengene values of the ME46:mediumpurple3 module were c) robustly increased and d) significantly correlated with lower FEV1 values in COPD adults compared with smoker controls, but e) not associated with cigarette smoke intensity (pack-years). For b), d) and e), p-values and correlations are computed using Pearson’s method and the regression lines are plotted for the overall data along with the 95% confidence intervals.
Enrichment and pathway analysis
Among the preserved and IUS exposure–COPD or lung function-associated modules, the ME34:purple module was enriched in several developmental processes and pathways, including Hippo, Wnt, mitogen-activated protein kinase (MAPK) and transforming growth factor (TGF)-β signalling as well as several transcription factors (supplementary table S10). The ME90:darkolivegreen module (supplementary figure S13) was primarily associated with positive regulation of telomerase RNA localisation to Cajal bodies (supplementary table S11). The ME100:skyblue module (supplementary figure S14 and supplementary table S11) was enriched for cell–cell communication and ERBB4 signalling with members of the Hippo pathway and cadherin gene family. Fetal age–COPD-associated modules (ME79:salmon, ME13:orange, ME30:black, ME4:yellow and ME18:saddlebrown) were mostly large and demonstrated significant enrichment (supplementary tables S12–S16 and supplementary figure S15) related to development, proliferation, differentiation and morphogenesis. Significantly enriched pathways included regulation of the phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT) network, ERBB receptor family and Wnt signalling, and signalling by receptor tyrosine kinases and MAPK. Interestingly, most interconnected and enriched terms in the ME13:orange module included Wnt signalling and the PI3K/AKT signalling network (supplementary table S15 and figure 4). The ME46:mediumpurple3 module (supplementary table S11 and supplementary figure S16) was enriched for viral pathways (FDR adjusted p=6.58×10−7), ribonucleoprotein biogenesis (FDR adjusted p=4.09×10−5), DNA repair (FDR adjusted p=8.44×10−4) and PI3K/AKT signalling (FDR adjusted p=1.81×10−3).
FIGURE 4.

Reactome analysis reveals phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT) and Wnt signalling pathway networks for the ME13:orange module (false discovery rate <0.05). PDGF: platelet-derived growth factor; PIP3: phosphatidylinositol 3,4,5-trisphosphate; MAPK: mitogen-activated protein kinase; PI5P: phosphatidylinositol 5-phosphate; PP2A: protein phosphatase 2A; IER3: immediate early response 3; NCAM: neural cell adhesion molecule. Node size corresponds to the number of significant genes in the enriched term (supplementary table S15). Node colour corresponds to the significance of the term (false discovery rate <0.05). Edge thickness corresponds to the number of genes that overlap between two connected terms/gene sets.
Adult lung network projection onto fetal lung methylation
To assess the robustness of our findings, we repeated our analyses using the overlapping set of 349 325 CpGs from the adult LT-COPD dataset projecting the adult lung methylation network structure as the starting reference on the fetal lung methylation data (test) and identified 55 co-methylation modules (supplementary figures S17 and S18, and supplementary table S17). Most modules were associated with fetal age, lung function and COPD, but none significantly associated with both IUS exposure and COPD. Enriched pathways for those modules included epidermal growth factor receptor tyrosine kinase inhibitor resistance, ERBB signalling downregulation and PI3K/AKT. This analysis further supports that adult lung network perturbations might recapitulate and have origins in fetal development-associated epigenetic perturbations, especially with enrichment in the PI3K/AKT signalling pathway.
Integration with prior gene expression and GWAS associations
We identified 14 236 CpGs at an MM subthreshold of ⩾0.70 annotated to 7364 unique genes across all modules (supplementary table S18). Of those, 100 CpGs from the ME34:purple, ME79:salmon, ME30: black, ME13:orange and ME4:yellow modules mapped to 62 unique differentially expressed genes (FDR <0.05) in the LT-COPD dataset. The highest MM was associated with the CpG site cg07495027 mapped to EDNRB in the ME4:yellow module (supplementary table S18). The strongest correlation between CpG methylation and gene expression was observed for EEF1B2 (correlation −0.42, p=9.9×10−8) and ABL1 (correlation 0.34, p=2.4×10−5). Interestingly, 38 unique high MM genes had an overlap with the 82 previously identified COPD GWAS associations (supplementary table S18). Several genes were also shared between modules with maximum presence for ASAP2, IER3 and THRA across modules.
Discussion
Network analyses may reveal deeper insights into the developmental origins of disease. Addressing the fetal origins hypothesis, we utilised a network-driven approach to focus on smaller gene clusters in relevant functional pathways that may influence risk for adult COPD. The network analysis suggested that the early origins of COPD may be related to both the developmental- and exposure-related perturbations in gene networks, not single genes working in isolation.
Pathway analysis of IUS exposure, fetal age and COPD-associated module genes (ME46:mediumpurple3) as well as the most highly preserved module genes (ME13:orange) included annotation to the PI3K pathway. The PI3K/AKT pathway not only plays a crucial role in human disease [55] but also represents a therapeutic target for ageing [56], lung dysfunction, asthma pathogenesis [57] and treatment of COPD [58]. Most of the mentioned Hippo pathway components interact, including Mst1/2, Sav1, Lats1/2, Mob1 or Yap. Their homozygous deletion causes embryonic lethality [59, 60]. The Hippo signalling pathway regulates lung development/alveolarisation [61, 62], and closely interacts with TGF-β [63], Wnt, Notch and Hedgehog signalling pathways. Furthermore, the Hippo pathway is implicated in progression of lung diseases, including asthma and COPD [64, 65], and may therefore be a crucial link for fetal origins of COPD. The Wnt pathway is annotated to almost all of the highlighted modules. Wnt/β-catenin is known for its critical role in embryonic and fetal lung development [66]. IUS exposure has been reported to be associated with downregulation of genes in Wnt signalling and impaired lung growth in the developing fetus [67]. We have previously identified Wnt signalling genes associated with lower lung function in asthmatic subjects [68]. The Wnt/β-catenin pathway has also been implicated in COPD [69–71] and is regulated by FAM13A, a COPD susceptibility gene [72]. These findings further support that the disruption of the Wnt pathway in early life may be a prominent contributor to fetal origins of COPD. The role for Wnt in lung ageing has been proposed [73], suggesting this pathway together with the PI3K/AKT pathway may link fetal exposure and adult lung disease through perturbations in ageing pathways. Highlighted modules revealed pathways to consider for further investigation for fetal origins of COPD. Genes with the highest MM may represent functional COPD susceptibility hubs perturbed in early life. FOXB1, the gene with the highest MM in the ME34:purple module, is a transcription factor that has been implicated in embryonic development and was associated with childhood asthma symptoms in a GWAS in Latin American children [74]. The ME34:purple module included multiple enriched pathways (Hippo, PI3K, MAPK, Wnt and TGF-β). The ME90:darkolivegreen module enriched in positive regulation of telomerase activity catalysed by telomerase reverse transcriptase enzyme has often been linked to chromosome maintenance [75, 76]. Its negative association with pack-years and sex may point to sex-specific activation of repair mechanisms after smoking cessation. The ME100:skyblue module was enriched for several members of the cadherin family implicated in lung diseases such as asthma [77] and COPD [78]. ERBB4 signalling highlighted through the ME100:skyblue module included the YAP1 gene, a component of the Hippo pathway [60]. ERBB4 deletion is associated with delayed fetal development [79]. ERBB4 demonstrated increased gene expression from nonsmokers to smokers with and without COPD and inverse association with FEV1, suggesting links to COPD severity [80]. Integration with previously reported gene expression and GWAS associations highlighted candidate genes such as EDNRB and IER3. Increased EDNRB expression has been associated with emphysema, FEV1/FVC [81] and COPD severity [82]. Ier3 knockout in mouse models was associated with immune dysfunction and interference with several pathways, including PI3K/AKT [83].
The biologically plausible and statistically significant pathways we identified using correlation networks are in contrast to the limited findings of the single CpG site analysis. Our findings of many differentially methylated CpGs with a small magnitude of effect is, however, consistent with previously published large EWASs associated with maternal smoking [84]. Notably, the γ-aminobutyric acid (GABA) and adenosine receptor signalling pathways were identified from overlapping fetal and adult lung EWAS results. Nicotine exposure has been reported previously to increase GABA signalling in airway epithelium [85], while adenosine receptors are known for their anti-inflammatory properties and therapeutic potential for the treatment of asthma and COPD [86].
A key strength of the study includes interrogating epigenetic profiles in the fetal lung tissue ascertained during airway branching morphogenesis. These samples are particularly relevant to test the developmental origins of COPD hypothesis. Abnormalities in bronchial airways during this stage can lead to decreased airflow and slowed growth of airways, limiting maximally attained lung function. Analytical strengths of our study include methylation profiles in both fetal and adult lung tissue using the same platform and leveraging a network-based approach for both relevant tissues. Due to complex underlying mechanisms, it remains a challenge to identify significant single-site associations while retaining statistical power, often with effect sizes of small magnitude, which is consistent with previously published large epigenome-wide studies [84]. Previous studies have identified and compared differentially methylated CpGs associated with COPD in whole blood and lung tissue. In light of the considerable heterogeneity across studies and varying statistical analyses [87], our systematic evaluation of previously validated COPD EWAS associations found in blood [48, 88–90] did not identify any consistent overlap with our high MM CpGs.
We have therefore provided extensive details about the network modules to facilitate future research and collaboration in this less explored area. We acknowledge that our study has several limitations, including the sample sizes of the datasets and the use of lung homogenates. Moreover, prospective studies with expanded coverage of methylation sites such as from the Illumina EPIC array and bisulfite sequencing may support validation and improve our understanding of the identified associations significantly. Furthermore, dichotomising continuous variables such as cotinine facilitates interpretation and is consistent with IUS exposure as measured in many studies. However, this may lead to loss of information, especially with small datasets. Therefore, to be consistent with the literature, but also to more precisely model exposure, we reported module–trait associations using both dichotomous and continuous measures of cotinine. In addition, we have limited phenotype data associated with the fetal lung samples. To address this, we directly measured cotinine and given the timing of sample collection presumed elevated cotinine is related to maternal smoking and not nicotine replacement therapy or e-cigarette use. Efforts linking fetal tissue findings with cord blood will increase the opportunity to fully leverage cord blood datasets and understand the fetal origins of adult lung disease. Future research on the developmental contributions to COPD should focus on single-cell analyses to further understand fetal programming in the lung relevant to adult tissue.
Conclusions
In summary, our co-methylation network evaluation resulted in the identification of gene modules linking fetal lung tissue IUS exposure and fetal age to adult lung function and COPD. Additional studies and functional validation will support causal links between fetal methylation alterations in pathways including Hippo, Wnt and PI3K/AKT and risk for and development of COPD. As we move towards precision medicine, understanding the impact of early-life molecular trajectories will provide unprecedented insight into complex adult lung diseases and reveal ways to leverage the plasticity of the epigenome towards primordial prevention, lung health and resilience despite early-life programming.
Supplementary Material
Support statement:
This work was supported by US National Institutes of Health grants P01 HL132825, K25 HL136846, P01 HL 105339, P01 HL114501, R21 HL107927 and R01 HL097144. Funding information for this article has been deposited with the Crossref Funder Registry.
Conflict of interest:
P. Kachroo has nothing to disclose. J.D. Morrow reports grants from the National Institutes of Health, during the conduct of the study. A.T. Kho has nothing to disclose. C.A. Vyhlidal reports grants from the National Institutes of Health, during the conduct of the study. E.K. Silverman reports grants from the National Institutes of Health, during the conduct of the study; grants and travel support from GlaxoSmithKline and grants from Bayer, outside the submitted work. S.T. Weiss reports personal fees from UpToDate, outside the submitted work. K.G. Tantisira reports grants from the National Institutes of Health, during the conduct of the study. D.L. DeMeo reports grants from the National Institutes of Health, during the conduct of the study; and grants from Bayer, outside the submitted work.
Footnotes
This article has supplementary material available from erj.ersjournals.com
References
- 1.Nurmagambetov T, Kuwahara R, Garbe P. The economic burden of asthma in the United States, 2008–2013. Ann Am Thorac Soc 2018; 15: 348–356. [DOI] [PubMed] [Google Scholar]
- 2.Guarascio AJ, Ray SM, Finch CK, et al. The clinical and economic burden of chronic obstructive pulmonary disease in the USA. Clinicoecon Outcomes Res 2013; 5: 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease 2017 Report: GOLD Executive Summary. Arch Bronconeumol 2017; 53: 128–149. [DOI] [PubMed] [Google Scholar]
- 4.McGeachie MJ. Childhood asthma is a risk factor for the development of chronic obstructive pulmonary disease. Curr Opin Allergy Clin Immunol 2017; 17: 104–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bush A Lung development and aging. Ann Am Thorac Soc 2016; 13: Suppl. 5, S438–S446. [DOI] [PubMed] [Google Scholar]
- 6.Hollams EM, de Klerk NH, Holt PG, et al. Persistent effects of maternal smoking during pregnancy on lung function and asthma in adolescents. Am J Respir Crit Care Med 2014; 189: 401–407. [DOI] [PubMed] [Google Scholar]
- 7.Lange P, Celli B, Agusti A, et al. Lung-function trajectories leading to chronic obstructive pulmonary disease. N Engl J Med 2015; 373: 111–122. [DOI] [PubMed] [Google Scholar]
- 8.Regan EA, Lynch DA, Curran-Everett D, et al. Clinical and radiologic disease in smokers with normal spirometry. JAMA Intern Med 2015; 175: 1539–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stern DA, Morgan WJ, Wright AL, et al. Poor airway function in early infancy and lung function by age 22 years: a non-selective longitudinal cohort study. Lancet 2007; 370: 758–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perret JL, Walters EH, Abramson MJ, et al. The independent and combined effects of lifetime smoke exposures and asthma as they relate to COPD. Expert Rev Respir Med 2014; 8: 503–514. [DOI] [PubMed] [Google Scholar]
- 11.Tong VT, Dietz PM, Morrow B, et al. Trends in smoking before, during, and after pregnancy – Pregnancy Risk Assessment Monitoring System, United States, 40 sites, 2000–2010. MMWR Surveill Summ 2013; 62: 1–19. [PubMed] [Google Scholar]
- 12.Gould GS, Patten C, Glover M, et al. Smoking in pregnancy among indigenous women in high-income countries: a narrative review. Nicotine Tob Res 2017; 19: 506–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scherman A, Tolosa JE, McEvoy C. Smoking cessation in pregnancy: a continuing challenge in the United States. Ther Adv Drug Saf 2018; 9: 457–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drake P, Driscoll AK, Mathews TJ. Cigarette smoking during pregnancy: United States, 2016. NCHS Data Brief 2018; 305: 1–8. [PubMed] [Google Scholar]
- 15.Tager IB, Ngo L, Hanrahan JP. Maternal smoking during pregnancy. Effects on lung function during the first 18 months of life. Am J Respir Crit Care Med 1995; 152: 977–983. [DOI] [PubMed] [Google Scholar]
- 16.Wang X, Zuckerman B, Pearson C, et al. Maternal cigarette smoking, metabolic gene polymorphism, and infant birth weight. JAMA 2002; 287: 195–202. [DOI] [PubMed] [Google Scholar]
- 17.Rokoff LB, Rifas-Shiman SL, Coull BA, et al. Cumulative exposure to environmental pollutants during early pregnancy and reduced fetal growth: the Project Viva cohort. Environ Health 2018; 17: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ion R, Bernal AL. Smoking and preterm birth. Reprod Sci 2015; 22: 918–926. [DOI] [PubMed] [Google Scholar]
- 19.Pereira PdS, Da Mata FAF, Figueiredo ACG, et al. Maternal active smoking during pregnancy and low birth weight in the Americas: a systematic review and meta-analysis. Nicotine Tob Res 2017; 19: 497–505. [DOI] [PubMed] [Google Scholar]
- 20.Noakes PS, Hale J, Thomas R, et al. Maternal smoking is associated with impaired neonatal Toll-like-receptor-mediated immune responses. Eur Respir J 2006; 28: 721–729. [DOI] [PubMed] [Google Scholar]
- 21.Qiu F, Liang C-L, Liu H, et al. Impacts of cigarette smoking on immune responsiveness: up and down or upside down? Oncotarget 2017; 8: 268–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kupers LK, Xu X, Jankipersadsing SA, et al. DNA methylation mediates the effect of maternal smoking during pregnancy on birthweight of the offspring. Int J Epidemiol 2015; 44: 1224–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richmond RC, Simpkin AJ, Woodward G, et al. Prenatal exposure to maternal smoking and offspring DNA methylation across the lifecourse: findings from the Avon Longitudinal Study of Parents and Children (ALSPAC). Hum Mol Genet 2015; 24: 2201–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Isayama T, Shah PS, Ye XY, et al. Adverse impact of maternal cigarette smoking on preterm infants: a population-based cohort study. Am J Perinatol 2015; 32: 1105–1111. [DOI] [PubMed] [Google Scholar]
- 25.Morrow LA, Wagner BD, Ingram DA, et al. Antenatal determinants of bronchopulmonary dysplasia and late respiratory disease in preterm infants. Am J Respir Crit Care Med 2017; 196: 364–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bobolea I, Arismendi E, Valero A, et al. Early life origins of asthma: a review of potential effectors. J Investig Allergol Clin Immunol 2019; 29: 168–179. [DOI] [PubMed] [Google Scholar]
- 27.Beyer D, Mitfessel H, Gillissen A. Maternal smoking promotes chronic obstructive lung disease in the offspring as adults. Eur J Med Res 2009; 14: Suppl. 4, 27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Postma DS, Bush A, van den Berge M. Risk factors and early origins of chronic obstructive pulmonary disease. Lancet 2015; 385: 899–909. [DOI] [PubMed] [Google Scholar]
- 29.Martinez FD. Early-life origins of chronic obstructive pulmonary disease. N Engl J Med 2016; 375: 871–878. [DOI] [PubMed] [Google Scholar]
- 30.Vasquez MM, Zhou M, Hu C, et al. Low lung function in young adult life is associated with early mortality. Am J Respir Crit Care Med 2017; 195: 1399–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Savran O, Ulrik CS. Early life insults as determinants of chronic obstructive pulmonary disease in adult life. Int J Chron Obstruct Pulmon Dis 2018; 13: 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krishnan JK, Martinez FJ. Lung function trajectories and chronic obstructive pulmonary disease: current understanding and knowledge gaps. Curr Opin Pulm Med 2018; 24: 124–129. [DOI] [PubMed] [Google Scholar]
- 33.Foreman MG, Zhang L, Murphy J, et al. Early-onset chronic obstructive pulmonary disease is associated with female sex, maternal factors, and African American race in the COPDGene Study. Am J Respir Crit Care Med 2011; 184: 414–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joubert BR, Felix JF, Yousefi P, et al. DNA methylation in newborns and maternal smoking in pregnancy: genome-wide consortium meta-analysis. Am J Hum Genet 2016; 98: 680–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.den Dekker HT, Burrows K, Felix JF, et al. Newborn DNA-methylation, childhood lung function, and the risks of asthma and COPD across the life course. Eur Respir J 2019; 53: 1801795. [DOI] [PubMed] [Google Scholar]
- 36.Martinez FJ, Han MK, Allinson JP, et al. At the root: defining and halting progression of early chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2018; 197: 1540–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heijmans BT, Tobi EW, Lumey LH, et al. The epigenome: archive of the prenatal environment. Epigenetics 2009; 4: 526–531. [DOI] [PubMed] [Google Scholar]
- 38.Liang P, Song F, Ghosh S, et al. Genome-wide survey reveals dynamic widespread tissue-specific changes in DNA methylation during development. BMC Genomics 2011; 12: 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Faner R, Cruz T, Lopez-Giraldo A, et al. Network medicine, multimorbidity and the lung in the elderly. Eur Respir J 2014; 44: 775–788. [DOI] [PubMed] [Google Scholar]
- 40.Weiss ST. Biological network modeling and systems biology to advance our understanding of lung disease. Am J Respir Crit Care Med 2016; 194: 920–921. [DOI] [PubMed] [Google Scholar]
- 41.Vyhlidal CA, Riffel AK, Haley KJ, et al. Cotinine in human placenta predicts induction of gene expression in fetal tissues. Drug Metab Dispos 2013; 41: 305–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bibikova M, Barnes B, Tsan C, et al. High density DNA methylation array with single CpG site resolution. Genomics 2011; 98: 288–295. [DOI] [PubMed] [Google Scholar]
- 43.Aryee MJ, Jaffe AE, Corrada-Bravo H, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014; 30: 1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang C, Zhan X, Liang L, et al. Improved ancestry estimation for both genotyping and sequencing data using projection Procrustes analysis and genotype imputation. Am J Hum Genet 2015; 96: 926–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Iterson M, van Zwet EW, Heijmans BT, et al. Controlling bias and inflation in epigenome- and transcriptome-wide association studies using the empirical null distribution. Genome Biol 2017; 18: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kho AT, Chhabra D, Sharma S, et al. Age, sexual dimorphism, and disease associations in the developing human fetal lung transcriptome. Am J Respir Cell Mol Biol 2016; 54: 814–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chhabra D, Sharma S, Kho AT, et al. Fetal lung and placental methylation is associated with in utero nicotine exposure. Epigenetics 2014; 9: 1473–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morrow JD, Cho MH, Hersh CP, et al. DNA methylation profiling in human lung tissue identifies genes associated with COPD. Epigenetics 2016; 11: 730–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008; 9: 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Geeleher P, Hartnett L, Egan LJ, et al. Gene-set analysis is severely biased when applied to genome-wide methylation data. Bioinformatics 2013; 29: 1851–1857. [DOI] [PubMed] [Google Scholar]
- 51.Phipson B, Maksimovic J, Oshlack A. missMethyl: an R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics 2016; 32: 286–288. [DOI] [PubMed] [Google Scholar]
- 52.Reimand J, Arak T, Adler P, et al. g:Profiler – a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res 2016; 44: W83–W89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Merico D, Isserlin R, Stueker O, et al. Enrichment Map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One 2010; 5: e13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sakornsakolpat P, Prokopenko D, Lamontagne M, et al. Genetic landscape of chronic obstructive pulmonary disease identifies heterogeneous cell-type and phenotype associations. Nat Genet 2019; 51: 494–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell 2017; 169: 381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mendez-Pertuz M, Martinez P, Blanco-Aparicio C, et al. Modulation of telomere protection by the PI3K/AKT pathway. Nat Commun 2017; 8: 1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yoo EJ, Ojiaku CA, Sunder K, et al. Phosphoinositide 3-kinase in asthma: novel roles and therapeutic approaches. Am J Respir Cell Mol Biol 2017; 56: 700–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bozinovski S, Vlahos R, Hansen M, et al. Akt in the pathogenesis of COPD. Int J Chron Obstruct Pulmon Dis 2006; 1: 31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhao B, Li L, Guan K-L. Hippo signaling at a glance. J Cell Sci 2010; 123: 4001–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dai Y, Jablons D, You L. Hippo pathway in lung development. J Thorac Dis 2017; 9: 2246–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Makita R, Uchijima Y, Nishiyama K, et al. Multiple renal cysts, urinary concentration defects, and pulmonary emphysematous changes in mice lacking TAZ. Am J Physiol Renal Physiol 2008; 294: F542–F553. [DOI] [PubMed] [Google Scholar]
- 62.Mitani A, Nagase T, Fukuchi K, et al. Transcriptional coactivator with PDZ-binding motif is essential for normal alveolarization in mice. Am J Respir Crit Care Med 2009; 180: 326–338. [DOI] [PubMed] [Google Scholar]
- 63.Saito A, Nagase T. Hippo and TGF-beta interplay in the lung field. Am J Physiol Lung Cell Mol Physiol 2015; 309: L756–L767. [DOI] [PubMed] [Google Scholar]
- 64.Anderson GP. Advances in understanding COPD. F1000Res 2016; 5: 2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xie H, Wu L, Deng Z, et al. Emerging roles of YAP/TAZ in lung physiology and diseases. Life Sci 2018; 214: 176–183. [DOI] [PubMed] [Google Scholar]
- 66.Hussain M, Xu C, Lu M, et al. Wnt/beta-catenin signaling links embryonic lung development and asthmatic airway remodeling. Biochim Biophys Acta Mol Basis Dis 2017; 1863: 3226–3242. [DOI] [PubMed] [Google Scholar]
- 67.Blacquiere MJ, Timens W, van den Berg A, et al. Maternal smoking during pregnancy decreases Wnt signalling in neonatal mice. Thorax 2010; 65: 553–554. [DOI] [PubMed] [Google Scholar]
- 68.Sharma S, Tantisira K, Carey V, et al. A role for Wnt signaling genes in the pathogenesis of impaired lung function in asthma. Am J Respir Crit Care Med 2010; 181: 328–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Skronska-Wasek W, Gosens R, Konigshoff M, et al. WNT receptor signalling in lung physiology and pathology. Pharmacol Ther 2018; 187: 150–166. [DOI] [PubMed] [Google Scholar]
- 70.Huang P, Yan R, Zhang X, et al. Activating Wnt/beta-catenin signaling pathway for disease therapy: challenges and opportunities. Pharmacol Ther 2019; 196: 79–90. [DOI] [PubMed] [Google Scholar]
- 71.Qu J, Yue L, Gao J, et al. Perspectives on Wnt signal pathway in the pathogenesis and therapeutics of chronic obstructive pulmonary disease. J Pharmacol Exp Ther 2019; 369: 473–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jiang Z, Lao T, Qiu W, et al. A chronic obstructive pulmonary disease susceptibility gene, FAM13A, regulates protein stability of beta-catenin. Am J Respir Crit Care Med 2016; 194: 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lehmann M, Baarsma HA, Konigshoff M. WNT signaling in lung aging and disease. Ann Am Thorac Soc 2016; 13: Suppl. 5, S411–S416. [DOI] [PubMed] [Google Scholar]
- 74.Costa GNO, Dudbridge F, Fiaccone RL, et al. A genome-wide association study of asthma symptoms in Latin American children. BMC Genet 2015; 16: 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hebert MD, Poole AR. Towards an understanding of regulating Cajal body activity by protein modification. RNA Biol 2017; 14: 761–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhu Y, Tomlinson RL, Lukowiak AA, et al. Telomerase RNA accumulates in Cajal bodies in human cancer cells. Mol Biol Cell 2004; 15: 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Georas SN, Rezaee F. Epithelial barrier function: at the front line of asthma immunology and allergic airway inflammation. J Allergy Clin Immunol 2014; 134: 509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gohy ST, Hupin C, Fregimilicka C, et al. Imprinting of the COPD airway epithelium for dedifferentiation and mesenchymal transition. Eur Respir J 2015; 45: 1258–1272. [DOI] [PubMed] [Google Scholar]
- 79.Liu W, Purevdorj E, Zscheppang K, et al. ErbB4 regulates the timely progression of late fetal lung development. Biochim Biophys Acta 2010; 1803: 832–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Anagnostis A, Neofytou E, Soulitzis N, et al. Molecular profiling of EGFR family in chronic obstructive pulmonary disease: correlation with airway obstruction. Eur J Clin Invest 2013; 43: 1299–1306. [DOI] [PubMed] [Google Scholar]
- 81.Obeidat M, Nie Y, Fishbane N, et al. Integrative genomics of emphysema-associated genes reveals potential disease biomarkers. Am J Respir Cell Mol Biol 2017; 57: 411–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shi K, Chen X, Xie B, et al. Celastrol alleviates chronic obstructive pulmonary disease by inhibiting cellular inflammation induced by cigarette smoke via the Ednrb/Kng1 signaling pathway. Front Pharmacol 2018; 9: 1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Arlt A, Schafer H. Role of the immediate early response 3 (IER3) gene in cellular stress response, inflammation and tumorigenesis. Eur J Cell Biol 2011; 90: 545–552. [DOI] [PubMed] [Google Scholar]
- 84.Breton CV, Marsit CJ, Faustman E, et al. Small-magnitude effect sizes in epigenetic end points are important in children’s environmental health studies: the Children’s Environmental Health and Disease Prevention Research Center’s Epigenetics Working Group. Environ Health Perspect 2017; 125: 511–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fu XW, Wood K, Spindel ER. Prenatal nicotine exposure increases GABA signaling and mucin expression in airway epithelium. Am J Respir Cell Mol Biol 2011; 44: 222–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Polosa R, Blackburn MR. Adenosine receptors as targets for therapeutic intervention in asthma and chronic obstructive pulmonary disease. Trends Pharmacol Sci 2009; 30: 528–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Machin M, Amaral AFS, Wielscher M, et al. Systematic review of lung function and COPD with peripheral blood DNA methylation in population based studies. BMC Pulm Med 2017; 17: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sundar IK, Yin Q, Baier BS, et al. DNA methylation profiling in peripheral lung tissues of smokers and patients with COPD. Clin Epigenetics 2017; 9: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.de Vries M, van der Plaat DA, Nedeljkovic I, et al. From blood to lung tissue: effect of cigarette smoke on DNA methylation and lung function. Respir Res 2018; 19: 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bermingham ML, Walker RM, Marioni RE, et al. Identification of novel differentially methylated sites with potential as clinical predictors of impaired respiratory function and COPD. EBioMedicine 2019; 43: 576–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.