

Abstract
A primordial gut-epithelial innate defense response is the release of hydrogen peroxide by dual NADPH oxidase (DUOX). In inflammatory bowel disease (IBD), a condition characterized by an imbalanced gut microbiota-immune homeostasis, DUOX2 isoenzyme is the highest induced gene. Performing multiomic analyses using 2872 human participants of a wellness program, we detected a substantial burden of rare protein-altering DUOX2 gene variants of unknown physiologic significance. We identified a significant association between these rare loss-of-function variants and increased plasma levels of interleukin-17C, which is induced also in mucosal biopsies of patients with IBD. DUOX2-deficient mice replicated increased IL-17C induction in the intestine, with outlier high Il17c expression linked to the mucosal expansion of specific Proteobacteria pathobionts. Integrated microbiota/host gene expression analyses in patients with IBD corroborated IL-17C as a marker for epithelial activation by gram-negative bacteria. Finally, the impact of DUOX2 variants on IL-17C induction provided a rationale for variant stratification in case control studies that substantiated DUOX2 as an IBD risk gene. Thus, our study identifies an association of deleterious DUOX2 variants with a preclinical hallmark of disturbed microbiota-immune homeostasis that appears to precede the manifestation of IBD.
Keywords: Gastroenterology
Keywords: Genetic variation, Inflammatory bowel disease, Innate immunity

Introduction
A monolayer of epithelial cells covers the intestinal lumen preventing an overt immune response against the normal gut microbiota, while at the same time controlling infection with potentially life-threatening pathogens. A primordial innate defense response conserved in the gut epithelium of metazoans is the microbial-induced release of hydrogen peroxide (H2O2) by an epithelial NADPH oxidase, dual oxidase (DUOX), expressed at the apical membrane of enterocytes. In mammals, this function is executed by the heterodimeric DUOX2 isoenzyme (DUOX2 and DUOXA2 subunits). While the fundamental importance of DUOX-mediated host defense against microbial infection has been well established in invertebrate models (1, 2), the role of DUOX2 in maintaining immune homeostasis and dysbiosis-associated diseases in mammals is less clear.
In mice, DUOX2-generated H2O2 limits Helicobacter colonization of the gastric mucus layer (3, 4), and its absence in the gut leads to subtle activation of compensatory epithelial defense systems together with an increased translocation of Proteobacteria DNA into the mesenteric lymph nodes (5). In humans, overexpression of DUOX2, accompanied by an expansion of mucosa-associated Proteobacteria, is a hallmark of the gene expression changes found in intestinal biopsies of patients with inflammatory bowel disease (IBD) even in the absence of overt local inflammation (6). Thus, by its function and regulation, DUOX2 could be a susceptibility factor in IBD, a disease category that includes chronic disorders of the gastrointestinal tract resulting from the breakdown of the homeostatic relationship between the host immune system and the gut microbiota.
Results
We previously noted a substantial burden of rare protein-altering DUOX2 variants in the general population (7). To explore the phenotypic impact of such variants, we carried out a multiomic phenome-wide association study (PheWAS) with data from 2872 participants in a commercial lifestyle coaching program (Arivale) (Figure 1 and Table 1). Genetic variants falling within the DUOX2 and DUOXA2 (essential DUOX2 heterodimerization partner) exonic boundaries and passing quality filters were annotated with the Ensembl Variant Effect Predictor; only protein-altering variants were included in downstream analyses. In total, we identified 155 unique alleles with less than 1% frequency each (Supplemental Tables 1 and 2; supplemental material available online with this article; https://doi.org/10.1172/JCI141676DS1). Of the 357 individuals with rare variants, a majority (92%) carried only a single heterozygous variant.
Figure 1. Outline of DUOX2/DUOXA2-specific multiomic PheWAS.

We identified all rare (AF < 0.01) protein-altering DUOX2/DUOXA2 variants found in whole-genome sequencing data of 2872 participants of a lifestyle coaching program. Baseline phenotyping obtained for all participants comprised 124 clinical laboratory tests, 951 plasma metabolites, 266 plasma proteins related to inflammation and cardiovascular health, and 16S rRNA-based profiling data of the fecal microbiome. We used rare-variant test statistics (SKAT-O) to find statistical associations between the identified variants and the quantitative phenotypes.
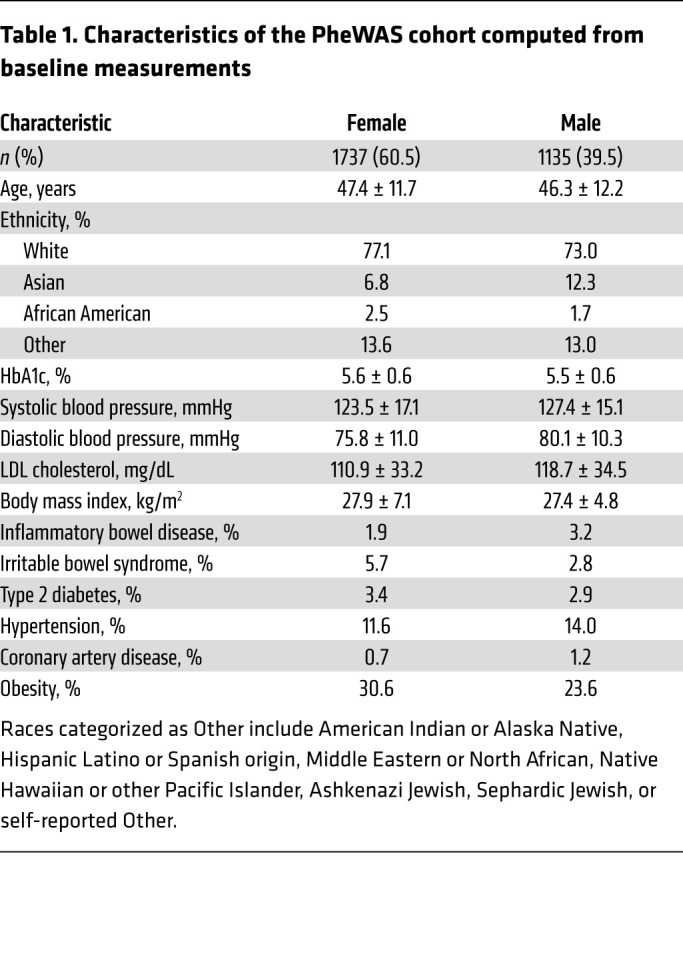
Table 1. Characteristics of the PheWAS cohort computed from baseline measurements.
We used optimal unified sequence kernel association (SKAT-O) tests to find statistical associations between the identified variants and quantitative phenotypes comprising 124 clinical laboratory tests, 951 plasma metabolites, 266 plasma proteins, and 16S rRNA-based profiling data of the fecal microbiome. We found that protein-altering DUOX2/DUOXA2 variants were most significantly associated with the plasma level of interleukin-17C (IL-17C; FDR = 2.6 × 10–5) (Figure 2A and Supplemental Table 3). The distributions of IL-17C values differed between variant-carriers and individuals without variant, with the former having a more right-tail heavy distribution (Figure 2B). For further analysis, we stratified carriers based on minor allele rarity, a strong predictor of deleteriousness (8). The prevalence of abnormally high plasma IL-17C levels indeed substantially increased with allele rarity in ancestry-matched control populations (Figure 2C and Supplemental Table 4).
Figure 2. Rare DUOX2 protein variants are associated with outlier high plasma IL-17C concentration in the general population.

(A) Manhattan plot of the PheWAS results. We used the FDR to correct for multiple testing across all combined phenotypes, with the dashed line indicating the FDR less than 0.05 significance level. (B) Plasma IL-17C baseline levels in study participants with or without DUOX2/DUOXA2 protein variants. Violin plot with quartiles indicated by the horizontal lines. Data are log2-scaled normalized protein expression units (NPX). 2-tailed Kolmogorov-Smirnov test. (C) Prevalence of high IL-17C level in subjects with or without DUOX2/DUOXA2 protein variants. We set the cut-off for outlier high IL-17C level (IL-17Chi) to Q3+2*IQR of the no-variant group and stratified variants by rarity according to ancestry-specific allele frequency (AF) data from gnomAD. Two-tailed Fisher’s exact test. (D) Enrichment of rare DUOX2 protein variants in IL-17Chi (99th percentile; n = 27) subjects. The plot depicts the proportion of individuals who carry rare DUOX2 protein variants of the indicated minor allele frequency. (E) IL-17Chi status is associated with specific alterations of the plasma protein profile that are not unique to carriers of DUOX2 protein variants. Plotted are the relative protein levels of 91 inflammation-related proteins in IL-17Chi subjects with (y axis; n = 13) or without (x axis; n = 14) rare DUOX2 protein variant(s). Protein levels are expressed as a geometric mean ratio (GMR) relative to the total study cohort. *P < 0.05.
To further define the phenotype in subjects with high plasma IL-17C concentration, we compared the plasma levels of 91 additional inflammation-related proteins between individuals with the highest IL-17C level (IL-17Chi; 99th percentile for IL-17C) and those with normal or low IL-17C level (<95th percentile for IL-17C). We found that the level of the chemokine CCL20 was the most robustly induced protein in IL-17Chi subjects (Supplemental Figure 1). CCL20, the ligand for the chemokine receptor CCR6, is a powerful chemoattractant for lymphocytes and dendritic cells, and thereby critical for the formation of mucosa-associated lymphoid tissues. The profile of additional proteins with weaker, but significant coregulation included FGF23, CXCL11, IL-17A, IL-6, and CXCL9, all of which play a role in intestinal mucosal immunity. We next examined whether any of these protein changes are unique to IL-17Chi subjects carrying DUOX2 protein variants. As expected, rare DUOX2 protein variants were highly enriched in the IL-17Chi group (3.7 fold, Figure 2D). However, stratification of IL-17Chi subjects by DUOX2 genotype revealed that the plasma protein profiles associated with IL-17Chi did not differ between those with or without rare DUOX2 protein variants (Figure 2E). Overall, this analysis indicates that IL-17Chi is part of a common response pattern that is not altered in individuals with underlying DUOX2 defect.
To formally assess IL-17C–associated DUOX2 variants for their impact on the enzyme’s activity, we tested 10 variants with the most significant contribution to the association signal (Figure 3A and Supplemental Table 5) in a heterologous expression system (9). We confirmed a significant functional impairment for the majority of tested alleles (Figure 3B). Except for R1039Q, which was correctly inserted in the plasma membrane, the functional loss could be accounted for by a defect in intracellular trafficking (Figure 3, C–E and Supplemental Figure 2). The variants analyzed and found to be defective were all very rare (allele frequency [AF] < 0.001) in independent population cohorts (gnomAD). Thus, carriers of deleterious DUOX2 variants are prone to have excessively high plasma IL-17C levels.
Figure 3. Rare DUOX2 protein variants linked to excessive plasma IL-17C levels impair the expression of a functional DUOX2/DUOXA2 enzyme complex.

(A) Identification of variants significantly contributing to the association with plasma concentration of IL-17C in the study cohort (Wald χ2 test). (B) Extracellular H2O2 production of DUOX2 protein variants expressed in a heterologous system (9). pcDNA: transfections with empty vector; DUOXA2 only: transfections with DUOXA2 only; DUOX2 only: transfections with DUOX2 only; all other transfections are cotransfections of the indicated DUOX2 plasmids (WT or variant) with DUOXA2. Data were obtained from 3 independent transfection experiments each with 3–4 (WT: 6–8) replicates and are mean ± SEM. One-way ANOVA with Dunnett’s multiple comparisons test. (C) Quantitation of DUOX2 cell-surface expression by flow cytometry (see Supplemental Figure 2 for details). Data represent means ± SEM from 3 independent transfection experiments, each including all variants and duplicate transfections of the reference DUOX2 plasmid. One-way ANOVA with Dunnett’s multiple comparisons test. (D) Summary of the functional assessment of rare DUOX2 protein variants. Data are mean ± SEM. (E) DUOX2 topology model depicting the location of tested variants. *P < 0.05; **P < 0.01; ****P < 0.0001.
In contrast to other members of the IL-17 family expressed in lymphoid cell populations, IL-17C appears to be inducibly expressed in barrier epithelial cells of the intestine, airway, and skin (10). Therefore, we assessed in mice whether DUOX2 inactivation is sufficient to trigger Il17c expression in the gut mucosa. Compared with WT littermates, both Duoxa–/– mice lacking whole-body DUOX2 activity (Figure 4, A and B) and intestinal epithelial-specific Duoxa–/– mice (Figure 4, F and G and Supplemental Figure 3) had significantly higher Il17c expression in the mucosa of the terminal ileum, but not the colon. The finding of outlier high Il17c expression levels in KO mice (Il17Chi; arrows in Figure 4A) was reminiscent of the positively skewed distribution of plasma IL-17C levels in DUOX2 variant carriers (Figure 2B). We found that Il17c induction was accompanied by significantly higher tissue expression of the chemokine Ccl20, but not of Il17a or Il17f (Figure 4, C–E), consistent with the elevated plasma CCL20 level observed in human subjects with high plasma IL-17C level (Figure 2E). This phenotype of mice lacking intestinal DUOX2 activity was completely T cell independent since it was conserved in a T (and B) cell–deficient Rag–/– background (Figure 4, H and I). The finding of abnormal Il17c expression in the ileum but not colon is consistent with the relatively higher baseline expression of Duox2 in the ileum of mice kept in a specific pathogen–free (SPF) environment (5). In the colon, the thick inner mucus layer is essentially sterile, whereas the thinner nonstratified mucus layer of the ileum is more readily penetrable by bacteria-sized particles, but important for the effectiveness of antimicrobial compounds by limiting their diffusion into the lumen (11). We found that impairment of this vertical compartmentalization by a dietary emulsifier, carboxymethyl cellulose, that causes supraepithelial mucus layer thinning (12), robustly induced Il17c in the colon of WT and Duoxa-deficient mice, indicating that its expression is remarkably sensitive to pathological exposure of the epithelium to microbiota. In the ileum, carboxymethylcellulose feeding equalized the Il17c mRNA level between WT and Duoxa–/– mice, consistent with the concept that the mucus layer — by retaining DUOX2 generated H2O2 — is critical for the protective function of DUOX2 in the gut.
Figure 4. Il17c induction in the gut epithelium of DUOX2 deficient mice is T cell–independent and mimicked by impairment of the supraepithelial mucus layer.

(A and B) Il17c mRNA expression in the terminal ileum and colon of Duoxa–/– (n = 26) and WT (n = 22) littermates. Arrows indicate samples with outlier high Il17c expression (Il17chi). Ccl20 (C), Il17a (D), and Il17f (E) expression in the terminal ileum. Two-tailed Mann-Whitney. (F and G) Expression of Il17c in the ileum and colon of intestinal epithelial-specific Duoxa–/– and floxed littermate control mice. We challenged the normal bacterial compartmentalization by chronically feeding the emulsifier CMC (1% wt/vol in drinking water for 8 weeks) that thins the mucus layer (12). n = 6 and n = 17 for floxed control mice without or with CMC treatment, respectively, and n = 5 and n = 20 for intestinal epithelial-specific Duoxa–/– mice without or with CMC treatment, respectively. Kruskal-Wallis and Dunn’s post hoc test. (H and I) Il17c expression is preserved in Rag1–/– mice lacking T cells as a major source of IL-17 family cytokines. n = 11 for Rag1–/–, Duoxafl/fl mice; n = 9 for Rag1–/–, Duoxafl/fl, Vil1-Cre mice. Two-tailed Mann-Whitney. *P < 0.05; **P < 0.01; ***P < 0.001. Error bars in A–I indicate 95% CI of geometric means.
IL-17C binds an epithelial-specific expressed receptor in auto/paracrine fashion to boost the production of chemokines for localized recruitment of immune cells, thereby linking epithelial and immune cell–mediated innate defense systems (10, 13). Its expression is acutely upregulated in epithelial cell lines by stimulation with the TLR5 ligand flagellin (14). We found that in vivo, colonization of germ-free WT mice with SPF microbiota or monocolonization with segmented filamentous bacteria (SFB; epithelial-attaching, gram-positive bacteria) failed to significantly induce Il17c (Figure 5A). This was in contrast to the well-known activation of other antimicrobial host defense systems (e.g., Duox2, Reg3g) and their cognate inducers, such as Il22 and Il17a, under these conditions (5, 15). Thus, Il17c is not substantially activated by any of the signaling pathways upregulated in response to the conventionalization of axenic animals (16). On the other hand, the upregulation of Il17c in mice deficient in DUOX2 enzyme was dependent on the gut microbiota since it was completely abolished following peroral treatment with an antibiotic cocktail that eradicates gram-negative bacteria (Figure 5B and Supplemental Figure 4). Consistent with a cell-autonomous regulation by microbiota, we found that in epithelial cell monolayers derived from mouse colonoids, Il17c expression was rapidly induced by direct exposure to gram-negative Enterobacteriaceae, but not the gram-positive bacteria tested (Figure 5C). Duoxa–/– and WT monolayers did not differ in their response to direct bacterial contact, suggesting that DUOX2 deficiency does not cause a cell-intrinsic abnormality in either recognition of microbial patterns or the signal transduction leading to Il17c expression.
Figure 5. High Il17c expression in the intestinal epithelium of DUOX2-deficient mice is linked to the expansion of gram-negative pathobionts.

(A) Differential microbiota-dependent regulation of Il17c, Il17a, and Reg3g (IL-22 target gene) in the mouse intestine. GF, germ-free; CONV, conventionalized (SPF); SFBmono, monocolonized with segmented filamentous bacteria. n = 5 animals per condition. Data represent median expression values with IQR. Kruskal-Wallis with Dunn’s post hoc test. (B) Mice were treated for 3 days with an antibiotics (Abx) regimen comprising ciprofloxacin and metronidazole that suppresses the gram-negative gut microbiota (see Supplemental Figure 4). n = 6 and n = 5 for control mice without or with Abx treatment, respectively, and n = 8 and n = 4 for intestinal epithelial-specific Duoxa–/– mice without or with Abx treatment, respectively. Data represent geometric means with 95% CI. Kruskal-Wallis test with Dunn’s post hoc test. (C) Acute cell-autonomous induction of Il17c expression in enteroid-derived epithelial monolayers directly exposed to bacteria. Each treatment was performed on 6 independent enteroid cultures derived from 3 Duoxa–/–/WT littermate pairs. Bars indicate median expression values. Kruskal-Wallis with Dunn’s post hoc test. (D) Cladogram (phylum to genus level) depicting results of LEfSe (54) analysis identifying taxa with distinct relative abundance (P < 0.01; LDA > 2) in ileal mucosa of Duoxa–/– (n = 26) compared with WT (n = 22) littermates. (E) Discriminative taxa in the ileal mucosal microbiota of Il17chi animals (marked with arrows in Figure 4A). (F) The relative mucosal abundance of genus Helicobacter. Data represent median values with IQR. Two-tailed Mann-Whitney. (G) The relative abundance of Proteobacterium otu0194 vs mucosal Il17c expression. Arrows in F and G indicate animals classified as Il17chi in Figure 4A. *P < 0.05; **P < 0.01; ***P < 0.001.
It is plausible that a defect in H2O2 release from the apical membrane of enterocytes increases access of susceptible gram-negative bacteria to the epithelium, for instance, due to reduced chemorepulsive, virulence-suppressing, or bactericidal effects (3, 4, 17). Furthermore, a stochastic shift in mucosal microbiota composition with an expansion of specific gram-negative pathobionts could underlie excessive Il17c levels found in a subset of Duoxa-deficient mice. Therefore, we profiled the composition of the mucosa-adherent ileal microbiota by 16S rDNA sequencing. Compared with WT littermates, Duoxa–/– mice had altered mucosal microbiota composition characterized by a relative loss of SFB with correspondingly higher abundance of the genera Helicobacter and Lactobacillus (Figure 5, D and F and Supplemental Tables 6 and 7). The most discriminative taxonomic feature in Il17chi mice (compare arrows in Figure 4A) was an unclassified Proteobacterium (Otu0194; Figure 5, E and G and Supplemental Table 8). Otu0194 was also the feature most significantly associated with Il17chi in multivariate association adjusted for the effect of Duoxa genotype (Duoxa–/– or WT; FDR = 0.0065; Supplemental Tables 9 and 10). The mucosal niche appeared to be its preferred habitat since it was not detected by sequencing of the corresponding luminal samples (Supplemental Figure 5).
Linking abnormally high Il17c expression to the mucosal expansion of specific gram-negative Proteobacteria species in mice lacking DUOX2 activity provides a likely explanation for the increased prevalence of excessive IL-17C plasma level in carriers of deleterious DUOX2 mutations. When we modeled the plasma IL-17C concentrations of the study participants on self-reported health history conditions involving potential sources of IL-17C (i.e., GI tract, lung, skin, chronic infectious disease), high IL-17C level appeared to be associated the strongest with IBD (FDR = 0.32; Figure 6A and Supplemental Table 11). Consistently, previous studies have found elevated blood and mucosa IL-17C concentrations among patients with IBD (14, 18). We found that ileal IL17C expression in treatment-naive patients with Crohn’s disease (CD) from the RISK cohort study (6, 19) was indeed more frequently induced compared with controls without IBD (Figure 6B). Analysis of gene expression profiles revealed that the pathways most strongly associated with IL17C induction were linked to antibacterial response, with the leading IL17C-correlated genes being the strongest implicated in gram-negative bacterial infections (Figure 6, C and D and Supplemental Tables 13–15).
Figure 6. IL17C induction observed in a subset of patients with IBD is a marker for abnormal epithelial stimulation by gram-negative bacteria.

(A) Positive associations of plasma IL-17C concentration with self-reported health history of study participants considering GI, skin, lung, and chronic infectious disease categories. Shown is the average difference in standardized plasma IL-17C for presence versus absence of a condition. We evaluated the nominal significance of effects using the Welch 2-sample test adjusted for age, sex, body mass index, season, and ancestry. See Supplemental Table 11 for detailed results. (B) Expression of IL17C in ileal mucosal biopsies from patients with CD (n = 174) and non-IBD controls (n = 42) from the RISK cohort. Error bars represent medians with IQR. Two-tailed Mann-Whitney. **P = 0.0027. (C) Gene set enrichment analysis using correlation with IL17C expression (rIL17C) as the rank metric to identify IL17C-correlated KEGG pathways in the mucosal biopsies of CD patients (FDR < 0.05). See Supplemental Tables 13 and 14 for additional information. (D) Overrepresentation of IL17C-coexpression signature (rIL17C > 0.5) in disease-associated gene sets from the GLAD4U database (59) (FDR < 0.05; Supplemental Table 15). (E) Multivariate association analysis using the expression of IL17C and proinflammatory cytokines (TNF, IL1B) in ileal CD biopsies (n = 135) as predictors and genus-level microbial abundance data of the mucosal microbiome as a response. Positive coefficients indicate a positive correlation between gene expression and compositional abundance of a bacterial genus (see Supplemental Tables 16 and 17 for input data and detailed results).
To further corroborate the potential of IL-17C as a marker of mucosal dysbiosis, we performed an integrated analysis of matched host transcriptome and microbial 16S rRNA sequencing data from the RISK cohort study (Supplemental Table 12). The mucosa-associated microbiota in the ileum of these patients with CD is primarily characterized by a higher relative abundance of Proteobacteria of the Enterobacteriaceae and Neisseriaceae families (19). Though these characteristic shifts in the ileal microbial composition are to some degree observed in colonic patients with CD without overt ileal inflammation (6), there is also a well-established interdependency between the bloom of Enterobacteriaceae and the inflammatory environment (20). Furthermore, at least in epithelial cell lines, treatment with the proinflammatory cytokines TNF-α and IL-1β directly induces IL-17C expression and secretion (10). Thus, to test whether the induction of IL17C is a predictor of epithelial activation by mucosal dysbiosis, we performed multivariate association analysis using IL17C, TNF, and IL1B as predictor variables and microbial abundance data as a response. We found that IL17C rather than TNF or IL1B had the strongest positive associations, comprising all major genera of the Enterobacteriaceae family (Figure 6E and Supplemental Tables 16 and 17). The link between IL17C expression and relative abundance of Enterobacteriaceae in human mucosal biopsies supports the concept that analogous to Duoxa-deficient mice, high IL-17C levels are indicative of a shift in the gram-negative microbiota at the mucosal surface.
Though 3 children with very-early-onset IBD and concomitant rare DUOX2 variants have been recently described (21, 22), these isolated cases have been insufficient to establish causality between DUOX2 genetics and IBD risk. To directly assess whether rare DUOX2 protein variants associated with abnormally high plasma IL-17C levels ultimately contribute to the population risk for IBD, variants detected in whole-genome sequencing data of 3 large IBD cohorts (IBD Exomes Portal) were classified by their predicted impact and stratified by ancestry-specific allele frequencies (Supplemental Tables 18–20). We defined high-impact DUOX2 variants as protein-altering variants with AF less than 0.001 since carriers of such variants had a significantly increased prevalence of outlier high plasma IL-17C concentrations in the PheWAS cohort (Figure 7, A and B). Using a meta-analysis of the 3 IBD cohorts, we found a significantly increased risk among DUOX2 variant carriers to develop IBD (pooled OR = 1.54 [95% CI: 1.09–2.18]; P = 0.0007; random-effects model, Figure 7C). With respect to the specific ancestry groups, there was a significant effect of DUOX2 variants on IBD risk in the Ashkenazi Jewish cohort with an OR estimate of 2.13 (95% CI: 1.427–3.187; P = 0.0002; 2-tailed Yates’s χ2 test). For the non-Finnish European cohorts, the calculated OR for IBD was 1.27, but the result did not pass the significance threshold (95% CI 0.977–1.67; P = 0.0741; 2-tailed Yates’s χ2 test). Note that for the Finnish cohort, the smaller size of this IBD cohort and genetic bottlenecks in this population leading to a lower rate of very rare variants (23) severely limited the statistical power of this analysis (IBD: OR = 1.3823 [0.5969–3.2013]; P = 0.4498). Concerning IBD subtypes, the risk associated with DUOX2 variants appeared to be similar for patients with CD and those with ulcerative colitis (UC) (Figure 7C). To check for internal consistency of these associations, we reviewed the small subset of predicted null variants (i.e., nonsense, frameshift, canonical splice donor, or acceptor site mutations) that should each confer the maximum possible risk for heterozygous DUOX2 variants (Figure 7D). Compared with the overall high impact variant selection, the distribution of null variants was indeed suggestive of even more pronounced enrichment among patients with IBD. Thus, high plasma IL-17C in carriers of DUOX2 loss-of-function variants is not only a potential biomarker for disturbed gut microbe-immune homeostasis, but appears to reflect an early stage of IBD pathogenesis.
Figure 7. High impact DUOX2 variants confer increased risk for IBD.

(A) Outline of the case-control study comparing the burden of high-impact DUOX2 protein variants in patients with IBD and ancestry-matched non-IBD control cohorts. We stratified variants using population-specific allele frequencies from the gnomAD database. (B) Contribution of individual high impact DUOX2 protein variants to the cumulative allele frequencies. NFE, non–Finnish European; ASJ, Ashkenazi Jewish; FIN, Finnish. Note that the low prevalence of very rare variant carriers in Finnish participants is due to multiple genetic bottlenecks in that isolated population (23). See Supplemental Tables 18–20 for detailed data and Supplemental Figure 6 for the distribution of variants with higher allele frequencies. (C) Carriers of high-impact DUOX2 protein variants are at increased risk for developing IBD. The Forest plot depicts estimated ORs with 95% CI for patients with UC and CD from the 3 ancestry cohorts. The combined OR was calculated using a random-effects model with the Mantel-Haenszel weighting method (Supplemental Table 21). Test of the null hypothesis that OR is equal to 1 (60). (D) Detailed view of DUOX2 variants with predicted complete loss-of-function (i.e., frameshift, stop gained, and splice donor or acceptor site variants) in IBD and control cohorts. Two-tailed Fisher’s exact test. ND, not detected. *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
Conceptually, IBD is a chronic inflammatory disease resulting from a loss of gut microbiota–host immune system homeostasis. A crucial host factor in maintaining a homeostatic relationship with the gut microbiota is DUOX2, an inducible, epithelial-specific NADPH oxidase releasing H2O2 into the supraepithelial mucus layer. Here, using multiomic PheWAS for rare DUOX2 protein variants, we linked partial defects in the DUOX2 system to the occurrence of excessive plasma IL-17C level (IL-17Chi) in the general population. Functional studies support the notion that IL-17Chi constitutes a preclinical hallmark of inappropriate stimulation of gut epithelial cells by the expansion of mucosa-associated pathobionts. Besides being a major genetic cause of IL-17Chi in the population, the burden of deleterious DUOX2 defects also constitutes a genetic risk factor for IBD. Taken together, the results of our study implicate mucosal dysbiosis as an early driver in the pathogenesis of IBD.
The seminal finding of our PheWAS was that carriers of heterozygous, deleterious DUOX2 variants were prone to manifest the IL-17Chi phenotype. This association was carried by the burden of very rare variants. In the PheWAS cohort, approximately 5.6% carried a very rare DUOX2 protein variant (i.e., variants with AF < 0.001 in the ancestry-matched gnomAD database) and approximately 1.5% carried an ultrarare variant (i.e., variants not found in the overall gnomAD database). Among individuals with the highest measured baseline IL-17C levels, almost half carried rare DUOX2 protein variants (Figure 2D), indicating that DUOX2 defects are a major genetic factor of the IL-17Chi phenotype in the general population.
In contrast to other members of the IL-17 family, IL-17C is exclusively expressed in barrier epithelial cells upon TLR stimulation (10, 24). The IL-17C receptor (IL-17RE/IL-17RA heterodimer) is expressed in the epithelium itself but also in Th17 cells (13). Mechanistically, IL-17C induction reinforces mucosal immunity by inducing the secretion of antimicrobial proteins and chemokines in an autocrine/paracrine fashion and by boosting Th17 cell function (25). In our study population, the IL-17Chi phenotype was associated with an elevated level of other inflammation-related plasma proteins. Of these, the chemokine CCL20, the unique ligand for CCR6-mediated recruitment of Th17 cells, was most consistently increased in concert with high IL-17C. In the healthy gut, CCL20 shows only weak constitutive expression in the surface epithelial layer, predominantly the follicle-associated epithelium in the small intestine. Bacterial contact triggers CCL20 expression either directly via TLR-dependent signaling (26) or indirectly by being an IL-17C downstream target (10). Apart from CCL20, IL-17Chi subjects had significantly higher mean plasma levels of CXCL9, CXCL11, FGF23, IL-6, and IL-17A (Supplemental Figure 1A). With respect to the latter proteins, it is noteworthy that they belong to a plasma protein signature that is commonly upregulated in the plasma of patients with CD (Supplemental Figure 1B) (27). The presence of IBD biomarkers in IL-17Chi subjects was not driven by the inclusion of individuals with self-reported IBD diagnosis. Thus, in IL-17Chi subjects without prior IBD diagnosis, the plasma protein profile is compatible with a concerted gut mucosal immune response. This specific chemokine/cytokine signature was not unique to carriers of DUOX2 variants but similarly found in IL-17Chi subjects without DUOX2 variant (Figure 2E). Therefore, disturbed gut immune homeostasis appeared to be the most plausible cause of the IL-17Chi phenotype in a general population.
Concerning the regulation of IL-17C by the microbiota in vivo, colonization of WT axenic mice with SPF microbiota only marginally induced Il17c expression, indicating that Il17c is essentially silenced under homeostatic conditions. This was in marked contrast to the strong production of Th17 cytokines (Il17a, Il22) and their respective epithelial targets (e.g., Duox2) (5, 15). Induction of intestinal Th17 cells by a healthy microbiota is driven by contact of specific gram-positive symbionts (SFB, Bifidobacterium species) (15, 28, 29) with epithelial cells thereby triggering STAT3-dependent expression and secretion of serum amyloid 1 (30). While barely detectable in WT mice, Il17c expression was markedly induced in the ileal mucosa of mice deficient in DUOX2 enzyme. This abnormal expression in KO mice was completely dependent on the presence of gram-negative microbiota (Figure 5B) and independent of T cells (Figure 4H). Monolayers derived from WT and Duoxa–/– enteroids did not differ in their acute response to direct bacterial exposure (Figure 5C), indicating that Duoxa–/– epithelial cells were not deficient in recognition of bacterial ligands or the downstream intracellular signal transduction. The in vitro model appeared to be not suitable to recapitulate the effect of DUOX2-generated H2O2 on epithelium-encroaching bacteria in vivo, likely because of the fundamental differences in the chronicity of exposure and overall redox environment, and the incomplete formation of a supraepithelial mucus layer in vitro. The latter restricts access of bacteria to the epithelium and limits outward diffusion of DUOX2-generated H2O2. In fact, treatment of WT and Duoxa–/– mice with mucus-thinning carboxymethylcellulose (CMC) obliterated the difference in Il17c expression in vivo (Figure 4F). Microbes potentially exposed to the DUOX2-generated H2O2 flux will include those for which the outer mucus layer constitutes the natural microhabitat (31) and any pathobionts invading that niche. Compared with WT littermates, the composition of the mucosa-associated microbiota in Duoxa–/– mice was characterized by the expansion of gram-negative pathobionts (Helicobacteriaceae, uncharacterized proteobacterial operational taxonomic unit [OTU]). The sensitivity of intestinal Helicobacteriaceae to DUOX2-generated H2O2 would be consistent with the profound effect of DUOX2 in restricting gastric colonization by Helicobacter species (3, 4) that robustly induce Il17c in vitro and in vivo (32).
In human subjects with heterozygous DUOX2 loss-of-function variants, haploinsufficiency is expected to manifest when DUOX2 gene expression is strongly induced. An illustration of this phenomenon is provided by our earlier studies in heterozygous (Duoxa+/–) mice colonized with Helicobacter felis (3). Haploinsufficiency of DUOX2 has also been identified as the most common genetic risk factor for transient congenital hypothyroidism, defined as elevated thyrotropin level at birth that spontaneously normalizes after the newborn period. We recently found that the risk for developing IBD in later life is higher in individuals with transient congenital hypothyroidism at birth (7), a finding congruent with the idea that DUOX2 mutations could be a hitherto unrecognized genetic risk factor in IBD. In the present study, we found that filtering of protein-altering variants by allele rarity sufficiently enriched for deleterious variants that are associated with the IL-17Chi phenotype. Employing the same variant stratification in IBD cohorts identified very rare DUOX2 variants as a significant disease susceptibility factor. From the odds ratios and estimated population exposure, the cumulative population attributable risk (PAR) for very rare (AF < 0.001) protein-altering DUOX2 variants can be estimated as 0.68% (IBD: CD 0.5%; UC 0.9%) among the Ashkenazi Jewish population. Similar attributable risk can be estimated for the non–Finnish European population (IBD: 0.46%), but the calculation is based on a nonsignificant (P = 0.07) odds ratio. For comparison, NOD2 p.Leu1007fsX1008, the variant most significantly linked to CD in Europeans (GWAS catalog; http://www.ebi.ac.uk/gwas/), has an estimated PAR of 1.6%. Thus, DUOX2 variants could be quite relevant in accounting for some of the missing heritability in IBD, i.e., the difference between the estimated heritability (from twin studies) and the disease liability explained by variants in risk loci identified by common variant association studies (33, 34).
While common changes in the microbiota composition have been described in patients with IBD (e.g., an expansion of E. coli and other facultative anaerobes), a dysbiotic microbiota may be both a consequence of the inflammation milieu as well as an important driver of the inflammation process. There is a need for early biomarkers of abnormal host-microbiota interaction to potentially identify at-risk individuals at an early, reversible stage of the disease pathogenesis. It appears that IL-17C combines characteristics in its expression and regulation that should render it a suitable plasma biomarker to monitor gut microbiota-immune system homeostasis. First, expression of IL-17C is specific to barrier epithelial cells, essentially silenced under homeostatic conditions yet highly inducible when homeostasis is disturbed. Second, as a basolaterally secreted protein, the level of IL-17C in the systemic circulation is expected to closely correlate with the tissue expression. Together, these factors would confer high sensitivity and specificity on IL-17Chi as a blood biomarker for disturbed homeostasis at the microbiota-mucosal interface.
In conclusion, our study provides a paradigm for the power of multiomic PheWAS and cumulative rare variant associations in yielding novel insights into pathophysiologic processes and the genetic underpinnings of complex diseases. We identified elevated plasma IL-17C as a hallmark of the abnormal microbiota-epithelial interaction associated with rare DUOX2 protein variants. The impact of DUOX2 variants on IL-17C induction informed the variant stratification in our case control study that substantiated DUOX2 as a novel genetic risk factor in the pathogenesis of IBD. By its features, IL-17C appears to be an excellent candidate plasma biomarker to monitor early changes in host-microbiota interaction in diseases associated with mucosal dysbiosis. The identification of at-risk individuals at a presymptomatic stage of disease will facilitate the development of preventive intervention strategies targeting the microbiota.
Methods
Collection of human blood samples.
The study was reviewed and approved by the Western IRB (study number 1178906). The research was performed entirely using deidentified and aggregated data of US residents who had signed a research authorization allowing the use of their anonymized data in research. Trained phlebotomists collected blood used for whole-genome sequencing, clinical laboratory tests, proteomics, and metabolomics in standard clinical facilities. Four days in advance of each blood draw, study participants were asked to discontinue nonprescription medications, including acetaminophen, ibuprofen, and over-the-counter cold remedies. For 24 hours in advance of each blood draw, participants were asked to avoid alcohol, vigorous exercise, and products containing aspartame or MSG. For 12 hours in advance of each blood draw, participants were asked to fast (no food or drink except water) until after the draw was completed. Nonfasting samples were excluded from the study.
Whole-genome sequencing and DUOX2/DUOXA2 variants annotation.
DNA was extracted from whole-blood samples for whole-genome sequencing in a lab approved by Clinical Laboratory Improvement Amendments (WuXi NextCODE) using Illumina HiSeq X technology with sequencing mode PE150 and 30 times target coverage. The sequenced reads were aligned to human reference GRCh37/hg19 using BWA 0.7.12. (35). Variant calling was performed with GATK 3.3.0, including indel local realignment followed by base quality recalibration (36). Variant calls were produced by GATK HaplotypeCaller. Only calls with DP greater than 8 and GQ greater than 20 were included in this study. The Ensembl GRCh37 annotation v75 was used to identify gene boundaries for DUOX2/DUOXA2. Variants passing quality filters were selected within these gene boundaries using custom Python scripts. The Ensembl Variant Effect Predictor REST API was used to assign the functional impact of each variant. The API query was defined as http://grch37.rest.ensembl.org/vep/human/region/{chr}:{start}-{end}:1/{allele}?CADD=1&Conservation=1&ExAC=1 The most severe consequence at each position was used to filter the variants. Variants were selected for downstream analysis if VEP consequence was one of the following: ‘missense_variant’, ‘frameshift_variant’, ‘splice_acceptor_variant’, ‘splice_donor_variant’, ‘stop_gained’.
Clinical laboratory tests.
Blood samples were analyzed at either LabCorp or Q2 Solutions. Clinical blood tests included diabetes markers, a lipid panel, complete blood cell counts, inflammation markers, liver function markers, kidney function markers, nutrition markers, and other markers, all of which were tested according to standard clinical procedures defined by the testing laboratories.
Plasma proteomics.
Plasma concentrations of proteins were measured using the ProSeek Cardiovascular II, Cardiovascular III, and Inflammation panels (Olink Biosciences) at Olink facilities. The ProSeek method is based on the highly sensitive and specific proximity extension assay, which involves the binding of distinct polyclonal oligonucleotide-labeled antibodies to the target protein, followed by quantification with real-time quantitative PCR (37). Samples were processed in several batches; potential batch effects were adjusted using pooled control samples included with each batch.
Plasma metabolomics.
Metabolon Inc. conducted the metabolomics assays on plasma samples. Data were generated using the Global Discovery platform. Samples were processed in several batches with pooled quality control samples included in each batch; potential batch effects for each metabolite were adjusted by dividing by the corresponding average value identified in the pooled quality control samples from the same batch.
Human fecal microbiome.
Individuals collected stool samples at home using the DNA Genotek OMNIGene GUT collection kit and shipped the samples at ambient temperature to the sequencing laboratory. Baseline gut microbiome sequencing data in the form of FASTQ files were provided by Second Genome or DNA Genotek based on 250 bp paired-end MiSeq profiling of the 16S v4 region. OTU abundances were calculated using the QIIME (38) pipeline and Greengenes database. PICRUSt (39) was used to infer metagenome functional content, and KEGG orthologies were collapsed into KEGG Pathways and KEGG Modules for analysis.
Phenome-wide association study.
Prior to performing the analyses, the highest and lowest 0.25% of values were winsorized. Highly skewed distributions (|skew| > 1.5) were log-transformed prior to analysis. To adjust for potential confounding effects, the non–time-varying covariates age, sex, body mass index, enrollment channel, whether or not the participant reported taking cholesterol medications, blood pressure medications, or diabetes medications, and genetic ancestry, as well as the time-varying covariates observation month and observation vendor (when multiple vendors were used) were included as fixed effects in all models. Genetic ancestry was represented by principal components (PCs) 1–8 from an analysis of 107,280 linkage disequilibrium pruned autosomal SNPs with minor allele frequency greater than 5% using the combined PC-AiR (40) and PC-Relate (41) approach as described by Conomos et al. (42). The GENESIS R package was used to perform SKAT-O tests using Madsen-Browning weights (43). Gaussian null models were used with test type Score.
Site-directed mutagenesis and heterologous expression of DUOX2 variants.
Individual DUOX2 variants were introduced into an N-terminal hemagglutinin epitope–tagged (HA-tagged) DUOX2 expression vector (44) by site-directed mutagenesis (QuikChange, Stratagene). All constructs were verified by bidirectional Sanger sequencing (Supplemental Figure 2A). The DUOXA2-EGFP expression vector was prepared as described (44). HEK293 cells were transfected at 50% to 60% confluence using FuGENE 6 reagent (Promega). DUOXA2-EGFP (controls: EGFP and empty vector) was cotransfected with an equal amount (105 ng/cm2 cell monolayer) of one of the DUOX2 plasmids (WT or variant, control: empty vector). Under these conditions, DUOXA2 is available in substantial excess and does not limit DUOX2/DUOXA2 heterodimerization (45). In all experiments, the total amount of DNA in each transfection was kept constant by adjusting with the empty vector.
DUOX2 enzymatic activity assay.
H2O2 released into the culture medium was measured using a peroxidase-independent homogenous bioluminescence detection system (ROS Glo H2O2, Promega). Briefly, cells were washed and incubated at 37°C for 1 hour in HBSS(Ca2+)/10 mM HEPES (pH 7.4)/10 mM glucose containing 1 μM ionomycin/200 nM PMA to stimulate DUOX2 intrinsic activity and 25 M ROS-Glo substrate that reacts with H2O2 to generate a luciferin precursor. Following incubation, aliquots of the culture medium were mixed with equal amounts of ROS-Glo Detection Solution containing recombinant luciferase, and luminescence was measured on a Synergy 2 plate reader (BioTek Instruments, Inc.). As an internal control for transfection efficiency, luciferase activity from cotransfected pGL3-Promoter (Promega) was determined in the remaining cells (Luciferase Assay, Biotium).
Quantitation of DUOX2 expression in the plasma membrane.
The flow cytometry assay to quantitate recombinant DUOX2 expression at the cell surface of HEK-293 cells (ATCC) has been previously described in detail (Supplemental Figure 2, B and C) (9). Briefly, exposure of the N-terminal HA epitope of HA-DUOX2 in nonpermeabilized cells was detected using rat anti-HA (clone 3F10, Roche) as primary and Alexa Fluor 647–conjugated anti-rat IgG as the secondary antibody, respectively. The intracellular EGFP moiety of the cotransfected DUOXA2-EGFP was used to select the population of transfected cells. Cytometry data were acquired on an Accuri C6 flow cytometer (BD Biosciences) (FL1: EGFP; FL4: AF647 nm) and analyzed using FlowJo v10.5.3 software. Relative DUOX2 surface expression (AUC of FL4 in EGFP-positive cells) was normalized for the number of EGFP-positive cells.
Acute microbial exposure of colonoid-derived monolayers.
Colonoids and colonoid-derived monolayers from Duoxa–/– mice (n = 3) and WT littermates (n = 3) were established following previously outlined protocols (46) and cultured for 48 hours in the presence of 50 ng/mL recombinant mouse IL-22 (R&D Systems) to induce DUOX2 and DUOXA2 gene expression (5). For acute exposure to bacteria, the culture medium was replaced by HBSS(Ca2+) supplemented with 20 mM HEPES, 10 mM glucose, and 1% FBS. Bacteria (Salmonella Typhimurium strain SL1344, Citrobacter rodentium strain DBS120, E. coli strain K12, Enterococcus faecalis (mouse cecum-derived isolate), Lactobacillus rhamnosus GG, Clostridium scindens, ref. 47) were washed in the same buffer and added at MOI approximately 10 to the apical compartment for 1 hour. For experiments under anaerobic conditions, cell monolayers and buffer were preequilibrated for 1 hour.
Animals.
Duoxa1/Duoxa2 floxed (Duoxafl/fl) mice on a C57BL/6N background were generated by inserting LoxP sites between Duoxa1 intron 3 and Duoxa2 intron 5 to delete Duoxa1 exons 4–6 and Duoxa2 exons 6–9, followed by Neo-cassette excision using FLP recombination, by the Mice Biology Program at the University of California, Davis (project number MBP-714). Duoxafl/fl homozygous were used for breeding. Intestinal epithelial-specific Duoxa1/Duoxa2 deletion (Duoxafl/fl, Vil1-Cre) was made by mating with Villin-Cre mice (B6.Cg-Tg(Vil1-cre)1000Gum/J, Jackson Laboratory). Duoxa–/– mice lacking functional DUOX enzymes have been previously described (48). Duoxa–/– mice and their WT littermates received drinking water supplemented with L-thyroxine to equalize thyroid hormone status (5). Rag1–/– (Rag1tm1Mom) in C57BL/6 background (Jackson Laboratory) were used to generate Duoxafl/fl mice deficient in T and B cells (49). All mice were housed in ventilated microisolator cages under specific pathogen–free (SPF) conditions. Food and water were supplied ad libitum.
Gut microbiota manipulations.
Germ-free (GF) mice were aseptically transferred to microisolator cages and housed in sterile laminar-flow hoods. GF mice were orally gavaged with a freshly prepared suspension of frozen cecal material from either mice monocolonized with SFB (50), SPF mice, or GF controls. Tissues were collected one week following treatment.
CMC (degree of substitution: 0.7; average MW ~250 kDa; Sigma-Aldrich) was dissolved at 1% (wt/vol) concentration in drinking water. Treatment was initiated at weaning and continued with weekly solution changes for 8 weeks (P21–P77).
To suppress the gram-negative gut microbiota, mice were treated for 3 days with a combination of ciprofloxacin and metronidazole (each at 50 mg/kg BW; twice daily) or PBS (controls) by oral gavage.
Tissue collection.
Animals were euthanized by isoflurane overdose. Intestinal segments were collected from the ileum (terminal portion) and colon (midportion). The isolated segments were opened longitudinally, rinsed thrice with PBS, and snap-frozen in liquid nitrogen.
Real-time reverse transcription PCR (RT-qPCR).
Total RNA extractions were prepared using TRIzol reagent, treated with deoxyribonuclease, and cleaned up on RNeasy spin columns (Qiagen). RNA was reverse transcribed with Superscript II (Life Technologies) using random hexamer priming. Concentration and purity of RNA preparations were determined on a NanoDrop ND-1000 UV spectrophotometer. PCR amplifications were performed using a C1000 Thermal Cycler (Bio-Rad) with SYBR Green dye (Molecular Probes) and Platinum Taq DNA polymerase (Invitrogen). Each reaction was performed in triplicate with the following conditions: 1 minute at 95°C, 40 cycles of 10 seconds at 95°C, and 1 minute at 65°C. Amplification specificity was confirmed by melting curve analysis of products and gene expression was normalized to Hprt1 mRNA using the 2−ΔΔCt method (51). Sequences of oligonucleotide primers are listed in Supplemental Table 22.
16S rDNA profiles from mouse mucosal samples.
Genomic DNA was extracted using a modified protocol of the Qiagen DNeasy Blood & Tissue kit that included an initial bead-beating step (0.7 mm garnet) for cell wall disruption. 16S rRNA gene libraries were constructed using primers specific to the V4 region and subjected to Illumina MiSeq 250 bp paired-end sequencing (University of Michigan Medical School Host Microbiome Initiative). FASTQ files have been deposited in the NCBI Sequence Read Archive under BioProject PRJNA590250. Sequences were curated using the mothur v1.40.5 (52) pipeline implemented in Nephele (v2.2.8) (53). Sequences were assigned to OTUs using a dissimilarity cutoff equal to 0.03 and classified against the nonredundant SILVA v128 ribosomal RNA database.
Correlation of host gene expression level with microbial abundance data.
LEfSe (linear discriminant effect size) analysis (54) was used to identify taxa distinguishing IL-17Chi and IL-17Clo microbiota based on significance level and estimated effect size. Boosted additive general linear models between multiple host predictors and arcsin–square root transformed relative abundance data of the mucosal microbiome as a response were calculated using MaAsLin (55).
The burden of high-impact DUOX2 mutations in IBD.
DUOX2 variant frequency data for IBD (4970 non–Finnish European, 2641 Ashkenazi Jewish, 696 Finnish) and control cohorts (2770 non–Finnish European, 3044 Ashkenazi Jewish, 9930 Finnish) were obtained from the IBD Exomes Portal (http://ibd.broadinstitute.org). Genotype quality control and relatedness filter have been described (56). Protein-altering variants were selected using Ensembl VEP classifier. Ancestry-specific minor allele frequencies for stratification were obtained from gnomAD v2.1. ORs were calculated from cumulative allele frequency data. The combined effect size for all cohorts was estimated using a random effect model with Mantel-Haenszel weighting (57). The point estimate for the proportion of observed variance in OR between cohorts that reflects real OR differences (I2) was 36%.
Statistics.
As indicated, we evaluated group differences for statistical significance with 1-way ANOVA with Dunnett’s multiple comparisons test (more than 2 groups; data follow Gaussian distribution as determined by D’Agostino-Pearson normality test), Kruskal-Wallis test with Dunn’s post hoc test (more than 2 groups; nonparametric), Mann-Whitney (2 groups; nonparametric), or Fisher’s exact test (contingency tables). Data were analyzed with GraphPad Prism 8.0. We used Meta-Essentials (57) to assess genetic risk from allele count data and WebGestalt 2017 (58) for gene set enrichment and overrepresentation analyses. P values were adjusted for multiple testing with the Benjamini-Hochberg procedure to control the false discovery rate (FDR). Results with a P value less than 0.05 were considered significant.
Study approval.
The human studies were reviewed and approved by the Western IRB (study number 1178906). The research was performed entirely using deidentified and aggregated data of individuals who had signed a research authorization allowing the use of their anonymized data in research. All animal studies were approved by the University of Michigan IACUC (PRO-00007922) and the IACUC of the Greater West Los Angeles Veterans Affairs Hospital.
Author contributions
HG, ATM, and JYK conceived and supervised the study. HG and ATM analyzed the data and wrote the manuscript with input from all authors. ES and MPC were involved in data curation and statistical analysis. MZ, LSG, GH, and MEZ contributed to animal studies. NK and SB contributed to the interpretation of results. NK, HNK, S Kitamoto, RWS, YA, S Kugathasan, JK, YH, LAD, and GSO provided essential research materials and resources.
Supplementary Material
Acknowledgments
We would like to thank the Helmsley IBD Exomes Program; a full list of contributing groups can be found at http://ibd.broadinstitute.org/about. We also like to acknowledge the Crohn’s and Colitis Foundation’s support of the RISK cohort study. This study was supported by NIH grants R01 DK117565-01 (to HG and JYK), P30 DK034933 and UL1TR002240 (to JYK), R01 DK054221-17 (to JK); DVA Merit Review Award I01 BX001245 (to JK); the Mouse Biology Program (MBP) at UC Davis grants U24CA210967 and P30ES017885 (to GSO); and by work performed by The University of Michigan Microbial Systems Molecular Biology Laboratory.
Version 1. 03/02/2021
In-Press Preview
Version 2. 05/03/2021
Electronic publication
Footnotes
Conflict of interest: LAD received research funding from FrieslandCampina, Glycosyn, and Janssen.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(9):e141676.https://doi.org/10.1172/JCI141676.
See the related Commentary at Decoding the matrix: multiomics reveals host-microbe biomarker for inflammatory bowel disease.
Contributor Information
Helmut Grasberger, Email: helmut@umich.edu.
Andrew T. Magis, Email: andrewmagis@gmail.com.
Elisa Sheng, Email: elisasheng@gmail.com.
Matthew P. Conomos, Email: mconomos@uw.edu.
Min Zhang, Email: minzhang@med.umich.edu.
Lea S. Garzotto, Email: glea@umich.edu.
Guoqing Hou, Email: hougqing@umich.edu.
Shrinivas Bishu, Email: bishus@umich.edu.
Hiroko Nagao-Kitamoto, Email: khiroko@umich.edu.
Mohamad El-Zaatari, Email: mohamade@med.umich.edu.
Sho Kitamoto, Email: kitamoto@med.umich.edu.
Nobuhiko Kamada, Email: nkamada@med.umich.edu.
Ryan W. Stidham, Email: ryanstid@med.umich.edu.
Yasutada Akiba, Email: yasuakiba@hotmail.com.
Jonathan Kaunitz, Email: jake@ucla.edu.
Yael Haberman, Email: yael.haberman@cchmc.org.
Subra Kugathasan, Email: skugath@emory.edu.
Lee A. Denson, Email: lee.denson@cchmc.org.
Gilbert S. Omenn, Email: gomenn@med.umich.edu.
John Y. Kao, Email: jykao@umich.edu.
References
- 1.Ha EM, et al. A direct role for dual oxidase in Drosophila gut immunity. Science. 2005;310(5749):847–850. doi: 10.1126/science.1117311. [DOI] [PubMed] [Google Scholar]
- 2.Chavez V, et al. Ce-Duox1/BLI-3 generates reactive oxygen species as a protective innate immune mechanism in Caenorhabditis elegans. Infect Immun. 2009;77(11):4983–4989. doi: 10.1128/IAI.00627-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grasberger H, et al. Dual oxidases control release of hydrogen peroxide by the gastric epithelium to prevent Helicobacter felis infection and inflammation in mice. Gastroenterology. 2013;145(5):1045–1054. doi: 10.1053/j.gastro.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collins KD, et al. Chemotaxis allows bacteria to overcome host-generated reactive oxygen species that constrain gland colonization. Infect Immun. 2018;86(5):e00878-17. doi: 10.1128/IAI.00878-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grasberger H, et al. Increased expression of DUOX2 Is an epithelial response to mucosal dysbiosis required for immune homeostasis in mouse intestine. Gastroenterology. 2015;149(7):1849–1859. doi: 10.1053/j.gastro.2015.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haberman Y, et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J Clin Invest. 2014;124(8):3617–3633. doi: 10.1172/JCI75436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grasberger H, et al. Increased risk for inflammatory bowel disease in congenital hypothyroidism supports the existence of a shared susceptibility factor. Sci Rep. 2018;8(1):10158. doi: 10.1038/s41598-018-28586-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kryukov GV, et al. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am J Hum Genet. 2007;80(4):727–739. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levine AP, et al. Genetic complexity of Crohn’s disease in two large Ashkenazi Jewish families. Gastroenterology. 2016;151(4):698–709. doi: 10.1053/j.gastro.2016.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramirez-Carrozzi V, et al. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat Immunol. 2011;12(12):1159–1166. doi: 10.1038/ni.2156. [DOI] [PubMed] [Google Scholar]
- 11.Johansson ME, et al. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc Natl Acad Sci U S A. 2011;108 Suppl 1:4659–4665. doi: 10.1073/pnas.1006451107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chassaing B, et al. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature. 2015;519(7541):92–96. doi: 10.1038/nature14232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Song X, et al. IL-17RE is the functional receptor for IL-17C and mediates mucosal immunity to infection with intestinal pathogens. Nat Immunol. 2011;12(12):1151–1158. doi: 10.1038/ni.2155. [DOI] [PubMed] [Google Scholar]
- 14.Im E, et al. Toll-like receptor 5 engagement induces interleukin-17C expression in intestinal epithelial cells. J Interferon Cytokine Res. 2012;32(12):583–591. doi: 10.1089/jir.2012.0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ivanov II, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139(3):485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Larsson E, et al. Analysis of gut microbial regulation of host gene expression along the length of the gut and regulation of gut microbial ecology through MyD88. Gut. 2012;61(8):1124–1131. doi: 10.1136/gutjnl-2011-301104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pircalabioru G, et al. Defensive mutualism rescues NADPH oxidase inactivation in gut infection. Cell Host Microbe. 2016;19(5):651–663. doi: 10.1016/j.chom.2016.04.007. [DOI] [PubMed] [Google Scholar]
- 18.Friedrich M, et al. Intestinal neuroendocrine cells and goblet cells are mediators of IL-17A-amplified epithelial IL-17C production in human inflammatory bowel disease. Mucosal Immunol. 2015;8(4):943–958. doi: 10.1038/mi.2014.124. [DOI] [PubMed] [Google Scholar]
- 19.Gevers D, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15(3):382–392. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Winter SE, et al. The dynamics of gut-associated microbial communities during inflammation. EMBO Rep. 2013;14(4):319–327. doi: 10.1038/embor.2013.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayes P, et al. Defects in NADPH oxidase genes NOX1 and DUOX2 in very early onset inflammatory bowel disease. Cell Mol Gastroenterol Hepatol. 2015;1(5):489–502. doi: 10.1016/j.jcmgh.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parlato M, et al. First identification of biallelic inherited DUOX2 inactivating mutations as a cause of very early onset inflammatory bowel disease. Gastroenterology. 2017;153(2):609–611. doi: 10.1053/j.gastro.2016.12.053. [DOI] [PubMed] [Google Scholar]
- 23.Chheda H, et al. Whole-genome view of the consequences of a population bottleneck using 2926 genome sequences from Finland and United Kingdom. Eur J Hum Genet. 2017;25(4):477–484. doi: 10.1038/ejhg.2016.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kusagaya H, et al. Toll-like receptor-mediated airway IL-17C enhances epithelial host defense in an autocrine/paracrine manner. Am J Respir Cell Mol Biol. 2014;50(1):30–39. doi: 10.1165/rcmb.2013-0130OC. [DOI] [PubMed] [Google Scholar]
- 25.Nies JF, Panzer U. IL-17C/IL-17RE: emergence of a unique axis in TH17 biology. Front Immunol. 2020;11:341. doi: 10.3389/fimmu.2020.00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Skovdahl HK, et al. Expression of CCL20 and its corresponding receptor CCR6 is enhanced in active inflammatory bowel disease, and TLR3 mediates CCL20 expression in colonic epithelial cells. PLoS One. 2015;10(11):e0141710. doi: 10.1371/journal.pone.0141710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andersson E, et al. Subphenotypes of inflammatory bowel disease are characterized by specific serum protein profiles. PLoS One. 2017;12(10):e0186142. doi: 10.1371/journal.pone.0186142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Atarashi K, et al. Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell. 2015;163(2):367–380. doi: 10.1016/j.cell.2015.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan TG, et al. Identifying species of symbiont bacteria from the human gut that, alone, can induce intestinal Th17 cells in mice. Proc Natl Acad Sci U S A. 2016;113(50):E8141–E8150. doi: 10.1073/pnas.1617460113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sano T, et al. An IL-23R/IL-22 circuit regulates epithelial serum amyloid A to promote local effector Th17 responses. Cell. 2015;163(2):381–393. doi: 10.1016/j.cell.2015.08.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H, et al. The outer mucus layer hosts a distinct intestinal microbial niche. Nat Commun. 2015;6:8292. doi: 10.1038/ncomms9292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanaka S, et al. Interleukin-17C in human helicobacter pylori gastritis. Infect Immun. 2017;85(10):e00389-17. doi: 10.1128/IAI.00389-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu JZ, Anderson CA. Genetic studies of Crohn’s disease: past, present and future. Best Pract Res Clin Gastroenterol. 2014;28(3):373–386. doi: 10.1016/j.bpg.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cleynen I, Halfvarsson J. How to approach understanding complex trait genetics — inflammatory bowel disease as a model complex trait. United European Gastroenterol J. 2019;7(10):1426–1430. doi: 10.1177/2050640619891120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lundberg M, et al. Homogeneous antibody-based proximity extension assays provide sensitive and specific detection of low-abundant proteins in human blood. Nucleic Acids Res. 2011;39(15):e102. doi: 10.1093/nar/gkr424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caporaso JG, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Langille MG, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31(9):814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Conomos MP, et al. Robust inference of population structure for ancestry prediction and correction of stratification in the presence of relatedness. Genet Epidemiol. 2015;39(4):276–293. doi: 10.1002/gepi.21896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Conomos MP, et al. Model-free estimation of recent genetic relatedness. Am J Hum Genet. 2016;98(1):127–148. doi: 10.1016/j.ajhg.2015.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Conomos MP, et al. Genetic diversity and association studies in US Hispanic/Latino populations: applications in the hispanic community health study/study of Latinos. Am J Hum Genet. 2016;98(1):165–184. doi: 10.1016/j.ajhg.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Madsen BE, Browning SR. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009;5(2):e1000384. doi: 10.1371/journal.pgen.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grasberger H, Refetoff S. Identification of the maturation factor for dual oxidase. Evolution of an eukaryotic operon equivalent. J Biol Chem. 2006;281(27):18269–18272. doi: 10.1074/jbc.C600095200. [DOI] [PubMed] [Google Scholar]
- 45.Grasberger H, et al. Missense mutations of dual oxidase 2 (DUOX2) implicated in congenital hypothyroidism have impaired trafficking in cells reconstituted with DUOX2 maturation factor. Mol Endocrinol. 2007;21(6):1408–1421. doi: 10.1210/me.2007-0018. [DOI] [PubMed] [Google Scholar]
- 46.Fernando EH, et al. A simple, cost-effective method for generating murine colonic 3D enteroids and 2D monolayers for studies of primary epithelial cell function. Am J Physiol Gastrointest Liver Physiol. 2017;313(5):G467–G475. doi: 10.1152/ajpgi.00152.2017. [DOI] [PubMed] [Google Scholar]
- 47.Buffie CG, et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature. 2015;517(7533):205–208. doi: 10.1038/nature13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grasberger H, et al. Mice deficient in dual oxidase maturation factors are severely hypothyroid. Mol Endocrinol. 2012;26(3):481–492. doi: 10.1210/me.2011-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mombaerts P, et al. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68(5):869–877. doi: 10.1016/0092-8674(92)90030-G. [DOI] [PubMed] [Google Scholar]
- 50.Umesaki Y, et al. Segmented filamentous bacteria are indigenous intestinal bacteria that activate intraepithelial lymphocytes and induce MHC class II molecules and fucosyl asialo GM1 glycolipids on the small intestinal epithelial cells in the ex-germ-free mouse. Microbiol Immunol. 1995;39(8):555–562. doi: 10.1111/j.1348-0421.1995.tb02242.x. [DOI] [PubMed] [Google Scholar]
- 51.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 52.Schloss PD, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75(23):7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weber N, et al. Nephele: a cloud platform for simplified, standardized and reproducible microbiome data analysis. Bioinformatics. 2018;34(8):1411–1413. doi: 10.1093/bioinformatics/btx617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Segata N, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morgan XC, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13(9):R79. doi: 10.1186/gb-2012-13-9-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rivas MA, et al. Insights into the genetic epidemiology of Crohn’s and rare diseases in the Ashkenazi Jewish population. PLoS Genet. 2018;14(5):e1007329. doi: 10.1371/journal.pgen.1007329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Suurmond R, et al. Introduction, comparison, and validation of Meta-Essentials: a free and simple tool for meta-analysis. Res Synth Methods. 2017;8(4):537–553. doi: 10.1002/jrsm.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang J, et al. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017;45(W1):W130–W137. doi: 10.1093/nar/gkx356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jourquin J, et al. GLAD4U: deriving and prioritizing gene lists from PubMed literature. BMC Genomics. 2012;13 Suppl 8:S20. doi: 10.1186/1471-2164-13-S8-S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sheskin DJ, ed. Handbook Of Parametric And Nonparametric Statistical Procedures. Chapman & Hall/CRC; 2007. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.