

Keywords: acute pancreatitis, capillary leak syndrome, multiple organ dysfunction syndrome, multiple organ failure, systemic inflammatory response syndrome
Abstract
Severe acute pancreatitis (SAP) includes persistent systemic inflammation (SIRS) and multiorgan failure (MOF). The mechanism of transition from SIRS to MOF is unclear. We developed a fluid compartment model and used clinical data to test predictions. The model includes vascular, interstitial and “third-space” compartments with variable permeability of plasma proteins at the capillaries. Consented patients from University of Pittsburgh Medical Center Presbyterian Hospital were studied. Preadmission and daily hematocrit (HCT), blood urea nitrogen (BUN), creatine (Cr), albumin (Alb), and total protein (TP) were collected, and nonalbumin plasma protein (NAPP = TP minus the Alb) was calculated. Subjects served as their own controls for trajectory analysis. Of 57 SAP subjects, 18 developed MOF (5 died), and 39 were non-MOF (0 died). Compared with preadmission levels, admission HCT increased in MOF +5.00 [25%-75% interquartile range, IQR] versus non-MOF −0.10 [−1.55, 1.40] (P < 0.002) with HCT > +3 distinguishing MOF from non-MOF (odds ratio 17.7, P = 0.014). Preadmission Alb fell faster in MOF than non-MOF (P < 0.01). By day 2, TP and NAPP dropped in MOF but not non-MOF (P < 0.001). BUN and Cr levels increased in MOF (P = 0.001), but BUN-to-Cr ratios remained constant. Pancreatic necrosis was more common in MOF (56%) than non-MOF (23%). Changing capillary permeability to allow loss of NAPP in this model predicts loss of plasma oncotic pressure and reduced vascular volume, hypotension with prerenal azotemia and acute kidney dysfunction, pancreas necrosis, and pulmonary edema from capillary leak in the lung with acute respiratory distress syndrome. Sequential biomarker analysis in humans with or without MOF is consistent with this model. This study is registered on https://clinicaltrials.gov at NCT03075605.
NEW & NOTEWORTHY Acute pancreatitis is a sudden inflammatory response to pancreatic injury that may spread to systemic inflammation, multiorgan failure, and death in some patients. With the use of the predictions of a new mechanistic model, we compared patients with severe acute pancreatitis with or without multiorgan failure. All biomarkers of capillary leak and clinical features of multiorgan failure were accurately predicted. This provides a new paradigm for understanding and developing new treatments for patients with severe acute pancreatitis.
INTRODUCTION
Acute pancreatitis (AP) is an inflammatory condition of the pancreas with a sudden onset, variable clinical course, and an increasing incidence over the last several decades (5, 27, 61). Among gastrointestinal diseases, AP is among the top 3 causes for hospitalization per year with >290,000 admissions and costs exceeding 2.7 billion dollars in the United States (47). The majority of patients with AP will experience a mild clinical course, whereas 10–20% of patients will develop severe AP with systemic inflammatory response syndrome (SIRS; Ref. 36), persistent SIRS (lasting ≥48 h; Ref. 8), single-organ failure or multiorgan failure (MOF, also called multiorgan dysfunction syndrome; Refs. 5, 54), and ≤30% mortality (5). Acute pancreatitis also has long-standing consequences for the patient (58, 59) and is responsible for a significant financial burden on the healthcare systems.
Acute pancreatitis is a rapidly evolving condition with sequential states of injury, proinflammatory cascade, an anti-inflammatory counterresponse, and resolution/healing. However, multiple risk factors can alter the magnitude of the response or trigger secondary complications that contribute to pathology. A hyperproinflammatory response results in a “cytokine storm” and SIRS with potential secondary effects of MOF. A hyper-anti-inflammatory response can lead the compensatory anti-inflammatory response syndrome with susceptibility to sepsis and infections. Most of the morbidity and mortality in AP is associated with MOF, yet the mechanism linking SIRS to MOF is poorly understood.
A model is a simplified representation of a complex system that is used to understand the mechanisms leading to an observed phenomenon. Although SIRS is, by definition, an inflammatory phenomenon, MOF is more complex and defined by clinical signs and symptoms rather than by the underlying mechanisms leading to the phenomenon. Furthermore, MOF is a syndrome with a defined set of organs that fail in characteristic ways. Additionally, the only clinically effective way to mitigate evolving MOF is fluid resuscitation, suggesting that fluid dynamics underly some of the pathophysiology. Furthermore, the loss of plasma from the blood requires a change in the permeability of blood vessels, likely in the capillary bed, and the cells that regulate this function are endothelial cells.
Under normal conditions, endothelial cells control the flux of molecules and fluids between the vascular and interstitial compartments through locally and systemically regulated mechanisms. Increased but locally restricted permeability between endothelial cells of capillaries and small blood vessels may occur in a regulated, reversible way after injury or immune activation. In contrast, severe endothelial injury, dysfunction, or death may allow small, medium, and large plasma proteins to freely move from the plasma into the interstitial space.
In severe AP, endothelial permeability may increase systemically and lead to the pathological capillary leak syndrome (CLS, also called vascular leak syndrome, VLS) resulting in tissue edema and intravascular hypovolemia (11, 19, 41, 45, 63). Mechanistically, the loss of plasma proteins diminishes the colloid osmotic gradient in the postcapillary venules, resulting in inadequate resorption of fluid from the tissues. Failure of postcapillary tissue fluid resorption and abnormal accumulation of protein-rich fluid in the interstitial spaces or other fluid compartments (e.g., 3rd space, compartments not directly linked to the lymphatic drainage of extravascular proteins) result in continued loss of intravascular proteins and fluid. Eventually, the loss of fluid and protein into the tissues and 3rd spaces results in intravascular hypovolemia.
The effects of plasma loss include hemoconcentration and MOF manifest in the cardiovascular system as hypotension and hypovolemic shock, in the kidney as prerenal azotemia and acute kidney injury, in the pancreas as pancreatic necrosis, in the gut as intestinal mucosal injury, and in other organ systems (8, 42, 45, 54, 63). In addition to organ hypoperfusion, the expanded interstitial fluid compartment results in tissue edema and contributes to organ dysfunction (20, 33). This mechanism is especially important in the lung, where the process leads to pulmonary edema, hypoxemia, and acute respiratory distress syndrome (17, 46). The factors determining the type, extent, and importance of CLS leading to MOF in AP are poorly defined.
Clinical observations suggest that about half of the patients who experience SIRS will not progress to MOF, even though many of these patients have some degree of hypovolemia and may benefit from early intravenous fluids (13, 60, 65). Other patients will experience SIRS, CLS, and MOF with significantly higher morbidity and mortality (36). The importance of missing variables between SIRS and MOF is reflected by the very poor positive predictive value (PPV) of all commonly used clinical severity scoring systems (i.e., <0.60; Ref. 37). We hypothesize that the difference between patients with MOF and those without MOF (non-MOF) is the dysregulated and irreversible capillary leakage of both small and large plasma proteins between damaged endothelial cells.
The initial hours of AP are critical to minimize the morbidity and mortality with specific intervention (23). Since the inflammatory process amplifies and evolves over time, there may be a window of opportunity to identify patients who are destined to develop CLS and MOF and intervene, thus preventing much of the morbidity and mortality of severe AP. However, to target and minimize the phenomenon, we must understand the underlying mechanisms linking SIRS to MOF. Furthermore, biomarkers are needed as dynamic indicators of the pathogenic mechanisms, processes, and magnitude of SIRS, CLS, and MOF. These biomarkers can be used to predict the overall disease trajectory within the pre-MOF window and would allow for earlier treatment, leading to better outcomes for patients with AP (18).
The aim of this study was to develop an evidence-based, dynamic model of MOF development and to describe early, clinically available biomarkers from a series of patients with AP that were ascertained very early in their clinical course. We focused on a dynamic modeling of variable capillary permeability of smaller and larger plasma proteins in patients with MOF and non-MOF patients. We hypothesize that the dynamic changes in levels of plasma albumin (Alb), total protein (TP), and nonalbumin plasma protein (NAPP; i.e., TP minus the Alb) could be used as surrogates of capillary permeability, with changes in hematocrit (HCT) inversely related to loss of plasma from the vascular compartment. In addition, we used blood urea nitrogen (BUN) and creatinine (Cr) to estimate the type and severity of renal dysfunction and contrast enhanced computed tomography (CT) scans to detect pancreatic necrosis (PNec). With the use of this model, we identified clinically available biomarkers that could be tracked in individual patients and could be used to provide a more accurate assessment of their status than the traditional approach of using population-defined cutoff values for calculation of various severity scores (37).
METHODS
Compartment Model
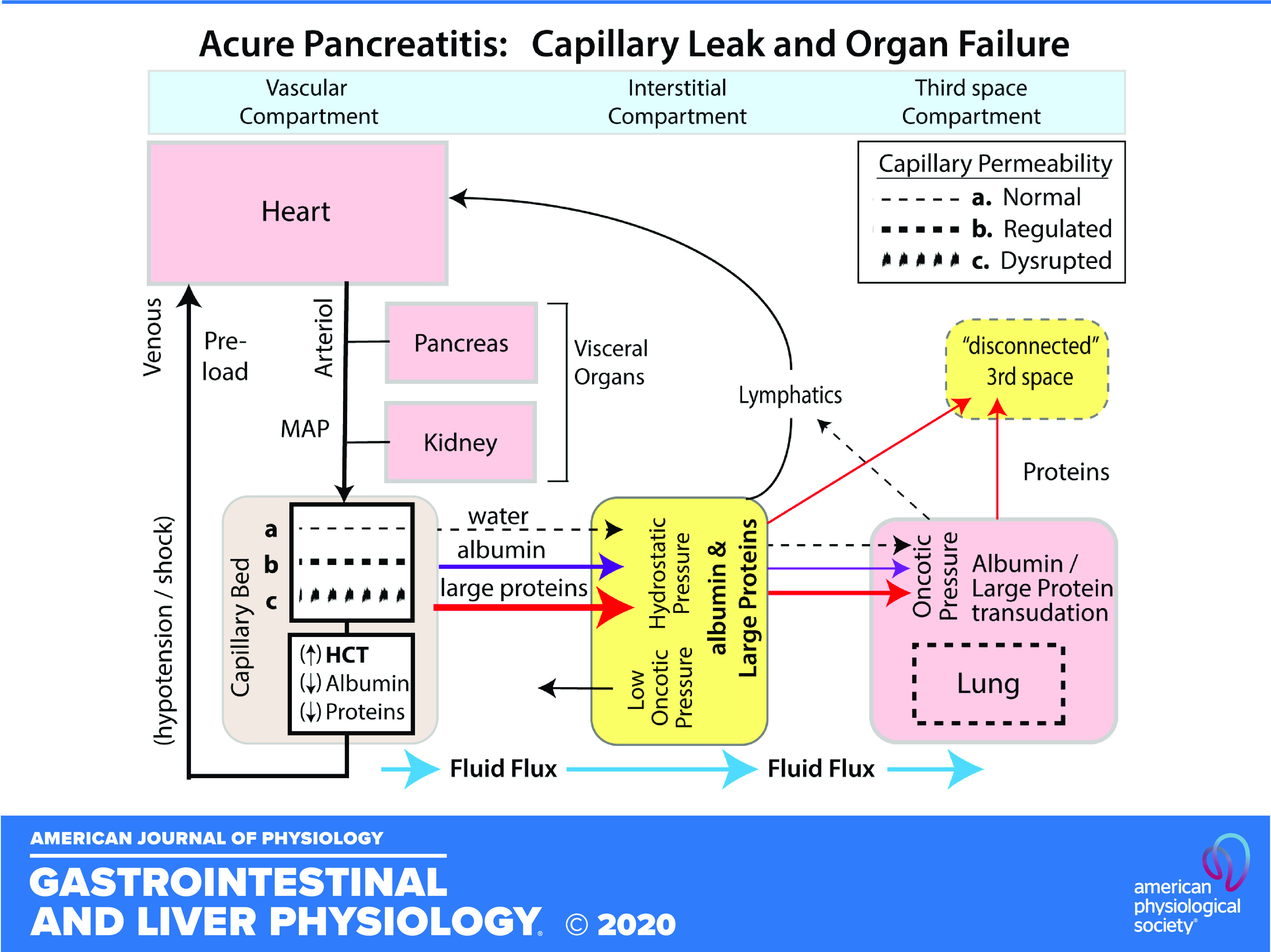
A compartment model was constructed to organize the major organs linked to the vascular system as outlined in Fig. 1 to track the consequence of CLS (44). The model organized multiple fluid compartments and defined permeability of characteristics of the capillary beds to test the hypothesis that the primary driver to organ dysfunction is capillary leak in the nonsinusoidal nonfenestrated blood capillaries of the skin, muscle, adipose tissue, intestinal mesentery, and lungs (53). When viewed as a descriptive influence diagram with a change in the permeability variable (Fig. 1B), progressive loss of oncotic proteins into the interstitial and third spaces will result in both cardiovascular hypovolemia and pulmonary edema. Loss of fluid return to the heart (venous preload) results in arterial hypotension, with reduced perfusion to the pancreas (increased susceptibility to pancreatic necrosis) and the kidney (prerenal azotemia and risk of acute kidney injury). The compartment models (Fig. 1, A and B) were used to assess the effects of altering permeability in the capillary bed with normal periendothelial pore size of 2 nm, cytokine-driven and regulated increased effective periendothelial pore size of ≤7 nm [i.e., the size of albumin (53)], and finally larger relative pore size (i.e., ≫ 7 nm) representing unregulated breakdown of the transendothelial barrier allowing free movement of large plasma proteins between the vascular and interstitial compartments. The model was used to envision new steady states and the predicted effects of altered endothelial permeability on the plasma concentration of albumin and larger plasma proteins, on hematocrit levels with concurrent hypovolemia, on cardiovascular physiology, and on fluid shifting into the lung (i.e., pulmonary edema) and the effects of decreased blood perfusion on the kidney and pancreas. In this version of the model, we did not vary the extent of pathogenic capillary leak, calculate rate constants, or predict the effect of resuscitation with fluids (e.g., lactated Ringer solution) or proteins (e.g., albumin or plasma).
Fig. 1.
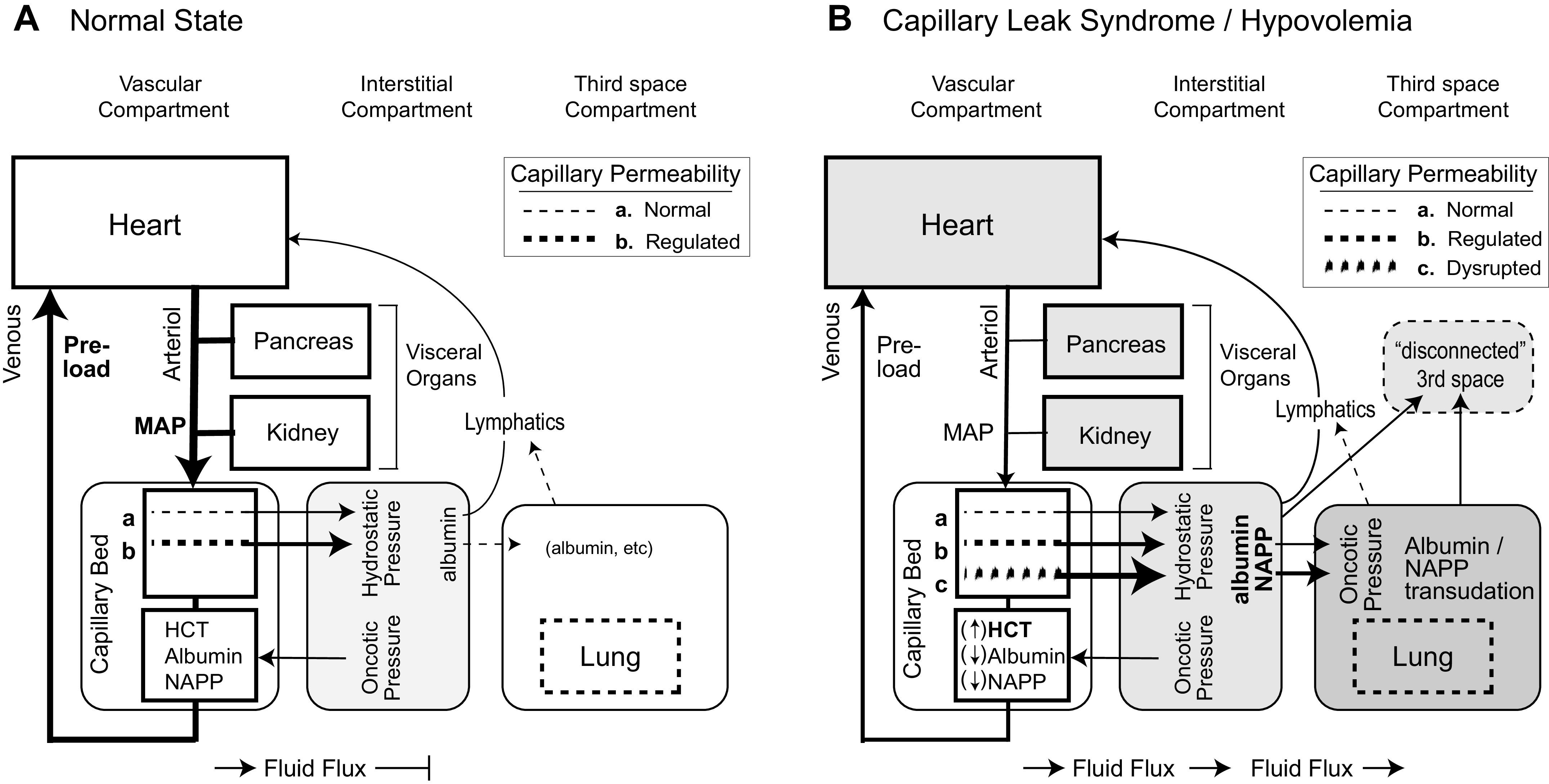
The pathophysiology of fluid sequestration: normal physiology (A) and vascular leak syndrome and hypovolemia (B). Three fluid compartments are organized as an influence diagram with the lymphatic fluid eventually returning to the heart. The key variable in B is addition of increased, large-molecule capillary permeability from disrupted endothelial barrier (c). The shaded nodes reflect sequential dysfunction resulting from disrupted capillary endothelial barrier with tissue edema in the interstitial compartment with direct effects on the lung (pulmonary edema), progressive hypovolemia with reduced blood return to the heart and hypotension, and reflex vasoconstriction and hypoperfusion of the pancreas and kidney. Albumin and proteins are lost into the 3rd space compartment, with either delayed lymphatic return or an irreversible loss to the system (disconnected 3rd space). HCT, hematocrit; MAP, mean arterial pressure; NAPP, nonalbumin plasma protein.
For the experimental design, the model is predictive of the effects of permeability changes. Clinical biomarkers that are directly relevant to the predictions of the model were used to test the model.
Patients
All patients were recruited at the University of Pittsburgh Medical Center under institutional review board (IRB)-approved research protocols. Patients with AP were ascertained from the Pancreatitis-associated Risk Of Organ Failure (PROOF; https://clinicaltrials.gov Identifier NCT03075605) study from 2009 to 2019 as described previously (24, 37, 63). Patients with severe AP were also recruited to the Mechanism of Systemic Inflammation-Associated Endothelial and Epithelial Cell Dysfunction Following Acute Pancreatitis, Trauma, and Burns (IRB PRO17080710), the Acute Pancreatitis as a Model to Predict Organ Failure (IRB PRO14060166), and Pancreatic Rest in Acute Pancreatitis (IRB PRO07080011; NCT00580749), studies that included more detailed protocols and sample collections in patients with severe AP. This study represented secondary analysis of prospective study data supplemented with existing electronic health records.
Definitions
The definitions and diagnosis of acute pancreatitis and SIRS (12) followed the revised Atlanta criteria (RAC) with cumulative SIRS scores ≥ 2 considered positive or severe AP (5). Organ failure was diagnosed according to the modified Marshall score as described in the RAC for respiratory failure, renal failure, and cardiovascular failure (5), with MOF indicating more than one organ system (5) and non-MOF indicating one or no organ failure. Organ failure was subdivided into transient and persistent if it lasted less or more than 48 h, respectively. The term severe acute pancreatitis is defined in the RAC as persistent organ failure caused by SIRS but does not distinguish single-organ failure from MOF (5).
CLS reflects pathologically increased capillary permeability due to changes in the endothelial cell barrier allowing proteins and fluids to move into the tissue interstitial space (2, 10). We used a clinical diagnosis of CLS based on pulmonary edema or pleural effusions (in the absence of heart failure, fluid overload, or lung disease) and hemoconcentration. Supportive clinical evidence of CLS included new-onset peripheral tissue edema. Hypovolemia was defined as a decrease in circulatory blood volume of >10% (48) and measured clinically by a drop in normal blood pressure, increase in heart rate, and beneficial responses to fluid resuscitation. Orthostatic blood pressure measurements were not included in the PROOF protocol (34).
Data Elements
Detailed case report forms were available for all study patients (24, 37, 63). The IRB included permission to maintain the identity of the patients and review their electronic health records, including baseline laboratory values at any time before the onset of acute pancreatitis. Specific laboratory values that were collected for modeling included HCT, Alb, TP, Cr, and BUN. NAPP was calculated from TP and Alb. Laboratory values were collected from baseline before AP attack, admission to 24 h, and for each 24-h period thereafter up to 96 h after admission.
Statistical Methods and Trajectory Analysis
Laboratory biomarker trajectories were calibrated based on date of pain onset: defined as day “−1” for any measurement before AP onset and 1 for any measurement up to 24 h after pain onset, and the value was incremented by 1 for each 24-h period thereafter in our analysis. Subjects with available data on ≥3 days of −1, 1, 2, 3, and 4 were deemed eligible for statistical analysis.
Biomarker time courses were assessed using a linear mixed model with random intercepts for each subject and fixed effects controlling for age and sex. Visual inspection of residual plots did not reveal any obvious deviations from homoscedasticity or normality. P values were obtained by likelihood ratio tests of the full model with the effect in question against the model without the effect in question. Wilcoxon rank-sum test was applied to compare earliest HCT values. All statistical tests were run using R (49) and the lme4 package (6) to perform a linear mixed-effects analysis.
All authors had access to the study data and reviewed and approved the final manuscript.
RESULTS
A total of two hundred eighteen charts of patients with AP were screened for inclusion in this analysis. One hundred sixty-one patients were excluded for missing baseline albumin and other baseline characteristics. Ultimately, a total of fifty-seven subjects met inclusion criteria with a minimum of preadmission, day of pain, and 24 h after pain onset and preferably continuing until 96 h of pain onset.
Median age of our studied cohort was 57 yr, and 54% were female. Three main etiologies were biliary (54%), idiopathic (12%), and postendoscopic retrograde cholangiopancreatography pancreatitis (14%). Eighteen subjects developed MOF, eight had single-organ failure, and seventeen subjects did not develop any organ failure. Five patients with severe AP died during the course of hospitalization. Baseline characteristics of the study population are summarized in Table 1.
Table 1.
Baseline characteristics of the study cohort, multiorgan failure subset, and patients with single-organ failure or without organ failure
| Non-MOF, n = 39 | MOF, n = 18 | P Value | |
|---|---|---|---|
| Age, yr | 55 [36, 63] | 63 [56.2, 70.8] | 0.078 |
| BMI, kg/m2 | 31 [26.4, 36.6] | 30.8 [26, 34] | 0.668 |
| Sex, female | 24 (61.5) | 7 (38.9) | 0.190 |
| Race | |||
| White | 36 (92.3) | 15 (83.3) | |
| Black | 3 (7.7) | 3 (16.7) | 0.574 |
| Etiology | |||
| Biliary | 19 (48.7) | 12 (66.7) | |
| Idiopathic | 4 (10.3) | 3 (16.7) | |
| Post-ERCP | 7 (17.9) | 1 (5.6) | |
| Alcohol | 4 (10.3) | 0 (0.0) | |
| Other | 3 (7.7) | 0 (0.0) | |
| HTG | 2 (5.1) | 2 (11.1) | 0.274 |
| Active drinking | 18 (46.2) | 7 (38.9) | 0.821 |
| Active smoking | 10 (25.6) | 3 (16.7) | 0.681 |
| ICU admission | 10 (25.6) | 18 (100.0) | <0.001 |
| Total length of stay, days | 7 [5.5, 11.0] | 28.5 [12.5, 45.5] | 0.002 |
| Death | 0 | 6 | <0.001 |
| Pancreatic necrosis | |||
| No | 16 (41.0) | 0 (0.0) | |
| Yes | 9 (23.1) | 10 (55.6) | |
| No CECT | 14 (35.9) | 6 (33.3) | |
| Unknown | 0 (0.0) | 2 (11.1) | 0.001 |
| SIRS-positive within 48 h | 17 (43.6) | 18 (100.0) | <0.001 |
Values are totals (percentage) or median [IQR]. BMI, body mass index; CECT, contrast-enhanced computed tomography scan; ERCP, endoscopic retrograde cholangiopancreatography; HTG, hypertriglyceridemia; ICU, intensive care unit; MOF, multiorgan failure; non-MOF, single-organ failure or without organ failure; SIRS, systemic inflammatory response syndrome.
Consequences of Hypovolemia and Hypotension
Hemoconcentration.
Hemoconcentration describes the increase in the fraction of red blood cells in the blood as a function of loss of plasma. This phenomenon is well known in severe AP and is included in some severity scores [e.g., Acute Physiology and Chronic Health Examination (APACHE)-II, Harmless Acute Pancreatitis Score (HAPS), Panc 3, Ranson, summarized in Mounzer et al. (37)]. However, the patient’s HCT at the time of AP evaluation reflects the combined effects of their baseline HCT and hemoconcentration. Patients with MOF had a slightly higher, although nonsignificant, HCT on admission, with median hematocrit 44.4 [43.3–45.4] versus 39.9 [37.3–42.1] (P = 0.08).
We evaluated the change of HCT in subjects comparing pre-AP data and data within 24 h of pain onset. Patients with MOF had incremental increases in hematocrit from baseline of 5.00 [25%-75% interquartile range, IQR], which was significantly higher than incremental changes in non-MOF of −0.20 [−1.55, 1.40] (P < 0.002). Using a rise in HCT > 3 from baseline in individual patients in this study distinguishes MOF from non-MOF [odds ratio (OR) 17.7, P = 0.014].
We compared the results calculated using individual patient trajectories with the results using typical population-based, cutoff values for hemoconcentration of HCT ≥ 40% (22), HCT > 43 for men and HCT > 39.6 for women (26), HCT > 44 (7), or HCT > 47 (3). None of these HCT cutoff values was significant in distinguishing MOF from non-MOF, including HCT > 43 in all subjects (OR 6.2 P = 0.10), HCT > 43 in men (OR 0.71, P = 1), and HCT > 40 in women (OR 2.1, P = 1). These data suggest that population-based cutoff values for HCT used to evaluate hemoconcentration are of limited value in individual patients, whereas individual patient trends over time, beginning with baseline data, are better indicators of hemoconcentration as previously hypothesized (64).
Visceral organ hypotension.
Hypotension, as a clinical term, is used to describe a decrease in the systemic blood pressure below normal levels resulting in abnormal clinical signs and symptoms, arbitrarily defined as a systolic blood pressure < 100 mmHg or mean arterial pressure < 70 mmHg (39). The origin of hypotension can be low cardiac output, which includes heart failure and hypovolemia, or systemic vasodilatation with low peripheral resistance as in sepsis or dysautonomia. Our compartment model of CLS (Fig. 1) predicts that hypotension is a result of hypovolemia, causing reducing blood return to the heart (low cardiac ventricular preload) and a reflex peripheral vasoconstriction. The role of severe abdominal pain with hypovolemia, as is typical for severe AP, was modeled to reflect the baroreceptor-mediated vasoconstriction reflex with visceral organs being more severely affected compared with systemic vascular beds making visceral organs such as the pancreas, intestinal mucosa, and kidneys selectively more severely hypotensive (9, 50). Thus we analyzed biomarkers from two different visceral organ systems that are typically evaluated during AP, the kidneys (modeled as a visceral organ) and the pancreas.
Kidney dysfunction in MOF.
We tested the hypothesis that MOF was associated with hypotension from hypovolemia by evaluating the changes in BUN (linked to renal nitrogen clearance) and creatinine (linked to kidney perfusion and filtration). In our study, the MOF subset of patients with AP had higher levels of BUN and creatinine at the baseline and also at the subsequent time points as seen in the results of our linear mixed-effects model in Table 2 and illustrated in Fig. 2. Multiorgan failure subset had a sharper increase in BUN (P < 0.001) and Cr (P < 0.001) compared with non-MOF subset. Despite distinctive BUN and Cr slopes, BUN-to-Cr ratio did not change and did not show significant difference between MOF and non-MOF subsets. This indicates that the primary early effect of MOF on the kidney is perfusion and filtration rather than dysfunction of the renal epithelial cells indicating prerenal azotemia (28, 40).
Table 2.
Results from mixed linear model: Cr, BUN, and BUN-to-Cr ratio
| BUN |
Cr |
BUN-to-Cr Ratio |
||||
|---|---|---|---|---|---|---|
| Coefficient | Estimates (95% CI) | P | Estimates (95% CI) | P | Estimates ( 95% CI) | P |
| Intercept | 11.53 (7.66–15.40) | <0.001 | 0.66 (0.42–0.90) | <0.001 | 16.15 (13.65–18.65) | <0.001 |
| Day from pain onset | −0.10 (−0.77 to 0.58) | 0.782 | 0.01 (−0.05 to 0.07) | 0.639 | −0.38 (−0.85 to 0.08) | 0.105 |
| Multiple organ failure = true | 11.48 (5.44–17.53) | <0.001 | 0.55 (0.16–0.95) | 0.006 | 1.59 (−2.34 to 5.52) | 0.427 |
| Male | 4.78 (−0.44 to 10.01) | 0.073 | 0.63 (0.31–0.94) | <0.001 | −1.87 (−5.23 to 1.49) | 0.275 |
| Day-by-MOF interaction | 4.15 (2.91–5.39) | <0.001 | 0.46 (0.34–0.57) | <0.001 | −0.18 (−1.03 to 0.67) | 0.672 |
| Random effects | ||||||
| σ2 | 47.65 | 0.39 | 22.55 | |||
| τ00 | 86.34 subject | 0.25 subject | 34.81 subject | |||
| ICC | 0.64 | 0.38 | 0.61 | |||
| n | 60 subject | 60 subject | 60 subject | |||
| Observations | 205 | 205 | 205 | |||
| Marginal r2/conditional r2 | 0.446/0.803 | 0.558/0.728 | 0.026/0.617 | |||
Values are means (95% CI). Significant P values shown in bold. The models do not show any significant changes over time for the non-multiorgan failure (non-MOF) group, although men show significantly higher creatinine (Cr) levels compared with women. The MOF group showed increased mean blood urea nitrogen (BUN) and Cr levels as well as an increase in BUN and Cr levels over time as shown in the interaction term “Day-by-MOF interaction.” There was no significant difference across any explanatory variable for BUN-to-Cr ratio. proof.id is the subject identifier. σ2, Fixed-effects variance; τ00, between-subject variance; ICC, intraclass correlation coefficient.
Fig. 2.
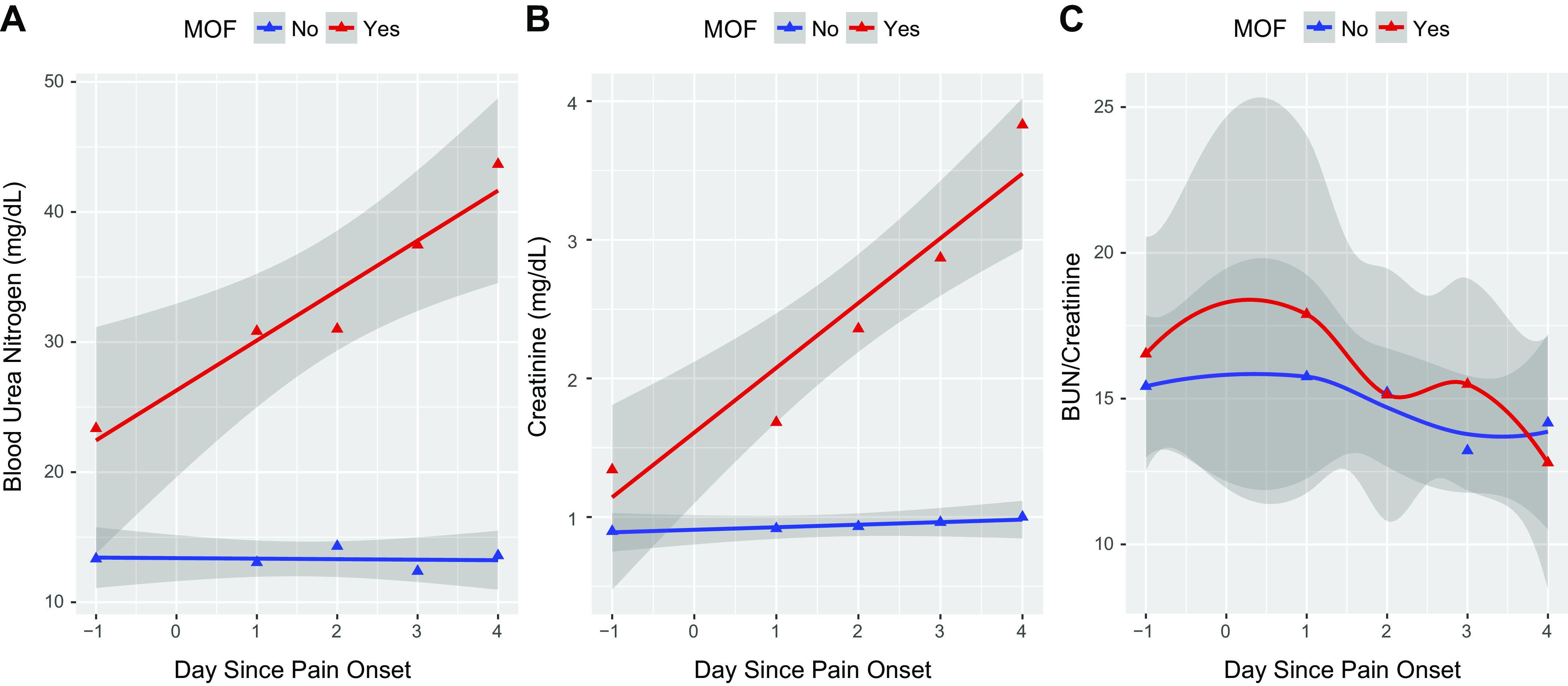
The trajectories with 95% confidence intervals (shaded gray) of biomarkers blood urea nitrogen (BUN; A), creatinine (Cr; B), and BUN-to-Cr ratios (C) over time. The larger confidence intervals are associated with multiorgan failure (MOF; red line). Day −1 represents baseline values acquired before acute pancreatitis onset (see main text). Time points 0–4 are days after pain onset. Comparisons between subsets with (red) and without (blue) MOF are shown.
Pancreatic necrosis.
Pancreatic necrosis (PNec) is a complication of AP that is usually diagnosed with a contrast-enhanced CT scan (CECT) and demonstrating nonperfusion of a section of the pancreas. The severity of PNec is typically graded as <30, 30–50, and >50% of the gland (4). PNec was observed in 9 of 39 non-MOF subjects (23%) and 10 of 18 subjects with MOF (56%). Among subjects with MOF, 6 of the 18 patients did not receive a CECT scan and 2 had having an inconclusive CT scan because they were done without a contrast agent. Thus 10 of 10 (100%) of subjects with MOF with a CECT had significant PNec. Among non-MOF subjects, 14 of the 39 patients did not have AP at a severity level for the managing physicians to obtain a CECT scan so occult PNec could not be excluded. Among non-MOF subjects with a CECT, 36% had PNec. Our interpretation is that PNec, an organ perfusion deficit, is present in the majority of patients with MOF.
Mechanisms of Hypovolemia and Hypotension
A critical but unanswered question concerning AP-associated hypovolemia is whether increased capillary permeability in non-MOF and MOF is regulated or unregulated. Our hypothesis is that regulated increase in permeability would favor transudation of Alb over larger proteins (i.e., NAPP) and that regulated increased permeability would begin resolving by 48 h, whereas unregulated permeability would persist. To test these hypotheses, we measure the trajectory of plasma Alb, TP, and NAPP at pre-AP time points, on admission and daily for 4 days.
Albumin, Total Protein, and Albumin-to-Total Protein Ratio
To evaluate the dynamic changes in plasma proteins, we used linear mixed-effects models, the results of which can be seen in Table 3. Preadmission Alb and TP values were similar between the two subsets. We noted a consistent decline in Alb, TP, and NAPP levels throughout study time points in both MOF and non-MOF subsets. In the non-MOF subgroup, there were slight but nonsignificant differences in Alb, TP, and NAPP trends in patients with SIRS (n = 17) or without SIRS (n = 22), so the two subgroups were combined. However, in comparison with non-MOF subset, patients with MOF demonstrated a sharper decline in Alb (P < 0.001), TP (P < 0.001), and NAPP (P < 0.05; Fig. 3). On day 1, there appeared to be a slight increase in NAPP in the MOF group, possibly reflecting albumin loss. However, beginning on day 2, the albumin-to-NAPP ratio in the non-MOF began decreasing, whereas the MOF ratio increased, reflecting a proportionately larger loss of NAPP in MOF (Fig. 3C). Assuming the NAPP is mostly large-molecular-weight proteins (e.g., immunoglobulins), it appears that capillary leak in non-MOF subjects is regulated (loss of proteins the size of albumin), whereas the capillary leak in MOF is initially regulated but becomes unregulated by the end of day 1, with continued and indiscriminate loss of plasma proteins into interstitial and third spaces over the duration of the observations. Thus it follows that systemic dysfunction of endothelial cells and CLS is a proximal cause of MOF.
Table 3.
Results from mixed linear model: albumin, total protein, albumin-to-total protein ratio, and nonalbumin plasma protein
| Albumin |
Total Protein |
Albumin/Total Protein |
Nonalbumin Plasma Protein |
|||||
|---|---|---|---|---|---|---|---|---|
| Coefficient | Estimates (95% CI) | P | Estimates | P | Estimates | P | Estimates | P |
| Intercept | 3.76 (3.61–3.91) | <0.001 | 6.87 (6.61–7.13) | <0.001 | 0.55 (0.53–0.57) | <0.001 | 3.11 (2.89–3.33) | <0.001 |
| Day from pain onset | −0.17 (−0.21 to −0.13) | <0.001 | −0.29 (−0.35 to −0.23) | <0.001 | −0.00 (−0.01 to 0.00) | 0.564 | −0.12 (−0.17 to −0.08) | <0.001 |
| Multiple organ failure = true | −0.10 (−0.35 to 0.14) | 0.415 | 0.00 (−0.42 to 0.42) | 0.995 | −0.01 (−0.05 to 0.02) | 0.510 | 0.10 (−0.26 to 0.45) | 0.599 |
| Male | 0.15 (−0.05 to 0.34) | 0.138 | 0.23 (−0.11 to 0.57) | 0.177 | 0.00 (−0.03 to 0.03) | 0.886 | 0.08 (−0.22 to 0.38) | 0.599 |
| Day-by-MOF interaction | −0.13 (−0.20 to −0.05) | 0.001 | −0.23 (−0.34 to −0.12) | <0.001 | −0.00 (−0.01 to 0.01) | 0.789 | −0.10 (−0.18 to −0.01) | 0.022 |
| Random effects | ||||||||
| σ2 | 0.16 | 0.40 | 0.00 | 0.23 | ||||
| τ00 | 0.09 subject | 0.31 proof.id | 0.00 proof.id | 0.26 proof.id | ||||
| ICC | 0.35 | 0.43 | 0.51 | 0.53 | ||||
| n | 60 proof.id | 60 proof.id | 60 proof.id | 60 proof.id | ||||
| Observations | 208 | 207 | 207 | 207 | ||||
| Marginal r2/conditional r2 | 0.411/0.619 | 0.418/0.671 | 0.011/0.514 | 0.148/0.600 | ||||
Values are means (95% CI). Significant P values marked in bold, and range is actually (95% CI). The models show significant reductions in albumin, total protein, and nonalbumin plasma protein in the non-multiorgan failure (non-MOF) group equal to the negative value for “day from pain onset” for each unit of day. In addition, the interaction term between MOF and day from pain onset shows the additional significant decrease for those with MOF for each day. There was no significant difference for any explanatory variable for albumin-to-total protein ratio. proof.id is the subject identifier for within-subject variance. σ2, Fixed-effects variance; τ00, between-subject variance; ICC, intraclass correlation coefficient.
Fig. 3.

The trajectories with 95% confidence intervals (shaded gray) of biomarkers albumin (A), total protein (TP; B), nonalbumin plasma protein (TP minus albumin; C), and albumin-to-TP ratio (D) over time. Day −1 represents baseline values acquired before acute pancreatitis onset (see main text). Time points 0–4 are days after pain onset. Comparisons between subsets with (red) and without (blue) multiorgan failure subset (MOF) are shown.
DISCUSSION
The compartment model with variable capillary permeability could be used to describe the mechanism and the characteristics of organ systems that typically fail during the transition from SIRS to MOF. The key variable in the model is the inclusion of the unregulated loss of large plasma proteins in the MOF case. We tested this hypothesis focusing on NAPP as a biomarker and demonstrating for the first time, to our knowledge, that exudation of large plasma proteins from the vascular compartment during the initial phases of AP is a critical variable determining progression of SIRS to MOF.
Our approach differs from epidemiological and cohort-based studies that depend on subgroup classifiers and population averages to define pathological cutoff values for various biomarkers. We used each subject’s pre-AP biomarker values as baseline and determined post-AP trajectories to track mechanism-specific processes and then summarized the quantitative effects between patients with similar features (i.e., SIRS) and different outcomes (i.e., MOF). The importance of modeling AP in this way is highlighted by comparison of HCT levels using the subject’s baseline level, which showed dramatic changes, versus population-based average cutoff values from various cohort studies that could not accurately distinguish MOF from non-MOF.
Our compartment model/influence diagram allowed key clinical features of MOF to be organized and help visualize the effect of progressive loss of Alb and NAPP and corresponding fluid shifts on multiple systems of importance in AP. Of note, the half-life of albumin and immunoglobulins (e.g., IgG, a major component of large-molecular-weight proteins) is ∼3 wk (1, 32), making their concentrations ideal for short-term dynamic studies. Our findings of incremental increase in HCT only in patients with MOF with loss of both Alb and NAPP and strong clinical evidence of hypoperfusion of the kidneys (prerenal azotemia) and pancreas (high frequency of PNec) fit this model and further support the hypothesis that systemic dysfunction of endothelial cells with increased permeability to both small and large proteins (i.e., pathological CLS) is a proximal cause of MOF. Thus these data clarify a major pathological mechanism of MOF in AP and possibly other disorders with SIRS and MOF such as multiple trauma, severe burns, and sepsis (55).
The Importance of SIRS
Systemic inflammation (i.e., SIRS) is a systemic response to pancreatic injury that has been characterized as a cytokine storm (31, 55). SIRS appears to be the bridge between local pancreatic inflammation to MOF with 100% of our patients with MOF having SIRS. Furthermore, the absence of SIRS predicts no MOF in multiple previous studies (e.g., negative predictive value of 0.88–0.96; Ref. 37). However, the PPV of SIRS to predict MOF is poor (e.g., 0.11–0.43; Ref. 37), indicating that other critical variables exist between the states associated with SIRS and MOF.
We found non-MOF AP subjects had a significant loss of Alb from the plasma, suggesting that factors associated with the innate immune system during SIRS affect capillary permeability. However, the effect of SIRS alone likely reflects a regulated increase in permeability, since the trajectory of Alb loss in non-MOF was significantly less than in MOF and the organ systems that were measured appeared to compensate for this effect and the progression to MOF required a further factor causing pathological responses in the endothelial cells. The cytokine storm as associated factors clearly has many deleterious effects on various body functions such as cardiac function. However, patients with and without MOF have SIRS, suggesting that these factors alone are not sufficient to cause pathogenic CLS and/or MOF.
Regulation of Capillary Permeability
Normally, capillary integrity depends on the binding of adherens junctions and tight junctions between endothelial cells (55). A variety of cytokines increase capillary permeability through a regulated disruption of adherens junctions (14, 15, 30). In trauma, interleukin (IL)-1 and tumor necrosis factor (TNF) have been associated with directly increasing capillary permeability (56). Trauma, sepsis, burns, and certain inflammatory conditions share the sequence of SIRS with variable CLS with hypotension, edema, and other issues (55).
Capillary permeability at the endothelial level is also regulated by angiopoietin-1 (ANG I) and angiopoietin-2 (ANG II). These antagonistic autocrine peptides are expressed exclusively in the endothelial cells (51). ANG I inhibits capillary leakage by stabilizing the endothelium, whereas ANG II enhances capillary leak by priming the endothelium to respond to inflammatory cytokines. High levels of ANG II are present in patients with sepsis and CLS (63). We (63) previously found that ANG II levels from peripheral blood were significantly elevated early in the course of AP in patients that developed MOF. Although systemically elevated ANG II levels could cause systemic capillary leak, these data fail to distinguish ANG release by a regulated versus unregulated mechanism and fail to directly link the cytokine storm of SIRS with MOF.
Association of Albumin and NAPP Levels With Severe Acute Pancreatitis
Plasma Alb levels have been identified as biomarkers associated with MOF and poor outcomes in AP (21, 29, 52). Albumin is a negatively charged plasma protein that is synthesized in the liver and excreted into the bloodstream (16). It is a flexible, ellipsoid-shaped molecule with a molecular mass of 66.5 kDa and a diameter of 3.8 by 15 nm (57), typically modeled as a diameter of ∼7 nm (53). Alb is a significant contributor to total plasma proteins and represents a protein of intermediate size that is above the typical periendothelial cell filtration cutoff size in nonsinusoidal nonfenestrated blood capillaries of ∼5 nm as seen in skin, muscle, adipose tissue, intestinal mesentery, and lung (53). About 30–40% of Alb is found in the vascular compartment, whereas the remaining Alb is distributed in the much larger extravascular compartment so that the concentration of Alb is higher in the blood. The Alb in the interstitial space normally returns to circulation via the lymphatics system to establish a steady state. Much of the Alb found in interstitial spaces is transferred directly through the endothelial cells by a transcellular pathway involving caveolae via an absorptive (receptor-mediated) or fluid-phase pathways that are also highly regulated (35). The fact that plasma Alb but not NAPP levels dropped in non-MOF AP (Fig. 3) suggests that the increased permeability is regulated, especially since non-MOF patients tend to recover quickly unless they develop complications such as sepsis or infected PNec.
In contrast to non-MOF, the rapid drop in Alb and NAPP in MOF suggests severe damage to the endothelial cells (e.g., endothelial cell toxicity) resulting in unregulated capillary leak with increases in Alb-to-TP or Alb-to-NAPP ratio on days 2–4 with continued loss of larger plasma proteins. In addition, there appears to be a breakdown in the intestinal epithelial barrier with leakage of intestinal bacteria and/or bacterial products from the gut lumen into the mesenteric lymphatics during MOF that may be responsible for the persistence of SIRS (25). Whether the CLS and gut leak are affected by the same upstream pathological agent(s) is unknown.
Kidney and Pancreas
We focused on two intraabdominal organs, the kidney and the pancreas, that are affected by hypovolemia and reflex vasoconstriction resulting in diminished perfusion. BUN and Cr are common clinical laboratory analytes that are used to assess kidney function and can be used to estimate intravascular volume depletion with a proportional rise in BUN and Cr (i.e., prerenal azotemia; Ref. 18). Evidence of prerenal azotemia was demonstrated in this cohort (Fig. 2). Furthermore, we identified a very high rate of PNec in the pancreas of MOF (100% with CECT) versus non-MOF (<40% with CECT), although data were not available in all patients. These data also support our previous hypothesis of parallel mechanisms of kidneys and pancreas damage as a rise in Cr was predictive of PNec (38).
Study Strengths and Weaknesses
This study has numerous strengths. First, we were able to select a cohort of patients that were rapidly ascertained, within <24 h of pain onset, so that early biomarkers of pancreatitis were captured. Second, we used mechanistic modeling to test our hypothesis rather than agnostic data-driven models. Mechanistic models are more useful in this type of study because each variable is defined by known location and effect within the model, and the effects of multiple organized and structured variables can be applied and tested in individual cases. Mechanistic models also provide insights into causes of phenomenon rather than measuring the association between variables, and they require far fewer subjects because limited number of variables avoids penalties of multiple testing and false discovery (62). Third, we used pre-AP laboratory values of each patient as their own baseline, providing a more accurate assessment of dynamic changes in biomarkers rather than using population averages as in typical scoring systems as previously recommended by leading experts (44). Fourth, the longitudinal measurements of biomarkers allowed analysis of the complex behavior of each element via generalized linear mixed model, which is less sensitive to missing data. Furthermore, our data set encompassed the detailed chronology of AP and the endpoints. Fifth, the use of compartment/influence models also provides rationale for designing the study and understanding sequential events and risks for dysfunction of the cardiovascular system, the lungs, the kidney, and the pancreas in severe AP. Another important feature of this study is the use of common clinical biomarkers or examinations so that the application of new insights can be rapidly applied to patient care.
There were several limitations to this study. First, pre-AP data and other data were obtained from existing medical records so that analysis was limited to 57 of 218 previously ascertained patients. As a result, some of the patients from our prospective studies had to be excluded in this analysis because laboratory tests such as Alb and TP were not ordered at the time of AP admission; because there were no prior medical records in the electronic health record to obtain preadmission laboratory values; or because some patients were transferred to our center several days after the onset of pain and laboratory samples and key measures were not acquired at the outside hospital. Nevertheless, the approach and sample size were sufficient to provide statistically significant differences between the patients with MOF and non-MOF patients and provide critical new insights into the nature of CLS in MOF. Second, CECT scans were only performed in a subset of patients based on clinical indications, so a more accurate estimate of PNec in non-MOF and MOF was not possible. Third, the permeability of endothelial cells was not directly measured, nor were the concentrations of plasma proteins in the interstitial and other compartments. However, the compartment/influence model was based on known physical and biological principles. The clinical measures of total protein and albumin provide a reasonable estimate of different capillary permeabilities that are associated with well-established endothelial cell barrier functions. Thus we believe that this approach is clinically useful, and the results and interpretation are compelling.
Future directions include studies to determine how human microvascular endothelial cells respond to serum of patients with SIRS with or without MOF and if the response is dramatic, to determine the mechanisms and mitigation. Second, studies focusing on the use of fresh-frozen plasma in patients with early signs of NAPP loss are needed. Traditional clinical studies of AP often result in ascertainment of patients after 72 h from onset (23); thus new technology needs to be developed to automatically screen the electronic health records and alert physicians and researchers to these conditions within 6 h of arriving at the emergency department (44).
Conclusions
Trajectory analysis of individual patients using changes in their own biomarker levels suggests that a regulated increase in epithelial permeability in most patients with AP and SIRS that is likely associated with a cytokine storm. The dramatic, progressive, and sustained loss of both Alb and NAPP from the plasma in patients with MOF suggests widespread dysfunction or loss of the endothelial cells with unregulated CLS. These data are consistent with a mechanistic model in which transudation of large and small proteins and fluids into the lungs results in pulmonary edema and lung failure. Loss of intravascular proteins results in tissue edema and third spacing within extravascular compartments and hypovolemia and hemoconcentration within the vascular compartment. Intravascular hypovolemia leading to systemic hypotension with visceral organs suffering disproportional effects, possibly aggravated by reflex vasoconstriction of the mesenteric and/or renal arterioles or other mechanisms. Further evaluation of the agents causing epithelial cell dysfunction or loss in AP and other diseases with SIRS and CLS and therapeutic interventions to prevent MOF is needed.
GRANTS
This research was funded by the US Department of Defense Awards W81XWH-14-1-0376 and W81XWH-17-1-0502 (both to D. C. Whitcomb).
DISCLOSURES
D. C. Whitcomb is cofounder and consultant to Ariel Precision Medicine. Commercial use of compartment and other modeling approaches in acute inflammation may be protected by Ariel Precision Medicine (patent pending). The other authors report no conflict of interest related to this work.
AUTHOR CONTRIBUTIONS
G.I.P. and D.C.W. conceived and designed research; N.L.K. and P.P. performed experiments; N.L.K., P.P., P.J.G., A.S.W., and D.C.W. analyzed data; P.P., P.J.G., A.S.W., C.B., and D.C.W. interpreted results of experiments; P.J.G. and D.C.W. prepared figures; N.L.K., P.P., and D.C.W. drafted manuscript; N.L.K., P.P., P.J.G., A.S.W., C.B., G.I.P., and D.C.W. edited and revised manuscript; N.L.K., P.P., P.J.G., A.S.W., C.B., G.I.P., and D.C.W. approved final version of manuscript.
ACKNOWLEDGMENTS
Present address of G. I. Papachristou: Div. of Gastroenterology, Hepatology and Nutrition, Dept. of Medicine, The Ohio State Univ., Columbus, OH.
REFERENCES
- 1.Andersen JT, Dalhus B, Viuff D, Ravn BT, Gunnarsen KS, Plumridge A, Bunting K, Antunes F, Williamson R, Athwal S, Allan E, Evans L, Bjørås M, Kjærulff S, Sleep D, Sandlie I, Cameron J. Extending serum half-life of albumin by engineering neonatal Fc receptor (FcRn) binding. J Biol Chem 289: 13492–13502, 2014. doi: 10.1074/jbc.M114.549832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Assaly R, Olson D, Hammersley J, Fan PS, Liu J, Shapiro JI, Kahaleh MB. Initial evidence of endothelial cell apoptosis as a mechanism of systemic capillary leak syndrome. Chest 120: 1301–1308, 2001. doi: 10.1378/chest.120.4.1301. [DOI] [PubMed] [Google Scholar]
- 3.Baillargeon JD, Orav J, Ramagopal V, Tenner SM, Banks PA. Hemoconcentration as an early risk factor for necrotizing pancreatitis. Am J Gastroenterol 93: 2130–2134, 1998. doi: 10.1111/j.1572-0241.1998.00608.x. [DOI] [PubMed] [Google Scholar]
- 4.Balthazar EJ, Robinson DL, Megibow AJ, Ranson JH. Acute pancreatitis: value of CT in establishing prognosis. Radiology 174: 331–336, 1990. doi: 10.1148/radiology.174.2.2296641. [DOI] [PubMed] [Google Scholar]
- 5.Banks PA, Bollen TL, Dervenis C, Gooszen HG, Johnson CD, Sarr MG, Tsiotos GG, Vege SS; Acute Pancreatitis Classification Working Group . Classification of acute pancreatitis–2012: revision of the Atlanta classification and definitions by international consensus. Gut 62: 102–111, 2013. doi: 10.1136/gutjnl-2012-302779. [DOI] [PubMed] [Google Scholar]
- 6.Bates D, Mächler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. J Stat Softw 67: 2015. doi: 10.18637/jss.v067.i01. [DOI] [Google Scholar]
- 7.Brown A, Orav J, Banks PA. Hemoconcentration is an early marker for organ failure and necrotizing pancreatitis. Pancreas 20: 367–372, 2000. doi: 10.1097/00006676-200005000-00005. [DOI] [PubMed] [Google Scholar]
- 8.Buter A, Imrie CW, Carter CR, Evans S, McKay CJ. Dynamic nature of early organ dysfunction determines outcome in acute pancreatitis. Br J Surg 89: 298–302, 2002. doi: 10.1046/j.0007-1323.2001.02025.x. [DOI] [PubMed] [Google Scholar]
- 9.Ceppa EP, Fuh KC, Bulkley GB. Mesenteric hemodynamic response to circulatory shock. Curr Opin Crit Care 9: 127–132, 2003. doi: 10.1097/00075198-200304000-00008. [DOI] [PubMed] [Google Scholar]
- 10.Clarkson B, Thompson D, Horwith M, Luckey EH. Cyclical edema and shock due to increased capillary permeability. Am J Med 29: 193–216, 1960. doi: 10.1016/0002-9343(60)90018-8. [DOI] [PubMed] [Google Scholar]
- 11.Cuthbertson CM, Christophi C. Disturbances of the microcirculation in acute pancreatitis. Br J Surg 93: 518–530, 2006. doi: 10.1002/bjs.5316. [DOI] [PubMed] [Google Scholar]
- 12.Davies MG, Hagen PO. Systemic inflammatory response syndrome. Br J Surg 84: 920–935, 1997. doi: 10.1002/bjs.1800840707. [DOI] [PubMed] [Google Scholar]
- 13.de-Madaria E, Herrera-Marante I, González-Camacho V, Bonjoch L, Quesada-Vázquez N, Almenta-Saavedra I, Miralles-Maciá C, Acevedo-Piedra NG, Roger-Ibáñez M, Sánchez-Marin C, Osuna-Ligero R, Gracia Á, Llorens P, Zapater P, Singh VK, Moreu-Martín R, Closa D. Fluid resuscitation with lactated Ringer’s solution vs normal saline in acute pancreatitis: a triple-blind, randomized, controlled trial. United European Gastroenterol J 6: 63–72, 2018. doi: 10.1177/2050640617707864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci 121: 2115–2122, 2008. doi: 10.1242/jcs.017897. [DOI] [PubMed] [Google Scholar]
- 15.Dejana E, Tournier-Lasserve E, Weinstein BM. The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Dev Cell 16: 209–221, 2009. doi: 10.1016/j.devcel.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 16.Doweiko JP, Nompleggi DJ. Role of albumin in human physiology and pathophysiology. JPEN J Parenter Enteral Nutr 15: 207–211, 1991. doi: 10.1177/0148607191015002207. [DOI] [PubMed] [Google Scholar]
- 17.Fei Y, Gao K, Li WQ. Prediction and evaluation of the severity of acute respiratory distress syndrome following severe acute pancreatitis using an artificial neural network algorithm model. HPB (Oxford) 21: 891–897, 2019. doi: 10.1016/j.hpb.2018.11.009. [DOI] [PubMed] [Google Scholar]
- 18.Fisher JM, Gardner TB. The “golden hours” of management in acute pancreatitis. Am J Gastroenterol 107: 1146–1150, 2012. doi: 10.1038/ajg.2012.91. [DOI] [PubMed] [Google Scholar]
- 19.Ghim M, Alpresa P, Yang SW, Braakman ST, Gray SG, Sherwin SJ, van Reeuwijk M, Weinberg PD. Visualization of three pathways for macromolecule transport across cultured endothelium and their modification by flow. Am J Physiol Heart Circ Physiol 313: H959–H973, 2017. doi: 10.1152/ajpheart.00218.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hahn RG. Adverse effects of crystalloid and colloid fluids. Anaesthesiol Intensive Ther 49: 303–308, 2017. doi: 10.5603/AIT.a2017.0045. [DOI] [PubMed] [Google Scholar]
- 21.Hong W, Lin S, Zippi M, Geng W, Stock S, Basharat Z, Cheng B, Pan J, Zhou M. Serum albumin is independently associated with persistent organ failure in acute pancreatitis. Can J Gastroenterol Hepatol 2017: 5297143, 2017. doi: 10.1155/2017/5297143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jinno N, Hori Y, Naitoh I, Miyabe K, Yoshida M, Natsume M, Kato A, Asano G, Sano H, Hayashi K. Predictive factors for the mortality of acute pancreatitis on admission. PLoS One 14: e0221468, 2019. doi: 10.1371/journal.pone.0221468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson CD, Kingsnorth AN, Imrie CW, McMahon MJ, Neoptolemos JP, McKay C, Toh SK, Skaife P, Leeder PC, Wilson P, Larvin M, Curtis LD. Double blind, randomised, placebo controlled study of a platelet activating factor antagonist, lexipafant, in the treatment and prevention of organ failure in predicted severe acute pancreatitis. Gut 48: 62–69, 2001. doi: 10.1136/gut.48.1.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koutroumpakis E, Slivka A, Furlan A, Dasyam AK, Dudekula A, Greer JB, Whitcomb DC, Yadav D, Papachristou GI. Management and outcomes of acute pancreatitis patients over the last decade: a US tertiary-center experience. Pancreatology 17: 32–40, 2017. doi: 10.1016/j.pan.2016.10.011. [DOI] [PubMed] [Google Scholar]
- 25.Landahl P, Ansari D, Andersson R. Severe acute pancreatitis: gut barrier failure, systemic inflammatory response, acute lung injury, and the role of the mesenteric lymph. Surg Infect (Larchmt) 16: 651–656, 2015. doi: 10.1089/sur.2015.034. [DOI] [PubMed] [Google Scholar]
- 26.Lankisch PG, Weber-Dany B, Hebel K, Maisonneuve P, Lowenfels AB. The harmless acute pancreatitis score: a clinical algorithm for rapid initial stratification of nonsevere disease. Clin Gastroenterol Hepatol 7: 702–705, 2009. doi: 10.1016/j.cgh.2009.02.020. [DOI] [PubMed] [Google Scholar]
- 27.Lee PJ, Papachristou GI. New insights into acute pancreatitis. Nat Rev Gastroenterol Hepatol 16: 479–496, 2019. doi: 10.1038/s41575-019-0158-2. [DOI] [PubMed] [Google Scholar]
- 28.Levy M, Geller R, Hymovitch S. Renal failure in dogs with experimental acute pancreatitis: role of hypovolemia. Am J Physiol Renal Physiol 251: F969–F977, 1986. doi: 10.1152/ajprenal.1986.251.6.F969. [DOI] [PubMed] [Google Scholar]
- 29.Li S, Zhang Y, Li M, Xie C, Wu H. Serum albumin, a good indicator of persistent organ failure in acute pancreatitis. BMC Gastroenterol 17: 59, 2017. [Erratum in BMC Gastroenterol 17: 86, 2017.] doi: 10.1186/s12876-017-0615-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.London NR, Zhu W, Bozza FA, Smith MC, Greif DM, Sorensen LK, Chen L, Kaminoh Y, Chan AC, Passi SF, Day CW, Barnard DL, Zimmerman GA, Krasnow MA, Li DY. Targeting Robo4-dependent Slit signaling to survive the cytokine storm in sepsis and influenza. Sci Transl Med 2: 23ra19, 2010. doi: 10.1126/scitranslmed.3000678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Makhija R, Kingsnorth AN. Cytokine storm in acute pancreatitis. J Hepatobiliary Pancreat Surg 9: 401–410, 2002. doi: 10.1007/s005340200049. [DOI] [PubMed] [Google Scholar]
- 32.Mankarious S, Lee M, Fischer S, Pyun KH, Ochs HD, Oxelius VA, Wedgwood RJ. The half-lives of IgG subclasses and specific antibodies in patients with primary immunodeficiency who are receiving intravenously administered immunoglobulin. J Lab Clin Med 112: 634–640, 1988. [PubMed] [Google Scholar]
- 33.Marik P, Bellomo R. A rational approach to fluid therapy in sepsis. Br J Anaesth 116: 339–349, 2016. doi: 10.1093/bja/aev349. [DOI] [PubMed] [Google Scholar]
- 34.McGee S, Abernethy WB 3rd, Simel DL. The rational clinical examination. Is this patient hypovolemic? JAMA 281: 1022–1029, 1999. doi: 10.1001/jama.281.11.1022. [DOI] [PubMed] [Google Scholar]
- 35.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev 86: 279–367, 2006. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 36.Mofidi R, Duff MD, Wigmore SJ, Madhavan KK, Garden OJ, Parks RW. Association between early systemic inflammatory response, severity of multiorgan dysfunction and death in acute pancreatitis. Br J Surg 93: 738–744, 2006. doi: 10.1002/bjs.5290. [DOI] [PubMed] [Google Scholar]
- 37.Mounzer R, Langmead CJ, Wu BU, Evans AC, Bishehsari F, Muddana V, Singh VK, Slivka A, Whitcomb DC, Yadav D, Banks PA, Papachristou GI. Comparison of existing clinical scoring systems to predict persistent organ failure in patients with acute pancreatitis. Gastroenterology 142: 1476–1482, 2012. doi: 10.1053/j.gastro.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 38.Muddana V, Whitcomb DC, Khalid A, Slivka A, Papachristou GI. Elevated serum creatinine as a marker of pancreatic necrosis in acute pancreatitis. Am J Gastroenterol 104: 164–170, 2009. doi: 10.1038/ajg.2008.66. [DOI] [PubMed] [Google Scholar]
- 39.Napolitano LM. Sepsis 2018: definitions and guideline changes. Surg Infect (Larchmt) 19: 117–125, 2018. doi: 10.1089/sur.2017.278. [DOI] [PubMed] [Google Scholar]
- 40.Nassar TI, Qunibi WY. AKI associated with acute pancreatitis. Clin J Am Soc Nephrol 14: 1106–1115, 2019. doi: 10.2215/CJN.13191118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Orfanos SE, Kotanidou A, Glynos C, Athanasiou C, Tsigkos S, Dimopoulou I, Sotiropoulou C, Zakynthinos S, Armaganidis A, Papapetropoulos A, Roussos C. Angiopoietin-2 is increased in severe sepsis: correlation with inflammatory mediators. Crit Care Med 35: 199–206, 2007. doi: 10.1097/01.CCM.0000251640.77679.D7. [DOI] [PubMed] [Google Scholar]
- 42.Pandol SJ, Saluja AK, Imrie CW, Banks PA. Acute pancreatitis: bench to the bedside. Gastroenterology 132: 1127–1151, 2007. doi: 10.1053/j.gastro.2007.01.055. [DOI] [PubMed] [Google Scholar]
- 44.Paragomi P, Spagnolo DM, Breze CR, Gougol A, Haupt M, Whitcomb DC, Papachristou GI. Dynamic analysis of patients with acute pancreatitis: validation of a new predictive tool for severity assessment in a large prospective cohort (Abstract). Gastroenterology 156: S122–S123, 2019. doi: 10.1016/S0016-5085(19)37098-2. [DOI] [Google Scholar]
- 45.Parikh SM, Mammoto T, Schultz A, Yuan HT, Christiani D, Karumanchi SA, Sukhatme VP. Excess circulating angiopoietin-2 may contribute to pulmonary vascular leak in sepsis in humans. PLoS Med 3: e46, 2006. doi: 10.1371/journal.pmed.0030046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pastor CM, Matthay MA, Frossard JL. Pancreatitis-associated acute lung injury: new insights. Chest 124: 2341–2351, 2003. doi: 10.1378/chest.124.6.2341. [DOI] [PubMed] [Google Scholar]
- 47.Peery AF, Crockett SD, Murphy CC, Lund JL, Dellon ES, Williams JL, Jensen ET, Shaheen NJ, Barritt AS, Lieber SR, Kochar B, Barnes EL, Fan YC, Pate V, Galanko J, Baron TH, Sandler RS. Burden and cost of gastrointestinal, liver, and pancreatic diseases in the United States: update 2018. Gastroenterology 156: 254–272.e11, 2019. [Erratum in Gastroenterology 156: 1936, 2019.] doi: 10.1053/j.gastro.2018.08.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perner A. Diagnosing hypovolemia in the critically ill. Crit Care Med 37: 2674–2675, 2009. doi: 10.1097/CCM.0b013e3181ad77d8. [DOI] [PubMed] [Google Scholar]
- 49.R Core Team . R: A Language and Environment for Statistical Computing (Online). Vienna, Austria: R Foundation for Statistical Computing, 2018. https://www.r-project.org. [Google Scholar]
- 50.Reilly PM, Wilkins KB, Fuh KC, Haglund U, Bulkley GB. The mesenteric hemodynamic response to circulatory shock: an overview. Shock 15: 329–343, 2001. doi: 10.1097/00024382-200115050-00001. [DOI] [PubMed] [Google Scholar]
- 51.Ricciuto DR, dos Santos CC, Hawkes M, Toltl LJ, Conroy AL, Rajwans N, Lafferty EI, Cook DJ, Fox-Robichaud A, Kahnamoui K, Kain KC, Liaw PC, Liles WC. Angiopoietin-1 and angiopoietin-2 as clinically informative prognostic biomarkers of morbidity and mortality in severe sepsis. Crit Care Med 39: 702–710, 2011. doi: 10.1097/CCM.0b013e318206d285. [DOI] [PubMed] [Google Scholar]
- 52.Robert JH, Frossard JL, Mermillod B, Soravia C, Mensi N, Roth M, Rohner A, Hadengue A, Morel P. Early prediction of acute pancreatitis: prospective study comparing computed tomography scans, Ranson, Glasgow, Acute Physiology and Chronic Health Evaluation II scores, and various serum markers. World J Surg 26: 612–619, 2002. doi: 10.1007/s00268-001-0278-y. [DOI] [PubMed] [Google Scholar]
- 53.Sarin H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J Angiogenes Res 2: 14, 2010. doi: 10.1186/2040-2384-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schepers NJ, Bakker OJ, Besselink MG, Ahmed Ali U, Bollen TL, Gooszen HG, van Santvoort HC, Bruno MJ; Dutch Pancreatitis Study Group . Impact of characteristics of organ failure and infected necrosis on mortality in necrotising pancreatitis. Gut 68: 1044–1051, 2019. doi: 10.1136/gutjnl-2017-314657. [DOI] [PubMed] [Google Scholar]
- 55.Siddall E, Khatri M, Radhakrishnan J. Capillary leak syndrome: etiologies, pathophysiology, and management. Kidney Int 92: 37–46, 2017. doi: 10.1016/j.kint.2016.11.029. [DOI] [PubMed] [Google Scholar]
- 56.Stein DM, Scalea TM. Capillary leak syndrome in trauma: what is it and what are the consequences? Adv Surg 46: 237–253, 2012. doi: 10.1016/j.yasu.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 57.Tojo A, Kinugasa S. Mechanisms of glomerular albumin filtration and tubular reabsorption. Int J Nephrol 2012: 481520, 2012. doi: 10.1155/2012/481520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Umapathy C, Raina A, Saligram S, Tang G, Papachristou GI, Rabinovitz M, Chennat J, Zeh H, Zureikat AH, Hogg ME, Lee KK, Saul MI, Whitcomb DC, Slivka A, Yadav D. Natural history after acute necrotizing pancreatitis: a large US tertiary care experience. J Gastrointest Surg 20: 1844–1853, 2016. doi: 10.1007/s11605-016-3264-2. [DOI] [PubMed] [Google Scholar]
- 59.Vipperla K, Somerville C, Furlan A, Koutroumpakis E, Saul M, Chennat J, Rabinovitz M, Whitcomb DC, Slivka A, Papachristou GI, Yadav D. Clinical profile and natural course in a large cohort of patients with hypertriglyceridemia and pancreatitis. J Clin Gastroenterol 51: 77–85, 2017. doi: 10.1097/MCG.0000000000000579. [DOI] [PubMed] [Google Scholar]
- 60.Warndorf MG, Kurtzman JT, Bartel MJ, Cox M, Mackenzie T, Robinson S, Burchard PR, Gordon SR, Gardner TB. Early fluid resuscitation reduces morbidity among patients with acute pancreatitis. Clin Gastroenterol Hepatol 9: 705–709, 2011. doi: 10.1016/j.cgh.2011.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Whitcomb DC. Acute pancreatitis. N Engl J Med 354: 2142–2150, 2006. doi: 10.1056/NEJMcp054958. [DOI] [PubMed] [Google Scholar]
- 62.Whitcomb DC, Aoun E, Vodovotz Y, Clermont G, Barmada MM. Evaluating disorders with a complex genetics basis. The future roles of meta-analysis and systems biology. Dig Dis Sci 50: 2195–2202, 2005. doi: 10.1007/s10620-005-3033-7. [DOI] [PubMed] [Google Scholar]
- 63.Whitcomb DC, Muddana V, Langmead CJ, Houghton FD Jr, Guenther A, Eagon PK, Mayerle J, Aghdassi AA, Weiss FU, Evans A, Lamb J, Clermont G, Lerch MM, Papachristou GI. Angiopoietin-2, a regulator of vascular permeability in inflammation, is associated with persistent organ failure in patients with acute pancreatitis from the United States and Germany. Am J Gastroenterol 105: 2287–2292, 2010. doi: 10.1038/ajg.2010.183. [DOI] [PubMed] [Google Scholar]
- 64.Working Group IAP/APA Acute Pancreatitis Guidelines . IAP/APA evidence-based guidelines for the management of acute pancreatitis. Pancreatology 13, Suppl 2: e1–e15, 2013. doi: 10.1016/j.pan.2013.07.063. [DOI] [PubMed] [Google Scholar]
- 65.Wu BU, Hwang JQ, Gardner TH, Repas K, Delee R, Yu S, Smith B, Banks PA, Conwell DL. Lactated Ringer’s solution reduces systemic inflammation compared with saline in patients with acute pancreatitis. Clin Gastroenterol Hepatol 9: 710–717.e1, 2011. doi: 10.1016/j.cgh.2011.04.026. [DOI] [PubMed] [Google Scholar]