

Focused ultrasound–mediated delivery of RNA-loaded nanoparticles increases brain tumor cell apoptosis through oncogene targeting.
Abstract
RNA-based therapies offer unique advantages for treating brain tumors. However, tumor penetrance and uptake are hampered by RNA therapeutic size, charge, and need to be “packaged” in large carriers to improve bioavailability. Here, we have examined delivery of siRNA, packaged in 50-nm cationic lipid-polymer hybrid nanoparticles (LPHs:siRNA), combined with microbubble-enhanced focused ultrasound (MB-FUS) in pediatric and adult preclinical brain tumor models. Using single-cell image analysis, we show that MB-FUS in combination with LPHs:siRNA leads to more than 10-fold improvement in siRNA delivery into brain tumor microenvironments of the two models. MB-FUS delivery of Smoothened (SMO) targeting siRNAs reduces SMO protein production and markedly increases tumor cell death in the SMO-activated medulloblastoma model. Moreover, our analysis reveals that MB-FUS and nanoparticle properties can be optimized to maximize delivery in the brain tumor microenvironment, thereby serving as a platform for developing next-generation tunable delivery systems for RNA-based therapy in brain tumors.
INTRODUCTION
RNA-based therapeutics using RNA interference, including small interfering and short hairpin RNAs (siRNA/shRNA), adenosine deaminase acting on RNA (ADAR), and CRISPR RNAs (1, 2), can be designed to selectively target core tumor signaling pathways (3). While minimally invasive systemic delivery of RNA to intracranial malignancies offers several advantages over conventional invasive approaches (e.g., Ommaya reservoir) (4), effective delivery remains a major challenge. This is due, in part, to degradation of unmodified nucleic acids in biological fluids, poor brain accumulation, and poor cancer cell uptake or escape from the endosomal lipid bilayer barrier (3, 5, 6). Although incorporation of an RNA into a nanoparticle can prolong circulation time and facilitate cellular uptake (2, 7), its accumulation in the brain tumor microenvironment (TME) remains particularly poor (8, 9) due to the low nanoparticle permeability across the blood-brain/blood-tumor barriers (BBB/BTB) (10) and limited interstitial transport (11). The requirement to concurrently surpass these rate-limiting factors, which consistently hinder clinically effective nanomedicine delivery against primary brain tumors (12–15), underscores the need for safe and more robust RNA-based drug delivery strategies.
Low-intensity focused ultrasound (FUS) combined with ultrasound (vascular) contrast agents called “microbubbles” (MBs) provides a physical method to transiently modulate the brain TME and improve the delivery of anticancer agents in the brain (16, 17). Recent clinical trials have demonstrated the safety of MB-FUS and provided evidence of its efficacy using small chemotherapeutic agents (18–21). While the improved drug delivery in brain TME is primarily attributed to transient changes in the BBB/BTB permeability (16, 22), recent investigations have alluded to the ability of MB-FUS to also increase the interstitial fluid flow in the brain TME (23). These observations both explained and underscored its potential to improve the delivery of therapeutic nanoparticles in brain tumors (24–31), including DNA (luciferase plasmid)–loaded nanoparticles (32). While all studies to date have used 50- to 100-nm polydisperse, anionic (−25- to −5-mV surface charge) nanoparticles, it is still unclear how to best combine these technologies. Moreover, there is a lack of evidence for selective and effective delivery of the nanoparticle and its load, including RNA, in the cellular compartment of brain tumors, which is required for demonstrating successful delivery (33, 34). As a result, the relationship among the changes in BBB/BTB permeability, nanoparticle properties and uptake, and tumor cell death (i.e., effective nucleic acid–based therapeutic delivery) in the brain TME is yet to be established.
We hypothesize that the combined abilities of cationic nanoparticles to prolong RNA circulation and augment cell uptake with those of MB-FUS to alleviate vascular and interstitial barriers to transport can lead to a robust strategy for the systemic delivery of RNA-based therapeutics in brain tumors. Here, we test this hypothesis by fabricating and assessing the MB-FUS delivery of a fluorescently labeled lipid-polymer hybrid (LPH) nanoparticle loaded with a test siRNA (RohB-LPH loaded with Cy5-siRNA, LPH:Cy5-siRNA) and a therapeutic Smoothened (SMO) targeting siRNA (LPH:SMO-siRNA) in preclinical models of glioma and medulloblastoma, the most common malignant brain tumors in adults and children, respectively. The SMO-siRNA targets the SMO-activated sonic hedgehog (SHH) subgroup of medulloblastoma (35). Subsequently, we investigate the abilities of the proposed therapeutic strategy to concurrently surpass the rate-limiting factors to the effective delivery of siRNA by directly assessing the LPH and SMO-siRNA delivery and activation of apoptotic pathways in the brain TME using single-cell image analysis. Last, by combining quantitative imaging and mathematical modeling, we evaluate and quantify every step of the proposed strategy and establish design rules for the delivery of RNA therapeutics against brain tumors.
RESULTS
MB-FUS mediates enhanced penetration of LPH:siRNA nanoparticles in the brain of immunocompetent mice
In this study, the basic design specifications for fabricating the siRNA nanoparticles for targeting the brain TME have been shaped by the median size of the brain extracellular space (pore size: 30 to 60 nm) (36) along with evidence suggesting that positively charged nanoformulations (>+5 mV) display high transvascular flux and cell uptake (2, 7, 37). With this in mind, we fabricated cationic (surface charge > +10 mV) LPH nanoparticles (38) with a median size of 40 nm. This nanoparticle technology is scalable, produces nanoparticles with narrow size distributions, and can be used to deliver a wide range of therapeutic compounds (39). We fabricated the LPH nanoparticles with rhodamine B (RhoB-LPH), using a swirling microvortex reactor, and loaded them with nontargeting Cy5-siRNA (5:1, w/w). After the Cy5-siRNA loading, the median particle size increases from 40 to 50 nm, and surface charge decreases from +26 to +11 mV (Fig. 1, A and B, and fig. S1, A and B). These changes in size and charge kept the LPH:siRNA complex within our design specifications (>+5 mV and ≤60 nm) while retaining narrow size distribution. Incubation of the double-labeled LPH:siRNA with murine GL261 glioma cancer cells showed significant LPH:siRNA complex uptake with cell viability higher than 95% (Fig. 1, C to E).
Fig. 1. In vitro and in vivo characterization of transcellular and transvascular (BBB) penetration of cationic lipid-polymer hybrid nanoparticles loaded with siRNA.
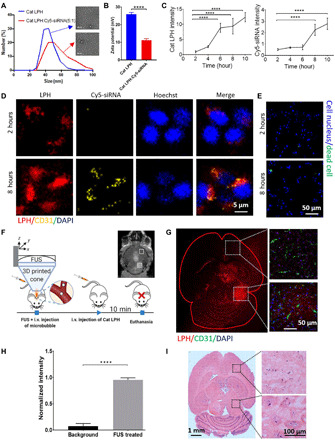
(A) Size distribution and morphology of the cationic LPH and LPH:Cy5-siRNA nanoparticles. (B) Surface charge of the cationic (Cat) LPH and LPH:Cy5-siRNA. P values were determined by unpaired t test. (C) Quantification of the LPH and Cy5-siRNA uptake by GL261 glioma cells using fluorescence microscopy. P values were determined by one-way analysis of variance (ANOVA). (D) In vitro cellular uptake kinetics of the cationic LPH:Cy5-siRNA into GL261 glioma cells. (E) Live and dead cell staining at 2 and 8 hours after LPH:Cy5-siRNA exposure. (F) In vivo experimental protocol for the delivery of cationic LPH in the brain of healthy, immunocompetent mice. Inset: Representative contrast-enhanced T1-weighted MR image after MB-FUS. i.v., intravenous. (G) Representative fluorescent microscopy data of LPH extravasation in the brain of healthy mouse at 10 min after LPH administration for non-FUS region (top) and FUS-treated region (bottom). (H) Quantification of the LPH extravasation in non–FUS-treated region and FUS-treated region 10 min after treatment (12-fold; P < 0.0001, unpaired t test). (I) Representative H&E staining images at 10 min after LPH administration. Plots show means ± SEM (N = 3). ****P ≤ 0.0001.
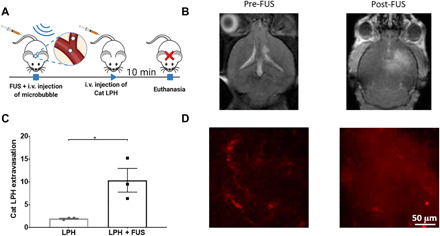
Following the fabrication of the LPH nanoparticles, we assessed their accumulation in the brain of healthy mice with and without the application of MB-FUS. To account for immune-related reactions and nanoparticle clearance, in the present study, our in vivo experiments were performed using the C57BL/6J immunocompetent mice (40). Our experimental protocol, using a custom-built magnetic resonance–guided FUS (MRgFUS) system, is summarized in Fig. 1F. For these investigations, we used (i) a 0.5-MHz transducer with standard exposure settings (10-ms bursts, every 1 s for 1 min, at 475-kPa peak negative pressure in water), (ii) Optison (GE Healthcare) MBs with a dosage of 100 μl/kg, and (iii) RhoB-LPH nanoparticles (5 mg/kg). The in vivo dosage of nanoparticle was decided on the basis of the dosages used in previous studies that showed that this nanoparticle dose led to minimal side effects (41–43). Fluorescence imaging of the excised brain 10 min after sonication indicated a 12-fold increase in the extravasation of LPH in the FUS-treated region as compared to the non-FUS region (P < 0.0001) (Fig. 1, G and H). Notably, contrast-enhanced MR imaging (MRI) was able to both confirm the areas targeted by MB-FUS and track the LPH extravasation (Fig. 1F, inset), potentially providing a noninvasive way to predict LPH delivery and distribution. By measuring the intensity of cationic LPH in blood from immunocompetent mice (C57BL/6J mice), we estimated the elimination half-life (t1/2) to be 30 min (fig. S1C). These preliminary findings suggest that the circulation time of nucleic acid has been doubled after loading to cationic LPH (44). Last, histological analysis of the brain slices of immunocompetent mice showed that the delivery of cationic LPH in the FUS-targeted brain areas did not result in significant hematoxylin and eosin (H&E)–evident adverse effects (Fig. 1I). Together, these data demonstrated that MB-FUS can be used to substantially improve (>10-fold) the delivery of cationic nanoparticles in the FUS-targeted brain areas of immunocompetent mice without significant adverse effects.
MB-FUS promotes cationic LPH:Cy5-siRNA extravasation, penetration, and uptake in GL261 orthotopic brain tumors
Following the successful and robust delivery of the LPH nanoparticle in healthy brain using MB-FUS, we assessed the delivery of the nontargeting Cy5-siRNA–loaded LPH complex in brain tumors. Here, we used the established orthotopic GL261 glioma syngeneic murine model, where GL261 glioma cells are injected intracranially in C57BL/6J mice (the location of the tumor is shown in fig. S2). Fourteen days after tumor implantation, we sonicated four nonoverlapping areas to cover the entire tumor using our MRgFUS system and the same exposure settings and protocol with the one used in healthy mice (Fig. 2A). Fluorescence microscopy indicated a fivefold increase in the LPH accumulation in FUS-treated tumors as compared to control (P = 0.032) (Fig. 2, B to D). It also showed higher variability in the observed accumulation as compared to healthy brains, which is consistent with the heterogeneous properties of the brain TME.
Fig. 2. Improved cationic LPH extravasation in the GL261 glioma mouse tumors using MB-FUS.
(A) In vivo experimental protocol for the delivery of cationic LPH in GL261 glioma mouse tumor model. LPH distribution is analyzed at 10 min after LPH administration. (B) Representative contrast-enhanced T1-weighted MR images before and after MB-FUS treatment. (C) Quantification of the LPH extravasation in non–FUS-treated and FUS-treated tumor at 10 min after treatment (5.4-fold, P = 0.032). (D) Representative fluorescent microscopy data of LPH extravasation in tumor at 10 min after LPH administration. Plots show means ± SEM (N = 3). P values were determined by unpaired t tests. *P ≤ 0.05.
In a separate cohort, we administered LPH:siRNA nanoparticles and sacrificed the mice 8 hours after administration (instead of 10 min) (Fig. 3A). This time point was dictated by our in vitro investigations that showed robust LPH:siRNA uptake by the GL261 glioma cells (Fig. 1, C and D). Fluorescence microscopy indicated a 13-fold increase in the LPH accumulation in the FUS-targeted tumor as compared to the non–FUS-treated tumor (P = 0.044) (Fig. 3, B and C). Crucially, there was a 10-fold increase in siRNA uptake after MB-FUS compared to control (P = 0.0364) (Fig. 3, E and G, and fig. S3), demonstrating that LPH and siRNA delivery track each other very well (i.e., reliable siRNA loading). Moreover, the observed improvement in delivery was also accompanied by a fivefold increase in siRNA penetration as compared to control LPH:siRNA only (P = 0.0045) (Fig. 3, B and D). Up to 60% of the LPH:siRNA nanoparticles were taken up by cancer cells (F-Luc–positive cells) among all cell types [4′,6-diamidino-2-phenylindole (DAPI)–positive cells] (Fig. 3, F and G), demonstrating good uptake by the GL261-Luc2 glioma cancer cells. Together, the above experimental data demonstrate the combined abilities of 50-nm cationic LPH:siRNA nanoparticles and MB-FUS to attain a marked improvement (i.e., 10-fold) in the delivery of siRNA in brain TME.
Fig. 3. Improved cationic LPH:siRNA extravasation, penetration, and cellular uptake in the GL261 glioma mouse tumors using MB-FUS.
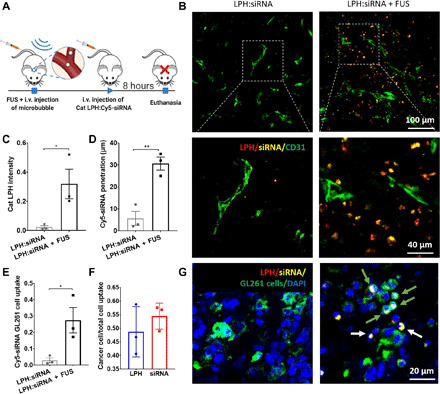
(A) In vivo experimental protocol for LPH:siRNA delivery in a GL261 glioma mice tumor model. LPH:siRNA distribution is analyzed at 8 hours after nanoparticle administration. (B) Representative fluorescent microscopy data of LPH:siRNA extravasation and penetration in tumor at 8 hours after LPH:siRNA administration. (C) Quantification of the LPH extravasation in tumor with and without FUS at 8 hours after treatment (13.7-fold, P = 0.044). (D) Quantification of the siRNA penetration in tumor with and without FUS at 8 hours after treatment (5.4-fold, P = 0.0045). (E) Quantification of siRNA delivery to cancer cells with and without FUS at 8 hours after treatment (9.5-fold, P = 0.0364). (F) Quantification of the ratio of LPH (blue) and Cy5-siRNA (red) delivery to cancer cells to total cell uptake at 8 hours after FUS treatment. (G) Representative fluorescent microscopy data of LPH:Cy5-siRNA cellular uptake in tumor at 8 hours after LPH:siRNA administration. Green arrows show the LPH:siRNA uptake by cancer cells, and white arrows show the LPH:siRNA uptake by brain cells. Plots show means ± SEM (N = 3). P values were determined by unpaired t tests. *P ≤ 0.05 and **P ≤ 0.01
MB-FUS promotes LPH:SMO-siRNA delivery, markedly reduces tumor SMO protein expression, and induces effective and specific apoptosis in SHH-activated medulloblastoma
To assess the robustness and effectiveness of the proposed therapeutic strategy in brain TME, we loaded a therapeutic SMO-siRNA to 40-nm cationic RhoB-LPH. This siRNA has already been validated in vitro (45). This siRNA was now used to inhibit the SMO-activated SHH signaling pathway in an established transgenic mouse model representative of the SHH-activated subgroup of medulloblastoma [Neurod2:SmoA1–green fluorescent protein (GFP) mice, expressing a constitutively activated form of Smo and a GFP reporter in the cerebellar granule neuron precursor cells; the location of the tumor is shown in fig. S6], which has an intact BBB (46). To ensure that the SMO-siRNA remained fully functional, we did not label it with fluorescence molecule; hence, we assessed its ex vivo distribution using the fluorescence in situ hybridization (FISH) assay. This assay can detect and localize single RNA molecules at subcellular resolution (47, 48), thereby allowing to confirm the presence of SMO-siRNA in the brain TME. For the experiments with these animals, we used a custom-built portable ultrasound-guided FUS (USgFUS) system (Fig. 4A and fig. S4A) and used the experimental protocol shown in Fig. 4D. This system allows submillimeter precision targeting along with real-time monitoring of MB dynamics via passive detection of the MB acoustic emissions during the sonication. Our measurements indicate strong harmonic emissions (up to 22 dB) with less than 1% (20 of 2040) broadband emissions during sonications, suggesting that our exposure settings were within the safety levels of MB-FUS (Fig. 4, B and C, and fig. S4, B to E). Immediately after sonication, we administered the LPH:SMO-siRNA and sacrificed the mice 30 hours after administration. This time point is a conservative estimation on the time required for SMO knockdown and the indication of cell apoptosis (49). Like the LPH:Cy5-siRNA delivery in glioma tumors, we found a 10-fold increase in the LPH accumulation in the FUS-targeted tumor as compared to the non–FUS-treated tumor (P = 0.0472; Fig. 4, E and F, and fig. S7). To confirm that the observed RhoB-LPH delivery led to SMO-siRNA delivery in the medulloblastoma cells, we performed FISH. As evidenced in Fig. 4G, the FISH assay confirmed the colocalization of the RohB-LPH and unlabeled siRNA in the medulloblastoma cells (GFP-positive cells). Together, these data demonstrate the robustness of the proposed strategy and confirm the successful delivery of the SMO-siRNA in the medulloblastoma cancer cells.
Fig. 4. Enhanced delivery of siRNA into the SHH-activated medulloblastoma tumor using MB-FUS and cationic LPH:SMO-siRNA.
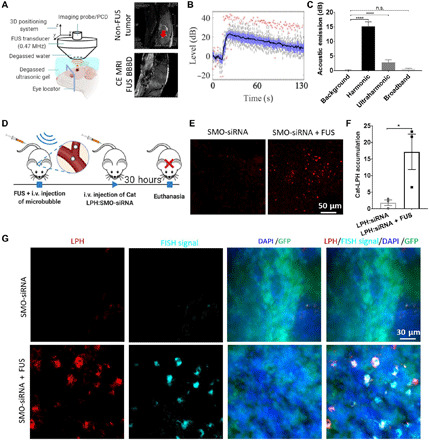
(A) In vivo experiment setup using a custom-built USgFUS system (left). Schematic graph is created with BioRender.com. Representative contrast-enhanced T2-weighted MR images of medulloblastoma tumor–bearing mice (right, top) and contrast-enhanced T1-weighted MR images of healthy mice after FUS BBB disruption (BBBD) targeted at the location of medulloblastoma tumor (right, bottom). (B) Harmonic emission from the MB-mediated BTB disruption using FUS. (C) Quantification of the acoustic emissions. (D) In vivo experimental protocol for the delivery of cationic LPH:SMO-siRNA in a medulloblastoma mouse tumor model. (E) Representative fluorescent microscopy data of LPH accumulation in tumor for non–FUS-treated group (left) and FUS-treated group (right). (F) Quantification of the LPH accumulation in tumor with and without FUS at 30 hours after treatment (9.4-fold, P = 0.0472). (G) Representative fluorescent data of FISH assay on non–MB-FUS group (top) and MB-FUS group (bottom). Plots show means ± SEM (N = 3). P values were determined by unpaired t tests. n.s., no statistical significance; *P ≤ 0.05; ****P ≤ 0.0001.
Next, we assessed whether the delivered therapeutic siRNA can induce SMO knockdown and promote cell apoptosis in the medulloblastoma brain tumors (Fig. 5A). Immunohistochemistry revealed reduced expression of constitutively activated SMO protein that drives the SHH signaling pathway and tumorigenesis in this model of SHH medulloblastoma, indicating effective SMO knockdown (Fig. 5, B and C). Using immunofluorescence staining, we detected a 16-fold increase in tumor cell apoptosis in the MB-FUS–treated tumor as compared to non–FUS-treated tumors (P = 0.0013; Fig. 5, D and E). Crucially, apoptotic cells [terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) positive] colocalized with RhoB-LPH nanoparticles, demonstrating that the SMO-siRNA delivery led to cancer cell death (Fig. 5D). To confirm that the observed apoptosis was not related to RhoB-LPH toxicity, we analyzed the GL261 glioblastoma tumors where we delivered the nontargeting Cy5-siRNA and found comparable levels of cell apoptosis between FUS-treated tumor and non–FUS-treated tumor at 8 hours after LPH:Cy5-siRNA nanoparticle administration (Fig. 5, F and G). For these analyses, we validated the TUNEL method for the detection of apoptosis by performing cleaved caspase-3 (CC3) staining, as it provides a more specific indication of apoptosis as compared to TUNEL staining, which may reflect other means of DNA damage. Collectively, our data support the hypothesis that combining MB-FUS with cationic LPH:siRNA can overcome the barriers to the delivery of siRNA therapeutics to primary brain tumors and effectively target an oncogene that is driving a primary brain tumor.
Fig. 5. MB-FUS, in combination with cationic LPH:SMO-siRNA, induces cell apoptosis in SHH-activated subgroup of medulloblastoma.
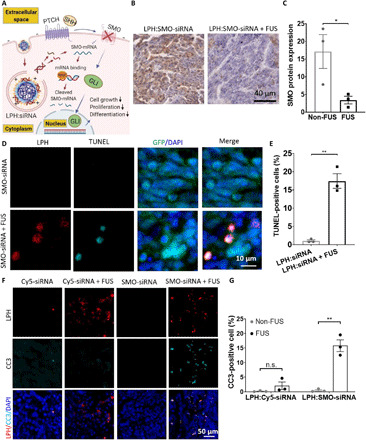
(A) Schematic illustration of LPH:siRNA-induced cell apoptosis. Schematic graph is created with BioRender.com. (B) Representative microscopy data of SMO protein immunostaining non–FUS-treated group (left) and FUS-treated group (right) demonstrating substantial SMO protein knockdown. SMO protein is shown in brown, and nucleus staining is shown in purple. (C) Quantification of SMO protein level in medulloblastoma tumor (fivefold, P = 0.0483). (D) Representative fluorescent microscopy data of SMO-siRNA–induced cell apoptosis in tumor at 30 hours after LPH:SMO-siRNA administration for non–FUS-treated group (top) and FUS-treated group (bottom) using TUNEL assay. (E) Quantification of tumor cell apoptosis (TUNEL-positive signal) that is LPH positive with and without FUS at 30 hours after treatment (16.6-fold, P = 0.0013). (F) Representative fluorescent microscopy data of SMO-siRNA–induced cell apoptosis in tumor at 30 hours after LPH:SMO-siRNA administration for non–FUS-treated group (top) and FUS-treated group (bottom) using CC3 assay. (G) Quantification of tumor cell apoptosis (CC3-positive signal) that is LPH positive 8 hours after nontherapeutic LPH:Cy5-siRNA delivery administration (n.s.) and 30 hours after LPH:SMO-siRNA delivery administration with and without FUS (34-fold, P = 0.0017). Plots show means ± SEM (N = 3). P values were determined by unpaired t tests. n.s., P > 0.05; *P ≤ 0.05; **P ≤ 0.01.
Forty-nanometer cationic nanoparticles combined with MB-FUS attain a fine balance between transvascular, interstitial, and transcellular transport in the brain TME
We postulated that quantitative image analysis and physiologically based pharmacokinetic (PBPK) modeling could be used to extrapolate findings to refine the design of the LPH and better understand its transport dynamics in the TME when combined with MB-FUS (e.g., BBB permeability, interstitial transport, and cell uptake), which would otherwise require extensive biological experiments. We used a well-established PBPK model that captured the diffusive and convective transport of nanoparticles from the blood vessel, across the BBB, in the interstitial space, and into the cells (37, 50). To reduce the extrapolation error, we tuned the model to the specific experimental conditions using parameter identification procedures that we developed based on experiment-specific objective function (Fig. 6A). The substantial overlap between model output and reference solution (Fig. 6A) indicated convergence between the model and the experimental data (RhoB-LPH in GL261 glioma tumors) and potentially reduced extrapolation error. The identified model parameters showed significant differences between the vessel effective diffusion coefficient, Dv (20-fold increase, P = 0.04), and the interstitial hydraulic conductivity, Ki (40-fold increase, P = 0.006), for the FUS and non-FUS group (Fig. 6B). These data, which corroborate our previous investigations using small anticancer agents (23), provide a quantitative assessment on the abilities of MB-FUS to promote acute changes in the LPH transport dynamics in the GL261 glioma TME without any major adverse effects.
Fig. 6. Integrated quantitative microscopy and PBPK modeling guides the integration of LPH nanoparticles and MB-FUS technologies.
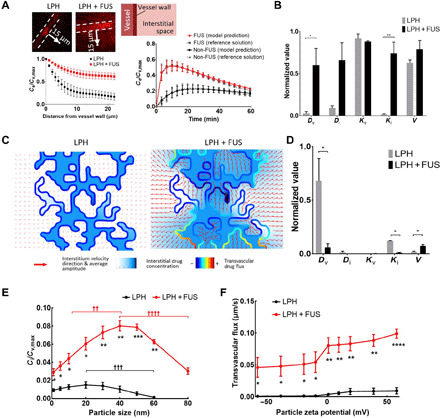
(A) Parameter identification procedures to recover LPH pharmacokinetics from the experimentally determined RhoB-LPH penetration (line profile perpendicular to vessel wall, left) in the GL261 glioma tumor model using 2D tumor cord geometry. The model output and the reference solutions agreed (right). (B) Normalized parameter fit for non–FUS-treated and FUS-treated groups using 2D tumor cord PBPK model. (C) Structurally heterogeneous modeling of LPH transport in TME. (D) Sensitivity analysis of the model parameters. (E) Cellular uptake of LPH with different sizes for non-FUS and FUS. (F) Transvascular flux with different surface charge LPH for non-FUS and FUS. †Difference between different LPH sizes for non-FUS and FUS (one-way ANOVA). *Difference between non-FUS and FUS for each LPH size or surface charge (unpaired t tests). Extracellular LPH concentration (Ce) and intracellular LPH concentration (Ci) normalized to maximum LPH concentration inside the vessel (Cv). Dv, vessel diffusion coefficient; Di, interstitium diffusion coefficient; Kv, vessel hydraulic conductivity; Ki, interstitium hydraulic conductivity; V, rate of endocytosis. The plots show means ± SEM (N = 3). In (B) and (C), the P values were determined by unpaired t tests. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, ††P ≤ 0.01, †††P ≤ 0.001, ††††P ≤ 0.0001.
To assess the relative importance of parameters that define the transport dynamics in the TME and identify the optimum LPH physical properties for maximum nucleic acid delivery to cancer cells, we performed a parametric sensitivity analysis. Specifically, we locally adjusted (one at a time) the identified modeling parameters (±25%) and subsequently recorded the resulting impact on LPH uptake by tumors at 1 hour after injection (LPH clearance time). To take into account the influence of tumor vascular heterogeneity on interstitial drug transport, we expanded the tumor cord geometry (i.e., a cylindrical geometry of a vessel surrounded by tumor; Fig. 6A) to vascular network geometry (Fig. 6C). Following this additional refinement, the model revealed a considerably higher LPH transvascular transport, extravasation, and penetration after MB-FUS as compared with the non-FUS group (Fig. 6C). This was consistent with experimental observations (Figs. 2 and 3). The LPH-only group confirms that the BBB/BTB is the most important rate-limiting factor in nanomedicine delivery in brain tumors (i.e., the highest normalized value in Fig. 6D). The FUS-LPH group shows that MB-FUS significantly reduces the relative importance of the BBB permeability (described by parameter Dv in Fig. 6D) and interstitial flow (described by parameter Ki in Fig. 6D), providing mechanistic understanding on the observed improved delivery of LPH in brain TME. It also indicates that the rate of endocytosis (described by parameter V in Fig. 6D) becomes a rate-limiting factor in the LPH delivery after MB-FUS, presumably due to higher LPH availability in the brain TME.
Notably, evaluation of the impact of nanoparticle size on the transport dynamics indicated that, under MB-FUS, 40-nm particles could lead to much higher uptake as compared to 10- or 80-nm particles (Fig. 6E). The optimum nanoparticle size to deliver nucleic acid therapeutics without disrupting the BBB in brain tumors is around 20 nm, which is consistent with the physical barriers to their transport imposed by the BBB (10), whereas with MB-FUS the optimum size is around 40 nm. These data also emphasize the relative importance of convective transport, which is critical for larger nanoparticles, and it is significantly improved by MB-FUS. Likewise, positively charged particles lead to a notable increase in the transvascular flux as compared to negatively charged ones (Fig. 6F). In summary, the proposed quantitative framework that is based on single-cell image analysis and mathematical modeling allowed us to quantify the FUS-mediated changes in the LPH transport dynamics in the TME and establish design rules (i.e., LPH size and charge) for robust siRNA delivery in brain tumors with MB-FUS.
DISCUSSION
The goal of this investigation is to identify strategies to overcome the transport barriers to the effective delivery of siRNA therapeutics in the brain TME. Although both viral and nonviral gene vectors can be used as a delivery vehicle for nucleic acids, viral vectors are limited by toxicity, immunogenicity, low loading capacity, and high production costs (51). Thus, in this study, we focused on siRNA nanoparticle formulations. As we alluded to, one of the key aspects of our study is the concurrent assessment of the nanoparticle penetration and payload (siRNA) delivery in the brain TME at subcellular resolution using fluorescence microscopy (Cy5-siRNA) and FISH (SMO-siRNA). The latter allowed us to directly show, in an immunocompetent animal, that MB-FUS siRNA delivery can reduce the oncogene driving the tumor and lead to a marked increase in cancer cell death and thus establish a causative relationship among changes in BBB/BTB permeability, nanoparticle/siRNA penetration and uptake, and tumor cell death. This is a major advancement as compared to previous investigations that have so far provided macroscopic evidence of successful transfection in the brain TME using luciferase plasmid DNA loaded to anionic nanoparticles (32). Moreover, the demonstrated high LPH:siRNA uptake by cancer cells into two separate preclinical models of malignant glioma and medulloblastoma, in combination with the high penetration across the BBB and BTB, underscores the potential of the proposed therapeutic strategy to target the tumor core and infiltrating margin, which currently remains inaccessible to therapy (15).
In addition, our combined experimental and modeling investigations revealed that balancing the improved transvascular and interstitial transport attained by MB-FUS with the high penetration and cancer cell uptake by 40-nm weakly cationic nanoparticles is critical for robust delivery and uptake in the brain TME. These findings, which depart significantly from current approaches that are based on 50- to 100-nm polydisperse, anionic nanoparticles (<−5 mV), provide a paradigm shift in drug delivery in brain tumors, where physical methods and nanotechnology are tuned together to systematically identify optimal FUS drug combinations and develop rational strategies for the effective delivery of nucleic acids in brain tumors. While additional experimental data using nanoparticles of different size and surface charge will further consolidate the observed trends, our findings (Fig. 5) provide a plausible explanation for the lower drug delivery observed in past investigations using larger nanoparticles (50 to 100 nm), as compared to the present data (>10-fold versus 3.7-fold median increase in delivery after MB-FUS compared to non-FUS; table S1). These findings can potentially be improved, including cell specificity, by surface functionalization. Increasing the sample size of each experimental group (table S2) will allow to assess further the reproducibility of our findings and better assess how tumor heterogeneity affects the proposed therapeutic strategy. Although past biodistribution and toxicity investigations of lipid nanoparticles with similar dosage to the one that we used in this study reported negligible toxicity (41–43), future studies in evaluating treatment outcomes (tumor growth and survival) in patient-derived tumors with and without known driver mutations, combined with extended toxicologic analysis under different LPH:siRNA doses, are warranted. Testing additional FUS exposures (e.g., lower harmonic emissions), ideally under closed-loop control (52–55), combined with a more detailed assessment of the BBB phenotype (i.e., structure and function) and brain/tumor tissue (56, 57) will allow to further define and refine the MB-FUS treatment window for safe and effective siRNA delivery in the brain TME.
Collectively, our findings demonstrate the potential of our work to lead to the development of rational strategies for the effective delivery of nucleic acids in brain tumors and provide a unified framework for prospective, quantitative, and mechanistic investigation of siRNA delivery in brain TME. The principles established here can also be extended to target more than one part of a signaling pathway such that its knockdown affects survival/growth (58, 59), overcome problems with drug resistance (60), and create unique opportunities for delivering shRNA, antisense RNA, or CRISPR-Cas9 (2, 61) and enhancing immunotherapy.
MATERIALS AND METHODS
LPH:siRNA fabrication and characterization
Cationic LPH (40 nm diameter) was prepared using a slightly modified swirling microvortex reactor (38). Briefly, a solution containing poly(d,l-lactic-co-glycolic) acid (PLGA) (0.3 mg/ml) in acetonitrile and another solution containing 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) (0.018 mg/ml), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (0.015 mg/ml) (Liss Rhod PE), N1-[2-((1S)-1-[(3-aminopropyl)amino]-4-[di(3-amino-propyl)amino]butylcarboxamido)ethyl]-3,4-di[oleyloxy]-benzamide (MVL-5) (0.02 mg/ml), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG) (0.02 mg/ml) in 4% ethanol were introduced into the swirling microvortex reactor at a Reynolds number of 250. All these parameters were adjusted to yield 40-nm-sized particles. The prepared particles were filtered and centrifuged at 2900 rpm for 15 min three times to remove any impurities.
The technique used for loading siRNA onto LPH is similar, although the precursors used are not the same as the siRNA is a charged molecule. The polymer core used is PLGA, while the lipid precursors used are DPPC, Liss Rhod PE, DSPE-PEG, and MVL5. The cationic LPHs were mixed with the siRNA at a ratio of 5:1 (w/w) under vigorous vortexing, which allowed optimal siRNA loading onto the LPHs. Two different siRNAs used in this study were purchased from Horizon Discovery (Dharmacon, Lafayette, CO): Cy5-siRNA (siSTABLE nontargeting; catalog no.: D-001700-01) and SMARTpool (a mixture of four siRNAs; catalog no.: L-041026-00-0020) ON-TARGETplus SMO-targeting siRNA. siRNA sequences are provided in the Supplementary Materials.
The hydrodynamic volumes and surface charge of the cationic LPH and LPH:siRNA was obtained with Zetasizer Nano (Malvern Instrument, Malvern, UK). The morphology of samples was taken from a transmission electron microscope (Hitachi 7700, Hitachi, Japan) at 120 kV coupled with a Digital Micrograph camera and software suite from Gatan. The samples were negatively stained with 2% uranyl acetate solution for 30 s.
Culture of GL261 cells
GL261-luc2 glioma cells (Caliper Life Sciences) were cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin at 37°C and 5% CO2.
Quantification of cellular uptake kinetics of nanoparticle into GL261 glioma cells
GL261 cells were seeded at a density of 20,000 cells/ml in a glass bottom dish (MatTek, catalog no. P35GCOL-1.5-14-C) over a period of 24 hours, which resulted in 60% confluency. At that point, the cells were incubated with cationic LPH:Cy5-siRNA at a concentration of 0.02 mg/ml and the intracellular fluorescent signal of the nanoparticles was measured at five different time points (2, 4, 6, 8, and 10 hours) using fluorescent microscopy. At the end of each time point, cell viability was determined by counting total versus dead cells using a cell viability kit according to the manufacturer’s instructions (ReadyProbes Cell Viability Imaging Kit, Blue/Green, Thermo Fisher Scientific, catalog no. R37609). The quantification of the fluorescence intensity of LPH and Cy5-siRNA at different time points was determined using ImageJ.
GL261 glioma cell inoculation
All animal procedures were performed according to the guidelines of the Public Health Policy on the Humane Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of Georgia Institute of Technology. GL261 cells (105 cells), genetically modified to express firefly luciferase, were stereotactically implanted into the brain at 1-mm anterior and 1 mm to the right of the bregma of 6- to 8-week-old female C57BL/6J mice (The Jackson Laboratory) (15 mice). After cell implantation, tumor growth was monitored using T2-weighted MRI (PharmaScan 7T, Bruker), and when tumors reached a size of ~20 to 40 mm3, BBB disruption was performed using a custom-built MRgFUS. To minimize differences in the (baseline) BBB permeability across different experimental arms, related to differences in tumor sizes, before each experiment, the tumors were measured with MRI and spread equally between control and FUS-treated groups.
Medulloblastoma tumor model
We used the SmoA1 transgenic mouse model of human SHH-type medulloblastoma (008831, The Jackson Laboratory). SmoA1-Math1-GFP mice were generated by crossing SmoA1 mice with Math1-driven GFP reporter mice. The mice were maintained in Emory University animal facilities, approved by the American Association for Accreditation of Laboratory Animal Care. When the mice reached 10 to 12 weeks old, we confirmed the growth of the tumor using MRI and enrolled the mice in the study.
MRgFUS system and sonication of GL261 tumor
The MRgFUS system is composed of an air-backed spherically curved transducer (frequency: 0.5 MHz, F-number: 0.70, focal distance: 25 mm) that is attached to a water-filled three-dimensionally (3D) printed cone with an exit window made of thin Mylar membrane. The system is mounted with a surface coil on a manual 3D positioning system that, under MRI guidance, allows to sonicate different brain targets with millimeter targeting precision. At the sonication experiments, the following exposure settings were used: 10-ms bursts, every 1 s for 1 min, with concurrent intravenous administration of clinical-grade MBs (100 μl/kg; Optison). A 475-kPa peak negative pressure (based on free-field measurements—water) was used throughout the present study. To cover the entire tumor and its periphery, we performed four nonoverlapping sonications (X-Y directions). Before the sonications, the tumor location was identified using T2-weighted MRI of the entire brain. Immediately after the sonication, the animals were injected with gadolinium contrast agent (Magnevist, 0.4 ml/kg) and T1-weighted contrast-enhanced MRIs were acquired to confirm BBB disruption at the tumor and surrounding healthy tissue. After BBB disruption was confirmed, the nanoparticles were intravenously administered, and the animals were euthanized at 10-min (for LPH) or 8-hour (LPH:siRNA) nanoparticle injection. Last, the brain was harvested for further processing.
USgFUS system and sonication of medulloblastoma tumor
To perform the BBB disruption in medulloblastoma tumors, we used a custom-built portable USgFUS system with high targeting accuracy and real-time monitoring of MB dynamics via passive cavitation detection (PCD). First, the USgFUS system creates a 2D contour of the mouse head by raster scanning a single-element imaging transducer (3.5 MHz) mounted on a 3D positioning system. This image is then used to estimate the locations of the eyes, which is then compared with an MRI image to locate the tumor. The system then allows to perform sonication at four nonoverlapping areas to cover the entire tumor with a coaxial therapeutic FUS transducer (0.5 MHz). During sonication, the imaging transducer is switched to passive mode to capture MB’s response, whose frequency spectrum is then normalized to the mean of spectrum recorded before MB arrival to remove unnecessary emissions other than the MB response (fig. S4). Harmonic, ultraharmonic, and broadband levels were obtained by taking the mean of ±5 frequency bins from each harmonic frequency—3f0, 2.5f0, and 6.72f0, respectively. PCD during BBB disruption experiments allowed us to capture the onset of harmonic emissions and ensure safe and effective disruption.
Brain tissue processing
In the group of animals that were euthanized at 10 min after nanoparticle administration, the brains were harvested without transcardial perfusion. The animals that were euthanized at 8 hours (glioma tumor–bearing mice) and 30 hours (medulloblastoma tumor–bearing mice) after nanoparticle administration were transcardially perfused with 20 ml of saline before harvesting the brains. The brains were fixed with 4% paraformaldehyde overnight at 4°C followed by 30% sucrose solution (4°C) until it sunk to the bottom of the container. The brains were placed in O.C.T. (optimal cutting temperature) compound and rapidly frozen to −80°C. Subsequently, 30-μm sections were cut using a cryostat (Leica 3050 S Cryostat).
Immunofluorescence staining and microscopy
Tissues were first prepared for staining by fixing in 4% paraformaldehyde at room temperature for 10 min. After washing with phosphate-buffered saline (PBS), the sections were first blocked for 1 hour at room temperature (2% bovine serum albumin and 5% goat serum in PBS) and then incubated with primary antibody diluted in 1% bovine serum albumin (1:100) for 12 hours at 4°C. Rabbit anti-mouse CD31 (ab28364, Abcam Inc.) was used for vessel staining, and rabbit anti-firefly luciferase (ab21176, Abcam Inc.) was used for the staining of the GL261 cells. Next, the sections were incubated with goat anti-rabbit Alexa Fluor 488 secondary antibody diluted in 1% bovine serum albumin (1:250; A31556, Invitrogen) for 1 hour at room temperature. To stain the cell nucleus, samples were incubated with DAPI diluted in PBS (1:1000; 62248, Invitrogen) for 10 min after washing. Last, the sections were rinsed with PBS to remove excess antibody, mounted with mounting medium (Prolong Glass Antifade Mountant, lot no. 2018752, Invitrogen), and covered with coverslips. Samples were cured with a mounting medium for 24 hours in the dark at room temperature before imaging. At the same time, H&E staining was also performed to confirm the location of the tumor (fig. S2).
The sections were imaged with a 20× objective using a laser scanning confocal microscope system (LSM 700, Zeiss). The excitation wavelengths used for cell nucleus, vessel/cancer cell, cationic LPH, and siRNA are 405, 488, 555, and 639 nm, respectively. The quantification of the fluorescence images was performed using ImageJ. The penetration of LPH and siRNA was quantified by integrating the fluorescence signal using thin layers (2 μm) perpendicular to the vessels using a custom-built MATLAB code.
FISH assay
Four DNA oligo probes with Cy5 fluorophore were used to detect SMO-siRNA. One DNA oligo probe with Cy3 fluorophore was used to detect nontargeting siRNA. The probes were dissolved in Tris-EDTA (TE) buffer at pH 8.0 to make 100 μM stock solution.
The fixed tissue samples were permeabilized in 70% ethanol at −20°C for 24 hours. After permeabilization, the samples were first scanned using Nikon Ti2 microscope with 20× objective lenses, and small areas were scanned with 60× objective lenses. The samples were then washed with 2× SSC and then incubated in wash buffer (30% formamide) at room temperature for 5 min. After the wash buffer was aspirated, the samples were then incubated in 1 ml of hybridization solution (hybridization buffer: 30% formamide and 10% dextran sulfate; dilute probe stock solution in hybridization buffer to prepare hybridization solution; 1 μl of each probe stock for SMO-siRNA and 2 μl of probe stock for nontargeting siRNA) in humidification chamber at room temperature for 24 hours. Then, the samples were incubated in wash buffer for 5 min. To stain the nucleus, the samples were incubated in DAPI dilution in 2× SSC (1:500) for 5 min. The samples were then washed with 2× SSC three times to remove excess probes. Last, the samples were mounted in mounting buffer (tris-HCl, 8% glucose, 1:100 catalase, Pyox mixed at a ratio of 7:1:1:1) and imaged using Nikon Ti2 microscope. The excitation wavelength of DAPI, GFP-positive cells, LPH, and FISH probe for SMO-siRNA was 395, 470, 555, and 640 nm, respectively. The excitation wavelength of the nontargeting siRNA probe was 555 nm. More detailed information is provided in the Supplementary Materials.
Cell apoptosis detection
To access the ex vivo cell death, we used frozen brain tissue and then stained for the cell death marker CC3 (9661, Cell Signaling Technology) and TUNEL assay (C10619, Invitrogen) following the protocols provided by the manufacturers. The fraction of CC3- and TUNEL-positive cells that are also LPH positive in the tumor (normalized to DAPI staining) is quantified using custom-built MATLAB code.
Immunohistochemical staining
Tissues were first prepared for staining by fixing in 4% paraformaldehyde at room temperature for 10 min and then washed with PBS. After neutralization of the endogenous peroxidase with 3% H2O2 for 10 min, the sections were incubated with protein blocking buffer for 10 min before undergoing incubation with the primary antibody. Anti-SMO (E-5: sc-166685, Santa Cruz Biotechnology Inc., Dallas, TX) staining was developed using DAB (3,3′-Diaminobenzidine) (Vector Laboratories, Burlingame, CA) followed by hematoxylin counterstaining (MilliporeSigma, St. Louis, MO). H&E staining was also performed to access the location of the tumor (fig. S6). The SMO protein intensity inside the brain tumor was quantified using ImageJ.
Mathematical modeling of nanoparticle transport in brain TME
To quantify the transport parameters before and after FUS treatment, we used a PBPK model using 2D tumor cord geometry (Fig. 6A). This model consists of a luminal area, vascular wall, and interstitial space, as shown before (23). Briefly, the model fully couples the diffusive and convective transport of the nanoparticles through the bloodstream and across the endothelium into the interstitial space along with their uptake by tumor cells. The blood inside the vessel is simulated as a laminar flow with the Stokes equation. The fluid flow through the vascular wall and interstitial space, which are considered as a porous medium, is modeled using the Brinkman equation. The transport of nanoparticles is modeled as a convection-diffusion problem in the luminal subdomain and a reaction-convection-diffusion problem in the interstitial subdomains. We define the extracellular concentration of any agent as a continuous scalar field relative to a peak concentration in the bloodstream inside the vessel. Blood and interstitial fluid are assumed to be homogeneous, Newtonian, and incompressible fluids with constant viscosity.
Experimentally measured LPH concentration is used as a boundary concentration at the luminal inlet (Fig. 2D), and outflow with Neumann boundary conditions is applied to the rest of the boundaries of the computational domain. The parameters of the mathematical model, including vessel and interstitial diffusion coefficient (Dv and Di), vessel and interstitial hydraulic conductivity (Kv and Ki), and cellular transmembrane transport (V), were fitted using a numerical optimization procedure based on initial reference values taken from the literature (table S3) and experiment-specific objective function.
Because of the unavailability of experimentally measured LPH pharmacokinetics, we devised a procedure to recover LPH pharmacokinetics in the interstitium based on the experimentally determined LPH penetration data (Fig. 6A). For each experiment, we determined the relative extracellular LPH concentration at 15 μm from the vessel and 10 min after LPH administration based on the experimental data of LPH delivery in the GL261 glioma model (Fig. 6A). The objective function for each experiment is generated on the basis of the average of LPH penetration at three randomly chosen locations inside tumor. We assumed that the dynamics are governed by the analytic solution of a 1D advection-diffusion problem with only one transport parameter, D, that describes the overall rate of LPH transport as follows: , where C0 is the experimentally measured LPH concentration in the bloodstream. D is determined using the LPH concentration at 15 μm from the vessel and 10 min after administration (Fig. 6A). Overall, six models were fitted comprising three repetitions of each class of experiment: non-FUS versus FUS.
To study the influence of the spatial structural heterogeneity of the brain TME on the interstitial drug transport, we used a synthetic tumor-like vascular network geometry. Briefly, the transvascular fluid transport is modeled using Starling’s law, assuming no osmotic pressure difference (50), while the flow inside interstitial space is modeled with Darcy’s law. The anticancer agent transport in the luminal subdomain is modeled as a convection-diffusion problem and a reaction-convection-diffusion problem in the interstitial subdomain, as described for the 2D tumor cord model. To study the effect of size and surface charge on LPH cancer cell uptake and transvascular flux, we adjusted hindrance to both diffusive and convective transport across the vessel wall (Supplementary Materials). Last, we incorporated the size-dependent rate of endocytosis based on the experimental data from the literature (62–64).
Using this model and the identified parameters using the procedures described above, we performed a sensitivity analysis to determine the rate-limiting factors in the nanoparticle transport and cellular uptake with and without FUS (three per group). This is performed by numerically approximating the derivative of the intracellular agent concentration C with respect to any transport parameter Pj, i.e., . To be able to compare the sensitivities to different parameters, we used the following normalized measure of sensitivity , where σj is the SD of Pj across the four repetitions of each experiment class and max(C) is the peak intracellular concentration measured. S should be interpreted as the relative change in C for a given change of Pj that is equally likely for all j. To study the effect of particle size and surface charge on the intracellular uptake and transvascular flux, we varied the hindrance factor. A more rigorous description of the PBPK model and the method used is provided in the Supplementary Materials. All the simulations were performed using the commercial finite element software, COMSOL (version 5.3a, Burlington, MA, USA), where necessary equations were added using the Mathematics module.
Statistical analysis
Results are expressed as means ± SEM. All statistical analyses were performed using GraphPad Prism. P < 0.05 was considered statistically significant.
Acknowledgments
We thank Georgia Institute of Technology Parker H. Petit Institute Optical Microscopy Core and Histology core for the instrumental support and A. Asberry and A. Shaw for technical assistance. We also thank J. Leisen at Georgia Institute of Technology’s Magnetic Resonance Imaging (MRI) core facility for technical support during MRI-guided FUS studies. Funding: This study was supported by the CURE Foundation and NIH grants R00EB016971 (NIBIB) and R37CA239039 (NCI). Research support for S.S. is from the Harold C. Schott Endowed Chair and the Pam and Tom Mischell Foundation. Author contributions: Y.G., D.P.K., S.S., T.J.M., and C.A. designed research; Y.G., H.L., A.V., J.L., M.B.T., and C.A. performed research; J.K., A.F.C., R.G.A., and Y.K. contributed new reagents/analytic tools; Y.G., H.L., and Z.F. analyzed data; Y.G. and C.A. wrote the paper; and D.P.K., S.S., T.J.M., and C.A. provided insightful comments in the overall project and edited the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/18/eabf7390/DC1
REFERENCES AND NOTES
- 1.Katrekar D., Chen G., Meluzzi D., Ganesh A., Worlikar A., Shih Y.-R., Varghese S., Mali P., In vivo RNA editing of point mutations via RNA-guided adenosine deaminases. Nat. Methods 16, 239–242 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheng Q., Wei T., Farbiak L., Johnson L. T., Dilliard S. A., Siegwart D. J., Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–Cas gene editing. Nat. Nanotechnol. 15, 313–320 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dowdy S. F., Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 35, 222–229 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Triarico S., Maurizi P., Mastrangelo S., Attinà G., Capozza M. A., Ruggiero A., Improving the brain delivery of chemotherapeutic drugs in childhood brain tumors. Cancer 11, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Juliano R. L., The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 44, 6518–6548 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whitehead K. A., Langer R., Anderson D. G., Knocking down barriers: Advances in siRNA delivery. Nat. Rev. Drug Discov. 8, 129–138 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akinc A., Maier M. A., Manoharan M., Fitzgerald K., Jayaraman M., Barros S., Ansell S., Du X., Hope M. J., Madden T. D., Mui B. L., Semple S. C., Tam Y. K., Ciufolini M., Witzigmann D., Kulkarni J. A., van der Meel R., Cullis P. R., The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 14, 1084–1087 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Wilhelm S., Tavares A. J., Dai Q., Ohta S., Audet J., Dvorak H. F., Chan W. C. W., Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 1, 16014 (2016). [Google Scholar]
- 9.Wong H. L., Wu X. Y., Bendayan R., Nanotechnological advances for the delivery of CNS therapeutics. Adv. Drug Deliv. Rev. 64, 686–700 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Arvanitis C. D., Ferraro G. B., Jain R. K., The blood–brain barrier and blood–tumour barrier in brain tumours and metastases. Nat. Rev. Cancer 20, 26–41 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kodack D. P., Askoxylakis V., Ferraro G. B., Fukumura D., Jain R. K., Emerging strategies for treating brain metastases from breast cancer. Cancer Cell 27, 163–175 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hau P., Fabel K., Baumgart U., Rümmele P., Grauer O., Bock A., Dietmaier C., Dietmaier W., Dietrich J., Dudel C., Hübner F., Jauch T., Drechsel E., Kleiter I., Wismeth C., Zellner A., Brawanski A., Steinbrecher A., Marienhagen J., Bogdahn U., Pegylated liposomal doxorubicin-efficacy in patients with recurrent high-grade glioma. Cancer 100, 1199–1207 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Beier C. P., Schmid C., Gorlia T., Kleinletzenberger C., Beier D., Grauer O., Steinbrecher A., Hirschmann B., Brawanski A., Dietmaier C., Jauch-Worley T., Kölbl O., Pietsch T., Proescholdt M., Rümmele P., Muigg A., Stockhammer G., Hegi M., Bogdahn U., Hau P., RNOP-09: Pegylated liposomal doxorubicine and prolonged temozolomide in addition to radiotherapy in newly diagnosed glioblastoma—A phase II study. BMC Cancer 9, 308 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ananda S., Nowak A. K., Cher L., Dowling A., Brown C., Simes J., Rosenthal M. A., Phase 2 trial of temozolomide and pegylated liposomal doxorubicin in the treatment of patients with glioblastoma multiforme following concurrent radiotherapy and chemotherapy. J. Clin. Neurosci. 18, 1444–1448 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Sarkaria J. N., Hu L. S., Parney I. F., Pafundi D. H., Brinkmann D. H., Laack N. N., Giannini C., Burns T. C., Kizilbash S. H., Laramy J. K., Swanson K. R., Kaufmann T. J., Brown P. D., Agar N. Y. R., Galanis E., Buckner J. C., Elmquist W. F., Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-Oncology 20, 184–191 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aryal M., Arvanitis C. D., Alexander P. M., McDannold N., Ultrasound-mediated blood–brain barrier disruption for targeted drug delivery in the central nervous system. Adv. Drug Deliv. Rev. 72, 94–109 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meng Y., Hynynen K., Lipsman N., Applications of focused ultrasound in the brain: From thermoablation to drug delivery. Nat. Rev. Neurol. 17, 7–22 (2021). [DOI] [PubMed] [Google Scholar]
- 18.Carpentier A., Canney M., Vignot A., Reina V., Beccaria K., Horodyckid C., Karachi C., Leclercq D., Lafon C., Chapelon J.-Y., Capelle L., Cornu P., Sanson M., Hoang-Xuan K., Delattre J.-Y., Idbaih A., Clinical trial of blood-brain barrier disruption by pulsed ultrasound. Sci. Transl. Med. 8, 343re2 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Mainprize T., Lipsman N., Huang Y., Meng Y., Bethune A., Ironside S., Heyn C., Alkins R., Trudeau M., Sahgal A., Perry J., Hynynen K., Blood-brain barrier opening in primary brain tumors with non-invasive MR-guided focused ultrasound: A clinical safety and feasibility study. Sci. Rep. 9, 321 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lipsman N., Meng Y., Bethune A. J., Huang Y., Lam B., Masellis M., Herrmann N., Heyn C., Aubert I., Boutet A., Smith G. S., Hynynen K., Black S. E., Blood–brain barrier opening in Alzheimer’s disease using MR-guided focused ultrasound. Nat. Commun. 9, 2336 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Idbaih A., Canney M., Belin L., Desseaux C., Vignot A., Bouchoux G., Asquier N., Law-Ye B., Leclercq D., Bissery A., De Rycke Y., Trosch C., Capelle L., Sanson M., Hoang-Xuan K., Dehais C., Houillier C., Laigle-Donadey F., Mathon B., André A., Lafon C., Chapelon J.-Y., Delattre J.-Y., Carpentier A., Safety and feasibility of repeated and transient blood-brain barrier disruption by pulsed ultrasound in patients with recurrent glioblastoma. Clin. Cancer Res. 25, 3793–3801 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Hynynen K., McDannold N., Vykhodtseva N., Jolesz F. A., Noninvasive MR imaging–guided focal opening of the blood-brain barrier in rabbits. Radiology 220, 640–646 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Arvanitis C. D., Askoxylakis V., Guo Y., Datta M., Kloepper J., Ferraro G. B., Bernabeu M. O., Fukumura D., McDannold N., Jain R. K., Mechanisms of enhanced drug delivery in brain metastases with focused ultrasound-induced blood–tumor barrier disruption. Proc. Natl. Acad. Sci. U.S.A. 115, E8717–E8726 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang F.-Y., Wang H.-E., Liu R.-S., Teng M.-C., Li J.-J., Lu M., Wei M. C., Wong T. T., Pharmacokinetic analysis of 111in-labeled liposomal doxorubicin in murine glioblastoma after blood-brain barrier disruption by focused ultrasound. PLOS ONE 7, e45468 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang F.-Y., Teng M.-C., Lu M., Liang H.-F., Lee Y.-R., Yen C.-C., Liang M.-L., Wong T.-T., Treating glioblastoma multiforme with selective high-dose liposomal doxorubicin chemotherapy induced by repeated focused ultrasound. Int. J. Nanomedicine 7, 965–974 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang F.-Y., Wong T.-T., Teng M.-C., Liu R.-S., Lu M., Liang H.-F., Wei M. C., Focused ultrasound and interleukin-4 receptor-targeted liposomal doxorubicin for enhanced targeted drug delivery and antitumor effect in glioblastoma multiforme. J. Control. Release 160, 652–658 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Aryal M., Park J., Vykhodtseva N., Zhang Y.-Z., McDannold N., Enhancement in blood-tumor barrier permeability and delivery of liposomal doxorubicin using focused ultrasound and microbubbles: Evaluation during tumor progression in a rat glioma model. Phys. Med. Biol. 60, 2511–2527 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aryal M., Vykhodtseva N., Zhang Y.-Z., Park J., McDannold N., Multiple treatments with liposomal doxorubicin and ultrasound-induced disruption of blood–tumor and blood–brain barriers improve outcomes in a rat glioma model. J. Control. Release 169, 103–111 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Timbie K. F., Afzal U., Date A., Zhang C., Song J., Wilson Miller G., Suk J. S., Hanes J., Price R. J., MR image-guided delivery of cisplatin-loaded brain-penetrating nanoparticles to invasive glioma with focused ultrasound. J. Control. Release 263, 120–131 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coluccia D., Figueiredo C. A., Wu M. Y. J., Riemenschneider A. N., Diaz R., Luck A., Smith C., Das S., Ackerley C., O’Reilly M., Hynynen K., Rutka J. T., Enhancing glioblastoma treatment using cisplatin-gold-nanoparticle conjugates and targeted delivery with magnetic resonance-guided focused ultrasound. Nanomedicine 14, 1137–1148 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Shen Y., Pi Z., Yan F., Yeh C.-K., Zeng X., Diao X., Hu Y., Chen S., Chen X., Zheng H., Enhanced delivery of paclitaxel liposomes using focused ultrasound with microbubbles for treating nude mice bearing intracranial glioblastoma xenografts. Int. J. Nanomedicine 12, 5613–5629 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Curley C. T., Mead B. P., Negron K., Kim N., Garrison W. J., Miller G. W., Kingsmore K. M., Thim E. A., Song J., Munson J. M., Klibanov A. L., Suk J. S., Hanes J., Price R. J., Augmentation of brain tumor interstitial flow via focused ultrasound promotes brain-penetrating nanoparticle dispersion and transfection. Sci. Adv. 6, eaay1344 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wittrup A., Ai A., Liu X., Hamar P., Trifonova R., Charisse K., Manoharan M., Kirchhausen T., Lieberman J., Visualizing lipid-formulated siRNA release from endosomes and target gene knockdown. Nat. Biotechnol. 33, 870–876 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gilleron J., Querbes W., Zeigerer A., Borodovsky A., Marsico G., Schubert U., Manygoats K., Seifert S., Andree C., Stöter M., Epstein-Barash H., Zhang L., Koteliansky V., Fitzgerald K., Fava E., Bickle M., Kalaidzidis Y., Akinc A., Maier M., Zerial M., Image-based analysis of lipid nanoparticle–mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol. 31, 638–646 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Rimkus T. K., Carpenter R. L., Qasem S., Chan M., Lo H.-W., Targeting the sonic hedgehog signaling pathway: Review of smoothened and GLI inhibitors. Cancer 8, 22 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thorne R. G., Nicholson C., In vivo diffusion analysis with quantum dots and dextrans predicts the width of brain extracellular space. Proc. Natl. Acad. Sci. U.S.A. 103, 5567–5572 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stylianopoulos T., Soteriou K., Fukumura D., Jain R. K., Cationic nanoparticles have superior transvascular flux into solid tumors: Insights from a mathematical model. Ann. Biomed. Eng. 41, 68–77 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Toth M. J., Kim T., Kim Y., Robust manufacturing of lipid-polymer nanoparticles through feedback control of parallelized swirling microvortices. Lab Chip 17, 2805–2813 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim Y. T., Chung B. L., Ma M., Mulder W. J. M., Fayad Z. A., Farokhzad O. C., Langer R., Mass production and size control of lipid–polymer hybrid nanoparticles through controlled microvortices. Nano Lett. 12, 3587–3591 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mouse Genome Sequencing Consortium, Waterston R. H., Lindblad-Toh K., Birney E., Rogers J., Abril J. F., Agarwal P., Agarwala R., Ainscough R., Alexandersson M., An P., Antonarakis S. E., Attwood J., Baertsch R., Bailey J., Barlow K., Beck S., Berry E., Birren B., Bloom T., Bork P., Botcherby M., Bray N., Brent M. R., Brown D. G., Brown S. D., Bult C., Burton J., Butler J., Campbell R. D., Carninci P., Cawley S., Chiaromonte F., Chinwalla A. T., Church D. M., Clamp M., Clee C., Collins F. S., Cook L. L., Copley R. R., Coulson A., Couronne O., Cuff J., Curwen V., Cutts T., Daly M., David R., Davies J., Delehaunty K. D., Deri J., Dermitzakis E. T., Dewey C., Dickens N. J., Diekhans M., Dodge S., Dubchak I., Dunn D. M., Eddy S. R., Elnitski L., Emes R. D., Eswara P., Eyras E., Felsenfeld A., Fewell G. A., Flicek P., Foley K., Frankel W. N., Fulton L. A., Fulton R. S., Furey T. S., Gage D., Gibbs R. A., Glusman G., Gnerre S., Goldman N., Goodstadt L., Grafham D., Graves T. A., Green E. D., Gregory S., Guigó R., Guyer M., Hardison R. C., Haussler D., Hayashizaki Y., Hillier L. D. W., Hinrichs A., Hlavina W., Holzer T., Hsu F., Hua A., Hubbard T., Hunt A., Jackson I., Jaffe D. B., Johnson L. S., Jones M., Jones T. A., Joy A., Kamal M., Karlsson E. K., Karolchik D., Kasprzyk A., Kawai J., Keibler E., Kells C., Kent W. J., Kirby A., Kolbe D. L., Korf I., Kucherlapati R. S., Kulbokas E. J., Kulp D., Landers T., Leger J. P., Leonard S., Letunic I., Levine R., Li J., Li M., Lloyd C., Lucas S., Ma B., Maglott D. R., Mardis E. R., Matthews L., Mauceli E., Mayer J. H., Carthy M. M., McCombie W. R., Laren S. M., Lay K. M., Pherson J. D. M., Meldrim J., Meredith B., Mesirov J. P., Miller W., Miner T. L., Mongin E., Montgomery K. T., Morgan M., Mott R., Mullikin J. C., Muzny D. M., Nash W. E., Nelson J. O., Nhan M. N., Nicol R., Ning Z., Nusbaum C., O’Connor M. J., Okazaki Y., Oliver K., Overton-Larty E., Pachter L., Parra G., Pepin K. H., Peterson J., Pevzner P., Plumb R., Pohl C. S., Poliakov A., Ponce T. C., Ponting C. P., Potter S., Quail M., Reymond A., Roe B. A., Roskin K. M., Rubin E. M., Rust A. G., Santos R., Sapojnikov V., Schultz B., Schultz J., Schwartz M. S., Schwartz S., Scott C., Seaman S., Searle S., Sharpe T., Sheridan A., Shownkeen R., Sims S., Singer J. B., Slater G., Smit A., Smith D. R., Spencer B., Stabenau A., Stange-Thomann N., Sugnet C., Suyama M., Tesler G., Thompson J., Torrents D., Trevaskis E., Tromp J., Ucla C., Ureta-Vidal A., Vinson J. P., Von Niederhausern A. C., Wade C. M., Wall M., Weber R. J., Weiss R. B., Wendl M. C., West A. P., Wetterstrand K., Wheeler R., Whelan S., Wierzbowski J., Willey D., Williams S., Wilson R. K., Winter E., Worley K. C., Wyman D., Yang S., Yang S.-P., Zdobnov E. M., Zody M. C., Lander E. S., Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Shi B., Keough E., Matter A., Leander K., Young S., Carlini E., Sachs A. B., Tao W., Abrams M., Howell B., Sepp-Lorenzino L., Biodistribution of small interfering RNA at the organ and cellular levels after lipid nanoparticle-mediated delivery. J. Histochem. Cytochem. 59, 727–740 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fonseca-Gomes J., Loureiro J. A., Tanqueiro S. R., Mouro F. M., Ruivo P., Carvalho T., Sebastião A. M., Diógenes M. J., Pereira M. C., In vivo bio-distribution and toxicity evaluation of polymeric and lipid-based nanoparticles: A potential approach for chronic diseases treatment. Int. J. Nanomedicine 15, 8609–8621 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fisher D. G., Price R. J., Recent advances in the use of focused ultrasound for magnetic resonance image-guided therapeutic nanoparticle delivery to the central nervous system. Front. Pharmacol. 10, 1348 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park J., Park J., Pei Y., Xu J., Yeo Y., Pharmacokinetics and biodistribution of recently-developed siRNA nanomedicines. Adv. Drug Deliv. Rev. 104, 93–109 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khvorova A., Reynolds A., Jayasena S. D., Functional siRNAs and miRNAs exhibit strand bias. Cell 115, 209–216 (2003). [DOI] [PubMed] [Google Scholar]
- 46.Kim J., Dey A., Malhotra A., Liu J., Ahn S. I., Sei Y. J., Kenney A. M., MacDonald T. J., Kim Y. T., Engineered biomimetic nanoparticle for dual targeting of the cancer stem-like cell population in sonic hedgehog medulloblastoma. Proc. Natl. Acad. Sci. U.S.A. 117, 24205–24212 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lubeck E., Coskun A. F., Zhiyentayev T., Ahmad M., Cai L., Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods 11, 360–361 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raj A., van den Bogaard P., Rifkin S. A., van Oudenaarden A., Tyagi S., Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 5, 877–879 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dorsett Y., Tuschl T., siRNAs: Applications in functional genomics and potential as therapeutics. Nat. Rev. Drug Discov. 3, 318–329 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Stylianopoulos T., Jain R. K., Combining two strategies to improve perfusion and drug delivery in solid tumors. Proc. Natl. Acad. Sci. U.S.A. 110, 18632–18637 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yin H., Kanasty R. L., Eltoukhy A. A., Vegas A. J., Dorkin J. R., Anderson D. G., Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 5, 541–555 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Patel A., Schoen S. J., Arvanitis C. D., Closed-loop spatial and temporal control of cavitation activity with passive acoustic mapping. IEEE Trans. Biomed. Eng. 66, 2022–2031 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schoen S., Arvanitis C. D., Heterogeneous angular spectrum method for trans-skull imaging and focusing. IEEE Trans. Med. Imaging 39, 1605–1614 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun T., Zhang Y., Power C., Alexander P. M., Sutton J. T., Aryal M., Vykhodtseva N., Miller E. L., McDannold N. J., Closed-loop control of targeted ultrasound drug delivery across the blood–brain/tumor barriers in a rat glioma model. Proc. Natl. Acad. Sci. U.S.A. 114, E10281–E10290 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Arvanitis C. D., Crake C., McDannold N., Clement G. T., Passive acoustic mapping with the angular spectrum method. IEEE Trans. Med. Imaging 36, 983–993 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kovacs Z. I., Kim S., Jikaria N., Qureshi F., Milo B., Lewis B. K., Bresler M., Burks S. R., Frank J. A., Disrupting the blood–brain barrier by focused ultrasound induces sterile inflammation. Proc. Natl. Acad. Sci. U.S.A. 114, E75–E84 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gorick C. M., Mathew A. S., Garrison W. J., Thim E. A., Fisher D. G., Copeland C. A., Song J., Klibanov A. L., Miller G. W., Price R. J., Sonoselective transfection of cerebral vasculature without blood–brain barrier disruption. Proc. Natl. Acad. Sci. U.S.A. 117, 5644–5654 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cavalli F. M. G., Remke M., Rampasek L., Peacock J., Shih D. J. H., Luu B., Garzia L., Torchia J., Nor C., Morrissy A. S., Agnihotri S., Thompson Y. Y., Kuzan-Fischer C. M., Farooq H., Isaev K., Daniels C., Cho B.-K., Kim S.-K., Wang K.-C., Lee J. Y., Grajkowska W. A., Perek-Polnik M., Vasiljevic A., Faure-Conter C., Jouvet A., Giannini C., Rao A. A. N., Li K. K. W., Ng H.-K., Eberhart C. G., Pollack I. F., Hamilton R. L., Gillespie G. Y., Olson J. M., Leary S., Weiss W. A., Lach B., Chambless L. B., Thompson R. C., Cooper M. K., Vibhakar R., Hauser P., van Veelen M.-L. C., Kros J. M., French P. J., Ra Y. S., Kumabe T., López-Aguilar E., Zitterbart K., Sterba J., Finocchiaro G., Massimino M., Van Meir E. G., Osuka S., Shofuda T., Klekner A., Zollo M., Leonard J. R., Rubin J. B., Jabado N., Albrecht S., Mora J., Van Meter T. E., Jung S., Moore A. S., Hallahan A. R., Chan J. A., Tirapelli D. P. C., Carlotti C. G., Fouladi M., Pimentel J., Faria C. C., Saad A. G., Massimi L., Liau L. M., Wheeler H., Nakamura H., Elbabaa S. K., Perezpeña-Diazconti M., de León F. C. P., Robinson S., Zapotocky M., Lassaletta A., Huang A., Hawkins C. E., Tabori U., Bouffet E., Bartels U., Dirks P. B., Rutka J. T., Bader G. D., Reimand J., Goldenberg A., Ramaswamy V., Taylor M. D., Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell 31, 737–754.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pearson J. R. D., Regad T., Targeting cellular pathways in glioblastoma multiforme. Signal Transduct. Target. Ther. 2, 17040 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Targeting multidrug resistance in cancer—ProQuest; www.proquest.com/docview/223597030?accountid=11107.
- 61.Bajan S., Hutvagner G., RNA-based therapeutics: From antisense oligonucleotides to miRNAs. Cells 9, 137 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schmidt M. M., Wittrup K. D., A modeling analysis of the effects of molecular size and binding affinity on tumor targeting. Mol. Cancer Ther. 8, 2861–2871 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cho E. C., Xie J., Wurm P. A., Xia Y., Understanding the role of surface charges in cellular adsorption versus internalization by selectively removing gold nanoparticles on the cell surface with a I2/KI etchant. Nano Lett. 9, 1080–1084 (2009). [DOI] [PubMed] [Google Scholar]
- 64.Behzadi S., Serpooshan V., Tao W., Hamaly M. A., Alkawareek M. Y., Dreaden E. C., Brown D., Alkilany A. M., Farokhzad O. C., Mahmoudi M., Cellular uptake of nanoparticles: Journey inside the cell. Chem. Soc. Rev. 46, 4218–4244 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Baish J. W., Gazit Y., Berk D. A., Nozue M., Baxter L. T., Jain R. K., Role of tumor vascular architecture in nutrient and drug delivery: An invasion percolation-based network model. Microvasc. Res. 51, 327–346 (1996). [DOI] [PubMed] [Google Scholar]
- 66.Smith F. G., Deen W. M., Electrostatic effects on the partitioning of spherical colloids between dilute bulk solution and cylindrical pores. J. Colloid Interface Sci. 91, 571–590 (1983). [Google Scholar]
- 67.G. Truskey, F. Yuan, D. Katz, Transport Phenomena in Biological Systems (Pearson Prentice Hall, 2009), pp. 415–439. [Google Scholar]
- 68.M. Lim, V. Dharmaraj, B. Gong, T. Xu, Diffusive flux analysis of tumor vascular permeability for 3-helix-micelles in comparison to other nanoparticles. bioRxiv 708263 [Preprint]. 23 July 2019. 10.1101/708263. [DOI]
- 69.Pluen A., Boucher Y., Ramanujan S., Mc Kee T. D., Gohongi T., di Tomaso E., Brown E. B., Izumi Y., Campbell R. B., Berk D. A., Jain R. K., Role of tumor–host interactions in interstitial diffusion of macromolecules: Cranial vs. subcutaneous tumors. Proc. Natl. Acad. Sci. U.S.A. 98, 4628–4633 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.El-Kareh A. W., Secomb T. W., A mathematical model for comparison of bolus injection, continuous infusion, and liposomal delivery of doxorubicin to tumor cells. Neoplasia 2, 325–338 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thurber G. M., Weissleder R., A systems approach for tumor pharmacokinetics. PLOS ONE 6, e24696 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/18/eabf7390/DC1