

Profiling of active gene-regulatory DNA elements in brain cancer tissues illuminates disease biology and therapeutic targets.
Abstract
Molecular profiling of the most aggressive brain tumor glioblastoma (GBM) on the basis of gene expression, DNA methylation, and genomic variations advances both cancer research and clinical diagnosis. The enhancer architectures and regulatory circuitries governing tumor-intrinsic transcriptional diversity and subtype identity are still elusive. Here, by mapping H3K27ac deposition, we analyze the active regulatory landscapes across 95 GBM biopsies, 12 normal brain tissues, and 38 cell line counterparts. Analyses of differentially regulated enhancers and super-enhancers uncovered previously unrecognized layers of intertumor heterogeneity. Integrative analysis of variant enhancer loci and transcriptome identified topographies of transcriptional enhancers and core regulatory circuitries in four molecular subtypes of primary tumors: AC1-mesenchymal, AC1-classical, AC2-proneural, and AC3-proneural. Moreover, this study reveals core oncogenic dependency on super-enhancer–driven transcriptional factors, long noncoding RNAs, and druggable targets in GBM. Through profiling of transcriptional enhancers, we provide clinically relevant insights into molecular classification, pathogenesis, and therapeutic intervention of GBM.
INTRODUCTION
Glioblastoma (GBM) is the deadliest and most devastating brain cancer in adults (1). Despite aggressive multimodality treatment of maximal surgical resection, radiation, chemotherapy with temozolomide (TMZ), and adjuvant tumor-treating fields, tumor recurrence is nearly universal, and patient survival remains dismal (2–4). Thus, an urgent need exists to advance the molecular understanding of GBM and develop innovative diagnostic or therapeutic approaches to combat this lethal disease.
Multiomics characterization by The Cancer Genome Atlas Research Network (TCGA) has revealed biologically relevant alterations and pathways across GBM samples and subsequently guided the molecular classification of this heterogeneous disease (5–8). Transcriptional classification of bulk GBM has greatly improved the understanding of molecular pathobiology. Transcriptional subtypes demonstrate close association with prognosis, therapeutic response, genomic insults, and tumor microenvironment (8–10). One prevalent classification scheme that is based on unsupervised clustering of bulk tumor transcriptome involves four subtypes, namely, mesenchymal (MES), classical (CL), neural, and proneural (PN) (8). Further, by using GBM-intrinsic gene expression (GIE) signature to reduce the transcriptional noise from GBM-associated stroma, classification of isocitrate dehydrogenase (IDH)–wild-type tumors is refined as three GIE subtypes (GIE-MES, GIE-CL, and GIE-PN) (9). GIE subtypes reflect heterogeneous compositions of GBM cells in distinct states among bulk tumors (11). At the single-cell level, four cellular states have been identified in GBM cells, resembling neural progenitors, oligodendrocyte progenitors, astrocytes, and MES cells, respectively (11). Although genomic defects such as aberrations in cyclin-dependent kinase 4 (CDK4), platelet-derived growth factor receptor alpha (PDGFRA), epidermal growth factor receptor (EGFR), and neurofibromin 1 (NF1) have been shown to favor the development of individual cellular states and transcriptional subtypes, epigenetic mechanisms governing transcriptional diversity among GBM subtypes stay largely unknown. Besides, the regulatory networks that dictate GBM identity and malignant traits remain inadequately addressed.
Active regulatory elements (AREs), especially enhancers and large clustered enhancers [known as super-enhancers (SEs)] (12), recruit sequence-specific transcription factors (TFs), chromatin regulatory machinery, and transcriptional apparatus to regulate tissue/disease-specific gene expression. As exemplified by application of DNA methylation signatures to fine-tune GBM molecular subtypes (6), integration of epigenetic chromatin information will provide critical insights into transcriptional heterogeneity and classification. Seminal studies of GBM AREs mainly focused on ex vivo expanded GBM cells, including patient-derived GBM-propagating cells (GPCs) (13–18), while tissue-centric analysis in the light of tissue complexity and tumor heterogeneity is still lacking. To this end, we performed in-depth chromatin immunoprecipitation followed by massive parallel sequencing (ChIP-seq) analysis for H3K27ac, a histone modification to annotate regions of actively transcribed chromatin (19), in a large collection of samples comprising 95 GBM tissues, 1 oligoastrocytoma, 12 normal brain specimens, and 38 cell lines. Our efforts to build a comprehensive compendium of clinically relevant active regulatory regions in GBM facilitated discovery of tumor/subtype-related enhancer-gene regulation, identification of core TF circuitries, and exploration of previously unrecognized oncogenic addiction, thereby advancing the understanding of transcriptional diversity and disease heterogeneity.
RESULTS
Characteristics of sample cohort and data availability
H3K27ac signals are deposited across transcriptionally active chromatin. To map genome-wide AREs in human GBM, we first analyzed the H3K27ac ChIP-seq profiles in a series of 50 fresh-frozen tumor specimens (49 GBM and a grade 3 oligoastrocytoma), 20 patient-derived GPCs, and 5 established GBM cell lines, together with a control set of samples including 12 normal brain tissues, 12 neural stem/progenitor cells (NPCs), and 1 line of primary human astrocytes [see detailed sample information in table S1 (A and B)] (14–18, 20–27). To facilitate transcriptional classification and detection of disease-relevant mutations/aberrant transcripts, a cognate transcriptomic-sequencing [RNA sequencing (RNA-seq)] analysis was performed for 48 tumors (i.e., 47 GBM and a grade 3 oligoastrocytoma) and 3 normal brain tissues. Our discovery cohort of primary GBM specimens (defined as cohort 1) is inclusive of all three tumor-intrinsic gene expression subtypes (GIE-MES, n = 21; GIE-CL, n = 12; and GIE-PN, n = 14). Among those, 40 GBM cases (number of uniquely mapped RNA-seq reads, >15 million) were enrolled in the study of GBM/subtype-specific gene expression (table S1A).
Landscape of AREs in GBM
In total, 4,715,512 and 1,591,664 H3K27ac peaks were captured in the discovery cohort of tumor and nontumor tissues/cells, respectively. These actively regulated regions were classified into either proximal promoters (containing 855,318 peaks) or putative enhancers (containing 5,451,858 peaks). More than 88% of promoter-associated H3K27ac peaks were conserved between normal/tumor brain and their respective cell line counterparts, while enhancer-associated signals showed greater variations (fig. S1A). Unsupervised hierarchical clustering of the top 5867 most-varying loci (mean, ≥10; median absolute deviation, >1) among our dataset (n = 100) revealed six discrete clusters (fig. S1, B to D, and table S1, A and B). The majority (47/50) of tumor tissue samples were classified into three clusters, one of which coenriched normal brain samples. About 90% of cell line samples including established GBM cell lines, GPCs, NPCs, and primary astrocytes were classified into the other three clusters, while a few cell lines were also found in clusters that were mainly composed of either normal or tumor tissues (fig. S1D). In general, tissues samples and cell line samples showed a low degree of coclustering, highlighting the necessity of chromatin profiling of primary tumor biopsies to understand its dysregulation in the context of complex tissue/tumor microenvironment.
To extend current knowledge of GBM chromatin, which is primarily derived from cell line–based work, we next focused our analysis on primary tissues. To begin with, we performed a pairwise H3K27ac–to–RNA-seq data analysis across tissue samples in this study using either all detected H3K27ac sites or the signals pertaining to gene promoters. Both types of chromatin signals showed a stronger correlation with the transcriptome from the corresponding sample (self) than that from other samples (fig. S1E), suggesting a robust sample concordance and no sample mix-ups. Our tissue-centric analysis then refined the clustering of GBM and normal brain samples using top-variant AREs (mean, ≥10; median absolute deviation, >1; n = 1107) among them. On the basis of ARE consensus clustering, GBM samples in cohort 1 can be categorized into three groups, namely, AC1 (n = 32), AC2 (n = 8), and AC3 (n = 9) (Fig. 1, A and B; fig. S1, F and G; and table S1A). The AC1 subclass included most of the GBM cases belonging to GIE-MES and GIE-CL, while more than 80% of GIE-PN tumor samples were classified into either AC2 or AC3. Convergence of “transcriptionally different” GIE-MES and GIE-CL samples on AC1 subtype indicates (i) a relatively high degree of similarity in the top-variant H3K27ac loci between the two groups and (ii) a substantial remodeling of ARE topography, which separates them from normal brain and GIE-PN tumors.
Fig. 1. Landscape and clustering of active regulatory elements in GBM tissues.
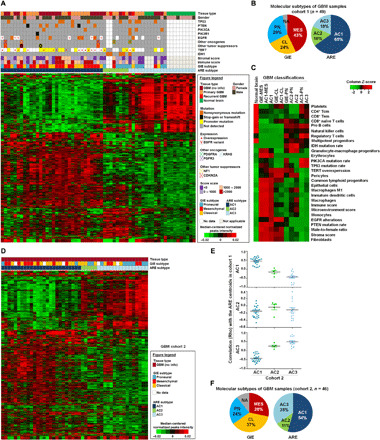
(A) Unsupervised hierarchical clustering of 1107 most-varying H3K27ac peaks (mean, ≥10; median absolute deviation, >1) across 49 GBM tissues (cohort 1) and 12 normal brain tissues. Top: The clinical, mutational, and molecular data of each sample. (B) Molecular classification of GBM samples in cohort 1 based on GIE subtypes and ARE subtypes. (C) Heatmap of differential enrichments of immune cell signatures, stromal cell signatures, gene mutations, and clinical parameters among normal brain tissues and various molecular subtypes of GBM. (D) ARE clustering of an independent cohort of 46 GBM samples (cohort 2) based on the same set of 1107 variant elements that were identified from cohort 1. (E) Spearman correlation of ARE status in cohort 2 samples with each subtype centroids of cohort 1. (F) Summary of molecular GIE subtypes and ARE subtypes among cohort 2. AC, active regulatory element-based cluster; ARE, active regulatory element; CL, classical subtype; GIE, GBM-intrinsic gene expression; MES, mesenchymal subtype; PN, proneural subtype.
Moreover, on the basis of the analysis of cohort 1, AC1 subtype showed higher microenvironment and stromal scores, male-to-female ratio, and alterations in telomerase reverse transcriptase (TERT), EGFR, and phosphatase and tensin homolog (PTEN) (Fig. 1, A and C, and table S1, A and C). In contrast, AC2 and AC3 subtypes enriched mutations of IDH1 and PDGFRA (Fig. 1, A and C, and table S1, A and C). Inspired by substantial interaction between the ARE and GIE subtypes, we sought to reconcile these two schemes and allocated further more than 85% of GBM cases (cohort 1) into four ARE-GIE integrative (AGI) subgroups (table S1A), inclusive of AC1-MES (n = 19), AC1-CL (n = 10), AC2-PN (n = 5), and AC3-PN (n = 6). Univariate analysis affirmed the correlation of IDH mutation with AC2-PN (P < 0.01, Fisher’s exact test) and EGFR alterations with AC1-CL (P = 0.023, Fisher’s exact test). In line with the notion of massive remodeling of GBM microenvironment (28), gene signature–based portraying of tissue cellular landscape (by xCell) revealed subtype-associated immune/stromal cell composition, such as enrichment of monocytes, macrophages, and immature dendritic cells in AC1-MES, pericytes in AC1-CL, and regulatory T cells in AC3-PN (Fig. 1C, fig. S1H, and table S1C) (29). Notably, GIE-MES tumors generally had lower sample purity than GIE-CL and GIE-PN (fig. S1I), while ARE clustering of MES and CL tumors was not influenced by variations in their purity. Hence, ARE subtype analysis and the AGI classification add a new layer of epigenomic information with interconnections to GBM clinical features, tumor microenvironment, genomic abnormalities, and transcriptional subclasses.
To verify our ARE classification scheme in cohort 1, we included additional 46 cases of GBM tissues as an independent cohort (cohort 2; table S1D), of which 41 cases had cognate ChIP-seq and RNA-seq data (GIE-MES, n = 13; GIE-CL, n = 17; and GIE-PN, n = 11) (18). Similar observations were made in cohort 2 (Fig. 1, D to F, and table S1D), supporting a conservative pattern of three ARE subtypes among GBM samples.
Rank ordering of clustered transcriptional enhancers in GBM subtypes
SE domains, characterized by excessive loading of H3K27ac on clustered enhancers, are shown to drive the expression of disease-promoting genes and cell identity regulators (30, 31). In total, we identified more than 85,000 SEs across 100 samples in the discovery cohort (table S1E; mean SE numbers: 848 in GBM tissues from cohort 1, 985 in normal brain tissues, 773 in GBM cells, and 966 in non-GBM cells). SE analysis substantially marked actively transcribed genes (fig. S2A). Inspired by seminal studies that small insertion variants in SE regions promote activation of oncogenes (32, 33), we sought to identify “recurrent enhancer-associated variants (RENVARs)” in primary GBM tissues based on variant discovery from H3K27ac reads inside both SE and typical enhancer (TE) regions (see Materials and Methods). We predicted 80 putative RENVARs (table S1F), of which 21 were recorded as noncoding somatic mutation/indels in the Catalogue of Somatic Mutations in Cancer (COSMIC) database (e.g., single nucleotide variants at SE of PHLDA1; chr12:76030506) and 59 were not reported previously in either COSMIC or dbSNP (e.g., dinucleotide variants at SE of TGIF1; chr18:3449064). This list of RENVARs represents a valuable resource for further exploration of noncoding genomic variants in regulating gene expression during GBM development.
Next, we investigated topographies of SE domains that were associated with diverse molecular entities of GBM. To compare relative loading of H3K27ac among various GBM subtypes, we compiled all H3K27ac peaks from the same subtype and ranked stitched meta-enhancers by average ChIP-seq signals (Fig. 2A). Meta-SE signals showed a stronger correlation with the expression levels of target genes than promoter-transcription start site (TSS) signals, although both types of signals showed positive Spearman rank correlations (fig. S2B). Subtype-associated meta-SE genes showed a generally higher expression in that particular subtype when compared with the rest of tumors (fig. S2C), while substantial overlaps of meta-SE domains among GBM subtypes compromised the pairwise comparison (fig. S2, C and D). In parallel, about half of nonrepetitive meta-SE–associated genes displayed subtype-specific engagement of SE domains, including those in proximity to signature genes of GIE subtypes such as TGFBI, SLC4A4, and EYA2 (fig. S2, D and E). To explore the connection between chromatin architecture and transcriptional heterogeneity, we focused our analysis on AGI subtypes. At the level of meta-SE–associated genes, 13% were commonly present in all AGI subtypes despite variations in enhancer ranking (fig. S2D). AC1-MES and AC1-CL showed the highest degree of similarity (45%; Fig. 2B). In contrast, AC2-PN and AC3-PN differed considerably at the SE landscape and SE-associated pathways (Fig. 2B and fig. S2, D and F), although both expressed a PN transcriptional signature. Subtype-specific meta-SE genes were overrepresented in key pathways underlying disease biology, including stimulus response, leukocyte activation and cell migration in AC1-MES, and angiogenesis in AC1-CL, as well as the neurogenesis pathway in AC3-PN (Fig. 2C). To complement the study of overall H3K27ac deposition, we interrogated frequency of individual SEs across each subtype. Our analysis identified 930 target genes that engaged subtype-enriched SE domains with statistical significance [odds ratio (OR), ≥2; P < 0.05, Fisher’s exact test; table S1G]. Intersection analysis further suggested activation of various phenotypic markers and druggable targets by subtype-enriched SEs (see details in table S1G). For instance, AC1-MES tumors showed biased enrichment of SE domains in two reported markers of MES GBM cells (34), namely, CD44 and CHI3L1, as well as several druggable targets such as LIMK2 and NAMPT (fig. S2G). Other potential actionable subtype-enriched SE targets included NUAK2 and WEE1 in AC1-CL, and FGFR2 in AC3-PN (fig. S2G and table S1G). Echoing the observations from meta-SE genes, ontological analysis of subtype-enriched SE targets in AC1-MES samples reaffirmed their unique and robust association with stimulus response, cell migration, and immune response (fig. S2H), suggesting hyperactivation of these pathways via subtype-specific enhancer reprogramming. Subtype-enriched SE genes in AC3-PN were strongly enriched in neurogenesis and nervous system development pathways (fig. S2H), suggesting that active and persistent engagement of neurogenic enhancers shapes AC3-PN identity. Together, our integrative analysis delineates differential contribution of enhancer domains to GBM heterogeneity and subtype identity.
Fig. 2. Subgroup-associated SE domains and disease pathways in GBM.
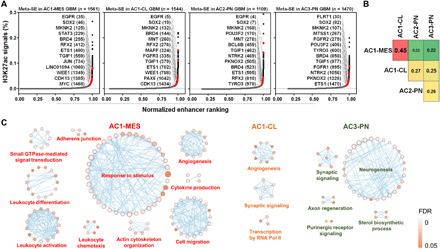
(A) Relative rank of stitched H3K27ac ChIP-seq signals in integrative subtypes of GBM samples. Representative meta-SE–associated genes and their rankings in parentheses are highlighted. (B) Pairwise comparison of similarity in meta-SE–associated genes among integrative subtypes of GBM samples. (C) Ontological enrichment of subtype-specific meta-SE–associated genes in GBM tissues. No significant pathways (Benjamini-Hochberg FDR, <0.05) were enriched in AC2-PN–specific meta-SE targets.
Asymmetrical engagement of tumor-specific SE domains in transcription regulators
To identify transcriptional enhancer domains that are associated with tumor specificity, we compared stitched H3K27ac signals between GBM (cohort 1) and normal brain tissues (Fig. 3A). Over those stitched enhancer regions showing hyperacetylation in GBM, preexisting H3K27ac signals were frequently detected in normal brain tissues. All of the 1806 GBM-associated meta-SEs were overlapped with either SE or TE domains in normal brain samples. At expression level, GBM- and normal-associated meta-SE genes showed biased expression in respective tumor and normal samples (fig. S3A). Genes driven by GBM-unique meta-SEs showed a greater degree of up-regulation in GBM than those associated with common meta-SEs (fig. S3B). These data suggest that GBM-associated SEs are developed from preexisting, developmental enhancers, prompting us to explore further enhancer remodeling during GBM development. More than 68% of meta-SE targets were exclusive to either GBM or normal brain tissues (Fig. 3B), suggesting a substantial reshaping of SE landscape during GBM development. In line with this, gene ontology (GO) and Reactome enrichment analysis of meta-SE–associated genes indicated sharp differences between GBM and normal brain samples. In GBM tissues, meta-SE–associated genes were strongly enriched in receptor tyrosine kinase signaling (e.g., EGFR) and regulatory region nucleic acid binding (e.g., RFX2 and TGIF1) pathways (Fig. 3, C and D, and fig. S3C). In normal brain tissues, functions of meta-SE–associated genes were preferentially implicated in nervous system development (fig. S3C).
Fig. 3. GBM-promoting transcription factors preferentially engage tumor-enriched super-enhancers.
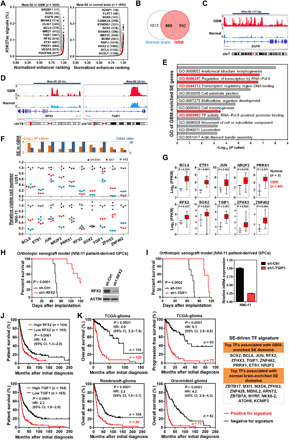
(A) Relative rank of stitched H3K27ac ChIP-seq signals in GBM (cohort 1) and normal brain tissues. Representative meta-SE–associated genes and their rankings in parentheses are highlighted. (B) Venn diagram showing common and sample type–specific meta-SE–associated genes in GBM and normal brain tissues. (C and D) Differential deposition of H3K27ac signals across regulatory regions of EGFR, RFX2, and TGIF1. (E) Ontological analysis of GBM-enriched SE genes. (F) Association of GBM-enriched SE domains with TFs and effect of shRNA-mediated silencing of top SE-associated TFs on cell viability of U251 GBM cells and NNI-11 GBM-propagating cells. (G) Elevated expression of TFs driven by tumor-enriched SEs in GBM samples (cohort 1). Student’s t test (two tailed) was applied. Boxplots represent the 25th and 75th percentiles, with midlines indicating the median values and whiskers extended to the lowest/highest values. (H and I) Effect of shRNA-mediated silencing of either RFX2 (n = 8) or TGIF1 (n = 7) in NNI-11 cells on survival of recipient mice with intracranial xenografts. Before intracranial implantation, Western blot and qPCR analyses were used to verify effective silencing of RFX2 and TGIF1, respectively. (J) Prognostic potentials of RFX2 and TGIF1 in Rembrandt cohort of glioma patients. (K) Prognostic potentials of an SE-driven TF signature in three independent cohorts of glioma patients. Log-rank test is applied in (H) to (K). HR, hazard ratio; CI, confidence interval.
Meanwhile, in light of disease heterogeneity, we surveyed SE topography of individual samples from cohort 1 and detected 766 GBM-associated SE domains (prevalence in GBM, >15%; OR, ≥2), of which 58% were absent in normal (table S1H). Top GBM-enriched SEs (n = 198; OR, ≥2; P < 0.05, Fisher’s exact test) were associated with many known Cancer Gene Census genes (table S1I) and prominent GBM drivers including EGFR and SOX2 (25, 35–37). Echoing the observations from the meta-SE approach, GO analysis of GBM-enriched SEs uncovered a robust enrichment of genes involved in transcriptional regulation (Fig. 3E), indicative of extensive rewiring of transcriptome. Next, to explore functional relevance of these transcriptional regulators, we performed a loss-of-function study of the top 10 GBM-enriched SE-driven TFs (OR, >10; P < 0.05, Fisher’s exact test), which showed concordantly overexpression in our discovery cohort (Fig. 3, F and G). Depletion of each TF attenuated the viability of both established GBM cells and GPCs (Fig. 3F and fig. S3, D and E). Expression of the majority of the abovementioned TFs (e.g., BCL6, ETS1, SOX2, JUN, PRRX1, and TGIF1) was consistently elevated in GBM samples relative to nontumor brain across three independent datasets (i.e., Gravendeel, Rembrandt, and TCGA-GBM) (6, 38, 39), implicative of a strong clinical relevance of SE-driven TFs (fig. S3, F to H). Orthotopic xenograft experiments using NNI-11 patient-derived GPCs demonstrated further unrecognized roles of RFX2 and TGIF1, two of the examined SE-driven TFs with high OR in tumor, in promoting GBM tumorigenicity (Fig. 3, H and I). Silencing of either RFX2 or TGIF1 in GPCs conferred a marked survival benefit on recipient immunocompromised animals. The deleterious impact of RFX2 and TGIF1 knockdown on tumor growth was independently verified in a subcutaneous xenograft model using U251 GBM cells (fig. S3I). In addition, transcriptional level of either RFX2 or TGIF1 was associated adversely with the prognosis of patients with glioma (Fig. 3J). To extend these findings, we generated an SE-regulated TF signature based on their frequency and enrichment in either GBM or normal tissues (Fisher’s exact test; see detailed criteria in table S1J). This signature consisted of top TFs with differential SE engagement (GBM enriched, n = 10; normal brain enriched, n = 12) and demonstrated its capability to stratify various clinical elements including age, pathology, and survival (Fig. 3K and table S1, J to L). Together, these observations suggest that GBM engages specific SE domains in productive expression of oncogenic TFs to foster malignant progression.
SE-driven core TFs and regulatory circuitries in GBM
Along with preferential engagement of GBM-specific SE domains in tumor-promoting TF genes (Fig. 3E), motifs of many SE-driven TFs (e.g., SOX2, JUN, and RFX2) were highly enriched in SE regions (fig. S4A). Consistently, SE-driven TFs in each GBM subtype had extensive binding sites across the meta-SE regions of respective subtypes (fig. S4B). To elucidate transcriptional homeostasis underlying subtype identity, we reversely engineered core TF network based on SE engagement and regulatory connectivity, using a model of core transcriptional regulatory circuitry (40–42). Our computational analysis uncovered groups of SE-driven master TFs, which collaborated on interconnected regulatory circuitries in each subtype (Fig. 4A and fig. S4, B to D). Notably, SOX2, NR2F1, IRF9, FOXO3, ETV6, and RARA were shared as common master TFs among all GBM subtypes and normal brain tissues, implicating their general association with lineage/tissue specificity. ETS1, RFX2, PRRX1, FOXK1, IRF1, NR2F2, and MEIS2 served as tumor-specific core TFs in at least three subtypes. PKNOX2, BCL6, MAZ, and POU3F2 occurred preferentially in circuitries among PN samples. Notably, targeting bromodomain and extraterminal domain (BET) proteins by dBET6 [a CRBN (cereblon)-dependent degrader of BET proteins] effectively disrupted the expression of AC1-MES core TFs (e.g., TEAD1, SOX9, MYC, and NFIL3) in an MES subtype model U87 cells (Fig. 4B) (43), suggesting a critical role of BET proteins in maintaining GBM transcriptional regulatory circuitries. Coexpression analysis further revealed that at least two cliques of closely correlated TFs in each subtype: a “stemness” module containing SOX2, NR2F1, and recurrent cooperating TFs that are involved in neuronal stem cell maintenance, and a “proliferation” module containing ETS family, AP-1 factors, and additional members with known functions in cell cycle and angiogenesis (Fig. 4, C and D, and fig. S5). These modular TFs co-operated with diverse core TFs to establish and maintain the GBM molecular subgroup identity. Together, our computational reconstruction depicts a blueprint of core TFs and their interconnected regulatory circuitry for each integrative molecular subtype.
Fig. 4. Subgroup-associated core regulatory circuitries in GBM.
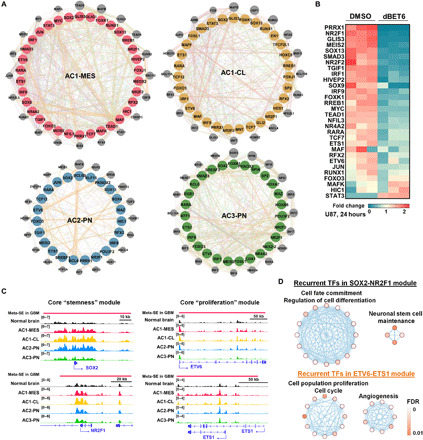
(A) Core interconnected transcriptional regulatory circuitries in each integrative subtype of GBM tissues. Core TFs are highlighted in colored circles. Key interactive partners of core TFs are indicated in gray circles. (B) Heatmap showing response of AC1-MES core TFs to a BET protein degrader (dBET6, 500 nM) treatment in U87 cells (GSE99181; transcripts per million, > 0.1). (C) Enhanced deposition of H3K27ac signals across regulatory regions of SOX2, NR2F1, ETV6, and ETS1 in GBM tissues. (D) Pathway enrichment of recurrent TFs in two coexpressed core modules across GBM subtypes.
Catalog of SE-associated long noncoding RNAs in GBM
Widespread transcription from the non–protein coding genome prompted us to explore potential involvement of SEs in the regulation of long noncoding RNAs (lncRNAs). At least 15% of GBM-associated SE domains were mapped primarily to annotated lncRNAs including antisense RNAs (fig. S6A). After filtering out those with either undetectable or low [mean fragments per kilobase of transcript per million mapped reads (FPKM), <0.5] expression in our discovery cohort, we identified a list of nine SE-associated lncRNAs whose expression was up-regulated selectively in GBM tissues (for SE: OR > 2, Fisher’s exact test; for expression: P < 0.05, Student’s t test) (Fig. 5A). Among these, LINC01094 and MIR99AHG (LINC00478) demonstrated elevated H3K27ac deposition in their regulatory regions in GBM samples except for the AC3-PN subtype (Fig. 5B). Genetic depletion of LINC01094 and MIR99AHG, both of which showed glioma-specific expression compared with other human malignancies, impaired GBM cell viability and tumorigenicity (Fig. 5, C to F, and fig. S6B). Defective expression of either LINC01094 or MIR99AHG in GBM cells compromised tumor growth in subcutaneous xenograft models (fig. S6B) and conferred a survival benefit to recipient animals in independent orthotopic xenograft models (Fig. 5E). Next, with a focus on LINC01094, we showed further that both BET proteins and disease-relevant core TFs (including MYC, ETV6, and NR2F2) contributed to the productive expression of this lncRNA in GBM cells (Fig. 5, G and H). Hence, LINC01094 represents an example of oncogenic lncRNA whose disease-specific expression is regulated through tumor-associated core TFs and SE domain. Together, these data functionally annotates LINC01094 and MIR99AHG as GBM-promoting lncRNAs with disease specificity. SE-associated lncRNAs represent an additional source of disease dependencies and biomarkers.
Fig. 5. SE-driven lncRNAs in GBM.
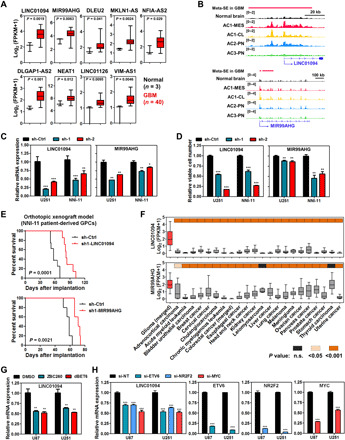
(A) Elevated expression of SE-driven lncRNAs in GBM samples (cohort 1). Student’s t test was applied. (B) Differential deposition of H3K27ac signals across regulatory regions of LINC01094 and MIR99AHG in GBM subtypes and normal brain tissues. (C to E) Effect of shRNA-mediated silencing of LINC01094 and MIR99AHG on target expression (C) and cell viability (D), and survival of recipient mice with intracranial transplantation of NNI-11 cells (E). (F) Differential expression of LINC01094 and MIR99AHG transcripts among various human cancers. (G) Effect of BET protein degraders on the expression of LINC01094. U87 and U251 GBM cells were treated with either ZBC260 or dBET6 (200 nM, 8 hours) before harvest. (H) Effect of siRNA-mediated silencing of core TFs on the expression of LINC01094. GBM cells were transfected with indicated siRNAs (48 hours) and subjected to qPCR analysis. Either one-way ANOVA or t test was used for analysis of significance. Error bars in (C), (D), (G), and (H) represent SEM, n = 3. Log-rank test was applied for survival analysis in (E). Boxplots in (A) and (F) represent the 25th and 75th percentiles, with midlines indicating the median values and whiskers extended to the lowest/highest values. n.s., not significant.
Engagement of GBM-associated SE domains in druggable genome
In line with the notion that GBM-specific and GBM-promoting genes are frequently associated with SE domains, we hypothesize that analysis of SE-associated druggable genes favors discovery of actionable cancer dependencies. To identify potential therapeutic targets whose expressions are regulated by GBM-associated SEs, we interrogated the common hits between SE-associated protein-coding genes and the druggable genome based on DGIdb (Drug Gene Interaction Database). More than 160 candidates were retrieved from this analysis, including the aforementioned EGFR and many others with actionable small molecules, e.g., BRD4, CDK13, WEE1 and MKNK2/MNK2 (Fig. 6A and table S1M). Immunoblot assay also verified the prevalent up-regulation of BRD4, CDK13, WEE1, and MNK2 proteins in GBM tissue lysates (fig. S6C). Given the profound function of BET bromodomain proteins in maintaining SE output and gliomagenesis (43), engagement of tumor-specific SE domains to BRD4 suggests a feed-forward mechanism of transcriptional dysregulation in a subset of GBM cases. A potent BET protein degrader, ZBC260 (44), exerted a picomolar to low nanomolar antiproliferative efficacy against both GPCs and established GBM cell lines (fig. S6D). In addition, we demonstrated the preclinical anti-GBM activities of THZ531 (CDK12/13 inhibitor), Adavosertib/AZD1775 (WEE1 inhibitor), and CGP57380 (MNK1/2 inhibitor) (fig. S6D). To strengthen translational potential of our findings, we tested a recently developed MNK inhibitor (ETC-168) with desirable potency and brain penetrance (Fig. 6, B to I) (45). ETC-168 treatment blocked both GBM cell proliferation and GPC self-renewal (Fig. 6, C to E). ETC-168 treatment induced cleavage of both caspase-3 and poly(ADP-ribose) polymerase 1 (PARP1) in GPCs, which was accompanied by a sharp down-regulation of p-EIF4E (Fig. 6F). Moreover, ETC-168 displayed an additive effect with TMZ to inhibit cell viability (Fig. 6, F and G). Similarly, ETC-168 treatment was able to reduce p-EIF4E in orthotopic xenografts and cooperated with TMZ to elicit cleavage of PARP1 in vivo (Fig. 6H). Despite limited survival benefit from ETC-168 as a single-agent regimen in vivo, combination of MNK inhibitor and TMZ not only prolonged the overall survival of tumor-bearing mice but also resulted in a cure of disease as evidenced by the tumor-free animals at the experimental end point (Fig. 6I). Promising anti-GBM activity of the drug combo of MNK inhibitor and TMZ encourages further clinical translation and repositioning of relevant compounds in trials (46, 47). In addition, genetic depletion of either MKNK1 (MNK1) or MKNK2 (MNK2) attenuated concordantly both GBM cell expansion and tumorigenicity, with MNK2 knockdown conferring a greater survival benefit on recipient mice (Fig. 6, J and K, and fig. S6, E and F). Together, these data suggest valid engagement of tumor-associated SE domains in druggable GBM addictions.
Fig. 6. Analysis of GBM-enriched SEs uncovers actionable drug targets.
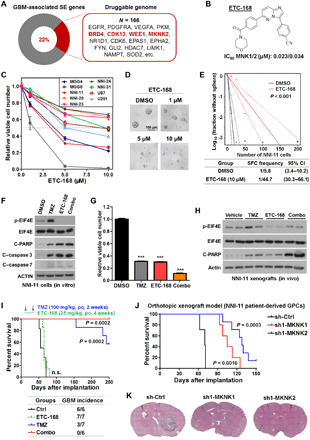
(A) Engagement of GBM-enriched SE domains in druggable genome. (B) Chemical structure and biochemical activity of an MNK kinase inhibitor ETC-168. (C) Effect of ETC-168 on viability of GBM cells and GBM-propagating cells. (D and E) Effect of ETC-168 treatment on sphere formation capability (SFC) of GBM-propagating cells (i.e., NNI-11). (F) Effect of ETC-168 (5 μM) and TMZ (100 μM) on expression of downstream targets in NNI-11 cells after 24-hour treatment. (G) Effect of ETC-168 (5 μM) and temozolomide (TMZ; 100 μM) on viability of NNI-11 cells after 72-hour treatment. One-way ANOVA was used for analysis of significance. (H) Effect of ETC-168 [25 mg/kg, orally (po), twice a day (bid)] and TMZ (100 mg/kg, po, once a day (qd)] on expression of indicated proteins in orthotopic xenograft tumors (NNI-11) after 24-hour treatment. (I) Effect of ETC-168 and TMZ treatment on both survival of recipient mice with intracranial transplantation of NNI-11 cells and the tumor incidence at the end point of experiment. Log-rank test was applied for survival analysis; n = 6 or 7. (J and K) Effect of shRNA-mediated silencing of either MKNK1 or MKNK2 in NNI-11 cells on survival of recipient mice (J) and orthotopic tumor expansion 2 months after implantation (K). Log-rank test was applied for survival analysis in (J); n = 5 or 7. Error bars in (C) and (G) represent SEM; n = 3. IC50, half maximal inhibitory concentration.
DISCUSSION
In this study, we have analyzed the landscape of transcriptionally active chromatin across a series of 95 fresh-frozen GBM specimens, 12 normal brain samples, and 38 cell lines. Integrative analysis of differentially regulated AREs, especially SE domains, informs on previously unrecognized oncogenic dependencies, molecular classification, and epigenetic regulation of subtype identity. Our data also serve as a valuable resource to the research community to explore epigenetic mechanisms underlying GBM and normal brain activities.
Through profiling of transcriptionally active chromatin, our study narrows the gap between epigenetic dysregulation and transcriptional diversity. Our unbiased analysis of AREs and gene expression in primary tissues supports a generally positive correlation between overall enhancer signals and expression of target genes. However, differences in clustering were observed between ARE classification and expressional classification, although both measured active gene expression. Our data reveal an additional layer of GBM heterogeneity at the level of AREs, complementing the current molecular classification, which is largely dependent on gene expressional signatures. Our integrative analysis classified GBM into four integrative molecular subtypes, namely, AC1-MES, AC1-CL, AC2-PN, and AC3-PN. Unexpectedly, AC1-MES and AC1-CL tumors had a similar ARE architecture including SE topography (Fig. 1, A and B), despite showing distinct gene expression clustering and tumor purity. Hence, H3K27ac-based clustering analysis of MES and CL tumors was unlikely confounded by their tumor purity. However, coclustering of samples upon unsupervised hierarchical clustering of AREs does not equal to being “epigenetically identical” between AC1-MES and AC1-CL. GO analysis of subtype-biased SE targets indicated a marked difference in pathway enrichment between AC1-MES and AC1-CL. Divergence of transcription between AC1-MES and AC1-CL is likely contributed by subtype-specific enhancers (Fig. 2C and fig. S2H), as well as additional mechanisms involving tumor microenvironment, cellular heterogeneity (11, 48), and posttranscriptional regulation of mRNA (49). In support of this notion, AC1-MES tumors showed the highest immune scores and selective enrichment of SE genes in immune response pathways, while AC1-CL tumors harbored gene signatures of pericytes and angiogenesis pathway. Single-cell transcriptome analyses further support strong immune cell infiltration and inflammatory reactions within MES tumors (48). AC2-PN and AC3-PN differed substantially in ARE topography, despite their resemblance in a PN transcriptional signature. The identification of “epigenetically divergent” PN samples implicates distinct pathobiology, which is masked by their similarity in transcriptional signatures. ARE topography of AC3-PN resembled generally that of normal brain (Fig. 1A). AC3 GBM and normal brain samples were also coclustered with one grade 3 oligoastrocytoma (fig. S1D and table S1A), suggesting a tendency toward lower malignancy and a better prognosis. SE-associated genes in AC3-PN were skewed toward stimulus response, cell projection, and cell motility pathways when compared with those in AC2-PN. Strong enrichment of neurogenesis pathways in AC3-PN tumors suggests inheritance of a neurogenic, active chromatin state during transformation. Hence, malignant traits of AC3-PN tumors are established by integration of PN enhancers and associated core TFs (e.g., NFIA, EGR1, CDX1, PKNOX2 and NKX2-2). Preferential enrichment of IDH mutations in AC2-PN may underpin its unique ARE features. Nevertheless, the existence of IDH–wild-type AC2-PN samples implicates that the H3K27ac landscape of AC2-PN can be established by mechanisms independent of IDH mutation. As IDH-mutant gliomas have been recognized by their distinct DNA methylation profiles, chromatin state, and aberrant chromosomal topology compared with IDH–wild-type counterparts (7, 50–52), IDH mutation induces both DNA hypermethylation and chromatin dysfunction and likely co-operates with dysregulated histone acetylation to shape AC2-PN subtype identity. These data, in turn, predict that combinatory targeting of IDH and histone acetylation axis (e.g., agents against HDAC and BET bromodomain proteins) is a promising strategy to impair epigenetic dependency of IDH-mutant gliomas.
Our ARE profiling of primary tumor biopsies provides clinically relevant chromatin information. Integrated classification of GBM revealed strong correlation of ARE status with various pathological and genomic features, implicating its strong clinical relevance. Study of differentially engaged SEs uncovered both tumor-specific and subtype-specific regulation of target genes, including TFs (e.g., RFX2 and TGIF1), lncRNAs (e.g., LINC01094 and MIR99AHG), and druggable targets (e.g., MKNK2, BRD4, and WEE1). Although the precise cell of origin of each GBM tumor remains unknown, various animal genetic studies have revealed oncogenic transformation of neural stem cells, early progenitors, astrocytes, and neurons into malignant glioma (53). Because neurons and glial cells make up the majority of cells in the adult brain, normal brain tissues from anatomical regions with a relatively higher incidence rate of GBM can serve as surrogate controls for GBM samples. TFs are overrepresented among GBM-specific SE targets, highlighting the extensive remodeling of transcriptional network. In line with this, TF-centric SE signature genes that were extracted from GBM can faithfully stratify patient survival, tumor grade, and other molecular features of gliomas in patient cohorts. Reverse-engineering of core transcriptional circuitry outlined at least two conserved core modules of master TFs governing either stemness or proliferation across all GBM. Hence, two cliques of master TFs collaborate on propagation of GBM cells. Additional cooperating SE-driven TFs then extend the core regulatory network and skew transcriptional output toward a particular cell state and disease subtype. Moreover, the conservation and modular composition of stemness and proliferation TFs among GBM samples coincide with the phenotypical dichotomy of GPCs (13). GBM cellular states ranging from PN to MES and from noncycling to cycling may reflect the gradient activities of two modular TFs. Our study suggests that therapeutic agents targeting BET bromodomain proteins are promising modalities to break core regulatory circuitries. We further report a strong anti-GBM activity of a BET protein degrader ZBC260.
In an effort to target additional SE-associated druggable targets, we tested an MNK inhibitor ETC-168 against GBM cells. Our data support the brain penetrance of ETC-168 based on the observation that ETC-168 reduced p-EIF4E in orthotopic tumors. However, we observed a stronger in vitro anti-GBM activity of ETC-168 than in vivo. This discrepancy is likely caused by reduced bioavailability of ETC-168 in the animal brain, as it exerted a relatively weaker inhibitory effect on p-EIF4E in GBM cells in vivo, when compared with in vitro treatment. Albeit future efforts are required to develop optimal delivery strategy by improving dosing and schedule of single-agent treatment, our data indicate a promising brain penetrance of ETC-168 and in vivo activity of ETC-168/TMZ combo in tumor-bearing mice. Notably, MNK silencing exerted a greater impact on GBM tumor growth than MNK inhibition by ETC-168. In addition to bioavailability, kinase-independent role of MNK proteins may account for such a difference. Strong deleterious effects of MNK silencing on cancer cells encourage further development of MNK-degrading agents.
We also uncover many SE-driven functional targets/pathways that are associated with subtype specificity, encouraging follow-up studies to explore actionable tumor dependencies. Considering the possibility that multiple genes can be regulated through a single broad SE domain, the number of SE-associated targets is very likely underestimated, especially for lncRNAs. Our ARE dataset serves as a fertile ground for future hypothesis testing and in-depth biologic exploration (especially for lncRNAs). On the basis of our data, more sophisticated scoring systems may be developed to fulfill optimal mapping of both subtype-specific engagement of SE domains and their targets. With recent insights from the function of extrachromosomal circular DNAs in driving oncogene expression, genomic abnormalities, especially amplification and/or reintegration of noncoding regulatory DNA elements, greatly enhance oncogenic transcription (54–56). We anticipate that integration of extra layers of genomic and epigenetic information, including DNA abnormalities/modification (57–59), chromatin accessibility (60), and chromatin conformation capture (61), will expand further our vision of GBM pathobiology and precision therapies.
MATERIALS AND METHODS
Cell culture
All cell lines tested negative for mycoplasma and have been authenticated by short tandem repeat analysis with the Geneprint 10 System Kit (Promega). Human embryonic kidney 293T [American Type Culture Collection (ATCC)], U87-MG (U87, ATCC), U251-MG (U251, Sigma-Aldrich), and U343-MG (U343, ATCC) cell lines were cultured in Dulbecco’s modified Eagle medium (DMEM; Biowest). DBTRG-05MG (DBTRG, ATCC) cells were maintained in Roswell Park Memorial Institute medium 1640 (Biowest). For the aforementioned cells, culture media were supplemented with 1% penicillin-streptomycin (Gibco) and 10% fetal bovine serum (Biowest).
Patient-derived GPCs including NNI-11, NNI-20, NNI-23, NNI-24, and NNI-31 were provided by the National Neuroscience Institute (NNI), Singapore. These GPC cell lines were established from patient tumors obtained with informed consent and deidentified in accordance with the SingHealth Centralised Institutional Review Board A and the National Healthcare Group Domain-Specific Review Board A. MGG4 and MGG8 were provided by H.W. All GPCs were expanded as tumor spheres that were maintained in growth media consisting of epidermal growth factor (20 ng/ml), basic fibroblast growth factor (20 ng/ml), heparin (5 μg/ml), B27 supplement (Gibco), and DMEM/F-12 mixture (3:1). All cell cultures were maintained in 5% CO2 in a humidified incubator at 37°C. Information of cell lines we used for H3K27ac ChIP-seq analysis can be found in table S1B.
Chemicals and plasmids
ZBC260 was provided by S.W., University of Michigan. dBET6 was provided by J. Bradner, Harvard Medical School. ETC-168 was shared from the Experimental Drug Development Centre, Agency for Science, Technology and Research, Singapore. Other key chemicals used in this study included THZ531 (ApexBio), CGP57380 (Abcam), Adavosertib (Medchem Express), TMZ (ApexBio), and formaldehyde (Calbiochem, Merck).
On-TARGETplus small interfering RNA (siRNA) pools including control (si-NT), si-ETV6, si-MYC, and si-NR2F2 were purchased from Dharmacon. RNAiMAX (Life Technologies) was used to transfect siRNAs. Stable knockdown cells were generated by lentiviral infection, followed by antibiotic (puromycin, Sigma-Aldrich) selection. SHC002 was used as a negative control (sh-Ctrl). All pLKO.1-based short hairpin RNA (shRNA) vectors are listed in table S1N. 293T cells were used for viral production.
Cell viability assay
MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was performed to measure the cell viability of U87 and U251 cells (62). Briefly, GBM cells were seeded into 96-well plates at 2000 cells per well and cultured under indicated treatment. MTT substrate was added into each well and incubated for 3 hours, followed by careful aspiration of medium and addition of 100 μl of MTT Stop solution. Plates were read on a Tecan microplate reader with the absorbance at 570 nm.
CellTiter-Glo assay (Promega) was applied to measure the cell viability of GPCs. Tumor spheres were dissociated by Accutase (Gibco) and subsequently seeded into a 96-well plate at either 2000 or 3000 cells per well. Following indicated drug treatment, luminescence signals were measured after 72-hour incubation. For cells with shRNA transduction, luminescence signals were detected at both days 1 and 5 after seeding.
In vitro limiting-dilution tumor sphere formation assay
GPCs were dissociated by Accutase, seeded into low-attachment 96-well plates (Corning) with decreasing numbers of cells per well, and cultured with indicated treatment. Tumor sphere forming capability was examined by Extreme Limiting Dilution Analysis (http://bioinf.wehi.edu.au/software/elda/).
Animal models
Both subcutaneous and intracranial xenograft models were used to evaluate the tumorigenicity of GBM cells either after genetic manipulation or in response to drug treatment, in compliance with ethical regulations of the National University of Singapore (NUS) Institutional Animal Care and Use Committee. Regarding the subcutaneous xenograft assays, half a million of indicated GBM cells were resuspended with 100 μl of Matrigel (Corning)/phosphate-buffered saline (PBS) solution (volume ratio, 1:1) and injected subcutaneously on the upper flanks of NOD/SCID gamma (NSG) mice (male, 6 to 8 weeks old). Tumors were dissected from animals and weighed at the experimental end point.
For orthotopic xenograft models, 6- to 8-week-old male NSG mice were used for stereotaxic cell implantation (0.2 million cells in 2 μl of PBS). In preparation for histological examination, mouse brains were harvested by transcardial perfusion with 4% paraformaldehyde when mice either reached experimental end points or fulfilled humane end point criterion (20% loss of peak body weight or presented with neurological defects, such as ataxia, cachexia, lethargy, or seizure). Animal survival was recorded on the basis of humane end point criterion.
For in vivo drug treatment, mice bearing intracranial tumors were randomly allocated into each treatment arm 1 week after implantation. Mice in the experimental arm received oral gavage of indicated compounds. TMZ was given at a dose of 100 mg/kg, once a day, 5 days per week for 2 weeks; ETC-168 was given at a dose of 25 mg/kg, once a day, 5 days per week for 5 weeks. Animals in the control arm received the same volume of vehicle (100 μl of Ora-Plus Oral Suspending Vehicle, Paddock Laboratories). For immunoblot analysis, fresh tumor tissues were harvested from these experimental animals.
No specific randomization methods were used to allocate mice into different treatment groups. The investigators were not blinded to allocation during either experiments or outcome assessment. Minimum sample sizes for individual experiments were determined on the basis of our experience (n ≥ 5). No animals were excluded from these experiments.
Cell/tissue lysate preparation and immunoblot assay
Whole-cell lysate was extracted by incubating cells in lysis buffer [50 mM tris (pH 8.0), 420 mM NaCl, 5% glycerol, 0.1% NP-40, and 0.1 mM EDTA] with fresh addition of 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 1× protease inhibitor cocktail (Roche), 1× phosphatase inhibitor cocktail (Roche), 1 mM MgCl2, and Benzonase (1:500, Novagen) for 20 min on ice. To extract proteins from tissues, the same lysis buffer was added to minced samples and subsequently passed through a 1-ml syringe with a 26-gauge needle for several times. Clear supernatant after centrifugation (13,000 rpm, 10 min) was carefully transferred to a new tube and subjected to protein concentration quantification by Bradford assay (Bio-Rad). Immunoblot analysis was conducted following standard protocol with the following antibodies: β-actin (Sigma-Aldrich, A1978), BCL6 (Sigma-Aldrich, HPA004899), BRD4 (Cell Signaling Technology,13440S), CDK13 (Genetex, GTX102408), cleaved caspase-3 (Cell Signaling Technology, 9661), cleaved caspase-7 (Cell Signaling Technology, 8438S), cleaved PARP (Cell Signaling Technology, 9661), EIF4E (Cell Signaling Technology, 2067), ETS1 (Cell Signaling Technology, 14069), JUN (Cell Signaling Technology, 9165S), MNK1 (Cell Signaling Technology, 2195), MNK2 (Sigma-Aldrich, HPA021875), p-EIF4E (Abcam, ab76256), RFX2 (Sigma-Aldrich, HPA048969), SOX2 (Cell Signaling Technology, 3579S), and WEE1 (Cell Signaling Technology, 13084S).
Chromatin immunoprecipitation
Pathologically reviewed either human GBM or normal brain samples were collected from the National University Hospital Tissue Repository, the First Affiliated Hospital of Soochow University, and the Cedars-Sinai Medical Center, with study approvals from their respective Institutional Review Boards.
ChIP procedures in this study were in line with a previously published protocol (63). Briefly, either Douncer-homogenized tissue samples or dissociated cells were fixed with 1% formaldehyde (10 min, room temperature), washed three times with cold PBS, and subjected to nuclei isolation. Enriched nuclei were then resuspended in lysis buffer [1% SDS, 10 mM EDTA, and 50 mM tris (pH 8.0)] for 10 min on ice and sonicated by Bioruptor (Diagenode). After sonication, chromatin DNA was sheared into 200 to 500 base pairs (bp), and cell debris was removed by centrifugation. Supernatant was then incubated with Dynabeads (Invitrogen), which were preconjugated with antibodies specific to H3K27ac (Active Motif, 39133). After overnight incubation in the cold room, magnetic beads were subjected to a stepwise wash process with low-salt wash buffer, high-salt wash buffer, LiCl wash buffer, and TE buffer. ChIP-ed DNA was reverse-crosslinked (65°) and purified by MinElute PCR (polymerase chain reaction) Purification Kit (Qiagen). Eluted DNA was quantified by Qubit dsDNA HS Assay (Thermo Fisher Scientific).
Genomic DNA extraction and Sanger sequencing
A small aliquot of either GBM tissue or GPC pellet was reserved for the extraction of genomic DNA (Qiagen DNeasy Blood and Tissue Kit). Genomic DNA from the normal brain was included as negative control. Target region spanning C228T and C250T mutations in the TERT promoter (64), corresponding to c.−124C>T and c.−146C>T of TERT ATG start site, was PCR amplified by GoTaq Green Master Mix (Promega) with the following primer set: 5′-GTCCTGCCCCTTCACCTT-3′ (forward) and 5′-AGCACCTCGCGGTAGTGG-3′ (reverse). IDH1 genomic DNA harboring the region corresponding to hotspot mutation R132 was amplified using the following primer set (65): 5′-AATGAGCTCTATATGCCATCACTG-3′ (forward) and 5′- TTCATACCTTGCTTAATGGGTGT-3′ (reverse). Amplicons were Sanger sequenced to identify the mutational status.
RNA preparation and quantitative reverse transcription PCR
Total RNA was extracted by RNeasy Kit (Qiagen) and treated with deoxyribonuclease. RevertAid RT Reverse Transcription Kit (Thermo Fisher Scientific) was used to produce complementary DNA library for quantitative reverse transcription PCR (qRT-PCR) assay. qPCR was performed with Kapa SYBR Fast qPCR Master Mix (KAPA Biosystems) on a 7500 Real-Time PCR System (Applied Biosystems). qRT-PCR primers are listed in table S1O (66).
ChIP-seq analysis
For our in-house samples, ChIP-seq libraries were constructed using a ThruPLEX DNA-seq Kit (Rubicon Genomics). Next-generation sequencing was performed by BGI Tech Solutions Co. Ltd. and NovogeneAIT Genomics Singapore Pte Ltd. Raw ChIP-seq files for published data with controlled access (table S1A) were downloaded via Sequence Read Archive (SRA) tools from the database of Genotypes and Phenotypes (dbGAP), with approvals from their respective Data Access Committees (DACs). Other publicly available ChIP-seq data were uniformly processed, and the quality control information can be found in table S1 (A, B, and D).
For ChIP-seq data analysis, some of the following methods are similar to those previously published (63). Raw reads were aligned to hg38 reference genome using bwa v0.7.13-r1126 aligner followed by removal of PCR duplicates with Picard v2.5.0 markDuplicates utility (67). Resulting bam files were used for peak calling with MACS2 v2.1.4 by extending reads to fragment size predicted by strand cross-correlation analysis from phantompeakqualtools v1.0 and parameter --nomodel (68, 69). ChIP signals (bedGraph) were simultaneously generated, and input signal was subtracted with MACS2 bdgcmp function. bedGraphs were later converted to bigwig files with the UCSC bedGraphTobigWig utility. Detected peaks were annotated with HOMER v4.10 annotatePeaks. The ROSE (Rank Ordering of Super-Enhancers) algorithm (https://bitbucket.org/young_computation/rose) was applied to identify SEs. Regions within ±1250 bp from TSS were excluded for this analysis. For each sample, input-subtracted ChIP-seq signals were stitched and ranked on the basis of intensity. A geometrical inflection point was used as cutoff to separate SEs from TEs. SE association was annotated to Ensembl genes with default setting. Regarding meta-SE analysis, H3K27ac signals of all samples within each subgroup were merged as single input and subjected to SE calling. Core transcriptional regulatory circuitries were computationally reconstituted by CRCmapper (40, 41) based on the frequency of TF motifs within SE domains. Top-ranked circuitry and key-predicted interactive factors were visualized in Cytoscape with GeneMANIA (70). Motif enrichment analysis of SE-driven TFs was performed using MEME software (71). GO analysis of SE target genes was calculated by g:Profiler (72) with significance threshold set to Benjamini-Hochberg false discovery rate (FDR) (0.05) and subsequently visualized in Cytoscape with Enrichment Map (73).
RENVARs were identified by adapting a variant calling workflow from DNA-seq. The ChIP-seq bam files were processed on the basis of the Genome Analysis ToolKit (GATK) best practice. Briefly, the bam files were subjected to base recalibration, and variants were called using the GATK v4.0.1 Mutect2 (74) on the recalibrated bam files. Only variants that passed all default filters of GATK FilterMutectCalls were retained for downstream analyses. The variants were annotated using ANNOVAR v2019-10-24 (75), and variants that were present in normal tissues, exonic, annotated as benign/likely benign, having (ExAC and gnomAD) population allele frequency ≥0.01, depth <50, or alternate allele depth <5 were removed. COSMIC89 and dbSNP150 (avsnp150) were interrogated to filter out reported variants unless listed as somatic noncoding mutations. The remaining variants were then intersected with the MACS2 called H3K27ac peaks using bedtools v2.29.2 to identify enhancer-associated hits. Table S1F shows the list of RENVARs (occurred in two or more samples).
RNA-seq and gene expression analysis
RNA-seq libraries were constructed by the TruSeq Library Prep Kit (Illumina) and sequenced at BGI Tech Solutions Co. Ltd. (HiSeq, paired-end reads of 100 bases). Paired-end reads were aligned to hg38 genome using STAR v2.5.3a and subjected to transcript quantification using RSEM v1.3 based on Gencode v24 annotation (76, 77).
Variant identification was performed using STAR v2.5.3a and GATK v3.8 based on the best practice guideline outlined (https://gatk.broadinstitute.org/hc/en-us/articles/360035531192-RNAseq-short-variant-discovery-SNPs-Indels-) (74, 78). Briefly, the RNA-seq data were mapped to human genome hg38 using STAR 2.5.3a two-pass workflow. Subsequently, upon marked duplication, the mapped alignments were subjected to recalibration. Last, we performed variant calling on the recalibrated alignment files and filtered out low-quality, nonexonic mutation, SNP whose population allele frequency is ≥0.01, and clinVar-annotated benign variants. EGFR variant calling was performed in the same practice as TCGA-GBM study (6).
Transcriptome data for lower-grade gliomas and GBM samples from the TCGA cohort (TCGA-LGG and TCGA-GBM) were downloaded using TCGABiolinks Bioconductor package via the National Cancer Institute Genomic Data Commons (6, 79). Gene expression matrix files for microarray dataset of Rembrandt and Gravendeel cohorts were downloaded through ArrayExpress (E-MTAB-3073) and Gene Expression Omnibus (GEO) (GSE16011), respectively (38, 80). Raw SRA files for published RNA-seq data with restricted access (table S1A) were fetched via SRA tools from the dbGAP with DAC approvals. Molecular subtypes of glioma samples from both in-house and published cohorts were determined by the GIE signatures (9). Expressional data of lncRNAs that were subjected to cross-tissue/tumor comparison were retrieved from the MiTranscriptome (81) by October 2017. The data used for prognostic analyses of RFX2 and TGIF1 were retrieved from expressional analysis of the Rembrandt and TCGA-GBM cohorts via Project Betastasis (http://betastasis.com). Patient stratification with SE gene signature was performed using Connectivity Map (82). Gene signature–based portraying of tissue cellular landscape was modeled by xCell (29).
To identify gene expression molecular subtype of our samples, we first combined and standardized our cohort in FPKM values with TCGA-GBM using ComBat (83). Once standardized, we extracted the centroid from the seminal study (9) and performed consensus clustering on the combined dataset. Our samples were then assigned the molecular subtype based on the highest similarity to the subtype centroid.
Statistical analysis
Unless otherwise stated, statistical significance was reported on the basis of either two-tailed Student’s t test (two-group comparison) or one-way analysis of variance (ANOVA) (multigroup comparison). Survival analysis was performed by log-rank test; Kaplan-Meier survival curves were plotted to show the survival of either patient groups or experimental groups. n.s., not significant; *P < 0.05, **P < 0.01, and ***P < 0.001. Sample sizes were not predetermined statistically. Center values, error bars, and number of replicates are described in the corresponding figures and/or figure legends. Replicates represent (i) separate tumors in xenograft assays, (ii) individual animals in orthotopic tumor model, and (iii) independent biological repeats for in vitro assays.
Acknowledgments
We thank T. Sanda, C. W. Joo, Y. Ang, A. L. A. Wong, J. Bradner, T. T. Boon, H.-Q. Shen, and F. Hu for suggestions and help. Funding: The discovery of ETC-168 (also known as AUM168 in AUM Biosciences) was financially supported by the Biomedical Sciences Institutes and Joint Council Office (JCO Project 11 03 FG 07 05), Agency for Science, Technology and Research, Singapore. This work is funded by the NIH (R01-CA200992-04 to H.P.K., and R35CA197628 and R01CA213138 to M.M.), the Howard Hughes Medical Institute (HHMI-55108547 to M.M.), the Singapore Ministry of Health’s National Medical Research Council (NMRC) under its Singapore Translational Research Investigator Award (NMRC/STaR/0021/2014 to H.P.K.), the Singapore Ministry of Education Academic Research Fund Tier 2 (MOE2017-T2-1-033 to H.P.K.), the NMRC Centre Grant Programme awarded to the National University Cancer Institute of Singapore (NMRC/CG/012/2013 and CGAug16M005), the National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centres of Excellence initiatives, the RNA Biology Center at the Cancer Science Institute of Singapore (MOE2014-T3-1-006), the NMRC Open Fund Young Individual Research Grants (MOH-OFYIRG18May-0001 to L.X. and MOH-OFYIRG19Nov-0016 to Y.C.), and the NMRC Translational and Clinical Research Flagship Programme grant (NMRC/TCR/016-NNI/2016 to B.T.A. and C.T.). In addition, this work is supported by the NUS Center for Cancer Research, Cancer Programme under Translational Research Programmes, Yong Loo Lin School of Medicine, NUS (NUHSRO/2020/122/MSC/07/Cancer), a Seed Funding Program within the NCIS Centre Grant, an NCIS Yong Siew Yoon Research grant through donations from the Yong Loo Lin Trust, and philanthropic donations from the Melamed family, and Valerie Baker Fairbank who also gave us encouragement. J.C. is supported by the Start-up Grant of HZNU (4125C5021820470), National Natural Science Foundation of China (81802338 and 82072646), and Zhejiang Provincial Natural Science Foundation of China for Distinguished Young Scholars (LR21H160001). Y.H. is supported by Jiangsu Province Commission of Health and Family Planning Research funding (H2017064) and Suzhou Science and Technology Development Plan (SS201864). M.M. is a Howard Hughes Medical Institute (HHMI) Faculty Scholar. Author contributions: Conceptualization: L.X. and Y.C. Methodology: L.X., Y.C., E.S., P.D., A.M., M.-L.H., B.T.A., C.T., T.Z.T., and H.P.K. Investigation: L.X., Y.C., Y.H., E.S., R.Y.-T.L., P.D., X.-Y.K., Y.K.C., L.K., A.M., S.G., S.W.L., Z.H., J.C., T.Z.T., and C.T. Writing—original draft: L.X., Y.C., T.Z.T. and H.P.K. Writing—review and editing: L.X., Y.C., Y.H., R.Y.-T.L., S.G., J.C., B.T.A., M.M., C.T., T.Z.T., and H.P.K. Visualization: L.X., C.Y., E.S., P.D., A.M., and T.Z.T. Funding acquisition: L.X., C.Y., Y.H., J.C., B.T.A., M.M., C.T., and H.P.K. Resources: Y.H., J.S.Y., K.N., J.H., Y.X., L.B., S.W., H.W., T.T.Y., B.T.A., M.M., C.T., and H.P.K. Supervision: L.X., M.M., C.T., and H.P.K. Competing interests: L.B. and S.W. are coinventors on a patent (U.S. patent no. 10,633,386), which covers ZBC260 used in this study. The patent (U.S. patent no. 10,633,386) is owned by the University of Michigan and has been licensed by Roivant Sciences. L.B. and S.W. are eligible to receive royalty payments from the University of Michigan per the University of Michigan policies. S.W. is also a paid consultant of Roivant Sciences, and the University of Michigan has received a research contract from Roivant Sciences for which S.W. is the principal investigator. In addition, S.W. receives a commercial research grant and has ownership interest (including patents) in Medsyn Biopharma. S.W. also owns stock in and serves as a consultant for the OncoFusion Therapeutics Inc., the Oncopia Therapeutics LLC., and Ascentage Pharma Group. The remaining authors declare that they have no competing interests. Data and materials availability: RNA-seq and ChIP-seq data generated in this study have been deposited in the National Center for Biotechnology Information GEO and are available under accession GSE145646. Additional data used in this study are available via the following accessions: E-MTAB-3073, GSE16011, GSE16256, GSE16368, GSE28876, GSE29611, GSE40465, GSE61852, GSE62193, GSE67978, GSE72468, GSE74529, GSE79736, GSE92469, GSE96178, GSE99181, GSE119776, and phs001389.v1.p1. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. ETC-168 can be provided by the Experimental Drug Development Centre (EDDC)–A*STAR pending scientific review and a completed material transfer agreement. Requests for the ETC-168 should be submitted to K.N. Requests for the MGG4 and MGG8 cells should be submitted to H.W. Requests for the NNI-11/20/23/24/31 cells should be submitted to C.T. and B.T.A.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/18/eabd4676/DC1
REFERENCES AND NOTES
- 1.Cloughesy T. F., Cavenee W. K., Mischel P. S., Glioblastoma: From molecular pathology to targeted treatment. Annu. Rev. Pathol. 9, 1–25 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Stupp R., Mason W. P., van den Bent M. J., Weller M., Fisher B., Taphoorn M. J. B., Belanger K., Brandes A. A., Marosi C., Bogdahn U., Curschmann J., Janzer R. C., Ludwin S. K., Gorlia T., Allgeier A., Lacombe D., Cairncross J. G., Eisenhauer E., Mirimanoff R. O., Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Perry J. R., Laperriere N., O’Callaghan C. J., Brandes A. A., Menten J., Phillips C., Fay M., Nishikawa R., Cairncross J. G., Roa W., Osoba D., Rossiter J. P., Sahgal A., Hirte H., Laigle-Donadey F., Franceschi E., Chinot O., Golfinopoulos V., Fariselli L., Wick A., Feuvret L., Back M., Tills M., Winch C., Baumert B. G., Wick W., Ding K., Mason W. P., Short-course radiation plus temozolomide in elderly patients with glioblastoma. N. Engl. J. Med. 376, 1027–1037 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Stupp R., Taillibert S., Kanner A., Read W., Steinberg D. M., Lhermitte B., Toms S., Idbaih A., Ahluwalia M. S., Fink K., di Meco F., Lieberman F., Zhu J. J., Stragliotto G., Tran D. D., Brem S., Hottinger A. F., Kirson E. D., Lavy-Shahaf G., Weinberg U., Kim C.-Y., Paek S.-H., Nicholas G., Bruna J., Hirte H., Weller M., Palti Y., Hegi M. E., Ram Z., Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA 318, 2306–2316 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.The Cancer Genome Atlas Research Network , Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brennan C. W., Verhaak R. G. W., McKenna A., Campos B., Noushmehr H., Salama S. R., Zheng S., Chakravarty D., Sanborn J. Z., Berman S. H., Beroukhim R., Bernard B., Wu C. J., Genovese G., Shmulevich I., Barnholtz-Sloan J., Zou L., Vegesna R., Shukla S. A., Ciriello G., Yung W. K., Zhang W., Sougnez C., Mikkelsen T., Aldape K., Bigner D. D., van Meir E. G., Prados M., Sloan A., Black K. L., Eschbacher J., Finocchiaro G., Friedman W., Andrews D. W., Guha A., Iacocca M., O’Neill B. P., Foltz G., Myers J., Weisenberger D. J., Penny R., Kucherlapati R., Perou C. M., Hayes D. N., Gibbs R., Marra M., Mills G. B., Lander E., Spellman P., Wilson R., Sander C., Weinstein J., Meyerson M., Gabriel S., Laird P. W., Haussler D., Getz G., Chin L., Benz C., Barnholtz-Sloan J., Barrett W., Ostrom Q., Wolinsky Y., Black K. L., Bose B., Boulos P. T., Boulos M., Brown J., Czerinski C., Eppley M., Iacocca M., Kempista T., Kitko T., Koyfman Y., Rabeno B., Rastogi P., Sugarman M., Swanson P., Yalamanchii K., Otey I. P., Liu Y. S., Xiao Y., Auman J.-T., Chen P. C., Hadjipanayis A., Lee E., Lee S., Park P. J., Seidman J., Yang L., Kucherlapati R., Kalkanis S., Mikkelsen T., Poisson L. M., Raghunathan A., Scarpace L., Bernard B., Bressler R., Eakin A., Iype L., Kreisberg R. B., Leinonen K., Reynolds S., Rovira H., Thorsson V., Shmulevich I., Annala M. J., Penny R., Paulauskis J., Curley E., Hatfield M., Mallery D., Morris S., Shelton T., Shelton C., Sherman M., Yena P., Cuppini L., DiMeco F., Eoli M., Finocchiaro G., Maderna E., Pollo B., Saini M., Balu S., Hoadley K. A., Li L., Miller C. R., Shi Y., Topal M. D., Wu J., Dunn G., Giannini C., O’Neill B. P., Aksoy B. A., Antipin Y., Borsu L., Berman S. H., Brennan C. W., Cerami E., Chakravarty D., Ciriello G., Gao J., Gross B., Jacobsen A., Ladanyi M., Lash A., Liang Y., Reva B., Sander C., Schultz N., Shen R., Socci N. D., Viale A., Ferguson M. L., Chen Q. R., Demchok J. A., Dillon L. A. L., Shaw K. R. M., Sheth M., Tarnuzzer R., Wang Z., Yang L., Davidsen T., Guyer M. S., Ozenberger B. A., Sofia H. J., Bergsten J., Eckman J., Harr J., Myers J., Smith C., Tucker K., Winemiller C., Zach L. A., Ljubimova J. Y., Eley G., Ayala B., Jensen M. A., Kahn A., Pihl T. D., Pot D. A., Wan Y., Eschbacher J., Foltz G., Hansen N., Hothi P., Lin B., Shah N., Yoon J. G., Lau C., Berens M., Ardlie K., Beroukhim R., Carter S. L., Cherniack A. D., Noble M., Cho J., Cibulskis K., DiCara D., Frazer S., Gabriel S. B., Gehlenborg N., Gentry J., Heiman D., Kim J., Jing R., Lander E. S., Lawrence M., Lin P., Mallard W., Meyerson M., Onofrio R. C., Saksena G., Schumacher S., Sougnez C., Stojanov P., Tabak B., Voet D., Zhang H., Zou L., Getz G., Dees N. N., Ding L., Fulton L. L., Fulton R. S., Kanchi K.-L., Mardis E. R., Wilson R. K., Baylin S. B., Andrews D. W., Harshyne L., Cohen M. L., Devine K., Sloan A. E., VandenBerg S. R., Berger M. S., Prados M., Carlin D., Craft B., Ellrott K., Goldman M., Goldstein T., Grifford M., Haussler D., Ma S., Ng S., Salama S. R., Sanborn J. Z., Stuart J., Swatloski T., Waltman P., Zhu J., Foss R., Frentzen B., Friedman W., McTiernan R., Yachnis A., Hayes D. N., Perou C. M., Zheng S., Vegesna R., Mao Y., Akbani R., Aldape K., Bogler O., Fuller G. N., Liu W., Liu Y., Lu Y., Mills G., Protopopov A., Ren X., Sun Y., Wu C.-J., Yung W.-K. A., Zhang W., Zhang J., Chen K., Weinstein J. N., Chin L., Verhaak R. G. W., Noushmehr H., Weisenberger D. J., Bootwalla M. S., Lai P. H., Triche T. J. Jr., van den Berg D. J., Laird P. W., Gutmann D. H., Lehman N. L., VanMeir E. G., Brat D., Olson J. J., Mastrogianakis G. M., Devi N. S., Zhang Z., Bigner D., Lipp E., McLendon R., The somatic genomic landscape of glioblastoma. Cell 155, 462–477 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Noushmehr H., Weisenberger D. J., Diefes K., Phillips H. S., Pujara K., Berman B. P., Pan F., Pelloski C. E., Sulman E. P., Bhat K. P., Verhaak R. G. W., Hoadley K. A., Hayes D. N., Perou C. M., Schmidt H. K., Ding L., Wilson R. K., van den Berg D., Shen H., Bengtsson H., Neuvial P., Cope L. M., Buckley J., Herman J. G., Baylin S. B., Laird P. W., Aldape K., Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17, 510–522 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Verhaak R. G. W., Hoadley K. A., Purdom E., Wang V., Qi Y., Wilkerson M. D., Miller C. R., Ding L., Golub T., Mesirov J. P., Alexe G., Lawrence M., O’Kelly M., Tamayo P., Weir B. A., Gabrie S., Winckler W., Gupta S., Jakkula L., Feiler H. S., Graeme Hodgson J., David James C., Sarkaria J. N., Brennan C., Kahn A., Spellman P. T., Wilson R. K., Speed T. P., Gray J. W., Meyerson M., Getz G., Perou C. M., Neil Hayes D.; The Cancer Genome Atlas Research Network , Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Q., Hu B., Hu X., Kim H., Squatrito M., Scarpace L., deCarvalho A. C., Lyu S., Li P., Li Y., Barthel F., Cho H. J., Lin Y.-H., Satani N., Martinez-Ledesma E., Zheng S., Chang E., Sauvé C.-E. G., Olar A., Lan Z. D., Finocchiaro G., Phillips J. J., Berger M. S., Gabrusiewicz K. R., Wang G., Eskilsson E., Hu J., Mikkelsen T., De Pinho R. A., Muller F., Heimberger A. B., Sulman E. P., Nam D.-H., Verhaak R. G. W., Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32, 42–56.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Phillips H. S., Kharbanda S., Chen R., Forrest W. F., Soriano R. H., Wu T. D., Misra A., Nigro J. M., Colman H., Soroceanu L., Williams P. M., Modrusan Z., Feuerstein B. G., Aldape K., Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9, 157–173 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Neftel C., Laffy J., Filbin M. G., Hara T., Shore M. E., Rahme G. J., Richman A. R., Silverbush D., Shaw M. L., Hebert C. M., Dewitt J., Gritsch S., Perez E. M., Nicolas Gonzalez Castro L., Lan X., Druck N., Rodman C., Dionne D., Kaplan A., Bertalan M. S., Small J., Pelton K., Becker S., Bonal D., Nguyen Q.-D., Servis R. L., Fung J. M., Mylvaganam R., Mayr L., Gojo J., Haberler C., Geyeregger R., Czech T., Slavc I., Nahed B. V., Curry W. T., Carter B. S., Wakimoto H., Brastianos P. K., Batchelor T. T., Stemmer-Rachamimov A., Martinez-Lage M., Frosch M. P., Stamenkovic I., Riggi N., Rheinbay E., Monje M., Rozenblatt-Rosen O., Cahill D. P., Patel A. P., Hunter T., Verma I. M., Ligon K. L., Louis D. N., Regev A., Bernstein B. E., Tirosh I., Suvà M. L., An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 178, 835–849.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whyte W. A., Orlando D. A., Hnisz D., Abraham B. J., Lin C. Y., Kagey M. H., Rahl P. B., Lee T. I., Young R. A., Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang L., Babikir H., Müller S., Yagnik G., Shamardani K., Catalan F., Kohanbash G., Alvarado B., di Lullo E., Kriegstein A., Shah S., Wadhwa H., Chang S. M., Phillips J. J., Aghi M. K., Diaz A. A., The phenotypes of proliferating glioblastoma cells reside on a single axis of variation. Cancer Discov. 9, 1708–1719 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hall A. W., Battenhouse A. M., Shivram H., Morris A. R., Cowperthwaite M. C., Shpak M., Iyer V. R., Bivalent chromatin domains in glioblastoma reveal a subtype-specific signature of glioma stem cells. Cancer Res. 78, 2463–2474 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie Q., Wu Q., Kim L., Miller T. E., Liau B. B., Mack S. C., Yang K., Factor D. C., Fang X., Huang Z., Zhou W., Alazem K., Wang X., Bernstein B. E., Bao S., Rich J. N., RBPJ maintains brain tumor-initiating cells through CDK9-mediated transcriptional elongation. J. Clin. Invest. 126, 2757–2772 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller T. E., Liau B. B., Wallace L. C., Morton A. R., Xie Q., Dixit D., Factor D. C., Kim L. J. Y., Morrow J. J., Wu Q., Mack S. C., Hubert C. G., Gillespie S. M., Flavahan W. A., Hoffmann T., Thummalapalli R., Hemann M. T., Paddison P. J., Horbinski C. M., Zuber J., Scacheri P. C., Bernstein B. E., Tesar P. J., Rich J. N., Transcription elongation factors represent in vivo cancer dependencies in glioblastoma. Nature 547, 355–359 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou D., Alver B. M., Li S., Hlady R. A., Thompson J. J., Schroeder M. A., Lee J.-H., Qiu J., Schwartz P. H., Sarkaria J. N., Robertson K. D., Distinctive epigenomes characterize glioma stem cells and their response to differentiation cues. Genome Biol. 19, 43 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mack S. C., Singh I., Wang X., Hirsch R., Wu Q., Villagomez R., Bernatchez J. A., Zhu Z., Gimple R. C., Kim L. J. Y., Morton A., Lai S., Qiu Z., Prager B. C., Bertrand K. C., Mah C., Zhou W., Lee C., Barnett G. H., Vogelbaum M. A., Sloan A. E., Chavez L., Bao S., Scacheri P. C., Siqueira-Neto J. L., Lin C. Y., Rich J. N., Chromatin landscapes reveal developmentally encoded transcriptional states that define human glioblastoma. J. Exp. Med. 216, 1071–1090 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Creyghton M. P., Cheng A. W., Welstead G. G., Kooistra T., Carey B. W., Steine E. J., Hanna J., Lodato M. A., Frampton G. M., Sharp P. A., Boyer L. A., Young R. A., Jaenisch R., Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U.S.A. 107, 21931–21936 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stein C., Bardet A. F., Roma G., Bergling S., Clay I., Ruchti A., Agarinis C., Schmelzle T., Bouwmeester T., Schübeler D., Bauer A., YAP1 exerts its transcriptional control via TEAD-mediated activation of enhancers. PLOS Genet. 11, e1005465 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The ENCODE Project Consortium , An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ziller M. J., Edri R., Yaffe Y., Donaghey J., Pop R., Mallard W., Issner R., Gifford C. A., Goren A., Xing J., Gu H., Cacchiarelli D., Tsankov A. M., Epstein C., Rinn J. L., Mikkelsen T. S., Kohlbacher O., Gnirke A., Bernstein B. E., Elkabetz Y., Meissner A., Dissecting neural differentiation regulatory networks through epigenetic footprinting. Nature 518, 355–359 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bernstein B. E., Stamatoyannopoulos J. A., Costello J. F., Ren B., Milosavljevic A., Meissner A., Kellis M., Marra M. A., Beaudet A. L., Ecker J. R., Farnham P. J., Hirst M., Lander E. S., Mikkelsen T. S., Thomson J. A., The NIH Roadmap Epigenomics Mapping Consortium. Nat. Biotechnol. 28, 1045–1048 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rada-Iglesias A., Bajpai R., Prescott S., Brugmann S. A., Swigut T., Wysocka J., Epigenomic annotation of enhancers predicts transcriptional regulators of human neural crest. Cell Stem Cell 11, 633–648 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu F., Hon G. C., Villa G. R., Turner K. M., Ikegami S., Yang H., Ye Z., Li B., Kuan S., Lee A. Y., Zanca C., Wei B., Lucey G., Jenkins D., Zhang W., Barr C. L., Furnari F. B., Cloughesy T. F., Yong W. H., Gahman T. C., Shiau A. K., Cavenee W. K., Ren B., Mischel P. S., EGFR mutation promotes glioblastoma through epigenome and transcription factor network remodeling. Mol. Cell 60, 307–318 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vermunt M. W., Reinink P., Korving J., de Bruijn E., Creyghton P. M., Basak O., Geeven G., Toonen P. W., Lansu N., Meunier C., van Heesch S., Clevers H., de Laat W., Cuppen E., Creyghton M. P., Large-scale identification of coregulated enhancer networks in the adult human brain. Cell Rep. 9, 767–779 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Vermunt M. W., Tan S. C., Castelijns B., Geeven G., Reinink P., de Bruijn E., Kondova I., Persengiev S., Bank N. B., Bontrop R., Cuppen E., de Laat W., Creyghton M. P., Epigenomic annotation of gene regulatory alterations during evolution of the primate brain. Nat. Neurosci. 19, 494–503 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Chen Z., Hambardzumyan D., Immune microenvironment in glioblastoma subtypes. Front. Immunol. 9, 1004 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aran D., Hu Z., Butte A. J., xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 18, 220 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hnisz D., Abraham B. J., Lee T. I., Lau A., Saint-André V., Sigova A. A., Hoke H. A., Young R. A., Super-enhancers in the control of cell identity and disease. Cell 155, 934–947 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lovén J., Hoke H. A., Lin C. Y., Lau A., Orlando D. A., Vakoc C. R., Bradner J. E., Lee T. I., Young R. A., Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mansour M. R., Abraham B. J., Anders L., Berezovskaya A., Gutierrez A., Durbin A. D., Etchin J., Lawton L., Sallan S. E., Silverman L. B., Loh M. L., Hunger S. P., Sanda T., Young R. A., Thomas Look A., Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 346, 1373–1377 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abraham B. J., Hnisz D., Weintraub A. S., Kwiatkowski N., Li C. H., Li Z., Weichert-Leahey N., Rahman S., Liu Y., Etchin J., Li B., Shen S., Lee T. I., Zhang J., Look A. T., Mansour M. R., Young R. A., Small genomic insertions form enhancers that misregulate oncogenes. Nat. Commun. 8, 14385 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Behnan J., Finocchiaro G., Hanna G., The landscape of the mesenchymal signature in brain tumours. Brain 142, 847–866 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taylor T. E., Furnari F. B., Cavenee W. K., Targeting EGFR for treatment of glioblastoma: Molecular basis to overcome resistance. Curr. Cancer Drug Targets 12, 197–209 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gangemi R. M. R., Griffero F., Marubbi D., Perera M., Capra M. C., Malatesta P., Ravetti G. L., Zona G. L., Daga A., Corte G., SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells 27, 40–48 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Ikushima H., Todo T., Ino Y., Takahashi M., Miyazawa K., Miyazono K., Autocrine TGF-β signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell 5, 504–514 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Gravendeel L. A. M., Kouwenhoven M. C. M., Gevaert O., de Rooi J. J., Stubbs A. P., Elza Duijm J., Daemen A., Bleeker F. E., Bralten L. B. C., Kloosterhof N. K., De Moor B., Eilers P. H. C., van der Spek P. J., Kros J. M., Sillevis Smitt P. A. E., van den Bent M. J., French P. J., Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 69, 9065–9072 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Gusev Y., Bhuvaneshwar K., Song L., Zenklusen J.-C., Fine H., Madhavan S., The REMBRANDT study, a large collection of genomic data from brain cancer patients. Sci. Data 5, 180158 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saint-André V., Federation A. J., Lin C. Y., Abraham B. J., Reddy J., Lee T. I., Bradner J. E., Young R. A., Models of human core transcriptional regulatory circuitries. Genome Res. 26, 385–396 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang M., Chen Y., Yang M., Guo A., Xu Y., Xu L., Phillip Koeffler H., dbCoRC: A database of core transcriptional regulatory circuitries modeled by H3K27ac ChIP-seq signals. Nucleic Acids Res. 46, D71–D77 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Y., Xu L., Lin R. Y.-T., Müschen M., Koeffler H. P., Core transcriptional regulatory circuitries in cancer. Oncogene 39, 6633–6646 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu L., Chen Y., Mayakonda A., Koh L., Chong Y. K., Buckley D. L., Sandanaraj E., Lim S. W., Lin R. Y.-T., Ke X.-Y., Huang M.-L., Chen J., Sun W., Wang L.-Z., Goh B. C., Dinh H. Q., Kappei D., Winter G. E., Ding L.-W., Ang B. T., Berman B. P., Bradner J. E., Tang C., Koeffler H. P., Targetable BET proteins- and E2F1-dependent transcriptional program maintains the malignancy of glioblastoma. Proc. Natl. Acad. Sci. U.S.A. 115, E5086–E5095 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bai L., Zhou B., Yang C. Y., Ji J., McEachern D., Przybranowski S., Jiang H., Hu J., Xu F., Zhao Y., Liu L., Fernandez-Salas E., Xu J., Dou Y., Wen B., Sun D., Meagher J., Stuckey J., Hayes D. F., Li S., Ellis M. J., Wang S., Targeted degradation of BET proteins in triple-negative breast cancer. Cancer Res. 77, 2476–2487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang H., Chennamaneni L. R., Ho M. W. T., Ang S. H., Tan E. S. W., Jeyaraj D. A., Yeap Y. S., Liu B., Ong E. H., Joy J. K., Wee J. L. K., Kwek P., Retna P., Dinie N., Nguyen T. T. H., Tai S. J., Manoharan V., Pendharkar V., Low C. B., Chew Y. S., Vuddagiri S., Sangthongpitag K., Choong M. L., Lee M. A., Kannan S., Verma C. S., Poulsen A., Lim S., Chuah C., Ong T. S., Hill J., Matter A., Nacro K., Optimization of selective mitogen-activated protein kinase interacting kinases 1 and 2 inhibitors for the treatment of blast crisis leukemia. J. Med. Chem. 61, 4348–4369 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Teneggi V., Novotny-Diermayr V., Lee L. H., Yasin M., Yeo P., Ethirajulu K., Gan S. B. H., Blanchard S. E., Nellore R., Umrani D. N., Gomeni R., Teck D. L. W., Li G., Lu Q. S., Cao Y., Matter A., First-in-human, healthy volunteers integrated protocol of ETC-206, an oral Mnk 1/2 kinase inhibitor oncology drug. Clin. Transl. Sci. 13, 57–66 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reich S. H., Sprengeler P. A., Chiang G. G., Appleman J. R., Chen J., Clarine J., Eam B., Ernst J. T., Han Q., Goel V. K., Han E. Z. R., Huang V., Hung I. N. J., Jemison A., Jessen K. A., Molter J., Murphy D., Neal M., Parker G. S., Shaghafi M., Sperry S., Staunton J., Stumpf C. R., Thompson P. A., Tran C., Webber S. E., Wegerski C. J., Zheng H., Webster K. R., Structure-based design of pyridone–aminal eFT508 targeting dysregulated translation by selective mitogen-activated protein kinase interacting kinases 1 and 2 (MNK1/2) inhibition. J. Med. Chem. 61, 3516–3540 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Yuan J., Levitin H. M., Frattini V., Bush E. C., Boyett D. M., Samanamud J., Ceccarelli M., Dovas A., Zanazzi G., Canoll P., Bruce J. N., Lasorella A., Iavarone A., Sims P. A., Single-cell transcriptome analysis of lineage diversity in high-grade glioma. Genome Med. 10, 57 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li F., Yi Y., Miao Y., Long W., Long T., Chen S., Cheng W., Zou C., Zheng Y., Wu X., Ding J., Zhu K., Chen D., Xu Q., Wang J., Liu Q., Zhi F., Ren J., Cao Q., Zhao W., N6-Methyladenosine modulates nonsense-mediated mRNA decay in human glioblastoma. Cancer Res. 79, 5785–5798 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Flavahan W. A., Drier Y., Liau B. B., Gillespie S. M., Venteicher A. S., Stemmer-Rachamimov A. O., Suvà M. L., Bernstein B. E., Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110–114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Turcan S., Rohle D., Goenka A., Walsh L. A., Fang F., Yilmaz E., Campos C., Fabius A. W. M., Lu C., Ward P. S., Thompson C. B., Kaufman A., Guryanova O., Levine R., Heguy A., Viale A., Morris L. G. T., Huse J. T., Mellinghoff I. K., Chan T. A., IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479–483 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Turcan S., Makarov V., Taranda J., Wang Y., Fabius A. W. M., Wu W., Zheng Y., el-Amine N., Haddock S., Nanjangud G., LeKaye H. C., Brennan C., Cross J., Huse J. T., Kelleher N. L., Osten P., Thompson C. B., Chan T. A., Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat. Genet. 50, 62–72 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Friedmann-Morvinski D., Bushong E. A., Ke E., Soda Y., Marumoto T., Singer O., Ellisman M. H., Verma I. M., Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science 338, 1080–1084 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu S., Turner K. M., Nguyen N., Raviram R., Erb M., Santini J., Luebeck J., Rajkumar U., Diao Y., Li B., Zhang W., Jameson N., Corces M. R., Granja J. M., Chen X., Coruh C., Abnousi A., Houston J., Ye Z., Hu R., Yu M., Kim H., Law J. A., Verhaak R. G. W., Hu M., Furnari F. B., Chang H. Y., Ren B., Bafna V., Mischel P. S., Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature 575, 699–703 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morton A. R., Dogan-Artun N., Faber Z. J., MacLeod G., Bartels C. F., Piazza M. S., Allan K. C., Mack S. C., Wang X., Gimple R. C., Wu Q., Rubin B. P., Shetty S., Angers S., Dirks P. B., Sallari R. C., Lupien M., Rich J. N., Scacheri P. C., Functional enhancers shape extrachromosomal oncogene amplifications. Cell 179, 1330–1341.e13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koche R. P., Rodriguez-Fos E., Helmsauer K., Burkert M., MacArthur I. C., Maag J., Chamorro R., Munoz-Perez N., Puiggròs M., Dorado Garcia H., Bei Y., Röefzaad C., Bardinet V., Szymansky A., Winkler A., Thole T., Timme N., Kasack K., Fuchs S., Klironomos F., Thiessen N., Blanc E., Schmelz K., Künkele A., Hundsdörfer P., Rosswog C., Theissen J., Beule D., Deubzer H., Sauer S., Toedling J., Fischer M., Hertwig F., Schwarz R. F., Eggert A., Torrents D., Schulte J. H., Henssen A. G., Extrachromosomal circular DNA drives oncogenic genome remodeling in neuroblastoma. Nat. Genet. 52, 29–34 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xie Q., Wu T. P., Gimple R. C., Li Z., Prager B. C., Wu Q., Yu Y., Wang P., Wang Y., Gorkin D. U., Zhang C., Dowiak A. V., Lin K., Zeng C., Sui Y., Kim L. J. Y., Miller T. E., Jiang L., Lee C. H., Huang Z., Fang X., Zhai K., Mack S. C., Sander M., Bao S., Kerstetter-Fogle A. E., Sloan A. E., Xiao A. Z., Rich J. N., N6-Nethyladenine DNA modification in glioblastoma. Cell 175, 1228–1243.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Frattini V., Trifonov V., Chan J. M., Castano A., Lia M., Abate F., Keir S. T., Ji A. X., Zoppoli P., Niola F., Danussi C., Dolgalev I., Porrati P., Pellegatta S., Heguy A., Gupta G., Pisapia D. J., Canoll P., Bruce J. N., McLendon R. E., Yan H., Aldape K., Finocchiaro G., Mikkelsen T., Privé G. G., Bigner D. D., Lasorella A., Rabadan R., Iavarone A., The integrated landscape of driver genomic alterations in glioblastoma. Nat. Genet. 45, 1141–1149 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Johnson K. C., Houseman E. A., King J. E., von Herrmann K. M., Fadul C. E., Christensen B. C., 5-Hydroxymethylcytosine localizes to enhancer elements and is associated with survival in glioblastoma patients. Nat. Commun. 7, 13177 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Corces M. R., Granja J. M., Shams S., Louie B. H., Seoane J. A., Zhou W., Silva T. C., Groeneveld C., Wong C. K., Cho S. W., Satpathy A. T., Mumbach M. R., Hoadley K. A., Robertson A. G., Sheffield N. C., Felau I., Castro M. A. A., Berman B. P., Staudt L. M., Zenklusen J. C., Laird P. W., Curtis C.; The Cancer Genome Atlas Analysis Network, Greenleaf W. J., Chang H. Y., The chromatin accessibility landscape of primary human cancers. Science 362, eaav1898 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Johnston M. J., Nikolic A., Ninkovic N., Guilhamon P., Cavalli F. M. G., Seaman S., Zemp F. J., Lee J., Abdelkareem A., Ellestad K., Murison A., Kushida M. M., Coutinho F. J., Ma Y., Mungall A. J., Moore R., Marra M. A., Taylor M. D., Dirks P. B., Pugh T. J., Morrissy S., St Croix B., Mahoney D. J., Lupien M., Gallo M., High-resolution structural genomics reveals new therapeutic vulnerabilities in glioblastoma. Genome Res. 29, 1211–1222 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu L., Chen Y., Dutra-Clarke M., Mayakonda A., Hazawa M., Savinoff S. E., Doan N., Said J. W., Yong W. H., Watkins A., Yang H., Ding L.-W., Jiang Y.-Y., Tyner J. W., Ching J., Kovalik J.-P., Madan V., Chan S.-L., Müschen M., Breunig J. J., Lin D.-C., Koeffler H. P., BCL6 promotes glioma and serves as a therapeutic target. Proc. Natl. Acad. Sci. U.S.A. 114, 3981–3986 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen Y., Xu L., Mayakonda A., Huang M.-L., Kanojia D., Tan T. Z., Dakle P., Lin R. Y. T., Ke X. Y., Said J. W., Chen J., Gery S., Ding L.-W., Jiang Y.-Y., Pang A., Puhaindran M. E., Goh B. C., Koeffler H. P., Bromodomain and extraterminal proteins foster the core transcriptional regulatory programs and confer vulnerability in liposarcoma. Nat. Commun. 10, 1353 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Horn S., Figl A., Rachakonda P. S., Fischer C., Sucker A., Gast A., Kadel S., Moll I., Nagore E., Hemminki K., Schadendorf D., Kumar R., TERT promoter mutations in familial and sporadic melanoma. Science 339, 959–961 (2013). [DOI] [PubMed] [Google Scholar]
- 65.Bleeker F. E., Lamba S., Leenstra S., Troost D., Hulsebos T., Peter Vandertop W., Frattini M., Molinari F., Knowles M., Cerrato A., Rodolfo M., Scarpa A., Felicioni L., Buttitta F., Malatesta S., Marchetti A., Bardelli A., IDH1 mutations at residue p.R132 (IDH1(R132)) occur frequently in high-grade gliomas but not in other solid tumors. Hum Mutat 30, 7–11 (2009). [DOI] [PubMed] [Google Scholar]
- 66.Wang X., Spandidos A., Wang H., Seed B., PrimerBank: A PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res. 40, D1144–D1149 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Langmead B., Trapnell C., Pop M., Salzberg S. L., Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nussbaum C., Myers R. M., Brown M., Li W., Liu X. S., Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Landt S. G., Marinov G. K., Kundaje A., Kheradpour P., Pauli F., Batzoglou S., Bernstein B. E., Bickel P., Brown J. B., Cayting P., Chen Y., DeSalvo G., Epstein C., Fisher-Aylor K. I., Euskirchen G., Gerstein M., Gertz J., Hartemink A. J., Hoffman M. M., Iyer V. R., Jung Y. L., Karmakar S., Kellis M., Kharchenko P. V., Li Q., Liu T., Liu X. S., Ma L., Milosavljevic A., Myers R. M., Park P. J., Pazin M. J., Perry M. D., Raha D., Reddy T. E., Rozowsky J., Shoresh N., Sidow A., Slattery M., Stamatoyannopoulos J. A., Tolstorukov M. Y., White K. P., Xi S., Farnham P. J., Lieb J. D., Wold B. J., Snyder M., ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 22, 1813–1831 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Franz M., Rodriguez H., Lopes C., Zuberi K., Montojo J., Bader G. D., Morris Q., GeneMANIA update 2018. Nucleic Acids Res. 46, W60–W64 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McLeay R. C., Bailey T. L., Motif Enrichment Analysis: A unified framework and an evaluation on ChIP data. BMC Bioinformatics 11, 165 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Raudvere U., Kolberg L., Kuzmin I., Arak T., Adler P., Peterson H., Vilo J., g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47, W191–W198 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Merico D., Isserlin R., Stueker O., Emili A., Bader G. D., Enrichment map: A network-based method for gene-set enrichment visualization and interpretation. PLOS ONE 5, e13984 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M. A., The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang K., Li M., Hakonarson H., ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li B., Dewey C. N., RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Harrow J., Frankish A., Gonzalez J. M., Tapanari E., Diekhans M., Kokocinski F., Aken B. L., Barrell D., Zadissa A., Searle S., Barnes I., Bignell A., Boychenko V., Hunt T., Kay M., Mukherjee G., Rajan J., Despacio-Reyes G., Saunders G., Steward C., Harte R., Lin M., Howald C., Tanzer A., Derrien T., Chrast J., Walters N., Balasubramanian S., Pei B., Tress M., Rodriguez J. M., Ezkurdia I., van Baren J., Brent M., Haussler D., Kellis M., Valencia A., Reymond A., Gerstein M., Guigo R., Hubbard T. J., GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cancer Genome Atlas Research Network , Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N. Engl. J. Med. 372, 2481–2498 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Madhavan S., Zenklusen J.-C., Kotliarov Y., Sahni H., Fine H. A., Buetow K., Rembrandt: Helping personalized medicine become a reality through integrative translational research. Mol. Cancer Res. 7, 157–167 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Iyer M. K., Niknafs Y. S., Malik R., Singhal U., Sahu A., Hosono Y., Barrette T. R., Prensner J. R., Evans J. R., Zhao S., Poliakov A., Cao X., Dhanasekaran S. M., Wu Y. M., Robinson D. R., Beer D. G., Feng F. Y., Iyer H. K., Chinnaiyan A. M., The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 47, 199–208 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lamb J., Crawford E. D., Peck D., Modell J. W., Blat I. C., Wrobel M. J., Lerner J., Brunet J.-P., Subramanian A., Ross K. N., Reich M., Hieronymus H., Wei G., Armstrong S. A., Haggarty S. J., Clemons P. A., Wei R., Carr S. A., Lander E. S., Golub T. R., The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 313, 1929–1935 (2006). [DOI] [PubMed] [Google Scholar]
- 83.Johnson W. E., Li C., Rabinovic A., Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/18/eabd4676/DC1