

Tim-3 adaptor protein Bat3 regulates T cell terminal differentiation and exhaustion in an mTORC2-dependent manner.
Abstract
T cell exhaustion has been associated with poor prognosis in persistent viral infection and cancer. Conversely, in the context of autoimmunity, T cell exhaustion has been favorably correlated with long-term clinical outcome. Understanding the development of exhaustion in autoimmune settings may provide underlying principles that can be exploited to quell autoreactive T cells. Here, we demonstrate that the adaptor molecule Bat3 acts as a molecular checkpoint of T cell exhaustion, with deficiency of Bat3 promoting a profound exhaustion phenotype, suppressing autoreactive T cell–mediated neuroinflammation. Mechanistically, Bat3 acts as a critical mTORC2 inhibitor to suppress Akt function. As a result, Bat3 deficiency leads to increased Akt activity and FoxO1 phosphorylation, indirectly promoting Prdm1 expression. Transcriptional analysis of Bat3−/− T cells revealed up-regulation of dysfunction-associated genes, concomitant with down-regulation of genes associated with T cell effector function, suggesting that absence of Bat3 can trigger T cell dysfunction even under highly proinflammatory autoimmune conditions.
INTRODUCTION
Bat3 is a ubiquitin-like protein that has a number of biological functions including regulation of apoptosis (1), protein acetylation (2), and major histocompatibility complex (MHC) class II expression (3). Furthermore, as a chaperone of the heat-shock protein Hsp70/Hsc70 (4), Bat3 is also involved in protein refolding and the quality control of protein synthesis (5). The Bat3 gene (Bag6) is located in the MHC III cluster adjacent to tumor necrosis factor (TNF) locus. Single-nucleotide polymorphisms in the Bat3 locus are linked to type I diabetes (6) and cancer (7–9), yet little is known about the role of Bat3 in regulating immune responses.
We have previously shown that Bat3 is an adaptor protein that binds to and negatively regulates Tim-3, a checkpoint inhibitor recognized experimentally, and in patients, as an important negative regulator of the immune response in autoimmunity, cancer, and chronic viral infection (10–13). However, the mechanism by which interaction of Bat3 and Tim-3 regulates T cell responses, including T cell activation or exhaustion, has not been fully elucidated. To address this, we generated both Bat3fl/fl and Tim-3fl/fl mice to study the interplay between Bat3 and Tim-3 in vivo using a disease model of autoimmunity. Here, we show that loss of Bat3 in T cells results in a reduction of inflammatory cytokines with a concomitant increase in Tim-3, KLRG1 (killer cell lectin-like receptor G1), and interleukin-10 (IL-10), which together results in reduced disease in a passive transfer model of experimental autoimmune encephalomyelitis (EAE). While Tim-3 signaling partially contributes to Bat3-mediated T cell exhaustion, further mechanistic studies led us to hypothesize that Bat3 may function as a critical regulator of mTOR (mammalian target of rapamycin). We found that Bat3 preferentially suppresses mTORC2 (mTOR complex 2) activity by limiting the role of Hsc70, which is known to enhance mTORC2 function. As a result, Bat3 deficiency leads to increased Akt activity and FoxO1 phosphorylation, thereby inhibiting its transcriptional activity and indirectly promoting Prdm1 expression. This increase in Prdm1/Blimp-1 promotes expression of a module of coinhibitory molecules including Tim-3 and IL-10, which we have previously identified in tumor-infiltrating T cells (14). In support of this, analysis of the differentially expressed (DE) genes from Bat3- and Tim-3–deficient T helper 1 (TH1) cells demonstrated a substantial overlap of DE genes from Bat3-deficient T cells with the exhaustion module observed in CD8 tumor-infiltrating lymphocytes (TILs) (15), further suggesting that Bat3 may play a critical role in curtailing T cell dysfunction.
Together, here, we identify a previously unidentified mechanism by which Bat3 not only directly suppresses Tim-3 signaling but also plays a critical role in controlling the mTORC2–Akt–Blimp-1 pathway, thereby suppressing the induction of Tim-3 and preventing terminal differentiation and dysfunction in T cells.
RESULTS
Tim-3 signaling partially contributes to Bat3-mediated T cell exhaustion
We have previously shown that Bat3 binds to the cytoplasmic tail of Tim-3 and inhibits its downstream signaling. However, the mechanism by which Tim-3:Bat3 interactions regulate T cell responses, including T cell activation or exhaustion, had not been fully elucidated. To gain insight into the role of Bat3 in the regulation T cell responses at the effector stages of encephalitogenic T cell responses, we generated Bat3 conditional knockout mice by crossing Bat3fl/fl × CD4Cre (Bat3cko) mice (design of Bat3fl/fl shown in fig. S1). Active immunization with MOG35–55/CFA (myelin oligodendrocyte glycoprotein35–55/complete Freund’s adjuvant) in Bat3cko mice showed attenuated disease severity compared with wild-type (WT) littermate mice (Bat3fl/fl) (fig. S2). However, because CD4cre can delete genes in multiple cell types including CD4+, CD8+, and FoxP3+ T cells, we wanted to determine the effect of loss of Bat3 exclusively on encephalitogenic effector T cells. Therefore, we generated Bat3cko × 2D2 mice by crossing Bat3cko mice with myelin antigen MOG35–55–specific T cell receptor (TCR) transgenic mice (16). As Tim-3 is preferentially expressed on TH1 and Tc1 cells (17), we isolated CD4 T cells from Bat3cko × 2D2 and Bat3fl/fl × 2D2 mice, in vitro differentiated the cells into TH1 cells, and transferred them into C57BL/6 recipient mice to follow development of EAE. We found that Bat3cko × 2D2 TH1 cell–transferred mice developed significantly less severe EAE compared with Bat3fl/fl × 2D2 (WT) TH1 cell–transferred mice, suggesting that loss of Bat3 diminishes the encephalitogenic potential of effector TH1 cells (Fig. 1A). When we transferred in vitro differentiated pathogenic TH17 cells from Bat3cko × 2D2 and Bat3fl/fl × 2D2 mice into B6 mice, no difference in disease progression or severity was found between these two experimental groups (fig. S3). Thus, the passive EAE results demonstrated a specific role of Bat3 in effector TH1 cells.
Fig. 1. Tim-3 signaling partially contributes to Bat3-mediated T cell exhaustion.
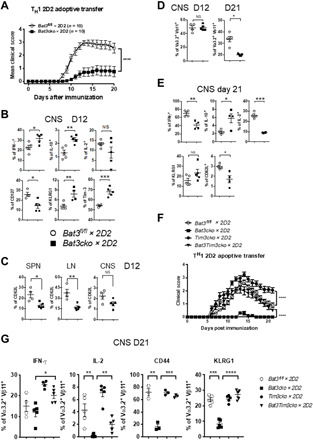
(A) CD4 T cells from Bat3cko × 2D2 and WT 2D2 mice were in vitro differentiated into TH1 cells, and 4 × 106 cells were transferred into C57BL/6 recipient mice to induce EAE. Disease progression was monitored on daily basis until the end of the experiment. Mean disease scores are shown as indicated. (B to E) Ex vivo analyses on transferred 2D2 cells (Va3.2+Vb11+) in the central nervous system (CNS), spleen (SPN), and lymph node (LN) on day 12 (D12) and day 21 (D21) were performed to determine the phenotype of Bat3-deficient CD4 T cells during EAE induction. Error bars indicate mean SEM [*P = 0.01, **P = 0.0085, and ***P = 0.006, unpaired two-tailed t test; ****P = 0.0001, two-way analysis of variance (ANOVA)]. NS, not significant. (F) Naïve CD4 T cells were isolated from Bat3fl/fl, Tim-3cko, Bat3cko × 2D2, and Tim-3×Bat3dko × 2D2 mice and were in vitro differentiated into TH1 cells; 4 × 106 cells were transferred into C57BL/6 recipient mice to induce EAE. Disease progression was monitored on daily basis till the end of experiment. Mean disease scores were shown as indicated. Statistical significance between Tim-3cko × 2D2 and Tim-3×Bat3dko × 2D2 was reached on D12 (**), increasing in significance until cessation of experiment. Statistical significance between Bat3cko × 2D2 and Tim-3 × Bat3dko × 2D2 mice was reached on D8 (*), increasing in significance until cessation of experiment. (G) Ex vivo analyses on transferred 2D2 cells (Va3.2+Vb11+) in the CNS on D21 was performed to determine the phenotype of Bat3fl/fl, Tim-3cko, Bat3cko × 2D2, and Tim-3 × Bat3dko–deficient CD4 T cells during EAE induction. Data represent three independent experiments. Error bars indicate mean SEM (*P = 0.01 and ***P = 0.0012, unpaired two-tailed t test; ****P = 0.0001, two-way ANOVA).
We next assessed the phenotype of the transferred Bat3fl/fl and Bat3cko 2D2 TH1 cells ex vivo at disease onset (day 12 after transfer) and found that the frequency of Tim-3–expressing cells in the central nervous system (CNS) was higher in Bat3cko T cells. However, Bat3cko 2D2 cells displayed a higher percentage of interferon-γ (IFN-γ) positivity, albeit concomitant with increased IL-10 (Fig. 1B). The unexpected increase in the IFN-γ population among Bat3cko TH1 led us to consider whether there were changes in the effector status of these cells. Therefore, we analyzed the CNS-infiltrating transferred 2D2+ TH1 cells for selected markers of effector T cells and found reduced expression of the memory marker IL-7R (CD127) but enhanced expression of the effector molecule KLRG1 in Bat3cko × 2D2 TH1 cells (Fig. 1B). In addition, CD62L expression on 2D2 T cells from peripheral lymphoid organs (lymph node and spleen) was significantly reduced in the absence of Bat3; however, no differences were observed in the CNS (Fig. 1C). Although equal numbers of Bat3cko × 2D2 and Bat3fl/fl × 2D2 TH1 cells were transferred into recipient C57BL/6 mice, and no difference in cell frequencies was detected on day 12, there was a marked reduction of Bat3cko × 2D2 TH1 cells within the CNS on day 21 after transfer, indicating a more rapid contraction of transferred TH1 cells with Bat3 deficiency (Fig. 1D). When cytokine production was measured on day 21 after transfer, we found significantly reduced IFN-γ and IL-2 yet sustained higher levels of IL-10 in Bat3fl/fl × 2D2 cells (Fig. 1E). While there was no difference in Tim-3 expression at this stage of the disease, we did observe a decreased expression of PD-1 (programmed cell death protein 1) on CNS infiltrating T cells (fig. S4). Collectively, these results suggest that Bat3cko × 2D2 TH1 cells accelerate terminal differentiation and are prone to develop a dysfunctional phenotype in vivo.
To confirm these findings, we used a well-established in vitro system in which naïve (CD62Lhi) CD4+ T cells are successively cultured and restimulated (polarized) under conditions that drive TH1 responses. Under these chronically activated conditions, TH1 cells express high levels of Tim-3 by the third successive polarization (17). Using this approach, we found that Bat3cko TH1 differentiated cells actually produced more IFN-γ than Bat3fl/fl TH1 cells during the first round of polarization. However, the Bat3-deficient TH1 cells lost the ability to produce IFN-γ after three rounds of polarization (fig. S5).
We have previously shown that deficiency of Bat3 in T cells not only increases Tim-3 expression but also results in enhanced Tim-3 function (11). However, it was not clear whether elevated Tim-3 expression contributed to the phenotype observed in Bat3cko CD4 T cells (Fig. 1A) or whether increased Tim-3 expression and signaling is simply a biological outcome of loss of Bat3 in T cells.
To better understand the molecular mechanism behind Bat3-mediated T cell dysfunction, we generated Bat3 and Tim-3 single and double conditional knockout mice in T cells (by crossing with CD4Cre transgenic mice). To focus on the role of Tim-3 and Bat3 on TH1 immunity, we further bred Tim-3fl/fl × CD4Cre (Tim-3cko) and Bat3fl/fl × Tim-3fl/fl × CD4Cre (Tim-3 × Bat3dko) mice onto 2D2 TCR transgenic background (16). Using CD4 T cells from these mice, we generated TH1 cells in vitro from all four genotypes and transferred the cells into C57BL/6 mice to induce EAE. Consistent with the results presented in Fig. 1A, Bat3cko × 2D2 TH1 cells induced very mild EAE, while Tim-3cko × 2D2 TH1 cells induced significantly more severe EAE than the WT Bat3fl/fl × 2D2 TH1 cells. Concomitant deletion of Tim-3 and Bat3 (Tim-3Bat3dko × 2D2 TH1 cells) completely reversed the encephalitogenic phenotype of Bat3cko 2D2 TH1 cells and showed induction of EAE similar to what was observed in the WT mice (Fig. 1F). Given their enhanced capacity to induce disease, we found that CNS-infiltrating Tim-3cko × 2D2 TH1 cells showed elevated IFN-γ and IL-2 production relative to controls. The Tim-3 × Bat3 dko × 2D2 [double knockout (DKO)] TH1 cells shared features with both Bat3cko × 2D2 TH1 cells and Tim-3cko × 2D2 TH1 cells. Examining IFN-γ production, the DKO behaved more like Tim-3cko. Yet, with regard to IL-2, the DKO cells looked were more comparable to the Bat3cko T cells.
However, overall, the frequency of the CNS-infiltrating effector TH1 cells (CD44+; KLRG1+) in Tim-3 × Bat3dko was similar to that of Tim-3cko and WT TH1 cells, indicating that deletion of Tim-3 rescued the phenotypic defects of Bat3 deficiency in TH1 cell differentiation (Fig. 1G). Together, these results suggested that Tim-3 signaling substantially contributes to the Bat3-deficient phenotype, and the deletion of Tim-3 is sufficient to rescue the pathogenicity of Bat3 deficient TH1 cells during passive EAE induction.
Tim-3+ TH1 cells represent terminally differentiated and dysfunctional T cells
Although high levels of Tim-3 can be detected on CNS-infiltrating T cells during EAE induction, there has been poor characterization of these Tim-3–expressing cells to date. To examine the role of Tim-3 expression in T cell expansion in vivo, we immunized C57BL/6 mice with MOG35–55/CFA to induce EAE. At the peak of the disease (day 10), we injected mice with 5-bromo-2′-deoxyuridine (BrdU) 1 day before isolating CNS-infiltrating effector CD4+ (FoxP3−) T cells. Using flow cytometry analysis, we found that BrdU-incorporated proliferating cells were predominantly Tim-3− CD4 T cells. In contrast, Tim-3+ cells were mostly BrdU negative, indicating that Tim-3+ CD4 T cells were not proliferating in vivo. In contrast, PD-1 expression was largely associated with BrdU incorporation, consistent with the data that PD-1 is expressed on recently activated and proliferating cells, a finding that has been noted in other studies (19). We also calculated the ratio (fold) differences between BrdU+PD-1+/BrdU+PD-1− and BrdU+Tim-3+/BrdU+Tim-3− cells. Predominantly, PD-1+ cells, but not PD-1−, were labeled with BrdU. In contrast, predominantly, Tim-3+ cells, but not Tim-3− cells, were BrdU negative (Fig. 2A). These results clearly suggest distinct roles between Tim-3 and PD-1 in controlling effector CD4 T cell responses during inflammation, and that Tim-3 expression may antagonize T cell proliferation in vivo.
Fig. 2. Tim-3+ TH1 cells represent terminal differentiated and dysfunctional T cells.
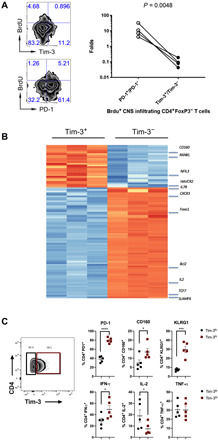
(A) WT (FoxP3-GFP KI) mice were immunized with MOG35–55/CFA to induce EAE. At the peak of the disease (day 10), the immunized mice were intraperitoneally injected with BrdU and were euthanized the following day to isolate CNS-infiltrating CD4+FoxP3− T cells for flow cytometry analysis. Flow cytometry data (to the left) represent results from four mice from the same experiment. Further, the ratio (fold) differences between Brdu+PD-1+/Brdu+PD-1− and Brdu+Tim-3+/Brdu+Tim-3− cells were analyzed by ratio pair t test and were shown to the right (P = 0.0048). (B) FoxP3-GFP KI mice were immunized with MOG35–55/CFA to induce EAE. At the peak of the disease (day 10), Tim-3+FoxP3− and Tim-3−FoxP3− CD4 T cells were isolated from the CNS of EAE mice by cell sorting. Total RNA samples were prepared for Nanostring analysis using in-house–designed Nanostring code set (14). Cells from three EAE mice were analyzed as indicated in each individual column. (C) WT mice were immunized with MOG35–55/CFA to induce EAE. On day 14, CNS-infiltrating T cells were isolated. CD4+ Foxp3−; Tim-3hi/lo cells were analyzed by flowcytometry for expression of PD-1, CD160, and KLRG1. Data shown (C) as mean ± SEM. *P < 0.05; ***P < 0.001; ****P < 0.0001 (Student two-tailed t test).
We next wanted to explore the impact of Tim-3 signaling on effector CD4 T cell function in the context of EAE. To exclude Tim-3+ Foxp3+ T cells, we immunized FoxP3–green fluorescent protein (GFP) knock-in mice with MOG35–55/CFA, subsequently isolated Tim-3+FoxP3− and Tim-3−FoxP3− CD4 T cells from the CNS of MOG-immunized animals, and performed NanoString analysis on these cells. As we had observed acquisition of a dysfunctional phenotype on Bat3-deficient T cells, we used a custom code set representing the dysfunctional CD8+ TIL gene signature (245 genes), to interrogate the potential role of Tim-3 in the regulation of effector CD4 T cells. Because there is an overlap between the IL-27–regulated coinhibitory module and the dysfunctional CD8+ TIL signature (14), and IL-27 is an important regulator of Tim-3 expression via transcription factors T-bet, NFIL3, and Blimp-1 (14, 19), we further included a custom gene set from the IL-27–driven gene signature for a total of 397 genes. We found 94 DE genes, with 26 more highly expressed in Tim-3+ cells and 68 more highly expressed in Tim-3− cells. Further analysis showed that the expression profile of Tim-3+ cells exhibited highly enriched signals for Prdm1 and Nfil3 (Fig. 2B), supporting our previous observations of the critical roles of these transcription factors in Tim-3 gene expression (14, 19). In contrast, Tim-3− CD4+ T cells demonstrated significantly elevated levels of Tcf7, Bcl2, and Foxo1, all of which have been shown to promote memory and stem-like T cells but suppress effector T cell differentiation and proliferative capacity (Fig. 2B). Tim-3+ T cells also exhibited elevated expression of other checkpoint inhibitors including Lag3 and Pdcd1 and lost their capability to express Il2, which is important for cell survival. Because these cells were still functionally capable of expressing Ifng and Tnfa, Tim-3+ T cells collected from mice at the peak of CNS inflammation phenotypically resembled terminally differentiated effector TH1 T cells.
We further analyzed CNS-infiltrating CD4 effector T cells from mice 12 days after active induction of EAE by MOG35–55 immunization. We found that almost 100% of Tim-3hi CD4 T cells coexpressed PD-1, but very few Tim-3lo cells were PD-1 negative. In addition, we investigated the expression levels of KLRG1 and CD160, both widely appreciated markers of terminal differentiation, and found increased expression of both on Tim-3hi cells compared with Tim-3lo cells (Fig. 2C). We further investigated the expression of cytokines within these two populations and found significantly elevated expression of IFN-γ, while TNF-α remained unchanged. IL-2 was significantly decreased, which is in line with our findings using Bat3cko T cells (Fig. 2C). Together, these data show that Tim-3hi TH1 cells exhibited cardinal features of terminally differentiated effector cells.
Bat3 deficiency results in increased Akt phosphorylation at S473
Our data thus far suggested that Bat3-deficient T cells, with elevated expression of Tim-3, may undergo accelerated contraction upon activation. We have previously shown that Tim-3 binds to the TCR-associated intracellular kinase Lck (11), which led us to question the role of Bat3 downstream of TCR activation. To further understand the role of Bat3 in T cell activation, we stimulated in vitro cultured CD4 T cells from Bat3cko and Bat3fl/fl mice with anti-CD3 and anti-CD28 to examine the impact of Bat3 deficiency on TCR-dependent signaling. We assessed transducers of the TCR pathway and found no notable differences in phosphorylation of protein kinase C or extracellular signal–regulated kinase 1/2 between Bat3cko and Bat3fl/fl CD4 T cells (Fig. 3). Although, an increased phosphorylation of phospholipase C, Y783 was found in Bat3fl/fl T cells, this transiently increased phosphorylation 1 hour after TCR stimulation was not sustained. As the Akt/mTOR pathway is a key driver of murine CD4+ T cell differentiation, we assessed and found significantly elevated Akt S473 phosphorylation in Bat3cko T cells after anti-CD3/CD28 costimulation (Fig. 3). In contrast, we did not detect a significant difference in Akt T308 phosphorylation between Bat3cko and Bat3fl/fl CD4 T cells, suggesting that phosphatidylinositol 3-kinase (PI3K)–PDK1 (pyruvate dehydrogenase kinase 1) activation was not affected by Bat3 deficiency. Phosphorylation of PI3K and PDK1 S241 showed little difference. We also detected a slight increase in 70S6K phosphorylation, suggesting that Bat3 deficiency may only affect kinase activity downstream of the PI3K-PDK1 pathway likely due to increased Akt activation (Fig. 3). Together, while there may be some subtle changes in response to TCR activation, Bat3 deficiency preferentially enhances Akt kinase activity without significantly affecting other downstream mediators of TCR activation.
Fig. 3. Bat3 deficiency results in increased Akt phosphorylation at S473.
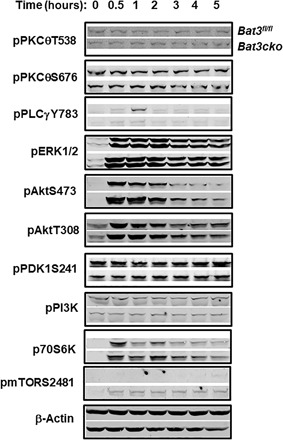
Total CD4 T cells from Bat3fl/fl or Bat3cko mice were activated with plate bound anti-CD3 and anti-CD28 antibodies for 2 days. Cells were rested for additional 2 days afterward. After overnight serum starvation, the cells were stimulated with anti-CD3 and anti-CD28 antibodies for indicated time points. Whole-cell lysates were prepared, and Western blot was performed to analyze the TCR and mTOR signaling pathways. Results represent at least three independent experiments.
As mTORC2 is the major regulator for Akt S473 phosphorylation (20), we analyzed phospho-mTOR and found that Bat3cko CD4 T cells have increased phosphorylation at mTOR S2481 after anti-CD3 and anti-CD28 stimulation (Fig. 3). Together, these data suggest that the function of Bat3 is preferentially involved in regulating the mTOR-Akt signaling axis through mTORC2.
Bat3 interacts with Rictor to regulate mTORC2 function
While regulatory-associated protein of mTOR (Raptor) is only found in mTOR complex 1 (mTORC1), rapamycin-insensitive companion of mammalian target of rapamycin (Rictor) specifically binds to mTORC2 (21). To understand the molecular mechanism of Bat3 in the regulation of mTORC2 activity, we performed Bat3 coimmunoprecipitations (co-IPs) from whole-cell lysates from EL4 T cell lymphomas using antibodies against Rictor, Raptor, and mTOR. We found that Bat3 was preferentially immunoprecipitated by anti-Rictor antibody, but not anti-Raptor, suggesting that Bat3 may be associated with the mTORC2 complex (Fig. 4A). This important finding of Bat3-Rictor interaction was confirmed in primary TH1 cells with Bat3cko T cells as a control (fig. S6A). As the interaction with Rictor is required for mTOR kinase activity, the regulation of Rictor binding to mTOR would be an important mechanism to control mTORC2 activity (22, 23). To investigate whether Bat3 may play a role in this regulation, we examined the interaction of mTOR and Rictor in the presence or absence of Bat3. We first established four cell lines transducing EL4 cells with Bat3-expressing retrovirus (Bat3-RV), empty vector (GFP-RV), Bat3-sh RNA-expressing RV (Bat3-sh-RV), and empty vector (ctrl-sh-RV), respectively (fig. S6B). We then performed anti-mTOR co-IP with cell lysates from the four established EL4 cell lines to examine the mTOR-Rictor interaction. We found that overexpression of Bat3 in EL4 cells reduced the interaction of mTOR with Rictor as compared with WT levels of Bat3 (Fig. 4B). In contrast, the Rictor-mTOR interaction was increased when Bat3 expression was knocked down (Fig. 4B). Thus, the interaction between mTOR and Rictor can be perturbed depending on intracellular Bat3 availability, indicating that the interaction between Bat3 and Rictor could represent a previously unidentified regulatory mechanism for mTORC2 function.
Fig. 4. Bat3 interacts with Rictor to regulate mTORC2 function.
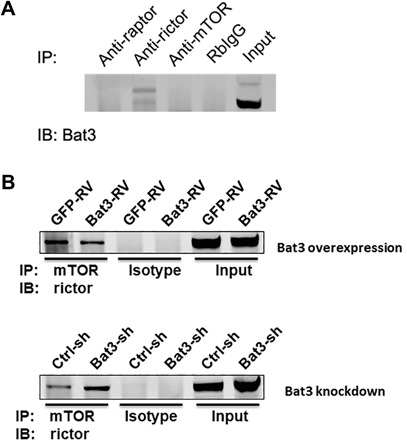
(A) Equal concentration of EL4 cell lysate was used in co-IP using antibodies to Rictor, Raptor, mTOR, and isotype rabbit immunoglobulin G (IgG), respectively. Protein samples were analyzed by Western blot to examine the signal for Bat3. IB, immunoblot. (B) Whole-cell lysates were prepared from EL4 cells transduced with Bat3-expressing retrovirus (Bat3-RV), empty vector (GFP-RV), Bat3-sh RNA-expressing RV (Bat3-sh-RV), and empty vector (ctrl-sh-RV), respectively. Cell lysates were incubated with anti-mTOR antibody (Ab), respectively, to immunoprecipitate mTOR complex to examine the signal of coprecipitated Rictor.
Bat3 suppresses Hsc70-mediated activation of mTORC2
Bat3 is known for its cochaperone function by interacting with Hsc70/Hsp70 as a nucleotide exchange factor (24). Interaction between Bat3 and Hsc70 facilitates the release of adenosine 5′-diphosphate and Hsc70 substrate (25). Hsc70 was recently found to directly bind Rictor to facilitate the Rictor-mTOR interaction and mTORC2 function. The absence of Hsc70 destabilizes the mTORC2 complex and attenuates Akt S473 phosphorylation (22). We therefore investigated the role of constitutively expressed Hsc70 in the regulation of mTORC2 function. By performing anti-Rictor and anti-mTOR co-IP, we demonstrated an interaction between Rictor and Hsc70, but not between mTOR and Hsc70. This interaction can be suppressed by addition of 1 mM adenosine 5′-triphosphate (ATP) (Fig. 5A). In addition, we found that the Rictor-Hsc70 interaction is significantly increased in the absence of Bat3 as detected in Bat3cko T cells. We could detect Hsc70-mTOR interaction in the absence of Bat3, which may be an indirect interaction mediated by enhanced binding of Hsc70 to Rictor in the absence of Bat3 (Fig. 5A). This suggests that Bat3 may limit the access of Hsc70 to mTORC2, thereby destabilizing this protein complex.
Fig. 5. Bat3 suppresses Hsc70-mediated activation of mTORC2.
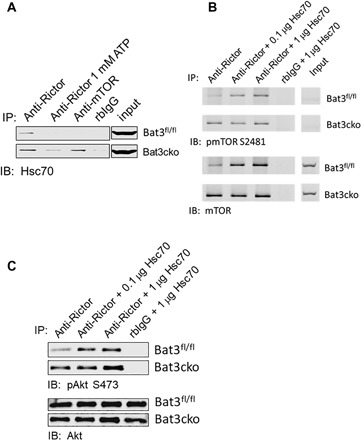
(A) CD4 T cells from Bat3fl/fl and Bat3cko mice were activated with plate-bound anti-CD3 and anti-CD28 antibodies for 2 days. Cells were rested for an additional 2 to 3 days, and whole-cell lysates were prepared for anti-Rictor and anti-mTOR co-IP. When anti-Rictor co-IP was performed, 1 mM adenosine 5′-triphosphate (ATP) was added in a control sample to trigger adenosine triphosphatase (ATPase) activity of Hsc70. Eluted protein samples were analyzed by Western blot to determine the binding of Hsc70 to the mTORC2 complex. (B) Bat3fl/fl and Bat3cko CD4 T cell whole-cell lysates were prepared for anti-Rictor or isotype IgG co-IP with or without the presence of indicated doses of Hsc70. Immunoblot was performed to analyze the phosphorylation at mTOR S2481 (top), and total mTOR coprecipitated by anti-Rictor (bottom). (C) Co-IP experiment was performed as described in (B), and Akt phosphorylation at S473 by the mTORC2 complex was analyzed by Western blot.
To investigate the functional consequence of the absence of Bat3, we performed anti-Rictor co-IP and compared phosphorylation at mTOR S2481, an indicator of autoactivation of mTOR between WT and Bat3cko T cells. We demonstrated that in the absence of Bat3, there was greater interaction between mTOR and Rictor demonstrated by anti-Rictor co-IP. This result was in line with the increased interaction of mTOR-Rictor interaction in Bat3 knockdown CD4 T cells (Fig. 4B). Further addition of exogenous Hsc70 could strengthen the mTOR-Rictor interaction in WT CD4 T cell–derived cell lysates to the comparable levels observed in Bat3cko CD4 T cell–derived cell lysates (Fig. 5B). This enhanced mTOR binding to Rictor also associated with increased phosphorylation of mTOR S2481. This result indicated that Bat3cko CD4 T cells have more active mTOR function than WT CD4 T cells. Exogenous Hsc70 could override the negative regulatory effect of Bat3. Our findings suggest that this elevated mTOR-Rictor interaction is likely due to enhanced Hsc70 binding to the mTORC2 complex in the absence of Bat3. We further conducted an in vitro kinase assay by adding recombinant Akt to mTORC2 complex purified from the anti-Rictor co-IP. Results showed a higher level of Akt S473 phosphorylation when recombinant Akt was incubated with Bat3-deficient mTORC2 complex than with Bat3-sufficient mTORC2 complex. Addition of exogenous recombinant Hsc70 increased Akt S473 phosphorylation (Fig. 5C). Together, our data suggest that Bat3 functions as a negative regulator of the mTORC2 complex by limiting the role of Hsc70 in mTORC2 activation.
Bat3 regulates FoxO1–Blimp-1 activity
A key phenotype of Bat3-deficient T cells is increased Tim-3 expression. We and others have recently demonstrated Blimp-1 to be a key transcription factor driving the expression of Tim-3 and a module of coinhibitory molecules in dysfunctional CD8 T cells in tumors (14, 26). To understand the mechanism behind the induction of Tim-3 in Bat3cko concomitant with the development of a dysfunctional phenotype in Bat3cko T cells, we assessed the expression of Blimp-1 (Prdm1). We found increased expression of Prdm1 in Bat3cko T cells compared to WT controls (Fig. 6A), and this was confirmed at the protein level by immunoblot (Fig. 6B). In contrast to Blimp-1, FoxO1 promotes memory T cells but suppresses effector T cell differentiation (27) and has been reported to suppress Prdm1 transcription through induction of Bcl6 (28).
Fig. 6. Bat3 regulates FoxO1–Blimp-1 activity.
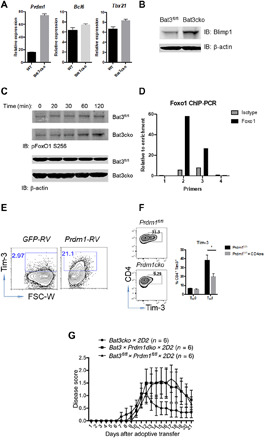
Naïve Bat3fl/fl and Bat3cko CD4+ T cells were in vitro differentiated into TH1 cells. (A) qPCR to determine the transcripts of Prdm1, Bcl6, and Tbx21 after 3 days. (B) Whole-cell lysates (WCLs) were prepared for Western blot to detect Blimp-1 expression. (C) Cells were activated with anti-CD3/CD28 for 2 days and rested for 2 days afterward. After overnight serum starvation, cells were stimulated with anti-CD3/CD28 for indicated time points. WCL were prepared and immunoblotted for pFoxO1 S256. (D) Four ChIP-PCR primer sets were used to confirm binding at selected sites of FoxO1 to Blimp-1 locus. (E) Naïve CD4+ T cells were activated with anti-CD3/CD28 for 1 day and retrovirally transduced with control (GFP-RV) or Blimp-1–expressing vector (Prdm1-RV). Tim-3 expression was analyzed 3 days later. (F) Naïve Prdm1fl/fl or Prdm1fl/fl cko CD4+ T cells were stimulated under TH0 and TH1 conditions with anti-CD3/CD28. Tim-3 expression was assessed after 72 hours. (G) CD4+ T cells from Bat3cko × 2D2, Bat3fl/fl × Prdm1fl/flcko × 2D2, and Bat3fl/fl × Prdm1fl/fl dko × 2D2 mice were differentiated into TH1 cells and transferred into C57BL/6 recipient mice. Disease progression was monitored daily; mean disease scores are shown as indicated. Results represent at least two independent experiments. Error bars indicate means ± SEM (*P = 0.01, unpaired two-tailed t test).
Because Akt (which was enhanced in Bat3cko T cells; Fig. 3) plays a critical role in the regulation of FoxO1 cellular distribution and transcriptional function, we therefore decided to investigate the role of Bat3 regulation of FoxO1. We first stimulated in vitro cultured WT and Bat3cko CD4 T cells with anti-CD3/anti-CD28 and found increased FoxO1 S256 phosphorylation in Bat3cko T cells (Fig. 6C). This result is in line with the increased Akt activity observed in Bat3cko T cells (Fig. 3). Next, we prepared nuclear and cytoplasmic proteins from Bat3fl/fl and Bat3cko CD4 T cells after 2-hour stimulation with anti-CD3 and anti-CD28. Western blot result showed that the total amount of FoxO1 (combination of nuclear and cytoplasmic) were similar between Bat3fl/fl and Bat3cko cells. However, when comparing protein subcellular distribution, we found that nuclear FoxO1 was greater in Bat3cko cells than in WT Bat3fl/fl cells, but the trend was opposite in cytoplasm, indicating that the increased FoxO1 phosphorylation affected the protein cellular distribution (fig. S7A).
The reduced nuclear localization of FoxO1 indicates attenuated transcriptional suppression. Therefore, to explore the possible regulatory role of Foxo1 in Blimp-1 transcription, we analyzed FoxO1 chromatin immunoprecipitation (ChIP) sequencing data (GSE60470) (29) and identified direct FoxO1 binding to the Prdm1 locus (fig. S7B), including one binding site in the proximal promoter region, which was confirmed by ChIP–quantitative polymerase chain reaction (qPCR) (Fig. 6D). To confirm the direct suppression of transcription initiation of Prdm1 by FoxO1, we generated a Blimp-1 promoter luciferase reporter construct. Cotransfection of the Prdm1-promoter reporter construct with the FoxO1 expression plasmid suppressed Prdm1 promoter-driven luciferase expression (fig. S7C).
Last, to investigate whether increased Blimp-1 expression contributes to the phenotype of Bat3-deficient T cells, we performed retroviral overexpression of Blimp-1 in naïve CD4 T cells, which led to enhanced Tim-3 expression (Fig. 6E). Conversely, consistent with our previous results (14), comparison of Tim-3 expression between Prdm1fl/fl × CD4-Cre and WT (Prdm1fl/fl) CD4 T cells demonstrated that Blimp-1–deficient cells expressed less Tim-3 than WT cells (Fig. 6F).
Collectively, our data suggested that FoxO1 transcription function is regulated by mTORC2 pathway. The absence of Bat3 results increases mTORC2/Akt activity and subsequently increases FoxO1 phosphorylation and redistribution of FoxO1 to cytoplasm. Hence, its transcriptional suppression of Blimp-1 is attenuated. The increased Blimp-1 expression contributes to increased Tim-3 expression, TH1 cell terminal differentiation, and dysfunction in Bat3cko CD4 (TH1) cells (illustrated in Fig. 8).
Fig. 8. Graphical abstract.
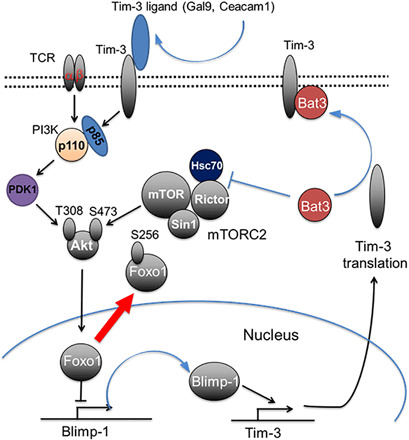
Model for Bat3-mediated mTORC2–Akt–Blimp-1–Tim-3 pathway regulation in T cell terminal differentiation and exhaustion.
To validate this hypothesis, we generated Bat3fl/fl × Prdm1fl/fl cko (Bat3 × Prdm1dko) × 2D2 mice to perform passive EAE induction with differentiated 2D2 TH1 cells derived from these mice together with control group cells from Bat3fl/fl × Prdm1fl/fl × 2D2 and Bat3cko × 2D2 mice. Results showed that codeletion of Blimp-1 in Bat3-deficient T cells was able to rescue the encephalitogenic potential of transferred 2D2 cells (Fig. 6G). We additionally performed an adoptive T cell transfer model whereby Bat3fl/fl, Bat3cko, Prdm1cko, and Bat3 × Prdm1dko were immunized on day 0 with MOG35–55 peptide. On day 8, spleens and lymph nodes were harvested and expanded in the presence of MOG+IL-12+ anti–IL-4. On day 3 after harvest, isolated CD4+ T cells were injected into C57BL/6 recipient mice. Using this experimental setting, we observed significantly worse disease in animals that received Blimp-1–deficient T cells, and that codeletion of Blimp-1 in Bat3-deficient T cells could rescue their encephalitogenic potential (fig. S7D). Together, these EAE data support the critical role of Blimp-1 transcriptional function downstream of Bat3/mTORC2/Akt pathway in the regulation of TH1 cells terminal differentiation and dysfunction.
Bat3 deficiency promotes an exhaustion/dysfunctional profile in T cells
Our data thus far suggested that unrestrained mTORC2/Akt/Blimp-1 pathway in Bat3cko cells drives TH1 cell terminal differentiation and exhaustion. From our earlier EAE studies (Fig. 1, F and G), we were able to demonstrate that Tim-3 is as a key effector molecule downstream of this pathway. To gain a deeper understanding of the role of Tim-3 and Bat3 pathways in the regulation of T cell function, we performed a whole genome RNA sequencing (RNA-seq) analysis of Bat3- and Tim-3–deficient CNS-infiltrating TH1 cells from mice undergoing EAE. We found that many genes were reciprocally regulated by Tim-3 and Bat3 (157 genes were up in Bat3KO and 132 genes were up in Tim-3KO; Fig. 7A and table S1), consistent with our earlier findings that Bat3 is a negative regulator of Tim-3 (Fig. 1F) (11). Analysis of the DE genes between Bat3- and Tim-3–deficient TH1 cells demonstrated that the genes up-regulated in Bat3cko cells were significantly associated with genes from the dysfunction_not_activation module that we have described in TILs (P = 0.0002) (Fig. 7, B and C) (15). The dysfunction_not_activation module is associated with T cell exhaustion in the tumor microenvironment, and the genes up-regulated in Tim-3cko T cells were not associated with this module (P = 0.31). Further analysis revealed that up-regulated genes in Bat3cko T cells were enriched in gene signatures from data generated in additional studies for anergy (30), tolerance (31, 32), and dysfunction_not_activation (15), but not with signatures associated directly with CD8 activation (32, 33) or effector cell function of CD8 T cells (Fig. 7C) (33, 34). This may be partly because of increased Blimp-1 expression observed in Bat3cko T cells. We (14) and others (26, 35) have previously shown that Blimp-1 is a key transcription factor that drives the expression of a whole module of coinhibitory gene in exhausted T cells, and other studies have shown that Blimp-1 can serve as a key transcriptional regulator balancing effector CD8 T cell function with T cell exhaustion, whereby high Blimp-1expression tips the balance toward T cell exhaustion/dysfunction. We investigated whether the up-regulated genes in Bat3cko T cells were enriched for a Blimp-1 signature. We found that the genes that are up in the Bat3cko, as compared to the Tim-3cko T cells, are significantly enriched for targets of Blimp-1 including coinhibitory genes Tim-3, TIGIT (T cell immunoreceptor with Ig and ITIM domains), and Lag-3, together with IL-10 (genes that are up- and/or down-regulated upon Prdm1 KO; P < 10−17). This is not true for genes up-regulated in the Tim-3KO compared to the Bat3KO (P = 0.20) (Fig. 7D). We have previously identified IL-27 as a key cytokine responsible for driving the expression of a module of checkpoint molecules in cancer, in part via Blimp-1. Using the single-cell RNA-seq (scRNA-seq) dataset of 316 CD4+ T cells from B16F10 melanoma, we defined three clusters identified as regulatory T (Treg) (0), dysfunctional CD4 (1), and naïve like (2). As a reference, we projected the IL-27 coinhibitory gene module signature onto this scRNA-seq data, and the signature highlighted clusters 0 (Tregs) and 2 (dysfunctional) CD4 (2) (Fig. 7E). Projection of the gene signature up-regulated in Bat3cko TH1 cells overlaps not only with that of IL-27 but also chronic viral infection signature crossed to IL-27 (Fig. 7E). The Bat3cko signature furthermore scored highly for overlap between several other states of T cell nonresponsiveness including anergy and tolerance (fig. S8). Together, our results show that Bat3 has a role in regulating the naïve effector versus memory T cell response in autoimmune disease setting and that loss of Bat3 promotes T cell dysfunction via the Akt/mTOR/Blimp-1 axis.
Fig. 7. Bat3 deficiency promotes an exhaustion/dysfunctional profile in T cells.
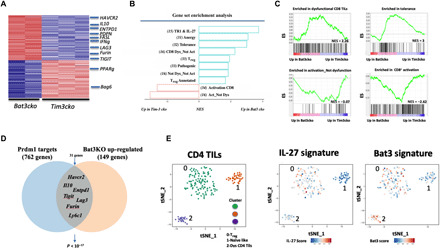
Naïve CD4 T cells were isolated from Bat3cko × 2D2 and Tim-3cko × 2D2 and in vitro differentiated into TH1 cells; 4 × 106 cells were transferred into C57BL/6 recipient mice to induce EAE. (A) Gene expression in Tim-3cko and Bat3cko 2D2 cells (Va3.2+Vb11+) isolated from CNS at peak of disease (day 14) for population RNA-seq. DE genes (289) are shown as a heatmap. (B) Enrichments of different signatures from literature in Bat3cko versus Tim-3cko cells, determined using GSEA preranked analysis. Selected signatures with an adjusted P < 0.05 are shown, and references for signatures are numbered in italics. (C) Descriptive leading-edge plots for some of the signatures. NES, normalized enrichment score. (D) Graphical representation of the selected overlap genes between the Prdm1cko and Bat3cko CD4 T cells. (E) A t-distributed stochastic neighbor embedding (t-SNE) plot of the 316 CD4+ TILs obtained from WT mice bearing B16F10 melanoma tumors (14), colored by the relative signature score for Bat3cko versus Tim-3cko module (36 genes; table S2) and the IL-27 TIL signature (14).
DISCUSSION
The function of Tim-3 as a checkpoint inhibitor in effector TH1 cells and CD8 T cells has been well characterized (36). The cytoplasmic tail of Tim-3 potentially binds to SH2 domain-containing proteins such as Fyn, PI3K adaptor p85 (37), and lymphocyte-specific protein tyrosine kinase (LCK) (11), and this potential signaling can be further regulated by the cytoplasmic protein Bat3 to prohibit premature Tim-3 inhibitory signal transduction (11, 38). Bat3 is a ubiquitin-like protein that has a variety of intracellular biological functions (1, 2, 5); however, our study provides previously unidentified insights into its immunologic role in the induction of T cell dysfunction, partly by regulating Tim-3 signaling (11).
Our previous studies on syngeneic 4T1 and CT26 mouse tumor model characterized dysfunctional Tim-3+PD-1+ TILs that produce much lower amounts of IFN-γ and TNF-α than Tim-3−PD-1+ TILs. These Tim-3+PD-1+ TILs expressed 50% lower amounts of Bat3 mRNA relative to Tim-3−PD-1+ cells. Similarly, we also found that Bat3 mRNA expression was significantly reduced in Tim-3+PD-1+-exhausted CD4+ cells isolated from untreated HIV-1–infected individuals relative to Tim-3−PD-1+CD4+ T cells (11). This observation indicates that reduced expression of Bat3 is associated with T cell dysfunction in cancer and chronic HIV infection. In this study, we further demonstrated that deficiency of Bat3 expression in 2D2 TH1 cells led to the development of an exhaustion-like phenotype, including increased Tim-3, PD-1, and IL-10 expression but reduced IFN-γ and TNF-α production. Together, these changes result in attenuated pathogenicity of encephalitogenic TH1 cells. Our study suggested a mechanism of which Bat3 plays a key role in the regulation of mTORC2-Akt–Blimp-1 pathway during effector T cell differentiation. This effect is profound enough to induce a dysfunctional phenotype in effector T cells even under highly inflammatory autoimmune neuroinflammation. In patients with multiple sclerosis (MS), while myelin antigen–specific CD4+Tim-3+ were shown to be augmented in patients with benign MS (BEMS), CD4+Bat3+ T cells were increased in primary progressive MS (PPMS) (40). Blockade of the galectin-9–Tim-3 interaction reversed Tim-3–mediated suppression in the BEMS, relapsing-remitting MS, and healthy control groups, but not in the PPMS group where Bat3 expression is high. Collectively, these data suggest that Tim-3 and Bat3 reciprocally control the encephalitogenic phenotype of T cells also in humans.
Here, we demonstrate that Bat3 deficiency in T cells results in a profound defect of effector TH1 cell differentiation in a murine EAE model. Phenotypically, Bat3-deficient TH1 cells showed compromised effector function and early contraction. In contrast, Tim-3 deficiency in T cells exacerbates EAE induction. Tim-3Bat3dko largely rescued the phenotypic defects of Bat3 deficiency in TH1 cell differentiation, including IFN-γ production, frequency of the CNS-infiltrating effector TH1 cells (CD44+; KLRG1+), and IL-7R+ cells. However, IL-2 production in the Tim-3Bat3dko mice was still low compared with WT T cells. This result indicates that, in addition to regulating the Tim-3 signaling pathway, Bat3 may have nuanced roles in the regulation of effector TH1 cells.
As a central regulator of growth and cellular metabolism, mTOR plays a critical role in T cell differentiation and effector/memory differentiation (21). mTOR exists in two multiprotein complexes, mTORC1 and mTORC2. While Raptor is only found in mTORC1, Rictor specifically binds to mTORC2 (21). The molecular mechanism by which mTORC1 is regulated has been extensively investigated, but knowledge of mTORC2 regulation is still limited (40). Here, we demonstrate a previously uncharacterized mechanism whereby Bat3 limits Hsc70-dependent activation of mTORC2. In line with this, we found that Bat3-deficient T cells preferentially increased mTORC2 function and downstream Akt kinase activity. A recent study suggested a potential role of Tim-3 signaling in activation of PI3K kinase pathway in myeloid cells (41). Thus, it is likely that Tim-3 expression could facilitate terminal TH1 differentiation by enhancing mTORC1 pathway. Hence, Bat3 deficiency expedites terminal differentiation and exhaustion (i) directly through unrestrained mTORC2 that enhances Akt signaling and (ii) indirectly through loss of gatekeeper function on Tim-3 downstream signaling, thus promoting mTORC1 activity.
On the basis of our study, we propose the following model for the role of Bat3 in T cell terminal differentiation and exhaustion. Bat3 binds Rictor, thereby inhibiting mTORC2 activity via an Hsc70-dependent mechanism. During effector T cell terminal differentiation and exhaustion, expression of Bat3 is reduced. Consequently, Tim-3 signaling is enhanced. At the same time, mTORC2 function is also activated, which subsequently activates Akt kinase and induces high level of Blimp-1 expression by phosphorylating FoxO1 and thereby excluding it from the nucleus. Increased expression of Tim-3 will further activate PI3K-PDK1 pathway to phosphorylate T308 on Akt (41), thereby fully activating Akt kinase and downstream events (Fig. 8). This regulatory loop will eventually dampen TH1 cell–mediated immune responses. Thus, increased expression of Tim-3 in Bat3cko T cells could provide a positive feedback loop and further strengthen Akt function, enhancing terminal effector T cell differentiation and exhaustion. Akt signaling regulates the expression of genes encoding T cell factor 1 (TCF1), IL-7Rα, CC-chemokine receptor 7 (CCR7), and L-selectin, molecules which are essential for stemness/memory T cell differentiation (42, 43). It has previously been proposed that forced expression of Tim-3 leads to generation of short-lived effector T cell responses with increased Akt/mTOR activity (44). However, in that system, the expression of Bat3 was not assessed, and we suggest cautious interpretation of these findings, as increased long-lived memory cell generation may be due to increased sequestration of Bat3 by Tim-3 in this overexpression model. In the same paper, the authors describe reduced Akt/mTOR signaling in Tim-3−/− mice. This could be explained by the greater availability of Bat3 that is in line with our observation that Bat3 interacts with, and limits, Rictor availability for mTORC2 complex formation. Further supporting this finding, we observed increased PI3K/Akt/mTOR activity in Bat3cko T cells.
The increased Akt activity in Bat3cko cells results in elevated pFoxO1, which has previously been shown to regulate expression of TCF1 (Tcf7). TCF1 has been shown to play an important role in subverting terminal differentiation in effector CD8 T cells, and FoxO1-driven TCF1 is associated with increased T cell stemness, diminished effector, and increased memory phenotype (45, 46). There is an antagonism between TCF1, which promotes stemness in CD4 and CD8 T cells, and Tim-3 expression. It has been shown that high expression of Tim-3 inversely correlates with Tcf7 expression, with low expression of Tim-3 often used to identify high TCF1 expressing populations in vivo (47). We predict that loss of Bat3 (unrestrained Tim-3) not only promotes generation of terminal effectors but also promotes induction of exhaustion and oppose stemness during T cell expansion. The potential mechanism by which Bat3 may mediate this effect is partly explained by the observation made in Bat3cKO mice that develop an “exhaustion”-like phenotype and whereby loss of Bat3 promotes contraction of effector T cells. Previous studies have shown that TCF1 can transactivate itself and promote self-expression in a feed forward loop (48). The reciprocal relationship of Tim-3 and Tcf1 could lead one to hypothesize that activation of Tim-3 may play a critical role in directly regulating Tcf7 expression. However, this remains to be experimentally determined.
As one of the key transcription factors that controls TH1 cell terminal differentiation and exhaustion, Blimp-1 directly regulates a number of effector molecules including Tim-3, IL-10, and KLRG1, at the same time, suppressing IL-7R (CD127) and IL-2 expression. While T cell exhaustion has been associated with poor prognosis in chronic viral infection and cancer, in the context of autoimmunity, exhaustion has actually been linked favorably to long-term clinical outcome in a number of autoimmune diseases including inflammatory bowel disease, type 1 diabetes, and psoriasis (49). We hypothesize that acquisition of an exhaustion-like phenotype by pathogenic autoimmune cells is likely a protective mechanism to curtail damage to the tissue in question. Relapse rate and response to immunomodulatory therapy in autoimmune diseases has been shown to be directly related to the expression of checkpoint molecules and development of T cell exhaustion (49). Our studies show that Bat3 may be a critical regulator of T cell exhaustion, even under proinflammatory autoimmune setting.
In our study, we found that Bat3-deficient T cells have elevated levels of Prdm1 and that up-regulated genes in Bat3cko T cells were significantly enriched for the Prdm1 and exhaustion gene signature, including expression of a module of coinhibitory molecules together with Il10. More recently, we have identified IL-27 as a key cytokine responsible for driving the expression of a module of checkpoint molecules in cancer, in part via Blimp-1. As we had also observed increased Blimp-1 expression in Bat3cko T cells, we were interested to see whether there was an overlapping gene expression patterns existing between IL-27 signaling and mTORC2-Akt pathways in the regulation of TH1 cell dysfunction during autoimmunity. Our data suggest that that Blimp-1 is the key node linking these two networks and may be partly responsible for reduced tissue inflammation and autoimmunity observed in Bat3cKo mice.
Our studies raise an important question of whether methods that promote T cell exhaustion can be exploited positively to alter the course of an autoimmune disease. Our studies suggest that an understanding of the processes governing T cell exhaustion in proinflammatory diseases may also advance therapies for autoimmunity, as principles of T cell exhaustion have been exploited in treatment of cancer.
MATERIALS AND METHODS
Mice
C57BL/6 mice were purchased from the Jackson laboratory. Tim-3fl/fl mice and Bat3fl/fl mice were generated as described in the Supplementary Materials. Prdm1fl/fl mice were acquired from the Jackson laboratory. Tim-3cko, Bat3cko, Prdm1cko, Tim-3 × Bat3dko, and Bat3 × Prdm1dko mice were generated by breeding with CD4-Cre tg mice from the Jackson laboratory. In addition, Tim-3cko, Bat3cko, Tim-3 × Bat3dko, and Bat3 × Prdm1dko mice were further bred with 2D2 mice (in-house). Animal experiments were done in accordance with the guidelines of the Institutional Animal Care and Use Committee at the Brigham and Women’s Hospital and Harvard Medical School.
Active and passive EAE induction
For active EAE induction, 6- to 8-week-old mice were immunized subcutaneously with 100 μg of MOG35–55 peptide (MEVGWYRSPFSRVVHLYRNGK; QCB BioSource International) in CFA [with Mtb (heat-killed Mycobacterium tuberculosis) extract H37-Ra (5 mg/ml; Difco)]. Mice received 150 ng of pertussis toxin (List Biological Laboratories) intraperitoneally on days 0 and 2. For passive EAE induction, 4 × 106 2D2 TH1 cells from different mutant mice were transferred intravenously to C57BL/6 recipient. The mice were monitored daily for development of EAE and scored using the following criteria: 0, no disease; 1, decreased tail tone; 2, hind limb weakness or partial paralysis; 3, complete hind limb paralysis; 4, front and hind limb paralysis; or 5, moribund state. Mice were euthanized at the indicated time points. All mice were housed in a specific pathogen–free animal facility. All breeding and experiments were reviewed and approved by the Harvard Medical Area Standing Committee on Animals and were performed in accordance with the U.S. National Institutes of Health guidelines for the use of live animals.
Antibodies and cytokines
The following antibodies conjugated with different fluorescent dyes for immune cells surface staining and flow cytometry were purchased from BioLegend: anti-CD4 (clone RM4-5), anti-CD62L (clone MEL-14), anti–IL-10 (clone JES5-16E3), anti-TNF (clone TN3-19.12), anti–IL-2 (clone JES6-5H4), anti–IFN-γ (clone XMG1.2), LAG3 (clone C9B7W), CD127, CD44, KLRG1, Va3.2, and Vb11. APC (allophycocyanin)–anti–Tim-3 (5D12) was prepared in house. All of the primary antibodies for Western blots were purchased from Cell Signaling Technologies. All of the IRDye secondary antibodies to detect primary antibody signals in LI-COR Odyssey Imaging system were purchased from LI-COR Biotechnology. All of the recombinant mouse cytokines for T cell in vitro culture and Tim-3 induction were purchased from R&D systems.
Cell isolation and culture
Total CD4+ T cells from different lines of mice were first enriched by positive selection using CD4+ T cell isolation reagent from Miltenyi Biotec. Cells were stained with CD25–fluorescein isothiocyanate, CD44-PE (phycoerythrin), and CD62L-APC. Naïve CD4+ (CD4+ CD44−CD62L+) T cells were subsequently sorted by BD FACSAria (BD Biosciences). The sorted naïve T cells were then activated with plate-bound anti-CD3 (1 μg/ml; 145-2C11) and anti-CD28 (1 μg/ml; PV-1) (both were made in-house) for 2 days. For TH1 conditions, cells were cultured under the presence of IL-12 (10 ng/ml) and anti–IL-4 (10 μg/ml).
Retroviral transduction
Retroviral expression plasmids for Blimp-1 and GFP control were packed in 293T cells and were used to transduce mouse naïve CD4+ T cells activated by plate-bound anti-CD3 and anti-CD28 antibodies.
Western blot
Cells were harvested, washed in ice-cold phosphate-buffered saline (PBS), and lysed in medium stringency lysis buffer for 30 min. The lysates were centrifuged at 13,000 rpm, the supernatants were harvested, and protein concentration was determined by a Pierce assay. The samples were diluted in 1× SDS loading buffer, boiled for 5 min in reducing conditions, followed by separation on 10% SDS–polyacrylamide gel electrophoresis (PAGE) gel. Gels were transferred to nitrocellulose membranes, blocked with tris-buffered saline plus 0.1% Tween 20 and 3% bovine serum albumin (BSA) or milk, and probed with indicated primary antibodies. Blots were analyzed by Odyssey Imaging System.
Intracellular cytokine staining
For in vitro experiments, naïve CD4+ T cells were activated by plate-bound anti-CD3 and anti-CD28 antibodies for 2 days. Cells were then rested for 3 days and restimulated with plate-bound anti-CD3 (0.1 μg/ml) and anti-CD28 for 24 hours before they were subjected to phorbol 12-myristate 13-acetate (PMA) and ionomycin stimulation (50 ng/ml and 1 μM, respectively) in the presence of Golgi Stop (BD Biosciences) for intracellular cytokine detection. All data were collected on LSR II (BD Biosciences) or Calibur (BD Biosciences) and analyzed by FlowJo software (Tree Star Inc.). For ex vivo experiments, CNS-infiltrating T cells and draining lymph node T cells were isolated as previously described (50). The cells were further stimulated with PMA and ionomycin for 3 hours and subsequently subjected to intracellular cytokine stain. Briefly, cells were stained on ice for 20 min with cell surface markers including (Va3.2+Vb11+) in the case of 2D2 cells. Cells were washed in fluorescence-activated cell sorting buffer (PBS, 0.5% BSA, and 2 mM EDTA) and fixed with 4% paraformaldehyde for 10 min on ice. Cells were then washed with BD perm/wash and stained with antibodies directed against indicated intracellular cytokines for 30 min on ice.
ChIP assays
CD4+ T cells from C57BL/6 mice were purified by a CD4+ T cell negative selection kit (Miltenyi Biotech) and were activated by plate-bound anti-CD3 and anti-CD28 (2 μg/ml each) under either neutral condition (TH0) for 2 days. Cells were rested for additional 3 days and were restimulated with plate-bound anti-CD3 and anti-CD28 (0.1 μg/ml) for 24 hours before they were subjected to chromatin preparation for the ChIP analysis. Chromatin fraction preparation and ChIP were performed using the SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling Technology). Antibody against Foxo1 was purchased from Santa Cruz Biotechnology.
Real-time PCR analysis
RNA was extracted with RNeasy Plus kits (Qiagen), and cDNA was made by Iscript (Bio-Rad). All of the gene expression was analyzed by quantitative real-time PCR on a ViiA7 System (Thermo Fisher Scientific) using TaqMan Fast Advanced Master Mix (Thermo Fisher Scientific) with the following primer/probe sets: Prdm1 Mm00476128_m1, Tbx21 Mm00450960_m1, Bcl6 Mm00477633_m1, and Actb (Applied Biosystems). Expression values were calculated relative to Actb detected in the same sample by duplex qPCR.
Nanostring analysis
We analyzed gene expression in CD4+ effector T cells (sorted on Tim-3+ or Tim-3−) from MOG immunized Foxp3GFP mice at peak of disease (day 14) using a custom nanostring code set of 397 genes representing both the IL-27–driven gene signature (245 genes) and the dysfunctional CD8+ TIL gene signature (245 genes) (14). Expression values were normalized by first adjusting each sample based on its relative value to all samples. This was followed by subtracting the calculated background (mean.2sd) from each sample with additional normalization by housekeeping geometric mean, where housekeeping genes were defined as Hprt, Gapdh, Actin, and Tubb5. To identify DE genes, P values of differential expression between the Tim-3_positive and Tim-3_negative CD4 cells were obtained with a t test and corrected for multiple hypothesis testing with the Bonferroni-Hochberg method (R function p.adjust with parameter “method” set to “BH”). Genes with a corrected P value smaller than 0.05 were considered DE.
RNA-seq analysis
Reads were aligned with RSEM. TPM (transcripts per million) values were computed from count data, followed by quantile normalization and log2-transformed. Three replicate samples were generated for each condition, but one of the Bat3cko samples was excluded because it is a definite outlier as compared to the other samples. Genes were kept for downstream analysis if their mean expression was greater than 1 and their variance larger than 0. Of the 10,289 genes passing these thresholds, genes were determined as DE if their mean and median expression change was greater than twofold, and the fold change between the largest and smallest value from each condition was greater than 1.3. Significance in Fig. 7B was assessed using the gene set enrichment analysis (GSEA) preranked analysis tool. Genes were ranked on the basis of their fold change. Enrichment plots that are output from the GSEA tool are used in Fig. 7C to illustrate signatures. Three hundred sixteen CD4 cells were plotted on a tSNE using the package Seurat in R. Contaminating populations were filtered out, and plots were colored using Seurat’s AddModuleScore function.
Acknowledgments
We thank M. Collins for insightful discussions and J. Xia, H. Stroh, and M. Weinstein for laboratory support. Funding: This work was supported by grants from the National Institutes of Health (P01 AI073748, R01NS045937, NS030843, P01NS038037, and P01AI056299 to V.K.), European Commission Excellent Science H2020 (#708658 and #10130984 to K.O.D.), and Career Transitional Fellowship from the National Multiple Sclerosis Society (C.W.). Author contributions: C.Z. and K.O.D. performed most of the experiments with help from G.G., S.X., S.Z., M.A.S., C.W., H.Z., K.G., and E.C. K.N. performed computational analysis with guidance from M.S. and A.R. O.R.-R., H.O., T.M., and M.R. provided conceptual input and other essential resources. V.K. and C.Z. designed the experimental setup and conceived the study. C.Z. and K.O.D. wrote the manuscript and prepared figures with conceptual input and edits from V.K. and all authors. Competing interests: V.K. has an ownership interest in and is a member of the scientific advisory board for Tizona Therapeutics and Astellas Global Pharma Development Inc. V.K. is a named inventor on a patent related to this work filed by the Brigham and Women’s Hospital (no. WO2011159877A2, filed 16 June 2011, published 22 December 2011).A.R. and V.K. are cofounders of and have an ownership interest in Celsius Therapeutics. A.R. is a cofounder and equity holder of Celsius Therapeutics and, until 31 August 2020, was a SAB member of Syros Pharmaceuticals, Neogene Therapeutics, Asimov, and Thermo Fisher Scientific. From 1 August 2020, A.R. is an employee of Genentech. V.K.’s interests were reviewed and managed by the Brigham and Women’s Hospital and Partners Healthcare in accordance with their conflict of interest policies. A.R.’s interests were reviewed and managed by the Broad Institute and HHMI in accordance with their conflict of interest policies. The authors declare that they have no other competing interests. Data and materials availability: Data have been uploaded to NCBI Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) under data repository accession number GSE168435. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/18/eabd2710/DC1
REFERENCES AND NOTES
- 1.Desmots F., Russell H. R., Lee Y., Boyd K., McKinnon P. J., The reaper-binding protein scythe modulates apoptosis and proliferation during mammalian development. Mol. Cell. Biol. 25, 10329–10337 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sasaki T., Gan E. C., Wakeham A., Kornbluth S., Mak T. W., Okada H., HLA-B-associated transcript 3 (Bat3)/Scythe is essential for p300-mediated acetylation of p53. Genes Dev. 21, 848–861 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kämper N., Franken S., Temme S., Koch S., Bieber T., Koch N., γ-Interferon-regulated chaperone governs human lymphocyte antigen class II expression. FASEB J. 26, 104–116 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Corduan A., Lecomte S., Martin C., Michel D., Desmots F., Sequential interplay between BAG6 and HSP70 upon heat shock. Cell. Mol. Life Sci. 66, 1998–2004 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Minami R., Hayakawa A., Kagawa H., Yanagi Y., Yokosawa H., Kawahara H., BAG-6 is essential for selective elimination of defective proteasomal substrates. J. Cell Biol. 190, 637–650 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Degli-Esposti M. A., Abraham L. J., McCann V., Spies T., Christiansen F. T., Dawkins R. L., Ancestral haplotypes reveal the role of the central MHC in the immunogenetics of IDDM. Immunogenetics 36, 345–356 (1992). [DOI] [PubMed] [Google Scholar]
- 7.Chen J., Zang Y.-s., Xiu Q., BAT3 rs1052486 and rs3117582 polymorphisms are associated with lung cancer risk: A meta-analysis. Tumour Biol. 35, 9855–9858 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Truong T., Sauter W., McKay J. D., Hosgood H. D. III, Gallagher C., Amos C. I., Spitz M., Muscat J., Lazarus P., Illig T., Wichmann H. E., Bickeböller H., Risch A., Dienemann H., Zhang Z.-F., Naeim B. P., Yang P., Zienolddiny S., Haugen A., Marchand L. L., Hong Y.-C., Kim J. H., Duell E. J., Andrew A. S., Kiyohara C., Shen H., Matsuo K., Suzuki T., Seow A., Ng D. P. K., Lan Q., Zaridze D., Szeszenia-Dabrowska N., Lissowska J., Rudnai P., Fabianova E., Constantinescu V., Bencko V., Foretova L., Janout V., Caporaso N. E., Albanes D., Thun M., Landi M. T., Trubicka J., Lener M., Lubinski J.; EPIC-lung, Wang Y., Chabrier A., Boffetta P., Brennan P., Hung R. J., International Lung Cancer Consortium: Coordinated association study of 10 potential lung cancer susceptibility variants. Carcinogenesis 31, 625–633 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y., Broderick P., Webb E., Wu X., Vijayakrishnan J., Matakidou A., Qureshi M., Dong Q., Gu X., Chen W. V., Spitz M. R., Eisen T., Amos C. I., Houlston R. S., Common 5p15.33 and 6p21.33 variants influence lung cancer risk. Nat. Genet. 40, 1407–1409 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakuishi K., Apetoh L., Sullivan J. M., Blazar B. R., Kuchroo V. K., Anderson A. C., Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 207, 2187–2194 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rangachari M., Zhu C., Sakuishi K., Xiao S., Karman J., Chen A., Angin M., Wakeham A., Greenfield E. A., Sobel R. A., Okada H., McKinnon P. J., Mak T. W., Addo M. M., Anderson A. C., Kuchroo V. K., Bat3 promotes T cell responses and autoimmunity by repressing Tim-3–mediated cell death and exhaustion. Nat. Med. 18, 1394–1400 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou Q., Munger M. E., Veenstra R. G., Weigel B. J., Hirashima M., Munn D. H., Murphy W. J., Azuma M., Anderson A. C., Kuchroo V. K., Blazar B. R., Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood 117, 4501–4510 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin H.-T., Anderson A. C., Tan W. G., West E. E., Ha S.-J., Araki K., Freeman G. J., Kuchroo V. K., Ahmed R., Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. U.S.A. 107, 14733–14738 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chihara N., Madi A., Kondo T., Zhang H., Acharya N., Singer M., Nyman J., Marjanovic N. D., Kowalczyk M. S., Wang C., Kurtulus S., Law T., Etminan Y., Nevin J., Buckley C. D., Burkett P. R., Buenrostro J. D., Rozenblatt-Rosen O., Anderson A. C., Regev A., Kuchroo V. K., Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature 558, 454–459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singer M., Wang C., Cong L., Marjanovic N. D., Kowalczyk M. S., Zhang H., Nyman J., Sakuishi K., Kurtulus S., Gennert D., Xia J., Kwon J. Y. H., Nevin J., Herbst R. H., Yanai I., Rozenblatt-Rosen O., Kuchroo V. K., Regev A., Anderson A. C., A distinct gene module for dysfunction uncoupled from activation in tumor-infiltrating T cells. Cell 166, 1500–1511.e9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bettelli E., Pagany M., Weiner H. L., Linington C., Sobel R. A., Kuchroo V. K., Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J. Exp. Med. 197, 1073–1081 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Monney L., Sabatos C. A., Gaglia J. L., Ryu A., Waldner H., Chernova T., Manning S., Greenfield E. A., Coyle A. J., Sobel R. A., Freeman G. J., Kuchroo V. K., Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 415, 536–541 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Li H., van der Leun A. M., Yofe I., Lubling Y., Gelbard-Solodkin D., van Akkooi A. C. J., van den Braber M., Rozeman E. A., Haanen J. B. A. G., Blank C. U., Horlings H. M., David E., Baran Y., Bercovich A., Lifshitz A., Schumacher T. N., Tanay A., Amit I., Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell 176, 775–789.e18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu C., Sakuishi K., Xiao S., Sun Z., Zaghouani S., Gu G., Wang C., Tan D. J., Wu C., Rangachari M., Pertel T., Jin H.-T., Ahmed R., Anderson A. C., Kuchroo V. K., An IL-27/NFIL3 signalling axis drives Tim-3 and IL-10 expression and T-cell dysfunction. Nat. Commun. 6, 6072 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sarbassov D. D., Ali S. M., Sengupta S., Sheen J.-H., Hsu P. P., Bagley A. F., Markhard A. L., Sabatini D. M., Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 22, 159–168 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Powell J. D., Pollizzi K. N., Heikamp E. B., Horton M. R., Regulation of immune responses by mTOR. Annu. Rev. Immunol. 30, 39–68 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martin J., Masri J., Bernath A., Nishimura R. N., Gera J., Hsp70 associates with Rictor and is required for mTORC2 formation and activity. Biochem. Biophys. Res. Commun. 372, 578–583 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sarbassov D. D., Ali S. M., Kim D.-H., Guertin D. A., Latek R. R., Erdjument-Bromage H., Tempst P., Sabatini D. M., Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 14, 1296–1302 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Thress K., Song J., Morimoto R. I., Kornbluth S., Reversible inhibition of Hsp70 chaperone function by Scythe and Reaper. EMBO J. 20, 1033–1041 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Esser C., Alberti S., Höhfeld J., Cooperation of molecular chaperones with the ubiquitin/proteasome system. Biochim. Biophys. Acta 1695, 171–188 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Sawant D. V., Yano H., Chikina M., Zhang Q., Liao M., Liu C., Callahan D. J., Sun Z., Sun T., Tabib T., Pennathur A., Corry D. B., Luketich J. D., Lafyatis R., Chen W., Poholek A. C., Bruno T. C., Workman C. J., Vignali D. A. A., Adaptive plasticity of IL-10+ and IL-35+ Treg cells cooperatively promotes tumor T cell exhaustion. Nat. Immunol. 20, 724–735 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hess Michelini R., Doedens A. L., Goldrath A. W., Hedrick S. M., Differentiation of CD8 memory T cells depends on Foxo1. J. Exp. Med. 210, 1189–1200 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang T. T.-L., Dowbenko D., Jackson A., Toney L., Lewin D. A., Dent A. L., Lasky L. A., The forkhead transcription factor AFX activates apoptosis by induction of the BCL-6 transcriptional repressor. J. Biol. Chem. 277, 14255–14265 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Liao W., Ouyang W., Zhang M. Q., Li M. O., Genome wide mapping of Foxo1 binding-sites in murine T lymphocytes. Genomics Data 2, 280–281 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mayo L., Cunha A. P. D., Madi A., Beynon V., Yang Z., Alvarez J. I., Prat A., Sobel R. A., Kobzik L., Lassmann H., Quintana F. J., Weiner H. L., IL-10-dependent Tr1 cells attenuate astrocyte activation and ameliorate chronic central nervous system inflammation. Brain 139, 1939–1957 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burton B. R., Britton G. J., Fang H., Verhagen J., Smithers B., Sabatos-Peyton C. A., Carney L. J., Gough J., Strobel S., Wraith D. C., Sequential transcriptional changes dictate safe and effective antigen-specific immunotherapy. Nat. Commun. 5, 4741 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qin H., Wang Z., Du W., Lee W.-H., Wu X., Riggs A. D., Liu C.-P., Killer cell Ig-like receptor (KIR) 3DL1 down-regulation enhances inhibition of type 1 diabetes by autoantigen-specific regulatory T cells. Proc. Natl. Acad. Sci. U.S.A. 108, 2016–2021 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sarkar S., Kalia V., Haining W. N., Konieczny B. T., Subramaniam S., Ahmed R., Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J. Exp. Med. 205, 625–640 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wherry E. J., Ha S.-J., Kaech S. M., Haining W. N., Sarkar S., Kalia V., Subramaniam S., Blattman J. N., Barber D. L., Ahmed R., Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27, 670–684 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Shin H., Blackburn S. D., Intlekofer A. M., Kao C., Angelosanto J. M., Reiner S. L., Wherry E. J., A role for the transcriptional repressor Blimp-1 in CD8+ T cell exhaustion during chronic viral infection. Immunity 31, 309–320 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu C., Anderson A. C., Kuchroo V. K., TIM-3 and its regulatory role in immune responses. Curr. Top. Microbiol. Immunol. 350, 1–15 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Lee J., Su E. W., Zhu C., Hainline S., Phuah J., Moroco J. A., Smithgall T. E., Kuchroo V. K., Kane L. P., Phosphotyrosine-dependent coupling of Tim-3 to T-cell receptor signaling pathways. Mol. Cell. Biol. 31, 3963–3974 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang Y.-H., Zhu C., Kondo Y., Anderson A. C., Gandhi A., Russell A., Dougan S. K., Petersen B.-S., Melum E., Pertel T., Clayton K. L., Raab M., Chen Q., Beauchemin N., Yazaki P. J., Pyzik M., Ostrowski M. A., Glickman J. N., Rudd C. E., Ploegh H. L., Franke A., Petsko G. A., Kuchroo V. K., Blumberg R. S., CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 517, 386–390 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saresella M., Piancone F., Marventano I., La Rosa F., Tortorella P., Caputo D., Rovaris M., Clerici M., A role for the TIM-3/GAL-9/BAT3 pathway in determining the clinical phenotype of multiple sclerosis. FASEB J. 28, 5000–5009 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Thomson A. W., Turnquist H. R., Raimondi G., Immunoregulatory functions of mTOR inhibition. Nat. Rev. Immunol. 9, 324–337 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prokhorov A., Gibbs B. F., Bardelli M., Rüegg L., Fasler-Kan E., Varani L., Sumbayev V. V., The immune receptor Tim-3 mediates activation of PI3 kinase/mTOR and HIF-1 pathways in human myeloid leukaemia cells. Int. J. Biochem. Cell Biol. 59, 11–20 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Kim M. V., Ouyang W., Liao W., Zhang M. Q., Li M. O., The transcription factor Foxo1 controls central-memory CD8+ T cell responses to infection. Immunity 39, 286–297 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Macintyre A. N., Finlay D., Preston G., Sinclair L. V., Waugh C. M., Tamas P., Feijoo C., Okkenhaug K., Cantrell D. A., Protein kinase B controls transcriptional programs that direct cytotoxic T cell fate but is dispensable for T cell metabolism. Immunity 34, 224–236 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Avery L., Filderman J., Szymczak-Workman A. L., Kane L. P., Tim-3 co-stimulation promotes short-lived effector T cells, restricts memory precursors, and is dispensable for T cell exhaustion. Proc. Natl. Acad. Sci. U.S.A. 115, 2455–2460 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou X., Yu S., Zhao D.-M., Harty J. T., Badovinac V. P., Xue H.-H., Differentiation and persistence of memory CD8+ T cells depend on T cell factor 1. Immunity 33, 229–240 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Delpoux A., Lai C.-Y., Hedrick S. M., Doedens A. L., FOXO1 opposition of CD8+ T cell effector programming confers early memory properties and phenotypic diversity. Proc. Natl. Acad. Sci. U.S.A. 114, E8865–E8874 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sade-Feldman M., Yizhak K., Bjorgaard S. L., Ray J. P., de Boer C. G., Jenkins R. W., Lieb D. J., Chen J. H., Frederick D. T., Barzily-Rokni M., Freeman S. S., Reuben A., Hoover P. J., Villani A.-C., Ivanova E., Portell A., Lizotte P. H., Aref A. R., Eliane J.-P., Hammond M. R., Vitzthum H., Blackmon S. M., Li B., Gopalakrishnan V., Reddy S. M., Cooper Z. A., Paweletz C. P., Barbie D. A., Stemmer-Rachamimov A., Flaherty K. T., Wargo J. A., Boland G. M., Sullivan R. J., Getz G., Hacohen N., Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 175, 998–1013.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weber B. N., Chi A. W.-S., Chavez A., Yashiro-Ohtani Y., Yang Q., Shestova O., Bhandoola A., A critical role for TCF-1 in T-lineage specification and differentiation. Nature 476, 63–68 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McKinney E. F., Lee J. C., Jayne D. R. W., Lyons P. A., Smith K. G. C., T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature 523, 612–616 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dixon K. O., Schorer M., Nevin J., Etminan Y., Amoozgar Z., Kondo T., Kurtulus S., Kassam N., Sobel R. A., Fukumura D., Jain R. K., Anderson A. C., Kuchroo V. K., Joller N., Functional anti-TIGIT antibodies regulate development of autoimmunity and antitumor immunity. J. Immunol. 200, 3000–3007 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kawazu M., Saso K., Tong K. I., McQuire T., Goto K., Son D.-O., Wakeham A., Miyagishi M., Mak T. W., Okada H., Histone demethylase JMJD2B functions as a co-factor of estrogen receptor in breast cancer proliferation and mammary gland development. PLOS ONE 6, e17830 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/18/eabd2710/DC1