

Abstract
Autophagy is a regulated mechanism that removes unnecessary or dysfunctional cellular components and recycles metabolic substrates. In response to stress signals in the tumour microenvironment, the autophagy pathway is altered in tumour cells and immune cells — thereby differentially affecting tumour progression, immunity and therapy. In this Review, we summarize our current understanding of the immunologically associated roles and modes of action of the autophagy pathway in cancer progression and therapy, and discuss potential approaches targeting autophagy to enhance antitumour immunity and improve the efficacy of current cancer therapy.
Autophagy is a tightly regulated and stress-induced catabolic pathway in all eukaryotes, during which double-membraned vesicles (called autophagosomes) are formed that engulf cellular targets — including damaged organelles, unfolded proteins and pathogens — and deliver them to the lysosome to be digested1. Selective forms of autophagy represent quality control and homeostatic mechanisms (BOX 1). The basic biology of autophagy has been extensively reviewed2–4. The canonical form of autophagy under discussion herein (more formally, macro-autophagy) is best understood under conditions of nutrient starvation5 (FIG. 1). In the non-canonical autophagy pathway, proteins also function to lipidate microtubule-associated protein 1A/1B-light chain 3 (LC3; also known as ATG8) family proteins on a single lipid bilayer. The most studied examples of this are LC3-associated phagocytosis (LAP)6 and the more recently described LC3-associated endocytosis (LANDO)7 (FIG. 1).
Box 1 | Overview of selective forms of autophagy in mammalian cells.
Mitophagy is the selective degradation of damaged and aged mitochondria via autophagy. Mitophagy is triggered by a loss of electrical potential across the inner mitochondrial membrane (ΔΨm). although several mechanisms exist, the best understood involves the proteins phosphatase and tensin homologue (PteN)-induced kinase 1 (PiNK1) and the e3 ubiquitin ligase Parkin189. when ΔΨm is disrupted, PiNK1 is stabilized at the mitochondrial outer membrane, where it phosphorylates ubiquitin. in turn, phospho-ubiquitin activates the function of Parkin to ubiquitinate outer mitochondrial membrane proteins, including Mitofusin ii. ubiquitinated Mitofusin ii is then targeted by adapters to bring the damaged mitochondria into a forming autophagosome.
Lipophagy targets lipid droplets and catabolizes their components into free fatty acids and glycerol by lysosomal acid lipase (LaL)99. Lipophagy is induced by nutrient deprivation and signalling via mtOr complex 1 (mtOrC1) and 5′-aMP-activated protein kinase (aMPK) during lengthy fasting190. soluble N-ethylmaleimide-sensitive factor attachment protein receptors (sNares), lipidate microtubule-associated protein 1a/1B light chain 3 (LC3), LaMP1, LaMP2B and LaMP2C as well as small GtPases (such as ras-related protein rab7a (REF.191)) are involved in the fusion of lipophagosome with the lysosome.
Endoplasmic reticulum-phagy is the selective clearance and degradation of endoplasmic reticulum components and membranes by the autophagic machinery. Six endoplasmic reticulum surface proteins have been identified as specific receptors through LC3/GaBaraP-interacting regions to recruit autophagy machinery in mammalian cells, including atlastin GtPase 3 (atL3)192, cell-cycle progression gene 1 (CCPG1)193, family with sequence similarity 134 member B (FaM134B)194, reticulon 3 long isoform (rtN3L)195, translocation protein seC62 (seC62)196 and testis-expressed protein 264 precursor (teX264)197,198. These receptors function to fragment the endoplasmic reticulum into suitable pieces that can be readily incorporated into autophagosomes. the COPii subunit, seC24-related protein C (seC24C), acts with autophagy receptors to deliver endoplasmic reticulum sheets and tubules to lysosomes for degradation199. Atlastin, an endoplasmic reticulum-resident GtPase, acts downstream of the endoplasmic reticulum-phagy receptors to remodel the endoplasmic reticulum membrane to separate pieces for efficient autophagosomal engulfment200.
Other forms of selective autophagy also exist in mammalian cells. Ferritinophagy is an alternative form of autophagy that selectively degrades ferritin to regulate iron homeostasis via nuclear receptor coactivator 4 (NCOa4)201,202. Nuclear fragile X mental retardation-interacting protein 1 (NuFiP1) is a receptor for starvation-induced ribophagy, which interacts with ribosomes and delivers them to autophagosomes by directly binding to LC3B (REF.203). in the nucleus, nucleophagy mediates degradation of nuclear lamin B1 which interacts with LC3 in mammals. LC3 assists lamin B1 in transferring from nuclear to cytoplasmic and degrading in the lysosome following ras signalling204. Peroxisomes are targeted to the autophagosome by p62/sQstM1, which is defined as pexophagy205. in addition, autophagy can regulate the rNa silencing machinery by selectively degrading DiCer and argonaute risC catalytic component 2 (aGO2)206.
Fig. 1 |. The machinery of canonical and non-canonical autophagy.
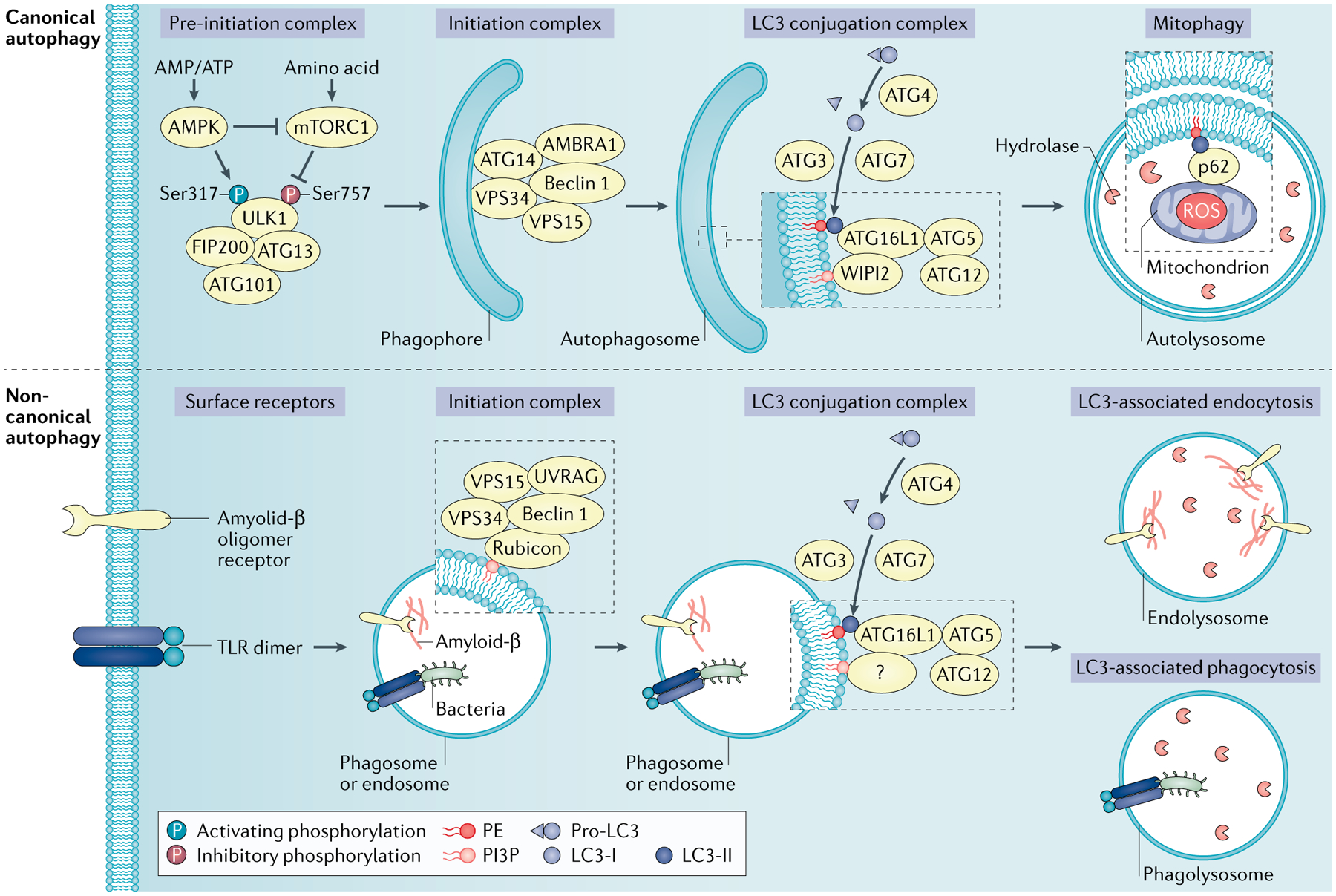
Canonical autophagy is normally induced in response to the cell undergoing an energy crisis. Autophagy-inducing signals, such as nutrient deprivation, trigger the activation of 5′-AMP-activated protein kinase (AMPK), whose kinase activity simultaneously inhibits mTOR and activates the pre-initiation complex (unc-51-like kinase 1 (ULK1), autophagy-related protein 13 (ATG13), ATG101 and FAK family interacting protein of 200 kDa (FIP200)). This complex, in turn, phosphorylates components in the autophagy initiation complex composed of vacuolar protein sorting 34 (VPS34), VPS15 and Beclin 1, along with ATG14 and AMBRA1. The initiation complex converts phosphoinositides in the endoplasmic reticulum to phosphatidylinositol-3-phosphate (PI3P), which recruits ligation machinery composed of the E1 ligase ATG7, the E2 ligase ATG3 and a complex of ATG16L1 and an ATG5–ATG12 conjugate that together act as an E3 ligase. The ligase machinery functions to ligate lipidate microtubule-associated protein 1A/1B light chain 3 (LC3) family proteins to phosphatidylethanolamine (PE) to form LC3–PE. The activity and coordination of the ATG5–ATG12 system and the LC3–PE system facilitate the curvature and sealing of the autophagosome, as well as the lipidation and embedding of LC3–PE into the autophagosomal membrane. The resulting autophagosomes then fuse to lysosomes (forming autolysosomes), resulting in digestion of the autophagosome contents loaded by the LC3 binding proteins. The non-canonical autophagy pathway also functions to lipidate LC3 family proteins on a single membrane. LC3-associated phagocytosis (LAP) and LC3-associated endocytosis (LANDO) process independently of the autophagy pre-initiation (ULK1) complex, and utilize a different initiation complex composed of VPS34, VPS15, Beclin 1, UVRAG and Rubicon without requirements for ATG14 or AMBRA1. mTORC1, mTOR complex 1; ROS, reactive oxygen species; TLR, Toll-like receptor; WIPI2, WD repeat domain phosphoinositide-interacting protein 2.
Here, we focus on the autophagy pathway in the tumour microenvironment (TME) where, in response to various stresses, the autophagy pathway in various cell types can be modulated, resulting in suppression or promotion of tumour progression. The effects of changes in autophagy in the tumour may be context-dependent, including variable tumour genetics and stages as well as host factors. The immune system plays a critical role in prevention of tumour occurrence, progress and metastasis, and shapes the tumour response to therapy. Immune surveillance provides a way to recognize, control and kill tumour cells. However, tumour cells evade tumour immunity via reducing their immunogenicity, forming immunosuppressive networks and inhibiting immune response8. Immunotherapies harness the immune system to fight against the tumour through evoking successful antitumour immune responses. For example, immune checkpoint blockade (ICB) unleashes the ability of T cells to restrain tumour growth9,10. Other immunotherapy approaches can also enhance antitumour immunity, including chimeric antigen receptor T cells (CAR T cells), dendritic cell vaccines and cytokine therapies.
Recent studies have demonstrated the involvement of the autophagy pathway in survival and apoptosis, differentiation, activation, effector function and tumour trafficking of immune cell subsets11. Meanwhile, tumour autonomous autophagy can alter tumour growth through modulating the immune response12,13. Furthermore, the pro-tumour role of autophagy may be abolished by combining immune checkpoint therapy with autophagy inhibitors14. Given our recent advances in understanding the multiple layers of the relationship between the autophagy pathway and the tumour immune responses, and the potential of targeting the autophagy pathway in cancer therapy, it is timely to summarize and discuss recent findings on the autophagy pathway in the context of tumour immunity and immunotherapy.
Autophagy induction in tumour cells
The TME, including the immunosuppressive networks8, plays a key role in cancer progression, metastasis and resistance to therapies. In the TME, autophagy in tumour cells can be induced by integrated intracellular and extracellular stress signals including metabolic stress, hypoxia, redox stress and immune signals.
Metabolic stress
Insufficient nutrient uptake from the TME affects the metabolic machinery, leading to intracellular metabolic stress. In response to metabolic stress, tumour cells rewire their own metabolic pathways via upregulating nutrient transporters15 and activating autophagy16. Mechanistically, 5′-AMP-activated protein kinase (AMPK) and mTOR complex 1 (mTORC1) are two opposing regulatory kinases that alter autophagy induction under nutrient deprivation. As an AMP sensor, AMPK is activated via the increased ratio of AMP and ATP, whereas the activity of mTORC1 is reduced due to limited amino acid availability. This leads to phosphorylation of the targets in the autophagy pre-initiation complex (AMPK at an activating site, mTORC1 at an inhibitory site). To initiate autophagy, AMPK phosphorylates unc-51-like kinase 1 (ULK1) at six different sites (S317, S467, S555, T574, S637 and S777)17,18 and at S91/S94 in Beclin 1 (REF.19). Moreover, AMPK targets Raptor, a subunit of mTORC1, and activates tuberous sclerosis complex 2 (TSC2), an upstream negative regulator of mTOR, to inhibit mTORC1 activity20,21. Unlike AMPK, mTORC1-induced phosphorylation of ULK1 on S757 inhibits its downstream activation of the vacuolar protein sorting 34 (VPS34) complex and autophagy17 (FIG. 1). In addition, AMPK activates the transcriptional factors forkhead box O3 (FoxO3) and transcription factor EB (TFEB) to amplify autophagy-related gene expression22. Activated autophagy promotes recycling of key metabolites — including nucleotides, amino acids and lipids — thereby supporting tumour cell survival and proliferation in the TME.
Hypoxic stress
Hypoxia is a hallmark of solid tumours. Hypoxia results in suppression of oxidative phosphorylation in the mitochondria, an increase in the ratio of AMP to ATP and activation of AMPK. As discussed above, AMPK then targets ULK1, Beclin 1 and mTORC1 to induce autophagy23. Furthermore, hypoxia restrains mTOR signalling and unleashes protein phosphatase 2A (PP2A) activity via regulated in development and DNA damage response 1 (REDD1; also known as DDIT4). PP2A directly dephosphorylates prolyl hydroxylase domain-containing protein 2 (PHD2) on S125, which restricts the ability of PHD2 to degrade hypoxia-induced factor 1α (HIF1α), thereby increasing HIF1α stabilization24. HIF1α is then translocated into the nucleus and induces the expression of downstream target genes, including B cell lymphoma 2 (BCL2) interacting protein 3 (BNIP3) and BNIP3L, which dissociates Beclin 1 from BCL2 and, consequently, activates autophagy25. In addition, hypoxia causes activation of activating transcription factor 4 (ATF4), which results in upregulation of LC3B and autophagy-related protein 5 (ATG5) expression, and maintenance of high levels of autophagic flux26. These events promote autophagy-mediated tumour cell survival under hypoxic conditions24.
Oxidative stress
Oxidative stress reflects an imbalance between free radicals and antioxidants. reactive oxygen species (ROS) are generated within cells through oxygen metabolism and other processes. Excessive ROS may increase the risk of DNA damage and promote tumorigenesis27. In response to elevated ROS, ataxia telangiectasia mutated (ATM) activates the TSC2 tumour suppressor via the liver kinase B1 (LKB1) and AMPK metabolic pathway in the cytoplasm to repress mTORC1 and induce autophagy28. Oxidative stress can also promote autophagy through nuclear factor-κB (NF-κB)-mediated upregulation of p62/SQSTM1 (REF.29). In return, activated autophagy can alleviate oxidative stress through p62-induced degradation of kelch-like ECH-associated protein 1 (KEAP1), resulting in the stabilization and accumulation of nuclear factor erythroid 2-related factor 2 (NRF2) and activation of antioxidant enzymes, such as glutathione peroxidase, superoxide dismutase and thioredoxin30. Furthermore, autophagy can mediate a selective degradation of damaged and dysfunctional mitochondria (mitophagy). By controlling oxidative stress, autophagy may inhibit tumorigenesis but may also promote tumour cell survival.
Immune signals
Immune signals can modulate the autophagy pathway in the TME. Damage-associated molecular patterns (DAMPs) and cytokines are major soluble mediators that modulate autophagic responses. Extracellular DAMPs — such as DNA complexes, ATP and high-mobility group box 1 protein (HMGB1)31 — activate innate immune cells and promote inflammation. These signals are perceived by extracellular or intracellular pattern recognition receptors including Toll-like receptors (TLRs), advanced glycosylation end product-specific receptor (AGER), purinergic P2RX7 receptors and absent in melanoma 2 (AIM2)31. Autophagy is stimulated by multiple cell death-associated DAMPs32. Various TLRs are proposed as autophagy inducers when they recognize DAMPs and activate downstream signalling. The E3 ligase TNF receptor-associated factor 6 (TRAF6)-mediated ubiquitination of Beclin 1 is critical for TLR4-triggered autophagy in macrophages, which releases Beclin 1 from its inhibitor BCL2 (REF.33). In non-small cell lung cancer (NSCLC) cells, activation of TLR3 and TLR4 with polyinosinic:polycytidylic acid (poly(I:C)) and lipopolysaccharide (LPS) induces autophagy and increases the production of certain cytokines related to tumour metastasis34. Moreover, TLR engagement activates NF-κB-dependent transcription and promotes autophagy activation through transcriptional upregulation of autophagy receptor p62, which mediates mitophagy and restricts NLR family pyrin domain containing 3 (NLRP3) inflammasome activation in LPS-treated macrophages35. In pancreatic and colon cancer cells, AGER is important for HMGB1-induced autophagy in a Beclin 1-dependent manner in vitro36. AGER also down-regulates the phosphorylation of mTOR and increases Beclin 1/VPS34-dependent autophagosome formation in pancreatic cancer cells37.
Cytokines regulate the autophagy pathway in a context-dependent fashion. In Drosophila, TNF and interleukin-6 (IL-6)-like signalling activates autophagy, thereby promoting early-stage tumour growth and invasion38. Transforming growth factor-β (TGFβ) increases the transcript levels of BECN1, ATG5 and ATG7 via both SMAD-dependent and SMAD-independent pathways, and enhances the degradation rate of long-lived proteins in human HuH7 hepatocellular carcinoma cells39. TGFβ activates autophagy to enhance its growth-inhibitory effect and postpones apoptosis in human hepatocellular and mammary carcinoma cells in vitro39. TGFβ2-induced autophagy also supports glioma invasion in mice via alteration of epithelial–mesenchymal transition and metabolism40. Additionally, type I and II interferons are autophagy inducers in multiple human cancer cell lines41. It seems that IFNγ-induced autophagy correlates with an inhibition of carcinogenesis in an inflammation-related gastric cancer model42.
Tumour cell autonomous autophagy
Function in tumorigenesis
Most human cancers develop over a long period of time as mutations accumulate in various proto-oncogenes, such as KRAS and BRAF43. The relationship between oncogenes and autophagy genes in tumorigenesis has been explored. There are more than 40 genes encoding ATGs, which are involved in the autophagy pathway44. Mutation of genes encoding ATGs may contribute to tumour initiation and immune system recognition45. However, the core autophagy machinery in human tumour tissues remains largely intact at the DNA level and is expressed at similar transcript levels to matched normal tissues, based on data available from The Cancer genome Atlas (TCGA)46. This observation is in line with studies showing that intact systemic autophagy is necessary for tumour growth, even if few mutated ATGs are observed in certain human tumour types47,48. Mosaic deletion of Atg5 in mice or Atg7 in mouse liver led to only benign hepatomas, suggesting that complete and specific autophagy deficiency promotes liver tumour initiation but restricts progression to malignant disease49. A similar phenomenon was observed in KrasG12D-driven pancreatic cancer models; loss of Atg5 or Atg7 in the mouse pancreas promoted benign pancreatic intraepithelial neoplasia formation, but also prevented progression of this pancreatic intraepithelial neoplasia to malignant disease50,51. Moreover, tissue-specific inactivation of Atg5 in mice markedly accelerated the initiation of but impaired the progression of KrasG12D-driven lung cancer52. Complete loss of ATG7 was also found to reduce the tumour burden in BrafV600E-driven melanoma53 and lung cancer54. The general observations in ATG-deficient cells include accumulated p62, damaged mitochondria, ROS and genome damage. The accumulation of ROS or p62 can promote genome instability and attenuate NF-κB signalling, which was shown to initiate tumour formation in autophagy-deficient tumour models55,56. However, the accumulation of damaged cellular components and ROS can induce cell senescence, which inhibits tumour growth by restraining the tumour cell proliferation57. In BrafV600E-driven melanoma, Atg7-deleted tumours showed increased oxidative stress and senescence. Thus, autophagy promotes BrafV600E-driven melanoma by limiting oxidative stress and overcoming senescence53. In line with this, reduced cell proliferation but increased cell senescence was observed in mice bearing Atg7−/− compared with wild-type BrafV600E-driven lung tumours54. Hence, autophagy deficiency-induced senescence prevents further tumour progression. Notably, a complete loss of autophagic function in cancer cells is rare in patients. Autophagy was first linked to human cancer based on the detection of monoallelic deletions of BECN1 in human breast cancer and ovarian cancer58,59. Similarly, heterozygous loss of ATG5 at chromosome band 6q21 was found to be a distinctive feature of human advanced melanomas60. Monoallelic loss of Atg5 enhanced melanoma metastasis and compromised the response to targeted therapy in mouse models60. In addition, although homozygous disruption of Atg5 did not lead to the development of any advanced tumour, heterozygous disruption of Atg5 in KrasG12D-driven pancreatic cancers with insufficient ATG5 levels led to an increased burden of tumours and metastases compared with wild-type mice61. In addition to its direct effect on tumour cells, inflammation may provide an indirect mechanism by which autophagy defects promote tumorigenesis62. It has been reported that functional loss of ATG16L1 based on the Thr300Ala (T300A) mutation induces chronic inflammation in Crohn’s disease, which predisposes patients to colorectal cancer development. ATG16L1 Thr300Ala mutation or Atg16l1 depletion in macrophages enhances endotoxin-induced inflammasome activation, which makes the ATG16L1-deficient mice highly susceptible to dextran sulfate sodium-induced acute colitis63,64. Interestingly, Atg16l1T300A knock-in mice have been shown to manifest an impaired antibacterial host defence, which in turn led to chronic inflammation, tissue damage and an elevated cancer risk65. Thus, it appears that a mild or intermediate defect in cancer cell autophagy could promote tumorigenesis and drive cancer progression, whereas complete loss of an autophagy component can trigger tumour cell senescence and slow down cancer progression.
Function in the immune response
Elevated autophagy in tumour cells plays a pro-tumour role in the TME12,38. The antitumour effect of autophagy deficiency on pancreatic tumour growth was observed in immunocompetent mice14, but not in immune-deficient mice66. This suggests a potential impact of tumour autonomous autophagy on antitumour immune response.
MHC class I-mediated tumour antigen presentation.
Major histocompatibility complex (MHC) molecules present antigens to T cells, which usually leads to initiation of an immune response. There are two types of MHC: MHC class I and MHC class II. MHC class I is expressed in all nucleated cell types, whereas MHC class II expression is limited to professional antigen-presenting cells (APCs), such as dendritic cells. MHC class I displays the peptides to prime and activate CD8+ cytotoxic T lymphocytes (CTLs). CTLs kill the targeted tumour cells via matched antigen–MHC class I complex recognition67. MHC class I-restricted antigens are generally derived from endogenous neosynthesized proteins, which are processed by the ubiquitin–proteasome system and transferred to the endoplasmic reticulum. Here, they are loaded onto newly translated MHC class I complexes. It has been reported that autophagy promotes MHC class I internalization and degradation in dendritic cells, and autophagy-deficient dendritic cells elevate presentation of different viral antigens to CD8+ T cells68,69.
Alterations in the genes encoding proteins of MHC class I, such as B2M and HLA genes, occur in certain types of cancer70,71. Tumour cells express low levels of MHC class I, which enables escape from CD8+ T cell-mediated killing. It has been reported that autophagy-related genes are enriched in MHC class I-negative pancreatic carcinoma cells that reside in liver metastasis72. Interestingly, the autophagy and lysosome system support pancreatic carcinoma metabolism and growth50. It is possible that autophagy mediates MHC class I lysosomal degradation in cancer cells. In support of this, in pancreatic cancer cells, selective autophagy has been shown to reroute MHC class I to lysosomes via the ubiquitin-binding receptor, neighbour of BRCA1 gene 1 protein (NBR1), leading to degradation, thereby precluding T cell recognition14. This MHC class I degradation depends on canonical autophagy, but not on LAP or LANDO14. Hence, tumour cells can evade immune surveillance via autophagy-mediated MHC class I degradation (FIG. 2a). Furthermore, it appears that basal autophagy flux varies in different pancreatic tumours, and this is associated with immunogenicity. Autophagy-low tumours exhibit higher levels of MHC class I expression and tumour-infiltrating CD8+ T cells compared with autophagy-high tumours14. In support of this observation, host autophagy — particularly hepatocyte autophagy — suppresses an antitumour T cell response in carcinogen-induced MB49 urothelial carcinoma tumour-bearing mice73. Thus, heterogeneous expression of autophagy genes can shape tumour immunogenicity14,74.
Fig. 2 |. Impact of tumour autonomous autophagy on immunity.
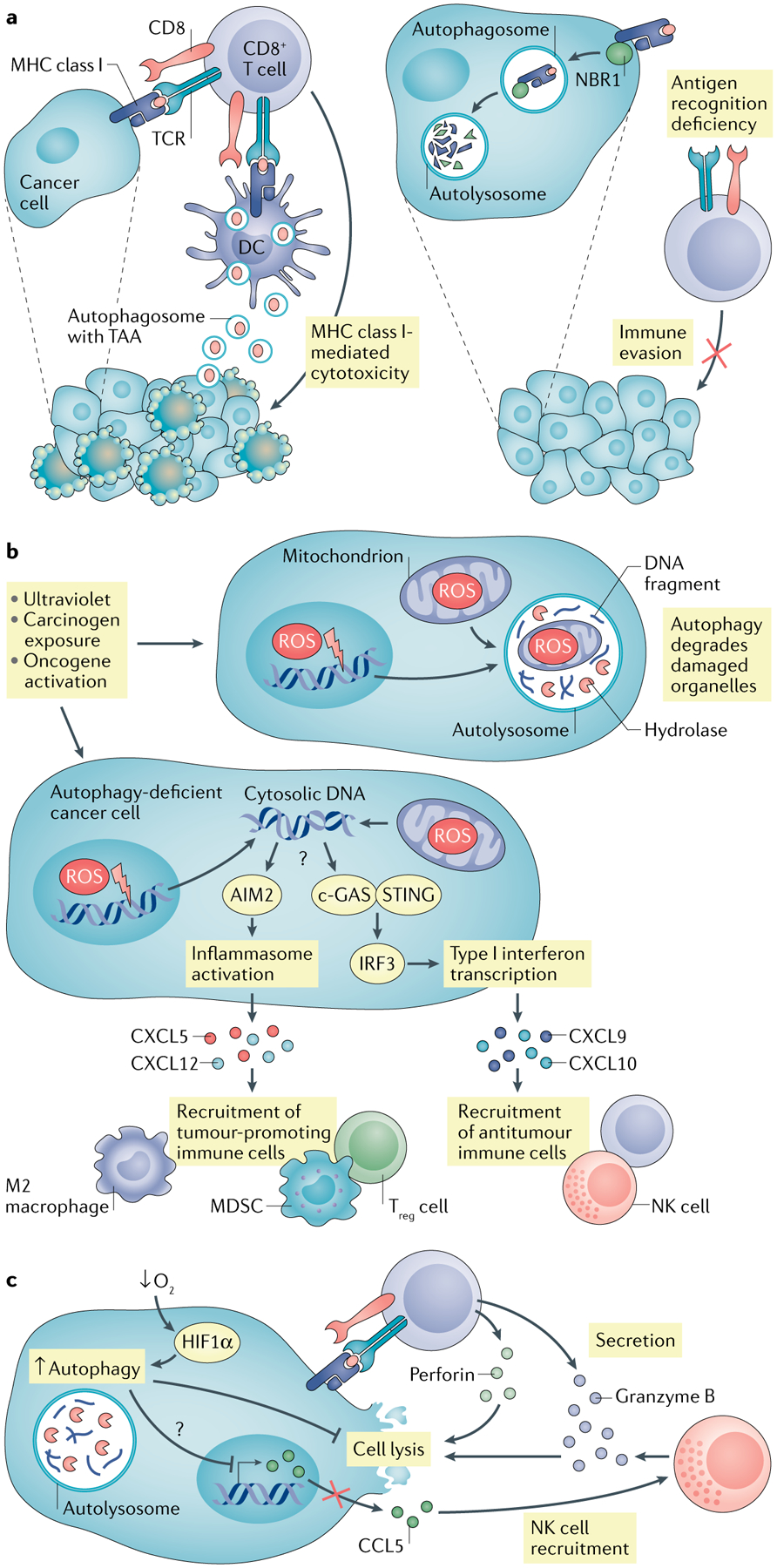
a | Autophagy in dead tumour cells promotes dendritic cell (DC)-mediated cross-presentation via increasing production of autophagosome with tumour antigen. In addition, neighbour of BRCA1 gene 1 protein (NBR1) mediates major histocompatibility complex (MHC) class I degradation via the autophagy pathway, resulting in reduced MHC class I surface expression in pancreatic cancer cells and abolished cytotoxic T lymphocyte (CTL) recognition. b | Autophagy blockade or deficiency results in accumulated reactive oxygen species (ROS) in tumour cells and subsequent mitochondria and genome damage, thereby releasing DNA into cytoplasm. Absent in melanoma 2 (AIM2) senses cytosolic DNA, activates inflammasome signalling and promotes tumorigenesis via recruiting immunosuppressive immune cells. c-GAS–stimulator of interferon genes (STING) can also sense cytosolic DNA, stimulate type I interferon responses and enhance antitumour immunity via recruiting CD8+ T and natural killer (NK) cells. c | Hypoxia induces autophagy in tumour cells through hypoxia-induced factor 1α (HIF1α). Elevated autophagy mediates degradation of granzyme B in tumour cells, which is secreted by activated CD8+ T cells and NK cells, and blocks CTL-mediated and NK cell-mediated tumour killing. Meanwhile, autophagy inhibits chemokine CCL5 expression, which recruits the NK cell migration to the tumour microenvironment (TME). MDSC, myeloid-derived suppressor cell; TAA, tumour-associated antigen; Treg cell, regulatory T cell.
In addition to cytosolic antigens, dendritic cell subsets, such as CD8α+ and CD103+ dendritic cells, can present exogenous antigens to CD8+ T cells via MHC class I — namely, antigen cross-presentation75. To this end, dendritic cells take up dead tumour cells, which are transported into the cytosol and processed by the proteasome. The processed antigens can then be loaded on MHC class I molecules in the endoplasmic reticulum or reimported into the phagosome to be loaded on MHC class I molecules for presentation to CD8+ T cells. Induction of autophagy in tumour cells can enhance their phagocytosis and MHC class I-mediated cross-presentation to CD8+ T cells. Indeed, the uptake of intact autophagosomes that comprise tumour antigens, released from dead tumour cells, is necessary for dendritic cell-mediated cross-presentation76,77 (FIG. 2a). Consistently, in autochthonous mouse models of sporadic intestinal tumour with signal transducer and activator of transcription 3 (STAT3) deficiency in intestinal epithelial cells (IECs), IEC mitophagy can enhance MHC class I-mediated tumour antigen presentation and augment CD8+ T cell-mediated antitumour immunity via dendritic cells78. Hence, the role of tumour autonomous autophagy in tumour antigen presentation is context-dependent.
Immune cell trafficking in the tumour.
Distinct chemokines mediate the trafficking of different immune cell subsets into the TME79. Autophagy can regulate the expression of chemokines in tumour cells and migration of immune cells to the tumour, in turn altering tumour immune responses. Atg5-inactivated KrasG12D-driven lung cancer cells expressed high protein levels of CXCL5, a pro-tumour chemokine for myeloid-derived suppressor cell (MDSC) migration, compared with control KrasG12D lung cancer cells52,79. Monoallelic Atg5-deleted KrasG12D-driven pancreatic cancer cells produced high levels of chemokines (including CXCL1, CXCL2 and CXCL12) compared with autophagy-competent KrasG12D pancreatic cancer cells, attracting more immune-suppressive macrophages into original pancreatic tumour tissues in these transgenic mice61. Interestingly, in the polyoma middle T antigen (PyMT)-driven spontaneous mouse model of breast cancer, FAK family interacting protein of 200 kDa (FIP200; also known as RB1CC1) ablation suppressed mammary tumour initiation and progression via elevated chemokine expression of CXCL9 and CXCL10, which mediates CD8+ T cell tumour trafficking80. Similarly, conditional inactivation of Atg7 in IECs inhibited the formation of precancerous lesions in Apc+/− mice. IECs with conditional Atg7 inactivation, but not those expressing wild-type Atg7, produced CXCL9 and CXCL10, and supported CD8+ T cell trafficking13. Accordingly, targeting autophagy genes or pharmacologically inhibiting autophagy induced the expression of CCL5 in melanoma cells, resulting in an increase of natural killer (NK) cell tumour infiltration81. These data suggest that autophagy may play an inhibitory role in the expression of pro-tumour chemokines as well as antitumour chemokines, thereby differentially affecting tumour progression (FIG. 2b).
Natural killer cell and CD8+ T cell effector function.
NK cells and CD8+ T cells are effector immune cells and mediate tumour killing. Tumour autonomous autophagy, induced by hypoxia, has been shown to degrade NK cell-derived granzyme B and weaken NK cell-mediated tumour lysis in B16-F10 and 4T1 mouse tumour models. Inhibition of autophagy restored the levels of granzyme B, recovered NK cell cytotoxicity and prevented tumour progression82. Similarly, hypoxia-induced tumour autophagy in IGR-Heu lung carcinoma cells has been shown to regulate tumour cell STAT3 phosphorylation and disable tumour cell sensitivity to CTL-mediated killing83. Therefore, hypoxia-induced autophagy can attenuate the antitumour effector function of NK cells and CTLs (FIG. 2c). In line with this, a genome-wide in vitro clustered regularly interspaced short palindromic repeats (CRISPR) screen across a panel of genetically diverse mouse cancer cell lines has shown that the autophagy pathway is a conserved mediator of tumour resistance to cytotoxicity induced by CTLs and the effector cytokines IFNγ and TNF. Thus, activated tumour autophagy may be a cancer cell-intrinsic immune evasion mechanism in the TME84.
CTL-mediated tumour ferroptosis.
Ferroptosis is a form of necrotic cell death caused by uncontrolled peroxidation of phospholipid membranes via an iron-dependent mechanism85. Nuclear receptor coactivator 4 (NCOA4) plays a role in degradation of ferritin (cellular iron storage protein) and supports cell ferroptosis. Blockade of autophagy or knockdown of NCOA4 abrogates the accumulation of ferroptosis-associated cellular labile iron and ROS, and prevents eventual ferroptotic cell death86,87. Tumour ferroptosis is a critical mechanism by which CD8+ T cells mediate tumour killing in tumour immunotherapy88 and radiotherapy89. Thus, autophagy activation may potentiate tumour ferroptosis and sensitize tumour immunotherapy.
Altogether, it seems that tumour autonomous autophagy not only supports tumour cell survival and proliferation but also alters tumour antigen presentation, immune cell trafficking and effector immune cell function in the TME.
Autophagy in tumour-infiltrating immune cells
T cells
Human tumour-infiltrating immune cells — including effector CD8+ T cells, regulatory T cells (Treg cells) and myeloid APCs90–92 — affect tumour progression and therapeutic response8,10. Apart from tumour cells, multiple stress signals affect the autophagy pathway in immune cell subsets, thereby shaping their survival, activation, differentiation and function in the TME11.
Effector T cell subsets.
CD8+ T cells recognize tumour-associated antigens (TAAs) and mediate tumour killing. CD4+ T helper cells (TH cells) help dendritic cells prime and activate CD8+ T cells. However, T cells (particularly naive T cells) in the tumour draining lymph nodes and TME undergo apoptosis in tumour-bearing mice and patients with cancer. It has been shown that tumour-infiltrating T cells in tumour-bearing mice manifest impaired autophagy along with a selectively reduced expression of FIP200 (which is essential for autophagosome formation)93. Mechanistically, tumour-derived lactate inhibited FIP200 expression and caused T cell apoptosis by disabling the balance between pro-apoptotic and anti-apoptotic BCL2 family members, thereby supporting tumour immune evasion93.
In addition to T cell survival and apoptosis, autophagy is also involved in the regulation of T cell activation, differentiation, stemness and effector function. After TCR engagement in vitro, autophagy in CD8+ T cells is induced, leading to degradation of BCL10 and protecting the cells from adverse consequences of unrestrained NF-κB activation94. Phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3; also known as VPS34) is an early key player in autophagy. Pik3c3-deficient T cells exhibit reduced levels of active mitochondria compared with wild-type controls and fail to differentiate into effector T cells upon T cell activation95. Autophagy-deficient effector CD4+ T cells with Atg7 depletion express minimal levels of IL-2 and IFNγ96. Human liver-resident memory CD8+ T cells manifest increased basal autophagy compared with CD8+ T cells in blood, which is critical for their proliferation and cytokine production. Blocking autophagy in IL-15-induced resident memory CD8+ T cells by MRT68921 dihydrochloride and 3-methyladenine (3MA) results in the accumulation of depolarized mitochondria, a feature of exhausted T cells97.
Furthermore, autophagy can regulate T cell metabolism to control T cell phenotypes. In the steady state, naive T cells largely rely on oxidative phosphorylation. Upon antigen stimulation, T cells undergo a metabolic transition from oxidative phosphorylation to glycolysis to support cellular proliferation and effector function. During memory formation, effector T cells revert to oxidative phosphorylation98. Although speculative, autophagy might be involved in promoting fatty acid β-oxidation during memory formation. This is because autophagy can contribute to the degradation of lipid droplets99, which can in turn support CD8+ T cell memory generation100. Autophagy-deficient effector CD8+ T cells are unable to establish effective memory to provide long lasting antiviral immunity101,102. Also, a persistent loss of mitochondrial function and mass has been observed in dysfunctional tumour-infiltrating CD8+ T cells103. In addition, decreased mitophagy leads to accumulated depolarized mitochondria in both human and mouse melanoma-infiltrating CD8+ T cells. These T cells display functional, transcriptomic and epigenetic characteristics of terminally exhausted T cells104. Hence, it is likely that a suitable level of autophagy is important for CD8+ T cell survival, differentiation and function.
Interestingly, it has been reported that potassium in the TME reduces the uptake and consumption of local nutrients in tumour-infiltrating T cells, which induces autophagy, enhances mitochondrially dominant metabolism and increases the depletion of intracellular acetyl-coenzyme A (AcCoA). Consumption of intracellular AcCoA in mitochondrial metabolism results in reduced histone acetylation on the promoters and enhancers of genes encoding effector molecules. Consequently, this causes impaired CD8+ T cell effector function, but sustains T cell stemness105. Blocking autophagy by disrupting Atg7 in potassium-treated T cells reduces T cell stemness, which is required for CD8+ T cell persistence in vivo, multipotency and tumour clearance105. However, it has also been shown that loss of ATG5 in T cells increases the number of tumour-infiltrating IL-9-expressing CD4+ T cells (TH9 cells)106, and CD8+ T cells isolated from Atg5-deficient mice show a profound shift to an effector memory phenotype compared with Atg5-proficient CD8+ T cells in tumour-bearing mice107. Thus, a balanced autophagy pathway shapes T cell immunity and alters tumour immune responses (FIG. 3a).
Fig. 3 |. autophagy in immune cells in the tumour microenvironment.
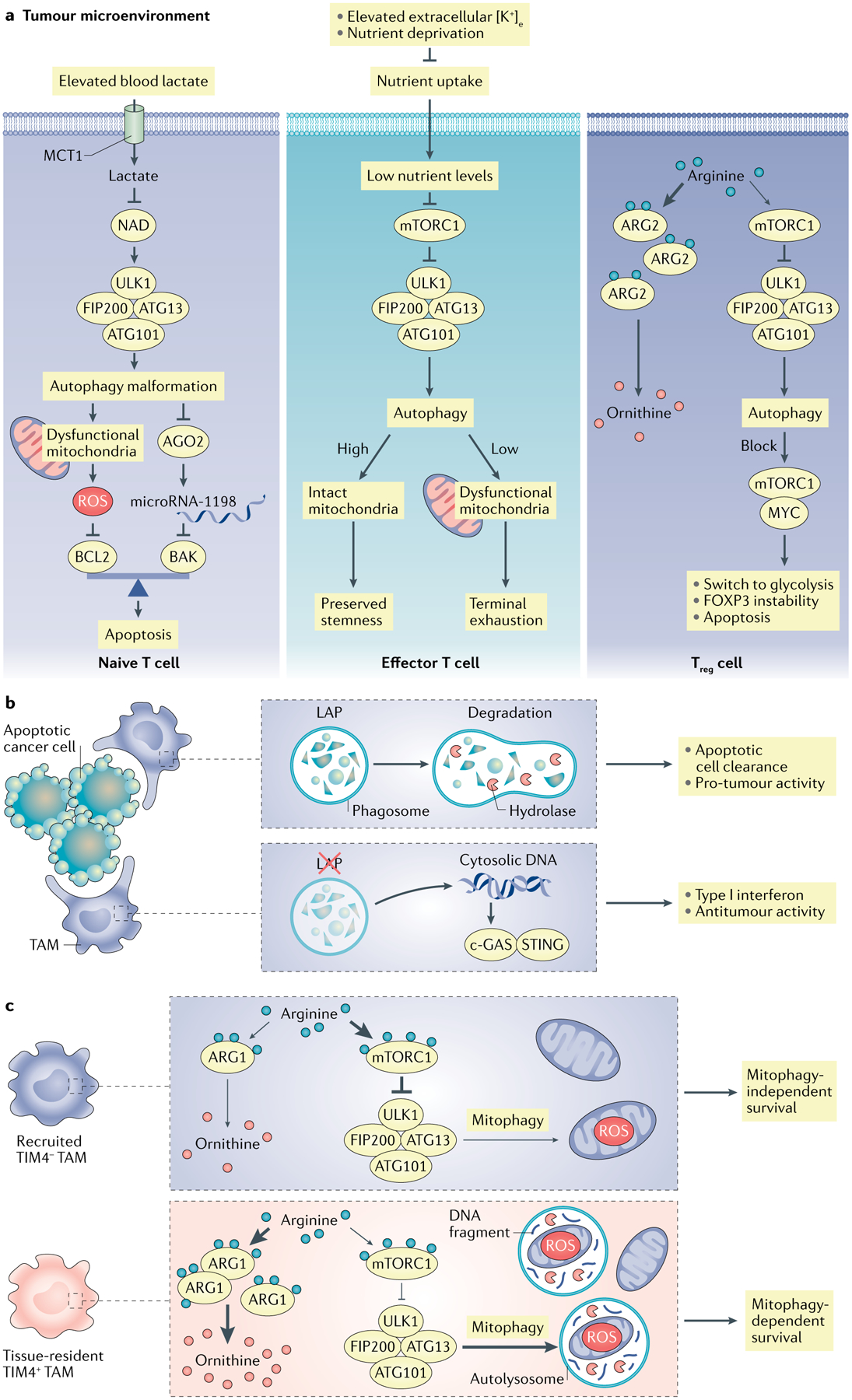
a | In the tumour microenvironment (TME), T cells rely on autophagy to support their survival and differentiation. Basal levels of autophagy in naive T cells maintain their quiescence and protect them from mitochondria-derived reactive oxygen species (ROS)-induced apoptosis. High levels of lactate in tumours disrupt autophagy of naive T cells and impair the antitumour response in mouse models. High levels of potassium or low levels of nutrients in the TME constrain the nutrient uptake in effector T cells and cause functional caloric restriction, which induces autophagy through mTOR complex 1 (mTORC1) inhibition. Autophagy is critical for mitochondrial integrity, which determines effector T cell differentiation via metabolic reprogramming. High levels of arginase 2 (ARG2) expression in tumour-infiltrating regulatory T cells (Treg cells) may maintain their high level of autophagy. The loss of autophagy in Treg cells switches their metabolism from oxidative phosphorylation (OXPHOS) to glycolysis with active mTORC1 and MYC, which leads to FOXP3 instability. Increased apoptosis and defective function in autophagy-deficient Treg cells contribute to greater tumour resistance. b | Tumour-associated macrophages (TAMs) take advantage of lipidate microtubule-associated protein 1A/1B light chain 3 (LC3)-associated phagocytosis (LAP) to degrade apoptotic cancer cells. LAP deficiency in TAMs leads to the release of mitochondrial DNA from apoptotic cancer cells, which induces type I interferon response through the cGAS–stimulator of interferon genes (STING) pathway and antitumour activity. c | In ovarian cancer metastasis, TIM4+ TAMs are embryonically originated and locally sustained, whereas TIM4− TAMs are replenished from circulating monocytes. Relative to TIM4− TAMs, TIM4+ TAMs manifest high levels of oxidative phosphorylation and adapt mitophagy to alleviate oxidative stress. High levels of ARG1 in TIM4+ TAMs contribute to potent mitophagy activities via weakened mTORC1 activation due to low arginine levels resulting from ARG1-mediated metabolism. Furthermore, genetic deficiency of autophagy element FAK family interacting protein of 200 kDa (FIP200) results in TIM4+ TAM loss via ROS-mediated apoptosis, and elevated T cell immunity and tumour inhibition in vivo. ATG13, autophagy-related protein 13; BCL2, B cell lymphoma 2; MCT1, monocarboxylate transporter 1; TIM4, T cell immunoglobulin and mucin domain-containing molecule 4; ULK1, unc-51-like kinase 1.
Of note, TAA-specific T cell activation is dependent on antigen processing and presentation. Autophagy is involved in the regulation of antigen processing and presentation in dendritic cells108. Efficient dendritic cell-mediated TAA presentation requires lysosomal proteolysis through autophagy109. Genetic knockout of Pik3c3 in dendritic cells results in a decrease of CD8α+ dendritic cells, impaired B16 melanoma-specific CTL activity and an increased incidence of lung metastasis in mice compared with controls69. In contrast to den dritic cells, MDSCs directly suppress T cell activation. MDSCs accumulate in the human cancer microenvironment, suppress antitumour immune responses, promote tumour progression and hinder cancer therapeutic efficacy110,111. MDSCs in patients with cancer exhibit increased levels of autophagy112, which may be induced by hypoxia113 and HMGB1 (REF.112). Ablation of autophagy in mono-cytic MDSCs results in elevated MHC class II expression, reduced suppressor activity and weakened tumour growth in a melanoma mouse model compared with controls114.
Regulatory T cell subsets.
CD4+ Treg cell-mediated suppression of antitumour immunity is a major mechanism of tumour immune evasion and immunotherapy resistance92,115,116. The autophagy pathway has been shown to be involved in Treg cell lineage commitment, thereby affecting antitumour immunity117. Indeed, specific deletion of genes encoding autophagy components, including ATG5, ATG7 and AMBRA1 (REFS118,119) in Treg cells, leads to T cell apoptosis and dysfunction in mice120–122 reg. On the molecular level, autophagy-deficient Treg cells have been shown to exhibit high levels of the mTORC1–MYC pathway and glycolytic activity121. In line with this, human melanoma-infiltrating Treg cells expressed high levels of arginase 2 (ARG2), which resulted in degradation of intracellular arginine and attenuation of arginine-mediated mTOR activation, thereby potentially leading to autophagy activation123. As a result of this interplay of metabolic and autophagy processes, Treg cell survival and function were enhanced in the TME122,123 (FIG. 3a).
Macrophages
Tumour-associated macrophages (TAMs) include peripheral migratory macrophages and long-lived, embryonic-derived and self-maintained residential macrophages124. Macrophages take up and process dead tumour cells, and present TAAs to T cells. When a cell dies, it is rapidly cleared by phagocytosis, which induces LAP in macrophages125. Clearance of apoptotic cells by macrophages through LAP induces anti-inflammatory cytokine production. However, when LAP is defective, such clearance induces the production of inflammatory cytokines and type I interferons125–127.
In the TME, death of cells and their clearance by TAMs are common events, and LAP can readily be detected in TAMs127. When LAP (but not canonical autophagy) was disrupted in TAMs by genetic depletion of various key components, antitumour T cell activity restricted Lewis lung carcinoma and B16-F10 tumour growth; this immunity was dependent on stimulator of interferon genes (STING; also known as TMEM173)-dependent type I interferon production127. As discussed above, disruption of autophagy in the TME has been shown to engage antitumour immunity; however, as components of autophagy and LAP are shared, it remains possible that it is LAP rather than autophagy that mediates these effects. This distinction may be particularly important in settings where lysosomal function is inhibited with chloroquine (CQ) or bafilomycin A1 as an adjunct to cancer therapy. Although such treatments block autophagic flux, they do not interfere with LC3 lipidation. Indeed, they are associated with accumulation of autophagosomes. By contrast, inhibition of the lysosomal V-ATPase blocks the lipidation of LC3 proteins at a single membrane, including the phagosome in LAP128. The ability of CQ treatment to improve anticancer immunity in preclinical and clinical settings might therefore be an effect of inhibiting LAP. LAP takes part in the MHC class II-mediated presentation of phagocytosed fungal antigens in human macrophages129. LAP in macrophages is required not only for efficient clearance of dead cells but also for determining their anti-inflammatory phenotype125,127. In line with this, defects in LAP induce pro-inflammatory gene expression, trigger STING-mediated type I interferon responses in TAMs and promote T cell-mediated antitumour immunity127 (FIG. 3b). Thus, non-canonical autophagy in TAMs may contribute to immune suppression in the TME.
Several human cancers, including ovarian cancer, often metastasize to the peritoneal cavity. The peritoneal cavity spaces contain a distinct population of cavity-resident macrophages. Deficiency of LAP-related components, such as Rubicon and ATG7, does not induce interferon responses in peritoneal residential macrophages130. Interestingly, loss of FIP200 in macrophages inhibits peritoneal ovarian cancer progression and results in a potent antitumour T cell response in an ID8 ovarian cancer-bearing mouse model. It appears that genetic knockout of Fip200 in macrophages causes high apoptosis of T cell immunoglobulin and mucin domain-containing molecule 4 (TIM4)-positive peritoneal residential macrophages in tumour-bearing mice due to the accumulation of mitochondria-related ROS. Relative to TIM4− TAMs, TIM4+ TAMs manifest high oxidative phosphorylation and adapt mitophagy to alleviate oxidative stress. Due to low arginine resultant from ARG1-mediated metabolism, high levels of ARG1 in TIM4+ TAMs contribute to potent mitophagy activities via weakened mTORC1 activation131. TIM4+ residential TAMs inhibit T cell function and promote tumour cavity metastasis131 (FIG. 3c). Thus, autophagy activation in tumour-associated residential macrophages supports TAM survival and enforces immunosuppression in the TME.
More recently, it has been recognized that LC3 proteins can also be lipidated at endosomal membranes when cell surface receptors are internalized — a process called LANDO7. LANDO functions in the recycling of endosomes from the cytosol to the plasma membrane and, similarly to LAP, also represses inflammatory cytokine production. To date, the molecules involved in LAP and LANDO are the same; therefore, it remains possible that the disruption of LANDO, rather than of LAP, contributes to antitumour immunity.
In summary, both canonical and non-canonical autophagy pathways participate in regulation of the tumour immune response in the TME. Further studies are essential to understand the distinct roles of these two autophagy pathways and their potential crosstalk in the context of tumour immunity and immunotherapy.
Autophagy in cancer therapy
Indirect modulation of tumour immunity
Treatment of cancer in a patient often includes cancer therapies such as chemotherapy, radiotherapy and/or immunotherapy. These therapies may take advantage of the immune system to eliminate tumour cells. In certain cancer therapies, autophagy is indispensable for the modulation of tumour immunity — which suggests that targeting autophagy in combination potentially improves these cancer therapies.
Re-establishing latent immune surveillance is important for inducing an antitumour response. The basis for tumour immune evasion is loss of immunogenicity. Immunogenic cell death (ICD), induced by some cancer therapies, renders tumours immunogenic and triggers antitumour immune responses. In response to ICD inducers, such as mitoxantrone and oxaliplatin, tumour autophagy was activated in mice bearing subcutaneous CT26 colon tumours, accompanied with increased tumour-infiltrating dendritic cells and T cells132. However, it remains unclear whether autophagy is directly induced by ICD inducers or is a consequence of ROS-based endoplasmic reticulum stress in the context of ICD in vivo133. Upon ICD, intact autophagosomes containing multiple tumour antigens and DAMPs from dying cells are released to the extracellular space and are taken up by APCs to initiate T cell immunity32. In addition, autophagy in CT26 cells was required for the preservation of lysosomal ATP stores and was related to the secretion of ATP during mitoxantrone treatment132. Extracellular ATP can function as a ‘find me’ signal to recruit APCs to dying cells and stimulate IL-1β production in APCs via NLRP3-mediated inflammasome activation, inducing antitumour adaptive immunity134. In addition, depletion of Atg5 or Atg7 in CT26 cells reduced chemotherapy-elicited ATP release from dying tumour cells and inhibited antitumour immunity132. In a similar vein, cisplatin failed to trigger ICD in prostate cancer cells due to weak autophagy induction by cisplatin compared with oxaliplatin135. Notably, dying B16-F10, MC38 or 3LL tumour cells released DAMPs in response to cisplatin, camptothecin or gemcitabine, thereby inducing TIM4 expression on tumour-associated myeloid cells. TIM4 mediated the phagocytosis of dying cells and activation of autophagy led to degradation of the ingested tumour cells, resulting in reduced antigen presentation and impaired CTL responses136. Because TIM4+ resident macrophages can sequester effector T cells and prevent CTL-mediated tumour killing137 (unpublished data), blocking the TIM4 and autophagy pathway with anti-TIM4 monoclonal antibody enhanced tumour antigen-specific CD8+ T cell infiltration and augmented the antitumour effect of chemotherapeutics136. This effect of TIM4 blockade on ICD induction was dispensable and may be useful to treat certain tumours without response to the ICD inducer136. Thus, autophagy can confer immunogenic properties to tumours in the context of some chemotherapies.
In addition to chemotherapy, radiotherapy can also induce ICD under specific doses and precise fractionation schedules. For instance, CT26 cells exposed to gamma-radiation or ultraviolet C light showed an increase in membrane calreticulin. Calreticulin serves as an ‘eat me’ signal for dying tumour cells138 and is a sign of early ICD, which can promote phagocytosis of tumour cells by dendritic cells139. Interestingly, multiple mouse and human tumour cells express high levels of stanniocalcin 1, which blocks calreticulin membrane exposure and abrogates membrane calreticulin-directed phagocytosis by APCs, thereby impairing tumour immunity140. Moreover, it has been reported that loss of autophagy in tumour cells reduces the efficacy of radiotherapy in immunocompetent mice, where autophagy-induced ATP released from dying tumour cells elicited an antitumour immune response during radiotherapy141. Hence, autophagy may be involved in the induction of tumour immunogenicity in radiotherapy.
Also in response to cancer targeted therapies, autophagy can modulate antitumour immune responses. CD47 on the tumour surface acts as a ‘don’t eat me’ signal and prevents macrophage-mediated phagocytosis through its direct interaction with signal regulatory protein-α (SIRPα)142. SIRPαD1–Fc, a fusion protein comprising the first extracellular domain of human SIRPα and the Fc fragment of human IgG1, blocked the CD47–SIRPα signalling pathway and induced autophagy in NSCLC cells. This led to a weakened macrophage-mediated phagocytosis of NSCLC cells and reduced SIRPαD1–Fc-mediated cytotoxicity against NSCLC cells. In line with the possibility that autophagy played a role in reducing cell clearing effectivity, simultaneous blockade of the CD47–SIRPα signalling and autophagy elicited potent antitumour effects in NSCLC xenograft models143. In addition, sunitinib, a receptor protein-tyrosine kinase inhibitor, downregulated tumour PDL1 expression by inducing p62-mediated selective autophagy, and increased antitumour immunity in a phase III trial in patients with metastatic breast cancer144. Thus, autophagy differentially modulates tumour immune responses in cancer therapy.
Targeting autophagy
Autophagy inhibitors are classified as early-stage inhibitors (SBI-0206965, 3MA and wortmannin), targeting ULK1/ULK2 or VPS34, and late-stage inhibitors (CQ, hydroxychloroquine (HCQ), bafilomycin A1 and monensin), targeting the lysosome. More specifically, CQ and HCQ inhibit autophagosome degradation via interfering with lysosome acidification. However, HCQ monotherapy failed to control tumour growth in patients with advanced pancreatic cancer in a clinical trial145. Given the effects of autophagy inhibition on the immune response as outlined above, autophagy inhibition in combination with other cancer therapies has been considered in order to improve therapy efficacy. For example, patients with resectable pancreatic adenocarcinoma treated with HCQ in combination with the chemotherapeutic agents nab-paclitaxel and gemcitabine exhibited an increase in immune cell tumour infiltration146. In addition, the combination of CQ and the mutated BRAF inhibitor vemurafenib improved tumour control in a patient with BRAF-mutant brain cancer who acquired resistance to vemurafenib147. Furthermore, multiple clinical trials have explored the antitumour efficacy of autophagy inhibitors in combination with different treatments (TABLE 1). As hinted above, HCQ mediates a modest lysosomal inhibition in patients with cancer. To enhance the potential of lysosomal inhibition and the antitumour effect, new lysosomotropic agents, such as Lys05 (REF.148) and DC661 (REFS149,150), are being developed using the CQ structure as a base. These drugs target palmitoyl-protein thioesterase 1 (PPT1) and manifest high tumour inhibition in preclinical studies.
Table 1 |.
Clinical trials targeting autophagy in cancer therapy
| Tumour type | Autophagy inhibitor | Clinical trial phase | Combination | Clinical response | Side effects | Ref. |
|---|---|---|---|---|---|---|
| Brain metastases | CQ | II | Radiation | ORR: 54% (controls: 55%) PFS of brain metastasis at 1 year: 83.9% (controls: 55.1%) |
Grade 1 or 2: headache, dizziness, nausea, vomiting, anorexia and myelosuppression Grade 3: nausea, constipation, headache and drowsiness |
174 |
| GBM | CQ | III | Radiation therapy and temozolomide | Median survival: 33 months (controls: 11 months) | Increased seizure frequency | 175 |
| GBM | HCQ | I/II | Radiation therapy and temozolomide | Median survival: 15.6 months | Neutropenia and thrombocytopenia | 176 |
| mCRPC | Pantoprazole | II | Docetaxel | Median OS: 15.7 months Median PFS: 5.3 months |
Grade 3 or 4: fatigue, anaemia, febrile neutropenia, grade 4 neutropenia and anorexia | 177 |
| NSCLC | HCQ | I/II | Carboplatin and paclitaxel | PFS: 3.3 months ORR: 33% |
Neutropenia, neuropathy and anaemia | 178 |
| NSCLC | HCQ | I | Erlotinib | 1 PR; 4 SD ORR: 5% |
Rash, nausea, diarrhoea and fatigue | 179 |
| PDAC | HCQ | I/II | Gemcitabine | Decreased CA 19–9: 61% Median OS: 34.8 months (control: 10.83 months) |
Grade 3: myelosuppression, hyponatraemia, elevated AST, hypoalbuminaemia, hyperbilirubinaemia rash and hyperglycaemia ileus | 180 |
| Pancreatic cancer | HCQ | II | Gemcitabine, nab-paclitaxel | Improved Evans grade histopathologic responses | None | 146 |
| Pancreatic cancer | HCQ | II | Gemcitabine, nab-paclitaxel | ORR: 38.2% (control: 21.1%) | Neutropenia, anaemia, fatigue, nausea, peripheral neuropathy, visual changes and neuropsychiatric symptoms | 145 |
| RCC | HCQ | I/II | Everolimus | Disease control: 67% PR: 6% PFS ≥6 months: 45% |
None | 181 |
| Refractory myeloma | HCQ | I | Bortezomib | Very good PR: 14% Minor response: 14% SD: 45% | Grade 1 or 2: myelosuppression fatigue, peripheral neuropathy, nausea, vomiting, diarrhoea and constipation Grade 3 or 4: nausea, constipation, diarrhoea, anorexia, myelosuppression and fatigue |
182 |
| Refractory myeloma | Ricolinostat | I/II | Bortezomib and dexamethasone | ORR: 37% CBR: 39% |
Thrombocytopenia, diarrhoea, anaemia, fatigue, nausea, hypokalaemia, vomiting, peripheral neuropathy, hyperglycaemia and renal insufficiency | 183 |
| Solid tumours | Pantoprazole | I | Doxorubicin | 2 patients with confirmed PR | Fatigue, neutropenia and leukopenia | 184 |
| Solid tumours | HCQ | I | MK-2206 | Overall response: 15% | Fatigue and maculo-papular rashes | 185 |
| Solid tumours including melanoma | HCQ | I | Temsirolimus | Solid tumours: SD (67%) Melanoma: SD (74%) |
Grade 1 or 2: fatigue, anorexia, nausea, stomatitis, rash and weight loss Grade 3 or 4: anorexia, fatigue and nausea |
186 |
| Solid tumours including melanoma | HCQ | I | Temozolomide | Solid tumour patients: PR (10%) and SD (27%) Melanoma: PR (14%) and SD (27%) |
Grade 2: fatigue, anorexia, nausea, constipation and diarrhoea | 187 |
| Solid tumours | HCQ | I | Vorinostat | 1 patient with renal cell carcinoma: durable PR; 2 patients with colorectal cancer: prolonged SD |
Grade 1–2: nausea, diarrhoea, fatigue, weight loss, anaemia and elevated creatinine Grade 3: fatigue and/or myelosuppression |
188 |
CBR, clinical benefit rate; CQ, chloroquine; GBM, glioblastoma multiforme; HCQ, hydroxychloroquine; mCRPC, metastatic castration-resistant prostate cancer; NSCLC, non-small cell lung cancer; ORR, overall response rate; OS, overall survival; PDAC, pancreatic adenocarcinoma; PFS, progression-free survival; PR: partial response; RCC, renal cell carcinoma; SD, stable disease.
Chemotherapy.
High autophagic flux in cancer correlates with reduced response to chemotherapy and is associated with poor survival in patients with cancer151. Preclinical studies have shown that autophagy inhibition overcomes chemotherapy resistance in NSCLC143,152, bladder cancer153, thyroid cancer154 and pancreatic cancer155,156. The results of several studies suggest that autophagy inhibition might synergize with inhibition of MEK–ERK signalling. In pancreatic ductal adenocarcinoma, inhibition of MEK1 (also known as MAPKK1) and MEK2 by trametinib leads to activation of autophagy. Interestingly, inhibition of MEK1 and MEK2 combined with autophagy inhibition by either CQ or the dominant-negative form of ATG4B synergistically reduced pancreatic ductal adenocarcinoma cell growth in vitro and in vivo155. In addition, inhibition of ERK in KRAS-driven human pancreatic ductal adenocarcinoma cells results in suppression of both glycolysis and mitochondrial function, and boosts autophagic activity156. In melanoma with BRAF mutation, BRAF and MEK inhibitors induced ERK reactivation and autophagy (via ERK-mediated phosphorylation of ATF4), which promoted tumour resistance to these inhibitors157,158. Expression of a dominant-negative ATF4 mutant in melanoma disrupted autophagy and sensitized tumours to BRAF and MEK inhibitors in vivo158. Therefore, autophagy inhibitors enhanced the ability of MEK, ERK and BRAF inhibitors to mediate antitumour activity155–158. However, there are other mechanisms of autophagy-mediated resistance to chemotherapy that are being uncovered. For example, autophagy induced by Fusobacterium nucleatum can protect human colon cancer cells from chemotherapy-induced apoptosis. Autophagy activation and abundant F. nucleatum were detected in colorectal cancer tissues in patients with recurrence post chemotherapy159. Therefore, novel clinical trials for chemotherapy will benefit from a comprehensive understanding of the nature of chemotherapy-induced autophagy.
Radiation therapy.
Autophagy plays a critical role in protecting tumour cells from cell death in response to radiation therapy. In breast cancer cells, radiation induced expression of autophagy-related genes, accompanied by accumulation of autophagosomes. Short-term inhibition of autophagy alongside radiotherapy led to enhanced cytotoxicity of radiotherapy in resistant cancer cells160. Similarly, hypoxia enhances radioresistance in A549 lung cancer cells by inducing autophagy, which regulates ROS production in vitro161. In glioblastoma, radiation therapy induced autophagy by increasing mammalian STE20-like protein kinase 4 (MST4) expression, which stimulates autophagy through phosphorylation of ATG4B at site S383 in a panel of patient-derived glioma stem-like cells in vitro. The small-molecule inhibitor NSC185058 (targeting ATG4B), in combination with radiotherapy, impairs intracranial xenograft growth of glioblastoma and extends the survival of treated mice162. Therefore, targeting tumour autophagy potentially enhances the efficacy of radiation therapy. Indeed, the effect of autophagy inhibitors in combination with radiation therapy has been exploited in clinical trials in patients with cancer (TABLE 1).
Immunotherapy.
Harnessing the immune system provides an important approach to combatting cancer. Inhibition of autophagy may impair systemic immunity as autophagy is involved in immune system development11 and effector T cell survival and function93. However, systemic inhibition of autophagy by CQ in a short time did not impair T cell function in preclinical models of melanoma and breast cancer. The data suggest that the immune system may be tolerant to a certain intensity of autophagy inhibition163. Yet, given that autophagy can regulate tumour immune response, targeting autophagy may promote the efficacy of immunotherapy and overcome immunotherapy resistance. For example, suppression of VPS34 kinase activity using its inhibitors SB02024 or SAR405 resulted in an increase in the levels of CCL5, CXCL10 and IFNγ in the TME, leading to high levels of NK cell and T cell tumour infiltration in models of melanoma and colorectal carcinoma. VPS34 inhibition also reversed either anti-PD1 or anti-PDL1 therapy resistance in these models164. In addition, treatment with CQ blocked autophagy-mediated MHC class I degradation, synergized with dual ICB therapy (anti-PD1 and anti-CTLA4 antibodies) and resulted in an enhanced antitumour immune response in a mouse model of pancreatic cancer14. Moreover, photodynamic therapy-induced tumour immunogenicity is counteracted by ROS-induced autophagy, and can be enhanced by treatment with the autophagy inhibitor 3MA, also leading to reduced PDL1 expression, in a model of osteosarcoma165. As an early immunotherapy regimen, high-dose IL-2 has been used to treat patients with renal cell carcinoma and melanoma166. High-dose IL-2 therapy shows an antitumour effect with broad stimulation of the immune system. However, high-dose IL-2 treatment can cause cytokine-induced systemic autophagic syndrome with increased serum levels of IFNγ, IL-6 and IL-18. Thus, pairing IL-2 with CQ limited toxicity and enhanced the immunotherapeutic efficacy of IL-2 in an advanced mouse model of metastatic liver cancer167. Hence, targeting autophagy could potentially enhance immunotherapy (FIG. 4). Accordingly, clinical trials using HCQ in combination with immunotherapy are ongoing to treat patients with different types of cancer (TABLE 2).
Fig. 4 |. Targeting autophagy for tumour immunotherapy.
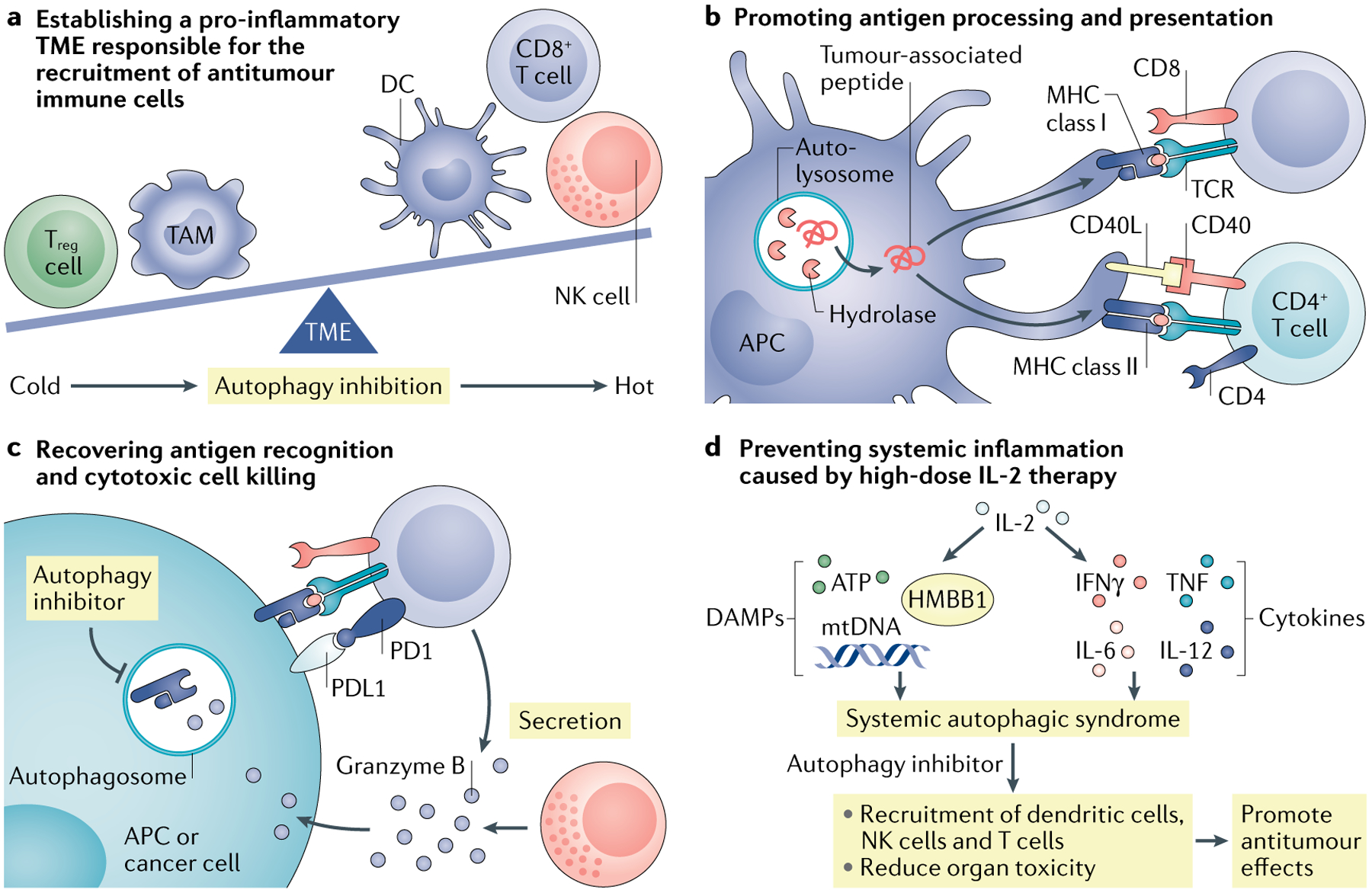
a | Targeting autophagy remodels the tumour microenvironment (TME). Tumour autophagy inhibition can upregulate production of the T helper 1 (TH1)-type chemokines, promoting effector immune cell tumour infiltration. Targeting tumour autophagy may be combined with adoptive transfer of engineered chimeric antigen receptor T cells (CAR T cells) and immune checkpoint blockade (ICB). b | Autophagy can mediate degradation of potential tumour antigens and major histocompatibility complex (MHC) components in antigen-presenting cells (APCs). Autophagy inhibition can enhance the antitumour T cell response through elevated antigen procession and presentation. c | Autophagy can mediate degradation of MHC class I and granzyme B to avoid cytotoxic T lymphocyte (CTL)-mediated and natural killer (NK) cell-mediated killing. Autophagy inhibition can result in increased levels of MHC class I on the cell surface and prevents the autophagic degradation of granzyme B, thereby allowing cell lysis. Autophagy inhibition can enhance ICB therapeutic efficacy. d | High-dose interleukin-2 (IL-2) treatment causes systemic autophagic syndrome. Autophagy inhibition protects patients with cancer from systemic damage and improves IL-2 therapy. DAMP, damage-associated molecular pattern; DC, dendritic cell; TAM, tumour-associated macrophage; Treg cell, regulatory T cell.
Table 2 |.
Ongoing clinical trials of combinations with immunotherapy
| Tumour type | Clinical trial phase | Treatment | Clinical trial identification number |
|---|---|---|---|
| Metastatic RCC | I/II | HCQ/aldesleukin (IL-2) | NCT01550367 |
| Metastatic melanoma | I/II | HCQ/nivolumab HCQ/nivolumab/Ipilimumab |
NCT04464759 |
| Gastrointestinal cancer | I/II | HCQ/cobimetinib/atezolizumab | NCT04214418 |
| Pancreatic cancer | II | HCQ/gemcitabine/nab-paclitaxel/avelumab | NCT03344172 |
Data are collected from US National Library of Medicine ClinicalTrials.gov. HCQ, hydroxychloroquine; RCC, renal cell carcinoma.
In addition, CAR T cells are a type of immunotherapy that involves T cells extracted from patients and genetically modified to express receptors that recognize cancer-specific antigens, followed by transfusion168. CAR T cell therapy has achieved clinical success in treating haematologic cancers, but failed in treating solid cancer169. Autophagy modulation may offer several benefits for patients with cancer treated with CAR T cells. It is well known that the TME presents a barrier for CAR T cell infiltration and function in solid tumours. Given that autophagy inhibition could remodel the TME and promote TH1-type chemokine production13,80, autophagy inhibition may facilitate CAR T cell tumour trafficking. Metabolic stress induces effector T cell apoptosis and impairs their function in the TME15. Elevated autophagy in CAR T cells may support T cell fitness and survival in the TME. Thus, prior to transfusion to patients with cancer, enforcing expression of autophagy components in vitro in CAR T cells may be a potential approach to improve their therapeutic efficacy. Furthermore, CAR T cells directly recognize antigens on the tumour cell surface, which can be degraded via the autophagy pathway77. Therefore, tumour autophagy inhibition may lead to increased expression of antigens, thereby enhancing CAR T cell-mediated tumour killing. Finally, CAR T cell therapy could cause cytokine release syndrome170. Because autophagic cell death may be involved in cytokine release syndrome, autophagy inhibition may ameliorate cytokine release syndrome and generate clinical benefit for patients. Overall, the potential for autophagy inhibition to increase the efficacy of immunotherapy is a promising area of continuous exploration, which will hopefully see patients benefitting in the future.
Conclusion
Autophagy is an important mechanism studied in multiple cancer fields, including cancer biology, immunology and therapy. Given that autophagy can be induced by different factors in different cells in the TME, its induction and activation can both promote and inhibit tumour progression. Autophagy in T cell subsets may play a positive role in the antitumour immune response11, whereas functional autophagy in tumour cells could support tumour antigen presentation and recognition, particularly in the setting of chemotherapy-induced ICD models132. In this context, inhibition of autophagy may be detrimental to antitumour immunity. By contrast, the autophagy pathway may be related to tumour cell survival, tumour antigen degradation, reduced TH1-type chemokines and enhanced Treg cells and MDSCs. Thus, systemically targeting autophagy as a cancer therapeutic approach is challenging.
This challenge may stem from multiple sources, such as our insufficient understanding of the different roles of the autophagy pathway in different tissues, organs and specific cells — including the immune system. For example, autophagy deficiency in the whole body, but not in specific immune cell subsets, leads to several systemic diseases including neurodegenerative disease, metabolic disease and cancer171. Liver-specific deletion of autophagy pathway components resulted in elevated circulating ARG1, causing a reduction of both serum and tumour arginine, slowing down arginine auxotroph tumour growth in a mouse model172 and inducing antitumour immunity in tumours with high mutational burden73. In order to target the autophagy pathway in the context of cancer immunotherapy, it is paramount to explore the biological activities of autophagy in the major immune cell subsets. Recent studies have started to assess the autophagy pathway in T cells, macrophages and dendritic cells. Future work may be extended to B cells, NK cells and NKT cells173. Additionally, following cancer progression, the autophagy pathway is dynamically altered in response to different stimuli in the TME. The role of autophagy has been largely studied in mouse models with specific genetic deletion of specific autophagy components. Therefore, developmental and functional compensation in murine models with specific genetic deficiency in autophagy components must be taken into account. Finally, it is well defined that autophagy degrades and recycles intracellular components to sustain metabolism and survival in response to stress signals, such as starvation. It is essential to understand how autophagy is simultaneously involved in fine-tuning the function and survival of immune cells, tumour cells and stromal cells in the TME, and whether systemic manipulation of the autophagy pathway eventually leads to increased clinical benefit in patients with cancer.
In fact, current clinical studies have found that systemically targeting tumour autophagy as a monotherapy has failed to yield reliable, objective responses in patients with cancer (TABLES 1 and 2). However, manipulation of autophagy remains an option in combinatorial therapy with conventional therapy and ICB (TABLES 1 and 2). Information on the tumour genetic status and immune infiltration profile may inform the optimal therapeutic combination. For example, if tumour genetic mutations result in autophagic adaptation, autophagy inhibition may potentially improve targeted therapy against specific mutated genes. As tumours with high genetic mutation burden may express relatively high levels of neoantigens alongside increased immune cell infiltration, compared with tumours with a low mutation burden, autophagy inhibition may maintain and boost the immune response via decreasing autophagy-mediated degradation of tumour antigen and MHC class I. Hence, immunotherapy in combination with autophagy inhibition may be a meaningful approach for tumours with a high mutation burden. Furthermore, autophagy manipulation in vitro and ex vivo through a dendritic cell-based cancer vaccine may be feasible. For example, inhibition of dendritic cell autophagy may enhance MHC class I-mediated tumour antigen presentation and T cell activation68,69. Autophagosomes derived from dead tumour cells contain tumour antigens and may be utilized for dendritic cell vaccines to induce cross-presentation76,77. Altogether, in order to pursue the next generation of autophagy modulators for cancer therapy, the immunological parameters should be considered. We expect that autophagy modulators can enhance tumour immunogenicity and antigen presentation, improve tumour-infiltrating T cell survival and function, and potentially disable the immunosuppressive networks in the TME. It is hopeful that targeting the autophagy pathway will enhance and/or synergize cancer therapy, including immunotherapy83,136.
Acknowledgements
The authors thank their trainees from the Zou and Green labs, as well as collaborators for their scientific input. This work was supported in part by research grants from the US National Institutes of Health (NIH) (AI40646 and CA231620 to D.R.G., and CA217648, CA123088, CA099985, CA193136 and CA152470 to W.Z.), the NCI Cooperative Human Tissue Network (CHTN) and the NIH through the University of Michigan Rogel Cancer Center Grant (P30 CA046592).
Glossary
- Immunogenicity
The ability to provoke an immune response in humans or other animals
- Chimeric antigen receptor T cells
(CAr T cells). Cells used in a type of cancer treatment in which the patient’s T cells are modified in the laboratory to carry CAr and reinfused back into the patient to attack cancer cells recognized by CAr
- Oxidative phosphorylation
The electron transfer chain driven by substrate oxidation that is coupled to the synthesis of ATP through an electrochemical transmembrane gradient
- Autophagic flux
The measure of autophagic degradation activity
- Reactive oxygen species
(roS). The highly reactive form of molecular oxygen formed as a by-product during the metabolism of oxygen
- Damage-associated molecular patterns
(DAMPs). Host-derived molecules that can initiate a non-infectious inflammatory response
- Epithelial–mesenchymal transition
A reversible cellular programme in which epithelial cells lose their cell polarity and cell–cell adhesion, gain migratory and invasive properties, and, finally, are transformed into mesenchymal cells
- The Cancer Genome Atlas
(TCgA). A project to catalogue genetic mutations responsible for cancer that takes advantage of genome sequencing and bioinformatics
- Senescence
A process by which cells irreversibly stop dividing and enter a state of permanent growth arrest without undergoing cell death
- Clustered regularly interspaced short palindromic repeats
(CriSPr). A family of repetitive DNA sequence used to detect and destroy DNA
- Glycolysis
The metabolic pathway that converts glucose into pyruvate independent of oxygen
- Fatty acid β-oxidation
The catabolic process by which fatty acid molecules are broken down to generate acetyl-coenzyme A (AcCoA) and utilized in the tricarboxylic acid (TCA) cycle
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Morishita H & Mizushima N Diverse cellular roles of autophagy. Annu. Rev. Cell Dev. Biol 35, 453–475 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Amaravadi RK, Kimmelman AC & Debnath J Targeting autophagy in cancer: recent advances and future directions. Cancer Discov 9, 1167–1181 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mizushima N, Yoshimori T & Ohsumi Y The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol 27, 107–132 (2011). [DOI] [PubMed] [Google Scholar]; This detailed review discusses the protein and membrane interactions required for autophagosome formation.
- 4.Marino G, Niso-Santano M, Baehrecke EH & Kroemer G Self-consumption: the interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol 15, 81–94 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dikic I & Elazar Z Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol 19, 349–364 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Galluzzi L & Green DR Autophagy-independent functions of the autophagy machinery. Cell 177, 1682–1699 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heckmann BL et al. LC3-associated endocytosis facilitates β-amyloid clearance and mitigates neurodegeneration in murine Alzheimer’s disease. Cell 178, 536–551.e14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zou W Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 5, 263–274 (2005). [DOI] [PubMed] [Google Scholar]; This comprehensive review discusses the interaction between host and tumour cells that creates an immunosuppressive TME, and describes how to target this microenvironment for anticancer therapy.
- 9.Sanmamed MF & Chen L A paradigm shift in cancer immunotherapy: from enhancement to normalization. Cell 175, 313–326 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zou W, Wolchok JD & Chen L PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci. Transl Med 8, 328rv4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This detailed review discusses the development and clinical application of ICB targeting the PDL1 and PD1 pathway in cancer therapy.
- 11.Clarke AJ & Simon AK Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat. Rev. Immunol 19, 170–183 (2019). [DOI] [PubMed] [Google Scholar]; This detailed review discusses the role of autophagy in the modulation of immune cell development and differentiation.
- 12.Yang A et al. Autophagy sustains pancreatic cancer growth through both cell-autonomous and nonautonomous mechanisms. Cancer Discov 8, 276–287 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article demonstrates that autophagy is critical for pancreatic tumour maintenance through tumour cell-autonomous and non-autonomous mechanisms.
- 13.Levy J et al. Intestinal inhibition of Atg7 prevents tumour initiation through a microbiome-influenced immune response and suppresses tumour growth. Nat. Cell Biol 17, 1062–1073 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Yamamoto K et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 581, 100–105 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article demonstrates that autophagy enables pancreatic cancer cells to evade an immune response through degrading MHC class I. Autophagy inhibition reverses this immune evasion in animal models of pancreatic cancer.
- 15.Bian Y et al. Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature 585, 277–282 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kimmelman AC & White E Autophagy and tumor metabolism. Cell Metab 25, 1037–1043 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim J, Kundu M, Viollet B & Guan KL AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol 13, 132–141 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Egan DF et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim J et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 152, 290–303 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inoki K, Zhu T & Guan KL TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Gwinn DM et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herzig S & Shaw RJ AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol 19, 121–135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu YL et al. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res 72, 1773–1783 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Di Conza G et al. The mTOR and PP2A pathways regulate PHD2 phosphorylation to fine-tune HIF1α levels and colorectal cancer cell survival under hypoxia. Cell Rep 18, 1699–1712 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mazure NM & Pouyssegur J Atypical BH3-domains of BNIP3 and BNIP3L lead to autophagy in hypoxia. Autophagy 5, 868–869 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Rouschop KM et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Invest 120, 127–141 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poillet-Perez L, Despouy G, Delage-Mourroux R & Boyer-Guittaut M Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol 4, 184–192 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alexander A et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl Acad. Sci. USA 107, 4153–4158 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song C et al. Oxidative stress-mediated NFκB phosphorylation upregulates p62/SQSTM1 and promotes retinal pigmented epithelial cell survival through increased autophagy. PLoS ONE 12, e0171940 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taguchi K et al. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc. Natl Acad. Sci. USA 109, 13561–13566 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gong T, Liu L, Jiang W & Zhou R DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol 20, 95–112 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Ma Y, Galluzzi L, Zitvogel L & Kroemer G Autophagy and cellular immune responses. Immunity 39, 211–227 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Shi CS & Kehrl JH TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci. Signal 3, ra42 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhan Z et al. Autophagy facilitates TLR4- and TLR3-triggered migration and invasion of lung cancer cells through the promotion of TRAF6 ubiquitination. Autophagy 10, 257–268 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhong Z et al. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell 164, 896–910 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang D et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 29, 5299–5310 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kang R et al. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ 17, 666–676 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katheder NS et al. Microenvironmental autophagy promotes tumour growth. Nature 541, 417–420 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kiyono K et al. Autophagy is activated by TGF-β and potentiates TGF-β-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res 69, 8844–8852 (2009). [DOI] [PubMed] [Google Scholar]
- 40.Zhang C et al. TGF-β2 initiates autophagy via Smad and non-Smad pathway to promote glioma cells’ invasion. J. Exp. Clin. Cancer Res 36, 162 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 41.Schmeisser H et al. Type I interferons induce autophagy in certain human cancer cell lines. Autophagy 9, 683–696 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tu SP et al. IFN-γ inhibits gastric carcinogenesis by inducing epithelial cell autophagy and T-cell apoptosis. Cancer Res 71, 4247–4259 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Strohecker AM & White E Targeting mitochondrial metabolism by inhibiting autophagy in BRAF-driven cancers. Cancer Discov 4, 766–772 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shibutani ST, Saitoh T, Nowag H, Munz C & Yoshimori T Autophagy and autophagy-related proteins in the immune system. Nat. Immunol 16, 1014–1024 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Li X, He S & Ma B Autophagy and autophagy-related proteins in cancer. Mol. Cancer 19, 12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lebovitz CB et al. Cross-cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy 11, 1668–1687 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kang MR et al. Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J. Pathol 217, 702–706 (2009). [DOI] [PubMed] [Google Scholar]
- 48.An CH, Kim MS, Yoo NJ, Park SW & Lee SH Mutational and expressional analyses of ATG5, an autophagy-related gene, in gastrointestinal cancers. Pathol. Res. Pract 207, 433–437 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Takamura A et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev 25, 795–800 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang A et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov 4, 905–913 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rosenfeldt MT et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 504, 296–300 (2013). [DOI] [PubMed] [Google Scholar]
- 52.Rao S et al. A dual role for autophagy in a murine model of lung cancer. Nat. Commun 5, 3056 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Xie X, Koh JY, Price S, White E & Mehnert JM Atg7 overcomes senescence and promotes growth of BrafV600E-driven melanoma. Cancer Discov 5, 410–423 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Strohecker AM et al. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov 3, 1272–1285 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Duran A et al. The signaling adaptor p62 is an important NF-κB mediator in tumorigenesis. Cancer Cell 13, 343–354 (2008). [DOI] [PubMed] [Google Scholar]
- 56.Mathew R et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 137, 1062–1075 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kwon Y, Kim JW, Jeoung JA, Kim MS & Kang C Autophagy is pro-senescence when seen in close-up, but anti-senescence in long-shot. Mol. Cell 40, 607–612 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aita VM et al. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics 59, 59–65 (1999). [DOI] [PubMed] [Google Scholar]
- 59.Liang XH et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402, 672–676 (1999). [DOI] [PubMed] [Google Scholar]; This study identifies that Beclin 1 functions as a putative tumour suppressor.
- 60.Garcia-Fernandez M et al. Metastatic risk and resistance to BRAF inhibitors in melanoma defined by selective allelic loss of ATG5. Autophagy 12, 1776–1790 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gorgulu K et al. Levels of the autophagy-related 5 protein affect progression and metastasis of pancreatic tumors in mice. Gastroenterology 156, 203–217.e20 (2019). [DOI] [PubMed] [Google Scholar]
- 62.Degenhardt K et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 10, 51–64 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Murthy A et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature 506, 456–462 (2014). [DOI] [PubMed] [Google Scholar]
- 64.Saitoh T et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 456, 264–268 (2008). [DOI] [PubMed] [Google Scholar]
- 65.Lassen KG et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc. Natl Acad. Sci. USA 111, 7741–7746 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Eng CH et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc. Natl Acad. Sci. USA 113, 182–187 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Blum JS, Wearsch PA & Cresswell P Pathways of antigen processing. Annu. Rev. Immunol 31, 443–473 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Loi M et al. Macroautophagy proteins control MHC class I levels on dendritic cells and shape anti-viral CD8+ T cell responses. Cell Rep 15, 1076–1087 (2016). [DOI] [PubMed] [Google Scholar]
- 69.Parekh VV et al. Autophagy-related protein Vps34 controls the homeostasis and function of antigen cross-presenting CD8α+ dendritic cells. Proc. Natl Acad. Sci. USA 114, E6371–E6380 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gettinger S et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov 7, 1420–1435 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grasso CS et al. Genetic mechanisms of immune evasion in colorectal cancer. Cancer Discov 8, 730–749 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pommier A et al. Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science 360, eaao4908 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Poillet-Perez L et al. Autophagy promotes growth of tumors with high mutational burden by inhibiting a T-cell immune response. Nat. Cancer 1, 923–934 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Perera RM et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 524, 361–365 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cruz FM, Colbert JD, Merino E, Kriegsman BA & Rock KL The biology and underlying mechanisms of cross-presentation of exogenous antigens on MHC-I molecules. Annu. Rev. Immunol 35, 149–176 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li Y et al. Tumor-derived autophagosome vaccine: mechanism of cross-presentation and therapeutic efficacy. Clin. Cancer Res 17, 7047–7057 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li Y et al. Efficient cross-presentation depends on autophagy in tumor cells. Cancer Res 68, 6889–6895 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ziegler PK et al. Mitophagy in intestinal epithelial cells triggers adaptive immunity during tumorigenesis. Cell 174, 88–101.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nagarsheth N, Wicha MS & Zou W Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol 17, 559–572 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wei H et al. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev 25, 1510–1527 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mgrditchian T et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc. Natl Acad. Sci. USA 114, E9271–E9279 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Baginska J et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc. Natl Acad. Sci. USA 110, 17450–17455 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Noman MZ et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res 71, 5976–5986 (2011). [DOI] [PubMed] [Google Scholar]
- 84.Lawson KA et al. Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nature 586, 120–126 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article demonstrates that the tumour cell-intrinsic autophagy pathway protects against CTL killing based on functional genomic screens using CRISPR–Cas9 approaches.
- 85.Friedmann Angeli JP, Krysko DV & Conrad M Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer 19, 405–414 (2019). [DOI] [PubMed] [Google Scholar]
- 86.Hou W et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gao M et al. Ferroptosis is an autophagic cell death process. Cell Res 26, 1021–1032 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang W et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569, 270–274 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lang X et al. Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of SLC7A11. Cancer Discov 9, 1673–1685 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang L et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med 348, 203–213 (2003). [DOI] [PubMed] [Google Scholar]
- 91.Curiel TJ et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat. Med 9, 562–567 (2003). [DOI] [PubMed] [Google Scholar]
- 92.Curiel TJ et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med 10, 942–949 (2004). [DOI] [PubMed] [Google Scholar]
- 93.Xia H et al. Suppression of FIP200 and autophagy by tumor-derived lactate promotes naive T cell apoptosis and affects tumor immunity. Sci. Immunol 2, eaan4631 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article demonstrates that tumour-derived lactate suppresses FIP200 expression and autophagy, thereby causing T cell apoptosis and attenuating antitumour immunity.
- 94.Paul S, Kashyap AK, Jia W, He YW & Schaefer BC Selective autophagy of the adaptor protein Bcl10 modulates T cell receptor activation of NF-κB. Immunity 36, 947–958 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yang G et al. Autophagy-related protein PIK3C3/VPS34 controls T cell metabolism and function. Autophagy 10.1080/15548627.2020.1752979 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hubbard VM et al. Macroautophagy regulates energy metabolism during effector T cell activation. J. Immunol 185, 7349–7357 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Swadling L et al. Human liver memory CD8+ T cells use autophagy for tissue residence. Cell Rep 30, 687–698 e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article reveals the importance of IL-15 in T cell autophagy in the liver, which provides a potential strategy to enhance immunotherapies.
- 98.Pearce EL & Pearce EJ Metabolic pathways in immune cell activation and quiescence. Immunity 38, 633–643 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Singh R et al. Autophagy regulates lipid metabolism. Nature 458, 1131–1135 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper uncovers the function of autophagy in regulating intracellular lipid stores (macrolipophagy).
- 100.O’Sullivan D et al. Memory CD8+ T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity 41, 75–88 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xu X et al. Autophagy is essential for effector CD8+ T cell survival and memory formation. Nat. Immunol 15, 1152–1161 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Puleston DJ et al. Autophagy is a critical regulator of memory CD8+ T cell formation. eLife 3, e03706 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Thommen DS & Schumacher TN T cell dysfunction in cancer. Cancer Cell 33, 547–562 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yu YR et al. Disturbed mitochondrial dynamics in CD8+ TILs reinforce T cell exhaustion. Nat. Immunol 21, 1540–1551 (2020). [DOI] [PubMed] [Google Scholar]; This article reveals that decreased autophagy in tumour-infiltrating T cells leads to accumulated depolarized mitochondria, which induces epigenetic reprogramming towards terminal exhaustion.
- 105.Vodnala SK et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science 363, eaau0135 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rivera Vargas T et al. Selective degradation of PU.1 during autophagy represses the differentiation and antitumour activity of TH9 cells. Nat. Commun 8, 559 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.DeVorkin L et al. Autophagy regulation of metabolism is required for CD8+ T cell anti-tumor immunity. Cell Rep 27, 502–513.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 108.Munz C Autophagy proteins in antigen processing for presentation on MHC molecules. Immunol. Rev 272, 17–27 (2016). [DOI] [PubMed] [Google Scholar]
- 109.Dorfel D et al. Processing and presentation of HLA class I and II epitopes by dendritic cells after transfection with in vitro-transcribed MUC1 RNA. Blood 105, 3199–3205 (2005). [DOI] [PubMed] [Google Scholar]
- 110.Cui TX et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity 39, 611–621 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li W et al. Aerobic glycolysis controls myeloid-derived suppressor cells and tumor immunity via a specific CEBPB isoform in triple-negative breast cancer. Cell Metab 28, 87–103 e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Parker KH, Horn LA & Ostrand-Rosenberg S High-mobility group box protein 1 promotes the survival of myeloid-derived suppressor cells by inducing autophagy. J. Leukoc. Biol 100, 463–470 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Corzo CA et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med 207, 2439–2453 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Alissafi T et al. Autophagy orchestrates the regulatory program of tumor-associated myeloid-derived suppressor cells. J. Clin. Invest 128, 3840–3852 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zou W Regulatory T cells, tumour immunity and immunotherapy. Nat. Rev. Immunol 6, 295–307 (2006). [DOI] [PubMed] [Google Scholar]
- 116.Maj T et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol 18, 1332–1341 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Delgoffe GM et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 30, 832–844 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nazio F et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol 15, 406–416 (2013). [DOI] [PubMed] [Google Scholar]
- 119.Fimia GM et al. Ambra1 regulates autophagy and development of the nervous system. Nature 447, 1121–1125 (2007). [DOI] [PubMed] [Google Scholar]
- 120.Le Texier L et al. Autophagy-dependent regulatory T cells are critical for the control of graft-versus-host disease. JCI Insight 1, e86850 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wei J et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat. Immunol 17, 277–285 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Becher J et al. AMBRA1 controls regulatory T-cell differentiation and homeostasis upstream of the FOXO3–FOXP3 axis. Dev. Cell 47, 592–607 e6 (2018). [DOI] [PubMed] [Google Scholar]
- 123.Lowe MM et al. Regulatory T cells use arginase 2 to enhance their metabolic fitness in tissues. JCI Insight 4, e129756 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.DeNardo DG & Ruffell B Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol 19, 369–382 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Martinez J et al. Microtubule-associated protein 1 light chain 3α (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc. Natl Acad. Sci. USA 108, 17396–17401 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]; This article demonstrates that LAP is required for degradation of ingested apoptotic cells and suppression of pro-inflammatory cytokine expression in macrophages.
- 126.Martinez J et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature 533, 115–119 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]; This article demonstrates that LAP defects, a non-canonical autophagy, are associated with impaired efferocytosis, secretion of pro-inflammatory cytokines and spontaneous onset of a systemic lupus erythematosus-like disorder in mice.
- 127.Cunha LD et al. LC3-associated phagocytosis in myeloid cells promotes tumor immune tolerance. Cell 175, 429–441 e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article demonstrates that TAMs take advantage of LC3-associated phagocytosis to degrade apoptotic tumour cells. Genetic deficiency of LC3-associated phagocytosis in TAM boosts antitumour immune response through the STING–type I interferon pathway.
- 128.Florey O, Gammoh N, Kim SE, Jiang X & Overholtzer M V-ATPase and osmotic imbalances activate endolysosomal LC3 lipidation. Autophagy 11, 88–99 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Romao S et al. Autophagy proteins stabilize pathogen-containing phagosomes for prolonged MHC II antigen processing. J. Cell Biol 203, 757–766 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wang YT et al. Select autophagy genes maintain quiescence of tissue-resident macrophages and increase susceptibility to Listeria monocytogenes. Nat. Microbiol 5, 272–281 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Xia H et al. Autophagic adaptation to oxidative stress alters peritoneal residential macrophage survival and ovarian cancer metastasis. JCI Insight 5, e141115 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Michaud M et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 334, 1573–1577 (2011). [DOI] [PubMed] [Google Scholar]; This article demonstrates that chemotherapy-associated autophagy is required for dying tumour cells to release ATP, and recruit and activate dendritic cells, thereby inducing tumour immunity.
- 133.Galluzzi L, Buque A, Kepp O, Zitvogel L & Kroemer G Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol 17, 97–111 (2017). [DOI] [PubMed] [Google Scholar]; This detailed review discusses the molecular and cellular mechanisms of cell death.
- 134.Ghiringhelli F et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β-dependent adaptive immunity against tumors. Nat. Med 15, 1170–1178 (2009). [DOI] [PubMed] [Google Scholar]
- 135.Shalapour S et al. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature 521, 94–98 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Baghdadi M et al. TIM-4 glycoprotein-mediated degradation of dying tumor cells by autophagy leads to reduced antigen presentation and increased immune tolerance. Immunity 39, 1070–1081 (2013). [DOI] [PubMed] [Google Scholar]
- 137.Chow A et al. Tim-4+ tissue-resident macrophages impair antitumor T-cell immunity [abstract 978]. Cancer Res 10.1158/1538-7445.AM2019-978 (2019). [DOI] [Google Scholar]
- 138.Obeid M et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med 13, 54–61 (2007). [DOI] [PubMed] [Google Scholar]
- 139.Obeid M et al. Calreticulin exposure is required for the immunogenicity of gamma-irradiation and UVC light-induced apoptosis. Cell Death Differ 14, 1848–1850 (2007). [DOI] [PubMed] [Google Scholar]
- 140.Lin H et al. Stanniocalcin 1 is a phagocytosis checkpoint driving tumor immune resistance. Cancer Cell 39, 1–14 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ko A et al. Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signalling. Cell Death Differ 21, 92–99 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Veillette A & Chen J SIRPα-CD47 immune checkpoint blockade in anticancer therapy. Trends Immunol 39, 173–184 (2018). [DOI] [PubMed] [Google Scholar]
- 143.Zhang X et al. Targeting CD47 and autophagy elicited enhanced antitumor effects in non-small cell lung cancer. Cancer Immunol. Res 5, 363–375 (2017). [DOI] [PubMed] [Google Scholar]
- 144.Li H et al. The beneficial role of sunitinib in tumor immune surveillance by regulating tumor PD-L1. Adv. Sci 8, 2001596 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Karasic TB et al. Effect of gemcitabine and nab-paclitaxel with or without hydroxychloroquine on patients with advanced pancreatic cancer: a phase 2 randomized clinical trial. JAMA Oncol 5, 993–998 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zeh HJ et al. A randomized phase II preoperative study of autophagy inhibition with high-dose hydroxychloroquine and gemcitabine/nab-paclitaxel in pancreatic cancer patients. Clin. Cancer Res 26, 3126–3134 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Levy JM et al. Autophagy inhibition improves chemosensitivity in BRAFV600E brain tumors. Cancer Discov 4, 773–780 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.McAfee Q et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc. Natl Acad. Sci. USA 109, 8253–8258 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Rebecca VW et al. A unified approach to targeting the lysosome’s degradative and growth signaling roles. Cancer Discov 7, 1266–1283 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Rebecca VW et al. PPT1 promotes tumor growth and is the molecular target of chloroquine derivatives in cancer. Cancer Discov 9, 220–229 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ma XH et al. Measurements of tumor cell autophagy predict invasiveness, resistance to chemotherapy, and survival in melanoma. Clin. Cancer Res 17, 3478–3489 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zou Y et al. The autophagy inhibitor chloroquine overcomes the innate resistance of wild-type EGFR non-small-cell lung cancer cells to erlotinib. J. Thorac. Oncol 8, 693–702 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kang M et al. Concurrent autophagy inhibition overcomes the resistance of epidermal growth factor receptor tyrosine kinase inhibitors in human bladder cancer cells. Int. J. Mol. Sci 18, 321 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wang W et al. Targeting autophagy sensitizes BRAF-mutant thyroid cancer to vemurafenib. J. Clin. Endocrinol. Metab 102, 634–643 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kinsey CG et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med 25, 620–627 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bryant KL et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med 25, 628–640 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ma XH et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J. Clin. Invest 124, 1406–1417 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ojha R et al. ER translocation of the MAPK pathway drives therapy resistance in BRAF-mutant melanoma. Cancer Discov 9, 396–415 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Yu T et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell 170, 548–563.e16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Apel A, Herr I, Schwarz H, Rodemann HP & Mayer A Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res 68, 1485–1494 (2008). [DOI] [PubMed] [Google Scholar]
- 161.Chen X et al. Autophagy enhanced the radioresistance of non-small cell lung cancer by regulating ROS level under hypoxia condition. Int. J. Radiat. Biol 93, 764–770 (2017). [DOI] [PubMed] [Google Scholar]
- 162.Huang T et al. MST4 phosphorylation of ATG4B regulates autophagic activity, tumorigenicity, and radioresistance in glioblastoma. Cancer Cell 32, 840–855.e8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Starobinets H et al. Antitumor adaptive immunity remains intact following inhibition of autophagy and antimalarial treatment. J. Clin. Invest 126, 4417–4429 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Noman MZ et al. Inhibition of Vps34 reprograms cold into hot inflamed tumors and improves anti-PD-1/PD-L1 immunotherapy. Sci. Adv 6, eaax7881 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article demonstrates that autophagy inhibition by targeting VPS34 reprogrammes the microenvironment of melanoma and colon cancer, and checkpoint blockade in combination with VPS34 inhibition enhances anticancer efficacy and prolongs mouse survival.
- 165.Yu W et al. Autophagy inhibitor enhance ZnPc/BSA nanoparticle induced photodynamic therapy by suppressing PD-L1 expression in osteosarcoma immunotherapy. Biomaterials 192, 128–139 (2019). [DOI] [PubMed] [Google Scholar]
- 166.Rosenberg SA IL-2: the first effective immunotherapy for human cancer. J. Immunol 192, 5451–5458 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Liang X et al. Inhibiting systemic autophagy during interleukin 2 immunotherapy promotes long-term tumor regression. Cancer Res 72, 2791–2801 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kershaw MH, Westwood JA & Darcy PK Gene-engineered T cells for cancer therapy. Nat. Rev. Cancer 13, 525–541 (2013). [DOI] [PubMed] [Google Scholar]
- 169.Newick K, O’Brien S, Moon E & Albelda SM CAR T cell therapy for solid tumors. Annu. Rev. Med 68, 139–152 (2017). [DOI] [PubMed] [Google Scholar]
- 170.Fajgenbaum DC & June CH Cytokine storm. N. Engl. J. Med 383, 2255–2273 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Mizushima N, Levine B, Cuervo AM & Klionsky DJ Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Poillet-Perez L et al. Autophagy maintains tumour growth through circulating arginine. Nature 563, 569–573 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sharonov GV, Serebrovskaya EO, Yuzhakova DV, Britanova OV & Chudakov DM B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat. Rev. Immunol 20, 294–307 (2020). [DOI] [PubMed] [Google Scholar]
- 174.Rojas-Puentes LL et al. Phase II randomized, double-blind, placebo-controlled study of whole-brain irradiation with concomitant chloroquine for brain metastases. Radiat. Oncol 8, 209 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Briceno E, Reyes S & Sotelo J Therapy of glioblastoma multiforme improved by the antimutagenic chloroquine. Neurosurg. Focus 14, e3 (2003). [DOI] [PubMed] [Google Scholar]; This clinical trial is the first to evaluate the effect of CQ on patients with glioblastoma multiforme during radiotherapy and chemotherapy.
- 176.Rosenfeld MR et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 10, 1359–1368 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Hansen AR et al. Pantoprazole affecting docetaxel resistance pathways via autophagy (PANDORA): phase II trial of high dose pantoprazole (autophagy inhibitor) with docetaxel in metastatic castration-resistant prostate cancer (mCRPC). Oncologist 24, 1188–1194 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Malhotra J et al. Phase Ib/II study of hydroxychloroquine in combination with chemotherapy in patients with metastatic non-small cell lung cancer (NSCLC). Cancer Treat. Res. Commun 21, 100158 (2019). [DOI] [PubMed] [Google Scholar]
- 179.Goldberg SB et al. A phase I study of erlotinib and hydroxychloroquine in advanced non-small-cell lung cancer. J. Thorac. Oncol 7, 1602–1608 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Boone BA et al. Safety and biologic response of pre-operative autophagy inhibition in combination with gemcitabine in patients with pancreatic adenocarcinoma. Ann. Surg. Oncol 22, 4402–4410 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Haas NB et al. Autophagy inhibition to augment mTOR inhibition: a phase I/II trial of everolimus and hydroxychloroquine in patients with previously treated renal cell carcinoma. Clin. Cancer Res 25, 2080–2087 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Vogl DT et al. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 10, 1380–1390 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Vogl DT et al. Ricolinostat, the first selective histone deacetylase 6 inhibitor, in combination with bortezomib and dexamethasone for relapsed or refractory multiple myeloma. Clin. Cancer Res 23, 3307–3315 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Brana I et al. A phase I trial of pantoprazole in combination with doxorubicin in patients with advanced solid tumors: evaluation of pharmacokinetics of both drugs and tissue penetration of doxorubicin. Invest. New Drugs 32, 1269–1277 (2014). [DOI] [PubMed] [Google Scholar]
- 185.Mehnert JM et al. A phase I trial of MK-2206 and hydroxychloroquine in patients with advanced solid tumors. Cancer Chemother. Pharmacol 84, 899–907 (2019). [DOI] [PubMed] [Google Scholar]
- 186.Rangwala R et al. Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 10, 1391–1402 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Rangwala R et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 10, 1369–1379 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Mahalingam D et al. Combined autophagy and HDAC inhibition: a phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 10, 1403–1414 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Harper JW, Ordureau A & Heo JM Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol 19, 93–108 (2018). [DOI] [PubMed] [Google Scholar]
- 190.Efeyan A, Comb WC & Sabatini DM Nutrient-sensing mechanisms and pathways. Nature 517, 302–310 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Schroeder B et al. The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology 61, 1896–1907 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Chen Q et al. ATL3 is a tubular ER-phagy receptor for GABARAP-mediated selective autophagy. Curr. Biol 29, 846–855.e6 (2019). [DOI] [PubMed] [Google Scholar]
- 193.Smith MD et al. CCPG1 is a non-canonical autophagy cargo receptor essential for ER-phagy and pancreatic ER proteostasis. Dev. Cell 44, 217–232.e11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Khaminets A et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522, 354–358 (2015). [DOI] [PubMed] [Google Scholar]
- 195.Grumati P et al. Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. eLife 6, e25555 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Fumagalli F et al. Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat. Cell Biol 18, 1173–1184 (2016). [DOI] [PubMed] [Google Scholar]
- 197.An H et al. TEX264 is an endoplasmic reticulum-resident ATG8-interacting protein critical for ER remodeling during nutrient stress. Mol. Cell 74, 891–908.e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Chino H, Hatta T, Natsume T & Mizushima N Intrinsically disordered protein TEX264 mediates ER-phagy. Mol. Cell 74, 909–921.e6 (2019). [DOI] [PubMed] [Google Scholar]
- 199.Cui Y et al. A COPII subunit acts with an autophagy receptor to target endoplasmic reticulum for degradation. Science 365, 53–60 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Liang JR, Lingeman E, Ahmed S & Corn JE Atlastins remodel the endoplasmic reticulum for selective autophagy. J. Cell Biol 217, 3354–3367 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Dowdle WE et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol 16, 1069–1079 (2014). [DOI] [PubMed] [Google Scholar]
- 202.Mancias JD, Wang X, Gygi SP, Harper JW & Kimmelman AC Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509, 105–109 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Wyant GA et al. NUFIP1 is a ribosome receptor for starvation-induced ribophagy. Science 360, 751–758 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Dou Z et al. Autophagy mediates degradation of nuclear lamina. Nature 527, 105–109 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Till A, Lakhani R, Burnett SF & Subramani S Pexophagy: the selective degradation of peroxisomes. Int. J. Cell Biol 2012, 512721 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Gibbings D et al. Selective autophagy degrades DICER and AGO2 and regulates miRNA activity. Nat. Cell Biol 14, 1314–1321 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]