

Watch a video presentation of this article
Answer questions and earn CME
Abbreviations
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- APC
antigen‐presenting cell
- AST
aspartate aminotransferase
- GGT
gamma‐glutamyltransferase
- GI
gastrointestinal
- GM‐CSF
granulocyte‐macrophage colony‐stimulating factor
- HLA
human leukocyte antigen
- IBD
inflammatory bowel disease
- IFN‐γ
interferon‐γ
- IL
interleukin
- MIP‐1
macrophage inflammatory protein‐1
- PBC
primary biliary cirrhosis
- PPI
proton pump inhibitor
- PSC
primary sclerosing cholangitis
- TNF‐α
tumor necrosis factor α
- UDCA
ursodeoxycholic acid
- ULN
upper limit of normal
Sarcoidosis is a multisystem inflammatory disorder characterized by noncaseating granulomas of unknown etiology. Histologically, granulomas are the main feature of this disease process and can be found in almost any organ, although the lung and hilar lymph nodes are most common. Hepatic granulomas are a common finding in systemic sarcoidosis, although most people remain asymptomatic. 1 Any part of the gastrointestinal (GI) tract, from oral cavity to colon, can also be affected. 2 The clinical spectrum of GI and hepatic sarcoidosis is broad, ranging from asymptomatic disease to fulminant liver failure requiring transplantation.
Epidemiology
The impact of sarcoidosis can be seen in all regions of the world. GI and hepatic sarcoidosis vary considerably in its incidence among ethnic groups but are well studied in northern European countries, Japan, and the United States, specifically in African American populations. 3 Prior studies have suggested that this was a disease process that affected young people, but more recent data have shown that the prevalence of this disease is greatest between ages 45 and 65 years. 4 It is estimated that the prevalence of sarcoidosis is 140 per 100,000 in African American individuals (even greater in African American females), an almost 3‐fold increase compared with Caucasians with a prevalence of 50 per 100,000, followed by Hispanic and then Asian individuals. 5 , 6 , 7 There is a higher prevalence of hepatic sarcoid compared with GI sarcoid, which is rare at less than 1%. 8 In patients with systemic sarcoidosis, clinically significant hepatic sarcoid is seen in 5% to 25% of those patients. 9 Asymptomatic disease, which is seen histologically, is present at a greater incidence rate of 50% to 80% of patients. 1 , 9
Pathogenesis
The hallmark of this condition is the development of epithelioid noncaseating granulomas. Although autopsy and imaging studies have shown widespread multiorgan involvement, these granulomas tend to have similar histological patterns. They consist of tightly organized collections of multinucleated giant cells and macrophage‐derived epithelioid cells encircled by lymphocytes and fibroblasts. 10 In the liver, these granulomas favor periportal and portal regions, whereas in the GI tract, they are located in the mucosa and submucosa.
Immunopathogenesis of Sarcoidosis
Immunologically, granuloma formation is guided by an inflammatory cascade. Antigen‐presenting cells (APCs), usually either macrophages or dendritic cells, process and present an antigen via human leukocyte antigen (HLA) class II molecule to T lymphocytes (TH1) and their receptors. 10 Given that the specific antigen is unknown and the many varied phenotypic presentations of this disease process, it is likely there are many antigens presented by HLA class II molecules to T cell receptors, thereby making the specific cause of sarcoidosis undetermined. 4 The interaction of APCs and T lymphocytes activates production of tumor necrosis factor α (TNF‐α), interleukin‐1 (IL‐1), IL‐6, and other cytokines, resulting in an inflammatory reaction. 10 This, in turn, results in the secretion of interferon‐γ (IFN‐γ), IL‐2, IL‐12, and other cytokines that activate and recruit monocytes and macrophages. These are then transformed into giant cells, which are important building blocks of the granuloma. 10
Diagnosis and Clinical Presentation
Sarcoidosis, involving the GI tract or liver, is a clinical diagnosis. Diagnostic criteria have not yet been formalized for GI or hepatic sarcoidosis. Typically, making a diagnosis involves obtaining a clinical history of systemic sarcoidosis, along with biopsy evidence of noncaseating granulomas (Fig. 1). 11
FIG 1.
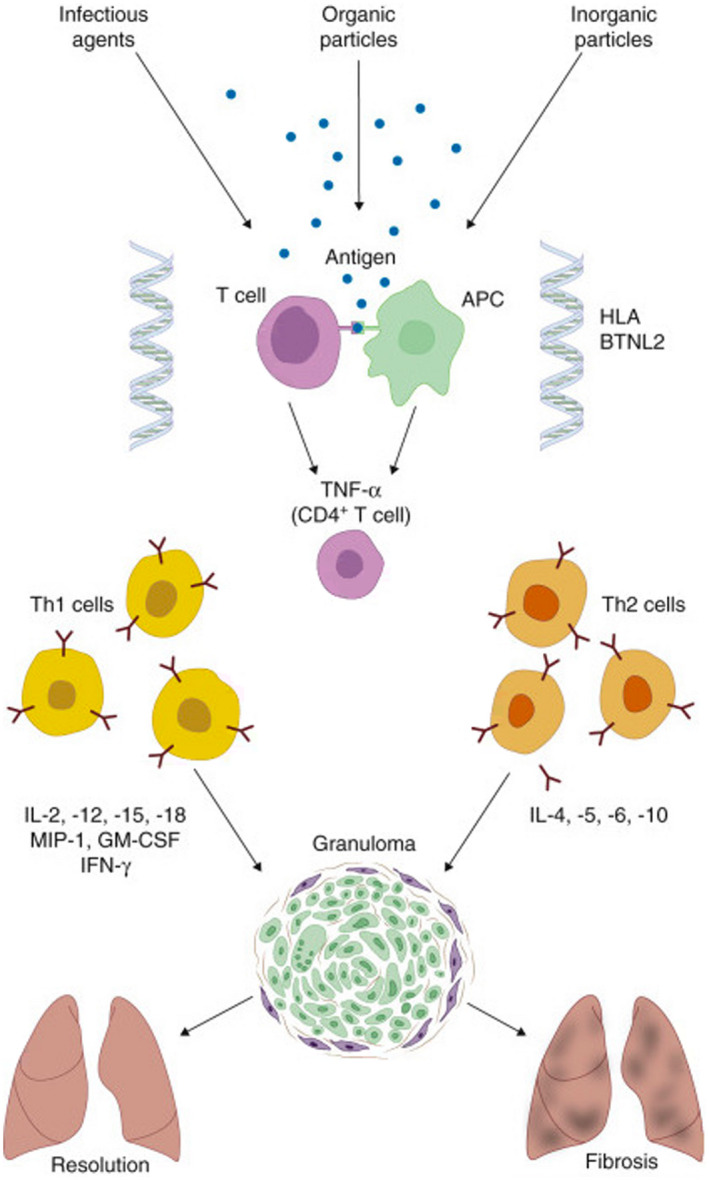
Immunopathogenesis of sarcoidosis. Reproduced with permission from Goldman‐Cecil Medicine, 26th ed. 10 Copyright 2020, Elsevier.
When symptomatic, GI sarcoidosis manifests with the presence of peptic ulcerations or narrowing of the gastric lumen as a result of granulomatous inflammation and/or associated fibrosis of the gastric wall. 12 In the latter case, diminished peristalsis subsequently results in early satiety or abdominal fullness. Epigastric pain is the predominant symptom in either circumstance and can be dull, burning, or cramping in nature and often postprandial, similar to dyspepsia. Nausea, vomiting, anorexia, heartburn, generalized abdominal discomfort, and diarrhea may also be reported. 12 , 13 Presence of multiple symptoms may be influenced by concomitant intestinal or rectal involvement.
Hepatic sarcoidosis is primarily asymptomatic even in the setting of abnormal liver function tests, but rarely can manifest with nonspecific symptoms, such as fever, night sweats, weight loss, malaise, and fatigue. These symptoms occasionally are associated with hepatic sarcoid but may be a sign of the systemic nature of the disease, presumably from cytokine release, rather than specific organ involvement. One retrospective study showed that the most common presenting symptom of hepatic sarcoid was fatigue, followed by pruritus, weight loss, and hepatomegaly. 11 Another clinical sequelae of hepatic sarcoid progression is the presence of portal hypertension, which can be cirrhotic or noncirrhotic and is seen in about 18% of patients. 14 Common complications of portal hypertension can be portal vein thrombosis, variceal bleeding, or splenomegaly and thrombocytopenia.
Clinical Spectrum of Hepatic Sarcoidosis
There are no specific laboratory derangements that occur with GI sarcoid; however, diagnosis is generally supported with endoscopic and/or histological evidence (Table 1). Measurement of the serum angiotensin‐converting enzyme levels can be useful; they have been reported to be elevated in about 60% of patients with active sarcoidosis. 1 However, the test lacks sensitivity and specificity, with low positive and negative predictive values (84% and 74%, respectively). 1 Therefore, an elevated test may help distinguish sarcoidosis from other granulomatous diseases, but a normal test does not necessarily rule out sarcoid.
TABLE 1.
Clinical Spectrum of Hepatic Sarcoidosis
| Feature | Presentation |
|---|---|
| Asymptomatic | 50%‐80% |
| Abnormal liver enzymes | 30% |
| Hepatomegaly or splenomegaly | On radiological examination, 50% |
| Detected clinically, <15%‐20% | |
| Symptoms | Abdominal pain, 15% |
| Pruritus and jaundice, <5% | |
| Cirrhosis | 6%‐8% |
| Portal hypertension severe liver dysfunction esophageal varices | 3%‐18% rare up to 78% of cases with portal hypertension |
| Advanced disease requiring transplantation | 0.012% of all liver transplants in the United States |
Reproduced with permission from Journal of Clinical and Translational Hepatology. 1 Copyright 2013, Creative Commons Attribution 4.0.
With regard to laboratory abnormalities in hepatic sarcoid, one population‐based study found that the liver chemistry findings were significant for elevated alkaline phosphatase (ALP) and gamma‐glutamyltransferase (GGT) usually up to three times the upper limit of normal. 15 Elevation in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels was less common than elevation in ALP. 4 , 16 This is suggestive of the infiltrative nature of sarcoid granulomas with resulting cholestatic liver injury and less so hepatocellular destruction. In addition, hyperbilirubinemia was uncommon, suggesting overall preserved synthetic function. The severity of liver test abnormalities was shown to be associated with the extent of granulomatous inflammation. 1 Given the chronic nature of this disease, it is estimated that 6% to 8% of patients progress to cirrhosis. 17 Eventually progression of the disease toward fulminant liver failure accounts for about 0.012% of all liver transplants. 1 Although there are no diagnostic imaging findings specific to hepatic sarcoid, signal intensity alone can be helpful in differentiating from infectious or neoplastic lesions. In addition, one study reviewing abdominal CT imaging for patients with hepatic sarcoid found extensive adenopathy in only 10% of patients, marked hepatomegaly was seen in only 8% of patients, and only 5% had liver nodules. 17
Histology of Sarcoid Granuloma
Once granulomas are discovered on biopsy of lesions in the GI tract, it is prudent to rule out inflammatory conditions such as inflammatory bowel disease (IBD), specifically Crohn’s disease (Fig. 2). In differentiating IBD from GI sarcoid, the latter will often have multivisceral lesions not usually seen in Crohn’s disease (i.e., interstitial lung disease, adenopathy, granulomatous hepatitis, hypercalcemia, central nervous system or heart involvement). 12 Sarcoidosis more often involves the upper GI tract (78% versus 6%; P = 0.0001), especially the stomach, whereas Crohn’s disease more often has macroscopic ileal or colonic involvement (95% versus 25%; P = 0.0001). 12 Similarly, for hepatic lesions, the differential diagnosis includes hypersensitivity drug reactions that result in granulomatous hepatitis, autoimmune conditions such as primary biliary cirrhosis (PBC) or primary sclerosing cholangitis (PSC), infections such as human immunodeficiency virus, amoebic liver abscess, infiltrative mycobacterium (tuberculosis or leprosy), or fungal infections (histoplasmosis, coccidiomycosis). 18 Other considerations would also include malignancy, such as lymphoma or malignant granuloma, and less commonly occupational or toxic exposure (i.e., beryllium). 19 Narrowing the differential can be done via stains and cultures of collected tissue samples prior to initiating immunosuppressive therapy.
FIG 2.
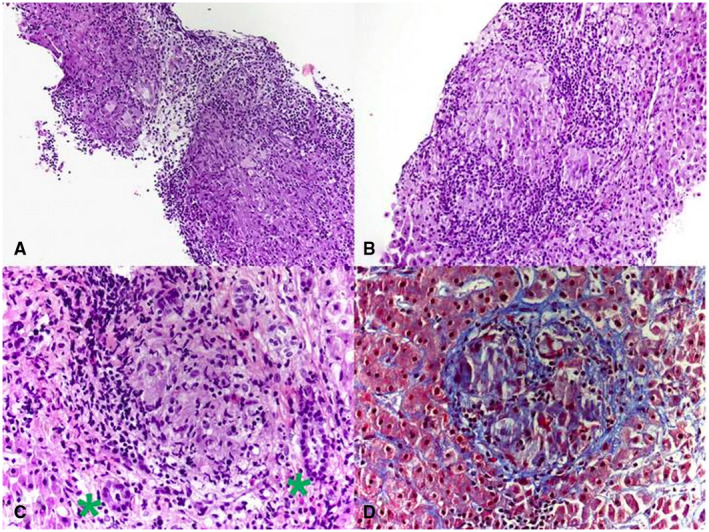
Histology of sarcoid granuloma. Photomicrograph from liver biopsy showing multiple coalescing nonnecrotizing granulomas (hematoxylin and eosin, original magnification 10×). (B) Lobular nonnecrotizing granuloma with tightly packed epithelioid cells surrounded by lymphocytes (hematoxylin and eosin, original magnification 20×). (C) Portal nonnecrotizing granuloma adjacent to uninvolved bile ducts (green asterisks) (hematoxylin and eosin, original magnification 40×). (D) Mild perigranulomatous fibrosis (Trichrome stain, original magnification 40×). Reproduced from Frontiers in Medicine. 11 Copyright 2019, Creative Commons Attribution 4.0.
Main Differential Diagnosis of Hepatic Sarcoidosis
Refer to Table 2 regarding the differentiating characteristics of autoimmune liver diseases, which can present similarly to hepatic sarcoid. Liver biopsy is the most direct means to diagnose hepatic sarcoid. 9 However, similar to PBC and PSC, sarcoidosis can lead to portal granulomas, lymphocytic cholangitis, bile duct injury and destruction, ductopenia, and chronic cholestasis; therefore, histopathology cannot be relied on alone. 20 , 21 Supportive laboratory workup, including negative anti‐mitochondrial antibody and negative anti‐neutrophil cytoplasmic autoantibody, along with rapid improvement with glucocorticoids, is supportive of sarcoidosis. 20 , 21
TABLE 2.
Main Differential Diagnosis of Hepatic Sarcoidosis
| Condition | Histology | Associated Findings |
|---|---|---|
| Sarcoidosis | Well‐differentiated noncaseating granulomas | Pulmonary adenopathy |
| Pulmonary interstitial disease | ||
| Primary biliary cholangitis | Poorly differentiated noncaseating granulomas | Positive anti‐mitochondrial antibody |
| No pulmonary hilar adenopathy | ||
| PSC | “Onion skin” periductal fibrosis | Association with IBD |
| Typical magnetic resonance cholangiography findings | ||
| Drug‐induced hepatic granulomas | Variable, depending on drug; presence of eosinophils in granulomas suggests diagnosis | Pertinent medication history, including recent antibiotics |
Reproduced from Clinics in Liver Disease. 17 Copyright 2019, Elsevier Inc.
Therapeutic Management
Treatment of GI sarcoid, given the low prevalence and likely presence of multivisceral lesions, is based on symptoms. Prior to discovering GI sarcoid, patients are often already receiving immunosuppressive therapy. Epigastric pain, abdominal fullness, and early satiety are typically managed with oral PPIs as seen in a majority of patients. 12 Blood transfusions and endoscopic intervention, such as pyloric stenosis dilatation or hemostatic intervention, are usually used in the setting of GI bleeding. 12
Suggested Approach to Hepatic Sarcoid
Treatment of hepatic sarcoid varies based on the degree of disease (Fig. 3). Initial treatment is guided primarily by extrahepatic manifestations that are more dominant. Thus far, steroids have been the mainstay of treatment for systemic and pulmonary sarcoidosis given that we do not have prospective or randomized controlled data specifically dedicated to hepatic sarcoid. 22 , 23 Hepatic sarcoidosis, as determined by the presence of noncaseating granulomas within the hepatic parenchyma, in the absence of any clinical or biochemical derangements, does not require treatment. 20 , 21 Ursodeoxycholic acid (UDCA) does appear to be effective in chronic cholestasis from hepatic sarcoid and is often used in steroid‐refractory disease. However, according to a current pilot study, given its excellent safety profile and minimal side effects, UDCA may be the consensus first‐line treatment for hepatic sarcoid in the presence of cholestatic injury, although this is still being investigated. In some circumstances, where the presence of extrahepatic disease is predominant, systemic glucocorticoids are used and typically decrease liver enzymes (ALP, GGT, AST, ALT, and bilirubin). Although steroids have been shown to improve clinical symptoms and liver enzymes and reduce hepatomegaly, they have not been shown to prevent the progression of hepatic sarcoidosis, as demonstrated by serial biopsies. 22 Considering the negative side effects of systemic corticosteroids (osteoporosis, hyperglycemia, edema, weight gain, depression, etc.) and the lack of evidence that they alter the natural history of hepatic sarcoidosis, their use is best reserved for patients who did not respond successfully to UDCA therapy, that is, no response after 3 months of therapy or patients with significant elevation in liver enzymes, portal hypertension, or unexplained hepatic decompensation with cirrhosis secondary to sarcoidosis. 22 Serial liver function test monitoring is reasonable once steroids are initiated but does not preclude the appearance of cholestasis or progression to portal hypertension and cirrhosis; therefore, liver biopsy remains prudent in disease follow‐up. 20 , 21 It is critical to rule out active infection or GI bleed prior to initiating steroids, and they must be discontinued if there is minimal or no clinical response. Although clinical and laboratory improvement can be seen with steroids, hepatic sarcoid can eventually progress to cause portal hypertension, bile duct depletion, and fibrotic changes, at which point steroids are unlikely to be of benefit. 21 Given the prolonged course of steroids required to induce clinical response, steroid‐sparing agents have been used as alternatives; however, the data are limited.
FIG 3.
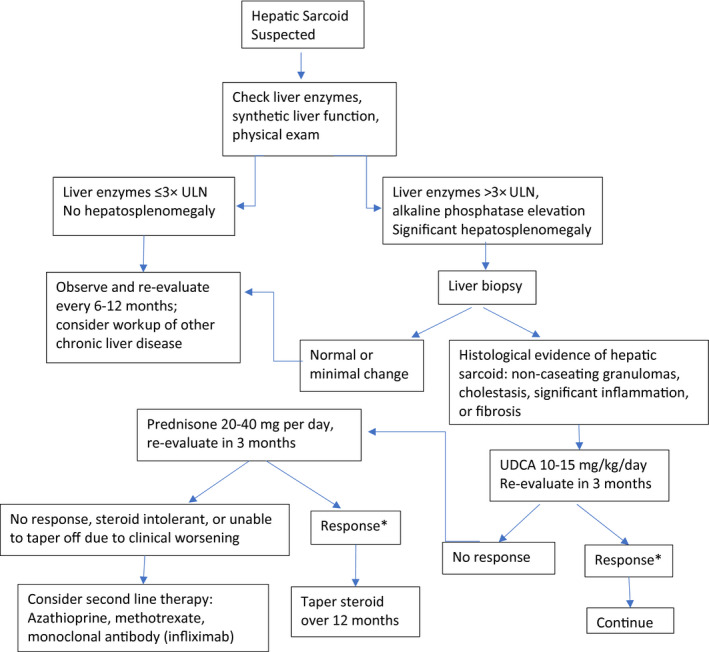
Suggested approach to hepatic sarcoid. *Response is defined as a biochemical response with improvement in liver chemistries and/or improvement in clinical symptoms. Reproduced with permission from Clinics in Liver Disease. 17 Copyright 2019, Elsevier Inc.
Alternative agents for those deemed to be steroid refractory or steroid dependent are not well studied, and their risk‐to‐benefit profile remains questionable. 24 Azathioprine has been reported to normalize aminotransferases but can also cause acute hepatitis. 1 Methotrexate, glutathione, chlorambucil, cyclosporine, cyclophosphamide, thalidomide, pentoxifylline, and infliximab have been reported to be beneficial. 17 Once portal hypertension and decompensated cirrhosis develop, liver transplantation may be indicated. The latter accounts for a very small fraction, approximately 0.012% of liver transplants. 1 Liver transplantation in sarcoidosis has resulted in graft and patient survival comparable with transplantation for other diseases. 21 Recurrence of sarcoid in transplanted livers has been reported and can occur anywhere between 8 months and 5 years after transplantation. 17
Summary
Hepatic sarcoid, a more common extrathoracic manifestation of systemic sarcoidosis compared with luminal GI sarcoid, is most often asymptomatic. Although there are no specific serological markers to detect the presence of hepatic or GI sarcoid, we often rely on clinical suspicion, physical examination findings, laboratory derangements, and histological findings. Clinically significant disease often presents with nonspecific symptoms and, in the case of hepatic sarcoid, signs of cholestasis. Treatment is guided by clinical severity through use of UDCA followed by systemic steroids, although it is prudent to rule out infection or drug‐induced disease. In certain cases, there can be progression with development of portal hypertension, decompensated cirrhosis, and potential indication for liver transplant evaluation.
Potential conflict of interest: Nothing to report.
References
- 1. Tadros M, Forouhar F, Wu GY. Hepatic sarcoidosis. J Clin Transl Hepatol 2013;1:87‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ungprasert P, Ryu JH, Matteson EL. Clinical manifestations, diagnosis, and treatment of sarcoidosis. Mayo Clin Proc Innov Qual Outcomes 2019;3:358‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dumas O, Abramovitz L, Wiley AS, et al. Epidemiology of sarcoidosis in a prospective cohort study of U.S. women. Ann Am Thorac Soc 2016;13:67‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Judson MA. Sarcoidosis. In: Kellerman RD, Rakel EP, eds. Conn's Current Therapy 2020. Philadelphia: Elsevier; 2020:871‐875. [Google Scholar]
- 5. Arkema EV, Cozier YC. Epidemiology of sarcoidosis: current findings and future directions. Ther Adv Chronic Dis 2018;9:227‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baughman RP, Field S, Costabel U, et al. Sarcoidosis in America. Analysis based on health care use. Ann Am Thorac Soc 2016;13:1244‐1252. [DOI] [PubMed] [Google Scholar]
- 7. Ennaifer R, Ayadi S, Romdhane H, et al. Hepatic sarcoidosis: a case series. Pan Afr Med J 2016;24:209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vahid B, Spodik M, Braun KN, et al. Sarcoidosis of gastrointestinal tract: a rare disease. Dig Dis Sci 2007;52:3316‐3320. [DOI] [PubMed] [Google Scholar]
- 9. Deutsch‐Link S, Fortuna D, Weinberg EM. A Comprehensive review of hepatic sarcoid. Semin Liver Dis 2018;38:284‐297. [DOI] [PubMed] [Google Scholar]
- 10. Iannuzzi MC. Sarcoidosis. In: Goldman‐Cecil Medicine. 26th ed. Philadelphia: Elsevier; 2020:585‐590.e2. [Google Scholar]
- 11. Sedki M, Fonseca N, Santiago P, et al. Hepatic sarcoidosis: natural history and management implications. Front Med (Lausanne) 2019;6:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ghrenassia E, Mekinian A, Chapelon‐Albric C, et al. Digestive‐tract sarcoidosis. Medicine (Baltimore) 2016;95:e4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Leeds JS, McAlindon ME, Lorenz E, et al. Gastric sarcoidosis mimicking irritable bowel syndrome: cause not association? World J Gastroenterol 2006;12:4754‐4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kennedy PTF, Zakaria N, Modawi SB, et al. Natural history of hepatic sarcoidosis and its response to treatment. Eur J Gastroenterol Hepatol 2006;18:721‐726. [DOI] [PubMed] [Google Scholar]
- 15. Baughman RP, Teirstein AS, Judson MA, et al. Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med 2001;164:1885‐1889. [DOI] [PubMed] [Google Scholar]
- 16. Ungprasert P, Crowson CS, Simonetto DA, et al. Clinical characteristics and outcome of hepatic sarcoidosis: a population‐based study 1976‐2013. Am J Gastroenterol 2017;112:1556‐1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kumar M, Herrera JL. Sarcoidosis and the liver. Clin Liver Dis 2019;23:331‐343. [DOI] [PubMed] [Google Scholar]
- 18. Coash M, Forouhar F, Wu CH, et al. Granulomatous liver diseases: a review. J Formosan Med Assoc 2012;111:3‐13. [DOI] [PubMed] [Google Scholar]
- 19. Rao DA, Dellaripa PF. Extrapulmonary manifestations of sarcoidosis. Rheum Dis Clin North Am 2013;39:277‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ebert EC. Gastrointestinal and hepatic manifestations of systemic diseases. In: Sleisenger and Fordtran’s Gastrointestinal and Liver Disease. 10th ed. Philadelphia: Elsevier/Saunders; 2016:579‐616.e14. [Google Scholar]
- 21. Ebert EC, Kierson M, Hagspiel KD. Gastrointestinal and hepatic manifestations of sarcoidosis. Am J Gastroenterol 2008;103:3184‐3192. [DOI] [PubMed] [Google Scholar]
- 22. Esfeh JM, Culver D, Plesec T, et al. Clinical presentation and protocol for management of hepatic sarcoidosis. Expert Rev Gastroenterol Hepatol 2015;9:349‐358. [DOI] [PubMed] [Google Scholar]
- 23. Karagiannidis A, Karavalaki M, Koulaouzidis A. Hepatic sarcoidosis: concise review. Ann Hepatol 2006;5:251‐256. [PubMed] [Google Scholar]
- 24. Ibrahim AM, Bhandari B, Soriano PK, et al. Hepatic involvement in systemic sarcoidosis. Am J Case Rep 2018;19:1212‐1215. [DOI] [PMC free article] [PubMed] [Google Scholar]