

Watch a video presentation of this article
Watch the interview with the author
Abbreviations
- Alb
albumin
- KF
Kayser‐Fleischer
- REC
relative exchangeable copper
- TTM
tetrathiomolybdate
- WD
Wilson’s disease
The growing new knowledge about Wilson’s disease (WD) has ramifications for understanding the pathophysiology and optimal clinical management of this rare condition. The aim of this review is to recapitulate the current understanding of the genetics of WD, explain the challenges in diagnosis and management, and appraise the pipeline of future diagnostic tests and therapies for WD.
The genetic bases of WD are the autosomal recessive variants involving the ATP7B gene. The consequent hepatic and systemic copper accumulation is due to deficient biliary copper excretion and ceruloplasmin maturation within hepatocytes. The number of described mutations affecting the ATP7B gene is rapidly increasing with more than 900 variants currently reported. 1 , 2 The most common variant in patients of European descent is the H1069Q missense mutation that, at a molecular level, has been shown to be associated with protein misfolding and localization of the mutated protein in the endoplasmic reticulum, aberrant phosphorylation of the P‐domain, and altered ATP binding orientation and affinity. 3 , 4
The clinical presentation of WD is varied and supports the fact that WD is a condition that affects primarily the liver and brain, but multiple organs, including heart, kidneys, and intestine, can be involved with a wide range of signs, symptoms, and tissue‐specific damage. 5 Liver disease ranges from steatosis and mild elevation of liver enzymes to cirrhosis and acute liver failure with hemolysis and hepatic necrosis. Neuropsychiatric findings include speech, gait and balance disturbances, drooling, involuntary movements, dystonia, Parkinsonism, psychosis, depression, and irritability. Kayser‐Fleischer (KF) rings, more commonly found in patients with neurological symptoms, have a strong association with WD and can resolve with copper‐lowering therapy. 5 , 6 Pediatric cases are usually diagnosed after the age of 3 years, and the most common manifestations are related to liver involvement, whereas neurological signs and symptoms before the age of 10 years are rare, although possible. 7 In general, WD should be considered and ruled out in any patient who is older than 3 years and presenting with unexplained liver disease of any level of severity.
The traditionally described WD prevalence is 1 in 30,000 individuals. However, in recent analyses using whole genome sequencing, the genetic prevalence is proposed to be as high as 1 in 7026 individuals 8 (Fig. 1). The genetic prevalence could be higher than the clinical prevalence because of incomplete penetrance or presence of yet to be identified modifier genes. The apparent discrepancy could also be explained by missed diagnoses because of a lack of clinical diagnostic gold standards.
FIG 1.
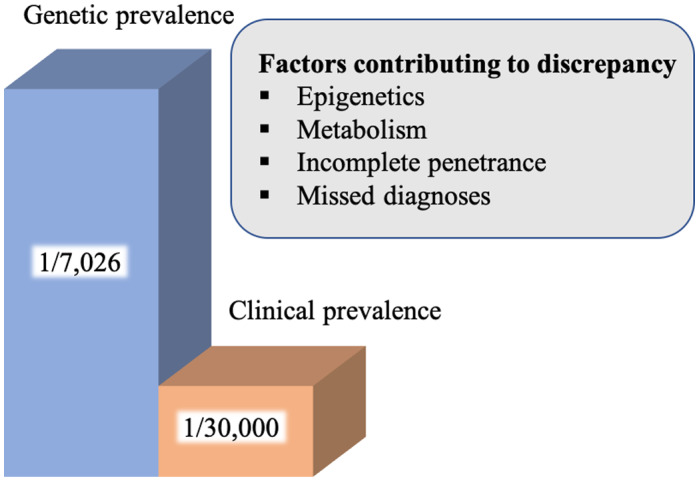
Genetic and clinical prevalence. Studies show genetic prevalence of WD is higher (1/7026), whereas the historical clinical prevalence is lower at 1/30,000. Several factors are potentially contributing to this discrepancy. 8
Despite the fact that the genetics of this disease have been elucidated, these genetics fail to predict the phenotypic variability of WD. This concept is supported by multiple studies showing a lack of correlation between genotype and phenotype. A recent, large study in a total of 1357 patients with WD attempted to identify an association between genotype and phenotype. However, the authors could not show evidence that the phenotype could be predicted on the basis of the genotype. 9 Therefore, it is likely that interacting epigenetic and metabolic factors are contributing to the varying phenotypes of WD. 10
The diagnosis of WD hinges on a combination of clinical, biochemical, histological, and genetic findings. However, currently available diagnostic tests often fail to detect the disease 11 because of low sensitivity and specificity (Table 1). Detection of disease‐causing ATP7B gene variants in a person with clinical and biochemical features (low ceruloplasmin, increased 24‐hour urinary copper excretion, and hepatic copper concentration) is confirmatory of the disease. Because the clinical presentation of WD is varied among those who are affected and not all who have ATP7B gene mutations exhibit the clinical manifestations, the diagnostic process is often arduous. Although the Leipzig score was developed to aid clinicians in determining WD diagnosis, and is recommended by European guidelines, 12 it lacks large‐scale validation. Within this scoring system, points are assigned for the presence of KF rings, neurological symptoms, and levels of urine, serum, and hepatic copper. A score of ≥4 establishes the diagnosis of WD, whereas a score of <2 excludes the diagnosis. The US guideline and diagnostic algorithm differ slightly in that there is lack of an assigned score and neurological symptoms are not accounted for. 13 Both the American Association for the Study of Liver Diseases and European Association for the Study of the Liver guidelines rely on liver biopsy and genetic testing to complete the diagnostic assessment.
TABLE 1.
Current Diagnostic Tools
| Parameter | Normal Range | WD Diagnosis |
|---|---|---|
| Serum ceruloplasmin | 20‐35 mg/dL | <20 mg/dL |
| 24‐Hour urinary copper | 20‐50 µg | >100 µg |
| Hepatic copper content | 15‐55 µg/g | >250 µg/g |
| Liver histology* | Steatosis (microvesicular and macrovesicular); portal and/or lobular inflammation; fibrosis; ballooning; Mallory hyaline bodies; glycogenated nuclei; necrosis; positive orcein and rhodamine staining |
All histology findings are nonspecific.
An additional major difficulty in understanding WD is the lack of widespread access to and interpretation of genetic analysis. Genetic testing modalities for WD include polymerase chain reaction detection of common point mutations, whole genome sequencing, next generation sequencing, and multiplex ligation‐dependent probe amplification, but many of these tools may not be widely available outside large academic centers. An additional obstacle may also be the need to have these data interpreted by a geneticist. Once the diagnosis of WD is established, testing of first‐degree relatives is indicated because the likelihood of homozygous mutation in siblings is 25% and among children of those affected is 0.5%. Parents of affected children should receive genetic counseling before attempting subsequent pregnancies. In cases where the diagnosis is still uncertain, radioactive copper incorporation is a highly sensitive and specific test for WD that is comparable with measuring 24‐hour urine copper. This nuclear medicine test relies on an intravenous bolus injection of 64Cu tracer and its incorporation into ceruloplasmin measured in the liver and serum at 2, 24, and 48 hours after infusion 14 (Table 2). Although the test is not commonly available, it is promising due to its high sensitivity, specificity, and diagnostic accuracy. Furthermore, compared with the liver biopsy, it offers the advantage of being noninvasive. Relative exchangeable copper (REC) is another noninvasive test that has been studied and found to be highly specific and sensitive for WD. It is calculated as the ratio of exchangeable copper to total serum copper. 15
TABLE 2.
New Diagnostic Tools
| Parameter | Description | WD Diagnosis |
|---|---|---|
| Radioactive copper (64Cu) ratio | 64Cu infused intravenously and measured within the liver and serum after 2, 24, and 48 hours 14 | |
| 24 hours/2 hours | <0.3 | |
| 48 hours/2 hours | <0.395 14 | |
| Genetic analysis | Polymerase chain reaction amplification of ATP7B mutations | Disease‐causing mutations |
| Whole‐genome sequencing: assesses all liver disease genes, not just WD | ||
| REC | Serum assay. REC = exchangeable copper/serum copper | >2.08 μmol/L for extrahepatic disease |
| >1.53 μmol/L for hepatic disease | ||
| Normal range: 0.62 and 1.15 µmol/L 15 |
The long‐term management of WD can be divided into an initial phase and a maintenance phase. Patients with prevalent hepatic manifestations are almost always started on chelating agents (penicillamine or trientine) in the initial phase, with the goal of achieving negative copper balance. In cases of acute liver failure related to WD, the only option is liver transplantation. The initial anticopper agent for neurological manifestations is controversial, but zinc salts tend to be the most common first choice. In the future, bis‐choline tetrathiomolybdate (TTM) could become the first‐line agent for patients with predominantly neurological phenotypes of WD 16 (Fig. 2). The lifelong maintenance phase attempts to preserve copper balance without resulting in copper deficiency. This is usually achieved by providing a lower dose of the chelating agent or by transitioning to zinc. There is limited research and knowledge regarding the timing and effective methods of transitioning between the various agents. 17 Current clinical practice includes frequent monitoring of liver enzymes and 24‐hour urine copper for a few months after the switch to zinc therapy, or until copper balance is stable. Patients with WD must remain on lifelong maintenance therapy, which is also a challenge due to potential adverse effects of anticopper agents, drug interactions with other medications, and financial constraints.
FIG 2.
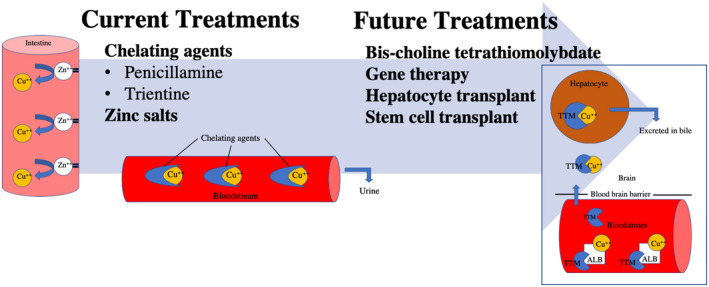
Current and future treatments. Current FDA‐approved treatments are chelating agents and zinc salts. Whereas zinc salts work at preventing copper absorption in the intestine, chelating agents bind copper in the bloodstream. Maintenance treatments include bis‐choline TTM, which works in multiple locations, including hepatocytes, brain, and the bloodstream. 16
An additional important aspect of WD pathogenesis is impairment of mitochondrial morphology and function, which has been described early in the disease progression. 18 Animal studies indicate that removal of copper from mitochondria results in amelioration of mitochondria dysfunction and liver pathology. 19 A challenge in both treating and monitoring WD is that there is a lack of anticopper agents that specifically act on mitochondria, as well as tools to monitor mitochondria bioenergetic improvement during treatment.
Gene therapy could be an important opportunity in the near future. The implementation of adeno‐associated viral 8 vector expressing ATP7B transgene has been shown to correct copper metabolism in the long term in a preclinical model of WD. 20 Administering this early in disease could potentially reduce liver damage in WD and prevent and improve its clinical manifestations. Although we look forward to the results of gene therapy in humans, the benefits of using this treatment option in WD, a condition that is now mostly managed by the available pharmaceutical agents and does not have complete penetrance, will have to be studied, and appropriate patient selection will have to be determined.
In conclusion, with improved understanding of WD, we are now facing growing opportunities and some challenges in optimizing its diagnosis and treatment. These challenges should motivate clinicians and researchers to improve the management of this condition, which is often viewed as too rare or too benign to be studied.
Potential conflict of interest: V.M. consults for and received grants from Ambys Medicines. V.M. also consults for Alexion Pharmaceuticals
References
- 1. Cooper DN, Ball EV, Stenson PD, et al.The Human Gene Mutation Database. Available at: http://www.hgmd.cf.ac.uk/ac/index.php. Published 2017. Accessed August 1, 2020.
- 2. Stenson PD, Ball EV, Mort M, et al. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat 2003;21:577‐581. [DOI] [PubMed] [Google Scholar]
- 3. Rodriguez‐Granillo A, Sedlak E, Wittung‐Stafshede P. Stability and ATP binding of the nucleotide‐binding domain of the Wilson disease protein: effect of the common H1069Q mutation. J Mol Biol 2008;383:1097‐1111. [DOI] [PubMed] [Google Scholar]
- 4. Huster D, Hoppert M, Lutsenko S, et al. Defective cellular localization of mutant ATP7B in Wilson's disease patients and hepatoma cell lines. Gastroenterology 2003;124:335‐345. [DOI] [PubMed] [Google Scholar]
- 5. Schilsky ML. Wilson disease: clinical manifestations, diagnosis, and treatment. Clin Liver Dis (Hoboken) 2014;3:104‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tooley AA, Sweetser S. Clinical examination: Eyes. Clin Liver Dis (Hoboken) 2016;7:154‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Socha P, Janczyk W, Dhawan A, et al. Wilson's disease in children: a position paper by the Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr 2018;66:334‐344. [DOI] [PubMed] [Google Scholar]
- 8. Coffey AJ, Durkie M, Hague S, et al. A genetic study of Wilson's disease in the United Kingdom. Brain 2013;136:1476‐1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ferenci P, Stremmel W, Czlonkowska A, et al. Age and sex but Not ATP7B genotype effectively influence the clinical phenotype of Wilson disease. Hepatology 2019;69:1464‐1476. [DOI] [PubMed] [Google Scholar]
- 10. Mordaunt CE, Kieffer DA, Shibata NM, et al. Epigenomic signatures in liver and blood of Wilson disease patients include hypermethylation of liver‐specific enhancers. Epigenetics Chromatin 2019;12:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Steindl P, Ferenci P, Dienes HP, et al. Wilson's disease in patients presenting with liver disease: a diagnostic challenge. Gastroenterology 1997;113:212‐218. [DOI] [PubMed] [Google Scholar]
- 12. European Association for Study of Liver . EASL Clinical Practice Guidelines: Wilson's disease. J Hepatol 2012;56:671‐685. [DOI] [PubMed] [Google Scholar]
- 13. Roberts EA, Schilsky ML;American Association for Study of Liver Diseases (AASLD) . Diagnosis and treatment of Wilson disease: an update. Hepatology 2008;47:2089‐2111. [DOI] [PubMed] [Google Scholar]
- 14. Czlonkowska A, Rodo M, Wierzchowska‐Ciok A, et al. Accuracy of the radioactive copper incorporation test in the diagnosis of Wilson disease. Liver Int 2018;38:1860‐1866. [DOI] [PubMed] [Google Scholar]
- 15. Poujois A, Poupon J, Woimant F. Direct determination of non‐ceruloplasmin‐bound copper in plasma. In: Nanda Kerkar EAR, ed. Clinical and Translational Perspectives on Wilson Disease. London: Academic Press; 2019:249‐255. [Google Scholar]
- 16. Weiss KH, Askari FK, Czlonkowska A, et al. Bis‐choline tetrathiomolybdate in patients with Wilson's disease: an open‐label, multicentre, phase 2 study. Lancet Gastroenterol Hepatol 2017;2:869‐876. [DOI] [PubMed] [Google Scholar]
- 17. Leung M, Wu Lanzafame J, Medici V. Switching pharmacological treatment in Wilson disease: case report and recommendations. J Investig Med High Impact Case Rep 2020;8:2324709619896876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sternlieb I. Electron microscopy of mitochondria and peroxisomes of human hepatocytes. Prog Liver Dis 1979;6:81‐104. [PubMed] [Google Scholar]
- 19. Einer C, Leitzinger C, Lichtmannegger J, et al. A high‐calorie diet aggravates mitochondrial dysfunction and triggers severe liver damage in wilson disease rats. Cell Mol Gastroenterol Hepatol 2019;7:571‐596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Murillo O, Luqui DM, Gazquez C, et al. Long‐term metabolic correction of Wilson's disease in a murine model by gene therapy. J Hepatol 2016;64:419‐426. [DOI] [PubMed] [Google Scholar]