

Abstract
Alzheimer's disease (AD) poses a significant global health concern over the next several decades. Multiple hypotheses have been put forth that attempt to explain the underlying pathophysiology of AD. Many of these are briefly reviewed here, but to‐date no disease‐altering therapy has been achieved. Despite this, recent work expanding on the role of noradrenergic system dysfunction in both the pathogenesis and symptomatic exacerbation of AD has shown promise. The role norepinephrine (NE) plays in AD remains complicated but pre‐tangle tau has consistently been shown to arise in the locus coeruleus (LC) of patients with AD decades before symptom onset. The current research reviewed here indicates NE can facilitate neuroprotective and memory‐enhancing effects through β adrenergic receptors, while α2A adrenergic receptors may exacerbate amyloid toxicity through a contribution to tau hyperphosphorylation. AD appears to involve a disruption in the balance between these two receptors and their various subtypes. There is also a poorly characterized interplay between the noradrenergic and cholinergic systems. LC deterioration leads to maladaptation in the remaining LC‐NE system and subsequently inhibits cholinergic neuron function, eventually leading to the classic cholinergic disruption seen in AD. Understanding AD as a dysfunctional noradrenergic system, provides new avenues for the use of advanced neural stimulation techniques to both study and therapeutically target the earliest stages of neuropathology. Direct LC stimulation and non‐invasive vagus nerve stimulation (VNS) have both demonstrated potential use as AD therapeutics. Significant work remains, though, to better understand the role of the noradrenergic system in AD and how electroceuticals can provide disease‐altering treatments.
Keywords: Alzheimer's disease, electroceuticals, neural stimulation, the locus coeruleus – norepinephrine (LC‐NE) system
In this review, we provide a brief overview of several of the most thoroughly researched pathogenic hypotheses for Alzheimer's disease (AD) and assess their clinical impact to‐date. We focus specifically on recent research into the role of the locus coeruleus norepinephrine (LC‐NE) system, in both AD pathogenesis and symptom exacerbation, as well as the potential to use advanced neural stimulation techniques as a novel therapeutic option in the earliest stages of neuropathology.
1. INTRODUCTION
The clinical syndrome of dementia is characterized by the slowly progressive decline of two or more cognitive domains, including but not limited to language, memory, executive function, personality, or behavior. 1 Alzheimer's disease (AD) plays a significant role in the clinical presentation of dementia, accounting for up to 80% of all dementia diagnoses. 2 In the United States alone, it is estimated that up to 4.7 million individuals over the age of 65 suffered from AD in 2010, and it is projected that 13.8 million Americans will be living with AD by 2050. 3 AD was also ranked as the sixth leading cause of death in the United States in 2010, with more than 84,000 deaths attributed to the disease, a 3.7% increase from the year prior. 4 The overall economic burden of AD is also significant, leading to an annual cost of $100 billion, third in the United States behind heart disease and cancer. 5 The impact is even more severe globally, with up to 24 million cases in 2012. That number is predicted to double every 20 years, until at least 2040. 6
In AD, dementia is currently viewed as the end stage of decades of neuropathological changes, which lead to a clinical manifestation that can range from asymptomatic, to a mild cognitive impairment (MCI), to a full amnestic syndrome with resultant loss of function. 7 , 8 The presentation and progression of this spectrum is highly variable between individuals, with a significantly higher prevalence in women, that has not been fully explained (for a review, see Nebel et al). 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 There is currently no effective method to prevent AD, and therapy is almost exclusively targeted towards symptomatic treatment. A large volume of work exists characterizing the underlying pathophysiology of AD, but a definitive consensus on the most promising therapeutic avenue remains elusive. 1
The purpose of this review article is to give a brief overview of some of the most thoroughly researched hypotheses and assess their clinical impact to‐date. The intent is not to provide a complete review of the progress in each, but to briefly highlight how they have contributed to an understanding of the disease. Most of the attention of this review will be focused on recent research into the role of the noradrenergic system in both AD pathogenesis and symptomatic exacerbation, as well as the potential to use advanced neural stimulation techniques to offer novel therapeutic options targeting the earliest stages of neuropathology.
2. UNDERSTANDING THE PATHOPHYSIOLOGICAL PROGRESSION OF ALZHEIMER'S DISEASE
The general brain abnormalities found in Alzheimer's disease (AD) were first described by Alois Alzheimer in 1906. 16 The disease is characterized by three major histopathological findings: brain atrophy, extracellular deposits of dense amyloid plaques, and intracellular cytoskeletal abnormalities such as accumulated neurofibrillary tangles. 17 The neuronal death in AD is diffuse and markedly more severe than that seen in the normal aging process. The deposits of amyloid, a fibrillar peptide arranged in sheets, are associated with evidence of nearby inflammation, such as swollen axons and dendrites, along with reactive processes of surrounding astrocytes and microglia. Another common finding is amyloid accumulation in the cerebral vasculature. In diseased neurons that are still alive, filamentous tangles are observed in cell bodies and proximal dendrites. These tangles contain helical and 15 nm straight filaments. 17
HIGHLIGHTS.
Locus coeruleus (LC) is the first brain structure to exhibit AD‐like pathology.
Severe LC degeneration is a ubiquitous feature of AD, and the LC degenerates much earlier than symptoms are clinically apparent.
Norepinephrine produced by the LC has an anti‐inflammatory influence in the brain.
Direct LC stimulation and non‐invasive vagus nerve stimulation have both demonstrated potential as AD therapeutics.
The plaques and tangles constituting AD have been identified as typically localizing to specific brain regions. 17 , 18 Areas of susceptibility in AD include the locus coeruleus (LC), the nucleus basalis of Meynert (NBM), and the neocortex. The entorhinal cortex and hippocampus are also significantly impacted and likely contribute to early problems with declarative memory. 19 The lateral entorhinal cortex, specifically, has been found to be dysfunctional in preclinical AD and can act as a source for the spread of disease to the parietal cortex and other regions of the brain. 19 The NBM is one of the most heavily researched neuroanatomic regions in AD and has been found to be particularly vulnerable to neurofibrillary degeneration.
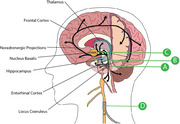
The efforts to build a complete understanding of the temporal evolution of AD progression have remained limited by the reliance on post‐mortem autopsies to characterize the disease, though numerous efforts are ongoing to discover biomarkers that can be used to further characterize how AD spreads and to aid in early clinical diagnosis. 1 , 8 , 20 , 21 , 22 , 23 , 24 , 25 To‐date, a stereotyped progression has not been entirely described, though a general outline for how AD pathology can be expected to progress is slowly being revealed. The earliest preclinical findings in AD appear to be amyloid accumulation and tauopathy in the brainstem, specifically the locus coeruleus (LC; Figure 1). 26 , 27 , 28 , 29 This accumulation can occur up to several decades before clinical manifestations, suggesting a threshold, second trigger, or loss of functional reserve are necessary for disease progression. As the Braak stage, a systematic method for staging AD, increases, a linear decrease in LC volume, not associated with normal aging, has been described, beginning as early as the fourth decade of life in sporadic AD patients. 28 From this early finding, tau pathology seems to spread to the dorsal raphe nucleus, entorhinal cortex, and perirhinal cortex, suggestive of anterior pathway susceptibility. 19 , 30 Layer II of the lateral entorhinal cortex appears to be the source of further progression to downstream synaptic circuitry, including the dentate gyrus and the cingulate cortex. 30 , 31
FIGURE 1.
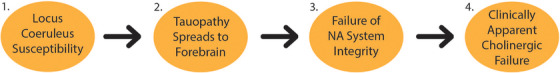
Pathway to noradrenergic dysregulation in Alzheimer's Disease: 1) Due to a combination of genetic and environmental factors, amyloid plaques and tauopathies occur in the Locus Coeruleus (LC) decades before symptom onset. 2) LC neuron count decreases and tauopathy spreads along anterior pathway to the forebrain and cortex during an asymptomatic period in middle‐age. Remaining noradrenergic (NA) neurons exhibit compensatory alterations. 3) NA system integrity is lost due to maladaptive LC changes. α and β AR expression alters across various brain regions, further disrupting connectivity. 4) Tauopathy spreads to the NBM. Hyperactive NA neurons may further inhibit the remaining cholinergic neurons. Widespread dysfunction occurs across multiple systems
Eventually, accumulation of toxic tauopathies in cholinergic neurons of the NBM precipitates dramatic neuronal degeneration, followed by the progression of a more rapid decline in cognitive function (Figure 1). 19 , 32 , 33 , 34 Though the evidence now suggests that the NBM is primarily implicated in symptomatic disease, it is still debated whether this pathology is primary or secondary. Multiple autopsy studies have shown that most cholinergic neurons in this region remain unaffected by tauopathy in mild AD but are extensively involved in severe AD, suggesting that NBM degeneration is likely a late‐stage finding following years of subclinical changes in other regions of the brain. 35 , 36 Following NBM degeneration, there is then spread of this tauopathy to the neocortical areas to which the NBM projects. 33 , 37 This final stage is associated with the classic cognitive deficits and bulk brain atrophy of AD.
Though the extracellular accumulation of amyloid plaques and the intracellular presence of neurofibrillary tangles were the first features identified in AD, it is now also recognized that synaptic degeneration, hippocampal neuronal loss, and aneuploidy are important features that may provide more primary contributions than originally suspected. 38 There is also still much debate about whether the amyloid plaque and neurofibrillary tangles themselves lead to the symptoms of dementia directly or are the result of a broader pathologic process. While plaque‐centered hypotheses were traditionally the most thoroughly researched, interest has turned towards the heavily phosphorylated tau protein that comprise the neurofibrillary tangles. A summary of this progression and the resulting clinical benefits is shown in Table S1 and expanded upon in the section below. To‐date, results remain inconclusive. 39
3. LEADING HYPOTHESES
3.1. Cholinergic hypothesis
The cholinergic hypothesis is the oldest theory underlying AD pathogenesis and builds off the observation that choline acetyltransferase (ChAT) activity is greatly reduced at synapses in the amygdala, hippocampus, and cortex of AD brains, 40 , 41 , 42 , 43 resulting in a corresponding cholinergic failure and impairment of memory, attention, and learning. 44 , 45 The projections to the cerebral cortex of presynaptic cholinergic neurons in the nucleus basalis of Meynert (NBM) appear to specifically undergo profound degeneration in late AD. 43 , 46 , 47 This leads to a subsequent loss of nicotinic receptors in the cerebral cortex and muscarinic M2 receptors, both of which are predominately pre‐synaptic. 48 , 49 , 50 Decreases in M1 receptors in the dentate gyrus and pyramidal neurons in layers III and V of the parahippocampal cortex have also been demonstrated in AD patients. 51 , 52 , 53 This body of research has led to the only clinically relevant treatment thus far for AD.
Cholinesterase inhibitors, pharmacological agents that reduce the degradation of synaptic acetylcholine, have been shown to have a consistent, though marginal, increase in functional outcomes over placebo. 48 , 54 It has been more than 20 years since the first cholinesterase, tacrine, was FDA approved, marking the last major milestone in AD therapeutics. 55 The current FDA‐approved cholinesterase inhibitors include rivastigmine, donepezil, and galantamine. Though they appear to result in some histological changes in AD patients, clinical trials have shown efficacy only in minor symptom improvement, with no underlying impact on disease course or progression. 48 Though disappointing in a clinical context, successful combination treatment in the future will likely include a cholinesterase inhibitor for patients that are already symptomatic.
Novel interventional approaches to halting or preventing cholinergic failure are in the early stages and are not currently being widely pursued. Researchers in Germany have performed small Phase I clinical trials using NBM bilateral, low frequency, deep brain stimulation (DBS) in six patients, four of which were considered responders on the basis of stable or improved primary outcome parameters 12 months after surgery. The study subsequently concluded NBM‐DBS is technically feasible and well‐tolerated. 56 Additional follow‐up results support disease stabilization. 57 Additional research on NBM‐DBS for other forms of dementia have shown results similar to those in AD patients in Germany. 56 , 57 , 58 , 59 , 60 , 61 , 62 , 63 , 64 , 65 , 66 , 67 , 68 , 69 , 70 , 71 , 72 , 73 A summary of the most relevant work is included in Table S2A. The mechanisms and overall impact of NBM‐DBS on disease pathophysiology are not well‐studied though, 63 , 74 and more investment is needed in this area to gain better control of the clinical response to NBM‐DBS. 75 Efforts in this area may prove limited for long‐term applicability, depending on whether NBM degradation proves to be a primary or secondary characteristic of clinically apparent AD. Given the importance of acetylcholine release in memory formation, however, electroceutical manipulation of a dysfunctional cholinergic system offers a promising avenue for further exploration in the pursuit of any clinically relevant treatment.
3.2. Amyloid cascade hypothesis
The extracellular plaques seen in AD primarily consist of Aβ protein, which is created by processing the parent amyloid precursor protein (APP). 76 , 77 , 78 , 79 At picomolar levels, APP and Aβ are involved in normal neuronal functioning and synaptic plasticity modulation. 79 , 80 , 81 , 82 The position of the APP gene on chromosome 21 led to an interest in the connection of the presenile cognitive decline and amyloid plaque pathology seen in Trisomy 21, 45 , 83 , 84 , 85 , 86 , 87 however, subsequent work has demonstrated the limitations of applying the pathophysiology of this form of cognitive decline to the later‐onset, sporadic form of AD. 79 , 88 , 89 This direction of research was important though because it led to the discovery of an autosomal dominant form of early‐onset AD, in which an APP gene mutation drives neurodegeneration. 90 Though distinctly different from the more common sporadic AD, work in this area has demonstrated the neurotoxicity of Aβ and has contributed to the formulation of the amyloid cascade hypothesis. 90 , 91 This theory postulates that toxic plaques are the earliest pathology of AD. 91 , 92 The aberrant form of amyloid plaque induces the phosphorylation of tau protein through an imbalance in cellular signaling (Figure 3), the latter of which then spreads to local neurons via microtubule transport. The corresponding buildup of hyperphosphorylated tau protein results in cell death and gradual cognitive dysfunction. 77 , 81 , 91 , 93 , 94 , 95
FIGURE 3.
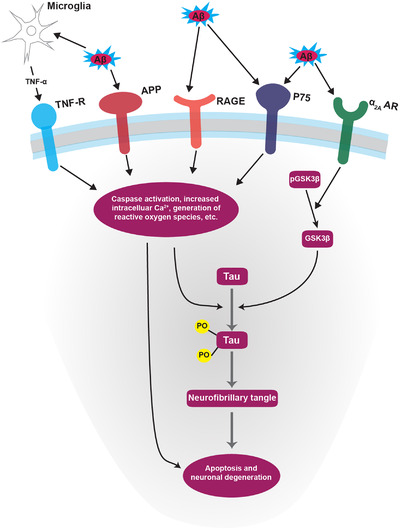
Alzheimer's disease signaling cascade. General signaling pathway underlying neuronal degeneration in Alzheimer's disease. Notably, adrenergic remodeling can influence pathologic activation of the α2AAR, which, coupled with the pro‐inflammatory loss of β‐AR integrity, may partially explain the accumulation of neurofibrillary tangles.
Therapeutic targets to‐date include several monoclonal antibody therapies that target and remove Aβ from the brain, though none have resulted in a major improvement in cognitive function. 96 , 97 Alternative approaches to reducing Aβ are ongoing, with recent focus turning specifically to the most toxic Aβ oligomers. 1 , 98 , 99 , 100 The amyloid cascade hypothesis continues to undergo serial iterations, and the most recent adaptation hypothesizes that Aβ accumulation is part of a broader alteration in the homeostasis of APP‐related functions. 101
3.3. Neurovascular hypothesis
The neurovascular hypothesis explores the vascular dysregulation that occurs in AD patients. 102 , 103 It has been known for nearly three decades that the cerebral microvasculature is damaged in AD, resulting in regional and laminar patterns of damage that follow neuronal degradation. 102 , 104 , 105 Subsequent research has shown that Aβ protein can interact with vascular endothelial cells to produce excessive superoxide radicals. 102 , 103 , 106 , 107 These superoxide radicals then produce a litany of degenerative alterations and inhibit the production of nitric oxide, contributing to increased vasoconstriction and a reduction in the local blood supply, exacerbating further pathologies. 108 , 109 , 110 , 111 , 112
The incomplete clearance of Aβ across the blood‐brain barrier (BBB) also contributes to eventual BBB degeneration and a chemical environment not conducive to cell survival. 107 , 110 Several studies have attempted to better understand the role of vascular function in AD, with some attention paid to the impact of hyperlipidemia and diabetes, which are also known to result in vascular endothelial damage, on AD prevalence and outcomes. 113 , 114 Results to‐date are not definitive, and no clinically relevant therapies addressing neurovascular dysfunction have been achieved, aside from a small risk reduction associated with preventative measures targeted at diabetes mellitus and artherosclerosis. 45 , 115 , 116 , 117
3.4. Mitochondrial cascade hypothesis
The idea behind the mitochondrial cascade hypothesis was first introduced in an attempt to explain a general pathophysiologic mechanism underlying the more common, sporadic form of AD. 118 It postulates that an individual's basal rate of production of reactive oxygen species (ROS) is genetically determined and sets the pace at which acquired mitochondrial damage occurs. 118 This eventually leads to three specifically defined events, termed a removal response, reset response, and replace response. Cellularly, this corresponds to an increase in Aβ generation due to the increase in ROS, compromised cells undergoing programmed cellular death, and neuronal progenitors unsuccessfully attempting to re‐enter the cell cycle leading to tau phosphorylation and aneuploidy. 118 , 119 , 120 , 121 , 122 , 123
There are several studies that seem to support this hypothesis, including findings that the middle‐aged children of AD mothers tend to utilize less glucose on fluorodeoxyglucose‐PET scans, exhibit more age‐associated brain atrophy, and perform worse on memory test performance than those of AD fathers. 124 There is also evidence that functional mitochondria are required to mediate cellular damage when Aβ plaques are available. Though this hypothesis has not been definitively proven in humans, mitochondrial dysfunction appears to be a crucial feature in a multi‐factorial view of AD. 125 , 126 , 127 Therapeutic trials targeting mitochondria have been mixed, with no AD‐approved drug yet appearing achievable, despite some mixed success with antioxidant treatments. 120 , 121 , 122 , 124 , 125 , 128 , 129 , 130 , 131 Future work is likely to focus on more targeted delivery with nanoparticle therapy. 132 , 133 , 134 , 135
3.5. Tau propagation hypothesis
Highly phosphorylated tau (p‐tau) proteins comprise the majority of the neurofibrillary tangles (NFTs) seen in AD. 30 , 93 , 95 , 136 , 137 , 138 These proteins typically bind and help stabilize microtubules in axons. 139 , 140 , 141 The function of these proteins is highly dependent on the phosphorylation state. 141 , 142 Hyperphosphorylation can expose a microtubule‐binding domain that enables self‐aggregation and oligomerization, eventually resulting in the formation of paired helical filaments, loss of axonal transport, and conversion to NFTs with further cellular disruption. 136 , 137 , 141 , 142 , 143 , 144 , 145 It is currently thought that the tau oligomer intermediate is more important for disease progression than the resulting NFTs. 146 Though the oligomerization process and formation of NFTs were originally viewed as a downstream effect of amyloid plaque accumulation, the levels of p‐tau correlate more closely with symptom severity and neuronal loss than Aβ plaque levels alone. 137 , 147 , 148 , 149 The pathological accumulation of p‐tau is now suspected to play a more primary role in disease progression with a direct, prion‐like spread and neuronal degeneration from the entorhinal cortex, to the perforant pathway, which links the cerebral cortex and hippocampus. 147 , 150 , 151 , 152 For these reasons, p‐tau targeted therapy has appealed as a promising avenue for the past decade.
Therapeutic investigation originally focused on microtubule stabilization, inhibition of the kinases responsible for phosphorylating tau, and direct inhibition of tau aggregation. These methods have mostly been abandoned due to their lack of efficacy or toxicity. 39 , 153 The approach to tau targeted treatment now centers on numerous immunotherapies that are still in the early stages of testing, but which have shown encouraging safety profiles thus far. 39 , 153 , 154
4. THE ROLE OF THE NORADRENERGIC SYSTEM IN ALZHEIMER'S DISEASE
As evidenced by the brief review above, none of the proposed mechanisms has yet provided a full explanation for the pattern and distribution of AD pathology. As a result, many researchers have begun looking for alternative theories. Recent interest has turned to the role of the noradrenergic system, and the LC specifically, in AD pathology and symptomology. Studies have indicated an average reduction of 60% in LC cells compared to similarly aged controls, with LC degeneration occurring early in the time course of AD progression. 24 , 26 , 27 , 155 , 156 , 157 , 158 , 159 , 160
4.1. Noradrenergic changes
The noradrenergic system in the brain is critical for the regulation of many normal brain functions. 161 , 162 , 163 , 164 , 165 , 166 , 167 , 168 The primary source of norepinephrine (NE) to the forebrain originates in the LC, and selective deterioration of this nucleus has been shown to significantly disrupt multiple cognitive processes. 169 , 170 The LC projects to a wide area, including the hippocampus, cerebral cortex, basal forebrain, preoptic area, and hypothalamus (Figure 2), meaning any disruption in the function of the LC has widespread implications. 171 , 172 Norepinephrine is released from presynaptic terminals, with extracellular levels largely determined by catechol‐O‐methyltransferase (COMT) and monoamine oxidase (MAO) degradation, or reuptake into the presynaptic terminal by norepinephrine transporter (NET). 173 , 174 , 175 , 176 , 177 The exact role of LC‐NE dysfunction in AD is not well understood, in part due to the numerous adrenergic receptor subtypes and their varying effects. A summary of what is known so far about how these receptors are altered in AD is presented in Table 1 (for a more thorough review, see Gannon et al). 170 , 178 Briefly, stimulation of the α2A receptor has been correlated with amyloidogenesis, while diseased brains have been routinely found to exhibit decreased levels of the α1A adrenergic receptor subtype in the prefrontal cortex. 179 , 180 , 181 , 182 Meanwhile, a decreased β/β2 receptor ratio has been observed in AD patients, with β2 blockade leading to a reduction in amyloidogenesis. 179 , 183 , 184 , 185 , 186 , 187 , 188
FIGURE 2.
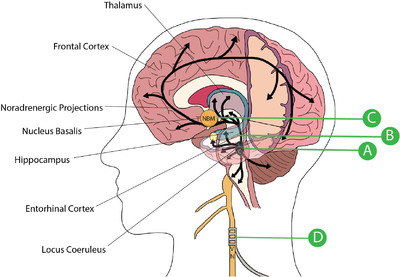
Role of electroceuticals in treating Alzheimer's disease. A, The Locus Coeruleus (LC) consistently exhibits the first pathology seen in AD and noradrenergic dysfunction exacerbates multiple aspects of disease progression. Chemogenetic manipulation has shown promising results. B, The entorhinal cortex is also significantly impacted in AD and likely contributes to early problems with declarative memory. Stimulation of the entorhinal cortex in animal models of AD have shown early promise in improving memory deficits. C, The Nucleus Basalis of Meynert (NBM) undergoes profound degeneration in late‐stage Alzheimer's disease (AD), leading to cholinergic dysfunction. Small clinical trials for NBM‐DBS have shown safety, tolerance, and modest levels of cognitive improvement or stabilization. D, Vagus Nerve Stimulation (VNS) is minimally invasive and has been proven to elevate NE and ACh levels in cortical and subcortical structures. Additionally, VNS alters microglial phenotypes and activates neuronal plasticity.
TABLE 1.
Currently understood role of noradrenergic receptor subtypes in Alzheimer's disease
| Receptor | Subtype | Location | Role in Cognition | Changes in Alzheimer's Disease | |
|---|---|---|---|---|---|
| α1 | α1A | Hippocampus | Improves spatial learning and memory 259 | ↓ α1A mRNA 180 | ↓ α1 non‐subtype selective radioligands in the hippocampus 260 and prefrontal cortex. 261 |
| α1B | Amygdala | Improves fear learning 262 |
|
||
| α1D | Hippocampus | Improves working memory and attention 263 | ↓ α1D mRNA 180 | ||
| α2 | α2A | Hippocampus | Impairs spatial and fear learning 264 , 265 , 266 | ↔ α2A levels unchanged 267 |
↑ α2 receptor density in remaining cortical membranes 268 and dentate gyrus granule cell layer 178 , 267 |
| Prefrontal cortex | Improves working memory | ↓ α2A mRNA in layer II 180 |
↑ α2 receptors in brain microvasculature innervated by LC 269 |
||
| Cerebellar cortex | – | ↑ α2 receptors in aggressive subgroup 270 | |||
| α2C | Hippocampus | – | ↓ α2C mRNA 267 | ||
| β | β1 | Hippocampus | Impairs spatial reference memory 178 | ↑ β1 receptors 186 | ↔ No consistent change in absolute level of β1 receptors in AD patients 271 |
| Prefrontal cortex | – | ↓ Decreased ratio of β/β2 186 | |||
| Putamen | – | ↓ β1 receptors 186 | |||
| β2 | Hippocampus | Impairs spatial reference memory 272 | ↑ β2 receptors 186 |
↓ β2 receptor density in cerebral microvessels 273 ↓ Decreased ratio of β/β2 186 |
|
| Prefrontal cortex | Improves memory retrieval 274 | ↓ Decreased ratio of β/β2 186 | |||
| Thalamus | – | ↓ β2 receptors 275 | |||
| Putamen | – | ↔ No consistent change 186 | |||
Tauopathy in the LC occurs as early as adolescence, with neuronal degeneration progressing over a number of decades. 29 , 31 , 152 In response to the overall loss of LC neurons, the surviving neurons display numerous compensatory changes, including sprouting additional axonal connections to the hippocampus and increasing the number of dendritic connections overall. 170 , 180 This keeps the number of connections relatively stable as AD pathology begins to progress but before symptoms are manifested clinically. It has also been shown that as AD develops, there is an associated increase in the enzyme tyrosine hydroxylase, which is responsible for the rate‐limiting step of norepinephrine synthesis, along with a decrease in the levels of norepinephrine transporter (NET). 180 , 189 As these disease processes continue there is mixed evidence on whether levels of norepinephrine (NE) are substantially altered in the brain. It is postulated that perhaps the regulatory mechanisms are able to compensate to keep extracellular levels of NE stable until late in the disease. 190 This increased noradrenergic tone helps maintain extracellular NE input to the cortex during the early disease stages, while the overall tissue levels of NE in the hippocampus, temporal lobe, thalamus, and LC decline anywhere from 12% to 65%. 170 At the time the most recent review on this topic was published, more consistent studies were needed to verify the extent of signaling dysfunction and adrenergic receptor involvement. 178 See Table S2B for a more thorough summary of the relevant literature.
The most significant new contribution to understanding the involvement of various adrenergic receptor subtypes has focused on the α2A adrenergic receptor (α2AAR), previously implicated in promoting amyloidogenesis (Figure 3). 181 This work found that Aβ oligomers (AβO) act as an allosteric ligand of α2AAR. AβO interaction aberrantly redirects NE‐induced α2AAR receptor signaling to activate the GSK3β/tau cascade, resulting in an increased intracellular signaling response and the subsequent hyperphosphorylation of tau (Figure 3). 190 The α2AAR is present in many CNS structures, especially the LC, but is also present in other regions of the brainstem, midbrain, hypothalamus, and hippocampus. 182 This supports the idea that AD‐specific neurodegeneration often begins with, and is a primary pathology of, the LC – before any other location. AD can then be thought of as affecting local neurons first and eventually spreading throughout the more classic regions of the brain. The disrupted cellular signal balance and dysregulation of microglial immune function create a complex interplay that eventually leads to the symptoms and pathology seen in AD.
Despite a limited understanding of the exact mechanisms underlying noradrenergic system dysregulation in the LC, several experiments have shown that a NE deficit exacerbates AD pathology, while NE supplementation appears to be beneficial. Ablation of noradrenergic neurons with DSP‐4 in AD transgenic mice increases Aβ deposition, alters adrenergic receptor subtype expression, and impairs spatial memory. 191 , 192 , 193 Much of this effect appears to be related to the ability of the noradrenergic system to modulate the immune system, with selective ablation of the LC significantly impairing the ability for microglia to phagocytose Aβ. 192 Additional experiments have shown that a DSP‐4 lesion of the LC also increases the levels of intracellular tau in the cortex of APP‐SL mice. 14 In contrast, peripheral administration of a NE precursor in these animals restored some microglia function and increased Aβ clearance. 194 NE appears to provide dose‐dependent protection to primary cortical and LC neurons from Aβ toxicity via the tropomyosin‐related kinase B (TrkB). 195
The full scope of these findings point to a spectrum of illness in which the resulting imbalance of adrenergic receptors, in combination with increased demands on the remaining noradrenergic neurons and time‐dependent synaptic remodeling, lead to a positive feedback of amyloid accumulation, increased tau phosphorylation, immune system dysfunction, and neuronal death. 169 , 170 , 173 , 178 , 196 , 197 , 198 , 199 , 200 More work should be pursued that can characterize the temporal and spatial characteristics of this disease process. Work that expands on the deleterious effects of a dysfunctional adrenergic system continues to be more compelling than that focusing on the late‐stage cholinergic changes mentioned previously. In fact, even when the disease has become clinically apparent, the most profound neuronal loss remains to be in the LC. 201
Contributing to the complex involvement of this neurotransmitter system in AD, the LC is unique in that its neurotransmitter adds to the oxidative stress of its corresponding neurons. 158 , 202 , 203 , 204 , 205 NE transporters reuptake the neurotransmitter after its release into the synaptic cleft which can result in cytoplasmic NE. 200 This NE can autoxidize or be converted into a toxic metabolite by MAOs. 200 , 206 Additional metabolic stress occurs because of the near continuous activation of the LC, resulting in the reliance on mitochondrial oxidative phosphorylation. 205 The human LC also synthesizes the granular pigment neuromelanin, which binds iron and other heavy metals from the blood; LC projections are exposed to an extensive microvascular surface area in the central nervous system. 207 Norepinephrine itself also plays an important role in maintaining the BBB, meaning any dysfunction in NE can result in increased toxin exposure. 198 , 208 , 209 In addition to the direct pathologies mentioned previously, dysfunction of LC neurons creates additional cerebral susceptibilities to further degradation. Therefore, an LC‐centric hypothesis would also build off the mitochondrial cascade hypothesis because the neurons in the LC are particularly prone to generating Aβ as a protective response. 200
It is also worth noting that there is a growing interest in the role of gut‐brain axis dysfunction in AD. 210 , 211 , 212 There has been some evidence to indicate that there is a link between the gut microbiota and the Aβ signaling pathway. 212 , 213 , 214 The vagus nerve provides extensive innervation to the visceral organs and is a key bi‐directional mediator of inflammation in both the central nervous system and peripheral tissue. 210 , 211 , 214 , 215 The extent of the role of the enteric neurotransmitter system, and NE in particular, is poorly understood in relation to cognitive function, but is of significant interest due to the role stimulation of the vagus nerve plays in regulation of the peripheral immune system. 211 , 215 , 216
4.2. Targeted noradrenergic therapies
The impact of noradrenergic neuron involvement in AD has generated much excitement at the prospect of utilizing NE‐targeted drugs to impede progression of the disease. 217 Several animal models have shown that increasing NE holds the potential to treat both the neuropathological and cognitive decline of AD. 170 , 190 , 194 , 195 , 206 , 217 , 218 , 219 , 220 In general, α2A antagonism has been shown to reverse memory deficits in mice, 181 and β receptor activation of the cAMP/protein kinase A signaling pathway reverses the toxic effects of AβO. 183 , 184 , 185 There has also been promise in experiments demonstrating that attenuation of NET activity can markedly reduce beta amyloid deposition. 221 Progress in building a successful theory behind the mechanisms responsible for improving memory performance remains complicated by the roles of the various adrenergic receptor antagonists (see Table 1). 187 , 222 , 223 A possible explanation for this wide array of findings is that the compensatory changes seen in the LC as neurons began to die results in some signaling pathways and regions of the brain becoming overactivated, while others become suppressed. Broadly targeted NE treatments likely do little to correct this misbalance, especially late in the disease. 158 , 202 , 224
Indeed, despite the growing body of evidence correlating disease severity most closely with noradrenergic neuron loss, drugs that act to increase synaptic NE have seen limited application in the clinical treatment in AD patients. MAO inhibitors, theoretically useful in increasing extracellular NE levels, have shown mixed results in clinical trials. 225 , 226 Selegiline, a MAO‐B inhibitor with proven neuroprotective benefits in Parkinson's, has exhibited very few significant treatment effects, though the studies have been small and a subsequent meta‐analysis has revealed a possible benefit on memory function. 226 MAO inhibitors also come with a range of undesirable side‐effects and non‐selectively alter multiple other neurotransmitter systems. 227 , 228 This limits the maximum therapeutic efficacy, but, given the promise of norepinephrine in improving Aβ clearance and limiting cognitive decline, a tremendous amount of research is now being done to formulate better MAO inhibitors and design more rigorous clinical trials. 225 It is likely, though, that these new efforts will continue to be plagued by the lack of MAO inhibitor specificity for the systems most strongly implicated in AD dysfunction, especially if NE misbalances between different regions of the brain prove clinically significant. This work does leave open, however, the idea that region‐specific NE stimulation, possibly through a combination of approaches to be discussed later in this review, may prove more effective.
Despite the current failure of MAO inhibitors, methylphenidate, a potent stimulant that acts as a norepinephrine‐dopamine reuptake inhibitor (NDRI) has been shown to slightly improve the attention and apathy of AD patients after symptom onset, demonstrating increased NE levels can result in a clinical benefit. 229 , 230 Studies are still extremely limited, and it has not been trialed as a preventative measure. 229 A more targeted noradrenergic therapy is atomoxetine, a norepinephrine reuptake inhibitor (NRI), that acts as a relatively selective inhibitor of NET in the central nervous system, especially the forebrain. 231 Multiple animal models have indicated possible therapeutic effects of atomoxetine, but clinical trials have not yet shown efficacy. 232 It is possible that the late‐stage increase in forebrain NE is not enough to reverse or significantly alter the underlying pathology. In essence, previous approaches have proven to be too little, too late.
4.3. Targeted locus coeruleus therapies
There is a large body of research demonstrating an important and primary role of the noradrenergic system and LC remodeling in AD. Coupled with the lack of tangible results from existing noradrenergic therapies, it is important to ask whether the LC itself can be directly stimulated. Animal studies have shown that chemogenetic stimulation of the LC in transgenic TgF344‐AD rats resulted in the rescue of impaired reversal learning in a Morris water maze task. 233 To‐date, no studies of direct LC stimulation have been performed in humans, but indirect modulation through non‐invasive brain stimulation (NIBS) techniques have been implemented in at least one clinical study (Figure 2). This pilot study using vagal nerve stimulation (VNS) was able to show that the treatment was well tolerated and that after one year 7 of 17 patients improved and 12 of 17 patients did not decline from baseline. 234 Despite such initial promise, the mechanism of brain stimulation in AD is not well understood, and treatments remain primarily open loop with no adjustment possible for an individual's response to stimulation. 235 Follow‐up clinical trials have not been widely pursued, and the mechanisms underlying the effect of VNS stimulation on AD, as well as the relationship with the LC, are not well understood. Work using VNS in animal models, however, has moved forward in exploring a variety of proposed mechanisms and will be discussed in the final section of this review (Supplementary Table 2C).
5. ADRENERGIC AND CHOLINERGIC RELATIONSHIP
An important connection that has been largely overlooked is the interaction between the noradrenergic and cholinergic centers of the brain. With the shift in AD focus from the NBM to the LC, there has been limited work understanding the impact of dysfunction of one of these systems on the other. One of the first studies to investigate this relationship, involved lesioning the fornix in rats, a structure important for transmitting acetylcholine to the hippocampus, and a subsequent neurochemical analysis of hippocampal tissue. It was found that choline acetyltransferase (ChAT) activity was reduced by 50%, correlating negatively with the number of errors the rats made in a maze task, while hippocampal NE also decreased by 50%. 236 Interestingly, there was no decrease, and in fact a slight increase in the noradrenergic metabolite methylhydroxyphenylglycol (MHPG), suggesting a net increase in NE turnover despite the decrease in noradrenergic cells. These researchers postulated that the negative correlation between ChAT activity and NE turnover suggested that hyperactivity in the remaining noradrenergic neurons inhibited proper functioning of the remaining cholinergic neurons. 236 This is a remarkably similar pathology to that seen in AD and warrants further exploration. It is important to note that the suspected mechanism of noradrenergic inhibition on the cholinergic system operated through the α2 receptors located on cholinergic terminals. 236 , 237 , 238 , 239
The dynamic relationship between these two neuromodulatory systems remains substantially uncharacterized, and limited work has been done explaining the precise mechanisms underlying adrenergic hyperactivation following LC disruption. There is evidence, however, that many of the mechanisms beneficial to memory storage occur with moderate levels of NE, while higher levels tend to impair memory storage. 240 Additional work has shown that more substantial and specific lesions to the LC do indeed result in an increase in cortical release of ACh. 239 This is likely not applicable to late‐stage AD when a significant portion of both the LC and NBM have degenerated and tau pathology has spread throughout the brain. It does raise interesting questions though about how this dysregulation can be targeted early in the course of AD.
Further compounding this relationship is additional evidence that decreased ACh activity possibly decreases noradrenergic tone. 241 This implies that when AD reaches the point of significant cholinergic destruction, there would be a rapid deterioration in the previously compensated noradrenergic system. Again, the deleterious mechanisms of NE appear to be regulated through α2 ARs, while the memory enhancing effects appear to be mediate through the β ARs. 240 This makes sense in the context of the relative affinities of each of these receptors. α2 ARs have the highest binding affinity to NE, while β ARs have the lowest. 173 Taken together with the information previously presented on the overall noradrenergic dysfunction seen in AD, this likely accounts for some of the contradictory effects of NE seen in experiments. Successful treatment approaches are likely to be ones that can stimulate healthy LC function before significant dendritic remodeling occurs, with an emphasis on immune regulation and perhaps selective α2 blockade.
6. THE FUTURE OF ELECTROCEUTICAL THERAPIES IN ALZHEIMER'S DISEASE
Direct neural stimulation offers many interesting and unique opportunities to halt or reverse AD pathology. 56 , 57 , 58 , 59 , 60 , 61 , 62 , 63 , 64 , 65 , 66 , 67 , 68 , 69 , 70 , 71 , 72 , 73 In addition to the need to further develop the NBM‐DBS approach mentioned above, there are also a variety of theoretically viable methods that can capitalize on a new AD therapeutic approach centered on the LC or entorhinal cortex (Figure 2). 242 , 243 Vagus nerve stimulation promises several unique, non‐invasive approaches to many of the hypotheses for AD, and can be applied at nearly every stage of the disease. The last clinically significant trial with this technology took place nearly two decades ago, and the control techniques available to researchers and physicians have improved substantially since that time. A tremendous amount of work remains to explore the underlying mechanisms and further develop the technology needed to address each of these promising avenues.
Vagal nerve stimulation (VNS) (Supplementary Table 2C) has been used for many years to treat various neurological and psychological conditions, including major depressive disorder and epilepsy. It is a safe and effective technique that allows a minimally invasive interface with the adrenergic system and, to a lesser known extent, the cholinergic system. 244 , 245 , 246 Experimental models of LC degeneration have shown that significantly decreased levels of NE can suppress microglial phagocytosis of beta amyloid (Figure 4). 191 , 192 , 193 , 194 VNS offers an opportunity to restore substantial microglial function in late stage AD, where NE levels may be substantially decreased in specific brain regions.
FIGURE 4.
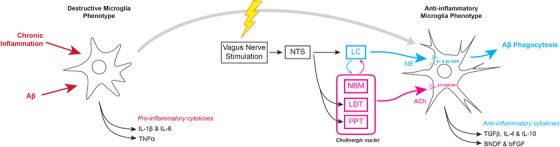
Anti‐inflammatory mechanisms of vagus nerve stimulation. Stimulation of the vagus nerve can result in the activation of anti‐inflammatory signaling cascades that shift microglia towards a phenotype more adept at clearing AD pathology.
Combined with early detection markers currently under development, VNS stands out as a relatively early intervention, when accumulating neurotoxic products elicit a generalized inflammatory response that further exacerbates neurodegeneration and dysfunction of the cerebrovascular endothelium. 247 , 248 VNS stimulation, in contrast to pure NE manipulation, has been shown to significantly reduce plasma levels of tumor necrosis factor‐α and prevent hippocampal microglial activation (Figure 4). 249 , 250 Much of this additional benefit from VNS may arise from simultaneous activation of what is known as the cholinergic anti‐inflammatory pathway, which is implicated in encouraging a neuroprotective microglial phenotype through activation of the α7 subtype of the nicotinic acetylcholine receptor. 250 , 251 Recent research has shown that microglial activation appears to be both a specific response to early Aβ plaque deposition as well as a nonspecific and late response to subsequent neurodegeneration. 252 VNS may be able to modulate the immune system in both early and late stages of the disease. This could perhaps delay symptomatic manifestations of AD to a point where they are no longer clinically relevant.
Given the dendritic remodeling discussed previously in this review, there is a need to better characterize the functional ability of VNS to either prevent or selectively reverse this change in AD patients. Previous work has shown that VNS is capable of modulating the dendritic cell profile outside of the central nervous system. 253 , 254 In the central nervous system, it is also known that VNS can induce neuronal plasticity. 255 , 256 Eventual control over these processes offers significant therapeutic potential, but the work is too pre‐mature to comment on in‐depth during this review.
7. CONCLUSION
The role NE plays in AD remains complicated. Both VNS and experiments with direct LC stimulation present opportunities to assess if the neuroprotective factors of NE remain true through all stages of AD. VNS is well documented to increase the extra‐cellular levels of NE in the hippocampus and cortex, depending on the intensity of stimulation. 257 This response can be used to evaluate an intervention that increases endogenous NE levels in a method that mimics natural LC communication.
VNS as an intervention in both early and late AD should be investigated more in‐depth. The invasive methods of directly stimulating the LC can also help differentiate the role of NE specifically, as opposed to the more general effect of VNS in the forebrain, thalamus, and reticular formation. 258 This non‐specific, generalized response to VNS may hold promise for reducing the cognitive deficits in AD patients through an entirely unrecognized set of mechanisms. It is imperative that researchers continue constructing methods to more accurately control the stimulation and response of both peripheral and central neural stimulation. A more nuanced understanding of the time‐dependent changes across AD in relation to the LC, with and without VNS, offers an opportunity to substantially increase our understanding of this devastating disease and to develop more effective therapeutics.
Supporting information
Table S1
Table S2
Slater C, Wang Qi. Alzheimer's disease: an evolving understanding of noradrenergic involvement and the promising future of electroceutical therapies. Clin Transl Med. 2021;11:e397. 10.1002/ctm2.397
REFERENCES
- 1. Weller J, Budson A. Current understanding of Alzheimer's disease diagnosis and treatment. F1000Res. 2018;7. 10.12688/f1000research.14506.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Crous‐Bou M, Minguillon C, Gramunt N, Molinuevo JL. Alzheimer's disease prevention: from risk factors to early intervention. Alzheimers Res Ther. 2017;9(1):71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010‐2050) estimated using the 2010 census. Neurology. 2013;80(19):1778‐1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Murphy SL, Xu J, Kochanek KD, Deaths: final data for 2010; 2013. [PubMed]
- 5. Dharmarajan TS, Gunturu SG. Alzheimer's disease: a healthcare burden of epidemic proportion. Am Health Drug Benefits. 2009;2(1):39‐47. [PMC free article] [PubMed] [Google Scholar]
- 6. Mayeux R, Stern Y. Epidemiology of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(8). 10.1101/cshperspect.a006239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Atri A. The Alzheimer's Disease Clinical Spectrum: diagnosis and Management. Med Clin North Am. 2019;103(2):263‐293. [DOI] [PubMed] [Google Scholar]
- 8. Jack CR Jr, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010;9(1):119‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dong A, Toledo JB, Honnorat N, et al. Heterogeneity of neuroanatomical patterns in prodromal Alzheimer's disease: links to cognition, progression and biomarkers. Brain. 2017;140(3):735‐747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lam B, Masellis M, Freedman M, Stuss DT, Black SE. Clinical, imaging, and pathological heterogeneity of the Alzheimer's disease syndrome. Alzheimers Res Ther. 2013;5(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mielke MM, Vemuri P, Rocca WA. Clinical epidemiology of Alzheimer's disease: assessing sex and gender differences. Clin Epidemiol. 2014;6:37‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vest RS, Pike CJ. Gender, sex steroid hormones, and Alzheimer's disease. Horm Behav. 2013;63(2):301‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carter CL, Resnick EM, Mallampalli M, Kalbarczyk A. Sex and gender differences in Alzheimer's disease: recommendations for future research. J Womens Health (Larchmt). 2012;21(10):1018‐1023. [DOI] [PubMed] [Google Scholar]
- 14. Oikawa N, Ogino K, Masumoto T, Yamaguchi H, Yanagisawa K. Gender effect on the accumulation of hyperphosphorylated tau in the brain of locus‐ceruleus‐injured APP‐transgenic mouse. Neurosci Lett. 2010;468(3):243‐247. [DOI] [PubMed] [Google Scholar]
- 15. Nebel RA, Aggarwal NT, Barnes LL, et al. Understanding the impact of sex and gender in Alzheimer's disease: a call to action. Alzheimers Dement. 2018;14(9):1171‐1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moller HJ, Graeber MB. The case described by Alois Alzheimer in 1911. Historical and conceptual perspectives based on the clinical record and neurohistological sections. Eur Arch Psychiatry Clin Neurosci. 1998;248(3):111‐122. [DOI] [PubMed] [Google Scholar]
- 17. Eric RK, Schwartz JH, Jessell TM, Siegelbaum SA, Hudspeth AJ. The Aging Brain. Principles of Neural Science. 5th ed.. McGraw‐Hill Education; 2014:1335‐1341.chap 59. [Google Scholar]
- 18. Fallon JH, Koziell DA, Moore RY. Catecholamine innervation of the basal forebrain. II. Amygdala, suprarhinal cortex and entorhinal cortex. J Comp Neurol. 1978;180(3):509‐532. [DOI] [PubMed] [Google Scholar]
- 19. Khan UA, Liu L, Provenzano FA, et al. Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer's disease. Nat Neurosci. 2014;17(2):304‐311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):270‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Badhwar A, Tam A, Dansereau C, Orban P, Hoffstaedter F, Bellec P. Resting‐state network dysfunction in Alzheimer's disease: a systematic review and meta‐analysis. Alzheimers Dement (Amst). 2017;8:73‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):263‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):280‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Theofilas P, Ehrenberg AJ, Dunlop S, et al. Locus coeruleus volume and cell population changes during Alzheimer's disease progression: a stereological study in human postmortem brains with potential implication for early‐stage biomarker discovery. Alzheimers Dement. 2017;13(3):236‐246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weiner MW, Veitch DP, Aisen PS, et al. 2014 Update of the Alzheimer's Disease Neuroimaging Initiative: a review of papers published since its inception. Alzheimers Dement. 2015;11(6):e1‐e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tomlinson BE, Irving D, Blessed G. Cell loss in the locus coeruleus in senile dementia of Alzheimer type. J Neurol Sci. 1981;49(3):419‐428. [DOI] [PubMed] [Google Scholar]
- 27. Grudzien A, Shaw P, Weintraub S, Bigio E, Mash DC, Mesulam MM. Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer's disease. Neurobiol Aging. 2007;28(3):327‐335. [DOI] [PubMed] [Google Scholar]
- 28. Theofilas P, Dunlop S, Heinsen H, Grinberg LT. Turning on the Light Within: subcortical Nuclei of the Isodentritic Core and their Role in Alzheimer's Disease Pathogenesis. J Alzheimers Dis. 2015;46(1):17‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70(11):960‐969. [DOI] [PubMed] [Google Scholar]
- 30. de Calignon A, Polydoro M, Suarez‐Calvet M, et al. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron. 2012;73(4):685‐697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Braak H, Del Tredici K. Alzheimer's pathogenesis: is there neuron‐to‐neuron propagation?. Acta Neuropathol. 2011;121(5):589‐595. [DOI] [PubMed] [Google Scholar]
- 32. Theofilas P, Ehrenberg AJ, Nguy A, et al. Probing the correlation of neuronal loss, neurofibrillary tangles, and cell death markers across the Alzheimer's disease Braak stages: a quantitative study in humans. Neurobiol Aging. 2018;61:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schmitz TW, Nathan Spreng R. Alzheimer's Disease Neuroimaging I. Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer's pathology. Nat Commun. 2016;7:13249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mesulam MM. Cholinergic circuitry of the human nucleus basalis and its fate in Alzheimer's disease. J Comp Neurol. 2013;521(18):4124‐4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gilmor ML, Erickson JD, Varoqui H, et al. Preservation of nucleus basalis neurons containing choline acetyltransferase and the vesicular acetylcholine transporter in the elderly with mild cognitive impairment and early Alzheimer's disease. J Comp Neurol. 1999;411(4):693‐704. [PubMed] [Google Scholar]
- 36. Grothe M, Heinsen H, Teipel SJ. Atrophy of the cholinergic Basal forebrain over the adult age range and in early stages of Alzheimer's disease. Biol Psychiatry. 2012;71(9):805‐813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Arendt T, Bigl V, Tennstedt A, Arendt A. Neuronal loss in different parts of the nucleus basalis is related to neuritic plaque formation in cortical target areas in Alzheimer's disease. Neuroscience. 1985;14(1):1‐14. [DOI] [PubMed] [Google Scholar]
- 38. Swerdlow RH. Pathogenesis of Alzheimer's disease. Clin Interv Aging. 2007;2(3):347‐359. [PMC free article] [PubMed] [Google Scholar]
- 39. Congdon EE, Sigurdsson EM. Tau‐targeting therapies for Alzheimer disease. Nat Rev Neurol. 2018;14(7):399‐415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Davies P, Maloney AJ. Selective loss of central cholinergic neurons in Alzheimer's disease. Lancet. 1976;2(8000):1403. [DOI] [PubMed] [Google Scholar]
- 41. Bowen DM, Smith CB, White P, Davison AN. Neurotransmitter‐related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain. 1976;99(3):459‐496. [DOI] [PubMed] [Google Scholar]
- 42. Perry EK, Perry RH, Blessed G, Tomlinson BE. Necropsy evidence of central cholinergic deficits in senile dementia. Lancet. 1977;1(8004):189. [DOI] [PubMed] [Google Scholar]
- 43. Muir JL. Acetylcholine, aging, and Alzheimer's disease. Pharmacol Biochem Behav. 1997;56(4):687‐696. [DOI] [PubMed] [Google Scholar]
- 44. Lemiere J, Van Gool D, Dom R. Treatment of Alzheimer's disease: an evaluation of the cholinergic approach. Acta Neurol Belg. 1999;99(2):96‐106. [PubMed] [Google Scholar]
- 45. Liu PP, Xie Y, Meng XY, Kang JS. Erratum: author Correction: history and progress of hypotheses and clinical trials for Alzheimer's disease. Signal Transduct Target Ther. 2019;4:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Whitehouse PJ, Price DL, Clark AW, Coyle JT, DeLong MR. Alzheimer disease: evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann Neurol. 1981;10(2):122‐126. [DOI] [PubMed] [Google Scholar]
- 47. Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR. Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215(4537):1237‐1239. [DOI] [PubMed] [Google Scholar]
- 48. Hampel H, Mesulam MM, Cuello AC, et al. The cholinergic system in the pathophysiology and treatment of Alzheimer's disease. Brain. 2018;141(7):1917‐1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nordberg A, Winblad B. Reduced number of [3H]nicotine and [3H]acetylcholine binding sites in the frontal cortex of Alzheimer brains. Neurosci Lett. 1986;72(1):115‐119. [DOI] [PubMed] [Google Scholar]
- 50. Mash DC, Flynn DD, Potter LT. Loss of M2 muscarine receptors in the cerebral cortex in Alzheimer's disease and experimental cholinergic denervation. Science. 1985;228(4703):1115‐1117. [DOI] [PubMed] [Google Scholar]
- 51. Anagnostaras SG, Murphy GG, Hamilton SE, et al. Selective cognitive dysfunction in acetylcholine M1 muscarinic receptor mutant mice. Nat Neurosci. 2003;6(1):51‐58. [DOI] [PubMed] [Google Scholar]
- 52. Shiozaki K, Iseki E, Hino H, Kosaka K. Distribution of m1 muscarinic acetylcholine receptors in the hippocampus of patients with Alzheimer's disease and dementia with Lewy bodies‐an immunohistochemical study. J Neurol Sci. 2001;193(1):23‐28. [DOI] [PubMed] [Google Scholar]
- 53. Atri A, Sherman S, Norman KA, et al. Blockade of central cholinergic receptors impairs new learning and increases proactive interference in a word paired‐associate memory task. Behav Neurosci. 2004;118(1):223‐236. [DOI] [PubMed] [Google Scholar]
- 54. Hansen RA, Gartlehner G, Lohr KN, Kaufer DI. Functional outcomes of drug treatment in Alzheimer's disease: a systematic review and meta‐analysis. Drugs Aging. 2007;24(2):155‐167. [DOI] [PubMed] [Google Scholar]
- 55. Davis KL, Powchik P. Tacrine. Lancet. 1995;345(8950):625‐630. [DOI] [PubMed] [Google Scholar]
- 56. Kuhn J, Hardenacke K, Lenartz D, et al. Deep brain stimulation of the nucleus basalis of Meynert in Alzheimer's dementia. Mol Psychiatry. 2015;20(3):353‐360. [DOI] [PubMed] [Google Scholar]
- 57. Hardenacke K, Hashemiyoon R, Visser‐Vandewalle V, et al. Deep Brain Stimulation of the Nucleus Basalis of Meynert in Alzheimer's Dementia: potential Predictors of Cognitive Change and Results of a Long‐Term Follow‐Up in Eight Patients. Brain Stimul. 2016;9(5):799‐800. [DOI] [PubMed] [Google Scholar]
- 58. Gratwicke J, Zrinzo L, Kahan J, et al. Bilateral nucleus basalis of Meynert deep brain stimulation for dementia with Lewy bodies: a randomised clinical trial. Brain Stimul. 2020;13(4):1031‐1039. [DOI] [PubMed] [Google Scholar]
- 59. Gratwicke J, Zrinzo L, Kahan J, et al. Bilateral Deep Brain Stimulation of the Nucleus Basalis of Meynert for Parkinson Disease Dementia: a Randomized Clinical Trial. JAMA Neurol. 2018;75(2):169‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lozano AM, Fosdick L, Chakravarty MM, et al. A Phase II Study of Fornix Deep Brain Stimulation in Mild Alzheimer's Disease. J Alzheimers Dis. 2016;54(2):777‐787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mirzadeh Z, Bari A, Lozano AM. The rationale for deep brain stimulation in Alzheimer's disease. J Neural Transm (Vienna). 2016;123(7):775‐783. [DOI] [PubMed] [Google Scholar]
- 62. Aldehri M, Temel Y, Alnaami I, Jahanshahi A, Hescham S. Deep brain stimulation for Alzheimer's Disease: an update. Surg Neurol Int. 2018;9:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Baldermann JC, Hardenacke K, Hu X, et al. Neuroanatomical Characteristics Associated With Response to Deep Brain Stimulation of the Nucleus Basalis of Meynert for Alzheimer's Disease. Neuromodulation. 2018;21(2):184‐190. [DOI] [PubMed] [Google Scholar]
- 64. Lee DJ, Lozano AM. Current Status of Deep Brain Stimulation for Alzheimer's Disease: from Chance Observation to Clinical Trials. Cold Spring Harb Symp Quant Biol. 2018;83:201‐205. [DOI] [PubMed] [Google Scholar]
- 65. Leoutsakos JS, Yan H, Anderson WS, et al. Deep Brain Stimulation Targeting the Fornix for Mild Alzheimer Dementia (the ADvance Trial): a Two Year Follow‐up Including Results of Delayed Activation. J Alzheimers Dis. 2018;64(2):597‐606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lv Q, Du A, Wei W, Li Y, Liu G, Wang XP. Deep Brain Stimulation: a Potential Treatment for Dementia in Alzheimer's Disease (AD) and Parkinson's Disease Dementia (PDD). Front Neurosci. 2018;12:360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mann A, Gondard E, Tampellini D, et al. Chronic deep brain stimulation in an Alzheimer's disease mouse model enhances memory and reduces pathological hallmarks. Brain Stimul. 2018;11(2):435‐444. [DOI] [PubMed] [Google Scholar]
- 68. Mao ZQ, Wang X, Xu X, et al. Partial improvement in performance of patients with severe Alzheimer's disease at an early stage of fornix deep brain stimulation. Neural Regen Res. 2018;13(12):2164‐2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Posporelis S, David AS, Ashkan K, Shotbolt P. Deep Brain Stimulation of the Memory Circuit: improving Cognition in Alzheimer's Disease. J Alzheimers Dis. 2018;64(2):337‐347. [DOI] [PubMed] [Google Scholar]
- 70. Scharre DW, Weichart E, Nielson D, et al. Deep Brain Stimulation of Frontal Lobe Networks to Treat Alzheimer's Disease. J Alzheimers Dis. 2018;62(2):621‐633. [DOI] [PubMed] [Google Scholar]
- 71. Huang C, Chu H, Ma Y, et al. The neuroprotective effect of deep brain stimulation at nucleus basalis of Meynert in transgenic mice with Alzheimer's disease. Brain Stimul. 2019;12(1):161‐174. [DOI] [PubMed] [Google Scholar]
- 72. Viana JNM, Gilbert F. Deep brain stimulation for people with Alzheimer's disease: anticipating potential effects on the tripartite self. Dementia (London). 2019;18(7‐8):2836‐2855. [DOI] [PubMed] [Google Scholar]
- 73. Lam J, Lee J, Liu CY, Lozano AM, Lee DJ. Deep Brain Stimulation for Alzheimer's Disease: tackling Circuit Dysfunction. Neuromodulation. 2020. 10.1111/ner.13305. Dec 30. [DOI] [PubMed] [Google Scholar]
- 74. Kumbhare D, Palys V, Toms J, et al. Nucleus Basalis of meynert stimulation for dementia: theoretical and technical considerations. Front Neurosci. 2018;12:614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Koulousakis P, Andrade P, Visser‐Vandewalle V, Sesia T. The Nucleus Basalis of Meynert and Its Role in Deep Brain Stimulation for Cognitive Disorders: a Historical Perspective. J Alzheimers Dis. 2019;69(4):905‐919. [DOI] [PubMed] [Google Scholar]
- 76. Kamenetz F, Tomita T, Hsieh H, et al. APP processing and synaptic function. Neuron. 2003;37(6):925‐937. [DOI] [PubMed] [Google Scholar]
- 77. Tyan SH, Shih AY, Walsh JJ, et al. Amyloid precursor protein (APP) regulates synaptic structure and function. Mol Cell Neurosci. 2012;51(1‐2):43‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yoshikai S, Sasaki H, Doh‐ura K, Furuya H, Sakaki Y. Genomic organization of the human amyloid beta‐protein precursor gene. Gene. 1990;87(2):257‐263. [DOI] [PubMed] [Google Scholar]
- 79. O'Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer's disease. Annu Rev Neurosci. 2011;34:185‐204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zheng H, Jiang M, Trumbauer ME, et al. beta‐Amyloid precursor protein‐deficient mice show reactive gliosis and decreased locomotor activity. Cell. 1995;81(4):525‐531. [DOI] [PubMed] [Google Scholar]
- 81. Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008;283(44):29615‐29619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Puzzo D, Privitera L, Leznik E, et al. Picomolar amyloid‐beta positively modulates synaptic plasticity and memory in hippocampus. J Neurosci. 2008;28(53):14537‐14545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Robakis NK, Ramakrishna N, Wolfe G, Wisniewski HM. Molecular cloning and characterization of a cDNA encoding the cerebrovascular and the neuritic plaque amyloid peptides. Proc Natl Acad Sci U S A. 1987;84(12):4190‐4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Glenner GG, Wong CW. Alzheimer's disease and Down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984;122(3):1131‐1135. [DOI] [PubMed] [Google Scholar]
- 85. Zigman WB, Lott IT. Alzheimer's disease in Down syndrome: neurobiology and risk. Ment Retard Dev Disabil Res Rev. 2007;13(3):237‐2346. [DOI] [PubMed] [Google Scholar]
- 86. Head E, Powell D, Gold BT, Schmitt FA. Alzheimer's Disease in Down Syndrome. Eur J Neurodegener Dis. 2012;1(3):353‐364. [PMC free article] [PubMed] [Google Scholar]
- 87. Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci. 1991;12(10):383‐388. [DOI] [PubMed] [Google Scholar]
- 88. Holmes C, Boche D, Wilkinson D, et al. Long‐term effects of Abeta42 immunisation in Alzheimer's disease: follow‐up of a randomised, placebo‐controlled phase I trial. Lancet. 2008;372(9634):216‐223. [DOI] [PubMed] [Google Scholar]
- 89. Giannakopoulos P, Herrmann FR, Bussiere T, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 2003;60(9):1495‐1500. [DOI] [PubMed] [Google Scholar]
- 90. Goate A, Chartier‐Harlin MC, Mullan M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349(6311):704‐706. [DOI] [PubMed] [Google Scholar]
- 91. Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184‐185. [DOI] [PubMed] [Google Scholar]
- 92. Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012;367(9):795‐804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Pooler AM, Polydoro M, Wegmann S, Nicholls SB, Spires‐Jones TL, Hyman BT. Propagation of tau pathology in Alzheimer's disease: identification of novel therapeutic targets. Alzheimers Res Ther. 2013;5(5):49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Reitz C. Alzheimer's disease and the amyloid cascade hypothesis: a critical review. Int J Alzheimers Dis. 2012;2012:369808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to beta ‐amyloid‐induced neurotoxicity. Proc Natl Acad Sci U S A. 2002;99(9):6364‐6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Tolar M, Abushakra S, Sabbagh M. The path forward in Alzheimer's disease therapeutics: reevaluating the amyloid cascade hypothesis. Alzheimers Dement. 2020;16(11):1553‐1560. [DOI] [PubMed] [Google Scholar]
- 97. Castellani RJ, Plascencia‐Villa G, Perry G, The amyloid cascade and Alzheimer's disease therapeutics: theory versus observation. Lab Invest . 2019;99(7):958‐970. [DOI] [PubMed] [Google Scholar]
- 98. Salloway S, Sperling R, Fox NC, et al. Two phase 3 trials of bapineuzumab in mild‐to‐moderate Alzheimer's disease. N Engl J Med. 2014;370(4):322‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Doody RS, Thomas RG, Farlow M, et al. Phase 3 trials of solanezumab for mild‐to‐moderate Alzheimer's disease. N Engl J Med. 2014;370(4):311‐321. [DOI] [PubMed] [Google Scholar]
- 100. Panza F, Lozupone M, Logroscino G, Imbimbo BP. A critical appraisal of amyloid‐beta‐targeting therapies for Alzheimer disease. Nat Rev Neurol. 2019;15(2):73‐88. [DOI] [PubMed] [Google Scholar]
- 101. Caselli RJ, Knopman DS, Bu G. An agnostic reevaluation of the amyloid cascade hypothesis of Alzheimer's disease pathogenesis: the role of APP homeostasis. Alzheimers Dement. 2020;16(11):1582‐1590. [DOI] [PubMed] [Google Scholar]
- 102. Zlokovic BV. Neurovascular mechanisms of Alzheimer's neurodegeneration. Trends Neurosci. 2005;28(4):202‐208. [DOI] [PubMed] [Google Scholar]
- 103. Buee L, Hof PR, Bouras C, et al. Pathological alterations of the cerebral microvasculature in Alzheimer's disease and related dementing disorders. Acta Neuropathol. 1994;87(5):469‐480. [DOI] [PubMed] [Google Scholar]
- 104. Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev Neurosci. 2004;5(5):347‐360. [DOI] [PubMed] [Google Scholar]
- 105. Nelson AR, Sweeney MD, Sagare AP, Zlokovic BV. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer's disease. Biochim Biophys Acta. 2016;1862(5):887‐900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Thomas T, Thomas G, McLendon C, Sutton T, Mullan M. beta‐Amyloid‐mediated vasoactivity and vascular endothelial damage. Nature. 1996;380(6570):168‐171. [DOI] [PubMed] [Google Scholar]
- 107. Provias J, Jeynes B. The role of the blood‐brain barrier in the pathogenesis of senile plaques in Alzheimer's disease. Int J Alzheimers Dis. 2014;2014:191863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Sutton ET, Hellermann GR, Thomas T. beta‐amyloid‐induced endothelial necrosis and inhibition of nitric oxide production. Exp Cell Res. 1997;230(2):368‐376. [DOI] [PubMed] [Google Scholar]
- 109. Tarantini S, Tran CHT, Gordon GR, Ungvari Z, Csiszar A. Impaired neurovascular coupling in aging and Alzheimer's disease: contribution of astrocyte dysfunction and endothelial impairment to cognitive decline. Exp Gerontol. 2017;94:52‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Montagne A, Barnes SR, Sweeney MD, et al. Blood‐brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85(2):296‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Stanimirovic DB, Friedman A. Pathophysiology of the neurovascular unit: disease cause or consequence?. J Cereb Blood Flow Metab. 2012;32(7):1207‐1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Sagare AP, Bell RD, Zlokovic BV. Neurovascular dysfunction and faulty amyloid beta‐peptide clearance in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(10). 10.1101/cshperspect.a011452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Anstey KJ, Cherbuin N, Budge M, Young J. Body mass index in midlife and late‐life as a risk factor for dementia: a meta‐analysis of prospective studies. Obes Rev. 2011;12(5):e426‐e4237. [DOI] [PubMed] [Google Scholar]
- 114. Biessels GJ, Strachan MW, Visseren FL, Kappelle LJ, Whitmer RA. Dementia and cognitive decline in type 2 diabetes and prediabetic stages: towards targeted interventions. Lancet Diabetes Endocrinol. 2014;2(3):246‐255. [DOI] [PubMed] [Google Scholar]
- 115. Li G, Mayer CL, Morelli D, et al. Effect of simvastatin on CSF Alzheimer disease biomarkers in cognitively normal adults. Neurology. 2017;89(12):1251‐1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Gold M, Alderton C, Zvartau‐Hind M, et al. Rosiglitazone monotherapy in mild‐to‐moderate Alzheimer's disease: results from a randomized, double‐blind, placebo‐controlled phase III study. Dement Geriatr Cogn Disord. 2010;30(2):131‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Wharton W, Stein JH, Korcarz C, et al. The effects of ramipril in individuals at risk for Alzheimer's disease: results of a pilot clinical trial. J Alzheimers Dis. 2012;32(1):147‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer's disease. Med Hypotheses. 2004;63(1):8‐20. [DOI] [PubMed] [Google Scholar]
- 119. Selfridge JE, E L, Lu J, Swerdlow RH. Role of mitochondrial homeostasis and dynamics in Alzheimer's disease. Neurobiol Dis. 2013;51:3‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Pantiya P, Thonusin C, Chattipakorn N, Chattipakorn SC. Mitochondrial abnormalities in neurodegenerative models and possible interventions: focus on Alzheimer's disease, Parkinson's disease, Huntington's disease. Mitochondrion. 2020;55:14‐47. [DOI] [PubMed] [Google Scholar]
- 121. Kandimalla R, Manczak M, Yin X, Wang R, Reddy PH. Hippocampal phosphorylated tau induced cognitive decline, dendritic spine loss and mitochondrial abnormalities in a mouse model of Alzheimer's disease. Hum Mol Genet. 2018;27(1):30‐40. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 122. Martins IV, Rivers‐Auty J, Allan SM, Lawrence CB. Mitochondrial Abnormalities and Synaptic Loss Underlie Memory Deficits Seen in Mouse Models of Obesity and Alzheimer's Disease. J Alzheimers Dis. 2017;55(3):915‐932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Hirai K, Aliev G, Nunomura A, et al. Mitochondrial abnormalities in Alzheimer's disease. J Neurosci. 2001;21(9):3017‐3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Swerdlow RH, Burns JM, Khan SM. The Alzheimer's disease mitochondrial cascade hypothesis: progress and perspectives. Biochim Biophys Acta. 2014;1842(8):1219‐1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Cenini G, Voos W. Mitochondria as Potential Targets in Alzheimer Disease Therapy: an Update. Front Pharmacol. 2019;10:902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Iturria‐Medina Y, Carbonell FM, Sotero RC, Chouinard‐Decorte F, Evans AC. Alzheimer's Disease Neuroimaging I. Multifactorial causal model of brain (dis)organization and therapeutic intervention: application to Alzheimer's disease. Neuroimage. 2017;152:60‐77. [DOI] [PubMed] [Google Scholar]
- 127. Toledo JB, Arnold M, Kastenmuller G, et al. Metabolic network failures in Alzheimer's disease: a biochemical road map. Alzheimers Dement. 2017;13(9):965‐984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Weyer G, Babej‐Dolle RM, Hadler D, Hofmann S, Herrmann WM. A controlled study of 2 doses of idebenone in the treatment of Alzheimer's disease. Neuropsychobiology. 1997;36(2):73‐82. [DOI] [PubMed] [Google Scholar]
- 129. Young AJ, Johnson S, Steffens DC, Doraiswamy PM. Coenzyme Q10: a review of its promise as a neuroprotectant. CNS Spectr. 2007;12(1):62‐68. [DOI] [PubMed] [Google Scholar]
- 130. McManus MJ, Murphy MP, Franklin JL. The mitochondria‐targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2011;31(44):15703‐15715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Grundman M, Grundman M, Delaney P. Antioxidant strategies for Alzheimer's disease. Proc Nutr Soc. 2002;61(2):191‐202. [DOI] [PubMed] [Google Scholar]
- 132. Luo Q, Lin YX, Yang PP, et al. A self‐destructive nanosweeper that captures and clears amyloid beta‐peptides. Nat Commun. 2018;9(1):1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Robert J, Stukas S, Button E, et al. Reconstituted high‐density lipoproteins acutely reduce soluble brain Abeta levels in symptomatic APP/PS1 mice. Biochim Biophys Acta. 2016;1862(5):1027‐1036. [DOI] [PubMed] [Google Scholar]
- 134. Vedagiri A, Thangarajan S. Mitigating effect of chrysin loaded solid lipid nanoparticles against Amyloid beta25‐35 induced oxidative stress in rat hippocampal region: an efficient formulation approach for Alzheimer's disease. Neuropeptides. 2016;58:111‐125. [DOI] [PubMed] [Google Scholar]
- 135. Reddy PH, Manczak M, Yin X, Reddy AP. Synergistic Protective Effects of Mitochondrial Division Inhibitor 1 and Mitochondria‐Targeted Small Peptide SS31 in Alzheimer's Disease. J Alzheimers Dis. 2018;62(4):1549‐1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Muralidar S, Ambi SV, Sekaran S, Thirumalai D, Palaniappan B. Role of tau protein in Alzheimer's disease: the prime pathological player. Int J Biol Macromol. 2020;163:1599‐1617. [DOI] [PubMed] [Google Scholar]
- 137. Medeiros R, Baglietto‐Vargas D, LaFerla FM. The role of tau in Alzheimer's disease and related disorders. CNS Neurosci Ther. 2011;17(5):514‐524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284(19):12845‐12852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Caceres A, Kosik KS. Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature. 1990;343(6257):461‐463. [DOI] [PubMed] [Google Scholar]
- 140. Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E. Overexpression of tau protein inhibits kinesin‐dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease. J Cell Biol. 1998;143(3):777‐794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol. 2002;156(6):1051‐1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Buee L, Bussiere T, Buee‐Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33(1):95‐130. [DOI] [PubMed] [Google Scholar]
- 143. Alonso AC, Grundke‐Iqbal I, Iqbal K. Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med. 1996;2(7):783‐787. [DOI] [PubMed] [Google Scholar]
- 144. Hampel H, Buerger K, Zinkowski R, et al. Measurement of phosphorylated tau epitopes in the differential diagnosis of Alzheimer disease: a comparative cerebrospinal fluid study. Arch Gen Psychiatry. 2004;61(1):95‐102. [DOI] [PubMed] [Google Scholar]
- 145. Alonso AC, Zaidi T, Grundke‐Iqbal I, Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91(12):5562‐5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Shafiei SS, Guerrero‐Munoz MJ, Castillo‐Carranza DL. Tau Oligomers: cytotoxicity, Propagation, and Mitochondrial Damage. Front Aging Neurosci. 2017;9:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Aschenbrenner AJ, Gordon BA, Benzinger TLS, Morris JC, Hassenstab JJ. Influence of tau PET, amyloid PET, and hippocampal volume on cognition in Alzheimer disease. Neurology. 2018;91(9):e859‐e866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Sperling RA, Mormino EC, Schultz AP, et al. The impact of amyloid‐beta and tau on prospective cognitive decline in older individuals. Ann Neurol. 2019;85(2):181‐193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Hanseeuw BJ, Betensky RA, Jacobs HIL, et al. Association of Amyloid and Tau With Cognition in Preclinical Alzheimer Disease: a Longitudinal Study. JAMA Neurol. 2019. 10.1001/jamaneurol.2019.1424. Jun 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Nelson PT, Jicha GA, Schmitt FA, et al. Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: neuritic plaques and neurofibrillary tangles “do count” when staging disease severity. J Neuropathol Exp Neurol. 2007;66(12):1136‐1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Soto C. In vivo spreading of tau pathology. Neuron. 2012;73(4):621‐623. [DOI] [PubMed] [Google Scholar]
- 152. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol. 1991;82(4):239‐259. [DOI] [PubMed] [Google Scholar]
- 153. Bittar A, Bhatt N, Kayed R. Advances and considerations in AD tau‐targeted immunotherapy. Neurobiol Dis. 2020;134:104707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Ng PY, Chang IS, Koh RY, Chye SM. Recent advances in tau‐directed immunotherapy against Alzheimer's disease: an overview of pre‐clinical and clinical development. Metab Brain Dis. 2020;35(7):1049‐1066. [DOI] [PubMed] [Google Scholar]
- 155. Heneka MT, Ramanathan M, Jacobs AH, et al. Locus ceruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. J Neurosci. 2006;26(5):1343‐1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Iversen LL, Rossor MN, Reynolds GP, et al. Loss of pigmented dopamine‐beta‐hydroxylase positive cells from locus coeruleus in senile dementia of Alzheimer's type. Neurosci Lett. 1983;39(1):95‐100. [DOI] [PubMed] [Google Scholar]
- 157. Zarow C, Lyness SA, Mortimer JA, Chui HC. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol. 2003;60(3):337‐341. [DOI] [PubMed] [Google Scholar]
- 158. Weinshenker D. Functional consequences of locus coeruleus degeneration in Alzheimer's disease. Current Alzheimer Research. 2008;5(3):342‐345. [DOI] [PubMed] [Google Scholar]
- 159. Liu L, Luo S, Zeng L, Wang W, Yuan L, Jian X. Degenerative alterations in noradrenergic neurons of the locus coeruleus in Alzheimer's disease. Neural regeneration research. 2013;8(24):2249‐2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Kelly SC, He B, Perez SE, Ginsberg SD, Mufson EJ, Counts SE. Locus coeruleus cellular and molecular pathology during the progression of Alzheimer's disease. Acta Neuropathol Commun. 2017;5(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Mather M, Harley CW. The Locus Coeruleus: essential for Maintaining Cognitive Function and the Aging Brain. Trends Cogn Sci. 2016;20(3):214‐226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Aston‐Jones G, Waterhouse B. Locus coeruleus: from global projection system to adaptive regulation of behavior. Brain Res. 2016;1645:75‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Berridge CW, Waterhouse BD. The locus coeruleus‐noradrenergic system: modulation of behavioral state and state‐dependent cognitive processes. Brain Res Brain Res Rev. 2003;42(1):33‐84. [DOI] [PubMed] [Google Scholar]
- 164. Rodenkirch C, Liu Y, Schriver BJ, Wang Q. Locus coeruleus activation enhances thalamic feature selectivity via norepinephrine regulation of intrathalamic circuit dynamics. Nat Neurosci. 2019;22(1):120‐133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Totah NK, Neves RM, Panzeri S, Logothetis NK, Eschenko O. The Locus Coeruleus Is a Complex and Differentiated Neuromodulatory System. Neuron. 2018;99(5):1055‐1068.e6. [DOI] [PubMed] [Google Scholar]
- 166. Liu Y, Rodenkirch C, Moskowitz N, Schriver B, Wang Q. Dynamic Lateralization of Pupil Dilation Evoked by Locus Coeruleus Activation Results from Sympathetic, Not Parasympathetic, Contributions. Cell Rep. 2017;20(13):3099‐3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Joshi S, Li Y, Kalwani RM, Gold JI. Relationships between Pupil Diameter and Neuronal Activity in the Locus Coeruleus, Colliculi, and Cingulate Cortex. Neuron. 2016;89(1):221‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Sara SJ, Bouret S. Orienting and reorienting: the locus coeruleus mediates cognition through arousal. Neuron. 2012;76(1):130‐141. [DOI] [PubMed] [Google Scholar]
- 169. Chamberlain SR, Robbins TW. Noradrenergic modulation of cognition: therapeutic implications. J Psychopharmacol. 2013;27(8):694‐718. [DOI] [PubMed] [Google Scholar]
- 170. Gannon M, Wang Q. Complex noradrenergic dysfunction in Alzheimer's disease: low norepinephrine input is not always to blame. Brain Res. 2019;1702:12‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Oleskevich S, Descarries L, Lacaille JC. Quantified distribution of the noradrenaline innervation in the hippocampus of adult rat. J Neurosci. 1989;9(11):3803‐3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Loughlin SE, Foote SL, Fallon JH. Locus coeruleus projections to cortex: topography, morphology and collateralization. Brain Res Bull. 1982;9(1‐6):287‐294. [DOI] [PubMed] [Google Scholar]
- 173. Hussain LS, Reddy V, Maani CV. Physiology, Noradrenergic Synapse. StatPearls. 2020. [PubMed] [Google Scholar]
- 174. Kaenmaki M, Tammimaki A, Myohanen T, et al. Quantitative role of COMT in dopamine clearance in the prefrontal cortex of freely moving mice. J Neurochem. 2010;114(6):1745‐1755. [DOI] [PubMed] [Google Scholar]
- 175. Axelrod J, Tomchick R. Enzymatic O‐methylation of epinephrine and other catechols. J Biol Chem. 1958;233(3):702‐705. [PubMed] [Google Scholar]
- 176. Karoum F, Chrapusta SJ, Egan MF. 3‐Methoxytyramine is the major metabolite of released dopamine in the rat frontal cortex: reassessment of the effects of antipsychotics on the dynamics of dopamine release and metabolism in the frontal cortex, nucleus accumbens, and striatum by a simple two pool model. J Neurochem. 1994;63(3):972‐979. [DOI] [PubMed] [Google Scholar]
- 177. Rivett AJ, Eddy BJ, Roth JA. Contribution of sulfate conjugation, deamination, and O‐methylation to metabolism of dopamine and norepinephrine in human brain. J Neurochem. 1982;39(4):1009‐1016. [DOI] [PubMed] [Google Scholar]
- 178. Gannon M, Che P, Chen Y, Jiao K, Roberson ED, Wang Q. Noradrenergic dysfunction in Alzheimer's disease. Front Neurosci. 2015;9:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Wang D, Fu Q, Zhou Y, et al. beta2 adrenergic receptor, protein kinase A (PKA) and c‐Jun N‐terminal kinase (JNK) signaling pathways mediate tau pathology in Alzheimer disease models. J Biol Chem. 2013;288(15):10298‐10307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Szot P, White SS, Greenup JL, Leverenz JB, Peskind ER, Raskind MA. Changes in adrenoreceptors in the prefrontal cortex of subjects with dementia: evidence of compensatory changes. Neuroscience. 2007;146(1):471‐480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Chen Y, Peng Y, Che P, et al. alpha(2A) adrenergic receptor promotes amyloidogenesis through disrupting APP‐SorLA interaction. Proc Natl Acad Sci U S A. 2014;111(48):17296‐17301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Saunders C, Limbird LE. Localization and trafficking of alpha2‐adrenergic receptor subtypes in cells and tissues. Pharmacol Ther. 1999;84(2):193‐205. [DOI] [PubMed] [Google Scholar]
- 183. Madrigal JL, Garcia‐Bueno B, Hinojosa AE, Polak P, Feinstein DL, Leza JC. Regulation of MCP‐1 production in brain by stress and noradrenaline‐modulating drugs. J Neurochem. 2010;113(2):543‐551. [DOI] [PubMed] [Google Scholar]
- 184. Gibbs ME, Maksel D, Gibbs Z, Hou X, Summers RJ, Small DH. Memory loss caused by beta‐amyloid protein is rescued by a beta(3)‐adrenoceptor agonist. Neurobiol Aging. 2010;31(4):614‐624. [DOI] [PubMed] [Google Scholar]
- 185. Li S, Jin M, Zhang D, et al. Environmental novelty activates beta2‐adrenergic signaling to prevent the impairment of hippocampal LTP by Abeta oligomers. Neuron. 2013;77(5):929‐941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Kalaria RN, Andorn AC, Tabaton M, Whitehouse PJ, Harik SI, Unnerstall JR. Adrenergic receptors in aging and Alzheimer's disease: increased beta 2‐receptors in prefrontal cortex and hippocampus. J Neurochem. 1989;53(6):1772‐1781. [DOI] [PubMed] [Google Scholar]
- 187. Tan KS, Nackley AG, Satterfield K, Maixner W, Diatchenko L, Flood PM. Beta2 adrenergic receptor activation stimulates pro‐inflammatory cytokine production in macrophages via PKA‐ and NF‐kappaB‐independent mechanisms. Cell Signal. 2007;19(2):251‐260. [DOI] [PubMed] [Google Scholar]
- 188. Yu NN, Wang XX, Yu JT, et al. Blocking beta2‐adrenergic receptor attenuates acute stress‐induced amyloid beta peptides production. Brain Res. 2010;1317:305‐310. [DOI] [PubMed] [Google Scholar]
- 189. Gulyas B, Brockschnieder D, Nag S, et al. The norepinephrine transporter (NET) radioligand (S,S)‐[18F]FMeNER‐D2 shows significant decreases in NET density in the human brain in Alzheimer's disease: a post‐mortem autoradiographic study. Neurochem Int. 2010;56(6‐7):789‐798. [DOI] [PubMed] [Google Scholar]
- 190. Zhang F, Gannon M, Chen Y, et al. beta‐amyloid redirects norepinephrine signaling to activate the pathogenic GSK3beta/tau cascade. Sci Transl Med. 2020;12(526). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Jardanhazi‐Kurutz D, Kummer MP, Terwel D, et al. Induced LC degeneration in APP/PS1 transgenic mice accelerates early cerebral amyloidosis and cognitive deficits. Neurochem Int. 2010;57(4):375‐382. [DOI] [PubMed] [Google Scholar]
- 192. Kalinin S, Gavrilyuk V, Polak PE, et al. Noradrenaline deficiency in brain increases beta‐amyloid plaque burden in an animal model of Alzheimer's disease. Neurobiol Aging. 2007;28(8):1206‐1214. [DOI] [PubMed] [Google Scholar]
- 193. Jardanhazi‐Kurutz D, Kummer MP, Terwel D, Vogel K, Thiele A, Heneka MT. Distinct adrenergic system changes and neuroinflammation in response to induced locus ceruleus degeneration in APP/PS1 transgenic mice. Neuroscience. 2011;176:396‐407. [DOI] [PubMed] [Google Scholar]
- 194. Heneka MT, Nadrigny F, Regen T, et al. Locus ceruleus controls Alzheimer's disease pathology by modulating microglial functions through norepinephrine. Proc Natl Acad Sci U S A. 2010;107(13):6058‐6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Liu X, Ye K, Weinshenker D. Norepinephrine Protects against Amyloid‐beta Toxicity via TrkB. J Alzheimers Dis. 2015;44(1):251‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Perry EK, Tomlinson BE, Blessed G, Perry RH, Cross AJ, Crow TJ. Neuropathological and biochemical observations on the noradrenergic system in Alzheimer's disease. J Neurol Sci. 1981;51(2):279‐287. [DOI] [PubMed] [Google Scholar]
- 197. Szabo ST, de Montigny C, Blier P. Progressive attenuation of the firing activity of locus coeruleus noradrenergic neurons by sustained administration of selective serotonin reuptake inhibitors. Int J Neuropsychopharmacol. 2000;3(1):1‐11. [DOI] [PubMed] [Google Scholar]
- 198. Kalinin S, Feinstein DL, Xu HL, Huesa G, Pelligrino DA, Galea E. Degeneration of noradrenergic fibres from the locus coeruleus causes tight‐junction disorganisation in the rat brain. Eur J Neurosci. 2006;24(12):3393‐3400. [DOI] [PubMed] [Google Scholar]
- 199. Espay AJ, LeWitt PA, Kaufmann H. Norepinephrine deficiency in Parkinson's disease: the case for noradrenergic enhancement. Mov Disord. 2014;29(14):1710‐1719. [DOI] [PubMed] [Google Scholar]
- 200. Weinshenker D. Long Road to Ruin: noradrenergic Dysfunction in Neurodegenerative Disease. Trends Neurosci. 2018;41(4):211‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Lyness SA, Zarow C, Chui HC. Neuron loss in key cholinergic and aminergic nuclei in Alzheimer disease: a meta‐analysis. Neurobiol Aging. 2003;24(1):1‐23. [DOI] [PubMed] [Google Scholar]
- 202. Amaral DG, Sinnamon HM. The locus coeruleus: neurobiology of a central noradrenergic nucleus. Prog Neurobiol. 1977;9(3):147‐196. [DOI] [PubMed] [Google Scholar]
- 203. Wang Q, Oyarzabal EA, Song S, Wilson B, Santos JH, Hong JS. Locus coeruleus neurons are most sensitive to chronic neuroinflammation‐induced neurodegeneration. Brain Behav Immun. 2020;87:359‐368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Salim S, Asghar M, Taneja M, et al. Potential contribution of oxidative stress and inflammation to anxiety and hypertension. Brain Res. 2011;1404:63‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Sanchez‐Padilla J, Guzman JN, Ilijic E, et al. Mitochondrial oxidant stress in locus coeruleus is regulated by activity and nitric oxide synthase. Nat Neurosci. 2014;17(6):832‐840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Kang SS, Liu X, Ahn EH, et al. Norepinephrine metabolite DOPEGAL activates AEP and pathological Tau aggregation in locus coeruleus. J Clin Invest. 2020;130(1):422‐437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Pamphlett R. Uptake of environmental toxicants by the locus ceruleus: a potential trigger for neurodegenerative, demyelinating and psychiatric disorders. Med Hypotheses. 2014;82(1):97‐104. [DOI] [PubMed] [Google Scholar]
- 208. MacKenzie ET, McCulloch J, O'Kean M, Pickard JD, Harper AM. Cerebral circulation and norepinephrine: relevance of the blood‐brain barrier. Am J Physiol. 1976;231(2):483‐488. [DOI] [PubMed] [Google Scholar]
- 209. MacKenzie ET, McCulloch J, Harper AM. Influence of endogenous norepinephrine on cerebral blood flow and metabolism. Am J Physiol. 1976;231(2):489‐494. [DOI] [PubMed] [Google Scholar]
- 210. Cryan JF, O'Riordan KJ, Cowan CSM, et al. The Microbiota‐Gut‐Brain Axis. Physiol Rev. 2019;99(4):1877‐2013. [DOI] [PubMed] [Google Scholar]
- 211. Doifode T, Giridharan VV, Generoso JS, et al. The impact of the microbiota‐gut‐brain axis on Alzheimer's disease pathophysiology. Pharmacol Res. 2021;164:105314. [DOI] [PubMed] [Google Scholar]
- 212. Kohler CA, Maes M, Slyepchenko A, et al. The Gut‐Brain Axis, Including the Microbiome, Leaky Gut and Bacterial Translocation: mechanisms and Pathophysiological Role in Alzheimer's Disease. Curr Pharm Des. 2016;22(40):6152‐6166. [DOI] [PubMed] [Google Scholar]
- 213. Generoso JS, Giridharan VV, Lee J, Macedo D, Barichello T. The role of the microbiota‐gut‐brain axis in neuropsychiatric disorders. Braz J Psychiatry. 2020. 10.1590/1516‐4446‐2020‐0987. Jul 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Ambrosini YM, Borcherding D, Kanthasamy A, et al. The Gut‐Brain Axis in Neurodegenerative Diseases and Relevance of the Canine Model: a Review. Front Aging Neurosci. 2019;11:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Bonaz B. Is‐there a place for vagus nerve stimulation in inflammatory bowel diseases?. Bioelectron Med. 2018;4:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Bonaz B, Bazin T, Pellissier S. The Vagus Nerve at the Interface of the Microbiota‐Gut‐Brain Axis. Front Neurosci. 2018;12:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Chalermpalanupap T, Kinkead B, Hu WT, et al. Targeting norepinephrine in mild cognitive impairment and Alzheimer's disease. Alzheimer's Research & Therapy. 2013;5(2):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Martignoni E, Blandini F, Petraglia F, Pacchetti C, Bono G, Nappi G. Cerebrospinal fluid norepinephrine, 3‐methoxy‐4‐hydroxyphenylglycol and neuropeptide Y levels in Parkinson's disease, multiple system atrophy and dementia of the Alzheimer type. J Neural Transm Park Dis Dement Sect. 1992;4(3):191‐205. [DOI] [PubMed] [Google Scholar]
- 219. Ross JA, McGonigle P, Van Bockstaele EJ. Locus Coeruleus, norepinephrine and Abeta peptides in Alzheimer's disease. Neurobiol Stress. 2015;2:73‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Jacobs HIL, Riphagen JM, Ramakers I, Verhey FRJ. Alzheimer's disease pathology: pathways between central norepinephrine activity, memory, and neuropsychiatric symptoms. Mol Psychiatry. 2019. 10.1038/s41380-019-0437-x. May 28. [DOI] [PubMed] [Google Scholar]
- 221. Wahle T, Thal DR, Sastre M, et al. GGA1 is expressed in the human brain and affects the generation of amyloid beta‐peptide. J Neurosci. 2006;26(49):12838‐12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Savitz SI, Erhardt JA, Anthony JV, et al. The novel beta‐blocker, carvedilol, provides neuroprotection in transient focal stroke. J Cereb Blood Flow Metab. 2000;20(8):1197‐1204. [DOI] [PubMed] [Google Scholar]
- 223. Wang J, Wright HM, Vempati P, et al. Investigation of nebivolol as a novel therapeutic agent for the treatment of Alzheimer's disease. J Alzheimers Dis. 2013;33(4):1147‐1156. [DOI] [PubMed] [Google Scholar]
- 224. Sara SJ. The locus coeruleus and noradrenergic modulation of cognition. Nat Rev Neurosci. 2009;10(3):211‐223. [DOI] [PubMed] [Google Scholar]
- 225. Manzoor S, Hoda N. A Comprehensive Review of Monoamine Oxidase Inhibitors as Anti‐Alzheimer's Disease Agents: a Review. Eur J Med Chem. 2020;206:112787. [DOI] [PubMed] [Google Scholar]
- 226. Birks J, Flicker L. Selegiline for Alzheimer's disease. Cochrane Database Syst Rev. 2003(1):Cd000442. 10.1002/14651858.Cd000442. [DOI] [PubMed] [Google Scholar]
- 227. Chaurasiya ND, Ibrahim MA, Muhammad I, Walker LA, Tekwani BL. Monoamine oxidase inhibitory constituents of propolis: kinetics and mechanism of inhibition of recombinant human MAO‐A and MAO‐B. Molecules. 2014;19(11):18936‐18952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Yamada M, Yasuhara H. Clinical pharmacology of MAO inhibitors: safety and future. Neurotoxicology. 2004;25(1‐2):215‐221. [DOI] [PubMed] [Google Scholar]
- 229. Lanctot KL, Chau SA, Herrmann N, et al. Effect of methylphenidate on attention in apathetic AD patients in a randomized, placebo‐controlled trial. Int Psychogeriatr. 2014;26(2):239‐246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Dolder CR, Davis LN, McKinsey J. Use of psychostimulants in patients with dementia. Ann Pharmacother. 2010;44(10):1624‐1632. [DOI] [PubMed] [Google Scholar]
- 231. Bymaster FP, Katner JS, Nelson DL, et al. Atomoxetine increases extracellular levels of norepinephrine and dopamine in prefrontal cortex of rat: a potential mechanism for efficacy in attention deficit/hyperactivity disorder. Neuropsychopharmacology. 2002;27(5):699‐711. [DOI] [PubMed] [Google Scholar]
- 232. Elmaleh DR, Farlow MR, Conti PS, Tompkins RG, Kundakovic L, Tanzi RE. Developing Effective Alzheimer's Disease Therapies: clinical Experience and Future Directions. J Alzheimers Dis. 2019;71(3):715‐732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Rorabaugh JM, Chalermpalanupap T, Botz‐Zapp CA, et al. Chemogenetic locus coeruleus activation restores reversal learning in a rat model of Alzheimer's disease. Brain. 2017;140(11):3023‐3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Merrill CA, Jonsson MA, Minthon L, et al. Vagus nerve stimulation in patients with Alzheimer's disease: additional follow‐up results of a pilot study through 1 year. J Clin Psychiatry. 2006;67(8):1171‐1178. [DOI] [PubMed] [Google Scholar]
- 235. Lin YC, Wang YP. Status of Noninvasive Brain Stimulation in the Therapy of Alzheimer's Disease. Chin Med J (Engl). 2018;131(24):2899‐2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Sara SJ, Dyon‐Laurent C, Guibert B, Leviel V. Noradrenergic hyperactivity after partial fornix section: role in cholinergic dependent memory performance. Exp Brain Res. 1992;89(1):125‐132. [DOI] [PubMed] [Google Scholar]
- 237. Levin ED, McGurk SR, Rose JE, Butcher LL. Cholinergic‐dopaminergic interactions in cognitive performance. Behav Neural Biol. 1990;54(3):271‐299. [DOI] [PubMed] [Google Scholar]
- 238. Beani L, Bianchi C, Tanganelli S, Antonelli T, Simonato M, Rando S. Inversion of the alpha‐2 and alpha‐1 noradrenergic control of the cortical release of acetylcholine and gamma‐aminobutyric acid in morphine‐tolerant guinea pigs. J Pharmacol Exp Ther. 1988;247(1):294‐301. [PubMed] [Google Scholar]
- 239. Vizi ES. Modulation of cortical release of acetylcholine by noradrenaline released from nerves arising from the rat locus coeruleus. Neuroscience. 1980;5(12):2139‐2144. [DOI] [PubMed] [Google Scholar]
- 240. Dalmaz C, Introini‐Collison IB, McGaugh JL. Noradrenergic and cholinergic interactions in the amygdala and the modulation of memory storage. Behav Brain Res. 1993;58(1‐2):167‐174. [DOI] [PubMed] [Google Scholar]
- 241. Mineur YS, Cahuzac EL, Mose TN, et al. Interaction between noradrenergic and cholinergic signaling in amygdala regulates anxiety‐ and depression‐related behaviors in mice. Neuropsychopharmacology. 2018;43(10):2118‐2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Xia F, Yiu A, Stone SSD, et al. Entorhinal Cortical Deep Brain Stimulation Rescues Memory Deficits in Both Young and Old Mice Genetically Engineered to Model Alzheimer's Disease. Neuropsychopharmacology. 2017;42(13):2493‐2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Zhang C, Hu WH, Wu DL, Zhang K, Zhang JG. Behavioral effects of deep brain stimulation of the anterior nucleus of thalamus, entorhinal cortex and fornix in a rat model of Alzheimer's disease. Chin Med J (Engl). 2015;128(9):1190‐1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Krahl SE, Clark KB, Smith DC, Browning RA. Locus coeruleus lesions suppress the seizure‐attenuating effects of vagus nerve stimulation. Epilepsia. 1998;39(7):709‐714. [DOI] [PubMed] [Google Scholar]
- 245. Hulsey DR, Hays SA, Khodaparast N, et al. Reorganization of Motor Cortex by Vagus Nerve Stimulation Requires Cholinergic Innervation. Brain Stimul. 2016;9(2):174‐181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Rodenkirch C, Wang Q. Rapid and transient enhancement of thalamic information transmission induced by vagus nerve stimulation. J Neural Eng. 2020;17(2):026027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Liu CH, Yang MH, Zhang GZ, et al. Neural networks and the anti‐inflammatory effect of transcutaneous auricular vagus nerve stimulation in depression. J Neuroinflammation. 2020;17(1):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. 2016;353(6301):777‐783. [DOI] [PubMed] [Google Scholar]
- 249. Huffman WJ, Subramaniyan S, Rodriguiz RM, Wetsel WC, Grill WM, Terrando N. Modulation of neuroinflammation and memory dysfunction using percutaneous vagus nerve stimulation in mice. Brain Stimul. 2019;12(1):19‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Kaczmarczyk R, Tejera D, Simon BJ, Heneka MT. Microglia modulation through external vagus nerve stimulation in a murine model of Alzheimer's disease. J Neurochem. 2017. 10.1111/jnc.14284. Dec 21. [DOI] [PubMed] [Google Scholar]
- 251. Zhang Q, Lu Y, Bian H, Guo L, Zhu H. Activation of the alpha7 nicotinic receptor promotes lipopolysaccharide‐induced conversion of M1 microglia to M2. Am J Transl Res. 2017;9(3):971‐985. [PMC free article] [PubMed] [Google Scholar]
- 252. Zou J, Tao S, Johnson A, et al. Microglial activation, but not tau pathology, is independently associated with amyloid positivity and memory impairment. Neurobiol Aging. 2020;85:11‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Morishita K, Costantini TW, Eliceiri B, Bansal V, Coimbra R. Vagal nerve stimulation modulates the dendritic cell profile in posthemorrhagic shock mesenteric lymph. J Trauma Acute Care Surg. 2014;76(3):610‐617.discussion 617–8. [DOI] [PubMed] [Google Scholar]
- 254. Lowry DM, Morishita K, Eliceiri BP, Bansal V, Coimbra R, Costantini TW. The vagus nerve alters the pulmonary dendritic cell response to injury. J Surg Res. 2014;192(1):12‐18. [DOI] [PubMed] [Google Scholar]
- 255. Biggio F, Gorini G, Utzeri C, et al. Chronic vagus nerve stimulation induces neuronal plasticity in the rat hippocampus. Int J Neuropsychopharmacol. 2009;12(9):1209‐1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Grimonprez A, Raedt R, Baeken C, Boon P, Vonck K. The antidepressant mechanism of action of vagus nerve stimulation: evidence from preclinical studies. Neurosci Biobehav Rev. 2015;56:26‐34. [DOI] [PubMed] [Google Scholar]
- 257. Roosevelt RW, Smith DC, Clough RW, Jensen RA, Browning RA. Increased extracellular concentrations of norepinephrine in cortex and hippocampus following vagus nerve stimulation in the rat. Brain Res. 2006;1119(1):124‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Henry TR, Votaw JR, Pennell PB, et al. Acute blood flow changes and efficacy of vagus nerve stimulation in partial epilepsy. Neurology. 1999;52(6):1166‐1173. [DOI] [PubMed] [Google Scholar]
- 259. Doze VA, Papay RS, Goldenstein BL, et al. Long‐term alpha1A‐adrenergic receptor stimulation improves synaptic plasticity, cognitive function, mood, and longevity. Mol Pharmacol. 2011;80(4):747‐758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Shimohama S, Taniguchi T, Fujiwara M, Kameyama M. Biochemical characterization of alpha‐adrenergic receptors in human brain and changes in Alzheimer‐type dementia. J Neurochem. 1986;47(4):1295‐1301. [PubMed] [Google Scholar]
- 261. Kalaria RN. Characterization of [125I]HEAT binding to alpha 1‐receptors in human brain: assessment in aging and Alzheimer's disease. Brain Res. 1989;501(2):287‐294. [DOI] [PubMed] [Google Scholar]
- 262. Nalepa I, Kreiner G, Bielawski A, Rafa‐Zablocka K, Roman A. alpha1‐Adrenergic receptor subtypes in the central nervous system: insights from genetically engineered mouse models. Pharmacol Rep. 2013;65(6):1489‐1497. [DOI] [PubMed] [Google Scholar]
- 263. Mishima K, Tanoue A, Tsuda M, et al. Characteristics of behavioral abnormalities in alpha1d‐adrenoceptors deficient mice. Behav Brain Res. 2004;152(2):365‐373. [DOI] [PubMed] [Google Scholar]
- 264. Gamache K, Pitman RK, Nader K. Preclinical evaluation of reconsolidation blockade by clonidine as a potential novel treatment for posttraumatic stress disorder. Neuropsychopharmacology. 2012;37(13):2789‐2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Zoladz PR, Fleshner M, Diamond DM. Differential effectiveness of tianeptine, clonidine and amitriptyline in blocking traumatic memory expression, anxiety and hypertension in an animal model of PTSD. Prog Neuropsychopharmacol Biol Psychiatry. 2013;44:1‐16. [DOI] [PubMed] [Google Scholar]
- 266. Warner TA, Drugan RC. Morris water maze performance deficit produced by intermittent swim stress is partially mediated by norepinephrine. Pharmacol Biochem Behav. 2012;101(1):24‐34. [DOI] [PubMed] [Google Scholar]
- 267. Szot P, White SS, Greenup JL, Leverenz JB, Peskind ER, Raskind MA. Compensatory changes in the noradrenergic nervous system in the locus ceruleus and hippocampus of postmortem subjects with Alzheimer's disease and dementia with Lewy bodies. J Neurosci. 2006;26(2):467‐478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Ruiz J, Martin I, Callado LF, Meana JJ, Barturen F, Garcia‐Sevilla JA. Non‐adrenoceptor [3H]idazoxan binding sites (I2‐imidazoline sites) are increased in postmortem brain from patients with Alzheimer's disease. Neurosci Lett. 1993;160(1):109‐112. [DOI] [PubMed] [Google Scholar]
- 269. Kalaria RN, Stockmeier CA, Harik SI. Brain microvessels are innervated by locus ceruleus noradrenergic neurons. Neurosci Lett. 1989;97(1‐2):203‐208. [DOI] [PubMed] [Google Scholar]
- 270. Russo‐Neustadt A, Cotman CW. Adrenergic receptors in Alzheimer's disease brain: selective increases in the cerebella of aggressive patients. J Neurosci. 1997;17(14):5573‐5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Lemmer B, Langer L, Ohm T, Bohl J. Beta‐adrenoceptor density and subtype distribution in cerebellum and hippocampus from patients with Alzheimer's disease. Naunyn Schmiedebergs Arch Pharmacol. 1993;347(2):214‐219. [DOI] [PubMed] [Google Scholar]
- 272. Lemon N, Aydin‐Abidin S, Funke K, Manahan‐Vaughan D. Locus coeruleus activation facilitates memory encoding and induces hippocampal LTD that depends on beta‐adrenergic receptor activation. Cereb Cortex. 2009;19(12):2827‐2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Kalaria RN, Harik SI. Increased alpha 2‐ and beta 2‐adrenergic receptors in cerebral microvessels in Alzheimer disease. Neurosci Lett. 1989;106(1‐2):233‐238. [DOI] [PubMed] [Google Scholar]
- 274. Introini‐Collison IB, Miyazaki B, McGaugh JL. Involvement of the amygdala in the memory‐enhancing effects of clenbuterol. Psychopharmacology (Berl). 1991;104(4):541‐544. [DOI] [PubMed] [Google Scholar]
- 275. Shimohama S, Taniguchi T, Fujiwara M, Kameyama M. Changes in beta‐adrenergic receptor subtypes in Alzheimer‐type dementia. J Neurochem. 1987;48(4):1215‐1221. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Table S2