

Abstract
Introduction
Synaptic dysfunction and degeneration is one of the earliest events in Alzheimer's disease (AD) and the best correlate of cognitive decline. Thus, identification and validation of biomarkers reflecting synaptic degeneration to be used as prognostic biomarkers are greatly needed.
Method
Solid‐phase extraction and parallel reaction monitoring mass spectrometry were used to quantify 17 synaptic proteins in CSF, in two cross‐sectional studies including AD (n = 52) and controls (n = 37).
Results
Increased concentrations of beta‐synuclein, gamma‐synuclein, neurogranin, phosphatidylethanolamine‐binding protein 1, and 14‐3‐3 proteins were observed in AD patients compared to controls, while neuronal pentraxin‐2 and neuronal pentraxin receptor were decreased.
Discussion
We have established a method with a novel panel of synaptic proteins as biomarkers of synaptic dysfunction. The results indicate that several of the proteins included in the panel may serve as synaptic biomarkers for AD.
Keywords: Alzheimer's disease, biomarkers, mass spectrometry, synaptic pathology
1. INTRODUCTION
Alzheimer's disease (AD) is clinically characterized by gradual memory dysfunction and other cognitive decline. Central to the disease pathogenesis is the abnormal aggregation and accumulation of extracellular amyloid beta (Aβ) plaques and intracellular neurofibrillary tau tangles. 1 The resulting synapse dysfunction from these pathological events is one of the earliest detectable changes in AD. 2 , 3 Synaptic loss at post mortem also correlates well with degree of cognitive decline and plays a significant role in the disease pathology and progression. 1 In the past 20 years, a multitude of drug trials have been conducted but only four symptomatic treatments have been approved. 4 The reason for this has been partially attributed to overlap in clinical manifestation between neurodegenerative diseases and thus poor diagnostic accuracy in the enrollment of trial participants. 5 To improve diagnostics and to monitor treatment effects, new, pathology‐specific biomarkers of AD are therefore in high demand.
Current methods for quantifying synaptic degeneration include measuring pre‐ and postsynaptic proteins in the cerebrospinal fluid (CSF) or by positron emission tomography ([11C]UCB‐J). 6 , 7 For example, CSF concentrations of both the presynaptic synaptosomal‐associated protein 25 (SNAP‐25) and synaptotagmin‐1 as well as postsynaptic proteins, such as neurogranin, have been shown to be altered in AD, 8 , 9 , 10 even at the mild cognitive impairment (MCI) stage. 11 CSF neurogranin has also been shown to increase soon after Aβ deposition—even in cognitively unimpaired adults. 3 Synaptic density, as determined by [11C]UCB‐J, is reduced in the medial temporal and neocortical brain regions of individuals with AD compared to healthy participants. 7 , 12 The detection and validation of new synaptic proteins as prognostic biomarkers of synaptic degeneration to improve our understanding and monitoring of cognitive decline in AD is of great importance.
In the field of biomarker discovery, mass spectrometry (MS) offers wide possibilities, both as a hypothesis‐generating tool that aims to find potential candidates, or as a confirmatory targeted approach for validation and quantification. With an explorative MS‐based proteomic approach, thousands of proteins can be identified and quantified in a single experiment without preselecting specific target proteins. Thus, potential candidates involved in the disease can be found in the absence of bias and, additionally, candidates that might otherwise have been overlooked could be investigated. For validation of candidate biomarkers, targeted MS is also advantageous as there is no reliance on antibodies compared to immunoassays, and the possibility of multiplexing both saves time and reduces cost.
In this study, we developed, based on a previous explorative proteomic study, 13 a multiplexed, robust, and high‐throughput method, simultaneously targeting 17 synaptic proteins with high sensitivity and specificity. These included syntaxins, vesicle‐associated membrane protein 2 (VAMP‐2), activating protein 2 (AP‐2) adaptor complex proteins, complexin‐2, synucleins, rab GDP dissociation inhibitor alpha (rab GDI alpha), neuronal pentraxins, phosphatidylethanolamine‐binding protein 1 (PEBP‐1), and members of the 14‐3‐3 protein family. Thus, parallel reaction monitoring (PRM) with high‐resolution MS coupled to liquid chromatography (LC) was used to quantify synaptic proteins to establish a synaptic panel assay for biomarker validation.
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the available scientific literature with a focus on biomarkers for synaptic dysfunction in Alzheimer's disease (AD). The detection and validation of new synaptic proteins as prognostic biomarkers of synaptic degeneration to improve our understanding and monitoring of cognitive decline in AD is of great importance. Therefore, in this study, we developed a biomarker panel of several synaptic proteins and quantified them in two cross‐sectional studies to be able to study their potential as biomarkers of synaptic dysfunction.
Interpretation: Our findings suggest that several proteins in the synaptic biomarker panel are potential biomarkers of AD, especially beta‐synuclein, gamma‐synuclein, neurogranin, phosphatidylethanolamine‐binding protein 1, 14‐3‐3 proteins, and the neuronal pentraxins.
Future directions: Further studies in larger cohorts will be needed to validate the synaptic biomarkers. These include other neurodegenerative diseases to assert the specificity of the biomarkers for AD.
HIGHLIGHTS
We established a novel panel assay to study 17 synaptic proteins in 100 µL CSF.
Several synaptic proteins were altered in AD compared with healthy controls.
14‐3‐3 proteins and β‐synuclein performed best at separating AD from controls.
Neuronal pentraxin‐2 in particular seems to correlate with cognitive decline.
2. METHOD
2.1. Biomarker candidate selection
In an explorative CSF proteomic study by Tijms et al. comparing AD to controls and using tandem mass tag (TMT) multiplex quantification, 2000 proteins were identified and quantified. 13 Based on the study by Tijms et al. and a literature review, 17 synaptic proteins were deemed to have potential as CSF biomarkers of synapse degeneration in AD and thus selected in our study for further evaluation (see Appendix A and Figure SA1 in supporting information, where the data are presented together with a brief method description and cohort demographics). In Table 1, the proteins and their respective proteotypic peptides selected for PRM‐MS analysis are shown.
TABLE 1.
The 17 synaptic proteins and their respective peptides selected for validation as potential synaptic biomarkers
| Protein | Accession | Sequence | Position a |
|---|---|---|---|
| 14‐3‐3 protein epsilon | P62258 | IISSIEQK | [62‐69] |
| LICCDILDVLDK | [95‐106] | ||
| 14‐3‐3 protein eta | Q04917 | AVTELNEPLSNEDR | [29‐42] |
| 14‐3‐3 protein theta | P27348 | AVTEQGAELSNEER | [28‐41] |
| 14‐3‐3 protein zeta/delta | P63104 | VVSSIEQK | [61‐68] |
| AP‐2 complex subunit beta | P63010 | NVEGQDMLYQSLK | [880‐892] |
| IQPGNPNYTLSLK | [905‐917] | ||
| Beta‐synuclein | Q16143 | EGVVQGVASVAEK | [46‐58] |
| Complexin‐2 | Q6PUV4 | AALEQPCEGSLTRPK | [84‐98] |
| Gamma‐synuclein | O76070 | ENVVQSVTSVAEK | [46‐58] |
| EQANAVSEAVVSSVNTVATK | [61‐80] | ||
| Neurogranin | Q92686 | KGPGPGGPGGAGVAR | [54‐68] |
| Neuronal pentraxin receptor | O95502 | NNYMYAR | [302‐308] |
| LVEAFGGATK | [479‐488] | ||
| Neuronal pentraxin‐1 | Q15818 | LENLEQYSR | [144‐152] |
| ETVLQQK | [63‐69] | ||
| CESQSTLDPGAGEAR | [89‐103] | ||
| LTPGEVYNLATCSTK | [385‐400] | ||
| Neuronal pentraxin‐2 | P47972 | VAELEDEK | [177‐184] |
| WPVETCEER | [419‐428] | ||
| ETVVQQK | [68‐74] | ||
| PEBP‐1 | P30086 | LYEQLSGK | [180‐187] |
| NRPTSISWDGLDSGK | [48‐62] | ||
| Rab GDI alpha | P31150 | QLICDPSYIPDR | [279‐290] |
| Syntaxin‐1B | P61266 | QHSAILAAPNPDEK | [56‐69] |
| Syntaxin‐7 | O15400 | EFGSLPTTPSEQR | [72‐84] |
| VAMP‐ 2 | P63027 | LQQTQAQVDEVVDIMR | [32‐47] |
The position reflects the amino acid sequence of the protein.
2.2. Patients and AD biomarker analysis
To study the panel proteins in relation to AD pathophysiology—that is, changed tau and Aβ levels—the pilot study consisted of biochemically defined AD (n = 20) and neurological controls (n = 20). The samples were selected based on concentrations of the AD CSF core biomarkers (tau phosphorylated at Thr181 [p‐tau181, cut‐off >60 pg/mL], total tau [t‐tau, cut‐off >380 pg/mL], and Aβ peptide 1‐42 [Aβ1‐42, cut‐off <550 pg/mL] as defined by the IWG‐2 biomarker criteria). 14 The AD core biomarker analysis and collection of the samples were performed at the Clinical Neurochemistry Laboratory, Mölndal, Sweden. The use of these patients’ samples was approved by the Ethics Committee at University of Gothenburg (EPN 140811).
The second cohort consisted of clinically diagnosed subjects and included AD patients (n = 32) and controls (n = 20) from a single‐center memory clinic, as presented previously. 15 In brief, patients were admitted for exploration of cognitive impairment while control subjects were recruited among spouses and through local advertisement. The healthy control subjects showed no symptoms of cognitive dysfunction and they were matched with the AD patients in terms of age, sex, and body mass index. AD was defined based on clinical evaluations according to National Institute of Neurological and Communicative Disorders and Stroke‐Alzheimer's Disease and Related Disorders Association. 16 Three controls were excluded from the analysis based on abnormally low Aβ1‐42 concentrations (cut‐off, Aβ1‐42 <550 pg/mL). All participants gave informed consent and ethical approval was retrieved from the ethical committee of the University of Gothenburg. The demographics of all cohorts are shown groupwise in Table 2 and in detail in Tables SB1 and SB2 in supporting information.
TABLE 2.
Cohort demographics
| Cohort | Group | n (F/M) | Age | MMSE a | APOE ε4 b (±/NA) | Aβ1‐42 c | P‐tau181 d | T‐tau e |
|---|---|---|---|---|---|---|---|---|
| Pilot | NC f | 20 (10/10) | 66 (19, 59‐80) | 772 (184, 679‐869) | 41 (8, 35‐44) | 253 (63, 231‐328) | ||
| AD | 20 (11/9) | 76 (9, 73‐80) | 442 (108, 321‐518) i | 72 (21, 66‐89) i | 535 (385, 475‐745) i | |||
| Clinical | HC h | 17 (8/9) | 75 (5, 70‐78) | 29 (2, 27‐30) | 4/13/0 | 993 (136, 908‐1046) | 61 (17, 45‐77) | 315 (87, 217‐351) |
| AD | 32 (17/15) | 75 (4, 71‐77) | 23 (4, 19‐25) i | 18/9/5 | 479 (116, 345‐565)i | 98 (33, 78‐113) i | 584 (233, 423‐764) i |
Mini‐Mental State Examination score.
Apolipoprotein E ε4 status.
Amyloid beta protein 1‐42.
Total tau.
Phosphorylated tau at amino acid Thr181.
Neurological controls.
Alzheimer's disease.
Healthy controls.
Two‐sided Mann‐Whitney test, P < .0001 compared to respective control group. Data presented as median (standard deviation, inter‐quartile interval) and in ng/L.
2.3. CSF samples
Lumbar puncture was used for CSF collection in polypropylene tubes. Samples were centrifuged at 2000 × g at 4°C for 10 minutes to remove insoluble material and cells. The supernatants were then aliquoted and stored at –80°C until use. The CSF AD core biomarker concentrations (p‐tau181, t‐tau, and Aβ1‐42) were quantified by commercially available enzyme‐linked immunosorbent assays (INNOTEST β‐AMYLOID [1‐42], INNOTEST hTAU Ag, and INNOTEST PHOSPHO‐TAU [181P]; Fujirebio Europe). Quality controls (QCs) consisted of pooled CSF samples, obtained from the Neurochemistry Laboratory at Sahlgrenska University Hospital, Mölndal, Sweden, and were aliquoted and stored in the same way as the samples. The collection and use are in accordance with the Swedish law on biobanks in healthcare (2002:297).
2.4. LC‐MS/MS analysis
Sample preparation was performed as described previously, 17 for details see Appendix C in supporting information. LC‐MS/MS analysis was performed using a microflow HPLC (Dionex Ultimate 3000, ThermoFisher Scientific) and an Orbitrap hybrid mass spectrometer (Q Exactive, ThermoFisher Scientific). Using a flow rate of 0.3 mL/min, sample injection volume of 45 μL, and a 50‐minute gradient (Figure SB1 in supporting information), separation was performed on a Hypersil Gold reversed phase column (dimension 100 × 2.1 mm, particle size 1.9 μm, ThermoFisher Scientific). Mobile phases consisted of 0.1% formic acid in water (v/v) (A) and 84% acetonitrile/0.1% formic acid in water (v/v) (B). Electrospray conditions were set as follows: spray voltage at +4100 V, capillary temperature at 320°C, sheath gas at 25, aux gas at 10, sweep gas at 0, probe heater temperature at 300°C, and S‐lens RF level to 55. Individually optimized collision energies were used for each peptide (Table SB3 in supporting information). The scheduled PRM method used retention time windows of 2 minutes for each peptide and a toggle limit of four different peptide pairs. For data acquisition, isolation window was set to 3 m/z units, automatic gain control target value to 3 × 106, and maximum injection time to 250 milliseconds with a matching resolution setting of 70,000. To test the repeatability of each peptide, a series of replicates (n = 8) of a QC sample was injected. QC samples were also injected at regular intervals during runs to monitor the performance of the assay over time.
2.5. Data processing and statistical analysis
Data processing was performed in Skyline 20.1 (MacCoss Lab Software). All peaks were visually inspected and adjusted if required for optimal peak area calculation (Figure SB2 in supporting information). Relative peptide abundance was obtained by summing all measured fragment peak areas for each peptide and dividing that by the sum of the fragment peak areas of the corresponding internal standard (IS). The group‐wise comparisons were assessed using two‐sided Mann‐Whitney test, whereas associations between continuous variables were tested with Spearman rank correlation analysis. The receiver operating characteristic curve (ROC) contrasting groups provided the area under the curve (AUC) to evaluate the discriminatory ability of the biomarkers. Statistical analysis and data visualization were performed with GraphPad Prism 8.3.0 (GrahpPad Software, Inc.).
3. RESULTS
3.1. Performance characteristics of PRM method
We have successfully developed a PRM assay to quantify 17 synaptic proteins in CSF, which were identified and selected using explorative TMT proteomics. 13 The method's performance characteristics can be found in Table SB3. The median coefficient of variation (CV) for all peptides was below 12%.
3.2. Pilot study
The CSF pilot study (n = 40) was performed on a cohort defined by the core AD biomarkers. Significant increases in AD compared to neurological controls were observed for several proteins: neurogranin (P ≤ .0001), complexin‐2, VAMP‐2, beta‐synuclein, rab GDI alpha, PEBP‐1, 14‐3‐3 protein epsilon, and 14‐3‐3 protein zeta/delta (all P ≤ .001), as shown in Figure 1. Significant increase was also observed for the syntaxins (P ≤ .01), while no significant difference was observed for the neuronal pentraxins (1, 2 and the receptor) and gamma‐synuclein. Figure SB3 in supporting information demonstrates the results for all peptides analyzed. For some of the proteins a correlation with age was found (Table SB4 in supporting information).
FIGURE 1.
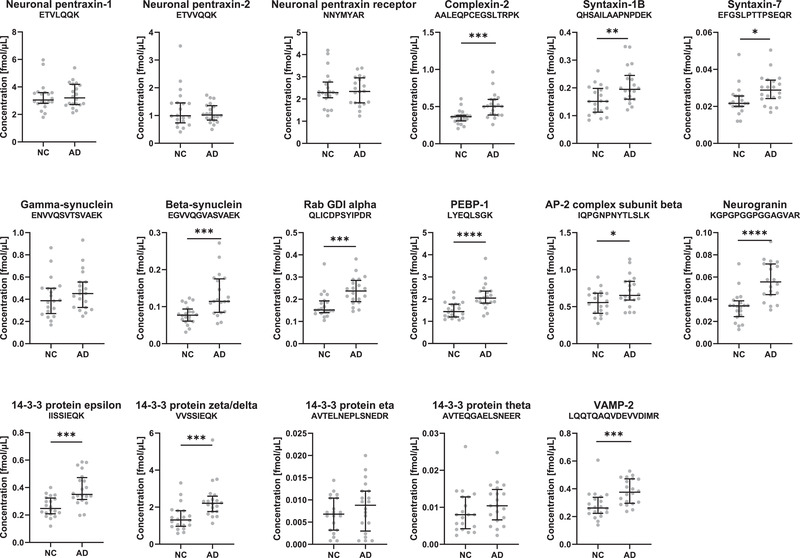
Levels of the synaptic panel proteins in the pilot cohort. Cerebrospinal fluid (CSF) concentrations obtained by parallel reaction monitoring (PRM) analysis of the synaptic panel proteins (one representative peptide for each protein) in the pilot cohort consisting of neurological controls (NC, n = 20) and Alzheimer's disease (AD, n = 20) of a biologically defined cohort. Statistical comparison was performed with Mann‐Whitney test, P‐values: * P ≤ .05, ** P ≤ .01, *** P ≤ .001, and **** P ≤ .0001. The bars indicate median with interquartile range
3.3. Clinical cohort
Next, we aimed to validate our pilot results using a clinical cohort from the memory clinic in Falköping, Sweden (Figure 2A). In support of the pilot study, beta‐synuclein, PEPB‐1, neurogranin, and 14‐3‐3 protein epsilon and zeta/delta were all significantly increased in AD patients compared to controls in the clinical cohort (Figure 2A). In addition, significant decrease in neuronal pentraxin‐2 and the neuronal pentraxin receptor levels were observed (P ≤ .05) as well as significant increase in gamma‐synuclein (P ≤ .05) and the 14‐3‐3 protein eta and theta. Four proteins had AUC above 0.75 (P ≤ .005), beta‐synuclein (AUC = 0.76, 95% confidence interval [CI] = 0.62–0.89), 14‐3‐3 zeta/delta, 14‐3‐3 theta (AUC = 0.77, 95% CI = 0.61–0.92), and 14‐3‐3 epsilon (AUC = 0.79, 95% CI = 0.66–0.92), where the highest AUC was found for 14‐3‐3 zeta/delta (AUC = 0.84, 95% CI = 0.72% to 0.96%, Figure 2B). Note that four peptides, including the only peptide for VAMP‐2, were excluded from the method and not quantified in the clinical cohort due to storage instability of the standards. For the proteins with more than one quantified peptide, one representative peptide is shown. Figure SB4 in supporting information displays the results of the additional peptides and Table SB5 in supporting information demonstrates the CVs of the QCs together with the AUC values for all peptides per protein.
FIGURE 2.
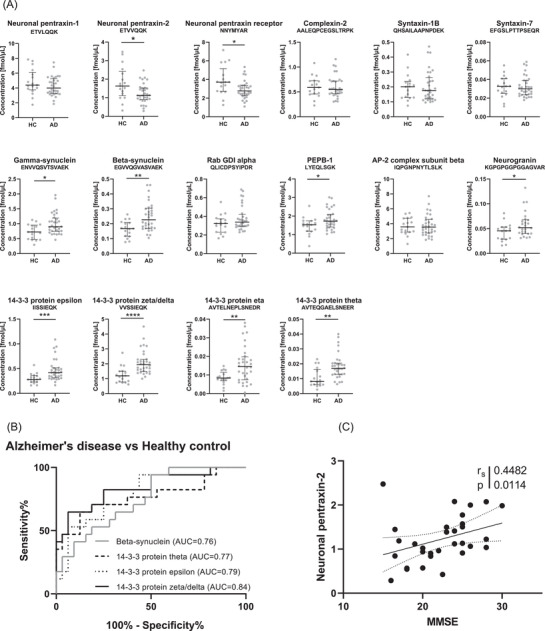
Levels of the synaptic panel proteins, diagnostic performance, and correlation to cognition in the clinical cohort. A, Cerebrospinal fluid (CSF) concentrations obtained by parallel reaction monitoring (PRM) analysis of the synaptic panel proteins (one representative peptide for each protein) in the clinical cohort consisting of healthy controls (HC, n = 17), and Alzheimer's disease (AD, n = 32). Statistical comparison was performed with Mann‐Whitney test, P‐values: * P ≤ .05, ** P ≤ .01, *** P ≤ .001, and **** P ≤ .0001. The bars indicate median with interquartile range. B, Receiver operating characteristic curves calculated for AD versus healthy controls for the four synaptic proteins with the highest area under the curve values. C, Association between neuronal pentraxin‐2 and Mini‐Mental State Examination (MMSE) with Spearman rank correlation coefficient and P‐value
The synaptic proteins included in the panel correlated well with each other (Spearman's correlation coefficient [rs] > 0.5, P ≤ .05, Figure SB5 in supporting information). Exceptions include the pentraxins and 14‐3‐3 protein theta in the AD samples and 14‐3‐3 protein eta in the control samples. Generally, a higher correlation was found in the control group than in the AD. Further, all peptides from the same protein had a strong association (rs > 0.7, P ≤ .0001, Figure SB6 in supporting information), except for neuronal pentraxin‐1 and the neuronal pentraxin receptor, which had a weak to moderate correlation between their peptides (rs = 0.3 to 0.7, P ≤ .05). The correlations with Mini‐Mental State Examination (MMSE) score and core CSF biomarkers are shown in Table SB5. It was shown that only neuronal pentraxin‐2 correlated significantly (rs = 0.45, P ≤ .05) with MMSE (Figure 2C). The pentraxins were also the only proteins that showed any correlation (rs = 0.3 to 0.5, P ≤ .05) with Aβ1‐42 levels. Interestingly most of the panel proteins correlated moderately (rs > 0.5, P ≤ .05) with both p‐tau181 and t‐tau with the exception of the neuronal pentraxins and 14‐3‐3 theta, which only showed a weak correlation (rs = 0.2 to 0.5, P ≤ .05). No correlation with age was found (Table SB4).
4. DISCUSSION
There is a need for biomarkers reflecting synaptic pathology that could be early indicators of AD, predict which patients are likely to suffer a rapid cognitive decline, and help monitor intensity of synaptic degeneration for clinical evaluation or drug monitoring. We have developed a biomarker panel of 17 synaptic proteins. Among these proteins are neurogranin, complexin‐2, VAMP‐2, neuronal pentraxins, synucleins, syntaxins, and several 14‐3‐3 proteins that collectively encompass the presynaptic and postsynaptic terminals as well as vesicle trafficking. Several of these proteins included in the synaptic panel in this study have also been implicated previously in relation to neurodegenerative diseases, in particular AD; for example, the 14‐3‐3 proteins, which have been genetically linked to AD and found to both interact with the key AD pathology protein tau and colocalize with it in neurofibrillary tangles. In connection with pathology, the 14‐3‐3 proteins are also of interest in relation with Creutzfeldt‐Jakob disease, for which they are established biomarkers. 18 They are a family of adaptor proteins that are highly expressed in the brain and notably enriched at the synapse, whose function is diverse but still largely unknown. However, the 14‐3‐3 proteins have been indicated at the presynaptic site to modulate N‐type voltage‐gated calcium channel (Cav2.2) activation and at the postsynaptic side, in the regulation of glutamate receptors, especially N‐methyl‐d‐aspartate receptors, leading to long‐term potentiation (Figure 3). 19 Further, the 14‐3‐3 gamma protein was recently quantified in the CSF of AD patients and found to be increased compared to controls. 20 In our study, the results indicate that this also is the case for other 14‐3‐3 proteins: zeta/delta, eta, theta, and epsilon. All the 14‐3‐3 proteins showed a good performance in discriminating AD and controls (AUC > 0.73). However, zeta/delta especially seems to have promise as an AD biomarker with the highest AUC out of all panel proteins (AUC = 0.84, 95% CI = 0.72% to 0.96%).
FIGURE 3.
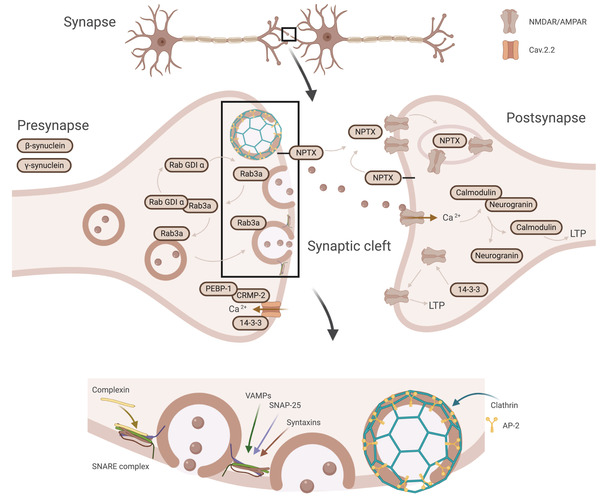
Schematic illustration of the synaptic proteins, their cellular location, and their synaptic processes. The synaptic proteins in the panel collectively encompass the presynaptic (beta‐synuclein and gamma‐synuclein) and postsynaptic terminals (neurogranin) as well as proteins involved in vesicle trafficking (complexin‐2, syntaxins, VAMP‐2, rab GDI alpha, and AP‐2). Further, proteins native to both the presynaptic and postsynaptic terminal (14‐3‐3 and NPTX) involved in, among others, postsynaptic receptor regulation (NPTX and 14‐3‐3). AMPAR, α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptor; AP‐2, activating protein 2; Cav2.2, N‐type voltage‐gated calcium channel; CRMP‐2, collapsin response mediator protein‐2; NPTX, neuronal pentraxins; NMDAR, N‐methyl‐D‐aspartate receptor; PEBP‐1, phosphatidylethanolamine‐binding protein 1; rab GDI alpha, rab GDP dissociation inhibitor alpha; Rab3a, ras‐related protein 3a; SNARE, soluble N‐ethylmaleimide‐sensitive factor attachment receptor; VAMP‐2, vesicle‐associated membrane protein 2. Created with BioRender.com
One synaptic protein that was characterized in relation to AD already in 1990, as a major component of amyloid plaques, apart from Aβ, is alpha‐synuclein. 21 The discovery was soon followed by that of two other family members, beta‐ and gamma‐synuclein, which also localize to presynaptic terminals. Their function is still unclear, but since then it has been suggested that all three synucleins might be involved in neurodegenerative diseases. For instance, alpha‐synuclein is well known to aggregate and form Lewy bodies in Lewy body dementia and Parkinson's disease. 22 Gamma‐ and beta‐synuclein also showed promising results in our study; beta‐synuclein in particular was the one together with the 14‐3‐3 proteins with the best ability to discriminate between AD and controls (AUC = 0.76). Interestingly, beta‐synuclein has been said to have neuroprotective abilities due to its inhibitory function on alpha‐synuclein aggregation. Oeckl et al. were the first to simultaneously quantify all three synucleins and they found increased concentrations for all three in CSF from AD patients compared to controls, 23 thus corroborating our results.
Further, complexin‐2, the syntaxins, neurogranin, rab GDI alpha, PEBP‐1, and AP‐2 complex subunit beta have also, similarly to the others, been implicated in relation to AD. 10 , 24 , 25 , 26 , 27 Additionally, their functions are all involved in the process of synapse vesicle exocytosis and neurotransmitter release at the synaptic cleft (Figure 3). The most important proteins involved in synapse vesicle exocytosis are so‐called SNARE (soluble N‐ethylmaleimide‐sensitive factor attachment receptor) proteins, to which the membrane‐bound syntaxins belong. Together with VAMPs and SNAP‐25, syntaxins form a trans‐ternary SNARE complex and regulate SNARE activity. 28 Other contributing proteins are SNARE accessory proteins such as the complexins, which bind and modulate the function of the SNARE complex. 29 The process of endocytosis is similarly regulated where clathrin coats the membrane to be endocytosed from the plasma membrane. However, to perform this function it needs adaptor proteins, which link clathrin to the membrane. Members of the family of adaptor proteins, such as AP‐2 adaptor complex proteins, thus fulfill the important function of mediating endocytosis. 30 Neurogranin is a postsynaptic protein, which in response to low calcium levels binds calmodulin and thus prolongs its availability in the postsynaptic space. This leads to sustained activation of signal transmission. 10 Further, the rab family together with its accessory regulatory proteins, including rab GDI alpha, is another protein family that regulates various steps of membrane trafficking such as synaptic vesicle docking (Rab3a). 24 Finally, PEBP‐1, which is a regulatory protein influencing a number of cellular processes, has been found to bind with collapsin response mediator protein‐2, which in turn interacts with and regulates Cav2.2 at the presynaptic site. 31 Complexin‐2, the syntaxins, neurogranin, rab GDI alpha, PEBP‐1, and AP‐2 complex subunit beta all showed a strong increase in levels in AD compared to controls in the pilot cohort. This difference, however, was not as apparent in the clinical cohort, most probably due to the fact that the patient inclusion was mainly based on clinical symptoms, differing from the first cohort, which was based solely on the core AD biomarkers. Of all these only neurogranin and PEBP‐1 still had a significant, albeit reduced, difference in the clinical cohort. PEBP‐1 CSF concentrations have to our knowledge never been studied in depth in relation to AD. On the other hand, as previously mentioned, neurogranin is one of the currently most established synaptic biomarkers, has extensively been studied, and has been found to be increased in AD as determined with both enzyme‐linked immunosorbent assays and mass spectrometric methods. 10 , 11 Our method differs from previously published mass spectrometric methods for neurogranin by using a tryptic peptide and not an endogenous peptide but still with similar results. Previous studies have reported an AUC for neurogranin of 0.70 to 0.73 comparing AD to controls, 32 , 33 , 34 compared to the AUC of 0.68 in our study. This suggests that the 14‐3‐3 proteins and beta‐synuclein are at least comparable to neurogranin as potential synaptic biomarkers in differentiating AD from controls, a finding that should be investigated in more heterogeneous populations with longitudinal cognitive data.
Last, both the two secreted neuronal pentraxin‐1 and ‐2 and the membrane‐bound receptor have received recent attention after the finding in several studies of decreased CSF levels in AD compared to controls. 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42 The neuronal pentraxins have been shown to be important for α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid‐type glutamate receptor recruitment during exocytosis. They bind and cluster the receptors and thus have an important function in modulating synaptic plasticity (Figure 3). 43 , 44 In this study, however, there was only a small decrease in AD compared to controls in the clinical cohort, which was not seen in the pilot study and not at all for neuronal pentraxin‐1. Even though the difference was small, it is very interesting that unlike the rest of the synaptic proteins the levels were decreased in AD. In a longitudinal study of the neuronal pentraxin receptor, Lim et al. hypothesize that the pentraxins in CSF may follow a biphasic pattern, rapidly increasing in MCI followed by a decrease in AD back to or past the normal levels. 42 This pattern has been observed in a study by Duits et al. for several synaptic proteins in which the MCI patients that later converted to AD exhibited this pattern in particular. 45 However, others have reported a constant decrease in neuronal pentraxin CSF concentrations from controls to MCI and further to AD. 35 Galasko et al. 35 found neuronal pentraxin‐2 to have a stronger correlation to cognitive decline than other synaptic biomarkers, such as SNAP‐25 and neurogranin, speculating that the reduced neuronal pentraxin‐2 levels might indicate a central role in the alteration of synaptic function, which is followed by the release of the more general synaptic markers during subsequent synapse degeneration. The association between neuronal pentraxin‐2 with MMSE in our study also corroborates that neuronal pentraxin‐2 in particular seems to correlate the best with cognitive decline. Future directions, therefore, involve replicating the results in a larger cohort that also should include a larger separate MCI group to follow the progression of the CSF levels of the various synaptic proteins along the different stages of the disease.
A major strength of simultaneously observing levels of proteins representing different parts of the synapse is that it makes it possible to distinguish and differentiate among general and specific pathological patterns. This will be particularly important when following disease progression and comparing different neurodegenerative diseases. An interesting example of this in the current study is that the levels of the neuronal pentraxins do not follow the same pattern as the rest of the investigated synaptic proteins. A limitation with these types of multiplex assays is that by using a relatively simple and nonspecific sample preparation the conditions are not perfect for any one of the targeted proteins. The analytical challenge is increased by the relatively broad concentration range among the targeted proteins and the substantial general protein background in CSF. However, it is possible to handle a large number of targets with great variability in concentration. Further, the small sample volume, the specificity of the method, and the fact that no antibodies are needed are all advantageous for verification of shotgun proteomics findings and in the search for new biomarkers. A limitation with the current study is the small sample numbers, thus no cut‐offs for the panel proteins were established for the ROC analysis and no multiple comparisons adjustments were performed for either cohort. However, the findings were confirmed in two independent cohorts, which strengthens the results. The discrepancies found between the cohorts could potentially be explained by the associations with age for some of the biomarkers found in the pilot study. Future perspectives include studying the synaptic panel in larger cohorts to verify the differences found in the current study and to establish cut‐offs for the synaptic panel proteins.
5. CONCLUSION
We have established a novel synaptic panel assay to study and validate 17 synaptic proteins as biomarkers of synaptic degeneration simultaneously in 100 μL of CSF. The results from this study indicate that beta‐synuclein, gamma‐synuclein, neurogranin, PEBP‐1, 14‐3‐3 proteins, and neuronal pentraxins are altered in AD compared to healthy controls and that they could be potential synaptic pathology biomarkers for AD. Their CSF levels may work as early indicators of AD and as complementing information to other CSF and imaging markers for guiding diagnostics.
CONFLICTS OF INTEREST
Henrik Zetterberg has served on scientific advisory boards for Denali, Roche Diagnostics, Wave, Samumed, Siemens Healthineers, Pinteon Therapeutics, and CogRx; has given lectures in symposia sponsored by Fujirebio, Alzecure, and Biogen; and is a co‐founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. Kaj Blennow has served as a consultant, on advisory boards, or on data monitoring committees for Abcam, Axon, Biogen, JOMDD/Shimadzu, Julius Clinical, Lilly, MagQu, Novartis, Roche Diagnostics, and Siemens Healthineers, and is a co‐founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. The other authors declare no conflicts of interest.
Supporting information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
This work was supported by the Eivind and Elsa K. Son Sylvan Foundation, the Märta and Gustaf Ågren Foundation, the Herbert and Karin Jacobsson Foundation, the Gun and Bertil Stohne Foundation, the Foundation for Gamla Tjänarinnor, the Felix Neubergh Foundation, Demensfonden, Rune and Ulla Almlöv Foundation, and an anonymous donor. HZ is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2018‐02532), the European Research Council (#681712), Swedish State Support for Clinical Research (#ALFGBG‐720931), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809‐2016862), the AD Strategic Fund and the Alzheimer's Association (#ADSF‐21‐831376‐C, #ADSF‐21‐831381‐C and #ADSF‐21‐831377‐C), the Foundation of Gamla Tjänarinnor, the Olav Thon Foundation, the Erling‐Persson Family Foundation, Hjärnfonden, Sweden (#FO2019‐0228), the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska‐Curie grant agreement No 860197 (MIRIADE), and the UK Dementia Research Institute at UCL. KB is supported by the Swedish Research Council (#2017‐00915), the Alzheimer Drug Discovery Foundation (ADDF), USA (#RDAPB‐201809‐2016615), the Swedish Alzheimer Foundation (#AF‐742881), Hjärnfonden, Sweden (#FO2017‐0243), the Swedish state under the agreement between the Swedish government and the County Councils, the ALF‐agreement (#ALFGBG‐715986), and European Union Joint Program for Neurodegenerative Disorders (JPND2019‐466‐236). JG is supported by Alzheimerfonden (AF‐930934) and the Foundation of Gamla Tjänarinnor.
Nilsson J, Gobom J, Sjödin S, et al. Cerebrospinal fluid biomarker panel for synaptic dysfunction in Alzheimer's disease. Alzheimer's Dement. 2021;13:e12179. 10.1002/dad2.12179
REFERENCES
- 1. Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572‐580. [DOI] [PubMed] [Google Scholar]
- 2. Palmqvist S, Insel PS, Stomrud E, et al. Cerebrospinal fluid and plasma biomarker trajectories with increasing amyloid deposition in Alzheimer's disease. EMBO Mol Med 2019;11:e11170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Milà‐Alomà M, Salvadó G, Gispert JD, et al. Amyloid beta, tau, synaptic, neurodegeneration, and glial biomarkers in the preclinical stage of the Alzheimer's continuum. Alzheimer's Dement. 2020;16(10):1358‐1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mehta D, Jackson R, Paul G, Shi J, Sabbagh M. Why do trials for Alzheimer's disease drugs keep failing? A discontinued drug perspective for 2010‐2015. Expert Opin Investig Drugs. 2017;26:735‐739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Salloway S, Sperling R, Fox NC, et al. Two phase 3 trials of bapineuzumab in mild‐to‐moderate Alzheimer's disease. N Engl J Med. 2014;370:322‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Camporesi E, Nilsson J, Brinkmalm A, et al. Fluid biomarkers for synaptic dysfunction and loss. Biomark Insights. 2020;15:1177271920950319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen M‐K, Mecca AP, Naganawa M, et al. Assessing synaptic density in Alzheimer disease with synaptic vesicle glycoprotein 2A positron emission tomographic imaging. JAMA Neurol. 2018;75:1215‐1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brinkmalm A, Brinkmalm G, Honer WG, et al. SNAP‐25 is a promising novel cerebrospinal fluid biomarker for synapse degeneration in Alzheimer's disease. Mol Neurodegener. 2014;9:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ohrfelt A, Brinkmalm A, Dumurgier J, et al. The pre‐synaptic vesicle protein synaptotagmin is a novel biomarker for Alzheimer's disease. Alzheimers Res Ther. 2016;8:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kvartsberg H, Duits FH, Ingelsson M, et al. Cerebrospinal fluid levels of the synaptic protein neurogranin correlates with cognitive decline in prodromal Alzheimer's disease. Alzheimer's Dement. 2015;11:1180‐1190. [DOI] [PubMed] [Google Scholar]
- 11. Mavroudis IA, Petridis F, Chatzikonstantinou S, Kazis D. A meta‐analysis on CSF neurogranin levels for the diagnosis of Alzheimer's disease and mild cognitive impairment. Aging Clin Exp Res. 2019;32(9):1639‐1646. [DOI] [PubMed] [Google Scholar]
- 12. Mecca AP, Chen MK, O'Dell RS, et al. In vivo measurement of widespread synaptic loss in Alzheimer's disease with SV2A PET. Alzheimer's Dement. 2020;16:974‐982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tijms BM, Gobom J, Reus L, et al. Pathophysiological subtypes of Alzheimer's disease based on cerebrospinal fluid proteomics. Brain. 2020;143(12):3776‐3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dubois B, Feldman HH, Jacova C, et al. Advancing research diagnostic criteria for Alzheimer's disease: the IWG‐2 criteria. Lancet Neurol. 2014;13:614‐629. [DOI] [PubMed] [Google Scholar]
- 15. Johansson P, Mattsson N, Hansson O, et al. Cerebrospinal fluid biomarkers for Alzheimer's disease: diagnostic performance in a homogeneous mono‐center population. J Alzheimer's Dis. 2011;24:537‐546. [DOI] [PubMed] [Google Scholar]
- 16. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA Work Group* under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939. [DOI] [PubMed] [Google Scholar]
- 17. Sjödin S, Brinkmalm G, Öhrfelt A, et al. Endo‐lysosomal proteins and ubiquitin CSF concentrations in Alzheimer's and Parkinson's disease. Alzheimer's Res Ther. 2019;11:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Foote M, Zhou Y. 14‐3‐3 proteins in neurological disorders. Int J Biochem Mol Biol. 2012;3:152. [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang J, Zhou Y. 14‐3‐3 proteins in glutamatergic synapses. Neural Plast. 2018;2018:8407609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Antonell A, Tort‐Merino A, Ríos J, et al. Synaptic, axonal damage and inflammatory cerebrospinal fluid biomarkers in neurodegenerative dementias. Alzheimer's Dement. 2019;16(2):262‐272. [DOI] [PubMed] [Google Scholar]
- 21. Uéda K, Fukushima H, Masliah E, et al. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:11282‐11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Burré J. The synaptic function of α‐synuclein. J Parkinson's Dis. 2015;5:699‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oeckl P, Metzger F, Nagl M, et al. Alpha‐, Beta‐, and Gamma‐synuclein quantification in cerebrospinal fluid by multiple reaction monitoring reveals increased concentrations in alzheimer′ s and creutzfeldt‐jakob disease but no alteration in Synucleinopathies. Mol Cell Proteomics. 2016;15:3126‐3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guadagno NA, Progida C. Rab GTPases: switching to human diseases. Cells. 2019;8:909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Honer WG, Ramos‐Miguel A, Alamri J, et al. The synaptic pathology of cognitive life. Dialogues Clin Neurosci. 2019;21:271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schoentgen F, Jonic S. PEBP1/RKIP behavior: a mirror of actin‐membrane organization. Cell Mol Life Sci. 2020;77:859‐874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nelson PT, Fardo DW, Katsumata Y. The MUC6/AP2A2 locus and its relevance to Alzheimer's disease: a review. J Neuropathol Exp Neurol. 2020;79:568‐584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Teng FYH, Wang Y, Tang BL. The syntaxins. Genome Biol. 2001;2:reviews3012. 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Trimbuch T, Rosenmund C. Should I stop or should I go? The role of complexin in neurotransmitter release. Nat Rev Neurosci. 2016;17:118. [DOI] [PubMed] [Google Scholar]
- 30. Royle SJ. The cellular functions of clathrin. Cell Mol Life Sci. 2006;63:1823‐1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ohi Y, Kato D, Mizuno M, et al. Enhancement of long‐term potentiation via muscarinic modulation in the hippocampus of HCNP precursor transgenic mice. Neurosci Lett. 2015;597:1‐6. [DOI] [PubMed] [Google Scholar]
- 32. Tarawneh R, D'Angelo G, Crimmins D, et al. Diagnostic and prognostic utility of the synaptic marker neurogranin in Alzheimer disease. JAMA Neurol. 2016;73:561‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mattsson N, Insel PS, Palmqvist S, et al. Cerebrospinal fluid tau, neurogranin, and neurofilament light in Alzheimer's disease. EMBO Mol Med. 2016;8:1184‐1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Blennow K, Diaz‐Lucena D, Zetterberg H, et al. CSF neurogranin as a neuronal damage marker in CJD: a comparative study with AD. J Neurol Neurosurg Psychiatry. 2019;90:846‐853. [DOI] [PubMed] [Google Scholar]
- 35. Galasko D, Xiao M, Xu D, et al. Synaptic biomarkers in CSF aid in diagnosis, correlate with cognition and predict progression in MCI and Alzheimer's disease. Alzheimers Dement (N Y). 2019;5:871‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xiao MF, Xu D, Craig MT, et al. NPTX2 and cognitive dysfunction in Alzheimer's Disease. Elife. 2017;6:e23798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Soldan A, Moghekar A, Walker KA, et al. Resting‐state functional connectivity is associated with cerebrospinal fluid levels of the synaptic protein NPTX2 in non‐demented older adults. Front Aging Neurosci. 2019;11:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brinkmalm G, Sjodin S, Simonsen AH, et al. A parallel reaction monitoring mass spectrometric method for analysis of potential CSF biomarkers for Alzheimer's disease. Proteomics Clin Appl. 2018;12. [DOI] [PubMed] [Google Scholar]
- 39. Spellman DS, Wildsmith KR, Honigberg LA, et al. Development and evaluation of a multiplexed mass spectrometry based assay for measuring candidate peptide biomarkers in Alzheimer's Disease Neuroimaging Initiative (ADNI) CSF. Proteomics Clin Appl. 2015;9:715‐731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Swanson A, Willette A, Initiative AsDN . Neuronal pentraxin 2 predicts medial temporal atrophy and memory decline across the Alzheimer's disease spectrum. Brain Behav Immun. 2016;58:201‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Begcevic I, Tsolaki M, Brinc D, et al. Neuronal pentraxin receptor‐1 is a new cerebrospinal fluid biomarker of Alzheimer's disease progression. F1000Res. 2018;7:1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lim B, Sando SB, Grøntvedt GR, Bråthen G, Diamandis EP. Cerebrospinal fluid neuronal pentraxin receptor as a biomarker of long‐term progression of Alzheimer's disease: a 24‐month follow‐up study. Neurobiol Aging. 2020;93:97.e1‐97. [DOI] [PubMed] [Google Scholar]
- 43. Xu D, Hopf C, Reddy R, et al. Narp and NP1 form heterocomplexes that function in developmental and activity‐dependent synaptic plasticity. Neuron. 2003;39:513‐528. [DOI] [PubMed] [Google Scholar]
- 44. Lee S‐J, Wei M, Zhang C, et al. Presynaptic neuronal pentraxin receptor organizes excitatory and inhibitory synapses. J Neurosci. 2017;37:1062‐1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Duits FH, Brinkmalm G, Teunissen CE, et al. Synaptic proteins in CSF as potential novel biomarkers for prognosis in prodromal Alzheimer's disease. Alzheimers Res Ther. 2018;10:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information