

SHOC2 is a prototypical leucine-rich repeat protein that promotes downstream receptor tyrosine kinase (RTK)/RAS signaling and plays important roles in several cellular and developmental processes. Gain-of-function germ line mutations of SHOC2 drive the RASopathy Noonan-like syndrome, and SHOC2 mediates adaptive resistance to mitogen-activated protein kinase (MAPK) inhibitors.
KEYWORDS: cancer, leucine-rich repeat, MAPK signaling, Noonan syndrome, PP1C, RAF, RAS, receptor tyrosine kinase, SHOC2
ABSTRACT
SHOC2 is a prototypical leucine-rich repeat protein that promotes downstream receptor tyrosine kinase (RTK)/RAS signaling and plays important roles in several cellular and developmental processes. Gain-of-function germ line mutations of SHOC2 drive the RASopathy Noonan-like syndrome, and SHOC2 mediates adaptive resistance to mitogen-activated protein kinase (MAPK) inhibitors. Similar to many scaffolding proteins, SHOC2 facilitates signal transduction by enabling proximal protein interactions and regulating the subcellular localization of its binding partners. Here, we review the structural features of SHOC2 that mediate its known functions, discuss these elements in the context of various binding partners and signaling pathways, and highlight areas of SHOC2 biology where a consensus view has not yet emerged.
INTRODUCTION
SHOC2 is a leucine-rich repeat protein that functions as a scaffold critical for RTK/RAS signaling. RAS proteins are a family of small GTPases that function as binary switches and are critical in development, multiple physiological processes, and diseases (1). The canonical RAS family members HRAS, KRAS, and NRAS have been shown to bind SHOC2 as well as a closely related RRAS subfamily member, MRAS, for muscle RAS homolog. RTKs are upstream regulators of RAS and drive RAS activation in response to growth factor binding (1, 2). Downstream effectors of RAS, including RAF and phosphatidylinositol 3-kinase (PI3K), bind GTP-loaded RAS and function as key mediators of RAS signaling. Upon activation, RAF kinases trigger a mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) signaling cascade (1, 3). Subsequently, activated ERK paralogs translocate to the nucleus to phosphorylate targets, such as transcription factors that upregulate cell proliferation and survival (1, 3).
Although RAS effectors have been extensively studied, the functional roles of ancillary regulatory proteins such as SHOC2 are less characterized. Most SHOC2 studies underscore its role in promoting MAPK signaling, best exemplified by gain-of-function mutations of SHOC2 that are causal in Noonan-like syndrome, a RASopathy (4, 5). Although aspects of SHOC2 biology and its binding have been previously discussed (6, 7), here we outline the function of SHOC2 through its structural properties that mediate protein-protein interactions in further depth, provide an extensive discussion of the current conflicting reports regarding SHOC2 function, and highlight how SHOC2 is essential for the RTK/RAS signaling circuit. We also discuss recent work pertaining to the role of SHOC2 in RAS mutant cancers and propose potential therapeutic strategies to target SHOC2.
SHOC2 DISCOVERY
SHOC2 was first discovered through a genetic screen designed to identify genes that facilitate fibroblast growth factor (FGF) receptor (FGFR) signaling in Caenorhabditis elegans (8). Mutations of genes that enhance FGFR signaling drive a clear phenotype in C. elegans, whereby the organisms appear transparent compared to the wild-type control (8). Using this clear phenotype, DeVore and colleagues performed a genetic screen for genes whose deletion acted as a suppressor of clear (soc) phenotype (8). Among these genes that they identified were orthologs of the RTK adaptor protein GRB2 (soc-1) and SHOC2 (soc-2) (8, 9). Subsequent genotype-phenotype mapping and yeast two-hybrid experiments placed SHOC2 directly downstream of let-60 RAS and upstream of RAF (lin-45), and SHOC2 was found to directly interact with both RAS and RAF (10, 11). These initial studies noted a striking similarity between the primary structures of C. elegans ortholog soc-2/sur-8 and human SHOC2, with the highest sequence similarities found within repeated leucine-rich motifs (9, 10).
SHOC2: A CANONICAL LRR PROTEIN
Repetitive amino acid sequences, known as protein tandem repeats, give rise to unique, complex tertiary structures (12). Leucine-rich repeat (LRR) proteins are among the most well-studied examples of tandem repeat proteins. The major characteristic of LRR proteins is the repetitive sequences of leucine or other hydrophobic residues. SHOC2 is a quintessential LRR protein with an unstructured N-terminal domain followed by 20 predicted LRRs (UniProtKB) and a short C-terminal domain (Fig. 1A). Many functionally important residues of SHOC2 map within its LRR domains; however, both the flanking N-terminal and C-terminal unstructured regions participate in maintaining key interactions as well (13). LRR domains of SHOC2 are 22 to 23 amino acids in length and start with a highly conserved segment (HCS) followed by a variable region with a reoccurring, medial proline residue (Fig. 1B). Each HCS consists of 11 to 12 amino acids with a sequence motif of LXXLXLXXN(X)1-2L (Fig. 1B), where L can be a leucine or any other hydrophobic residue and N is an asparagine (14).
FIG 1.

SHOC2 domains, sequence, and structural features. (A) Full-length SHOC2 (NCBI RefSeq NP_031399.2), depicting amino acid (AA) segments corresponding to domain regions and conserved leucine-rich repeats (LRRs). Conserved segments of SHOC2 from primary sequence analysis of several SHOC2 orthologs (COBALT) are annotated in red. (B) Primary sequence analysis of highly conserved SHOC2 LRRs (UniProtKB no. Q9UQ13-1) with consensus sequence logo (WebLogo v2.8.2) and sequence alignment (MAFFT v7.31). The canonical highly conserved leucine-rich repeat motif [LXXLXLXXN(X)2L] is outlined in blue. (C) 3D modeling of SHOC2 (SWISS-MODEL; template 3b2d.1.A) with predicted flexible hinge indicated. The inset represents a rotated view, highlighting parallel beta sheets.
Leucine-rich HCSs are known to form short beta-strands that arrange in parallel to one another through interdomain hydrogen bonding (14). The arrangement of repetitive parallel beta-strands concatenates each LRR domain, resulting in a solenoid structure (14). Although the SHOC2 structure has yet to be solved, homology modeling methods predict that SHOC2 forms a horseshoe shape, with the reoccurring leucine-rich HCS making up the concave surface (15–17) (Fig. 1C). Some structure modeling algorithms predict a flexible hinge region between N- and C-terminal LRRs (Fig. 1C), lending to speculation that SHOC2 undergoes conformational changes depending on its interactions with various binding partners (13, 17, 18). However, not all structural models anticipate this cleft but instead predict a more rigid solenoid with a greater helical turn along the LRR domains (15, 19). Further ambiguity exists with SHOC2 structure regarding the precise number of LRR domains, with estimates ranging from 18 to 21 (9, 10, 13, 16, 17, 20), where the purported 21st LRR motif at the C terminus serves as a cap to stabilize the inner domains (20) (Fig. 1C), but this putative LRR-like domain lacks an HCS and does not align with the SHOC2 LRR consensus sequence. We note that many of these features are based on predictive computational models, and further work to solve SHOC2 structure in the presence and absence of these effectors remains an important area of investigation.
SHOC2 is expressed in many tissue types and is conserved across phylogenetic domains, ranging from Cyanobacteria and Arabidopsis to nonhuman primates and humans (15). SHOC2 orthologs exhibit a high degree of sequence homology within their LRR domains (Fig. 1A), which are known to serve as scaffolding surfaces that mediate protein interactions. Accordingly, numerous functionally important segments of SHOC2 have been found to reside within the LRRs. In the following sections, we will discuss the catalog of SHOC2 binding proteins and complexes that regulate downstream RAS effector pathways. We will contextualize key SHOC2 gain-of-function (GOF)/loss-of-function (LOF) mutations and critical motifs that have been shown to mediate these interactions (Fig. 2).
FIG 2.
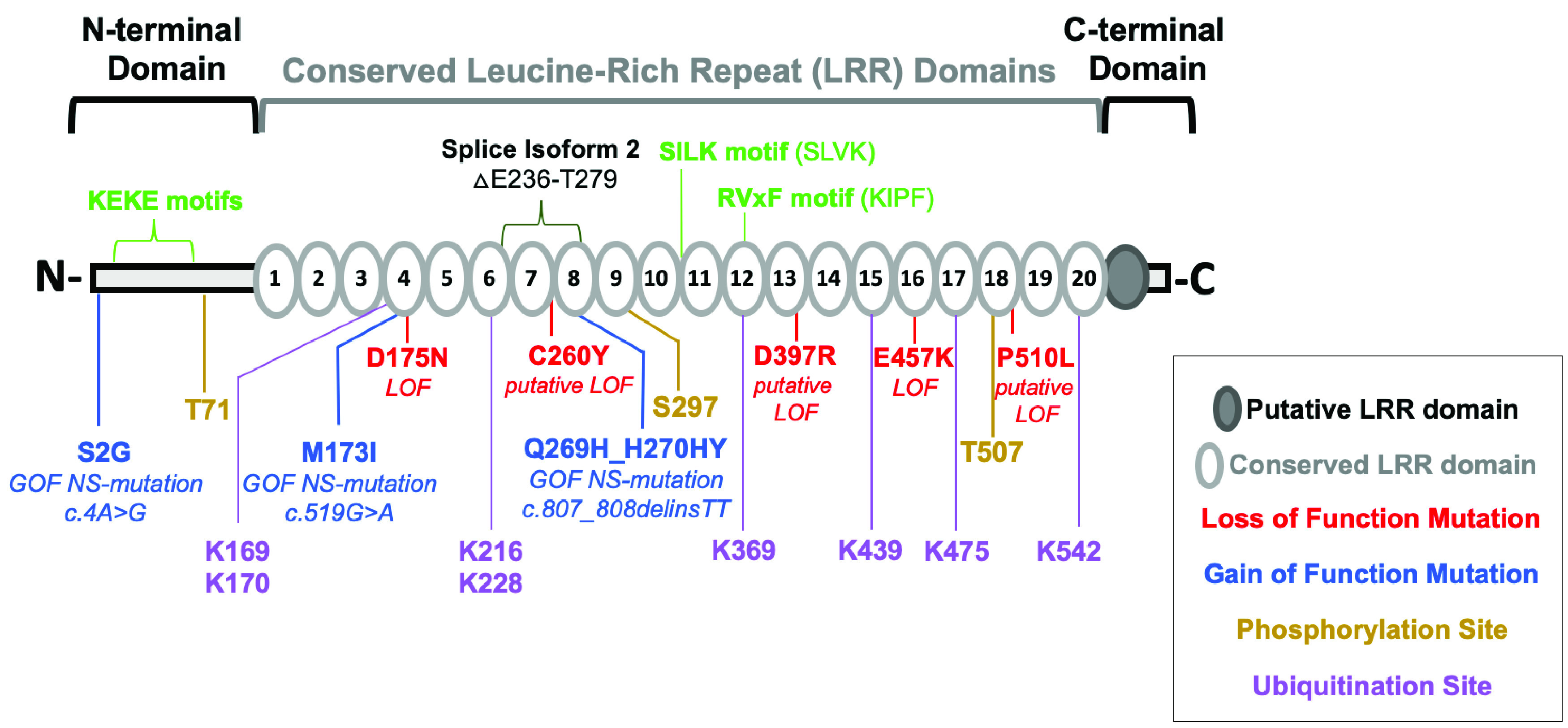
Critical SHOC2 sequence motifs and residues. Shown are a domain diagram of SHOC2 with the indicated domain regions, LRRs, and putative LRR and a domain diagram of full-length SHOC2 with motifs (green), deleted region of transcript variant 2 (536 aa) lacking exon 3 (NCBI RefSeq NR_136749.1), posttranslationally modified residues, and mutations reported to be functionally important in the literature, including gain-of-function (GOF) and loss-of-function (LOF) mutations. Putative mutations denote equivalent missense mutations of SHOC2 identified in C. elegans ortholog soc-2/sur-8.
THE MRAS/SHOC2/PP1 HOLOPHOSPHATASE
Initial C. elegans studies identified SHOC2 to be downstream of RAS but upstream of RAF, suggesting that SHOC2 facilitated proximal RAS-RAF interactions (10). Seminal work published by Rodriguez-Viciana and McCormick revealed that SHOC2 forms a holophosphatase with the catalytic subunit of phosphatase PP1 and MRAS, whereby SHOC2 serves as the linchpin for complex formation (21). Each of the three members plays indispensable roles in the proper function of the complex to enable RAS signaling.
Within the complex, PP1 serves as the enzymatic subunit for substrate serine/threonine dephosphorylation. PP1 is a class of serine/threonine phosphatases that has three isoforms (PP1α, PP1β, and PP1γ) (22). Similar to other phosphatases, PP1 requires regulatory subunits to control the specificity of target dephosphorylation, and several putative regulatory factors, including SHOC2, have been proposed (21, 22).
The best-known substrates of the MRAS/SHOC2/PP1 holoenzyme are the RAF kinases. MRAS/SHOC2/PP1 potentiates RAF activity through dephosphorylation of a critical regulatory site, S259, on RAF family members (S259 RAF1, S365 BRAF, and S214 ARAF) (16, 21) (Fig. 3A). During unstimulated conditions, inactive RAF kinases are found as monomers and are cytosolically sequestered by the binding of 14-3-3 to phosphorylated S259 and S621 on RAF (23, 24), which enforces a closed, inactive confirmation of RAF. Upon RAS activation, RAF associates with RAS-GTP at the plasma membrane, and S259 is dephosphorylated by the MRAS/SHOC2/PP1 complex (Fig. 3A), triggering an open RAF conformation (25). Active RAFs then dimerize, which is required for subsequent MAPK signaling (24). The regulation of RAF activity is intricate and has been extensively reviewed (24).
FIG 3.

SHOC2-regulated RAS effector pathways and dynamic functional interactions. Shown is a schematic pathway overview of SHOC2’s role in the MAPK/ERK pathway involving MRAS/PP1 holophosphatase complex (A), HUWE1/PSMC5/VCP (B), interactors that sequester SHOC2 from RAS (C), posttranslational modifications that result in SHOC2 degradation (D), and PIK3CA/p110α that mediates PI3K/AKT signaling (E). Direct SHOC2 interactors that result in diminished MAPK signaling are depicted in red, and those that enable MAPK signaling are in blue. Ubiquitin chains are represented in purple, and multivesicular bodies (MVB)/late endosome (LE) are indicated.
SHOC2 has two degenerate PP1 consensus binding motifs, SILK (SLVK) and RVxF (KIPF), found within medial LRRs (Fig. 2), and both sequence motifs cooperate in mediating PP1 binding (16). Although PP1α, PP1β, and PP1γ have been shown to bind SHOC2, only PP1α and PP1β dephosphorylate the inhibitory BRAF S365 phosphorylation site (16). Furthermore, PP1α/β-SHOC2 interactions are dependent on MRAS binding with SHOC2, but PP1γ is constitutively bound to SHOC2 irrespective of MRAS interaction (16), lending to the potential for differential SHOC2-PP1γ-specific substrates.
Although MRAS clearly interacts with SHOC2 (16, 21), several reports describe other RAS family interactions (10, 11, 26). MRAS, also known as R-RAS3, is a member of the RAS family and similarly functions as a binary switch dependent on its GTP/GDP bound state (1, 27). MRAS shares sequence similarity and an identical effector domain with the other more commonly studied RAS members, HRAS, NRAS, and KRAS, and also localizes to the plasma membrane (1, 25). MRAS-mediated localization of SHOC2 at the plasma membrane is critical for MAPK signaling, likely by increasing proximal interactions of PP1 to its substrates, ARAF, BRAF, and RAF1, that are bound to RAS (16, 28) (Fig. 3A).
RAS interacts with SHOC2 through its RAS effector domain upon GTP loading (11, 16, 21), and domain-mapping experiments identified the unstructured N-terminal region of SHOC2 to be particularly important in mediating MRAS interactions (10, 13, 15). Furthermore, SHOC2 mutations of specific residues that disrupt or enhance MRAS binding have been discovered within the fourth, seventh, and eighth conserved LRRs (Fig. 2). These mutations include SHOC2 GOF mutations discovered in Noonan-like syndrome, M173I and Q269_H270delinsHY, both leading to enhanced MRAS binding (18, 29).
C. elegans screens identified loss-of-function missense mutations of soc-2/sur8. Specifically, these mutants are equivalent to SHOC2 mutations D175N, C260Y, and E457K (Fig. 2) (8–10). Indeed, D175N and E457K SHOC2 mutants have disrupted MRAS binding, and the E457K mutant fails to bind PP1 (21, 28). Intriguingly, forced localization of SHOC2 to the plasma membrane by N-terminal myristylation in combination with E457K mutation rescues MRAS-PP1 interactions (28).
Initial studies suggested that SHOC2 serves as a scaffold that accelerates RAS-RAF interactions to enhance MAPK signaling (10, 11). However, a series of immunoprecipitation studies demonstrated that RAF1 interactions with SHOC2 are indirect and mediated by binding of MRAS (21). The model that has been proposed articulates that SHOC2 facilitates MAPK signaling through its RAF holophosphatase activity, resulting in enhanced RAF dimerization and activation.
THE MRAS/RAF1/SHOC2/HUWE1 COMPLEX
In addition to direct regulation of RAF dimerization, SHOC2 also regulates MAPK signaling through an additional complex that elicits a negative-feedback regulation of RAF1 activity following epidermal growth factor (EGF) receptor (EGFR) activation (20, 30). HUWE1 is an E3 ubiquitin-protein ligase previously reported to ubiquitinate several critical proteins, such as MCL1 (31), TP53 (32), and MYCN (33). HUWE1 was initially identified as a SHOC2 interactor through a yeast 2-hybrid screen where SHOC2 served as the bait (30). Structurally, LRRs 12 to 14 of SHOC2 are required for its interaction with the HECT domain of HUWE1 (30). Epidermal growth factor (EGF) stimulation results in SHOC2-dependent, acute MAPK activation (20). Shortly following this activation, SHOC2 is ubiquitinated by HUWE1, internalized, and localized to late endosomes (15, 20, 34) (Fig. 3B). SHOC2 is modified with K11, K48, and K63 ubiquitin linkages, and HUWE1 induces polyubiquitination of SHOC2 at numerous lysine residues (Fig. 2), resulting in subsequent RAF1 ubiquitination (20, 30, 34). This ubiquitination event does not impact the protein stability/half-life of SHOC2 but is a necessary step for RAF1 turnover, and RAF1 levels increase upon HUWE1 knockdown, while BRAF and ARAF stability are unaffected (30). Subsequently, HUWE1/SHOC2-mediated reduction of RAF1 expression results in diminished MAPK signaling (30). A total of 8 lysine residues on SHOC2 have been identified to be ubiquitinated (K169, K170, K216, K228, K369, K439, K475, and K542) (30) (Fig. 2). A version of SHOC2 that contained arginine substitutions that cannot be ubiquitinated at 7 of these lysine residues (K169, K170, K216, K228, K369, K475, and K542) led to reduced RAF1 ubiquitination and sustained ERK activation (30).
Following HUWE1-mediated feedback inhibition, the complex undergoes rearrangement for subsequent reactivation of MAPK signaling. The resetting of the complex is regulated by PSMC5 and VCP, which regulate complex localization and reorganization, respectively. PSMC5 and VCP are both AAA+ ATPases, a superfamily of proteins that are characterized by Walker A/B motifs that hydrolyze ATP for energy to perform a diverse array of functions, including protein remodeling and denaturation (35, 36). PSMC5 is predominately known for its role as part of the 26S proteasomal complex (37) and modulating complex disassembly (38, 39). However, in the case of SHOC2, PSMC5 facilitates SHOC2 subcellular localization (34). Upon EGF stimulation, PSMC5 binds to the C-terminal LRRs of SHOC2 through its ATPase domain and facilitates MRAS/RAF1/SHOC2/HUWE1 complex localization to late endosomes (34). Subsequently, VCP recognizes and binds to polyubiquitinated SHOC2, resulting in HUWE1 dissociation from the complex (40). The displacement of HUWE1 at the late endosomes causes decreased SHOC2/RAF1 ubiquitination and resets the complex for further MAPK reactivation (34, 40) (Fig. 3B). The MRAS/RAF1/SHOC2/HUWE1 complex adds additional spaciotemporal complexity to SHOC2-mediated control over MAPK signaling.
ERBIN/SCRIB: LAP PROTEINS CONTROL SHOC2-MEDIATED MAPK SIGNALING
ERBIN and SCRIB are a part of the conserved family of LRR and Psd95-Dlg-Zo1 (LAP) domain-containing proteins and are known to localize at the plasma membrane to facilitate apicobasal polarity (41–43). LAP proteins, as their name implies, are structurally characterized by a series of N-terminal LRR domains and C-terminal PDZ domains (44).
ERBIN, also known as Erbb2 interacting protein, is known to regulate ERBB2 signaling by facilitating interactions with β2-adrenergic receptor (β2-AR) through its PDZ domains (45). ERBIN directly binds SHOC2 through its LRR domains (46), and this interaction has been shown to be functionally important in developmental processes including cardiac development and keratinocyte differentiation (47–49). ERBIN inhibits MAPK signaling by competing SHOC2 away from RAS (46, 49) (Fig. 3C). ERBIN has also been reported to interact with PP1, but this interaction was not further functionally characterized (16).
SCRIB similarly has been found to directly bind SHOC2 and inhibit MAPK signaling. However, unlike ERBIN, SCRIB does not disrupt SHOC2/RAS interactions. Instead, SCRIB functions by sequestering PP1 from SHOC2 by binding to PP1 with higher affinity (16) (Fig. 3A). The interaction of SHOC2 with active MRAS enhances PP1 binding affinity, allowing for efficient holoenzyme formation by competing away PP1 from SCRIB (16). Furthermore, SCRIB LRR domains alone were not sufficient to enable its interaction with SHOC2. Rather, a segment of SCRIB between the N-terminal LRRs and C-terminal PDZ domains is necessary for SHOC2 binding (16). SCRIB binds to the C-terminal half of SHOC2, and LOF SHOC2 mutation E457K, found within conserved LRR16, results in hampered SCRIB binding (16, 28) (Fig. 2).
We note that both ERBIN and SCRIB are classic LAP proteins with similar structural features, yet they interact with SHOC2 through different domains. Further studies investigating the contact surfaces of LAP proteins that mediate SHOC2 interactions may reveal insights in LRR domain binding and define additional SHOC2 interactions.
NONCANONICAL ROLE OF RPTOR IN MAPK SIGNALING
SHOC2 has also been found to arbitrate cross talk between the MAPK pathway and mammalian target of rapamycin (mTOR) signaling (50). The mTOR kinase is a significant signaling hub downstream of various intracellular amino acid sensors and growth factor signaling and controls numerous signaling pathways designed to regulate nutrient homeostasis, metabolism, protein translation, and autophagy, among other cellular processes (51). Regulatory-associated protein of mTOR (RPTOR) is a well-known regulator of mTOR activity and functions by recruiting substrates to mTOR within the mTORC1 complex (52–54). RPTOR directly interacts with SHOC2 to inhibit SHOC2-RAS interactions and diminish MAPK signaling (50) (Fig. 3C). SHOC2/RPTOR interactions reciprocally lead to the dissociation of RPTOR from mTOR through competitive binding (50). The sequestration of RPTOR from the mTORC1 complex via SHOC2 binding decreases mTOR activity and increases autophagy (50), in line with the established role of mTOR as a negative regulator of autophagy (51). These findings highlight SHOC2 as a potential regulator of mTOR, which may serve as a mechanism that regulates both the MAPK and mTORC1 pathways.
SHOC2 PHOSPHORYLATION AND DEGRADATION CONTROL
SHOC2 protein degradation was first shown in neural progenitor cells, where differentiation resulted in a rapid decline of SHOC2 at the protein level (55). Recent studies have since identified key regulators of SHOC2 turnover and the phosphorylation of specific serine/threonine residues that trigger proteasomal degradation in the context of RTK signaling.
The first report of SHOC2 phosphorylation was discovered in the context of FGF/RTK signaling, whereby FGF2 stimulation stabilizes SHOC2 protein levels (56). Protein kinase C (PKC) family members PKC-α and PKC-δ phosphorylate SHOC2 at an N-terminal threonine residue (T71) and serine residue within LRR9 (S297), respectively (56) (Fig. 2 and 3D). Phosphorylation at either site is sufficient to cause SHOC2 polyubiquitination and proteasomal degradation, followed by diminished MAPK signaling (56). However, PKC protein levels are rapidly reduced upon FGF2 stimulation, resulting in SHOC2 stabilization (56).
In contrast, EGF stimulation was found to result in a MAPK negative feedback loop involving FBXW7-mediated SHOC2 degradation (50). FBXW7 is an F-box protein that is an integral member of the ubiquitin ligase Skp1–Cdc53/cullin–F-box protein (SCF/β-TrCP) complex and has been reported to function as a tumor suppressor (57). FBXW7 binds target substrates of SCF/β-TrCP complex to be marked for proteasomal degradation (58, 59). Upon EGF stimulation, SHOC2 is phosphorylated at a threonine residue in LRR18 (T507) in a MEK-dependent manner (50) (Fig. 2 and 3D). FBXW7 binds to the SHOC2 phosphorylation site through its F-box domain, leading to SHOC2 ubiquitination (50). In contrast to HUWE1 (30), FBXW7-dependent ubiquitination of SHOC2 results in proteasomal degradation (50). Although MEK inhibitors (MEKi) were found to be sufficient in reducing phosphorylation of T507, the kinase that directly phosphorylates SHOC2 was not identified (50). In contrast, other studies failed to observe changes in SHOC2 protein levels in response to MEKi treatment (60, 61), lending to the possibility of context-specific regulation of SHOC2 expression.
SHOC2 IN PI3K-AKT AND MTOR SIGNALING
Phosphatidylinositol 3-kinase (PI3K) is another important RAS effector that has been shown to be indispensable in the tumor growth of RTK/RAS-driven cancers (62–64). In addition to RAF, studies have identified a novel role of SHOC2 in regulating the PI3K-AKT signaling pathway. PI3Ks are membrane-associated lipid kinases and are composed of regulatory subunits (p55 and p85) as well as catalytic subunit PIK3C/p110. As a RAS effector, PIK3C binds active RAS through its effector domain, which activates PI3K, resulting in the phosphorylation of phosphatidylinositol 4,5-bisphosphate (PIP2) to produce PIP3 (65). PIP3 localizes AKT to the plasma membrane, where PDK1 phosphorylates and activates AKT (66). Subsequently, activated AKT phosphorylates numerous downstream substrates, resulting in proliferation, regulation of metabolism, and other cellular processes (65, 66). The presence of constitutively active HRAS or EGF treatment results in enhanced PIK3CA-SHOC2 interactions and activated AKT (67) (Fig. 3E). SHOC2 loss in colorectal cancer cells with mutant KRAS, BRAF, and PI3K also inhibited levels of active AKT (67). Although these findings pose a noncanonical role of SHOC2 beyond MAPK signaling, the mechanism of how PIK3CA-SHOC2 interaction enhances PI3K activity and whether these interactions are direct or indirect has not been fully explored. Furthermore, several independent studies have found SHOC2 to have a unique impact on MAPK signaling with no effect on the PI3K signaling axis (11, 21, 60, 61). Additional investigations are necessary to provide mechanistic insights on the determinants of differential SHOC2 regulation of the RAS effector pathways.
THE DYNAMICS OF SHOC2 SUBCELLULAR LOCALIZATION
Scaffold proteins serve as signaling hubs by coordinating binding partners and mediating proximal protein-protein interactions that would otherwise be stoichiometrically unfavorable (68). In addition, scaffold protein localization plays a vital role in signaling by facilitating spaciotemporal control of its interactors (68, 69). SHOC2 has been shown to be dynamically distributed across multiple intracellular compartments, including the plasma membrane, endosomes, multivesicular bodies, and the nucleus.
The importance of localization in SHOC2 biology is highlighted by a prominent GOF mutation discovered in Noonan-like syndrome. The SHOC2 missense mutation (c.4A>G) results in a glycine substitution at the second residue (S2G) (Fig. 2) and confers an aberrant N-terminal myristylation site (4). Myristylated SHOC2 S2G constitutively localizes to the plasma membrane, causing increased MAPK signaling in response to EGF stimulation (4, 17). The plasma membrane is dynamic and heterogeneous, with regions of ordered lipid rafts rich in saturated fatty acids as well as disordered domains (70). Different RAS family members have been found to cluster in particular types of lipid domains, and this membrane organization has been shown to be important in regulating RAS signaling (70). Intriguingly, SHOC2 S2G mutants, under serum-starved conditions, localize to both cholesterol-rich and cholesterol-free lipid domains (17). However, upon EGF treatment, SHOC2 S2G redistributes to cholesterol-independent regions, revealing another potential level of localized regulation (17). Unlike myristylated mutant SHOC2, wild-type SHOC2 requires MRAS binding for plasma membrane localization (Fig. 3). SHOC2 LOF mutants unable to bind MRAS (D175N and E457K) are retained within the cytoplasm (28). Recent work found SHOC2 colocalizes with active MRAS at cell-cell junctions (28). Here, SHOC2 promotes concentrated MAPK activation, resulting in the phosphoregulation of p120-catenin and subsequent cell migration/scattering through junctional E-cadherin turnover (28).
The activation cycle of RTKs is dynamic, and activated receptors are known to be internalized to facilitate intracellular signaling and recycling (71). SHOC2 has also been found to localize to endosomes and multivesicular bodies. In the context of the MRAS/RAF1/SHOC2/HUWE1 complex, EGF treatment triggers SHOC2 internalization in a clathrin-dependent manner (20). As discussed, this localization event was implicated in a stepwise resetting of the complex for subsequent MAPK reactivation (20). The C-terminal region of SHOC2 is necessary for SHOC2 localization at late endosomes and multivesicular bodies in response to EGF treatment (15, 34). Specifically, deletion of the putative LRR domain within the SHOC2 C-terminal region results in diffuse cytoplasmic distribution, presumably due to loss of PSMC5 interaction (15, 34).
Finally, SHOC2 has also been reported to be present within the nucleus (4, 15, 17, 20). Independent studies have found the N-terminal half of SHOC2 is required for both nuclear export and import (15, 17). The disordered N-terminal region of SHOC2 contains characteristic alternating lysine and glutamate residues (KEKE motifs) that are required for SHOC2 nuclear export and MAPK/ERK activity (13, 17). In addition, LRRs 2 to 5 are required for nuclear import, lending to speculation of a noncanonical nuclear localization sequence within this region (17). In initial studies, EGF stimulation of a panel of cell lines and primary patient fibroblasts was found to result in a robust translocation of SHOC2 into the nucleus (4). However, these findings conflict with reports involving the MRAS/RAF1/SHOC2/HUWE1 complex, whereby EGF treatment consistently results in SHOC2 detected in late endosomes and a clear absence of nuclear translocation (15, 20). Nuanced differences in EGF concentrations, treatment duration, and cell models are unlikely to explain this disparity, as the conflicting studies utilized similar EGF treatment conditions and reported disparate SHOC2 localization in the same COS-1 cell line (4, 15, 17, 20).
Nevertheless, these studies highlight the intricate role of SHOC2 within various compartments of the cell that affect RTK/MAPK signaling. Although the functions of SHOC2 within each compartment have been studied in isolation, we anticipate SHOC2 likely resides in multiple compartments, including at the plasma membrane, intracellular vesicles, and within the nucleus, undergoing constant dynamic trafficking.
SHOC2 IN RAS MUTANT CANCERS
Given the functional importance of SHOC2 in RAS signaling, it is not surprising that SHOC2 has been shown to play an instrumental role in transformation, metastasis, epithelial-to-mesenchymal transition (EMT), and MAPK pathway inhibitor resistance (16, 50, 56, 60, 61, 67, 72–74). RAS family proteins are known to be one of the most frequently mutated oncogenes, where ∼30% of all cancers possess constitutively active mutations in KRAS, HRAS, or NRAS (75, 76). RAS hot spot mutations have been found to drive many tumor types, and RAS mutant cancers display a clear addiction to oncogenic RAS (75, 76). For these reasons, RAS proteins have been long-sought-after therapeutic targets but have proven challenging to directly target (75). The development of KRAS G12C covalent inhibitors, which covalently bind to the sulfur atom found within the mutated cysteine residue (77, 78), has shown responses in early clinical trials.
Several RAS pathway germ line mutations have been described in Noonan and Noonan-like syndromes. These mutations cause constitutive MAPK activation, and, accordingly, these patients have been found to be predisposed to certain cancers (79, 80). Although many of the Noonan/Noonan-like syndrome-associated genes have also been found to be somatically mutated in spontaneously arising cancers, somatic mutations in SHOC2 have not been described to date. Specifically, no SHOC2 mutations were found in a sizable cohort of leukemia biopsy specimens (81). In addition, SHOC2 is rarely found to be amplified or mutated in numerous cancer types, with mutation rates ranging from <1% in glioblastoma to 2.9% in uterine cancers (cBioPortal) (Fig. 4A), and there are no apparent hot-spot mutations of SHOC2 (Fig. 4B). However, SHOC2 is ubiquitously and abundantly expressed in many cancer types (TCGA PanCan) and has been found to be significantly upregulated in metastatic lesions derived from colorectal cancer patients (56). Pancreatic cancers have been found to display increased expression of SHOC2 compared to normal adjacent tissue, and higher intratumoral SHOC2 protein levels are associated with a substantial reduction in overall survival (50). Finally, high expression of SHOC2 has also been clinically implicated in tyrosine kinase inhibitor resistance of EGFR mutant non-small-cell lung cancer (82).
FIG 4.

SHOC2 gene mutation, alterations, and dependency in cancer. (A) Percent occurrence of SHOC2 alterations found in various human cancers (TCGA, cBioPortal). SHOC2 mutation, amplification, and deletion frequencies are indicated in green, red, and blue, respectively. (B) Lollipop graph indicating SHOC2 mutations found across cancers (TCGA, cBioPortal). The x axis represents the span of full-length SHOC2 (582 aa), and the y axis represents the number of samples harboring mutations, with missense mutations colored green and nonsense mutations colored black. (C) Violin plot of SHOC2 depletion score (CERES) in WT NRAS and mutant NRAS across cell lines in genome-scale CRISPR-Cas9 screens (DepMap; 20Q2).
Functionally, SHOC2 has been identified as a potent synthetic lethal interactor of mutant K/NRAS in acute myeloid leukemia (83) and as a dependency in NRAS mutant cells (84) (Fig. 4C). The loss of SHOC2 also results in reduced proliferation of KRAS mutant carcinoma cell lines grown in low attachment (60, 61). However, SHOC2 is found to be dispensable for the proliferation of the same carcinoma cell lines grown in two dimensions, suggesting that three-dimensional culture conditions affect the observed growth dependence on SHOC2 expression (60, 61).
In addition, recent studies have highlighted SHOC2 as a potent synthetic lethal interactor with MEKi. MEK1/2 are key downstream effectors of KRAS, and several studies demonstrated that MEK inhibition elicits a potent antitumorigenic effect (85–87). Furthermore, MEK has been shown to play a vital role in the initiation and maintenance of RAS mutant cancers (87). However, MEKi as a single therapeutic agent has been shown to be limited due to both acquired and intrinsic resistance mechanisms, leading to cancer resurgence (88–91).
In genome-scale CRISPR/Cas9 fitness screens performed in multiple KRAS mutant cancer cells, SHOC2 emerged as a highly penetrant and consistent synthetic lethal interactor of MEKi (60). BRAF mutant cancer cells upregulate RTK signaling in response to RAF inhibitor treatment as a means of reactivating the MAPK pathway, leading to adaptive resistance (92). These findings were extended to additional MAPK inhibitors in cancer cells exhibiting compensatory RTK-mediated adaptive resistance and have led to efforts to inhibit adapter proteins critical for RTK signaling, including GRB2 and SHP2 inhibitors (93–97). SHOC2 plays a critical role in RTK-mediated reactivation of the MAPK pathway in the context of KRAS mutant cancer cells treated with MEKi (60, 61), whereby the loss of SHOC2 leads to diminished MEKi-induced RAF dimerization, blunting MAPK reactivation (61). Functionally, RAF1 and BRAF were found to be important in the context of MEKi adaptive resistance, whereas ARAF was dispensable (61). Small-molecule inhibitor screens also identified MEK inhibitors to be synergistic with SHOC2 loss in KRAS mutant lung adenocarcinoma (61).
SHOC2 AS A POTENTIAL THERAPEUTIC TARGET
Deletion of SHOC2 induces a penetrant phenotype in forward genetic screens of C. elegans (8, 10) as well as multiple genome scale, single-gene CRISPR/Cas9 fitness screens in human cells (60, 74, 83, 84, 98), suggesting a uniquely essential role of SHOC2 in regulating the RTK/RAS signaling circuit (Fig. 5). Thus, SHOC2 may serve as a unique vulnerability within the RTK/RAS/MAPK pathway.
FIG 5.

RTK/MAPK signaling circuitry. The receptor tyrosine kinase (RTK)/mitogen-activated protein kinase (MAPK) signaling pathway is represented as a circuit diagram, whereby RTKs are represented as parallel arranged power sources (green batteries) that transmit signal (e.g., electricity) downstream through functionally redundant paralogs, represented as parallel switches. (A) The loss of one paralog (single switch) still permits downstream signaling (MAPK signaling is represented as a lamp). (B) SHOC2 functions as a critical point of passage for RTK signal transduction to the MAPK pathway due to a lack of functionally redundant paralogs. The loss of SHOC2 alone is sufficient to abrogate the activation of the MAPK pathway.
SHOC2 has a relatively simplistic solenoid structure with no conventionally tractable enzymatic domains. Since SHOC2 serves as a scaffold, small-molecule ligands that disrupt essential interactions may be sufficient to elicit a therapeutic effect. For instance, SHOC2-RAS interactions are required for SHOC2 function, and the design of a small-molecule ligand that competitively or allosterically inhibits SHOC2 binding to RAS should efficiently blunt downstream signaling events. Alternatively, enhancing the interaction of SHOC2 with inhibitory proteins such as ERBIN or SCRIB that off-compete RAS or sequester PP1, respectively, may prove effective as well. However, this approach may be limited by context-specific interactions due to variable expression levels of each binding partner. Another promising therapeutic strategy may involve ligand-mediated proteasomal degradation. Protein degraders are bifunctional small molecules that eliminate the protein by recruiting E3 ligases to a target protein of interest, resulting in ubiquitin-linked proteasomal degradation (99, 100). However, not all proteins have been found to be amenable to this strategy (99). Utilizing a degrader model system, we demonstrated the feasibility of acute and efficient SHOC2 depletion by the degrader approach in KRAS mutant cancer cells, and SHOC2 degradation resulted in a profound MEKi sensitization (60).
FURTHER AREAS OF INTEREST IN SHOC2 BIOLOGY
Although it is clear that SHOC2 plays a key role in RTK/RAS signaling, several questions remain. First, the exclusivity of RAS family member interactions with SHOC2 remains unclear. Some studies have provided compelling evidence that SHOC2 exhibits preferential specificity to MRAS binding compared to the other RAS family members (15, 21, 28). As described above, there is both biochemical and genetic evidence for relevant interactions with MRAS, and clinical evidence of mutant MRAS, RAF1, and PPP1CB occurring in cases of Noonan-like syndrome provides strong support for this interaction (101–105). However, the exclusivity of SHOC2-MRAS interactions previously reported are in direct conflict with various independent groups demonstrating clear interactions between SHOC2 and KRAS (10, 11, 26). Furthermore, SHOC2 but not MRAS and PP1 scored in several genome-scale CRISPR/Cas9 fitness screens (60, 74, 83, 84, 98). The absence of PP1 in single-gene knockout screens may be explained by the pan-essential nature of PP1α and PP1β (16). One potential explanation could be that although SHOC2 binds MRAS to form a stable complex, transiently weak interactions with other RAS family proteins may be sufficient to localize SHOC2 to the membrane to properly regulate MAPK signaling. Detection of these transient interactions may require different assays than the previously reported immunoprecipitation studies. Indeed, prior work demonstrated clear SHOC2 interactions with H/K/NRAS proteins in yeast two-hybrid assays (10, 11), and bioluminescence resonance energy transfer assay displayed weak interactions between SHOC2 and mutant KRAS (28).
SHOC2 regulates MAPK signaling by recruiting partners that posttranslationally modify RAF kinases, and two distinct models of SHOC2-dependent regulation of RAF activity have been identified. In one model, SHOC2 complexes with PP1 to dephosphorylate the inhibitory “S259” site on RAF proteins at the cytoplasmic membrane, enabling its activation. In the second, SHOC2 binds HUWE1, which ubiquitinates and inhibits RAF1 signaling. How these two complexes interrelate and if they participate in the same subpools of SHOC2 to regulate MAPK signaling have not been well studied. One possibility is that the two complexes work in tandem: the SHOC2 holophosphatase enables initial RAF activation followed by negative feedback of MAPK signaling through SHOC2/HUWE1 ubiquitination of RAF1. However, further work investigating if SHOC2-mediated dephosphorylation and ubiquitination act in concert or in parallel to regulate the RAF proteins is needed.
Finally, SHOC2 is found to be expressed broadly with low tissue specificity (www.proteinatlas.org) (106), and binding partners that control complex disassembly or posttranslationally modify SHOC2 seem to impart context-specific regulation, raising the possibility that additional interactors mediate regulatory specificity over MAPK signaling. Given the expanding repertoire of SHOC2 interactions, systematically uncovering the binding partners of the SHOC2-regulome and elucidating their functions will be highly impactful in broadening our understanding of RTK/RAS signaling.
ACKNOWLEDGMENTS
This work was supported in part by funds from the National Cancer Institute (NCI) F32CA243290 (J.J.K.), Dana-Farber Cancer Institute Hale Center for Pancreatic Cancer Research (W.C.H.), P50CA127003 (W.C.H.), U01 CA224146 (W.C.H.), and U01 CA199253 (W.C.H.). W.C.H. is a consultant for ThermoFisher, Solasta, MPM Capital, iTeos, Frontier Medicines, Jubilant Therapeutics, Tyra Biosciences, KSQ Therapeutics, RAPPTA Therapeutics, and Paraxel.
We thank Alvaro Amor, Jonathan So, Josh Pan, and Justin Hwang for their input and suggestions.
J.J.K. and W.C.H. conceptualized and wrote the manuscript.
REFERENCES
- 1.Simanshu DK, Nissley DV, McCormick F. 2017. RAS proteins and their regulators in human disease. Cell 170:17–33. doi: 10.1016/j.cell.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lemmon MA, Schlessinger J. 2010. Cell signaling by receptor tyrosine kinases. Cell 141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Terrell EM, Morrison DK. 2019. Ras-mediated activation of the Raf family kinases. Cold Spring Harb Perspect Med 9:a033746. doi: 10.1101/cshperspect.a033746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cordeddu V, Di Schiavi E, Pennacchio LA, Ma'ayan A, Sarkozy A, Fodale V, Cecchetti S, Cardinale A, Martin J, Schackwitz W, Lipzen A, Zampino G, Mazzanti L, Digilio MC, Martinelli S, Flex E, Lepri F, Bartholdi D, Kutsche K, Ferrero GB, Anichini C, Selicorni A, Rossi C, Tenconi R, Zenker M, Merlo D, Dallapiccola B, Iyengar R, Bazzicalupo P, Gelb BD, Tartaglia M. 2009. Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat Genet 41:1022–1026. doi: 10.1038/ng.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leach NT, Wilson Mathews DR, Rosenblum LS, Zhou Z, Zhu H, Heim RA. 2019. Correction: comparative assessment of gene-specific variant distribution in prenatal and postnatal cohorts tested for Noonan syndrome and related conditions. Genet Med 21:1670. doi: 10.1038/s41436-018-0128-z. [DOI] [PubMed] [Google Scholar]
- 6.Jang ER, Galperin E. 2016. The function of Shoc2: a scaffold and beyond. Commun Integr Biol 9:e1188241. doi: 10.1080/19420889.2016.1188241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jang H, Stevens P, Gao T, Galperin E. 17 June 2020. The leucine-rich repeats signaling scaffolds Shoc2 and Erbin: cellular mechanism and role in disease. FEBS J doi: 10.1111/febs.15450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeVore DL, Horvitz HR, Stern MJ. 1995. An FGF receptor signaling pathway is required for the normal cell migrations of the sex myoblasts in C. elegans hermaphrodites. Cell 83:611–620. doi: 10.1016/0092-8674(95)90101-9. [DOI] [PubMed] [Google Scholar]
- 9.Selfors LM, Schutzman JL, Borland CZ, Stern MJ. 1998. soc-2 encodes a leucine-rich repeat protein implicated in fibroblast growth factor receptor signaling. Proc Natl Acad Sci U S A 95:6903–6908. doi: 10.1073/pnas.95.12.6903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sieburth DS, Sun Q, Han M. 1998. SUR-8, a conserved Ras-binding protein with leucine-rich repeats, positively regulates Ras-mediated signaling in C. elegans. Cell 94:119–130. doi: 10.1016/S0092-8674(00)81227-1. [DOI] [PubMed] [Google Scholar]
- 11.Li W, Han M, Guan KL. 2000. The leucine-rich repeat protein SUR-8 enhances MAP kinase activation and forms a complex with Ras and Raf. Genes Dev 14:895–900. [PMC free article] [PubMed] [Google Scholar]
- 12.Kajava AV. 2012. Tandem repeats in proteins: from sequence to structure. J Struct Biol 179:279–288. doi: 10.1016/j.jsb.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 13.Young LC, Hartig N, Boned Del Rio I, Sari S, Ringham-Terry B, Wainwright JR, Jones GG, McCormick F, Rodriguez-Viciana P. 2018. SHOC2-MRAS-PP1 complex positively regulates RAF activity and contributes to Noonan syndrome pathogenesis. Proc Natl Acad Sci U S A 115:E10576–E10585. doi: 10.1073/pnas.1720352115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsushima N, Takatsuka S, Miyashita H, Kretsinger RH. 2019. Leucine rich repeat proteins: sequences, mutations, structures and diseases. Protein Pept Lett 26:108–131. doi: 10.2174/0929866526666181208170027. [DOI] [PubMed] [Google Scholar]
- 15.Jeoung M, Abdelmoti L, Jang ER, Vander Kooi CW, Galperin E. 2013. Functional integration of the conserved domains of Shoc2 scaffold. PLoS One 8:e66067. doi: 10.1371/journal.pone.0066067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Young LC, Hartig N, Muñoz-Alegre M, Oses-Prieto JA, Durdu S, Bender S, Vijayakumar V, Vietri Rudan M, Gewinner C, Henderson S, Jathoul AP, Ghatrora R, Lythgoe MF, Burlingame AL, Rodriguez-Viciana P. 2013. Rodriguez-Viciana P an MRAS, SHOC2, and SCRIB complex coordinates ERK pathway activation with polarity and tumorigenic growth. Mol Cell 52:679–692. doi: 10.1016/j.molcel.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 17.Motta M, Chillemi G, Fodale V, Cecchetti S, Coppola S, Stipo S, Cordeddu V, Macioce P, Gelb BD, Tartaglia M. 2016. SHOC2 subcellular shuttling requires the KEKE motif-rich region and N-terminal leucine-rich repeat domain and impacts on ERK signalling. Hum Mol Genet 25:3824–3835. doi: 10.1093/hmg/ddw229. [DOI] [PubMed] [Google Scholar]
- 18.Motta M, Giancotti A, Mastromoro G, Chandramouli B, Pinna V, Pantaleoni F, Di Giosaffatte N, Petrini S, Mazza T, D'Ambrosio V, Versacci P, Ventriglia F, Chillemi G, Pizzuti A, Tartaglia M, De Luca A. 2019. Clinical and functional characterization of a novel RASopathy-causing SHOC2 mutation associated with prenatal-onset hypertrophic cardiomyopathy. Hum Mutat 40:1046–1056. doi: 10.1002/humu.23767. [DOI] [PubMed] [Google Scholar]
- 19.Jang H, Oakley E, Forbes-Osborne M, Kesler MV, Norcross R, Morris AC, Galperin E. 2019. Hematopoietic and neural crest defects in zebrafish shoc2 mutants: a novel vertebrate model for Noonan-like syndrome. Hum Mol Genet 28:501–514. doi: 10.1093/hmg/ddy366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galperin E, Abdelmoti L, Sorkin A. 2012. Shoc2 is targeted to late endosomes and required for Erk1/2 activation in EGF-stimulated cells. PLoS One 7:e36469. doi: 10.1371/journal.pone.0036469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodriguez-Viciana P, Oses-Prieto J, Burlingame A, Fried M, McCormick F. 2006. A phosphatase holoenzyme comprised of Shoc2/Sur8 and the catalytic subunit of PP1 functions as an M-Ras effector to modulate Raf activity. Mol Cell 22:217–230. doi: 10.1016/j.molcel.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 22.Peti W, Nairn AC, Page R. 2013. Structural basis for protein phosphatase 1 regulation and specificity. FEBS J 280:596–611. doi: 10.1111/j.1742-4658.2012.08509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tzivion G, Luo Z, Avruch J. 1998. A dimeric 14–3-3 protein is an essential cofactor for Raf kinase activity. Nature 394:88–92. doi: 10.1038/27938. [DOI] [PubMed] [Google Scholar]
- 24.Lavoie H, Therrien M. 2015. Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol 16:281–298. doi: 10.1038/nrm3979. [DOI] [PubMed] [Google Scholar]
- 25.Young LC, Rodriguez-Viciana P. 2018. MRAS: a close but understudied member of the RAS family. Cold Spring Harb Perspect Med 8:a033621. doi: 10.1101/cshperspect.a033621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsunaga-Udagawa R, Fujita Y, Yoshiki S, Terai K, Kamioka Y, Kiyokawa E, Yugi K, Aoki K, Matsuda M. 2010. The scaffold protein Shoc2/SUR-8 accelerates the interaction of Ras and Raf. J Biol Chem 285:7818–7826. doi: 10.1074/jbc.M109.053975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohba Y, Mochizuki N, Yamashita S, Chan AM, Schrader JW, Hattori S, Nagashima K, Matsuda M. 2000. Regulatory proteins of R-Ras, TC21/R-Ras2, and M-Ras/R-Ras3. J Biol Chem 275:20020–20026. doi: 10.1074/jbc.M000981200. [DOI] [PubMed] [Google Scholar]
- 28.Kota P, Terrell EM, Ritt DA, Insinna C, Westlake CJ, Morrison DK. 2019. M-Ras/Shoc2 signaling modulates E-cadherin turnover and cell-cell adhesion during collective cell migration. Proc Natl Acad Sci U S A 116:3536–3545. doi: 10.1073/pnas.1805919116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hannig V, Jeoung M, Jang ER, Phillips JA, III, Galperin E. 2014. A novel SHOC2 variant in rasopathy. Hum Mutat 35:1290–1294. doi: 10.1002/humu.22634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jang ER, Shi P, Bryant J, Chen J, Dukhande V, Gentry MS, Jang H, Jeoung M, Galperin E. 2014. HUWE1 is a molecular link controlling RAF-1 activity supported by the Shoc2 scaffold. Mol Cell Biol 34:3579–3593. doi: 10.1128/MCB.00811-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhong Q, Gao W, Du F, Wang X. 2005. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 121:1085–1095. doi: 10.1016/j.cell.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 32.Yoon SY, Lee Y, Kim JH, Chung AS, Joo JH, Kim CN, Kim NS, Choe IS, Kim JW. 2005. Over-expression of human UREB1 in colorectal cancer: HECT domain of human UREB1 inhibits the activity of tumor suppressor p53 protein. Biochem Biophys Res Commun 326:7–17. doi: 10.1016/j.bbrc.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 33.Zhao X, Heng JI, Guardavaccaro D, Jiang R, Pagano M, Guillemot F, Iavarone A, Lasorella A. 2008. The HECT-domain ubiquitin ligase Huwe1 controls neural differentiation and proliferation by destabilizing the N-Myc oncoprotein. Nat Cell Biol 10:643–653. doi: 10.1038/ncb1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jang ER, Jang H, Shi P, Popa G, Jeoung M, Galperin E. 2015. Spatial control of Shoc2-scaffold-mediated ERK1/2 signaling requires remodeling activity of the ATPase PSMC5. J Cell Sci 128:4428–4441. doi: 10.1242/jcs.177543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hanson PI, Whiteheart SW. 2005. AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol 6:519–529. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 36.Sauer RT, Baker TA. 2011. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem 80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- 37.Tanahashi N, Suzuki M, Fujiwara T, Takahashi E, Shimbara N, Chung CH, Tanaka K. 1998. Chromosomal localization and immunological analysis of a family of human 26S proteasomal ATPases. Biochem Biophys Res Commun 243:229–232. doi: 10.1006/bbrc.1997.7892. [DOI] [PubMed] [Google Scholar]
- 38.Ferdous A, Kodadek T, Johnston SA. 2002. A nonproteolytic function of the 19S regulatory subunit of the 26S proteasome is required for efficient activated transcription by human RNA polymerase II. Biochemistry 41:12798–12805. doi: 10.1021/bi020425t. [DOI] [PubMed] [Google Scholar]
- 39.Sulahian R, Sikder D, Johnston SA, Kodadek T. 2006. The proteasomal ATPase complex is required for stress-induced transcription in yeast. Nucleic Acids Res 34:1351–1357. doi: 10.1093/nar/gkl012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jang H, Jang ER, Wilson PG, Anderson D, Galperin E. 2019. VCP/p97 controls signals of the ERK1/2 pathway transmitted via the Shoc2 scaffolding complex: novel insights into IBMPFD pathology. Mol Biol Cell 30:1655–1663. doi: 10.1091/mbc.E19-03-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Borg JP, Marchetto S, Le Bivic A, Ollendorff V, Jaulin-Bastard F, Saito H, Fournier E, Adelaide J, Margolis B, Birnbaum D. 2000. ERBIN: a basolateral PDZ protein that interacts with the mammalian ERBB2/HER2 receptor. Nat Cell Biol 2:407–414. doi: 10.1038/35017038. [DOI] [PubMed] [Google Scholar]
- 42.Metais JY, Navarro C, Santoni MJ, Audebert S, Borg JP. 2005. hScrib interacts with ZO-2 at the cell-cell junctions of epithelial cells. FEBS Lett 579:3725–3730. doi: 10.1016/j.febslet.2005.05.062. [DOI] [PubMed] [Google Scholar]
- 43.Choi J, Troyanovsky RB, Indra I, Mitchell BJ, Troyanovsky SM. 2019. Scribble, Erbin, and Lano redundantly regulate epithelial polarity and apical adhesion complex. J Cell Biol 218:2277–2293. doi: 10.1083/jcb.201804201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bilder D, Birnbaum D, Borg JP, Bryant P, Huigbretse J, Jansen E, Kennedy MB, Labouesse M, Legouis R, Mechler B, Perrimon N, Petit M, Sinha P. 2000. Collective nomenclature for LAP proteins. Nat Cell Biol 2:E114. doi: 10.1038/35017119. [DOI] [PubMed] [Google Scholar]
- 45.Shi M, Zhao M, Hu M, Liu D, Cao H, Qian L, Yang Z, Hu Y, Yu M, Yang S, Ma Y, Guo N. 2013. beta2-AR-induced Her2 transactivation mediated by Erbin confers protection from apoptosis in cardiomyocytes. Int J Cardiol 167:1570–1577. doi: 10.1016/j.ijcard.2012.04.093. [DOI] [PubMed] [Google Scholar]
- 46.Dai P, Xiong WC, Mei L. 2006. Erbin inhibits RAF activation by disrupting the sur-8-Ras-Raf complex. J Biol Chem 281:927–933. doi: 10.1074/jbc.M507360200. [DOI] [PubMed] [Google Scholar]
- 47.Yi J, Chen M, Wu X, Yang X, Xu T, Zhuang Y, Han M, Xu R. 2010. Endothelial SUR-8 acts in an ERK-independent pathway during atrioventricular cushion development. Dev Dyn 239:2005–2013. doi: 10.1002/dvdy.22343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rachmin I, Tshori S, Smith Y, Oppenheim A, Marchetto S, Kay G, Foo RS, Dagan N, Golomb E, Gilon D, Borg JP, Razin E. 2014. Erbin is a negative modulator of cardiac hypertrophy. Proc Natl Acad Sci U S A 111:5902–5907. doi: 10.1073/pnas.1320350111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harmon RM, Simpson CL, Johnson JL, Koetsier JL, Dubash AD, Najor NA, Sarig O, Sprecher E, Green KJ. 2013. Desmoglein-1/Erbin interaction suppresses ERK activation to support epidermal differentiation. J Clin Investig 123:1556–1570. doi: 10.1172/JCI65220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xie CM, Tan M, Lin XT, Wu D, Jiang Y, Tan Y, Li H, Ma Y, Xiong X, Sun Y. 2019. The FBXW7-SHOC2-Raptor Axis Controls the Cross-Talks between the RAS-ERK and mTORC1 Signaling Pathways. Cell Rep 26:3037–3050. doi: 10.1016/j.celrep.2019.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu GY, Sabatini DM. 2020. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol 21:183–203. doi: 10.1038/s41580-019-0199-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. 2002. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110:163–175. doi: 10.1016/S0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 53.Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K. 2002. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110:177–189. doi: 10.1016/S0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- 54.Nojima H, Tokunaga C, Eguchi S, Oshiro N, Hidayat S, Yoshino K, Hara K, Tanaka N, Avruch J, Yonezawa K. 2003. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem 278:15461–15464. doi: 10.1074/jbc.C200665200. [DOI] [PubMed] [Google Scholar]
- 55.Moon BS, Kim HY, Kim MY, Yang DH, Lee JM, Cho KW, Jung HS, Choi KY. 2011. Sur8/Shoc2 involves both inhibition of differentiation and maintenance of self-renewal of neural progenitor cells via modulation of extracellular signal-regulated kinase signaling. Stem Cells 29:320–331. doi: 10.1002/stem.586. [DOI] [PubMed] [Google Scholar]
- 56.Lee KH, Jeong WJ, Cha PH, Lee SK, Min DS, Choi KY. 2017. Stabilization of Sur8 via PKCalpha/delta degradation promotes transformation and migration of colorectal cancer cells. Oncotarget 8:115596–115608. doi: 10.18632/oncotarget.23313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yeh CH, Bellon M, Nicot C. 2018. FBXW7: a critical tumor suppressor of human cancers. Mol Cancer 17:115. doi: 10.1186/s12943-018-0857-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hao B, Oehlmann S, Sowa ME, Harper JW, Pavletich NP. 2007. Structure of a Fbw7-Skp1-cyclin E complex: multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Mol Cell 26:131–143. doi: 10.1016/j.molcel.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 59.Nateri AS, Riera-Sans L, Da Costa C, Behrens A. 2004. The ubiquitin ligase SCFFbw7 antagonizes apoptotic JNK signaling. Science 303:1374–1378. doi: 10.1126/science.1092880. [DOI] [PubMed] [Google Scholar]
- 60.Sulahian R, Kwon JJ, Walsh KH, Pailler E, Bosse TL, Thaker M, Almanza D, Dempster JM, Pan J, Piccioni F, Dumont N, Gonzalez A, Rennhack J, Nabet B, Bachman JA, Goodale A, Lee Y, Bagul M, Liao R, Navarro A, Yuan TL, Ng RWS, Raghavan S, Gray NS, Tsherniak A, Vazquez F, Root DE, Firestone AJ, Settleman J, Hahn WC, Aguirre AJ. 2019. Synthetic lethal interaction of SHOC2 depletion with MEK inhibition in RAS-driven cancers. Cell Rep 29:118–134. doi: 10.1016/j.celrep.2019.08.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jones GG, Del Rio IB, Sari S, Sekerim A, Young LC, Hartig N, Areso Zubiaur I, El-Bahrawy MA, Hynds RE, Lei W, Molina-Arcas M, Downward J, Rodriguez-Viciana P. 2019. SHOC2 phosphatase-dependent RAF dimerization mediates resistance to MEK inhibition in RAS-mutant cancers. Nat Commun 10:2532. doi: 10.1038/s41467-019-10367-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Asati V, Mahapatra DK, Bharti SK. 2016. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: structural and pharmacological perspectives. Eur J Med Chem 109:314–341. doi: 10.1016/j.ejmech.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 63.Ramjaun AR, Downward J. 2007. Ras and phosphoinositide 3-kinase: partners in development and tumorigenesis. Cell Cycle 6:2902–2905. doi: 10.4161/cc.6.23.4996. [DOI] [PubMed] [Google Scholar]
- 64.Hervieu A, Kermorgant S. 2018. The role of PI3K in Met driven cancer: a recap. Front Mol Biosci 5:86. doi: 10.3389/fmolb.2018.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fruman DA, Rommel C. 2014. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov 13:140–156. doi: 10.1038/nrd4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hemmings BA, Restuccia DF. 2012. PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol 4:a011189. doi: 10.1101/cshperspect.a011189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kaduwal S, Jeong WJ, Park JC, Lee KH, Lee YM, Jeon SH, Lim YB, Min do S, Choi KY. 2015. Sur8/Shoc2 promotes cell motility and metastasis through activation of Ras-PI3K signaling. Oncotarget 6:33091–33105. doi: 10.18632/oncotarget.5173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Good MC, Zalatan JG, Lim WA. 2011. Scaffold proteins: hubs for controlling the flow of cellular information. Science 332:680–686. doi: 10.1126/science.1198701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dhanasekaran DN, Kashef K, Lee CM, Xu H, Reddy EP. 2007. Scaffold proteins of MAP-kinase modules. Oncogene 26:3185–3202. doi: 10.1038/sj.onc.1210411. [DOI] [PubMed] [Google Scholar]
- 70.Parker JA, Mattos C. 2015. The Ras-membrane interface: isoform-specific differences in the catalytic domain. Mol Cancer Res 13:595–603. doi: 10.1158/1541-7786.MCR-14-0535. [DOI] [PubMed] [Google Scholar]
- 71.Bergeron JJ, Di Guglielmo GM, Dahan S, Dominguez M, Posner BI. 2016. Spatial and temporal regulation of receptor tyrosine kinase activation and intracellular signal transduction. Annu Rev Biochem 85:573–597. doi: 10.1146/annurev-biochem-060815-014659. [DOI] [PubMed] [Google Scholar]
- 72.Kaplan FM, Kugel CH, III, Dadpey N, Shao Y, Abel EV, Aplin AE. 2012. SHOC2 and CRAF mediate ERK1/2 reactivation in mutant NRAS-mediated resistance to RAF inhibitor. J Biol Chem 287:41797–41807. doi: 10.1074/jbc.M112.390906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee YM, Kaduwal S, Lee KH, Park JC, Jeong WJ, Choi KY. 2016. Sur8 mediates tumorigenesis and metastasis in colorectal cancer. Exp Mol Med 48:e249. doi: 10.1038/emm.2016.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Han K, Pierce SE, Li A, Spees K, Anderson GR, Seoane JA, Lo YH, Dubreuil M, Olivas M, Kamber RA, Wainberg M, Kostyrko K, Kelly MR, Yousefi M, Simpkins SW, Yao D, Lee K, Kuo CJ, Jackson PK, Sweet-Cordero A, Kundaje A, Gentles AJ, Curtis C, Winslow MM, Bassik MC. 2020. CRISPR screens in cancer spheroids identify 3D growth-specific vulnerabilities. Nature 580:136–141. doi: 10.1038/s41586-020-2099-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. 2014. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov 13:828–851. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. 2011. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer 11:761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, Chen Y, Babbar A, Firdaus SJ, Darjania L, Feng J, Chen JH, Li S, Li S, Long YO, Thach C, Liu Y, Zarieh A, Ely T, Kucharski JM, Kessler LV, Wu T, Yu K, Wang Y, Yao Y, Deng X, Zarrinkar PP, Brehmer D, Dhanak D, Lorenzi MV, Hu-Lowe D, Patricelli MP, Ren P, Liu Y. 2018. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 172:578–589. doi: 10.1016/j.cell.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 78.O'Bryan JP. 2019. Pharmacological targeting of RAS: recent success with direct inhibitors. Pharmacol Res 139:503–511. doi: 10.1016/j.phrs.2018.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Villani A, Greer MC, Kalish JM, Nakagawara A, Nathanson KL, Pajtler KW, Pfister SM, Walsh MF, Wasserman JD, Zelley K, Kratz CP. 2017. Recommendations for cancer surveillance in individuals with RASopathies and other rare genetic conditions with increased cancer risk. Clin Cancer Res 23:e83–e90. doi: 10.1158/1078-0432.CCR-17-0631. [DOI] [PubMed] [Google Scholar]
- 80.Jongmans MC, van der Burgt I, Hoogerbrugge PM, Noordam K, Yntema HG, Nillesen WM, Kuiper RP, Ligtenberg MJ, van Kessel AG, van Krieken JH, Kiemeney LA, Hoogerbrugge N. 2011. Cancer risk in patients with Noonan syndrome carrying a PTPN11 mutation. Eur J Hum Genet 19:870–874. doi: 10.1038/ejhg.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Komatsuzaki S, Aoki Y, Niihori T, Okamoto N, Hennekam RC, Hopman S, Ohashi H, Mizuno S, Watanabe Y, Kamasaki H, Kondo I, Moriyama N, Kurosawa K, Kawame H, Okuyama R, Imaizumi M, Rikiishi T, Tsuchiya S, Kure S, Matsubara Y. 2010. Mutation analysis of the SHOC2 gene in Noonan-like syndrome and in hematologic malignancies. J Hum Genet 55:801–809. doi: 10.1038/jhg.2010.116. [DOI] [PubMed] [Google Scholar]
- 82.Terai H, Hamamoto J, Emoto K, Masuda T, Manabe T, Kuronuma S, Kobayashi K, Masuzawa K, Ikemura S, Nakayama S, Kawada I, Suzuki Y, Takeuchi O, Suzuki Y, Ohtsuki S, Yasuda H, Soejima K, Fukunaga K. 26 October 2020. SHOC2 is a critical modulator of sensitivity to EGFR-TKIs in non-small cell lung cancer cells. Mol Cancer Res doi: 10.1158/1541-7786.MCR-20-0664. [DOI] [PubMed] [Google Scholar]
- 83.Wang T, Yu H, Hughes NW, Liu B, Kendirli A, Klein K, Chen WW, Lander ES, Sabatini DM. 2017. Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic Ras. Cell 168:890–903. doi: 10.1016/j.cell.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Behan FM, Iorio F, Picco G, Goncalves E, Beaver CM, Migliardi G, Santos R, Rao Y, Sassi F, Pinnelli M, Ansari R, Harper S, Jackson DA, McRae R, Pooley R, Wilkinson P, van der Meer D, Dow D, Buser-Doepner C, Bertotti A, Trusolino L, Stronach EA, Saez-Rodriguez J, Yusa K, Garnett MJ. 2019. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 568:511–516. doi: 10.1038/s41586-019-1103-9. [DOI] [PubMed] [Google Scholar]
- 85.Sebolt-Leopold JS, Herrera R. 2004. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat Rev Cancer 4:937–947. doi: 10.1038/nrc1503. [DOI] [PubMed] [Google Scholar]
- 86.Samatar AA, Poulikakos PI. 2014. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov 13:928–942. doi: 10.1038/nrd4281. [DOI] [PubMed] [Google Scholar]
- 87.Collisson EA, Trejo CL, Silva JM, Gu S, Korkola JE, Heiser LM, Charles RP, Rabinovich BA, Hann B, Dankort D, Spellman PT, Phillips WA, Gray JW, McMahon M. 2012. A central role for RAF–>MEK–>ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer Discov 2:685–693. doi: 10.1158/2159-8290.CD-11-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Little AS, Balmanno K, Sale MJ, Newman S, Dry JR, Hampson M, Edwards PA, Smith PD, Cook SJ. 2011. Amplification of the driving oncogene, KRAS or BRAF, underpins acquired resistance to MEK1/2 inhibitors in colorectal cancer cells. Sci Signal 4:ra17. doi: 10.1126/scisignal.2001752. [DOI] [PubMed] [Google Scholar]
- 89.Sun C, Hobor S, Bertotti A, Zecchin D, Huang S, Galimi F, Cottino F, Prahallad A, Grernrum W, Tzani A, Schlicker A, Wessels LF, Smit EF, Thunnissen E, Halonen P, Lieftink C, Beijersbergen RL, Di Nicolantonio F, Bardelli A, Trusolino L, Bernards R. 2014. Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep 7:86–93. doi: 10.1016/j.celrep.2014.02.045. [DOI] [PubMed] [Google Scholar]
- 90.Halilovic E, She QB, Ye Q, Pagliarini R, Sellers WR, Solit DB, Rosen N. 2010. PIK3CA mutation uncouples tumor growth and cyclin D1 regulation from MEK/ERK and mutant KRAS signaling. Cancer Res 70:6804–6814. doi: 10.1158/0008-5472.CAN-10-0409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dai B, Meng J, Peyton M, Girard L, Bornmann WG, Ji L, Minna JD, Fang B, Roth JA. 2011. STAT3 mediates resistance to MEK inhibitor through microRNA miR-17. Cancer Res 71:3658–3668. doi: 10.1158/0008-5472.CAN-10-3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lito P, Pratilas CA, Joseph EW, Tadi M, Halilovic E, Zubrowski M, Huang A, Wong WL, Callahan MK, Merghoub T, Wolchok JD, de Stanchina E, Chandarlapaty S, Poulikakos PI, Fagin JA, Rosen N. 2012. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 22:668–682. doi: 10.1016/j.ccr.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mainardi S, Mulero-Sanchez A, Prahallad A, Germano G, Bosma A, Krimpenfort P, Lieftink C, Steinberg JD, de Wit N, Goncalves-Ribeiro S, Nadal E, Bardelli A, Villanueva A, Bernards R. 2018. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat Med 24:961–967. doi: 10.1038/s41591-018-0023-9. [DOI] [PubMed] [Google Scholar]
- 94.Fedele C, Ran H, Diskin B, Wei W, Jen J, Geer MJ, Araki K, Ozerdem U, Simeone DM, Miller G, Neel BG, Tang KH. 2018. SHP2 inhibition prevents adaptive resistance to MEK inhibitors in multiple cancer models. Cancer Discov 8:1237–1249. doi: 10.1158/2159-8290.CD-18-0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nichols RJ, Haderk F, Stahlhut C, Schulze CJ, Hemmati G, Wildes D, Tzitzilonis C, Mordec K, Marquez A, Romero J, Hsieh T, Zaman A, Olivas V, McCoach C, Blakely CM, Wang Z, Kiss G, Koltun ES, Gill AL, Singh M, Goldsmith MA, Smith JAM, Bivona TG. 2018. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat Cell Biol 20:1064–1073. doi: 10.1038/s41556-018-0169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gay B, Suarez S, Caravatti G, Furet P, Meyer T, Schoepfer J. 1999. Selective GRB2 SH2 inhibitors as anti-Ras therapy. Int J Cancer 83:235–241. doi:. [DOI] [PubMed] [Google Scholar]
- 97.Yu Y, Nie Y, Feng Q, Qu J, Wang R, Bian L, Xia J. 2017. Targeted covalent inhibition of Grb2-Sos1 interaction through proximity-induced conjugation in breast cancer cells. Mol Pharm 14:1548–1557. doi: 10.1021/acs.molpharmaceut.6b00952. [DOI] [PubMed] [Google Scholar]
- 98.Whittaker SR, Cowley GS, Wagner S, Luo F, Root DE, Garraway LA. 2015. Combined Pan-RAF and MEK inhibition overcomes multiple resistance mechanisms to selective RAF inhibitors. Mol Cancer Ther 14:2700–2711. doi: 10.1158/1535-7163.MCT-15-0136-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mullard A. 6 March 2019. First targeted protein degrader hits the clinic. Nat Rev Drug Discov doi: 10.1038/d41573-019-00043-6. [DOI] [PubMed] [Google Scholar]
- 100.Chamberlain PP, Hamann LG. 2019. Development of targeted protein degradation therapeutics. Nat Chem Biol 15:937–944. doi: 10.1038/s41589-019-0362-y. [DOI] [PubMed] [Google Scholar]
- 101.Higgins EM, Bos JM, Mason-Suares H, Tester DJ, Ackerman JP, MacRae CA, Sol-Church K, Gripp KW, Urrutia R, Ackerman MJ. 2017. Elucidation of MRAS-mediated Noonan syndrome with cardiac hypertrophy. JCI Insight 2:e91225. doi: 10.1172/jci.insight.91225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Motta M, Sagi-Dain L, Krumbach OHF, Hahn A, Peleg A, German A, Lissewski C, Coppola S, Pantaleoni F, Kocherscheid L, Altmuller F, Schanze D, Logeswaran T, Chahrokh-Zadeh S, Munzig A, Nakhaei-Rad S, Cave H, Ahmadian MR, Tartaglia M, Zenker M. 2019. Activating MRAS mutations cause Noonan syndrome associated with hypertrophic cardiomyopathy. Hum Mol Genet 29:1772–1783. doi: 10.1093/hmg/ddz108. [DOI] [PubMed] [Google Scholar]
- 103.Kobayashi T, Aoki Y, Niihori T, Cave H, Verloes A, Okamoto N, Kawame H, Fujiwara I, Takada F, Ohata T, Sakazume S, Ando T, Nakagawa N, Lapunzina P, Meneses AG, Gillessen-Kaesbach G, Wieczorek D, Kurosawa K, Mizuno S, Ohashi H, David A, Philip N, Guliyeva A, Narumi Y, Kure S, Tsuchiya S, Matsubara Y. 2010. Molecular and clinical analysis of RAF1 in Noonan syndrome and related disorders: dephosphorylation of serine 259 as the essential mechanism for mutant activation. Hum Mutat 31:284–294. doi: 10.1002/humu.21187. [DOI] [PubMed] [Google Scholar]
- 104.Gripp KW, Aldinger KA, Bennett JT, Baker L, Tusi J, Powell-Hamilton N, Stabley D, Sol-Church K, Timms AE, Dobyns WB. 2016. A novel rasopathy caused by recurrent de novo missense mutations in PPP1CB closely resembles Noonan syndrome with loose anagen hair. Am J Med Genet A 170:2237–2247. doi: 10.1002/ajmg.a.37781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lin CH, Lin WD, Chou IC, Lee IC, Fan HC, Hong SY. 2018. Epileptic spasms in PPP1CB-associated Noonan-like syndrome: a case report with clinical and therapeutic implications. BMC Neurol 18:150. doi: 10.1186/s12883-018-1157-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Ponten F. 2015. Proteomics. Tissue-based map of the human proteome. Science 347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]