

Rothmund-Thomson syndrome (RTS) is an autosomal recessive disorder characterized by defects in the skeletal system, such as bone hypoplasia, short stature, low bone mass, and an increased incidence of osteosarcoma. RTS type 2 patients have germ line compound biallelic protein-truncating mutations of RECQL4.
KEYWORDS: Rothmund-Thomson syndrome, RECQL4, RecQ, osteoporosis, osteosarcoma, mouse models, familial cancer syndrome, myeloid cells, osteoblast, tumor suppressor
ABSTRACT
Rothmund-Thomson syndrome (RTS) is an autosomal recessive disorder characterized by defects in the skeletal system, such as bone hypoplasia, short stature, low bone mass, and an increased incidence of osteosarcoma. RTS type 2 patients have germ line compound biallelic protein-truncating mutations of RECQL4. As existing murine models employ Recql4 null alleles, we have attempted to more accurately model RTS by generating mice with patient-mimicking truncating Recql4 mutations. Truncating mutations impaired the stability and subcellular localization of RECQL4 and resulted in homozygous embryonic lethality and a haploinsufficient low-bone mass phenotype. Combination of a truncating mutation with a conditional Recql4 null allele demonstrated that the skeletal defects were intrinsic to the osteoblast lineage. However, the truncating mutations did not promote tumorigenesis. We utilized murine Recql4 null cells to assess the impact of human RECQL4 mutations using an in vitro complementation assay. While some mutations created unstable protein products, others altered subcellular localization of the protein. Interestingly, the severity of the phenotypes correlated with the extent of protein truncation. Collectively, our results reveal that truncating RECQL4 mutations in mice lead to an osteoporosis-like phenotype through defects in early osteoblast progenitors and identify RECQL4 gene dosage as a novel regulator of bone mass.
INTRODUCTION
Rothmund-Thomson syndrome (RTS) (Online Mendelian Inheritance in Man [OMIM] no. 268400) is a rare autosomal recessive disorder that presents with skin rash (poikiloderma; areas of hypopigmentation, hyperpigmentation, telangiectasias, and atrophy of the skin), sparse or absent hair, juvenile cataracts, and gastrointestinal and skeletal complications (1, 2). Approximately 75% of patients have skeletal abnormalities, including bone hypoplasia, short stature, polydactyly, and low bone mass (3). Furthermore, this syndrome is frequently associated with osteosarcoma (bone cancer) and other malignancies (1, 2, 4, 5). RTS is classified into two forms, RTS type 1, where patients present with juvenile cataracts and have frequent mutations of ANAPC1 but do not have increased incidence of osteosarcoma (6), and RTS type 2, where the majority of patients harbor biallelic mutations of RECQL4 and have a significantly increased incidence of osteosarcoma (2).
RECQL4 is located on the long arm of chromosome eight (8q24.3) (7). The reported mutational spectrum includes the introduction of early stop codons, frameshift mutations, and deletions within numerous short introns (<100 bp) that can impair RNA splicing, resulting in protein truncations and loss-of-function alleles (8). The RECQL4 gene encodes a protein of 1,208 amino acids (aa) that has three well-characterized domains. The N-terminal region shares sequence homology to the essential yeast DNA replication factor Sld2. In higher eukaryotes, this Sld2-like homology domain is unique to RECQL4 (9, 10). Studies have shown that this region has roles in DNA replication and DNA repair and that it is critical for viability (11–13). The highly conserved central RecQ helicase domain contains an ATPase core with seven motifs that couple ATP hydrolysis to double-stranded DNA (dsDNA) separation (14). This ATP-dependent helicase activity was presumed to be critical for the function of RECQL4. However, using mice with a knock-in mutation (K525A) that inactivates the ATP-dependent helicase function, we recently reported that homozygous mice displayed normal embryonic development, body weight, hematopoiesis, B and T cell development, and physiological DNA damage repair (15). Finally, the C-terminal region harbors both the R4ZBD domain, in place of an RQC domain seen in other RecQ helicases, and a small C-terminal domain (CTD; 1,117 to 1,208 aa) associated with DNA-binding affinity (16). Regarding intracellular localization, RECQL4 is primarily localized in the nucleus but has also been reported in the cytoplasm (17, 18). Its distribution within these compartments, however, depends on the cell type and the phase of the cell cycle (17).
RTS type 2 patients have a high incidence of skeletal abnormalities and osteosarcoma. In a clinical cohort study evaluating 41 RTS patients, 32% developed osteosarcoma (2). Furthermore, an independent cohort study reported that the median age at diagnosis in RTS patients was 10 years of age (19). This is significantly younger than the median age of sporadic osteosarcoma, which is 16 years (20). Importantly, we and others have previously reported that in mice, the deletion of Recql4 in preosteoblasts or limb bud progenitors caused shorter bones and reduced bone volume (21, 22). However, neither model developed osteosarcoma, even in combination with Tp53 deficiency (22). In contrast to these models that generated null alleles, RTS patients predominantly have compound heterozygous RECQL4 mutations that are predicted to generate truncated protein products (4). More than half of these truncate the protein before or within the helicase domain and result in a substantially increased risk of developing osteosarcoma compared to nontruncating mutations (5). Therefore, it is critical to determine the in vivo effects of germ line truncating Recql4 mutations in normal homeostasis and tumor development.
To more faithfully model the RTS-relevant RECQL4 mutation spectrum beyond existing null alleles, we have generated mice bearing truncating mutations that map closely to those reported in RTS patients (15). Here, we show that these truncating mutations affected stability and subcellular localization of RECQL4, which translated to a homozygous developmental lethality and a haploinsufficient low-bone-mass phenotype through defects in early osteoblast progenitors. Additionally, we observed that the severity of the defect was related to the degree of the truncation, suggesting that gene dosage is an important determinant of the bone phenotype. However, unlike in RTS type 2 patients, these RECQL4 mutations were not sufficient in isolation to initiate tumorigenesis in mice, even after exposure to irradiation. This would suggest that additional molecular and cellular changes are required for the full spectrum of RTS phenotypes to develop.
RESULTS
Truncating mutations of RECQL4 affect protein expression levels and cause developmental lethality in homozygotes.
To understand the in vivo impact of truncating RECQL4 mutations, we generated two novel mouse Recql4 alleles (15). These new mutations were similar to common mutations seen in RTS type 2 patients (Fig. 1A). The p.Gly522GlufsTer43 (G522Efs) mutation, comparable to the human p.Cys525AlafsX33 (C525Afs) mutation, was created by a two-base-pair insertion (c.1646_1647insGA). The frameshift caused a premature stop codon 44 aa downstream, resulting in a predicted protein of 566 aa lacking the majority of the helicase domain and all of the C-terminal domain. The second allele was a p.Arg347* (R347X) mutation, a nonsense mutation (c.1122C>T) identified from an N-ethyl-N-nitrosourea (ENU) mutagenesis collection, similar to p.Arg350GlyfsX21 (R350Gfs) in RTS patients. This mutation yielded a predicted 347 aa protein lacking both the helicase and C-terminal domains entirely (Fig. 1B). To verify the genotypes of these mice, we used PCR-restriction fragment length polymorphism (PCR-RFLP) for the G522Efs allele and a competitive allele-specific PCR (KASP) assay for the R347X allele, both of which confirmed the correct genotype (see Fig. S1 in the supplemental material).
FIG 1.
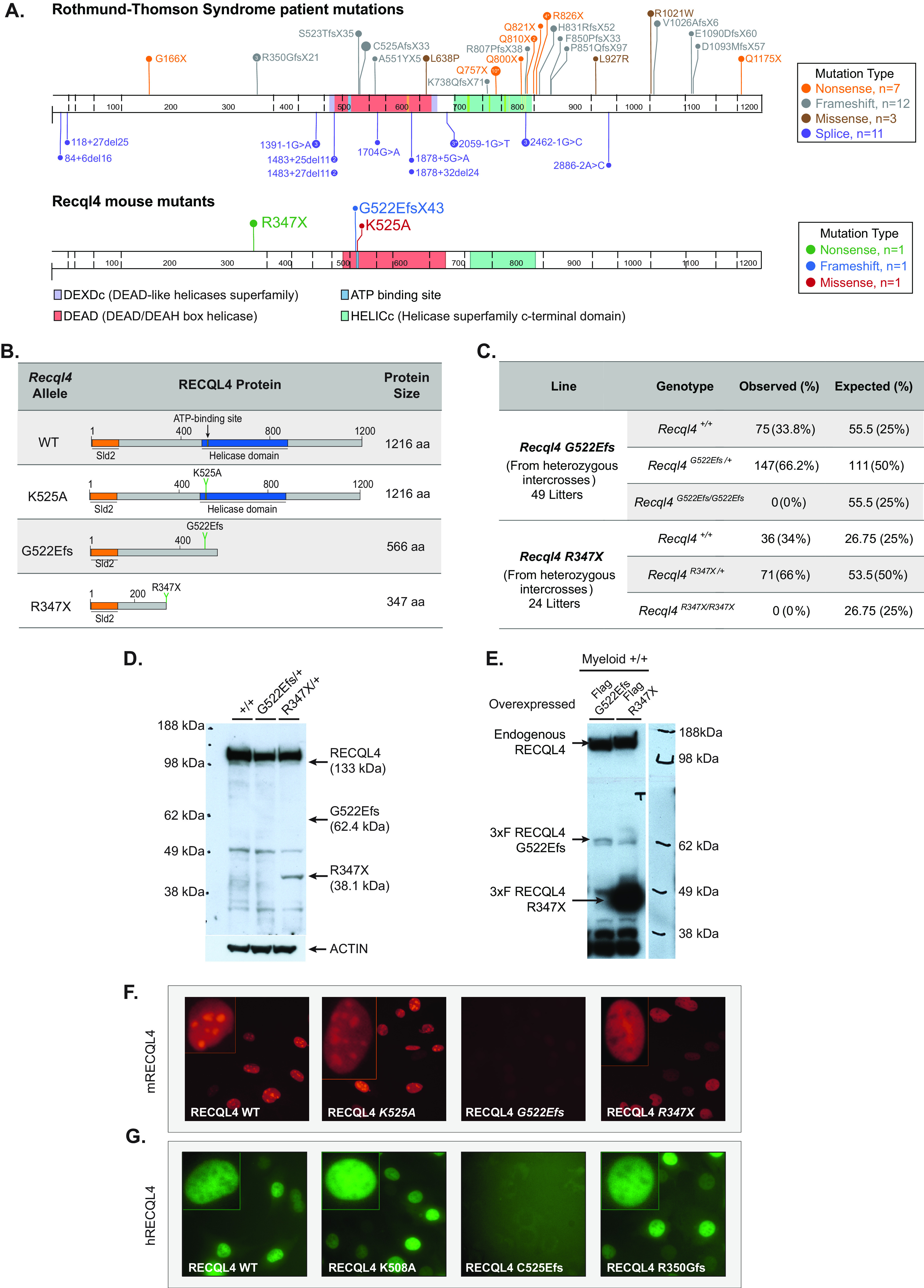
Truncating RECQL4 mutations G522Efs and R347X affect protein expression and localization differently and are homozygous embryo lethal. (A) Schematic illustration of RECQL4 mutations reported in RTS patients and murine mutations used in this study. Image generated using Protein Painter (PeCan portal, St Jude’s). (B) Recql4 mutations and their corresponding predicted protein products. (C) Breeding data from 49 litters of Recql4G522Efs/+ and 24 litters of Recql4R347X/+ intercrosses. Observed and expected Mendelian frequencies of the indicated genotypes are shown. No statistical significance was achieved. (D) Western blot of thymocyte lysates from Recql4+/+, Recql4G522Efs/+, and Recql4R347X/+ probed with anti-mouse RECQL4 (clone 3B10; top). The same blot reprobed with antiactin (bottom). (E) Western blot of lysates from HoxB8 immortalized R26-CreERT2 Recql4+/+ infected with MSCV puro 3×Flag-RECQL4 and probed with anti-RECQL4 (clone 3B10; top) and M2 anti-Flag antibody (bottom). (F and G) Fluorescent microscopy of RECQL4 expression in Kusa 4b10 cells with murine mCherry-fused WT, K525A, G522Efs, and R347X (F) and human EGFP-fused WT, K508A, C525Efs, and R350Gfs (G).
The heterozygous Recql4R347X/+ and Recql4G522Efs/+ mice were viable and fertile. To determine if individual homozygous truncating mutants were viable, the respective heterozygous mice were inbred. We did not recover any Recql4R347X/R347X or Recql4G522Efs/G522Efs pups at genotyping (days 7 to 10 after birth), indicating that the homozygous mutants were developmentally lethal (Fig. 1C). We have not established the time point in development at which the respective mutants are no longer viable.
Next, we investigated the in vivo expression of the predicted truncated proteins. We prepared lysates from the thymus of germ line heterozygous mutants of each respective allele and probed them with a monoclonal antibody raised against the first 200 aa of murine RECQL4 by Western blotting (15). We found a truncated protein product of the predicted size for the R347X mutant in thymocyte extracts, though at a much lower intensity than the wild-type (WT) band (Fig. 1D). In contrast, the G522Efs mutant protein could not be detected (Fig. 1D), and even when ectopically overexpressed as a cDNA with an N-terminal 3×Flag tag, its expression was significantly lower than the R347X (Fig. 1E).
To assess whether the truncated proteins had altered cellular localization, we generated N-terminal mCherry-tagged mouse RECQL4 fusion constructs. These were retrovirally infected into the murine osteoblastic Kusa4b10 cell line. Protein localization was analyzed by qualitative fluorescence microscopy. The full-length wild-type murine RECQL4 protein (WT) was predominantly localized in the nucleus as expected, with an apparent enrichment in the nucleolus. The ATP-dependent helicase inactive p.Lys525Ala (K525A) mutation, which we recently reported (15), had a similar localization to WT RECQL4 (Fig. 1F). In contrast, the R347X protein, while also localized to the nucleus, was poorly incorporated in the nucleoli (Fig. 1F). Interestingly, consistent with the weak protein expression in vivo (Fig. 1D), the G522Efs protein was poorly expressed and difficult to detect (Fig. 1F).
We also assessed the cellular localization patterns of similar human RTS-associated RECQL4 mutations. For this purpose, we utilized a human N-terminal enhance green fluorescent protein (EGFP)-tagged WT, an ATP-helicase inactive K508A mutant, and created the C525Afs and R350Gfs mutations. These constitute the human homologues of, or map closely to, the murine mutations K525A, G522Efs, and R347X, respectively. We found similar qualitative localization results between the human and murine proteins (Fig. 1G).
Finally, it has been suggested that RECQL4 could localize to the mitochondria (23, 24). Using the fluorescent fusion proteins, we could not detect mouse or human RECQL4 (WT or mutant) in the cytoplasm or overlapping with the mitochondria (Fig. S2A). To evaluate this result functionally, we assessed mitochondrial function using the Seahorse bioenergetic assay. To enable comparison of the different point mutations, we used HoxB8 immortalized myeloid progenitor cells (25) derived from R26-CreER Recql4fl/+, R26-CreER Recql4fl/K525A, R26-CreER Recql4fl/R347X, and R26-CreER Recql4fl/G522Efs mice and exposed them to tamoxifen for 4 days to delete the wild-type Recql4 floxed allele. We found no difference in either the basal or maximal oxygen consumption rate (OCR) between the non-tamoxifen- and tamoxifen-treated groups (Fig. S2B to E), demonstrating that mutations in RECQL4 that significantly impact protein stability and function do not measurably affect mitochondrial respiration. Taken together, our results demonstrate that the murine RECQL4 mutants behave similarly to their human counterparts, and while the specific mutations impact their level of expression and subcellular localization differently, they do not measurably affect mitochondrial respiration.
Recql4R347X/+ and Recql4G522Efs/+ heterozygosity leads to a reduced bone mass phenotype.
We previously reported that complete deletion of Recql4 in the osteoblast lineage resulted in mice with shorter bones and reduced bone volume (22). RTS patients, however, present with compound heterozygous mutations that result in truncating proteins rather than null alleles. Therefore, to assess the skeletal/bone phenotypes associated with RTS type 2-relevant RECQL4 mutations, we measured skeletal growth in the two viable heterozygous mutants. We could not evaluate homozygous mutants for either allele due to the developmental lethality previously described. The germ line heterozygous animals are therefore most similar to the parents of RTS patients. For all genotypes, 10-week-old male mice were analyzed. The heterozygous Recql4R347X/+ and Recql4G522Efs/+ mice had a reduced body weight that did not reach statistical significance within the cohort assessed (Fig. 2A). We utilized echo-magnetic resonance imaging (Echo-MRI) to investigate a change in fat/lean mass proportion and found no differences (Fig. S3). We then evaluated the tibial length by micro-computed tomography (micro-CT). Overall tibial length in both Recql4R347X/+ and Recql4G522Efs/+ mutants was not statistically different compared to either wild-type (littermate) controls or K525A homozygous males (Fig. 2B). However, the mediolateral and anteroposterior widths measured by micro-CT in the midshaft tibia were significantly lower in both Recql4R347X/+ and Recql4G522Efs/+ mutants, indicating narrower tibiae in both genotypes (Fig. 2C and D). For comparison, tibiae from the helicase-inactive K525A homozygous mice did not show differences in any of these parameters compared to the WT controls.
FIG 2.
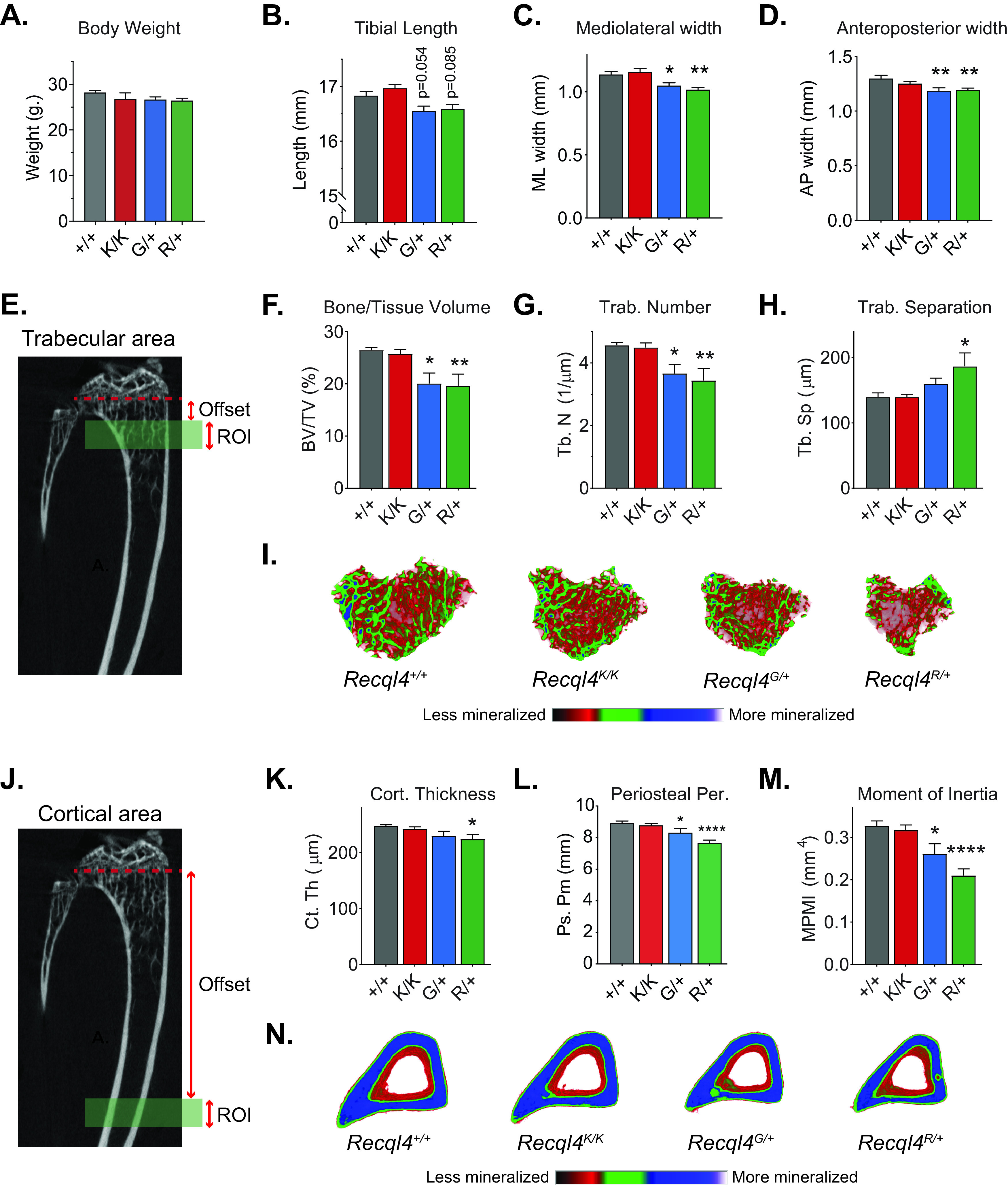
Germ line truncating mutants G522Efs and R347X cause low bone mass and narrow bones. (A) Gross body weights of 10-week-old male Recql4+/+, Recql4G522Efs/+, and Recql4R347X/+ mice. (B to D) Micro-CT measurements of (B) tibial length, (C) mediolateral width, and (D) anteroposterior width from 10-week-old male Recql4+/+, Recql4K525A/K525A, Recql4G522Efs/+, and Recql4R347X/+ mice. (E) Trabecular region of interest beginning at 3.5% (offset) distal to the growth plate and extending for 5% (ROI) of the total tibia length. (F) Trabecular bone volume. (G) Trabecular number. (H) Trabecular separation. (I) Representative images (axial plane) of reconstructed trabecular bone with color-coded quantitative mineralization from germ line Recql4 mutants. (J) Cortical region of interest beginning at 40% (offset) distal to the growth plate and extending for 5% (ROI) of the total tibia length. (K) Cortical thickness. (L) Periosteal perimeter. (M) Moment of inertia. (N) Representative images (axial plane) of reconstructed cortical bone with color-coded quantitative mineralization from germ line Recql4 mutants. Data expressed as mean ± SEM; ordinary one-way analysis of variance (ANOVA). *, P < 0.05; **, P < 0.01; ****, P < 0.0001. +/+, n = 7; K/K, n = 10; G/+, n = 6, R/+, n = 7. Experiments were independently executed on separate cohorts, with results pooled for presentation. K, K525A; G, G522Efs; R, R347X.
We further looked at possible bone changes in trabecular microarchitecture and cortical morphology of WT and mutant 10-week-old male mice. For trabecular analysis, we selected a region corresponding to the secondary spongiosa in the proximal metaphysis of the tibia (Fig. 2E). The trabecular bone volume of Recql4R347X/+ and Recql4G522Efs/+ mice was significantly lower by 24% and 26%, respectively (Fig. 2F). The trabecular number was also lower by 25% in the Recql4R347X/+ and 20% in the Recql4G522Efs/+ mice (Fig. 2G). The trabecular separation was 25% greater in the Recql4R347X/+ mice but not different in the Recql4G522Efs/+ mice (Fig. 2H). For cortical analysis, we assessed a region corresponding to the middiaphysis of the tibia (Fig. 2J). Cortical thickness was 10% lower in the Recql4R347X/+ mutants, whereas there was a slight (7%) but nonsignificant decrease in the Recql4G522Efs/+ mice compared to controls (Fig. 2K). For both the Recql4R347X/+ and Recql4G522Efs/+, the periosteal perimeter showed a reduction of 14% and 7%, respectively, compared with littermate controls (Fig. 2L), which was reflected in a lower moment of inertia for both groups (Fig. 2M). This suggested a lower torsional rigidity and increased fracture risk in the germ line heterozygous truncating mutant mice. In contrast, the nontruncating but ATP-binding helicase-inactive Recql4K525A/K525A mutants did not show any change in any trabecular or cortical parameter compared to the WT control (Fig. 2E to H and J to M). All morphological changes could be visualized in the color-coded three-dimensional (3D) reconstructions (Fig. 2I and N). Additional micro-CT parameters are provided in Fig. S4A to C. Collectively, these results demonstrate that heterozygous truncating mutations of RECQL4 in mice resulted in narrow bones and skeletal dysplasia.
Given that several studies have reported RTS patients with hematopoietic defects (26–28) and that there is an established reciprocal relationship between bone and hematopoiesis (29), we assessed whether the changes in skeletal parameters seen in the germ line heterozygous mutant mice would impact hematopoiesis. Results showed a decrease in the cell hierarchy involved in myeloid development, which did not affect mature granulocytes or macrophages (Fig. S5). The remaining parameters assessed were normal. Therefore, a single copy of a truncating mutation in the presence of a retained WT allele is not sufficient to cause marked changes in hematopoiesis and is consistent with the reports from RTS patients and the apparent normality of hematopoiesis in their heterozygous parents. Taken together, these observations suggest that heterozygous truncating RECQL4 mutations cause a haploinsufficient low bone mass phenotype similar to that reported in RTS patients. Interestingly, the expression of a single copy of a full-length wild-type RECQL4 is sufficient to maintain hematopoiesis, which indicates cellular differences between osteoblast lineage cells and blood-forming cells in sensitivity to Recql4 gene dosage.
Intrinsic defects in osteoblast lineage cells cause the low-bone mass phenotype of RECQL4 truncating mutants.
To determine whether the skeletal phenotypes seen in the germ line heterozygous mutant mice were caused by defects in the osteoblast lineage, we crossed all our mutant models (Recql4K525A/+, Recql4R347X/+, and Recql4G522Efs/+) to the Osx-Cre Recql4fl/fl mice (22, 30). This allowed us to delete the wild-type Recql4 allele from the osteoblastic lineage, leaving only the mutant protein expressed. These osteoblast-restricted point mutant models were compared to Osx-Cre+ Recql4fl/+ mice to control for the known effects of the Osx-Cre transgene on bone homeostasis (31, 32), allowing comparison across all Cre+ models. Furthermore, this approach also bypassed the lethality of homozygous mutant mice and the assessment of cells only expressing truncated proteins in adult mice.
The analysis showed that only the Osx-Cre Recql4Δ/R347X mice had a lower body weight and tibial length than Osx-Cre Recql4Δ/+ littermates (Fig. 3A and B). The Osx-Cre Recql4Δ/G522Efs mice had a reduction in body weight, but it did not reach statistical significance (P = 0.09) (Fig. 3A). Additionally, trabecular analysis showed lower trabecular bone volumes by 13% for Osx-Cre Recql4Δ/G522Efs and 25% for Osx-Cre Recql4Δ/R347X (Fig. 3C). Correspondingly, there was a 12% and 23% reduction in trabecular number, respectively, compared to controls (Fig. 3D). Trabecular separation was 17% greater in the Osx-Cre Recql4Δ/R347 mice only (Fig. 3E). For cortical bone parameters, only mice carrying the R347X mutation showed an 8% lower periosteal perimeter and 25% lower mean polar moment of inertia consistent with an 8% reduction in mediolateral width and no change in anteroposterior width compared to controls (Fig. 3G to K). Again, the K525A helicase-inactive mice did not show any detectable phenotypes compared to controls. All morphological changes are illustrated in the 3D reconstructed images (Fig. 3F and L). Additional micro-CT parameters are provided in Fig. S4D to F. In summary, these data demonstrate that truncating mutations of RECQL4 disrupt bone microstructure through defects intrinsic to the osteoblast lineage, with the more severe phenotype seen in mice expressing the shortest truncated protein (R347X).
FIG 3.
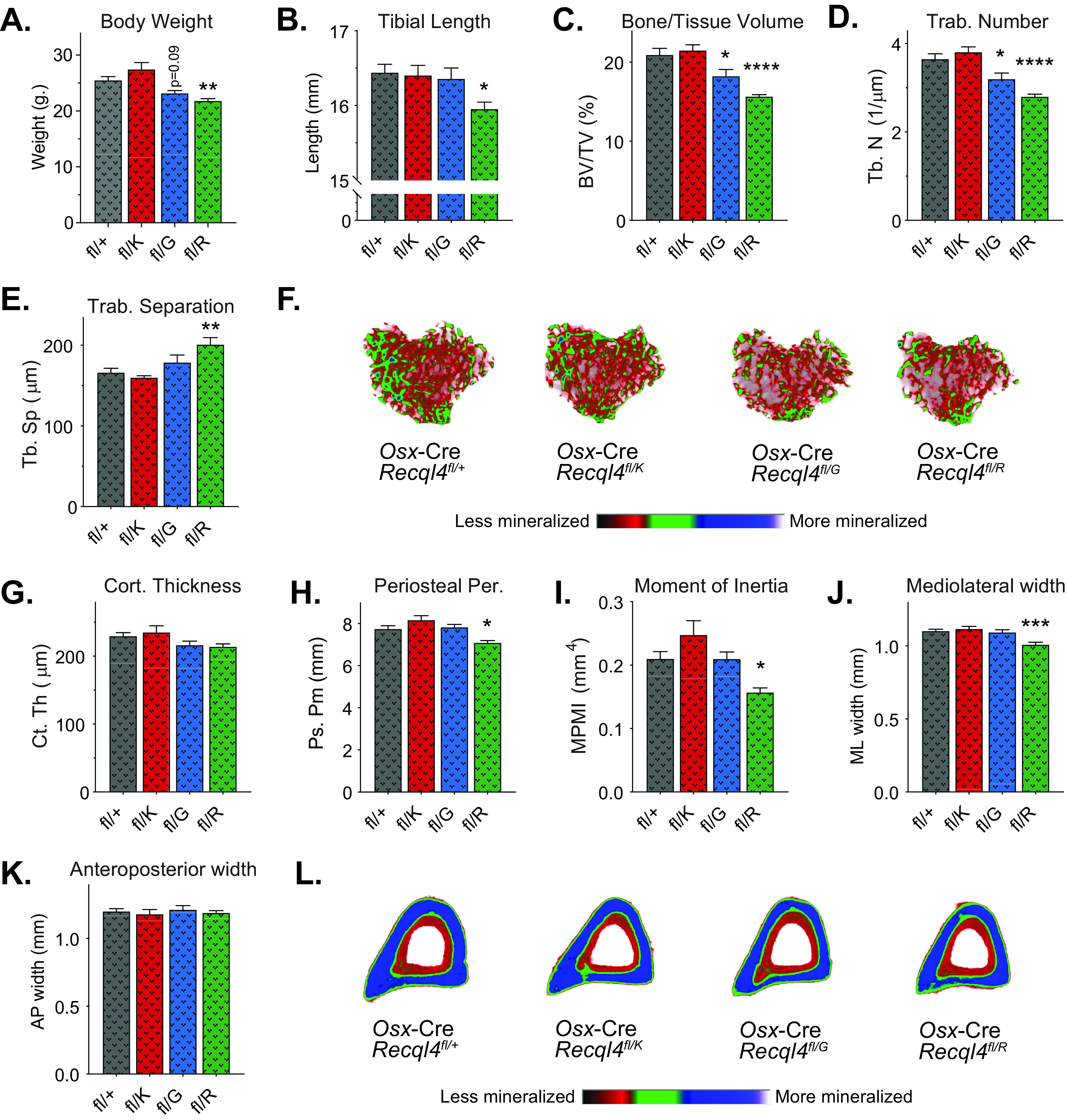
Expression of only the truncating mutations in preosteoblasts results in low bone mass. (A) Gross body weights of 10-week-old male Osx-Cre Recql4fl/+, Osx-Cre Recql4fl/K525A, Osx-Cre Recql4fl/G522Efs, and Osx-Cre Recql4fl/R347X mice. (B) Tibial length. (C) Trabecular bone volume. (D) Trabecular number. (E) Trabecular separation. (F) Representative images (axial plane) of reconstructed trabecular bone with color-coded quantitative mineralization from Osx-Cre Recql4 mutants. (G) Cortical thickness. (H) Periosteal perimeter. (I) Moment of inertia. (J) Mediolateral width. (K) Anteroposterior width. (L) Representative images (axial plane) of reconstructed cortical bone with color-coded quantitative mineralization from Osx-Cre Recql4 mutants. Data expressed as mean ± SEM; ordinary one-way ANOVA. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. fl/+, n = 9; fl/K, n = 6; fl/G, n = 7, fl/R, n = 7. Experiments were independently executed on separate cohorts, with results pooled for presentation. fl, floxed; K, K525A; G, G522Efs; R, R347X.
Compound heterozygous Recql4 mutants tolerate ionizing radiation and do not develop osteosarcoma.
Based on previous studies that show that RTS patients have compound heterozygous mutations with one allele more severely truncated than the other (4, 5), we generated compound heterozygous mouse lines combining the G522Efs or R347X mutations with the K525A. Although this approach does not fully mimic RTS, it allowed us to determine the in vivo effects of a truncated allele and a helicase-inactive allele. Surprisingly, the compound heterozygous Recql4G522Efs/K525A and Recql4R347X/K525A mice were viable, and pups were born at the expected Mendelian ratio (Fig. 4A). Moreover, monitoring of aged cohorts demonstrated that Recql4G522Efs/K525A and Recql4R347X/K525A mice had a normal life span compared to WT mice, without increased tumor incidence (Fig. 4B). All genotypes developed a small number of spontaneous tumors affecting the liver, spleen, thymus, and intestine; however, no osteosarcoma was detected in compound heterozygous mice (Table S1).
FIG 4.
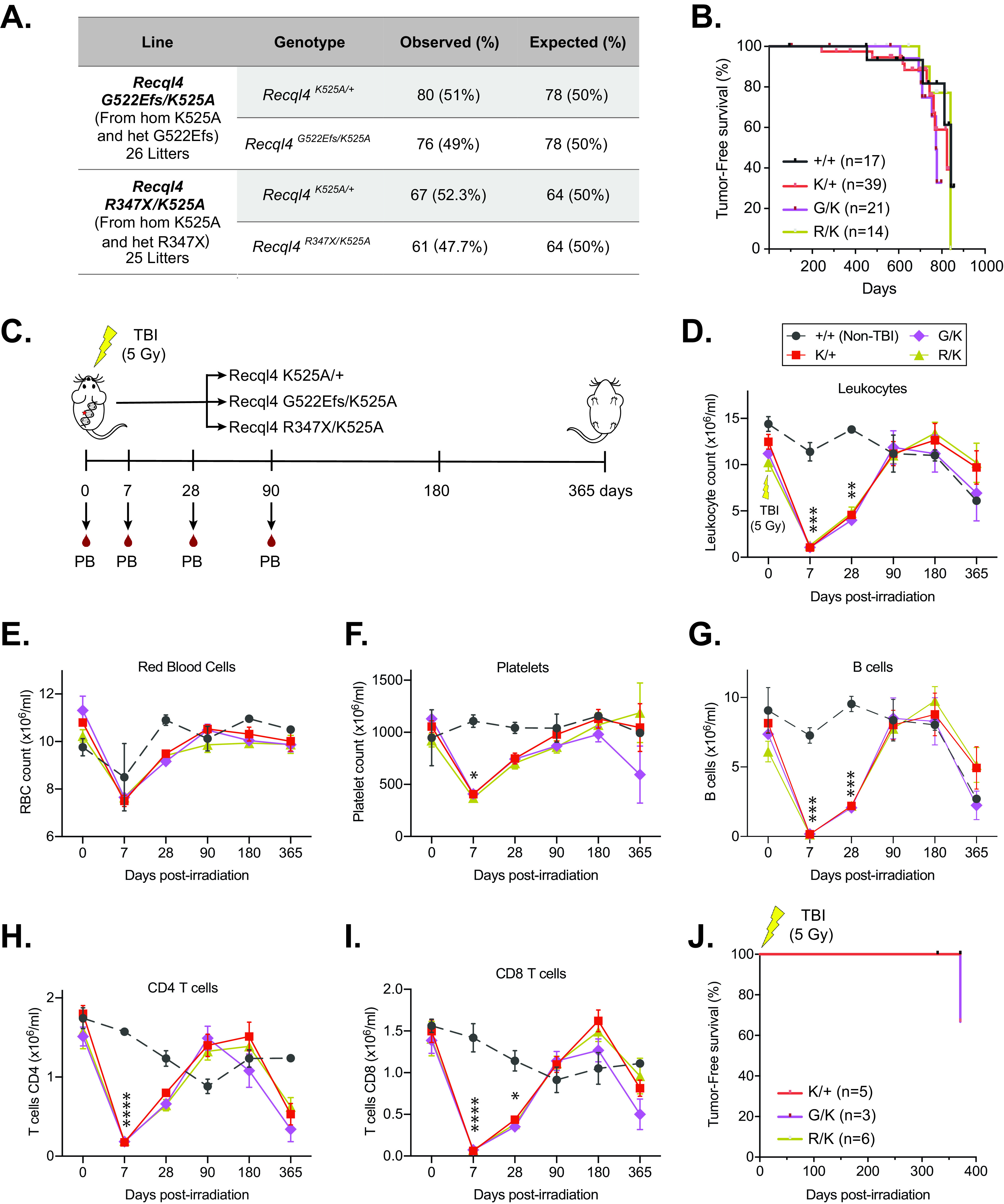
Compound heterozygous Recql4 mutants tolerate a sublethal dose of ionizing radiation and do not develop osteosarcoma. (A) Breeding data from 26 litters of Recql4G522/K525A and 25 litters of Recql4R347X/K525A intercrosses. Observed and expected Mendelian rates of the indicated genotypes are shown. No statistical significance was achieved. (B) Kaplan-Meier tumor-free survival plots of the indicated genotypes. +/+, n = 17; K/+, n = 39; G/K, n = 21; R/K, n = 14. (C) Schematic representation of the experimental setup. Compound heterozygous mutants received a single dose of 5-Gy gamma irradiation, peripheral blood was assessed, and the animals were monitored for tumor formation. Mice were euthanized at the last time point. (D to I) Peripheral blood cell counts following irradiation: leukocytes (D), red blood cells (E), platelets (F), B cells (G), CD4 T cells (H), and CD8 T cells (I). (J) Kaplan-Meier tumor-free survival plots of the irradiated mice. K/+, n = 5; G/K, n = 3; R/K, n = 6. Data expressed as mean ± SEM; ordinary one-way ANOVA. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. TBI, total body irradiation; K, K525A; G, G522Efs; R, R347X.
Previous studies have reported increased sensitivity of RECQL4 mutant cell lines to ionizing radiation (33, 34). To assess the in vivo response of compound heterozygous mutant mice and whether this increased susceptibility to cancer formation, we treated a small cohort of mice with whole-body ionizing radiation. A single sublethal dose of 5 Gy gamma irradiation was administered to 9-week-old Recql4G522Efs/K525A, Recql4R347X/K525A, and Recql4K525A/+ mice as controls. We monitored the cohorts for 1 year, assessing peripheral blood parameters at several time points to evaluate hematologic recovery (Fig. 5C). The efficacy of the radiation was demonstrated by a similar transient reduction in blood cell populations across all genotypes (Fig. 4D to I). After 12 months, all mice were euthanized, and autopsies were performed. We found that ionizing radiation did not result in abnormal/delayed hematopoietic recovery or failure, nor did it result in tumor development, with only one intestinal tumor found in a Recql4G522Efs/K525A mouse (Fig. 4J). Collectively, these results demonstrate that a full-length, even helicase-inactive, RECQL4 is sufficient to rescue the lethality caused by truncating mutations. They further demonstrate that these compound heterozygous Recql4 mutations are not sufficient to sensitize murine models to ionizing radiation or to accelerate cancer initiation.
FIG 5.
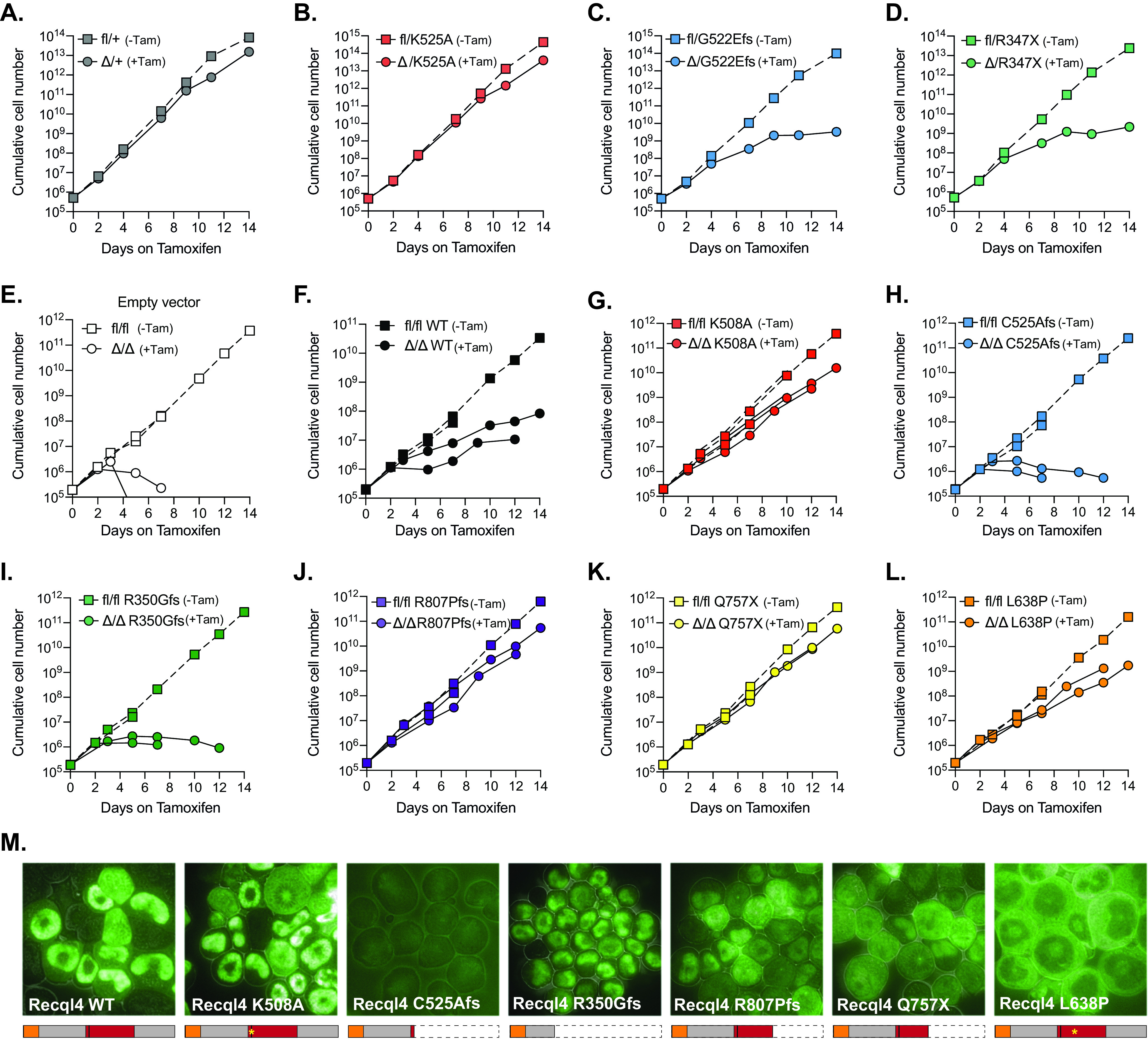
The truncating human RECQL4 mutations C525Afs and R350Gfs fail to rescue Recql4 deletion and impede proliferation, similar to their murine homologs, while mutations that conserve the protein are better tolerated. (A to D) Proliferation curves of HoxB8 immortalized R26-CreERT2 myeloid cells without (fl) and with (Δ) tamoxifen-mediated Recql4 deletion in the following cell lines: fl/+ (A), fl/K525A (B), fl/G522Efs (C), and fl/R347X (D). (E to L) Proliferation curves of HoxB8 immortalized R26-CreERT2 Recql4fl/fl control myeloid cells (E) in the presence or absence of tamoxifen and EGFP hRECQL4 overexpressing cells: wild type (F), K508A (G), C525Afs (H), R350Gfs (I), R807Pfs (J), Q757X (K), and L638P (L). Dotted lines represent individual controls not treated with tamoxifen. (M) Microscopy of EGFP-hRECQL4 expression in HoxB8 cells with WT, K508A, C525Afs, R350Gfs, R807Pfs, Q747X, and L638P. A schematic illustration of the expected protein products is outlined below each figure. The orange box represents the Sld2-like region, and the red box represents the helicase region. Cell proliferation assays using murine mutations were repeated two times using independent cell lines (replicate experiment plotted in Fig. S6). Retroviral complementation assays with human constructs were performed two times using the same parental cell line; data from each replicate are plotted separately.
Truncating human RECQL4 mutations fail to rescue Recql4 deletion, while mutations that conserve the protein are better tolerated.
To analyze the effects of different human RECQL4 mutations and how they compare to our murine models, we utilized the Hoxb8 immortalized myeloid progenitor cells. This is a relevant cell type, given the requirement of RECQL4 for maintenance of this cell population in vivo (35). First, we used this system to determine the effects of truncating murine Recql4 mutations in vitro. Hoxb8 immortalized myeloid progenitor cell lines from R26-CreERT2 Recql4fl/+, R26-CreERT2 Recql4fl/K525A, R26-CreERT2 Recql4fl/R347X, and R26-CreERT2 Recql4fl/G522Efs mice were treated with 400 nM/ml of 4-hydroxy-tamoxifen to induce Cre recombinase activity and deletion of the floxed wild-type Recql4 allele, leaving only the mutant allele expressed. We achieved successful deletion by day 4, as confirmed by PCR for genomic recombination (Fig. S5). The presence of a single Recql4 wild-type allele (fl/+ cells treated with 4-hydroxy-tamoxifen to become Δ/+) did not interfere with the proliferation rates (Fig. 5A), nor did the expression of a single helicase-inactive K525A allele (Fig. 5B). However, when the floxed allele was deleted in the cells carrying the truncating mutations G522Efs and R347X, a marked decrease of cell proliferation was observed, consistent with the previously described in vivo phenotype (15) and demonstrating an essential requirement of the helicase and C-terminal domains deleted in these mutations (Fig. 5C and D).
Next, we used this cell line model to determine the capacity of different human RECQL4 mutations to rescue HoxB8 R26-CreER Recql4Δ/Δ myeloid cells, where both alleles of the endogenous murine Recql4 were deleted. As expected, control fl/fl cells started dying at day 4 posttamoxifen as they became Recql4 null (Fig. 5E and Fig. S6). Interestingly, when we expressed the wild-type human RECQL4 protein, cell proliferation was not wholly restored, suggesting that overexpression of wild-type RECQL4 can be detrimental (Fig. 5F). We further tested the ability of frequent human RTS-associated RECQL4 mutations, including the human equivalents to our murine germ line mutations, to rescue Δ/Δ cell proliferation and viability. To do this, we used the same EGFP-fused human RECQL4 mutations, K508A, C525Afs, and R350Gfs (Fig. 5G and H), described previously and engineered the p.Arg807ProfsTer38 (R807Pfs), p.Gln757* (Q757X), and p.Leu638Pro (L638P) mutations based on recurrent mutations described in RTS patients (4). The analysis showed that the helicase-inactive K508A mutant successfully rescued the Δ/Δ cells (Fig. 5G), while both the C525Afs and R350Gfs human mutants did not (Fig. 5H and I), similar to endogenous murine G522Efs and R347X (Fig. 5C and D). Both R807Pfs and Q757X mutations rescued the Recql4 deletion (Fig. 5J and K), whereas the L638P achieved only partial rescue (Fig. 5L).
Finally, we visualized the EGFP-RECQL4 localization in HoxB8 R26-CreER Recql4fl/fl. Similar to what we saw in Kusa4b10 cells (Fig. 1G), the wild-type human RECQL4, the K508A, and the R350Gfs proteins were localized in the nucleus, while the C525Afs was poorly expressed (Fig. 5M). On the other hand, the R807Pfs and Q757Pfs mutants displayed both nuclear and cytoplasmic localization, while the L638P mutant was located primarily in the cytoplasm. The sizes and expression levels of the predicted protein products were confirmed by Western blotting (Fig. S7). Collectively, these data demonstrate that not all mutations of RECQL4 are functionally equivalent. Mutations that result in severely truncated protein products, due to early stop codons or frameshifts, are more likely to affect transcript stability and localization and are detrimental to both cellular and organismal health. In contrast, mutations that conserve most of the protein are largely tolerable.
DISCUSSION
Since Kitao et al. first cloned the RECQL4 gene more than 20 years ago (36), some inroads have been made in the understanding of these mutations and their contribution to RTS. It is now known that the majority of mutations reported in RTS patients are compound heterozygous mutations, containing at least one truncated allele and mainly impacting the helicase domain (4, 5). This mutational spectrum implied that defects in the helicase region might be the main reason for the phenotypes of RTS. However, we recently showed that mice with a homozygous knock-in mutation that specifically inactivates RECQL4 ATP-dependent helicase activity were strikingly normal in terms of embryonic development, hematopoiesis, and DNA damage repair (15). In the same study, by using a conditional deletion model that allowed the assessment of the effects of mutations after deleting the wild-type Recql4 allele, we found that mice carrying truncating, but not helicase-inactive, mutations developed bone marrow failure (15). This confirmed the deleterious effects of truncating mutations, which led us to investigate the impact of these mutations in other systems.
Assessing the reported RECQL4 mutations (1, 4, 5), we found RTS patients presenting with two severe truncating mutations to be extremely rare. Here, we generated two Recql4 mutant models with truncating mutations that closely map to those reported in RTS patients, affecting the helicase and C-terminal domain. We did not recover viable homozygous germ line mutant pups from either allele. These results are consistent with the human data and suggest that having two severely truncating mutations of RECQL4 is not tolerated and that there is an essential developmental role for the deleted domains. Furthermore, in addition to lacking essential domains, there is the potential for these mutations to yield aberrantly expressed and localized protein, further contributing to the phenotypes we observed. When we assessed protein levels of the truncating mutants, we were not able to detect the predicted truncating protein product from the G522Efs mutation. This could be the consequence of either nonsense-mediated mRNA decay or proteasomal degradation. The C525Afs human mutation, which maps closely to the murine G522Efs mutation, showed similar results. On the other hand, the R347X mutation produced a short but stable protein that localized to the nucleus, albeit with reduced relative nucleolar intensity compared to WT or K525A protein. The R350Gfs human mutation (mapping closely to R347X) had qualitatively similar expression and localization. We assessed the nucleolar localization signals/sequences in the murine and human RECQL4 using the NoD program (nucleolar localization sequence detector; University of Dundee). The analysis indicated that while murine RECQL4 has two putative signals between 253 to 278 aa and 359 to 386 aa, human RECQL4 has two predicted signals at 10 to 33 aa and 370 to 390 aa, consistent with previous analysis (37). Results also showed that the score for the murine sequences is higher than that for the human protein. Given that the R347X mutant truncates the protein prior to the second signal (conserved with the human protein) and that we fail to see accumulation from this protein in the nucleolus, it is suggested that this is the primary functional nucleolar localization sequence. However, we cannot exclude the possibility that differences in the tags (EGFP for human compared to mCherry for the murine proteins) or the cell lines used impacted the qualitative assessment of localization (38). Taking these results together, we have shown that Recql4-truncating mutations affect protein stability and subcellular localization differently and that this phenotype is reproduced using comparable human mutations.
The skeletal system is severely impacted in the majority of RTS patients. A study of 28 RTS subjects examined by radiologic survey found that up to 75% had some form of skeletal abnormality, including abnormal metaphyseal trabeculation, brachymesophalangy, thumb or radial agenesis, or hypoplasia (3). Several additional studies have associated a loss of RECQL4 with a more systemic skeletal involvement, with a high proportion of patients reporting low bone density (3, 39–42). However, to our knowledge, no studies have mapped skeletal defects to specific RECQL4 mutations. We found that heterozygous expression of the truncating alleles was sufficient to cause low trabecular bone mass, impaired growth of cortical bone, and narrower bones compared to WT controls. Furthermore, we found that truncating mutations affect normal skeletal formation by causing defects in the osteoblast lineage. These findings were comparable to findings we previously reported in an osteoblast lineage restricted knockout, which showed that complete deletion of Recql4 in the osteoblast lineage led to reduced bone volume and defects in osteoblast proliferation and maturation (22). This suggested that at least for bone development, having a truncated RECQL4 protein is equivalent to having no RECQL4 at all, which highlights the critical function of the deleted domains in bone homeostasis. Interestingly, an osteochondral-lineage-specific mouse model reported more severe findings than ours. Cao et al. used Prx1-Cre to delete Recql4 in early mesenchymal progenitor cells of the limb buds and described a 50% reduction in bone volume and cortical bone area (41). While RECQL4 is clearly needed in the earlier skeletal cell populations, it is important to consider that our models use truncating heterozygous mutations, instead of null alleles. Therefore, any remaining RECQL4 protein in the preosteoblast population of our mutants might be sufficient for partial function at this stage and explain the phenotypic differences between models. Lastly, we observed that the R347X mutation, which produced a stable yet the shortest predicted protein, led to a more severe bone phenotype than the poorly expressed G522Efs mutation. This suggested that the severity of the defects was proportional to the severity of the truncation, irrespective of protein expression. Furthermore, the fact that the largest truncation caused the most severe bone phenotype suggests that RECQL4 gene dosage is a critical regulator of bone mass, something relatively unknown until now. Collectively, these results demonstrate that heterozygous truncating mutations of RECQL4 cause a haploinsufficient low-bone-mass phenotype through defects in the osteoblast lineage. Although RTS patients generally present with compound heterozygous mutations, these results highlight the importance of having a full-length protein for bone development. Furthermore, they raise concerns regarding the osteoporosis status of the parents of RTS patients, which warrant further investigation.
A characteristic feature of syndromes associated with mutations in RECQL4 is cancer predisposition, particularly osteosarcoma, cutaneous epithelial tumors, and hematological malignancies (2, 43, 44). A recent study analyzed pediatric patients with cancer and identified a significant enrichment in heterozygous RECQL4 loss-of-function variants in those who presented with osteosarcoma (45). This raised the question of whether the presence of compound heterozygous Recql4 mutations in mice is sufficient to initiate tumorigenesis. We found no differences in tumor burden or spectrum in our compound heterozygous mutants compared to wild-type controls, even after exposure to gamma irradiation. Furthermore, although previous studies have reported ionizing radiation as a risk factor for the development of sarcomas (46, 47), and truncated RECQL4 products have been associated with hypersensitivity to this agent (48), we did not observe either hematopoietic failure or osteosarcoma development in our irradiated cohort. It is possible that the nontruncating but helicase-inactive allele is sufficient to rescue these phenotypes and that truncating mutations in both alleles are necessary for tumor initiation. However, this could not be addressed in this study given the developmental lethality seen in our homozygous truncating mice. Another possibility is that larger numbers of mice and longer times are necessary for this phenotype to develop. It is also possible that additional genes are involved in the development of these RTS phenotypes. All these limitations should be addressed in future studies.
Lastly, we established a tractable in vitro cell line model, which allowed us to examine the cellular consequences of different RECQL4 mutations. We found that cells carrying the murine truncating G522Efs and R347X mutations developed a proliferation defect after deletion of the floxed wild-type Recql4 allele. Similarly, the closely related human C525Afs and R350Gfs mutations failed to rescue the lethality caused by Recql4 deletion. On the other hand, the murine ATP-dependent helicase-inactive mutation (K525A) did not demonstrate a proliferation defect, and its human counterpart (K508A) successfully rescued Recql4 deletion. These results confirm that truncating, but not helicase-inactive, mutations are pathogenic and that our murine mutations are useful surrogates for understanding the functions of human disease-associated RECQL4 mutations. When using this system to analyze other human mutations, we found that cells overexpressing the human wild-type protein could not completely rescue Recql4 deletion, suggesting that overexpression of RECQL4 is not well tolerated. In fact, several studies have correlated overexpression of RECQL4 with the development of malignancies (49–54). We also found that the L638P mutation, which resulted in a stable full-length product with cytoplasmic localization, could not fully rescue the Recql4 deletion. This demonstrates that in order for RECQL4 to function effectively, it needs to be located within the nucleus. Finally, the R807Pfs and Q757X mutations, which have been found in RTS patients with osteosarcoma and lymphomas (4), were able to rescue the Recql4 deletion. This indicates that small C-terminal deletions do not severely affect viability, unlike larger deletions that include both the helicase and the C-terminal domains. However, their association with malignancies remains unknown. By comparing different mutations in the same genetic context, this system allowed us to conclude that the different mutations have distinct consequences for RECQL4. While some mutations create unstable proteins, some alter its localization without grossly affecting protein stability. Overall, the level of the truncation appeared to show the strongest correlation with the severity of the phenotype. Given the increasing number of somatic RECQL4 mutations reported in sporadic cancers, this in vitro system can serve as a platform to assess the impact of RECQL4 mutations at a cellular level.
In conclusion, truncating RECQL4 mutations affect protein stability and localization, contributing to the development of an osteoporosis-like phenotype through defects in early osteoblast progenitors in mice. However, they are not sufficient to promote tumorigenesis, even after exposure to irradiation. Future studies should focus on the identification of genes that cooperate with RECQL4 in normal development and tumorigeneses. These will allow a better understanding of the genetic landscape of RTS and permit the generation of more comprehensive models.
MATERIALS AND METHODS
Ethics statement.
All animal experiments conducted for this study were approved by the Animal Ethics Committee of St. Vincent’s Hospital, Melbourne, Australia (no. 007/14, 011/15, and 015/17). Animals were euthanized by cervical dislocation or CO2 asphyxiation.
Mice.
The chemically (N-ethyl-N-nitrosourea [ENU]) induced Recql4R347X point mutation was provided by the Australian Phenomics Facility (APF, Canberra, Australia; allele IGL01809). Recql4G522Efs mice were identified during CRISPR/Cas9 targeting to generate the previously described Recql4K525A mutation (15) by the Mouse Engineering at Garvan/ABR (MEGA) service (Garvan Institute, Darlinghurst, Australia). This allele is on a C57BL/6 background and carried a 2-bp insertion (GA) after the T521 codon. The Osx-Cre, Rosa26-eYFP, and Recql4K525A mutant animals have been previously described (15, 22, 30). Rosa26-CreERT2 mice were originally purchased from The Jackson Laboratory [B6.129-Gt(ROSA)26Sortm1(cre/ERT2)Tyj/J, stock no. 008463] and have been previously described (35). The ENU-derived mutant (Recql4R347X) was backcrossed to C57BL/6 at least six times and evaluated across multiple generations. All lines were on a C57BL/6 background. All animals were housed at the BioResources Centre (BRC) at St. Vincent’s Hospital. Mice were maintained and bred under specific pathogen-free conditions with food and water provided ad libitum.
All mouse lines are available from the Australian Phenome Bank (APB; https://pb.apf.edu.au/). The strain identification numbers/names are R347X (APB ID no. 7986), R26-CreER Recql4fl/fl (APB ID no. 7263). Osx-Cre R26-eYFP Recql4fl/fl (APB ID no. 7886), K525A (strain name C57BL/6-Recql4<tm4Crw>), and G522Efs (strain name C57BL/6-Recql4<tm5Crw>).
Cloning of mCherry and EGFP RECQL4 proteins and retroviral production.
Human N-terminal EGFP-fused RECQL4 and EGFP-fused RECQL4K508A (provided by T. Enomoto, Musashino University, Tokyo, Japan [55]) were cloned into MSCV-puro (35). The human mutations R807Pfs, Q757X, L638P, C525Afs, and R350Gfs, were created by gBlock (IDT) replacement of a wild-type fragment of EGFP-RECQL4 in the plasmid MSCV-puro with a mutant fragment. Murine mCherry-fused RECQL4 was assembled from a codon-optimized synthetic mRecql4 cDNA (GeneArt, Life Technologies) placed in-frame with an N-terminal mCherry cDNA (gBlock, IDT) in MSCV-puro. Mouse mutations were created by gBlock (IDT) replacement of the required fragment of Recql4. All constructs contain full-length cDNAs, including those coding for truncating mutations. All mutations were confirmed by Sanger sequencing. Retrovirus was produced by transient transfection of 293T cells using calcium phosphate-mediated transfection with an ecotropic packaging plasmid (35).
Genotyping.
Genotyping of the G522Efs mutants was determined by PCR using the following primers: mRecql4 K525A MO36-R3 (5′-AGAACATTGGGCATTCGGC-3′) and mRecql4 K525A MO36-F9 (5′-TAGACCTTATGAAACCTCAAAGCC-3′) to obtain a 591-bp product, which was then digested with MslI (New England Biolabs [NEB]) to generate three fragments of 347, 175, and 71 bp for the G522Efs mutant or two fragments of 416 and 175 bp for the WT. Genotyping of the K525A mutants has been previously described and used the same primers and restriction enzyme as the G522Efs mutation with the difference that this resulted in three fragments of 361, 175, and 55 bp (15). The presence of the R347X mutation was determined by KASP (competitive allele-specific PCR) technology (LGC) with facility-provided primers (5′-GAAGGTGACCAAGTTCATGCTAAAGCGTTTGTTTTTCATGTTGAGTCG-3′, 5′-GAAGGTCGGAGTCAACGGATTCAAAGCGTTTGTTTTTCATGTTGAGTCA-3′, and reverse primer 5′-GCTTCCCTAGACAGAGGGAACTATA-3′) used according to the manufacturer’s instructions.
Protein extraction and Western blotting.
Thymocyte lysates from Recql4R347X/+ and Recql4G522Efs/+ germ line were prepared in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, pH 8.0) plus cOmplete protease inhibitor (Roche) and PhosSTOP (Roche) tablets. Protein was quantified using the Pierce bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific) on an EnSpire multimode plate reader (Perkin Elmer). Lysates from HoxB8 immortalized (25) R26-CreERT2 Recql4fl/G522Efs, R26-CreERT2 Recql4fl/R347X, and R26-CreERT2 Recql4+/+ cells transduced with murine stem cell virus (MSCV) puro 3×Flag-RECQL4 were prepared in sample buffer (2 × 106 cells in 100 μl). Then, 50 μg of whole-protein extracts from thymocytes and 50 μl from myeloid cells were loaded on precast NuPAGE BOLT 8% Bis-Tris polyacrylamide gels (Invitrogen) and transferred onto Immobilon-P polyvinylidene difluoride (PVDF) membranes (Merck Millipore). Membranes were blocked with 5% milk in Tris-buffered saline with Tween (TBST) and incubated at 4°C overnight with rat monoclonal anti-mouse RECQL4 antibody (clone 3B10 [detects mouse] or clone 3B1 [detects both human and mouse]) (15), mouse antiactin (Sigma-Aldrich, A1978), or anti-Flag antibody (Sigma-Aldrich). Membranes were then probed with horseradish peroxidase (HRP)-conjugated goat anti-rat (Thermo Fisher Scientific; 31470) or anti-mouse (Thermo Fisher Scientific; 31444) secondary antibodies and visualized using ECL prime reagent for chemiluminescent detection on Hyperfilm ECL (Amersham). The predicted molecular weight for the truncated proteins is 62.4 kDa for G522Efs, 65.6 kDa for 3×Flag-G522Efs, 38.1 kDa for R347X, and 41.3 kDa for 3×Flag-R347X.
Live cell imaging and image processing of RECQL4 fusion proteins.
Transduced osteoblast-like Kusa4b10 cells were plated in 10-cm dishes, selected with puromycin, and grown to subconfluence. Single-plane images were acquired on an inverted fluorescence microscope (Olympus IX81) with a ×40 objective (LUCPLFLN 40X) and were recorded with a Retiga-EXi 12 Bit charge-couple-device (CCD) camera (QImaging). Image processing and analysis were done using MetaMorph (Molecular Devices) and Adobe Photoshop. HoxB8 immortalized myeloid cells were concentrated by centrifugation, and 5 μl of cell suspension was dispensed on a slide before image acquisition with a ×60 objective (UPLANAPO ×60 water immersion). For mitochondrial staining, 250 μl of mitochondrial staining solution (CytoPainter MitoBlue indicator reagent [ab219940], 1:500 diluted in Hanks' balanced salt solution plus 20 mM HEPES buffer [HHBS]) was added to Kusa4b10 cells grown on coverslips in 250 μl Dulbecco’s modified Eagle’s medium (DMEM) in a 24-well plate and incubated for 30 min to 2 h in a 37°C/5% CO2 incubator. Coverslips were washed twice with HHBS, and live cells were imaged as described above.
Seahorse XF24 extracellular flux analyzer.
Hoxb8 immortalized (25) R26-CreERT2 Recqlfll/+ (control), R26-CreERT2 Recql4fl/K525A, R26-CreERT2 Recql4fl/G522Efs, and R26-CreERT2 Recql4fl/R347X myeloid cells were maintained in Iscove’s modified Dulbecco’s medium (IMDM), 10% fetal bovine serum (FBS) (non-heat inactivated), and 1% granulocyte-macrophage colony-stimulating factor (GM-CSF)-containing medium (BHK-HM5 cell-conditioned medium). The cells were treated for 4 days with 400 nM 4-hydroxy tamoxifen (Merck Millipore) and then genotyped to confirm complete recombination. To adhere myeloid (suspension) cells to the XF24 cell culture plate, wells were first coated with 100 μl of RetroNectin solution (32 μg/ml in phosphate-buffered saline [PBS; TaKaRa Bio]), incubated for 2 h at room temperature, blocked with 200 μl of 2% bovine serum albumin (BSA) in PBS, and washed with PBS. Cells were then seeded at 120,000 cells/well and spun at 1,100 × g for 20 s. Cell culture medium was replaced with nonbuffered DMEM base medium (Seahorse Bioscience) containing 25 mM glucose, 1 mM sodium pyruvate, and glutamine at pH 7.4 and incubated for 1 h at 37°C in a non-CO2 incubator. The oxygen consumption rate (OCR) was measured in a Seahorse XF24-3 flux analyzer. Cells were assayed with a 2-min mix/2-min wait/5-min measurement cycle for three baseline measurements followed by three cycles after each injection of the following four compounds affecting bioenergetics: 0.5 μM oligomycin (complex V inhibitor; Sigma, St. Louis, MO), 0.7 μM carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP; ΔΨm uncoupler; Sigma), 3.6 μM antimycin A (complex III inhibitor; Sigma), and 6 μM rotenone (complex I inhibitor; Sigma). After completion of the analysis, CyQUANT (Life Technologies) was used to normalize measurements to cell number in the corresponding wells.
Micro-computed tomography (micro-CT) 3D analysis of tibiae.
Tibiae were collected from mutant mice and their littermate controls; the attached soft tissue was removed carefully, and tibiae were fixed in 2% paraformaldehyde overnight, which was then replaced by 70% ethanol. Tibia morphology and microarchitecture was analyzed by ex vivo micro-CT on the left tibia wrapped in 70% ethanol-soaked gauze within a cryovial by using a Skyscan 1076 system (Bruker MicroCT, Kontich, Belgium). Images were acquired at 9-μm pixel size, 0.5-mm aluminum filter, 44-kV voltage, 220-μA current, 2300-ms exposure time, and 0.5° rotation, with a 1-frame average. Image slices were reconstructed with NRecon (Bruker; version 1.6.10.2) using the following settings: 36% beam-hardening correction, 6 ring artifact correction, 1 smoothing, and no defect pixel masking. The reconstructed images were analyzed with the software programs DataViewer (Bruker; version 1.4.4), CTAn (Bruker; version 1.15.4.0), and CTvox (Bruker; version 2.4.0). The trabecular and cortical region of interest (ROI) was determined by identifying the start of the mineralized zone of the proximal growth plate and calculating a distance equal to 3.5% and 40% of the total tibial length, respectively. From that point, a further 5% of the total tibial length was analyzed as the secondary spongiosa trabecular ROI and a 5% cortical ROI. Analysis of bone structure was completed by adaptative thresholding in CTAn, which was determined by performing automatic thresholding on 3 samples from each experimental group, resulting in threshold values of 50 to 255 for trabecular bone and 90 to 255 for cortical bone. Representative images of reconstructed trabecular and cortical bone with color-coded quantitative mineralization were made of the specimen whose value was closest to the group mean using the trabecular bone volume and cortical thickness parameters.
Peripheral blood analysis.
Peripheral blood (approximately 100 μl) was obtained via retro-orbital bleeding. Then, 25 μl of blood was mixed with 75 μl of PBS to obtain cell counts on a hematological analyzer (Sysmex KX-21N; Roche Diagnostics). The remaining blood was red blood cell-depleted using hypotonic lysis buffer (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM Na2EDTA, pH 7.3) and resuspended in 150 μl of fluorescence-activated cell sorter (FACS) buffer for flow cytometry analysis.
Flow cytometry analysis.
Bones were flushed, spleens and thymus were crushed, and single-cell suspensions were prepared in FACS buffer. Antibodies against murine Ter119, CD71, B220, IgM, CD43, CD19, CD21, CD23, Mac-1, Gr1, F4/80, CD4, CD8, TCRβ, CD25, CD44, Sca-1, c-Kit, CD34, FLT3, FcγRII/III (CD16/32), CD41, CD105, and CD150, either biotinylated or conjugated with phycoerythrin, phycoerythrin-Cy7, peridinin chlorophyll protein-Cy5.5, allophycocyanin, allophycocyanin eFluor780, eFluor660, or eFluor450 were all obtained from eBioscience, BioLegend, or BD Pharmingen (see Table S2 in the supplemental material) (15, 35, 56, 57). Biotinylated antibodies were detected with streptavidin conjugated with brilliant violet-605. Then, 30,000 to 500,000 cells were acquired on a BD LSRIIFortessa and analyzed with FlowJo software version 9 or 10.0 (Treestar).
Cell proliferation assays.
Hoxb8 immortalized (25) R26-CreERT2 Recq4lfl/+, R26-CreERT2 Recql4fl/G522Efs, and R26-CreERT2 Recql4fl/R347X cells were maintained in IMDM, 10% FBS (non-heat inactivated), 1% penicillin-streptomycin (Pen-Strep), 1% l-glutamine, and 1% GM-CSF-containing medium (BHK-HM5 cell-conditioned medium). The cells were treated for 14 days with 400 nM 4-hydroxy tamoxifen (Merck Millipore) and then genotyped to confirm complete recombination. Cells were counted with Trypan blue using a Countess II automated counter (Thermo Fisher Scientific) and then split every two-3 days.
Retroviral transduction and complementation.
Hoxb8 immortalized R26-CreERT2 Recql4fllfl cells were maintained in IMDM, 10% FBS (non-heat inactivated), 1% Pen-Strep, 1% l-glutamine, and 1% GM-CSF-containing medium (BHK-HM5 cell-conditioned medium). Exponentially growing Hoxb8 R26-CreERT2 Recql4fl/fl cells (100,000 cells/ml) were spin infected with EGFP-RECQL4 retrovirus in a 1:1 ratio at 1,100 × g for 90 min in 8 μg/ml Polybrene. Two days after infection, cells were selected with puromycin (0.25 μg/ml) for 4 days and then expanded. Cells were then treated for 14 days with 400 nM 4-hydroxy tamoxifen (Merck Millipore) and genotyped to confirm complete recombination. Cells were counted with Trypan blue using a Countess II automated counter (Thermo Fisher Scientific) and then split every 2 to 3 days.
Statistical analysis.
To determine statistical significance, Kaplan-Meier survival plots and ordinary one-way ANOVA tests were conducted in Prism software version 8 (GraphPad, San Diego, CA, USA). Throughout this study, significance is indicated as noted in the figure legends; data are presented as the mean ± standard error of the mean (SEM). Furthermore, the number of samples used for each experiment is described in the corresponding figure legends.
Supplementary Material
ACKNOWLEDGMENTS
We thank R. Brink and the Mouse Engineering Garvan/ABR (MEGA) Facility (Garvan Institute, Sydney, Australia) for the generation of the G522Efs allele, the Australian Phenomics Facility and the Australian National University (Canberra, Australia) for their technical expertise and provision of the R347X allele, S. Galic and L. Murray-Segal (St. Vincent’s Institute) for training on Echo-MRI, T. Enomoto (Musashino University, Tokyo, Japan) for providing human EGFP-RECQL4 WT and K508A constructs, M. Kamps (University of California San Diego, USA) for providing the Hoxb8 vectors used to generate cell lines, D. Thorburn (Murdoch Children’s Research Institute and University of Melbourne, Australia), and J. Heierhorst (St. Vincent’s Institute) for comments on the manuscript.
Author contributions: Conceptualization: Wilson Castillo-Tandazo, Monique F. Smeets, Carl R. Walkley. Funding acquisition: Carl R. Walkley. Investigation: Wilson Castillo-Tandazo, Ann E. Frazier, Monique F. Smeets, Carl R. Walkley. Methodology: Wilson Castillo-Tandazo, Ann E. Frazier, Natalie A. Sims, Monique F. Smeets, Carl R. Walkley. Project administration: Monique F. Smeets, Carl R. Walkley. Supervision: Monique F. Smeets, Carl R. Walkley. Visualization: Wilson Castillo-Tandazo, Monique F. Smeets, Carl R. Walkley. Writing – original draft: Wilson Castillo-Tandazo, Monique F. Smeets, Carl R. Walkley. Writing – review and editing: Wilson Castillo-Tandazo, Ann E. Frazier, Natalie A. Sims, Monique F. Smeets, Carl R. Walkley.
This work was supported by the Office of the Assistant Secretary of Defense for Health Affairs through the Peer Reviewed Cancer Research under award no. W81XWH-15-1-0315 (to C.R.W.). Funding also came from the National Health and Medical Research Council (NHMRC) Australia project grant (to C.R.W.; APP1102004); a Melbourne Research Scholarship (to W.C.-T.; University of Melbourne); a Victorian Cancer Agency research fellowship (to CRW, MCRF15015); and the Mito Foundation (to A.E.F.). This work was enabled by the Australian Phenomics Network and partly supported by funding from the Australian government’s National Collaborative Research Infrastructure Strategy and the Super Science Initiative through the Education Investment Fund (to the Australian Phenomics Network) and in part supported by the Victorian State Government Operational Infrastructure Support (to St. Vincent’s Institute and the Murdoch Children’s Research Institute).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense (USA).
We declare no conflicts of interest.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Larizza L, Roversi G, Volpi L. 2010. Rothmund-Thomson syndrome. Orphanet J Rare Dis 5:2. doi: 10.1186/1750-1172-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang LL, Levy ML, Lewis RA, Chintagumpala MM, Lev D, Rogers M, Plon SE. 2001. Clinical manifestations in a cohort of 41 Rothmund-Thomson syndrome patients. Am J Med Genet 102:11–17. doi:. [DOI] [PubMed] [Google Scholar]
- 3.Mehollin-Ray AR, Kozinetz CA, Schlesinger AE, Guillerman RP, Wang LL. 2008. Radiographic abnormalities in Rothmund-Thomson syndrome and genotype-phenotype correlation with RECQL4 mutation status. AJR Am J Roentgenol 191:W62–W66. doi: 10.2214/AJR.07.3619. [DOI] [PubMed] [Google Scholar]
- 4.Siitonen HA, Sotkasiira J, Biervliet M, Benmansour A, Capri Y, Cormier-Daire V, Crandall B, Hannula-Jouppi K, Hennekam R, Herzog D, Keymolen K, Lipsanen-Nyman M, Miny P, Plon SE, Riedl S, Sarkar A, Vargas FR, Verloes A, Wang LL, Kaariainen H, Kestila M. 2009. The mutation spectrum in RECQL4 diseases. Eur J Hum Genet 17:151–158. doi: 10.1038/ejhg.2008.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang LL, Gannavarapu A, Kozinetz CA, Levy ML, Lewis RA, Chintagumpala MM, Ruiz-Maldanado R, Contreras-Ruiz J, Cunniff C, Erickson RP, Lev D, Rogers M, Zackai EH, Plon SE. 2003. Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothmund-Thomson syndrome. J Natl Cancer Inst 95:669–674. doi: 10.1093/jnci/95.9.669. [DOI] [PubMed] [Google Scholar]
- 6.Ajeawung NF, Nguyen TTM, Lu L, Kucharski TJ, Rousseau J, Molidperee S, Atienza J, Gamache I, Jin W, Plon SE, Lee BH, Teodoro JG, Wang LL, Campeau PM. 2019. Mutations in ANAPC1, encoding a scaffold subunit of the anaphase-promoting complex, cause Rothmund-Thomson syndrome type 1. Am J Hum Genet 105:625–630. doi: 10.1016/j.ajhg.2019.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kitao S, Shimamoto A, Goto M, Miller RW, Smithson WA, Lindor NM, Furuichi Y. 1999. Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nat Genet 22:82–84. doi: 10.1038/8788. [DOI] [PubMed] [Google Scholar]
- 8.Wang LL, Worley K, Gannavarapu A, Chintagumpala MM, Levy ML, Plon SE. 2002. Intron-size constraint as a mutational mechanism in Rothmund-Thomson syndrome. Am J Hum Genet 71:165–167. doi: 10.1086/341234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohlenschlager O, Kuhnert A, Schneider A, Haumann S, Bellstedt P, Keller H, Saluz HP, Hortschansky P, Hanel F, Grosse F, Gorlach M, Pospiech H. 2012. The N-terminus of the human RecQL4 helicase is a homeodomain-like DNA interaction motif. Nucleic Acids Res 40:8309–8324. doi: 10.1093/nar/gks591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaggioli V, Zeiser E, Rivers D, Bradshaw CR, Ahringer J, Zegerman P. 2014. CDK phosphorylation of SLD-2 is required for replication initiation and germline development in C. elegans. J Cell Biol 204:507–522. doi: 10.1083/jcb.201310083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keller H, Kiosze K, Sachsenweger J, Haumann S, Ohlenschlager O, Nuutinen T, Syvaoja JE, Gorlach M, Grosse F, Pospiech H. 2014. The intrinsically disordered amino-terminal region of human RecQL4: multiple DNA-binding domains confer annealing, strand exchange and G4 DNA binding. Nucleic Acids Res 42:12614–12627. doi: 10.1093/nar/gku993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shamanna RA, Singh DK, Lu H, Mirey G, Keijzers G, Salles B, Croteau DL, Bohr VA. 2014. RECQ helicase RECQL4 participates in non-homologous end joining and interacts with the Ku complex. Carcinogenesis 35:2415–2424. doi: 10.1093/carcin/bgu137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu X, Rochette PJ, Feyissa EA, Su TV, Liu Y. 2009. MCM10 mediates RECQ4 association with MCM2-7 helicase complex during DNA replication. EMBO J 28:3005–3014. doi: 10.1038/emboj.2009.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fairman-Williams ME, Guenther UP, Jankowsky E. 2010. SF1 and SF2 helicases: family matters. Curr Opin Struct Biol 20:313–324. doi: 10.1016/j.sbi.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castillo-Tandazo W, Smeets MF, Murphy V, Liu R, Hodson C, Heierhorst J, Deans AJ, Walkley CR. 2019. ATP-dependent helicase activity is dispensable for the physiological functions of Recql4. PLoS Genet 15:e1008266. doi: 10.1371/journal.pgen.1008266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaiser S, Sauer F, Kisker C. 2017. The structural and functional characterization of human RecQ4 reveals insights into its helicase mechanism. Nat Commun 8:15907. doi: 10.1038/ncomms15907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petkovic M, Dietschy T, Freire R, Jiao R, Stagljar I. 2005. The human Rothmund-Thomson syndrome gene product, RECQL4, localizes to distinct nuclear foci that coincide with proteins involved in the maintenance of genome stability. J Cell Sci 118:4261–4269. doi: 10.1242/jcs.02556. [DOI] [PubMed] [Google Scholar]
- 18.Woo LL, Futami K, Shimamoto A, Furuichi Y, Frank KM. 2006. The Rothmund-Thomson gene product RECQL4 localizes to the nucleolus in response to oxidative stress. Exp Cell Res 312:3443–3457. doi: 10.1016/j.yexcr.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 19.Hicks MJ, Roth JR, Kozinetz CA, Wang LL. 2007. Clinicopathologic features of osteosarcoma in patients with Rothmund-Thomson syndrome. J Clin Oncol 25:370–375. doi: 10.1200/JCO.2006.08.4558. [DOI] [PubMed] [Google Scholar]
- 20.Ferrari S, Smeland S, Mercuri M, Bertoni F, Longhi A, Ruggieri P, Alvegard TA, Picci P, Capanna R, Bernini G, Müller C, Tienghi A, Wiebe T, Comandone A, Böhling T, Del Prever AB, Brosjö O, Bacci G, Sæter G, Italian, Scandinavian Sarcoma Groups. 2005. Neoadjuvant chemotherapy with high-dose Ifosfamide, high-dose methotrexate, cisplatin, and doxorubicin for patients with localized osteosarcoma of the extremity: a joint study by the Italian and Scandinavian Sarcoma Groups. J Clin Oncol 23:8845–8852. doi: 10.1200/JCO.2004.00.5785. [DOI] [PubMed] [Google Scholar]
- 21.Lu L, Harutyunyan K, Jin W, Wu J, Yang T, Chen Y, Joeng KS, Bae Y, Tao J, Dawson BC, Jiang MM, Lee B, Wang LL. 2015. RECQL4 regulates p53 function in vivo during skeletogenesis. J Bone Miner Res 30:1077–1089. doi: 10.1002/jbmr.2436. [DOI] [PubMed] [Google Scholar]
- 22.Ng AJ, Walia MK, Smeets MF, Mutsaers AJ, Sims NA, Purton LE, Walsh NC, Martin TJ, Walkley CR. 2015. The DNA helicase Recql4 is required for normal osteoblast expansion and osteosarcoma formation. PLoS Genet 11:e1005160. doi: 10.1371/journal.pgen.1005160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chi Z, Nie L, Peng Z, Yang Q, Yang K, Tao J, Mi Y, Fang X, Balajee AS, Zhao Y. 2012. RecQL4 cytoplasmic localization: implications in mitochondrial DNA oxidative damage repair. Int J Biochem Cell Biol 44:1942–1951. doi: 10.1016/j.biocel.2012.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Croteau DL, Rossi ML, Canugovi C, Tian J, Sykora P, Ramamoorthy M, Wang ZM, Singh DK, Akbari M, Kasiviswanathan R, Copeland WC, Bohr VA. 2012. RECQL4 localizes to mitochondria and preserves mitochondrial DNA integrity. Aging Cell 11:456–466. doi: 10.1111/j.1474-9726.2012.00803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang GG, Calvo KR, Pasillas MP, Sykes DB, Häcker H, Kamps MP. 2006. Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8. Nat Methods 3:287–293. doi: 10.1038/nmeth865. [DOI] [PubMed] [Google Scholar]
- 26.Broom MA, Wang LL, Otta SK, Knutsen AP, Siegfried E, Batanian JR, Kelly ME, Shah M. 2006. Successful umbilical cord blood stem cell transplantation in a patient with Rothmund-Thomson syndrome and combined immunodeficiency. Clin Genet 69:337–343. doi: 10.1111/j.1399-0004.2006.00592.x. [DOI] [PubMed] [Google Scholar]
- 27.De Somer L, Wouters C, Morren MA, De Vos R, Van Den Oord J, Devriendt K, Meyts I. 2010. Granulomatous skin lesions complicating Varicella infection in a patient with Rothmund-Thomson syndrome and immune deficiency: case report. Orphanet J Rare Dis 5:37. doi: 10.1186/1750-1172-5-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rudilla F, Franco-Jarava C, Martinez-Gallo M, Garcia-Prat M, Martin-Nalda A, Riviere J, Aguilo-Cucurull A, Mongay L, Vidal F, Solanich X, Irastorza I, Santos-Perez JL, Tercedor Sanchez J, Cusco I, Serra C, Baz-Redon N, Fernandez-Cancio M, Carreras C, Vagace JM, Garcia-Patos V, Pujol-Borrell R, Soler-Palacin P, Colobran R. 2019. Expanding the clinical and genetic spectra of primary immunodeficiency-related disorders with clinical exome sequencing: expected and unexpected findings. Front Immunol 10:2325. doi: 10.3389/fimmu.2019.02325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ho MS, Medcalf RL, Livesey SA, Traianedes K. 2015. The dynamics of adult haematopoiesis in the bone and bone marrow environment. Br J Haematol 170:472–486. doi: 10.1111/bjh.13445. [DOI] [PubMed] [Google Scholar]
- 30.Rodda SJ, McMahon AP. 2006. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development 133:3231–3244. doi: 10.1242/dev.02480. [DOI] [PubMed] [Google Scholar]
- 31.Davey RA, Clarke MV, Sastra S, Skinner JP, Chiang C, Anderson PH, Zajac JD. 2012. Decreased body weight in young Osterix-Cre transgenic mice results in delayed cortical bone expansion and accrual. Transgenic Res 21:885–893. doi: 10.1007/s11248-011-9581-z. [DOI] [PubMed] [Google Scholar]
- 32.Huang W, Olsen BR. 2015. Skeletal defects in Osterix-Cre transgenic mice. Transgenic Res 24:167–172. doi: 10.1007/s11248-014-9828-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin W, Liu H, Zhang Y, Otta SK, Plon SE, Wang LL. 2008. Sensitivity of RECQL4-deficient fibroblasts from Rothmund-Thomson syndrome patients to genotoxic agents. Hum Genet 123:643–653. doi: 10.1007/s00439-008-0518-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kohzaki M, Ootsuyama A, Sun L, Moritake T, Okazaki R. 2020. Human RECQL4 represses the RAD52-mediated single-strand annealing pathway after ionizing radiation or cisplatin treatment. Int J Cancer 146:3098–3113. doi: 10.1002/ijc.32670. [DOI] [PubMed] [Google Scholar]
- 35.Smeets MF, DeLuca E, Wall M, Quach JM, Chalk AM, Deans AJ, Heierhorst J, Purton LE, Izon DJ, Walkley CR. 2014. The Rothmund-Thomson syndrome helicase RECQL4 is essential for hematopoiesis. J Clin Invest 124:3551–3565. doi: 10.1172/JCI75334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kitao S, Ohsugi I, Ichikawa K, Goto M, Furuichi Y, Shimamoto A. 1998. Cloning of two new human helicase genes of the RecQ family: biological significance of multiple species in higher eukaryotes. Genomics 54:443–452. doi: 10.1006/geno.1998.5595. [DOI] [PubMed] [Google Scholar]
- 37.Burks LM, Yin J, Plon SE. 2007. Nuclear import and retention domains in the amino terminus of RECQL4. Gene 391:26–38. doi: 10.1016/j.gene.2006.11.019. [DOI] [PubMed] [Google Scholar]
- 38.Martin RM, Ter-Avetisyan G, Herce HD, Ludwig AK, Lattig-Tunnemann G, Cardoso MC. 2015. Principles of protein targeting to the nucleolus. Nucleus 6:314–325. doi: 10.1080/19491034.2015.1079680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barisonek KL, Protzman NM, Wobst GM, Brigido SA. 2016. Delayed union of a Jones fracture in a patient with Rothmund-Thomson syndrome: a case report and review of the literature. J Foot Ankle Surg 55:291–293. doi: 10.1053/j.jfas.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 40.Beckmann N. 2015. Multiple low energy long bone fractures in the setting of Rothmund-Thomson syndrome. Case Rep Med 2015:495164. doi: 10.1155/2015/495164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cao F, Lu L, Abrams SA, Hawthorne KM, Tam A, Jin W, Dawson B, Shypailo R, Liu H, Lee B, Nagamani SCS, Wang LL. 2017. Generalized metabolic bone disease and fracture risk in Rothmund-Thomson syndrome. Hum Mol Genet 26:3046–3055. doi: 10.1093/hmg/ddx178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carlson AM, Thomas KB, Kirmani S, Lindor NM. 2012. Chronic tibial nonunion in a Rothmund-Thomson syndrome patient. Am J Med Genet A 158A:2250–2253. doi: 10.1002/ajmg.a.35475. [DOI] [PubMed] [Google Scholar]
- 43.Simon T, Kohlhase J, Wilhelm C, Kochanek M, De Carolis B, Berthold F. 2010. Multiple malignant diseases in a patient with Rothmund-Thomson syndrome with RECQL4 mutations: case report and literature review. Am J Med Genet A 152A:1575–1579. doi: 10.1002/ajmg.a.33427. [DOI] [PubMed] [Google Scholar]
- 44.Stinco G, Governatori G, Mattighello P, Patrone P. 2008. Multiple cutaneous neoplasms in a patient with Rothmund-Thomson syndrome: case report and published work review. J Dermatol 35:154–161. doi: 10.1111/j.1346-8138.2008.00436.x. [DOI] [PubMed] [Google Scholar]
- 45.Maciaszek JL, Oak N, Chen W, Hamilton KV, McGee RB, Nuccio R, Mostafavi R, Hines-Dowell S, Harrison L, Taylor L, Gerhardt EL, Ouma A, Edmonson MN, Patel A, Nakitandwe J, Pappo AS, Azzato EM, Shurtleff SA, Ellison DW, Downing JR, Hudson MM, Robison LL, Santana V, Newman S, Zhang J, Wang Z, Wu G, Nichols KE, Kesserwan CA. 2019. Enrichment of heterozygous germline RECQL4 loss-of-function variants in pediatric osteosarcoma. Cold Spring Harb Mol Case Stud 5:a004218. doi: 10.1101/mcs.a004218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huvos AG, Woodard HQ. 1988. Postradiation sarcomas of bone. Health Phys 55:631–636. doi: 10.1097/00004032-198810000-00004. [DOI] [PubMed] [Google Scholar]
- 47.Virtanen A, Pukkala E, Auvinen A. 2006. Incidence of bone and soft tissue sarcoma after radiotherapy: a cohort study of 295,712 Finnish cancer patients. Int J Cancer 118:1017–1021. doi: 10.1002/ijc.21456. [DOI] [PubMed] [Google Scholar]
- 48.Kohzaki M, Chiourea M, Versini G, Adachi N, Takeda S, Gagos S, Halazonetis TD. 2012. The helicase domain and C-terminus of human RecQL4 facilitate replication elongation on DNA templates damaged by ionizing radiation. Carcinogenesis 33:1203–1210. doi: 10.1093/carcin/bgs149. [DOI] [PubMed] [Google Scholar]
- 49.Maire G, Yoshimoto M, Chilton-MacNeill S, Thorner PS, Zielenska M, Squire JA. 2009. Recurrent RECQL4 imbalance and increased gene expression levels are associated with structural chromosomal instability in sporadic osteosarcoma. Neoplasia 11:260–268. doi: 10.1593/neo.81384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saglam O, Shah V, Worsham MJ. 2007. Molecular differentiation of early and late stage laryngeal squamous cell carcinoma: an exploratory analysis. Diagn Mol Pathol 16:218–221. doi: 10.1097/PDM.0b013e3180d0aab5. [DOI] [PubMed] [Google Scholar]
- 51.Thomassen M, Tan Q, Kruse TA. 2009. Gene expression meta-analysis identifies chromosomal regions and candidate genes involved in breast cancer metastasis. Breast Cancer Res Treat 113:239–249. doi: 10.1007/s10549-008-9927-2. [DOI] [PubMed] [Google Scholar]
- 52.Buffart TE, Coffa J, Hermsen MA, Carvalho B, van der Sijp JR, Ylstra B, Pals G, Schouten JP, Meijer GA. 2005. DNA copy number changes at 8q11-24 in metastasized colorectal cancer. Cell Oncol 27:57–65. doi: 10.1155/2005/401607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Narayan G, Bourdon V, Chaganti S, Arias-Pulido H, Nandula SV, Rao PH, Gissmann L, Durst M, Schneider A, Pothuri B, Mansukhani M, Basso K, Chaganti RS, Murty VV. 2007. Gene dosage alterations revealed by cDNA microarray analysis in cervical cancer: identification of candidate amplified and overexpressed genes. Genes Chromosomes Cancer 46:373–384. doi: 10.1002/gcc.20418. [DOI] [PubMed] [Google Scholar]
- 54.Chen H, Yuan K, Wang X, Wang H, Wu Q, Wu X, Peng J. 2018. Overexpression of RECQL4 is associated with poor prognosis in patients with gastric cancer. Oncol Lett 16:5419–5425. doi: 10.3892/ol.2018.9318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abe T, Yoshimura A, Hosono Y, Tada S, Seki M, Enomoto T. 2011. The N-terminal region of RECQL4 lacking the helicase domain is both essential and sufficient for the viability of vertebrate cells. Role of the N-terminal region of RECQL4 in cells. Biochim Biophys Acta 1813:473–479. doi: 10.1016/j.bbamcr.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 56.Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR. 2015. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349:1115–1120. doi: 10.1126/science.aac7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Singbrant S, Russell MR, Jovic T, Liddicoat B, Izon DJ, Purton LE, Sims NA, Martin TJ, Sankaran VG, Walkley CR. 2011. Erythropoietin couples erythropoiesis, B-lymphopoiesis, and bone homeostasis within the bone marrow microenvironment. Blood 117:5631–5642. doi: 10.1182/blood-2010-11s-320564. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.